
Abstract
Background
Wild relatives of wheat play a significant role in wheat improvement as a source of genetic diversity. Stem rust disease of wheat causes significant yield losses at the global level and stem rust pathogen race TTKSK (Ug99) is virulent to most previously deployed resistance genes. Therefore, the objective of this study was to identify loci conferring resistance to stem rust pathogen races including Ug99 in an Aegilops umbelluata bi-parental mapping population using genotype-by-sequencing (GBS) SNP markers.
Results
A bi-parental F2:3 population derived from a cross made between stem rust resistant accession PI 298905 and stem rust susceptible accession PI 542369 was used for this study. F2 individuals were evaluated with stem rust race TTTTF followed by testing F2:3 families with races TTTTF and TTKSK. The segregation pattern of resistance to both stem rust races suggested the presence of one resistance gene. A genetic linkage map, comprised 1,933 SNP markers, was created for all seven chromosomes of Ae. umbellulata using GBS. A major stem rust resistance QTL that explained 80% and 52% of the phenotypic variations for TTTTF and TTKSK, respectively, was detected on chromosome 2U of Ae. umbellulata.
Conclusion
The novel resistance gene for stem rust identified in this study can be transferred to commercial wheat varieties assisted by the tightly linked markers identified here. These markers identified through our mapping approach can be a useful strategy to identify and track the resistance gene in marker-assisted breeding in wheat.
Similar content being viewed by others

Background
Wild relatives of wheat (Triticum aestivum L. and T. turgidum ssp. durum (Desf.)) have been used in breeding programs as sources of agronomically valuable traits. Several genes derived from wild relatives have been deployed in cultivated wheat varieties over the last century and played a significant role in wheat improvement worldwide [1]. Species of the genus Aegilops have been successfully used in wheat wide-crossing programs [2, 3]. Though there are many challenges to introgress alien chromatin into wheat, some of the Aegilops species have a genome homologous to one of the three T. aestivum subgenomes (A, B, and D) and transfer of a favorable trait by conventional crossing is possible [4]. However, introgression barriers for many other species requires the application of techniques such as chemical treatments, irradiation, cold treatment, absence of particular genes such as Ph1 or gametocidal genes, and bridging crosses [2]. These techniques have facilitated the creation of a range of addition, substitution, and translocation lines through chromatin introgression between wheat and Aegilops species such as Ae. comosa Sibth & Sm., Ae. umbellulata Zhuk., Ae. geniculata Roth and Ae. biuncialis Vis. [5]. Consequently, numerous studies have demonstrated that Aegilops species carry useful genes for traits such as disease and insect resistance [6], drought tolerance [7] and salt tolerance [8]. Resistance genes to leaf rust, stripe rust, stem rust and powdery mildew have been successfully transferred from Aegilops species to wheat [9–15].
Aegilops umbellulata, a Mediterranean-western Asiatic grass, is one of the 11 diploid species in the Aegilops genera [16] that possesses seven pairs of chromosomes (2n = 2x = 14, UU genome). From the standard chromosome karyotype that has been completed for Ae. umbellulata [17], six of the seven chromosomes had about the same chromosome size. Relatively, chromosome 1U is the shortest chromosome.
Aegilops umbellulata is the source of leaf rust resistance gene Lr9 that was transferred to hexaploid wheat [18]. This species has also been identified as a source of resistance to stem rust [19, 20], powdery mildew, Hessian fly and greenbug [21]. In addition, it is a source of high-molecular-weight (HMW) glutenin subunits [22, 23].
The search for new sources of resistance to stem rust, caused by Puccinia graminis f. sp. tritici (Pgt), from wild relatives of wheat has been intensified [24] due to the emergence of stem rust pathogen race TTKSK (the first isolate of this race was named Ug99). The cultivated wheat gene pool has a narrow genetic base for resistance to Ug99 and up to 90% of world’s wheat cultivars are considered Ug99 susceptible [25]. Since 2011, a total of five Ug99 resistance genes have been introgressed from Ae. tauschii Coss., Ae. searsii Feldman & Kislev ex K. Hammer, and Ae. geniculata into wheat; these are Sr51 [26], Sr53 [27], SrTA1662 [28], SrTA10171 [29] and SrTA10187 [29].
The development of new sequencing technologies has facilitated the discovery of a large number of SNP markers for many crop species such as hexaploid wheat [30], barley [31], rice [32] and maize [33]. Genotyping-By-Sequencing (GBS), a reduced representation genotyping platform, has been used effectively to create high density genetic maps for several cultivated crops such as hexaploid wheat [34, 35], barley [36], oats L. [36] and maize [37]. However, as far as we know, there is no SNP-based genetic map that can be used to map novel traits in Ae. umbellulata. Mapping of disease resistance genes in wild diploid progenitor species of wheat has been a successful strategy to aid the cloning of stem rust resistance genes Sr35 and Sr22 from T. monococcum L [38, 39]. Mapping in a diploid species allows for recombination to readily occur whereas recombination may be inhibited once a genomic region has been introgressed. Only a few U genome-specific markers are available [40, 41]. Therefore, in the present study, we report a new stem rust resistance QTL identified by scanning the entire genome of Ae. umbellulata with GBS SNP markers.
Methods
Biological materials and genotyping
A total of 140 F2 individuals were derived from a cross made between two accessions of Ae. umbellulata: PI 298905, resistant to Pgt race TTTTF (isolate 01MN84A-1-2) and TTKSK (isolate 04KEN156/04), and PI 542369, susceptible to both races. The two Ae. umbellulata accessions were obtained from the publically available National Small Grains Collection (NSGC), USDA-ARS (https://www.ars.usda.gov/pacific-west-area/aberdeen-id/small-grains-and-potato-germplasm-research/docs/national-small-grains-collection/). Leaf tissue was collected from each F2 individual and the two parents at the seedling stage, and DNA was extracted following a BioSprint protocol [42]. F3 seeds were harvested from each mature F2 plant and used for stem rust assays. A single GBS library was constructed for a pool of 142 samples following a GBS protocol with the two restriction enzymes PstI (CTGCAG) and MspI (CCGG) [26]. Two barcoded adaptors were used for each sample. The two parents were sequenced to a depth 6X the F2 individuals. The library was sequenced on the Illumina HiSeq 2000 platform.
SNP calling and linkage map construction
Raw sequence data were processed for SNPs discovery with the UNEAK algorithm [43] implemented in TASSEL 3.0 [44]. The UNEAK parameters were set as follows: maximum number of expected reads per sequence file 300,000,0000, restriction enzymes used for library construction PstI-MspI, minimum number of tags required for output five, maximum tag number in the merged tag counts 200,000,000, option to merge multiple sample per line yes, error tolerance rate 0.03, minimum/maximum minor allele frequencies (MAF) 0.01 and 0.5, and minimum/maximum call rates 0 and 1. The default parameter set was used except for the minimum number of tag counts. SNPs with up to 80% missing data points were retained for subsequent data analysis. However, only SNPs with no missing data, no heterozygous allele states and polymorphic for the two parents were converted into parental genotypes.
SNP data was first converted into parental genotypes for polymorphic SNPs with no missing or heterozygous genotypes for both parents. SNPs with minor allele frequency (MAF) less than 20%, percent heterozygosity greater than 80%, and proportion of missing data points greater than 50% were also removed. Finally, after removing markers with segregation distortion (p < 0.01), SNPs with missing data points of less than 10% were used for framework linkage map construction.
Linkage map construction was done in MSTMap software [45] with the following parameters: Distance function Kosambi, cut_off_p_value 10−10, no_map_dist 15, no_map_size 0, missing_threshold 0.10, estimate_before_clustering no, detect_bad_data yes and objective function ML. Map construction was done four times to stabilize the inflated linkage map size. After each map, double recombinants were changed into missing data guided by graphical genotypes. Markers with missing data points greater than 10% were removed at each step of linkage map reconstruction. Linkage groups from the same chromosome were merged together and reordered.
Stem rust assay and resistance mapping
The F2 individuals and both parents were evaluated against race TTTTF to determine the inheritance of stem rust resistance. F2:3 families derived from the same F2 plants from which DNA were extracted were assessed for reaction to both races TTTTF (01MN84A-1–2) and TTKSK (04KEN156/04) in the Spring of 2016, under controlled greenhouse conditions, in order to predict the resistance genotype of the corresponding F2 plants. Ten to twenty seedlings per family for 124 families and the two parents were assessed for reaction to race TTTTF. A total of 63 families were assessed for reaction to race TTKSK. Experimental procedures for inoculation, incubation, and disease assessment were conducted according to previously described methods [46]. Stem rust seedling infection types were scored based on the 0–4 scale [47] and plants with infection types 0–2 were considered resistant whereas plants with infection types 3–4 were considered susceptible. Each F2 plant was classified as homozygous resistant, heterozygous, or homozygous susceptible to each Pgt race depending on the segregation of stem rust response of the corresponding F2:3 seedlings. Phenotypic segregation pattern of the disease scores recorded on F2:3 families were tested for fit to expected segregation ratios using chi-square (x 2) tests. For the purpose of QTL mapping, F2 plants classified as homozygous susceptible, homozygous resistant, or heterozygous were coded as 0, 1, 2, respectively. Quantitative trait loci (QTL) analysis was conducted in R package RQTL with Single QTL mapping (SIM), composite interval mapping (CIM) and multiple QTL mapping (MQM) methods. In addition, we mapped the resistance locus using the assessment of stem rust reaction of F2 plants as a molecular marker. The disease score data was included into the genotypic data used to create frame work linkage map using the same linkage map parameters (see linkage map construction section).
Linkage group chromosome assignment
Chromosome assignment of the linkage groups of the framework map was accomplished using draft genome sequences of hexaploid wheat and barley. Chinese Spring Wheat draft genome sequence version 2 [48] and barley whole genome sequence (WGS) [49] were used to anchor the GBS SNP sequences. All 20,252 SNP tags were aligned to the draft sequences using BWA “aln” algorithm with default settings. Then all linkage maps were merged to the output of sequence alignments. Circos tool [50] was used to visualize the syntenic relationship of Ae. umbellulata with hexaploid wheat and barley.
Results
SNP calling and linkage map construction
From processing raw sequences using de novo SNP calling approach in TASSEL 3, a total of 20,252 SNPs were obtained with the default setting except that the minimum tag count was set to five. However, after removing monomorphic SNPs, and also SNPs outside of thresholds set for missing or heterozygous genotypes for any of the two parents, a total of 10,657 SNPs were converted into parental genotypes (A, B and H). After another round of filtering with MAF greater or equal to 20%, level of heterozygosity between 20% and 80%, missing data points up to 10% and segregation distortion P < 0.05, a total of 1,933 high-quality SNP markers were selected. These relatively higher quality SNPs were used to construct a framework linkage map using MSTmap software.
Initially, eight linkage groups were obtained from the 1,933 SNPs using MSTMap (Table 1). However, after assigning all linkage groups into chromosomes, two linkage groups were assigned to chromosome 1U, and the remaining six chromosomes had a single linkage group each. The two linkage groups for 1U were merged and the SNPs were reordered. The total linkage map size was 932.47 cM with the average gap size of 0.65 cM (Table 1). We observed chromosome 1U as the shortest linkage group with the largest gap size of 39.96 cM. It also had the least number of markers (47). With the exception of chromosome 3U (max gap 18.75 cM), maximum gap size was less than 7 cM for all of the remaining chromosomes. Chromosome 4U had the largest number of markers (442), and it was also the longest chromosome at 184 cM, followed by chromosome 6U at 172 cM. The chromosome position (cM) and parental genotypes of each SNP in the genetic map is presented in Additional file 1: Table S1.
Stem rust resistance mapping
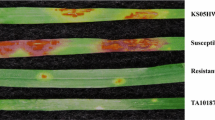
Accession PI 298905 was resistant to both Pgt races TTTTF and TTKSK with seedling infection type ‘2-’ whereas accession PI 542369 was susceptible to both races with seedling infection type ‘3+’ (Fig. 1). The segregation of disease reaction to races TTTTF and TTKSK significantly deviated from a 1:2:1 ratio for F2:3 families (Table 2). However, the segregation pattern across F2:3 families was in agreement with a 3:1 ratio for a single dominant gene model (TTTTF, X 2 = 0.46 p = 0.50; TTKSK, X 2 = 0.42 p = 0.52). Taken together, these results suggest that the resistance to each race is conferred by a single gene with dominant effect. In order to test if the gene conferring the resistance to each race is the same, the infection type to races TTTTF and TTKSK were compared. A total of seven F2:3 families were homozygous resistant to both races, 32 families were heterozygous to both races, seven families were homozygous susceptible to both races, and one family was susceptible to race TTKSK, but heterozygous to race TTTTF. This co-segregation pattern deviated from the expected ratio for independent resistant loci (X 2 = 70.3 p = 3.90 X 10−9). These results suggest that the resistance to races TTKSK and TTTTF resistances is conferred by the same gene or by tightly linked genes at the same locus.

Seedling infection types of PI 298905 and PI 542369 in response to Puccinia graminis f. sp. tritici race TTKSK. The two leaves on the left are PI 298905 (infection type 2-) whereas the two leaves on the right are PI 542369 (infection type 3+)
The disease resistance classification data for races TTTTF and TTKSK of the F2:3 families were used for QTL scanning across all seven chromosomes. On the basis of whole-genome scanning using CIM and MQM qtl mapping methods, a major resistance QTL for both races TTTTF and TTKSK was identified on chromosome 2U linked to markers Aeup1GBS11453 and Aeup1GBS13266 (Fig. 2, Table 3). This QTL explained close to 80% of the phenotypic variation for response to race TTTTF and more than 52% of the phenotypic variation for response to race TTKSK (Table 3). Using single qtl mapping methods (SIM) such as Haley-Knot regression (hk), extended Haley-Knot regression (ehk), expectation maximization (em) and non-parametric (np) methods (Additional file 2: Table S2), the QTL associated markers were located within 2–3 cM distance from the major QTL detected on chromosome 2U with multiple QTL methods. A minor QTL was also detected on chromosome 7U for response to race TTKSK (Additional file 2: Table S2, Fig. 2). In addition, the creation of a linkage map with the disease score as a marker placed the phenotype in the QTL region of chromosome 2U between marker Aeup1GBS16910 (89.4 cM) and Aeup1GBS2109/9405 (90.5 cM) (Fig. 3). From sequence similarity search in hexaploid genome sequence databases, best hits were found for marker Aeup1GBS16910 on chromosome 2AL and 2DL; for marker Aeup1GBS2109 on chromosome 2BL and 2DL; for marker Aeup1GBS9405 on chromosome 2DL indicating the QTL detected in this study is on the distal end of the centromere 2U.

Chromosome and logarithm of odds (LOD) profile of stem resistance QTL detected for races TTTTF and TTKSK in the Ae. umbellulata F2 population

Portion of chromosome 2U showing map location of disease score when mapped as a marker
Anchoring SNP tags into hexaploid wheat and barley genomes
Uniquely aligned SNP tags using BWA on genome assemblies of hexaploid wheat and barley were used to assign linkage groups to chromosomes. The most hits were found for hexaploid wheat followed by barley. The chromosome similarity of Ae. umbellulata with that of hexaploid wheat and barley varied depending on the chromosome. Good collinearity was observed between chromosomes 1U, 2U, 3U and 5U, and group 1, 2, 3 and 5 of hexaploid wheat, respectively (Fig. 4). On the contrary, the segments of the remaining three chromosomes of Ae. umbelullata (4U, 6U and 7U) were duplicated across different groups of wheat chromosomes. Segments of chromosome 4U were mainly found on group 1, 6 and 7 of hexaploid wheat whereas chromosome 6U segments were duplicated on chromosomes of group 4, 5, and 6 of hexaploid wheat. Segments of chromosome 7U were found on groups 3, 4 and 7 of hexaploid wheat. For all Ae. umbellulata chromosomes, the SNP tags were almost equally distributed across homoleogous chromosomes. A similar syntenic relationship with barley was observed (Fig. 5). With the exception of chromosomes 4U, 6U and 7U, the majority of SNP tags of Ae. umbellulata chromosomes were assigned to the corresponding chromosomes of barley. Although the stem rust resistance linked markers on 2U (94–95 cM) did not pass the threshold level used for blast search, other markers in the QTL region such as Aeu1GBS2874 (92.60 cM), Aeu1GBS6092 (96.24 cM) and Aeu1GBS17509 (99.72 cM) showed a match with scaffolds on the long arms of group2 chromosomes of wheat. Furthermore, majority of the markers within 20 cM on the proximal end of the QTL peak were consistently assigned to the long arms of group 2 chromosomes of wheat.

Synthenic relationship between Ae. umbellulata and hexaploid wheat using SNP tags of mapped GBS markers

Synthenic relationship between Ae. umbellulata and barley (H. vulgare) using SNP tags of mapped GBS markers
Discussion
The discovery of novel genes from alien sources and transfer to the domesticated gene pool is an efficient, cost-effective and environment-friendly strategy to combat rust epidemics including stem rust. In this line, many wild relatives of wheat have been used as a source of wheat rust resistance genes in wheat breeding programs [51]. Molecular markers have been found promising for introgression of favorable QTL/genes that confer disease resistance [52]. In the current study, we mapped a novel major stem rust resistance QTL from Ae. umbellulata, a diploid wild relative of cultivated wheat, using a bi-parental population genotyped with GBS technology. As there was no previously constructed genetic linkage map for Ae. umbellulata, a framework linkage map was created with a total of 1,933 SNPs. The total linkage map size across seven chromosomes of Ae. umbellulata was 932.47 cM with the average gap size of 0.65 cM. Although chromosome 1U was the shortest chromosome on the previously reported Ae. umbellulata chromosome karyotype [17], the short linkage group size obtained here for chromosome 1U was mainly due to the absence of markers residing in the centromeric region. This resulted in a gap of about 40 cM between the two arms of the chromosome. The poor marker coverage for chromosome 1U could be due to removal of a large number of markers due to segregation distortion, low minor allele frequency and high missing data. Overall, markers were fairly evenly distributed for the remaining six chromosomes. However, complete marker coverage for all chromosomes could be achieved by constructing a consensus map from two or more bi-parental populations.
The availability of the draft genome sequences of hexaploid wheat [47, 53] and barley [48, 54] greatly facilitated the construction of the linkage map in this work. Chromosome assignments and identification of inverted linkage groups were accomplished without anchor markers through the integration of 64 bp of GBS SNP tags into draft sequences of these species. As expected, more SNP tags were anchored to hexaploid wheat than barley. However, assignment of the linkage groups into their respective chromosomes still had high integrity with barley as there was a one-to-one relationship between Ae. umbellulata and barley chromosomes. The pattern of syntenic relationship observed between hexploid wheat and Ae. umbellulata in the current study is broadly similar with previously reported results based on wheat markers [40, 55, 56] except for chromosome 7U that shared segments with group 3 chromosomes of wheat instead of group 6. From the syntenic relationship between Ae. umbellulata and barley, chromosome 7U had common markers with 7H and 3H but none with 6H (Fig. 5). The relationship between 4U and group 1 of wheat is not known in previous observations. The previously reported syntenic relationships between wheat and Ae. umbellulata were established based on few markers per chromosome and only show macro-level synteny between the two species. However, generating large number of markers per chromosome as done in the current work may allow to access regions of chromosomes that not represented when sparse markers per chromosomes are used.
Seedling infection type data recorded after inoculating the F2:3 families allowed accurate resistance classification of F2 plants. With genome-wide QTL scanning methods, a major QTL region conferring resistance to stem rust pathogen races TTTTF and TTKSK was identified on chromosome 2U. When mapped as a qualitative trait, the linkage map position of the resistance gene also agreed with the detected QTL region. Group 2 chromosomes of hexaploid wheat also harbor 19 stem resistance QTL [57] including major genes such as Sr32 (chrs 2A, 2B and 2D), Sr21 (chr 2A), Sr39, Sr36, Sr47, Sr28 and Sr9 (chr 2B) and Sr46 (chr 2D). However, the newly mapped QTL in the current investigation and the Ae. umbellulata-derived leaf rust resistance gene (Lr9) are located on different chromosomes as the latter was derived from chromosome 6U and the introgressed segment was also mapped on chromosome arm 6BL of hexaploid wheat [58, 59]. This demonstrates that Ae. umbellulata is a source of untapped rust resistance genes that need to be exploited in the future.
Conclusions
This study presents the first QTL detected for stem rust resistance from Ae. umbellulata, a wild relative of wheat. The new QTL was mapped on chromosome 2U using an F2:3 bi-parental population with GBS markers. The stem resistance QTL-associated markers in the current study can facilitate a rapid selection of wheat-umbellulata recombinant events in alien gene introgression breeding programs and aid in the cloning of this gene.
Abbreviations
- cM:
-
Centimorgan
- GBS:
-
Genotype-by-sequencing
- LOD:
-
Logarithm of odds
- MQM:
-
Multiple QTL mapping
- QTL:
-
Quantitative trait loci
- RFLP:
-
Restriction fragment polymorphism
- SNP:
-
Single nucleotide polymorphism
- WGS:
-
Whole genome sequence
References
Wulf B, Moscou M. Strategies for transferring resistance into wheat: from wide crosses to GM cassettes. Front Plant Sci. 2014;5:1–11. doi:10.3389/fpls.2014.00692.
Zaharieva M, Monneveux P. Spontaneous hybridization between bread wheat (Triticum aestivum L.) and its wild relatives in Europe. Crop Sci. 2006;46:512–27.
Gong W, Li G, Zhou J, Li G, Liu C, et al. Cytogenetic and molecular markers in a wheat background. Genome. 2014;57:489–97.
Schneider A, Molnar I, Molnar-Lang M. Utilisation of Aegilops (goatgrass) species to widen the genetic diversity of cultivated wheat. Euphytica. 2008;163:1–19.
Friebe B, Jiang J, Raupp WJ, McIntosh RA, Gill BS. Characterization of wheat alien translocations conferring resistance to diseases and pests: current status. Euphytica. 1996;71:59–87.
Zaharieva M, Monneveux P, Henry M, Rivoal R, Valkoun J. Evaluation of a collection of wild wheat relative Aegilops geniculata Roth and identification of potential sources for useful traits. Euphytica. 2001;119:33–8.
Molnar I, Gaspar L, Sarvari E, Dulai S, Hoffmann B, et al. Physiological and morphological responses to water stress in Aegilops biuncialis and Triticum aestivum genotypes with differing tolerance to drought. Funct Plant Biol. 2004;31:1149–59.
Colmer TD, Flowers TJ, Munns R. Use of wild relatives to improve salt tolerance in wheat. J Exp Bot. 2006;57:1059–78.
Raupp WJ, Gill BS, Browder LE. Leaf rust resistance in Aegilops squarrosa L. its transfer and expression in common wheat (Triticum aestivum L.). Phytopathology. 1987;73:818.
Miller TE, Reader SM, Ainsworth CC, Summers RW. The introduction of a major gene for resistance to powdery mildew of wheat, Erysiphe graminis f. sp. tritici from Aegilops speltoides into wheat, T. aestivum. In: Jorna ML, Shootmaker LAJ (eds) Cereal breeding related to integrated cereal production: Proc Eucarpia Conf, Wageningen, The Netherlands: Backhuys Publishers; 1987. p. 179-183.
Kerber ER, Dyck PL. Transfer to hexaploid wheat of linked genes for adult-plant leaf rust and seedling stem rust resistance from an amphiploid of Aegilops speltoides X Triticum monococcum. Genome. 1990;33:530–7.
Hussien T, Bowden RL, Gill BS, Cox TS. Chromosomal location of leaf rust resistance gene Lr43 from Aegilops tauschii in common wheat. Crop Sci. 1997;37:1764–6.
Dhaliwal HS. Harjit-Singh, William M. Transfer of rust resistance from Aegilops ovata into bread wheat (Triticum aestivum L.) and molecular characterization of resistant derivatives. Euphytica. 2002;126:153–9.
Hsam SLK, Lapochkina IF, Zeller FJ. Chromosomal location of genes for resistance to powdery mildew in common wheat (Triticum aestivum L. em Thell.). 8. Gene Pm32 in a wheat-Aegilops speltoides translocation line. Euphytica. 2003;133:367–70.
Miranda LM, Murphy JP, Marshall D, Cowger C, Leath S. Chromosomal location of Pm35, a novel Aegilops tauschii derived powdery mildew resistance gene introgressed into common wheat (Triticum aestivum L.). Theor Appl Genet. 2007;114:1451–6.
Van Slageren MW. Wild wheats: a monograph of Aegilops L. and Amblyopyrum (Jaub. & Spach) Eig (Poaceae): a revision of all taxa closely related to wheat, excluding wild Triticum species, with notes on other genera in the tribe Triticcae, especially Triticum. Wageningen Agricultural University, Wageningen, The Netherlands; International Center for Agricultural Research in Dry Areas, Aleppo, Syria; 1994. p. 94-7
Friebe B, Jiang J, Tuleen N, Gill BS. Standard karyotype of Triticum umbellulatum and the characterization of derived chromosome addition and translocation lines in common wheat. Theor Appl Genet. 1995;90(1):150–6.
Sears ER. The transfer of leaf rust resistance from Aegilops umbellulata to wheat. Brookhaven Symp Biol. 1956;9:1–21.
Ozgen M, Yildiz M, Ulukan H, Koyuncu N. Association of gliadin protein pattern and rust resistance derived from Aegilops umbellulata Zhuk. In winter Triticum durum Desf. Breeding Sci. 2004;54:287–90.
Olivera PD, Jin Y. Stem rust resistance in Aegilops species. In: BGRI Technical Workshop. 2013. http://www.globalrust.org/sites/default/files/2013-BGRI-POSTERS-4-New Sources of Resistance.pdf . Accessed 6 July 2016.
Gill BS, Sharma HC, Raup WJ, Browder LE, Hatchet JH, Harvey TL, et al. Evaluation of Aegilops species for resistance to wheat powdery mildew, wheat leaf rust and Hessian fly and green bug. Plant Dis. 1985;69:314–6.
Payne PI, Corfield KG, Holt LM, Blackman JA. Correlation between the inheritance of certain high molecular weight subunits of glutenin and bread making quality in progenies of six crosses of bread wheat. J Sci Food Agric. 1981;31:229–41.
Castilho A, Miller TE, Heslop-Harrison J. Analysis of a set of homoeologous group1 wheat-Aegilops umbellulata recombinant chromosome lines using genetic markers. Theor Appl Genet. 1997;94:293–7.
Holubec V, Hanzalova A, Dumalasova V, Bartos P. Aegilops conservation and collection evaluation in the Czech Republic. J Syst Evol. 2014;52(6):783–9.
Singh RP, Hodson DP, Jin Y, Lagudah ES, Ayliffe MA, Bhavani S, et al. Emergence and spread of new races of wheat stem rust fungus: continued threat to food security and prospects of generic control. Phytopathology. 2015;105:872–84.
Liu WX, Jin Y, Rouse M, Friebe B, Gill BS, Pumphrey MO. Development and characterization of wheat-Ae. searsii Robertsonian translocations and a recombinant chromosome conferring resistance to stem rust. Theor Appl Genet. 2011;122:1537–45.
Liu W, Rouse M, Friebe B, Jin Y, Gill B, Pumphrey MO. Discovery and molecular mapping of a new gene conferring resistance to stem rust, Sr53, derived from Aegilops geniculata and characterization of spontaneous translocation stocks with reduced alien chromatin. Chromosome Res. 2011;19:669–82.
Olson EL, Rouse MN, Pumphrey MO, Bowden RL, Gill BS, Poland JA. Simultaneous transfer, introgression, and genomic localization of genes for resistance to stem rust race TTKSK (Ug99) from Aegilops tauschii to wheat. Theor Appl Genet. 2013;126:1179–88.
Olson EL, Rouse MN, Pumphrey MO, Bowden RL, Gill BS, Poland JA. Introgression of stem rust resistance genes SrTA10187 and SrTA10171 from Aegilops tauschii to wheat. Theor Appl Genet. 2013;126:2477–84.
Chapman J, Mascher M, Buluç A, Barry K, Georganas E, Session A, et al. A whole-genome shotgun approach for assembling and anchoring the hexaploid bread wheat genome. Genome Biol. 2015;16:26.
Mascher M, Muehlbauer GD, Rokhsar D, Chapman J, Schmutz J, Barry K, et al. Anchoring and ordering NGS contig assemblies by population sequencing (POPSEQ). Plant J. 2013;76:718–27.
Spindel J, Wright M, Chen C, Cobb J, Gage J, Harrington S, et al. Bridging the genotyping gap: using genotyping by sequencing (GBS) to add high-density SNP markers and new value to traditional bi-parental mapping and breeding populations. Theor Appl Genet. 2013;126:2699–716.
Romay M, Millard M, Glaubitz J, Peiffer J, Swarts K, Casstevens TM, et al. Comprehensive genotyping of the USA national maize inbred seed bank. Genome Biol. 2013;14:R55.
Poland J, Brown P, Sorrells M, Jannink JL. Development of high density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One. 2012;2:e32253.
Edae E, Bowden RL, Poland J. Application of Population Sequencing (POPSEQ) for Ordering and Imputing Genotyping-by-Sequencing Markers in Hexaploid Wheat. G3 (Bethesda). 2015;5(12):2547–53.
Huang Y, Poland A, Wight C, Jackson E, Tinker N. Using Genotyping-By-Sequencing (GBS) for Genomic Discovery in Cultivated Oat. PLoS One. 2014;9(7):e102448.
Chen Z, Wang B, Dong X, Liu H, Ren L, Chen J, et al. An ultra-high density bin-map for rapid QTL mapping for tassel and ear architecture in a large F2 maize population. BMC Genomics. 2014;15:433.
Saintenac C, Zhang W, Salcedo A, Rouse MN, Trick HN, Akhunov E, et al. Identification of wheat gene Sr35 that confers resistance to Ug99 stem rust race group. Science. 2013;341:783–6.
Steuernagel B, Periyannan SK, Hernandez-Pinzon I, Witek K, Rouse MN, Yu G, Hatta A, Ayliffe M, Bariana H, Jones JDG, Lagudah ES, Wulff BH. Rapid cloning of disease-resistance genes in plants using mutagenesis and sequence capture. Nat Biotechnol. 2016;34:652–5.
Zhang H, Jia J, Gale M, Devos K. Relationships between the chromosomes of Aegilops umbellulata and wheat. Theor Appl Genet. 1998;96:69–75.
Molnar I, Imkova H, Leverington-Waite M, Goram R, Cseh A, Vrana J, et al. Syntenic Relationships between the U and M Genomes of Aegilops, Wheat and the Model Species Brachypodium and Rice as Revealed by COS Markers. PLoS One. 2013;8(8):e70844.
BioSprint 96 DNA Plant Kit. www.qiagen.com/BioSprint 96 DNA plan kit. Accessed 15 May 2016.
Lu F, Lipka AE, Glaubitz J, Elshire R, Cherney JH, Casler MD, et al. Switchgrass Genomic Diversity, Ploidy, and Evolution: Novel Insights from a Network-Based SNP Discovery Protocol. PLoS Genet. 2013;9:e1003215. 33.
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ESTASSEL. software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–5.
Wu Y, Bhat PR, Close TJ, Lonardi S. Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. PLoS Genet. 2008;4:e1000212.
Rouse M, Wanyera R, Njau P, Jin Y. Sources of Resistance to Stem Rust Race Ug99 in Spring Wheat Germplasm. Plant Dis. 2011;95:762–6.
Stakman EC, Stewart DM, Loegering WQ. Identification of physiologic races of Puccinia graminis var. tritici. U. S. Dep Agric Agric Res Serv. 1962;E-617.
ftp://ftpmips.helmholtz-muenchen.de/plants/wheat/IWGSC/. Accessed 8 Aug 2015.
ftp://ftpmips.helmholtz-muenchen.de/plants/barley/public_data/. Accessed 8 Aug 2015.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an Information Aesthetic for Comparative Genomics. Genomics Res. 2009;19:1639–45.
Ellis J, Lagudah E, Spielmeyer W, Dodds P. The past, present and future of breeding rust wheat. Front Plant Sci. 2014;5:1–12.
Merchuk-Ovnat L, Barak V, Fahima T, Ordon F, Lidzbarsky GA, Krugman T, et al. Ancestral QTL alleles from wild emmer wheat improve drought resistance and productivity in modern wheat cultivars. Front Plant Sci. 2016;7:452.
IWGSC, The International Wheat Genome Sequencing Consortium. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 2014;345(6194):286.
IBGSC, International Barley Genome Sequencing Consortium. A physical, genetic and functional sequence assembly of the barley genome. Nature. 2012;491:711–6.
Molnar I, Vrana J, Buresova V, Capal P, Farkas A, Dark E, et al. Dissecting the U, M, S and C genomes of wild relatives of bread wheat (Aegilops spp.) into chromosomes and exploring their synteny with wheat. Plant J. 2016; dio:10.1111/tpj.13266.
Gale MD, Devos KM. Comparative genetics in the grasses. Proc Natl Acad Sci. 1998;95:1971–4.
Yu LX, Barbier LH, Rouse MN, Singh S, Singh RP, Bhavani S, et al. A consensus map for Ug99 stem rust resistance loci in wheat. Theor Appl Genet. 2014;127:1561–81.
Schachermayr G, Siedler H, Gale MD, Winzeler H, Winzeler M, Keller B, et al. Identification and localization of molecular markers linked to the Lr9 leaf rust resistance gene of wheat. Theor Appl Genet. 1994;88:110–5.
Gupta KS, Charpe A, Koul S, Prabhu KV, Haq QMR. Development and validation of molecular markers linked to an Aegilops umbellulata-derived leaf rust resistance gene, Lr9, for marker-assisted selection in bread wheat. Genome. 2005;48:823–30.
Acknowledgement
Computational resources for this project were through Minnesota Supercomputing Institute (MSI) high performance computing (HPC) at University of Minnesota. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture. USDA is an equal opportunity provider and employer.
Funding
This project was supported by the USDA-ARS Postdoctoral Research Program, the USDA-ARS National Plant Disease Recovery System and the USDA-ARS appropriated project 5062-21220-021-00-D “Cereal Rust Fungi: Genetics, Population Biology, and Host-Pathogen Interactions”.
Availability of data and materials
All the supporting data are included as Additional files 1 and 2. Raw GBS reads data supporting the results of this article are available in the NCBI Sequence Reads Archive (SRA) under submission SUB1955121 (http://www.ncbi.nlm.nih.gov/sra).
Authors’ contributions
EE, MR, PO and YJ conceived the study. PO and YJ developed the segregation population. EE, PO and MR performed disease screening. JP prepared GBS library. EE and MR performed QTL analysis. EE constructed linkage map and drafted the manuscript. All authors reviewed and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1: Table S1.
Chromosomal locations and genotypic data for GBS markers. (XLSX 924 kb)
Additional file 2: Table S2.
QTL detected by Single QTL mapping methods. (XLSX 17 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Edae, E.A., Olivera, P.D., Jin, Y. et al. Genotype-by-sequencing facilitates genetic mapping of a stem rust resistance locus in Aegilops umbellulata, a wild relative of cultivated wheat. BMC Genomics 17, 1039 (2016). https://doi.org/10.1186/s12864-016-3370-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-016-3370-2