
Abstract
Since the characterization of cytochrome c552 as a multiheme nitrite reductase, research on this enzyme has gained major interest. Today, it is known as pentaheme cytochrome c nitrite reductase (NrfA). Part of the NH4+ produced from NO2− is released as NH3 leading to nitrogen loss, similar to denitrification which generates NO, N2O, and N2. NH4+ can also be used for assimilatory purposes, thus NrfA contributes to nitrogen retention. It catalyses the six-electron reduction of NO2− to NH4+, hosting four His/His ligated c-type hemes for electron transfer and one structurally differentiated active site heme. Catalysis occurs at the distal side of a Fe(III) heme c proximally coordinated by lysine of a unique CXXCK motif (Sulfurospirillum deleyianum, Wolinella succinogenes) or, presumably, by the canonical histidine in Campylobacter jejeuni. Replacement of Lys by His in NrfA of W. succinogenes led to a significant loss of enzyme activity. NrfA forms homodimers as shown by high resolution X-ray crystallography, and there exist at least two distinct electron transfer systems to the enzyme. In γ-proteobacteria (Escherichia coli) NrfA is linked to the menaquinol pool in the cytoplasmic membrane through a pentaheme electron carrier (NrfB), in δ- and ε-proteobacteria (S. deleyianum, W. succinogenes), the NrfA dimer interacts with a tetraheme cytochrome c (NrfH). Both form a membrane-associated respiratory complex on the extracellular side of the cytoplasmic membrane to optimize electron transfer efficiency. This minireview traces important steps in understanding the nature of pentaheme cytochrome c nitrite reductases, and discusses their structural and functional features.
Graphical abstract

Similar content being viewed by others

Introduction
The focus of this minireview is on multiheme c-type cytochromes, written in honor of Isabel Moura and former SBIC president José Moura (2010–2012) from the Universidade Nova de Lisboa in Portugal on the occasion of their 70th birthday. In my view this topic seems well suited, considering the numerous important discoveries and contributions by Isabel and José to our understanding of transition metal enzymes, many of them key components of the global nitrogen cycle, carrying Fe, Cu, or Mo ions at the active site [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16]; for a complete list of their publications please visit https://sites.fct.unl.pt/biologicalchemistryatfctunl/pages/people accessed on 30 Nov 2021.
In c-type cytochromes the heme is covalently attached to the polypeptide backbone via two thioether (R–S–R′) bonds formed by the vinyl groups of heme and cysteine side chains in a Cys-X-X-Cys-His pentapeptide motif; “X” denotes a miscellaneous amino acid, and the histidine residue coordinates on the proximal binding site of the heme iron (Fig. 1). Cytochromes c possess a wide range of properties, they function as electron transfer proteins and are involved in many important redox processes [17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35].

Left: structure of heme c, with the covalently attached heme group present in cytochromes c. Right: structure of cytochrome c (PDB 1HRC), with Met80 and His18 ligated to iron [20]
In this contribution I will center on the pentaheme enzyme cytochrome c nitrite reductase (NrfA), a key player within the global nitrogen cycle [36,37,38,39,40,41,42,43,44,45,46]. It catalyses the six-electron reduction of nitrite (NO2−) to ammonium (NH4+) (Eq. 1), as part of the dissimilatory nitrate reduction to ammonium (DNRA) process, that competes with denitrification [47,48,49,50,51,52,53,54,55,56,57]. Notably, NrfA can also convert sulfite (SO32−) to hydrogen sulfide (H2S), an important reaction of the microbial sulfur cycle (Eq. 2), performed by dissimilatory sulfite reductase employing the coupled siroheme-[4Fe-4S] center for catalysis [58,59,60,61,62].
Unlike the process of denitrification, DNRA, also known as nitrate/nitrite ammonification, conserves bioavailable nitrogen in the system, producing soluble NH4+ rather than chemically unreactive dinitrogen gas (N2) (Scheme 1) [47,48,49,50,51,52,53,54,55,56,57].

Key processes of the global nitrogen cycle: dissimilatory nitrate reduction to ammonium (DNRA), denitrification, and nitrogen fixation; changes in the oxidation state of nitrogen, as well as in equations, are indicated by roman numerals
With the report by Fujita on soluble cytochromes in Enterobacteriaceae in 1966, followed by the purification and description of cytochrome c552 as hexaheme nitrite reductase in 1986 [63,64,65], the stage was set for an all-out attack on the problem of microbial nitrite to ammonia reduction by numerous pioneering researchers. Among them Isabel and José Moura, who applied advanced spectroscopic (EPR, Mössbauer) and electrochemical techniques to unravel structural and functional properties of these complex multi-centered metalloenzymes [1, 6, 8, 10], see also recent review entitled “How Biology Handles Nitrite” [54]. Here I will give an account of my personal experience in this exciting field of Bioinorganic Chemistry, and I will mention some of the great moments in this endeavour. In view of the vast literature in the field of c-type cytochromes, I suggest for introduction the monographs by Ambler [18], Pettigrew and Moore [19], and Salgueiro and Dantas [31], for deeper insight into the complex topic, the informative articles by experts are recommended [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35]. My apologies go to all colleagues who made significant contributions to our current knowledge of cytochrome c nitrite reductases and related enzymes that will not be discussed here. These omissions are not intentional, they are the consequence of time and space. Clearly, the emphasis is on the structural and functional properties of the pentaheme nitrite reductases from Sulfurospirillum deleyianum and Wolinella succinogenes. Yet this minireview will hopefully illustrate what one may learn about studying the structure and function of such an important and intensively studied enzyme.
Looking back: Tomar 1979—Jean Le Gall—cytochrome c 3—Desulfovibrio
I did my Ph.D. work under the supervision of Peter Hemmerich [66] at the newly founded University of Konstanz, in an area of research nowadays called Bioinorganic Chemistry [67,68,69,70,71,72,73,74]. Hemmerich had fruitful collaborations with scientists from around the world: Helmut Beinert (Madison), Anders Ehrenberg (Stockholm), Jean-Marc Lhoste (Paris), Vincent Massey (Ann Arbor), Israel Pecht (Rehovot), Jack Spence (Logan), and Cees Veeger (Wageningen), to name a few. Advanced spectroscopic techniques, among them magnetic resonance methods as well as stopped-flow and rapid quench kinetics in the millisecond range, were established in Konstanz and applied to investigate the structure and function of complex flavin and metal-dependent enzymes. I started my work with two plant proteins, the blue multi-copper enzyme ascorbate oxidase and the type 1 Cu protein mavicyanin together with Augusto Marchesini (Milan) [75,76,77].
In 1979 I attended a workshop in Tomar (Portugal) entitled “Metal Ions in Biology”, sponsored by NATO Advanced Study Institutes, and organized by António Xavier (Lisbon) and Allen Hill (Oxford) (Fig. 2). It was in Tomar where I met Isabel and José Moura for the first time. Both, together with Helena Santos and Isabel Coutinho, were working hard to keep us participants happy. Clearly, this meeting became a memorable milestone in the history of Bioinorganic Chemistry. Four years later Ivano Bertini (Florence), Harry Gray (Pasadena), Bo Malmström (Göteborg), and Helmut Sigel (Basel) initiated the International Conference on Biological Inorganic Chemistry (ICBIC) series in Florence [78, 79].

Participants of the workshop “Metal Ions in Biology”, sponsored by NATO Advanced Study Institutes, held September 16–28, 1979 in Tomar, Portugal [79]. The photograph shows António Xavier, Allen Hill, Isabel and José Moura, Helena Santos, Isabel Coutinho, Jean Le Gall, and numerous pioneers of Bioinorganic Chemistry mentioned in the text
For 2 weeks, leading researchers presented their work on metal-dependent enzymes in Tomar. In parallel, distinguished experts offered excellent—however quite exhausting—lectures on physical techniques, such as magnetic circular dichroism (MCD), Mössbauer, nuclear magnetic resonance (NMR), and electron paramagnetic resonance (EPR) spectroscopy, and not to forget the application of direct electrochemical methods to study electron transfer (ET) and catalysis [79]. I recall a crude three-dimensional model presented by Richard Haser and colleagues (Marseille) for the structure of the tetraheme ET protein cytochrome c3. The protein had been isolated from Desulfovibrio desulfuricans, and its structure was solved at 2.5 Å resolution [80,81,82]. The molecule consisted of a single polypeptide chain wrapped around a compact core of four non-parallel heme centers. Alignment of the amino acid sequences of cytochrome c3 from different sources suggested that the structure reported by Haser and colleagues was characteristic of the cytochrome c3 group which became the target of numerous NMR studies by Xavier and his associates [26, 83,84,85,86]. Notably, in the course of our early spectroscopic and structural studies of metal-dependent proteins and enzymes in sulfate reducing bacteria, we isolated cytochrome c3 from the periplasmic fraction of D. desulfuricans (strain Essex 6) and Oliver Einsle solved its three-dimensional structure. Its major physiological function appeared to be that of an electron carrier for the periplasmic hydrogenase, and Julia Steuber provided evidence that it interacted with the membrane-associated dissimilatory sulfite reductase (DSIR). In addition, a nonaheme cytochrome c from this organism could be structurally characterised by Günter Fritz in Konstanz [87,88,89,90]. Last but not least, the structural origin of nonplanar heme distortions in Fe(III) cytochromes c3 was investigated by resonance Raman spectroscopy together with John Shelnutt (Albuquerque), Isabel and José [91].
Next, I have to mention microbiologist Jean Le Gall from the Laboratoire de Chimie Bacterienne (Marseille) whom I met in Tomar for the first time. He also held a position as Research Professor of Biochemistry and Microbiology at the Department of Biochemistry, University of Georgia (Athens) [92, 93]. As a graduate student he discovered a new species of bacteria which he named Desulfovibrio gigas (gigas, latin, meaning gigantic). These bacteria are so-called anaerobes and extremely difficult to grow. Clearly, Jean Le Gall has to be regarded as one of the pioneering researchers in the field of Inorganic Microbial Sulfur Metabolism, and he turned into one of the most influential collaborators of Isabel and José Moura, and António Xavier and associates [94,95,96,97,98,99,100,101,102,103,104,105,106,107].
Admittedly, when I was trained as a chemist at the Universities of Basel and Konstanz in the 1960- and 70-ties (with a strong preference for transition metal coordination chemistry), anaerobic bacteria of the genus Desulfovibrio, microbial bioenergetics as well as biogeochemical cycles of the elements nitrogen and sulfur were not part of the program. It was not until 1986, when Andreas Zöphel finished his doctorate thesis on microbial sulfur respiration by “Spirillum 5175”. In Lisbon, supported by the Gulbenkian foundation, Andreas learned how to handle anaerobic bacteria and how to isolate and purify dioxygen sensitive enzymes. “Spirillum 5175” uses the reduction of elemental sulfur (S0) to hydrogen sulfide (H2S) for energy conservation (sulfur respiration). Together with Norbert Pfennig, Professor of Microbiology in Konstanz, and graduate student Wolfram Schumacher, “Spirillum 5175” was described as the type strain of the new genus and species S. deleyianum [108,109,110]. Thanks to Norbert Pfennig and his associates, Heribert Cypionka, Bernhard Schink, and Friedrich Widdel, and inspired by pioneering researchers of Microbiology and Bioinorganic Chemistry, either through experiments in the laboratory, or by fruitful discussions at international conferences, new and fascinating areas of research with exciting discoveries arose. Enzymes with novel transition metal centers (Fe, Cu, Mo, W) and unique catalytic properties could be purified from both anaerobic and aerobic microorganisms. These metal-dependent enzymes, among them two multi-centered key players of the global nitrogen and sulfur cycle, cytochrome c nitrite reductase and dissimilatory sulfite reductase, were structurally characterized and their mechanism of action was investigated by applying biochemical and spectroscopic methods [111, 112].
To finish my backward glance, close to four decades after the Tomar meeting, in 2015, both José and I were invited to participate in the workshop “Feeding the World in the twenty-first Century: Grand Challenges in the Nitrogen Cycle”. The workshop was initiated by Nicolai Lehnert (University of Michigan), with co-organizers Gloria Coruzzi (New York University), Eric Hegg (Michigan State University), Lance Seefeldt (Utah State University), and Lisa Stein (University of Alberta). In short, the purpose of this workshop was to identify ways that chemists can help the scientific community understand and manage the nitrogen cycle to improve agriculture and environmental quality [113].
Multiheme proteins and enzymes
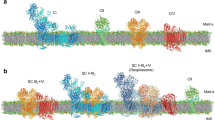
Heme proteins (Fe protoporphyrin IX complexes) exhibit an impressive range of biological functions, such as ET reactions, dioxygen (O2) transport and storage, O2 reduction to hydrogen peroxide (H2O2) or water (H2O), and the oxygenation of organic substrates (R–H → R–OH). The range of functions can be extended further by linking heme groups with other redox active cofactors and metal sites plus other heme centers. These combinations will permit heme cofactors to couple ET with other processes, such as the translocation of protons or the reduction/oxidation of molecules both inside and outside of the cell. In aerobic and anaerobic microbes, especially those of the biological nitrogen and sulfur cycles, there are many c-type cytochromes with multiple heme centers per polypeptide chain [2, 3, 5, 23, 25,26,27,28,29,30,31,32, 35, 55, 98, 103, 114,115,116,117,118,119,120,121,122]. Early examples include the octaheme hydroxylamine oxidoreductase (HAO) and related proteins [123,124,125,126,127,128,129], the tetraheme NapC/NirT/TorC family [130], the 16-heme-containing protein Hmc (high molecular mass cytochrome c) from sulfate-reducing bacteria [131, 132], the octaheme tetrathionate reductase from Shewanella oneidensis [133, 134], the tetraheme cytochrome c3 described in the previous section, and the pentaheme cytochrome c nitrite reductase, the central enzyme of this review. These multiheme proteins form structurally related families, in which the positions of the heme can often be overlaid, even when there is little sequence conservation between members of the family, e.g., pentaheme nitrite reductase NrfA, octaheme HAO, and flavocytochrome fumarate reductase [22, 133,134,135]. In 1999, Barker and Ferguson argued that only by fixing the hemes spatially via their thioether bonds can such clustering of hemes be achieved [22]. Furthermore, the dense packing of hemes allows rapid electron transfer between the heme centers [136], an essential part of the function of these cytochromes. This molecular arrangement is advantageous for enzymes such as NrfA, which catalyses the six-electron reduction of nitrite (Eq. 1), or the group of cytochrome P460 enzymes, which catalyse the oxidation of hydroxylamine (NH2OH) [45] (Fig. 3).

Structural comparison of pentaheme cytochrome c nitrite reductase of W. succinogenes (NrfAws) and octaheme hydroxylamine oxidoreductase of Nitrosomonas europaea (HAO). Left: NrfAws, functional homodimer; PDB 1FS7). Right: HAO, functional homotrimer; PDB 1FGJ). Below: Superposition of the heme groups of NrfAws (gray) and of HAO (black) numbered according to their attachment to the protein chain. With the exception of the active site heme (one in NrfA, four in HAO), all heme groups form so-called di-heme elbow motifs (circles), which are connected via a parallel stacking arrangement like in split-Soret cytochrome c (ovals) [104, 123, 163]
Finally, one of the recent important scientific discoveries in the global nitrogen cycle has to be brought forward, the anammox process (Anaerobic Ammonium Oxidation) described by Gijs Kuenen and associates (Delft) in 1995 [137, 138]. On a global scale, anammox bacteria significantly contribute to the removal of fixed nitrogen from the environment [139]. The process depends on multiheme proteins that structurally resemble the ones known from other microorganisms, but that exhibit new functions. In the three-step process, NH4+ becomes oxidized by anaerobic ammonium-oxidizing bacteria, they can oxidize ammonium with nitrite as the oxidant instead of O2 and form N2 as the end product (Eqs. 3–6).
Notably, substrate conversion proceeds through potentially toxic intermediates nitric oxide (NO) and hydrazine (N2H4). The anammox machinery resides in a special and unique cell organelle, the Anammoxosome. Here, energy released in the anammox reaction is used to drive ATP synthesis, powered by novel membrane-bound protein complexes [140, 141]. The end product N2 is produced from the oxidation of intermediate N2H4, with the octaheme protein hydrazine dehydrogenase (HDH) involved in catalysis which appears to be related to octaheme HAO. HDH is a soluble multi-protein complex (1.7 MDa) that is not spatially associated with the anammoxosome membrane. The enzyme of Kuenenia stuttgartiensis was characterized as a covalently cross-linked homotrimeric octaheme protein. The HDH trimers build an octameric architecture, with each octamer harbouring an amazing 192 (!) c-type heme centers. It is concluded that the multi-protein complex observed both X-ray crystallography and Cryo-Electron Microscopy, probably represents the functionally relevant oligomeric state of HDH [142,143,144,145,146].
Sulfur respiration and reduction of nitrite to ammonia in Sulfurospirillum deleyianum and Wolinella succinogenes
When asking myself how I, a coordination chemist by training, got into the field of metal-dependent proteins and enzymes in aerobic and anaerobic microorganisms, clearly all my excursions into microbiology are linked, from the early experiments on sulfur respiration to cytochrome c nitrite reductase and multiheme c-type cytochromes, with Norbert Pfennig. Together with Regina Bache, a survey was made of components of sulfur-reducing bacteria that can be detected by EPR spectroscopy around 10 K [147]. Among the organisms investigated was a small spirillum later described as S. deleyianum, which turned out to be a model organism for (1) studying sulfur respiration, that is the reduction of elemental sulfur (S0) to hydrogen sulfide (H2S), and (2) exploring the structural and functional characteristics of DNRA enzyme cytochrome c nitrite reductase (NrfA) [39, 43, 44, 108,109,110,111,112, 147,148,149,150,151,152,153,154,155,156,157]. Basically, DNRA is a short circuit that bypasses the processes of denitrification (NO3− → N2) and nitrogen fixation (N2 → NH3) (Scheme) [155]. Dihydrogen (H2) and formate (HCOO−) are the predominant electron donors for various nitrite-ammonifying bacteria including S. deleyianum and W. succinogenes. Sulfide (S2−) can serve as electron donor as well, thus connecting the biogeochemical cycles of nitrogen and sulfur [150]. In this case, the presence of a highly active nitrite reducing system might help to dispose toxic NO2−. When grown with NO3−, NO2−, or S0 as terminal electron acceptor, S. deleyianum expressed a red protein with intense EPR resonances centered at g ≈ 3.85 and 9.12 (recorded in perpendicular mode) and at g ≈ 9.8 (recorded in parallel mode), localized mainly in the membrane fraction. Under reducing conditions (Na+ dithionite) the prominent cytochrome in the membrane fraction exhibited absorption maxima at 553, 522.5 and 426 nm, and notably, both the soluble and the membrane fraction of S. deleyianum showed high nitrite reductase activity [108, 109, 147]. Following the intense EPR signal at g 3.85, Wolfram Schumacher isolated a heme-dependent nitrite reductase from both the soluble and membrane fraction of S. deleyianum and related microorganism W. succinogenes; it reduced NO2− to NH4+ with a specific activity of up to 1050 µmol NO2− (protein min)−1 (Fig. 4) which was key to build a nitrite sensor based on a highly sensitive nitrite reductase mediator-coupled amperometric detection [151,152,153]. The UV/Vis spectrum of the enzyme was typical for c-type cytochromes with absorption maxima at 280, 408, 532 and 610 nm (oxidized) and at 420, 523 and 553 nm (reduced). The EPR spectrum (perpendicular mode) revealed resonances at g 9.82, 3.85, 3.31, 2.95, 2.30, and 1.49, resulting from high-spin and low-spin Fe(III) heme centers, as reported for other nitrite reductases purified from D. desulfuricans, W. succinogenes, or Escherichia coli [151]. Early on, the characteristic signal at g 3.85, which could also be observed in EPR spectra of whole cells of S. deleyianum grown with S0, was suggested to originate from a magnetic interaction between high-spin and low-spin Fe(III) hemes [158].

Specific nitrite reductase activity and substrate spectrum of cytochrome c nitrite reductase of S. deleyianum (NrfAsd). With nitrogen monoxide (NO), hydroxylamine (NH2OH), and O-methyl hydroxylamine (NH2OCH3) ammonium (NH4+) was the product; in the case of nitrous oxide (N2O), dinitrogen (N2) was formed most likely; with sulfite (SO32−) hydrogen sulfide (H2S) was the only product [60, 156]
The view of the existence of a family of hexaheme nitrite reductases, based on their similar sizes, specific activities, UV/Vis and EPR spectral properties, had to be abandoned in 1993 when the amino acid sequence of the E. coli enzyme became available. During this time both Jeff Cole (University of Birmingham) and I had suggested, based on sequence and spectroscopic data, that NrfA of E. coli [159, 160] as well as of S. deleyianum and W. succinogenes hosted only four c-type hemes each [152]. Notably, the E. coli NrfA sequence revealed the presence of just ten cysteine residues, eight arranged as four conventional CXXCH heme-binding motifs. However, in a consecutive publication, Cole and associates provided convincing evidence for a fifth heme covalently attached to the remaining two cysteine residues within a novel cysteine-lysine Cys-Trp-Ser-Cys-Lys motif, and that the lysine residue is required for normal rates of nitrite to ammonia reduction [161]. Oliver Einsle, together with Albrecht Messerschmidt and Robert Huber (Max Planck Institute of Biochemistry, Martinsried), gave the final answer to the question about the number of hemes and nature of heme binding motifs by solving the three-dimensional structure of cytochrome c nitrite reductases of S. deleyianum (1.9 Å resolution) [162] and of W. succinogenes (1.6 Å) [163]. The molecular architecture showed a homodimer with five hemes anchored in each subunit, four of them bound to the protein with the conventional Cys-X-X-Cys-His motif, and one with the novel cysteine-lysine Cys-X-X-Cys-Lys motif (Fig. 5) [164]. These characteristic basic features of NrfA, detected in nitrite reductase of S. deleyianum and W. succinogenes, have been also found in the NrfA structures of E. coli [165, 166], D. desulfuricans ATCC27774 [167, 168], S. oneidensis [169], the NrfA4H2 complex of D. vulgaris [170, 171], and most recently in NrfA of the bacterium Geobacter lovleyi [172, 173].

Overall structure of the cytochrome c nitrite reductase dimer (NrfAsd PDB 1QD8; NrfAws PDB 1FS7); front view of the dimer, dimer formation is mediated by the central helical segments. The peptide chain packs into a compact, predominantly α-helical fold that can be subdivided into the central part, where long helices form the dimer interface, and the heme containing part, where the peptide chain wraps tightly around the cofactors; Fe ions in red, heme groups in green, Ca2+ in grey close to active site heme; the five hemes in the monomer are in close contact, with Fe–Fe distances of between 9.3 and 12.8 Å, and 11.7 Å for hemes at dimer interface [162,163,164]
However, recent investigations of the diversity and phylogeny of the NrfA enzyme, analysing 272 full-length NrfA protein sequences, distinguished 18 NrfA clades, with 3 clades having the conventional CysX-X-Cys-His motif in the first heme-binding domain, whereas all other clades had the Cys-X-X-Cys-Lys motif in this location [174, 175]. Earlier studies on the ε-proteo-bacterium Campylobacter jejeuni did already indicate that the putative nrfA gene carried the conventional Cys-X-X-Cys-His motif, and not the Cys-X-X-Cys-Lys motif present in E. coli, S. deleyianum, or W. succinogenes [176].
Structure of pentaheme cytochrome c nitrite reductase
Note that the main focus will be on the structural and functional properties of the cytochrome c nitrite reductase of ε-proteobacteria W. succinogenes (NrfAws) and S. deleyianum (NrfAsd); for a more comprehensive analysis the article by Oliver Einsle in Methods of Enzymology is recommended [164].
Overall structure and protein architecture
Both NrfAsd (PDB 1QD8) [162] and NrfAws (PDB 1FS7) [163] are pentaheme enzymes encoded by a single gene termed nrfA, with a protoporphyrin IX covalently linked to the protein backbone at the active site. The protein forms a homodimer with dimensions of ≈ 100 Å × 80 Å × 50 Å, in which the ten covalently attached heme groups are closely packed (Figs. 3, 5). It folds into one compact domain, with α-helices as the predominant secondary structural motif, ranging from short helical turns to four long helices at the carboxy-terminal end of the peptide chain; ß-sheet structures are only found in two short antiparallel strands, where one is part of a funnel-like cavity leading to the active site. The dimer interface is dominated by three long α-helices in each monomer, and although the area of this interface amounts to only ≈ 10% of the total surface area of the protein, the arrangement is highly conserved. The five heme groups in each monomer are sufficiently close to assure rapid intramolecular electron transfer that also bridges the dimer through the adjacent heme group 5 from each monomer. Heme center 1 is the active site of the enzyme, showing the typical scheme found in c-type cytochromes, with thioether bonds to the Cys residues of the heme binding motif. It has a Lys residue to replace the conventional His as a proximal ligand to the heme iron. The Fe atom is coordinated by the Nζ atom (sp3 N) of Lys (Fe–N distance 2.1 Å/NrfAws), and the distal axial position remains open for interaction with the substrate. As expected from amino acid sequence comparisons, the structures of NrfA from both S. deleyianum [162] and W. succinogenes [163] are very similar (root mean square displacement for Cα atoms 1.19 Å). The main differences in the peptide chain are two insertions in NrfAsd, which are absent in the NrfAws sequence. Apart from these insertions and the additional residues at the N terminus of the NrfAws polypeptide chain, a further major difference is the C-terminus of the chain, which forms an α-helix in NrfAsd and a short two-stranded β-sheet in NrfAws. The residues that surround the active site and form the substrate/product channel are completely conserved [162,163,164].
In many multiheme c proteins, heme groups pack into distinct and recurring motifs that were recognized early on in electron transfer proteins, e.g., cytochrome c554 from N. europaea [177] and enzymes, such as NrfA. They were classified in particular into a parallel and a perpendicular stacking interaction (Fig. 3). These two types of interactions are combined in the proteins to form efficient electron transfer chains. The five hemes in the NrfA monomer are in close contact, with Fe–Fe distances of between 9 and 12.8 Å. They are arranged as a group of three, almost coplanar hemes (1, 3, 4), with heme 1 forming the active site. Hemes 2 and 5 are farther apart and are not coplanar with hemes 1, 3 and 4. All hemes, with the exception of heme 1, are His/His coordinated; with edge-to-edge distances below 4 Å, hemes 1, 3 and 4 are close enough to allow direct π-electron interaction of the porphyrin rings. The propionate side chains of heme 1 form part of the active-site cavity, while those of heme 4 are exposed to the solvent and those of heme 3 are hydrogen-bonded inside the protein. All porphyrins show a slight distortion from planarity, most strongly in heme 2, and least in the active-site heme 1. Heme 2 could function as the entry point for electrons, see discussion on the complex between NrfA and its redox partner. The dimer interface is dominated by three long α-helices per NrfA monomer. Hemes 5 interact across the dimer interface which is even closer than hemes 2 and 3 within each monomer. The short Fe–Fe distance of 11.7 Å will allow efficient electron transfer across the NrfA dimer interface [136] which might be functionally relevant. Furthermore, both these hemes interact directly through hydrogen bonds between their propionate side chains. Note that the relative orientation of the five heme groups in NrfAsd corresponds exactly to the one observed in NrfAws, including the fact that heme 1 is the five-coordinate active site heme group, clearly all the structurally and functionally important features are conserved between both species. The five heme groups of NrfAws align with those of NrfAsd within a root mean square displacement of 0.12 Å for all atoms [162,163,164].
Fe–Fe distances between the heme groups are in a range commonly found in redox proteins and are short enough to allow for direct electron tunneling between the individual heme centers [136]. Whereas heme 1 is the site of nitrite binding, it is more difficult to define the entry point for electrons delivered by the physiological redox partner, the tetraheme c-type cytochrome NrfH [178, 179]. All heme groups cluster on one side of the dimer, and heme 2 as well as heme 5 have one edge of the porphyrin plane exposed to the solvent, although for heme 5, most of this area is in the dimer interface and is covered upon dimerization. Furthermore, the area where heme 2 reaches the protein surface is located within a patch of strong positive surface potential in the NrfAws structure. In S. deleyianum, the membranous nitrite reductase complex was described to be less stable than in W. succinogenes [152], and in accordance with this, the positive patch surrounding heme 2 is less pronounced. Thus, heme 2 has been proposed to be the most likely entry point for electrons into the nitrite reductase of both S. deleyianum and W. succinogenes [162,163,164]. The electropositive patch in the vicinity of heme 2 is conserved in the E. coli enzyme (NrfAec), however, it is significantly less pronounced than in NrfAsd and NrfAws. The size of the positive patch is reduced in the NrfAec structure by the substitution of an adjacent arginine residue (Arg 207/NrfAws; Arg 206/NrfAsd) for a glutamine residue (Gln 205). Although weak, this represents one of the few positive surface patches apart from that at the site of substrate entry to heme 1. This weakness may relate to the observation that, it has not been possible to isolate and crystallize stable complexes of NrfAsd, NrfAws, or NrfAec with its electron donors, either NrfH or NrfB, and to solve their three-dimensional structures [152, 165, 180]. A significant step forward in understanding the complex architecture of the complete cytochrome c nitrite reductase machinery came from the work by Inês Pereira, Margarida Archer, and associates (Lisbon) who solved the X-ray structure of the stable complex between the reductase NrfA and its electron donor NrfH from Desulfovibrio vulgaris. One NrfH molecule interacts with one NrfA dimer in an asymmetrical manner, forming a large membrane-bound complex with an overall α4ß2 quaternary arrangement. The menaquinol-interacting NrfH heme is pentacoordinated, bound by a methionine from the CXXCHXM sequence, with an aspartate residue occupying the distal position. The NrfH heme that transfers electrons to NrfA has a lysine residue from the closest NrfA molecule as distal ligand [170, 171].
Cytochrome c nitrite reductase does not only convert NO2− to NH4+, but also the potential reaction intermediates NO and NH2OH, as well as N2O, NH2OCH3, and SO32− (Fig. 4). However, intermediates are not released during nitrite turnover. Obviously, the active site cavity can accommodate both anions and uncharged molecules, and will release the NH4+ cation only after the full six-electron reduction of NO2−. The preference for anions is reflected by a positive electrostatic potential around and inside the active site cavity, induced by the residues forming the cavity, Tyr 127, His 282, Arg 113, Gln 281, and Lys 279 (numbering NrfAsd). These residues serve as stores for protons required for the reduction of NO2− to NH4+ (Eq. 1) and can be resupplied by water molecules. Considering the good accessibility of the active site for H2O and the presumably lower pH on the periplasmic side of the cytoplasmic membrane, the product of NO2− reduction will be the NH4+ cation rather than uncharged NH3. The cationic product might take advantage of a second channel leading to the protein surface opposite to the entry channel. This second channel is lined by His 406 and Tyr 95 and filled with coordinated water molecules, it branches before reaching the protein surface and ends with both arms in areas possessing a significantly negative electrostatic surface potential (Fig. 6) [162, 163].

The active-site channel of cytochrome c nitrite reductase of Sulfurospirillum deleyianum (NrfAsd PDB 1QD8). Apart from the channel (entrance) guiding NO2− from the protein surface to the catalytic heme 1 site, a second channel (exit) reaches the protein surface on the opposite side of the molecule. The whole channel is coloured according to the electrostatic surface potential, blue for a positive and red for a negative potential. In contrast to the immediate surroundings of the active-site heme group 1, the surface potential is drastically changed in the presumed exit channel for NH4+ [162]
Active site
The site of nitrite reduction is heme center 1, with the Nζ atom (sp3 N) of lysine, replacing a His (sp2 N) residue in the classical binding motif, and an oxygen atom of a bound SO42− anion detected in the first published structures NrfAsd (Lys 133; PDB 1QD8) and NrfAws (Lys 134; 1PDB 1FS8) (Fig. 7) [162, 163]. The SO42− anion binds to the iron with an oxygen atom, it further interacts with both a His and a Tyr via a single oxygen atom, and with a water molecule which in turn interacts with the two propionate side chains of heme 1. At low concentrations of SO42− it was replaced by H2O at heme 1. The structure of the water-bound form (1.6 Å resolution) of NrfAws showed the oxygen atom of H2O bound to the Fe at a distance of 2.05 Å. Without the bulky SO42− anion, the imidazole moiety of His 277 moved closer to the heme iron, and a hydrogen bond (length 2.88 Å) was formed between its Nε2 and the H2O molecule at the active site. The positions of both Tyr 218 and Arg 114 remained unchanged [163]. Although the bound SO42− originated from the crystallization buffer, its binding at the active heme center provided early information about substrate binding. As expected both SO42−, and structurally related PO43−, acted as weak inhibitors in the activity assay [156]. Azide, N3−, was expected to bind to the active site heme iron as a competitive inhibitor [181, 182] just like SO42−. However, the structure of the NrfAws-N3− complex (2.0 Å resolution) revealed a water bound to Fe at 2.05 Å, and in close proximity the N3− anion, bound to residues lining the active site entrance, with hydrogen bonds to Gln 276 (3.0 Å), Tyr 218 (2.8 Å) and two hydrogen bonds to Arg 114. With N3− bound in this fashion, the active site cavity can no longer accommodate SO42− [163].

The conserved Ca2+ site in cytochrome c nitrite reductase (NrfAsd) bridges two stretches of protein that host active site residues. Note the close proximity between heme 1 (SO42− bound), and a set of conserved tyrosine residues which was thought a role in radical stabilization during catalysis [162, 197]
A survey of NrfA (and variants) structures, including structures of NrfAws with H2O (PDB 1FS7), N3− (PDB 1FS9), NO2− (PDB 2E80), NH2OH (PDB 2E81), and SO32− (PDB 3BNF) bound at the active heme site, is compiled in [164]. The special feature of the active site heme 1 is its fivefold coordination, with the lysine ligand provided by the novel binding motif CXXCK, in contrast to the conventional bis-His coordination in the residual four heme centers. Early on it was suggested that the proximal lysine ligand is of crucial importance in promoting the binding of anions, and that the environment of this heme provides a reduction potential low enough to reduce the substrate NO2− to NH4+ [183]. Earlier studies on E. coli NrfA (cytochrome c552) showed that the lysine residue was required for normal rates of nitrite reduction, if it was altered to histidine, the enzyme was inactive [161]. Jörg Simon and co-workers (Darmstadt) [184] constructed the NrfAK134H variant from W. succinogenes; the specific nitrite reductase activity of the cell homogenate, the membrane fraction, as well as of the soluble fraction and the purified NrfA obtained from W. succinogenes strain K134H did not exceed 40% of that of the wild-type system.
An electron-density maximum close to the active site heme 1 was assigned to a Ca2+ ion, which could be confirmed by inductively coupled plasma atomic emission spectroscopy (1.0 ± 0.1 Ca2+ atoms/NrfA monomer). The Ca2+ion, first discovered in NrfAsd [162], is coordinated by the carboxy group of a glutamate in a bidentate manner and by glutamine, two peptide carbonyl oxygen atoms and two water molecules. The roof of the active-site cavity of NrfAsd is formed by Phe 91, Lys 279, Tyr 95, Ala 404 and Gln 281, which coordinates the Ca2+ by its carboxamide oxygen such that the amino group faces the active site (Fig. 7). The Ca2+ binding site appeared to be an essential structural feature in the overall architecture of the enzyme, and the region surrounding this site is one of the most highly conserved parts of the whole sequence. This is easily understood for Tyr 218, which is an active site residue that can directly interact with substrate. It can be rationalized that the Ca2+ ligands Lys 274 and Gln 276 immediately precede another active site residue, His 277, such that the Ca2+ ion bridges two stretches of protein that hold key residues for catalysis. Furthermore, both Lys 274 and Gln 276 take part in forming the active site cavity, whose electrostatic surface potential is presumably essential for guiding substrate influx and product efflux. Another remarkable feature close to the active site is a set of tyrosine residues, which are conserved in NrfA sequences. Tyr 219 follows directly on the active site residue Tyr 218, whose backbone carbonyl oxygen is a calcium ligand (Fig. 7).
In cytochrome c nitrite reductase from D. desulfuricans ATCC 27774, a second calcium site was discovered by Maria João Romão in collaboration with Isabel, José, and colleagues, featuring an octahedral geometry, coordinated to propionates of hemes 3 and 4, and caged by a loop absent in the NrfAsd and NrfAws structures. The highly negative electrostatic potential around hemes 3 and 4 suggests that the main role of this Ca2+ ion may be structural, namely to stabilize the conformation of the additional caging loop and to influence the solvent accessibility of heme 4 [168]. The NrfA active site is similar to that of peroxidases with a nearby Ca2+ at the heme distal side nearly in the same location as occurs in the class II and class III peroxidases. This finding suggests that the Ca2+ ion at the distal side of the active site in the NrfA enzymes may have a similar physiological role to that reported for the peroxidases [185]. On the other hand, Eric Hegg, Nicolai Lehnert, and associates characterized NrfA from bacterium G. lovleyi (NrfAgl) which had recently been recognized as one of the key drivers of DNRA in nature. The enzyme crystallized as a dimer, but dynamic light scattering characterization suggested that NrfA remained a monomer in solution even up to ≈ 300 μM. In this context one should recall that the interface of the NrfA dimer, which is dominated by three long α-helices in each monomer, only accounts to ≈ 10% of the total protein surface (Fig. 5). As a consequence, the state of NrfA in solution (monomer/dimer) is expected to vary depending on the protein source and experimental conditions [164]. Along these lines, nitrite reductase activity resided both in the soluble and the membrane fraction during protein purification, but the distribution of both pools of protein varied strongly. While in E. coli virtually all activity was found in the soluble fraction [165], in S. deleyianum and W. succinogenes, both the soluble and the membrane fractions showed nitrite reductase activity [151, 152]. In the case of D. vulgaris, however, the majority of enzyme resided in the membrane fraction, forming a stable membrane-bound complex with an overall NrfA4NrfH2 quaternary arrangement [170].
Notably, the crystal structure of NrfAgl (2.55 Å) revealed the presence of an arginine residue in the region otherwise occupied by the Ca2+ ion in canonical NrfA enzymes, such as NrfAsd or NrfAws. The presence of chelating agent EDTA did not affect the activity of NrfAgl, and site-directed mutagenesis of this arginine reduced enzymatic activity siginificantly. Furthermore, phylogenetic analysis revealed four separate emergences of Arg-containing NrfA enzymes. Thus, the Ca2+-independent, Arg-containing NrfAgl represents a new subclass of pentaheme cytochrome c nitrite reductase [172, 173].
Reaction mechanism of pentaheme cytochrome c nitrite reductase
Biological redox reactions classically involve the transfer of electrons one or two at a time. A limited group of reactions can be classified as multi-electron reductions, in that more than two electrons—in fact, as many as six—are transferred to an enzyme-bound substrate before product release from the site of reduction [186, 187]. Multi-electron reduction reactions occupy crucial positions in metabolism, being involved in the utilization of dioxygen as terminal electron acceptor by all organisms capable of aerobic metabolism, as well as in the biological conversion of inorganic sulfur and nitrogen compounds, e.g., SO32− and NO2−, for assimilatory (biosynthesis) and dissimilatory (energy conservation) purposes. For each of these reactions, enzymes exist that can catalyse the entire multi-electron transfer process without release of inorganic compounds of oxidation states intermediate between substrate and product. Furthermore, such novel types of redox reactions will be most likely associated with novel types of complex enzyme prosthetic groups. The enzymatic reductions of oxyanions SO32− and NO2− occur as part of two different physiological processes, (1) in plants, fungi, and many bacteria, the reductions of SO32− to H2S/S2− and NO2− to NH3/NH4+ are intermediate steps in the assimilation of sulfate (SO42−) and nitrate (NO3−), respectively, for the synthesis of S and N-containing cellular constituents, and (2) in microorganisms, the reductions of SO32− and NO2− are large-scale processes associated with anaerobic respiration utilizing SO42− and NO3− as terminal electron acceptors [111, 186, 187].
The six-electron reductions of nitrite to ammonia/ammonium and sulfite to hydrogen sulfide/sulfide (Eqs. 1, 2) are fundamental to early and contemporary life. These multi-electron, multi-proton transfer processes are catalysed by a group of diverse nitrite and sulfite reductases that provide a unique prosthetic group assembly in their active centers with structural features that are key for the catalytic mechanism [39, 46, 188,189,190,191,192]. Cytochrome c nitrite reductase (NrfA) catalyses the reduction of NO2− to NH4+ with high specific activity (Eq. 1), without the release of bound intermediates NO and NH2OH as discussed below (Fig. 8). Electrons usually are delivered from the membranous quinone pool, thereby generating a proton motive force [34].

Proposed reaction scheme for the six-electron reduction of nitrite to ammonia catalysed by cytochrome c nitrite reductase. When started from the water-bound resting state in either the oxidized (FeIII) or reduced (FeII) form, the binding of NO2− starts the reaction cycle, which, after a heterolytic cleavage of the first N–O bond, proceeds through two one-electron reductions and a protonation step to Fe(II)-HNO, which is readily reduced by two electrons to Fe(II)-H2NOH. A further reduction leads to the dissociation of the second water molecule, and subsequently, after the last reduction step, the product ammonia can dissociate [193]. Notably, N2O—in contrast to NO and NH2OH not an intermediate in the proposed cycle—is a substrate of NrfA, however with low activity (Fig. 4), and the reaction product has not been identified so far [156]
In addition to NO2−, NrfA converts NH2OH and NO to NH4+ (Fig. 4) [156], furthermore, the NO reductase activity of NrfA has been later shown to play an important role within the oxidative and nitrosative stress defense network of bacteria such as E. coli and W. succinogenes [54, 55]. Note that NH2OH and methyl derivatives reacted poorly with Fe(III)-NrfA, on the other hand, NH2NH2 acted as reductant. Photochemically reduced NrfA (using 5-deaza-10-methyl-3-sulfopropyl-isoalloxazine/ oxalate) was rapidly oxidized by NH2OH and derivatives in solution, but not by NH2NH2 [157]. Attempts to produce crystalline Fe(II)-NrfAsd from oxidized Fe(III) crystals, by reaction with either Na+ dithionite, or dihydrogen in the presence of traces of [Ni,Fe] hydrogenase from S. deleyianum [109], failed as the crystals began to disintegrate [156].
On the basis of crystallographic observations of the Fe(III)-NO2− adduct and potential reaction intermediates, and of density functional calculations, a first working hypothesis for the reaction mechanism of NrfA, which hosts the novel Lys coordinated heme group (Fe-Lys), was developed by Oliver Einsle and Frank Neese (Fig. 8) [193]. Reduction of NO2− started with the heterolytic cleavage of the N–O bond, facilitated by a pronounced back-bonding interaction of NO2− coordinated through nitrogen to the reduced Fe(II) but not the oxidized Fe(III) of the active heme site. This step led to the formation of an {FeNO}6 species and a water molecule and was further facilitated by a hydrogen bonding network that induced an electronic asymmetry in the NO2− molecule that weakened one N–O bond and strengthened the other. Subsequently, two rapid one-electron reductions led to an {FeNO}8 form and, by protonation, to an Fe(II)-HNO adduct. Hereafter, NH2OH, formed by a consecutive two-electron two-proton step, was dehydrated in the final two-electron reduction step to give NH3 and H2O. A single electron reduction of the active site closes the catalytic cycle [193]. In a set of consecutive theoretical studies, the NO2− reduction mechanism was analysed in greater detail by Dimytro Bykov and Frank Neese (Max-Planck Institut für Chemische Energiekonversion, Mülheim) [194,195,196,197]. The mechanism comprises five functional stages. In phase 1, the substrate binds via its N atom to the active site heme 1 followed by N−O bond cleavage, with His 277 acting as the proton donor. In this step, the N−O bond of NO2− is cleaved heterolytically through double protonation of the substrate. The second phase consists of two proton coupled electron-transfer events, leading to the HNO intermediate. Phase 3 involves the formation of NH2OH, where Arg 114 provides the necessary proton for the reaction. The second N−O bond is cleaved in phase 4 of the mechanism, again triggered by proton transfer from His 277. The Tyr 218 side chain governs the fifth and last phase of the mechanism, it consists of radical transfer and ammonia formation. In other words, this mechanism implies that all conserved active-site side chains work in a concerted way to achieve this complex chemical transformation from nitrite to ammonia. Interestingly, evidence for the active role of residue Tyr 218 was provided by earlier studies on the sulfite reductase activity of NrfAws and the active site variant Tyr218Phe. This NrfA variant exhibited an almost complete loss of nitrite reductase activity, while sulfite reduction remained unaffected [60]. According to the theoretical studies by Bykov and Neese, an intramolecular reaction with Tyr 218 in the final step of the nitrite reduction process led directly to the final product, NH3. Dissociation of the final product proceeds concomitantly with a change in heme Fe spin state [171, 197, 198].
Last but not least, recent reports surrounding the bioelectrochemical communication of enzymes, such as nitrogenase, nitrate reductase, or cytochrome c nitrite reductase, at electrode surfaces have demonstrated the ability to probe enzymatic mechanisms, to produce NH3 from a range of sources (e.g., NO3−, NO2−, N3−), and to detect biologically important N,O compounds, (e.g., NO2−, NO). Additionally, coupling of artificial cascade reactions have been utilized if a single catalyst is incapable of the complete substrate reduction to produce NH3 [199]. Electrochemical combined with spectroscopic techniques (UV/Vis, EPR, MCD) and site-directed mutagenesis, have been successfully applied to investigate multiheme enzymes by Julea Butt (Norwich), Sean Elliott (Boston), Kyle Lancaster (Ithaca), Andrew Pacheco (Milwaukee), and of course by Isabel, José Moura and their associates (Lisbon). The elegant studies by these leading researchers brought significant advances to our knowledge about important multi-electron, multi-proton transfer processes in biological systems [153, 166, 167, 173, 181, 200,201,202,203,204,205,206,207,208,209,210,211,212].
Outlook and conclusions
Despite several decades of intensive research since the report by Fujita on soluble cytochromes in Enterobacteriaceae and the characterization of cytochrome c552 as a hexaheme nitrite reductase [63,64,65], there remain many unresolved issues concerning Nature’s nitrite to ammonia reductases. Clearly, cytochrome c nitrite reductase has become a mature field. The basic catalytic mechanism is understood at the atomic level in the context of electronic changes leading to NO2− activation and its reduction to NH3/NH4+. Their catalytic versatility makes multiheme proteins and enzymes particularly valuable for numerous applications. Bioelectrochemical technology combined with various biophysical methods provide a new and powerful tool for a broad spectrum of biochemical and biophysical applications. Important structural and mechanistic information on multiheme proteins including extracellular electron transfer can be obtained by this technology, ranging from quaternary information, protein–protein and protein–surface recognition to highly resolved molecular pictures. In summary, c-type cytochromes will continue to be an important field of research. Determining and understanding their reaction mechanisms have greatly advanced a variety of fields and our understanding of activation of inorganic nitrogen and sulfur compounds as well as multi-electron and multi-proton transfer. Molecular dynamics simulations and quantum–mechanical/molecular-mechanical calculations will complement experiments and elucidate the choreography by which the cytochrome protein regulates the catalytic cycle [213,214,215,216,217,218,219,220].
Perhaps the exciting feature of the bacterial enzyme cytochrome c nitrite reductase (NrfA), first proposed by Jeff Cole and co-workers in 1998 on the basis of sequence studies for the nitrite reductase of E. coli, was the novel heme binding motif Cys-X-X-Cys-Lys (CXXCK) [161]. One year later, Oliver Einsle and associates presented the first high resolution NrfA structure of sulfur-reducing bacterium S. deleyianum, a homodimer with five covalently bound heme centers in each monomer, four of them carrying the conventional bis-His coordination (Cys-X-X-Cys-His motif), and the active site heme carrying the novel Cys-X-X-Cys-Lys motif, with a catalytically important Ca2+ cation above the distal side of the heme plane [162]. Today, we know from extensive investigations of the diversity and phylogeny of the NrfA enzyme including analysis of full-length NrfA protein sequences that there exist several clades carrying the conventional Cys-X-X-Cys-His motif in the first heme-binding domain [174,175,176], yet, detailed structural information on these enzymes by X-ray crystallography still has to come. The presence of the distal Lys residue was thought to be of crucial importance in promoting the binding of anions, and the design of the catalytic cavity around heme 1 was proposed to provide a reduction potential low enough for the reduction of nitrite to ammonium [183]. There exist naturally occurring heme proteins with Lys axially coordinated to Fe, such as the alkaline form of cytochrome c [221, 222], or the truncated hemoglobin THB1 of Chlamydomonas reinhardtii [223]. Furthermore, a Met100Lys variant of cytochrome c550 from Paracoccus versutus was produced, leading to a shift of the midpoint potential by − 329 mV compared to wild type [224, 225].
The His93Gly myoglobin cavity mutant was introduced by John Dawson and co-workers (University of South Carolina) as a valuable model system to investigate for endogenous Lys and terminal amine ligation in heme proteins. Replacement of proximal ligand His93 with the much smaller non-coordinating Gly residue left a cavity on the proximal side of the heme into which a wide variety of exogenous ligands could be delivered. The scaffold provided a remarkably versatile template for the preparation of model heme complexes and was used to mimic the heme iron coordination structure of native heme proteins, such as NrfA (Fe-NLys), the CooA transcription factor (Fe-NPro), or cytochrome f (Fe-NTyr) [226, 227]. Along these lines, Nicholas Watmough and associates (University of East Anglia) created the His93Lys variant of sperm whale myoglobin and studied its binding and reactivity with nitrite [228]. Substitution of the proximal His ligand with Lys led to an eightfold increase in the rate of NO2− → NO reduction relative to wild-type myoglobin. The binding of NO2− via the oxygen atom (O-nitrito mode) to the Fe(III) heme was retained in His93Lys myoglobin. Nitrite can coordinate to heme iron through either nitrogen (N-nitrito mode), e.g., in cytochrome c nitrite reductase or in assimilatory and dissimilatory sulfite reductase, or through oxygen as observed for myoglobin and hemoglobin. This is known as linkage isomerism [229, 230]. Based on site-directed mutagenesis studies, it was argued that the His residue on the distal heme side modulates this unique O-nitrito binding mode. This suggestion received strong support by the computation of spin Hamiltonian EPR parameters of different linkage isomers of NO2− bound myoglobin using wave function based “ab initio” and density functional theories [231].
Finally, the work by Tamara Tikhonova and colleagues (Russian Academy of Sciences) on related octaheme nitrite reductases should be mentioned here [232,233,234,235]. The first representative of this family of octaheme cytochrome c nitrite reductases was isolated from the haloalkaliphilic bacterium Thioalkalivibrio nitratireducens. The enzyme converted nitrite and hydroxylamine to ammonia without release of intermediates with high activity, as well as sulfite to sulfide. In solution, it exists as a stable hexamer, each subunit contains eight c-type hemes, seven of them are coordinated by the conventional CXXCH motif, while one, like in pentaheme NrfA, is bonded by the unique CXXCK motif.
In summary, the bioinorganic chemistry of multiheme c-type cytochromes is a vibrant field and will remain in the focus of active research in chemistry, biochemistry, microbiology and geochemistry, as impressively documented by discoveries of new enzymes with astounding chemical activities and novel active sites. Notably, the field of geomicrobiology has experienced an extraordinary growth in recent years, and microbes have been studied in all kinds of environments on Earth [113, 236]. In the context of c-type cytochromes, Geobacter bacteria are of high interest in the bioremediation and bioenergy fields, due to their ability to produce high current densities in microbial fuel cells, consequently, they are interesting targets for bioenergy applications. Closely related to these physiological features is the ability of Geobacter cells to produce conductive protein nanowires, a property that is currently being explored in the bionanotechnology field [122, 237].
Abbreviations
- DSIR:
-
Dissimilatory sulfite reductase
- EPR:
-
Electron paramagnetic resonance
- ET:
-
Electron transfer
- DNRA:
-
Dissimilatory nitrate reduction to ammonium
- HAO:
-
Octaheme hydroxylamine oxidoreductase
- HDH:
-
Hydrazine dehydrogenase
- MCD:
-
Magnetic circular dichroism
- NMR:
-
Nuclear magnetic resonance
- NrfA:
-
Pentaheme cytochrome c nitrite reductase
- NrfAec:
-
Nitrite reductase of Eschericia coli
- NrfAgl:
-
Nitrite reductase of Geobacter lovleyi
- NrfAsd:
-
Nitrite reductase of Sulfurospirillum deleyianum
- NrfAws:
-
Nitrite reductase of Wolinella succinogenes
- PDB:
-
Protein data bank; https://www.rcsb.org
- UV/Vis:
-
Ultraviolet/visible
References
Huynh BH, Liu M-C, Moura JJG, Moura I, Ljungdahl PO, Münck E, Payne WJ, Peck HD Jr, DerVartanian DV, LeGall J (1982) Mössbauer and EPR studies on nitrite reductase from Thiobacillus denitrificans. J Biol Chem 257:9576–9581. https://doi.org/10.1016/S0021-9258(18)34110-3
Moura I, Fauque G, Le Gall J, Xavier AV, Moura JJG (1987) Characterization of the cytochrome system of a nitrogen-fixing strain of a sulfate-reducing bacterium: Desulfovibrio desulfuricans strain Berre-Eau. Eur J Biochem 162:547–554. https://doi.org/10.1111/j.1432-1033.1987.tb10674.x
Liu M-C, Costa C, Coutinho IB, Moura JJG, Moura I, Xavier AV, Le Gall J (1988) Cytochrome components of nitrate- and sulfate-respiring Desulfovibrio desulfuricans ATCC 27774. J Bact 170:5545–5551. https://doi.org/10.1128/jb.170.12.5545-5551.1988
Fauque G, Peck HD Jr, Moura JJG, Huynh BH, Berlier Y, DerVartanian DV, Teixeira M, Przybyla AE, Lespinat PA, Moura I, Le Gall J (1988) The three classes of hydrogenases from sulfate-reducing bacteria of the genus Desulfovibrio. FEMS Microbiol Rev 54:299–344. https://doi.org/10.1111/j.1574-6968.1988.tb02748.x
Moura JJG, Costa C, Liu M-Y, Moura I, Le Gall J (1991) Structural and functional approach toward a classification of the complex cytochrome c system found in sulfate-reducing bacteria. Biochim Biophys Acta 1058:61–66. https://doi.org/10.1016/S0005-2728(05)80270-1
Moreno C, Costa C, Moura I, Le Gall J, Liu M-Y, Payne WJ, van Dijk C, Moura JJG (1993) Electrochemical studies of the hexaheme nitrite reductase from Desulfovibrio desulfuricans ATCC 27774. Eur J Biochem 212:79–86. https://doi.org/10.1111/j.1432-1033.1993.tb17635.x
Romão MJ, Archer M, Moura I, Moura JJG, Le Gall J, Engh R, Schneider M, Hof P, Huber R (1995) Crystal structure of the xanthine oxidase related aldehyde oxido-reductase from D. gigas. Science 270:1170–1176. https://doi.org/10.1126/science.270.5239.1170
Moura I, Bursakov SA, Costa C, Moura JJG (1997) Nitrate and nitrite utilization in sulfate-reducing bacteria. Anaerobe 3:279–290. https://doi.org/10.1006/anae.1997.0093
Brown K, Tegoni M, Prudêncio M, Pereira AS, Besson S, Moura JJG, Moura I, Cambillau C (2000) A novel type of catalytic copper cluster in nitrous oxide reductase. Nat Struct Biol 7:191–195. https://doi.org/10.1038/73288
Moura I, Moura JJG (2001) Structural aspects of denitrifying enzymes. Curr Opin Chem Biol 2:168–175. https://doi.org/10.1016/S1367-5931(00)00187-3
Pauleta SR, Carepo MSP, Moura I (2019) Source and reduction of nitrous oxide. Coord Chem Rev 387:436–449. https://doi.org/10.1016/j.ccr.2019.02.005
Brown K, Djinovic-Carugo K, Haltia T, Cabrito I, Saraste M, Moura JJG, Moura I, Tegoni M, Cambillau C (2000) Revisiting the catalytic CuZ cluster of nitrous oxide (N2O) reductase. Evidence of a bridging inorganic sulfur. J Biol Chem 275:41133–41136. https://doi.org/10.1074/jbc.M008617200
Chen P, De Beer GS, Cabrito I, Antholine WE, Moura JJG, Moura I, Hedman B, Hodgson KO, Solomon EI (2002) Electronic structure description of the μ4-sulfide bridged tetranuclear CuZ center in N2O reductase. J Am Chem Soc 124:744–745. https://doi.org/10.1021/ja0169623
Johnston EM, Dell’Acqua S, Ramos S, Pauleta SR, Moura I, Solomon EI (2014) Determination of the active form of the tetranuclear copper sulfur cluster in nitrous oxide reductase. J Am Chem Soc 136:614–617. https://doi.org/10.1021/ja411500p
Johnston EM, Carreira C, Dell’Acqua S, Dey SG, Pauleta SR, Moura I, Solomon EI (2017) Spectroscopic definition of the CuZ° intermediate in turnover of nitrous oxide reductase and molecular insight into the catalytic mechanism. J Am Chem Soc 139:4462–4476. https://doi.org/10.1021/jacs.6b13225
Pauleta SR, Carepo MSP, Moura I (2020) The tetranuclear copper-sulfide center of nitrous oxide reductase. Met Ions Life Sci 20:139–164. https://doi.org/10.1515/9783110589757-005
Dickerson RE (1980) Cytochrome c and the evolution of energy metabolism. Sci Am 242:136–153
Ambler RP (1982) The structure and classification of cytochrome c. In: Kaplan NO, Robinson A (eds) From cyclotrons to cytochromes. Essays in molecular biology and chemistry. Academic Press, London, pp 263–279
Pettigrew GW, Moore GR (1987) Resolution, characterisation and classification of c-type cytochromes. In: Cytochromes c. Springer series in molecular biology. Springer, Berlin, pp 1–28. https://doi.org/10.1007/978-3-642-72698-9_1
Bushnell GW, Louie GV, Brayer GD (1990) High-resolution three-dimensional structure of horse heart cytochrome c. J Mol Biol 214:585–595. https://doi.org/10.1016/0022-2836(90)90200-6
Chapman SK, Daft S, Munro AW (1997) Heme: the most versatile redox centre in biology? Struct Bond 88:39–70. https://doi.org/10.1007/3-540-62870-3_2
Barker PD, Ferguson SJ (1999) Still a puzzle: why is haem covalently attached in c-type cytochromes? Structure 7:R281–R290. https://doi.org/10.1016/S0969-2126(00)88334-3
Ferguson SJ (2001) Keilin’s cytochromes: how bacteria use them, vary them and make them. Biochem Soc Trans 29:629–640. https://doi.org/10.1042/bst0290629
Allen JWA, Daltrop O, Stevens JM, Ferguson SJ (2003) C-type cytochromes: diverse structures and biogenesis systems pose evolutionary problems. Phil Trans R Soc Lond B 358:255–266. https://doi.org/10.1098/rstb.2002.1192
Mowat CG, Chapman SK (2005) Multi-heme cytochromes—new structures, new chemistry. Dalton Trans. https://doi.org/10.1039/B505184C
Louro RO (2007) Proton thrusters: overview of the structural and functional features of soluble tetrahaem cytochromes c3. J Biol Inorg Chem 12:1–10. https://doi.org/10.1007/s00775-006-0165-y
Ascenzi P, Santucci R, Coletta M, Polticelli F (2010) Cytochromes: Reactivity of the “dark side” of the heme. Biophys Chem 152:21–27. https://doi.org/10.1016/j.bpc.2010.09.008
Simon J, Kern M, Hermann B, Einsle O, Butt JN (2011) Physiological function and catalytic versatility of bacterial multihaem cytochromes c involved in nitrogen and sulfur cycling. Biochem Soc Trans 39:1864–1870. https://doi.org/10.1042/BST20110713
Romão CV, Archer M, Lobo SA, Louro RO, Pereira IAC, Saraiva LM, Teixeira M, Matias PM (2012) Diversity of heme proteins in sulfate reducing bacteria, handbook of porphyrin science, with applications to chemistry, physics, materials science, engineering, biology and medicine. Volume 19: biochemistry of tetrapyrroles (Part II), pp 139–229. https://doi.org/10.1142/9789814335508_0016
Bewley KD, Ellis KE, Firer-Sherwood MA, Elliott SJ (2013) Multi-heme proteins: nature’s electronic multi-purpose tool. Biochim Biophys Acta 1827:938–948. https://doi.org/10.1016/j.bbabio.2013.03.010
Salgueiro CA, Dantas JM (2016) Multiheme cytochromes. Springer, Berlin. https://doi.org/10.1007/978-3-642-44961-1
Kleingardner JG, Bren KL (2015) Biological significance and applications of heme c proteins and peptides. Acc Chem Res 48:1845–1852. https://doi.org/10.1021/acs.accounts.5b00106
Bren KL (2016) Going with the electron flow: heme electronic structure and electron transfer in cytochrome c. Isr J Chem 56:693–704. https://doi.org/10.1002/ijch.201600021D
Raanan H, Pike DH, Moore EK, Falkowski PG, Nanda V (2018) Modular origins of biological electron transfer chains. Proc Natl Acad Sci USA 115:1280–1285. https://doi.org/10.1073/pnas.1714225115
Paquete CM, Rusconi G, Silva AV, Soares R, Louro RO (2019) A brief survey of the “cytochromome.” Adv Microb Physiol 75:69–135. https://doi.org/10.1016/bs.ampbs.2019.07.005
Gruber N, Galloway JN (2008) An earth-system perspective of the global nitrogen cycle. Nature 451:293–296. https://doi.org/10.1038/nature06592
Canfield DE, Glazer AN, Falkowski PG (2010) The evolution and future of earth’s nitrogen cycle. Science 330:192–196. https://doi.org/10.1126/science.1186120
Simon J, Klotz MG (2013) Diversity and evolution of bioenergetic systems involved in microbial nitrogen compound transformations. Biochim Biophys Acta 1827:114–135. https://doi.org/10.1016/j.bbabio.2012.07.005
Godfrey LV, Glass JB (2011) The geochemical record of the ancient nitrogen cycle, nitrogen isotopes, and metal cofactors. Meth Enzymol 486:483–506. https://doi.org/10.1016/B978-0-12-381294-0.00022-5
Ward BB, Jensen MM (2014) The microbial nitrogen cycle. Front Microbiol. https://doi.org/10.3389/fmicb.2014.00553
Stein LY, Klotz MG (2016) The nitrogen cycle. Curr Biol 26:R83–R101. https://doi.org/10.1016/j.cub.2015.12.021
Lehtovirta-Morley LE (2018) Ammonia oxidation: ecology, physiology, biochemistry and why they must all come together. FEMS Microbiol Lett 365:fny058. https://doi.org/10.1093/femsle/fny058
Lehnert N, Dong HT, Harland JB, Hunt AP, White CJ (2018) Reversing nitrogen fixation. Nat Rev Chem 2:278–289. https://doi.org/10.1038/s41570-018-0041-7
Zhang X, Ward BB, Sigman DM (2020) Global nitrogen cycle: critical enzymes, organisms, and processes for nitrogen budgets and dynamics. Chem Rev 120:5308–5351. https://doi.org/10.1021/acs.chemrev.9b00613
Ferousi C, Majer SH, DiMucci IM, Lancaster KM (2020) Biological and bioinspired inorganic N−N bond-forming reactions. Chem Rev 120:5252–5307. https://doi.org/10.1021/acs.chemrev.9b00629
Lehnert N, Musselman BW, Seefeldt LC (2021) Grand challenges in the nitrogen cycle. Chem Soc Rev 50:3640–3646. https://doi.org/10.1039/D0CS00923G
Cole JA, Brown CM (1980) Nitrite reduction to ammonia by fermentative bacteria: a short circuit in the biological nitrogen cycle. FEMS Microbiol Lett 7:65–72. https://doi.org/10.1016/S0378-1097(80)80001-2
Berks BC, Ferguson SJ, Moir JWB, Richardson DJ (1995) Enzymes and associated electron transport systems that catalyse the respiratory reduction of nitrogen oxides and oxyanions. Biochim Biophys Acta 1232:97–173. https://doi.org/10.1016/0005-2728(95)00092-5
Cole JA (1996) Nitrate reduction to ammonia by enteric bacteria: redundancy, or a strategy for survival during oxygen starvation? FEMS Microbiol Lett 136:1–11. https://doi.org/10.1111/j.1574-6968.1996.tb08017.x
Simon J (2002) Enzymology and bioenergetics of respiratory nitrite ammonification. FEMS Microbiol Rev 26:285–309. https://doi.org/10.1111/j.1574-6976.2002.tb00616.x
Einsle O, Kroneck PMH (2004) Structural basis of denitrification. Biol Chem 385:875–883. https://doi.org/10.1515/BC.2004.115
Lam P, Kuypers MMM (2011) Microbial nitrogen cycling processes in oxygen minimum zones. Annu Rev Mar Sci 3:317–345. https://doi.org/10.1146/annurev-marine-120709-142814
Thamdrup B (2012) New pathways and processes in the global nitrogen cycle. Annu Rev Ecol Evol Syst 43:407–428. https://doi.org/10.1146/annurev-ecolsys-102710-145048
Maia LB, Moura JJG (2014) How biology handles. Nitrite Chem Rev 114:5273–5357. https://doi.org/10.1021/cr400518y
Simon J, Kroneck PMH (2014) The production of ammonia by multiheme cytochromes c. Met Ions Life Sci 14:211–236. https://doi.org/10.1007/978-94-017-9269-1_9
Pandey B, Kumar U, Kaviraj UM, Minick KJ, Mishra AK, Singh JS (2020) DNRA: a short-circuit in biological N-cycling to conserve nitrogen in terrestrial ecosystems. Sci Total Environ 738:139710. https://doi.org/10.1016/j.scitotenv.2020.139710
Zhu J, He Y, Zhu Y, Huang M, Zhang Y (2018) Biogeochemical sulfur cycling coupling with dissimilatory nitrate reduction processes in freshwater sediments. Environ Rev 26:121–132. https://doi.org/10.1139/er-2017-0047
Pereira IC, Abreu IA, Xavier AV, Le Gall J, Teixeira M (1996) Nitrite reductase from Desulfovibrio desulfuricans (ATCC 27774), a heterooligomer heme protein with sulfite reductase activity. Biochem Biophys Res Commun 224:611–618. https://doi.org/10.1006/bbrc.1996.1074
Fritz G, Einsle O, Rudolf M, Schiffer A, Kroneck PMH (2005) Key bacterial multi-centered metal enzymes involved in nitrate and sulfate respiration. J Mol Microbiol Biotechnol 10:223–233. https://doi.org/10.1159/000091567
Lukat P, Rudolf M, Stach P, Messerschmidt A, Kroneck PMH, Simon J, Einsle O (2008) Binding and reduction of sulfite by cytochrome c nitrite reductase. Biochemistry 47:2080–2086. https://doi.org/10.1021/bi7021415
Kemp GL, Clarke TA, Marritt SJ, Lockwood C, Poock SR, Hemmings AM, Richardson DJ, Cheesman MR, Butt JN (2010) Kinetic and thermodynamic resolution of the interactions between sulphite and the pentahaem cytochrome NrfA from Escherichia coli. Biochem J 431:73–80. https://doi.org/10.1042/BJ20100866
Simon J, Kroneck PMH (2013) Microbial Sulfite Respiration. Adv Microb Physiol 62:45–117. https://doi.org/10.1016/B978-0-12-410515-7.00002-0
Fujita T (1966) Studies on soluble cytochromes in Enterobacteriaceae: I. Detection, purification, and properties of cytochrome c-552 in anaerobically grown cells. J Biochem 60:204–215. https://doi.org/10.1093/oxfordjournals.jbchem.a128420
Abou-Jaoudé A, Pascal M-C, Chippaux M (1979) Formate-nitrite reduction in Escherichia coli K12 2. Identification of components involved in the electron transfer. Eur J Biochem 95:315–321. https://doi.org/10.1111/j.1432-1033.1979.tb12967.x
Kajie S-I, Anraku Y (1986) Purification of a hexaheme cytochrome c 552 from Escherichia coli K 12 and its properties as a nitrite reductase. Eur J Biochem 154:457–463. https://doi.org/10.1111/j.1432-1033.1986.tb09419.x
Beinert H, Massey V (1982) Peter Hemmerich, 1929–1981. Trends Biochem Sci 7:43–44. https://doi.org/10.1016/0968-0004(82)90069-X
Holm RH, Kennepohl P, Solomon EI (1996) Structural and functional aspects of metal sites in biology. Chem Rev 96:2239–2314. https://doi.org/10.1021/cr9500390
Selverstone Valentine J, O’Halloran TV (1999) Bio-inorganic chemistry: what is it, and what’s so exciting ? Curr Op Chem Biol 3:129-l30. https://doi.org/10.1016/S1367-5931(99)80023-4
Beinert H (2002) Bioinorganic chemistry: a new field or discipline ? Words, meanings, and reality. J Biol Chem 277:37967–37972. https://doi.org/10.1074/jbc.X200002200
Gray HB (2003) Biological inorganic chemistry at the beginning of the 21st century. Proc Natl Acad Sci USA 100:3563–3568. https://doi.org/10.1073/pnas.0730378100
Holm RH, Solomon EI (2004) Biomimetic inorganic chemistry. Chem Rev 104:347–348. https://doi.org/10.1021/cr0206364
Lippard SJ (2006) The inorganic side of chemical biology. Nat Chem Biol 2:504–507. https://doi.org/10.1038/nchembio1006-504
Crichton B, Hagen F (2018) Professor Cees Veeger and the early years of bioinorganic chemistry. FEBS J 285:1560–1562. https://doi.org/10.1111/febs.14470
Reedijk J (2020) Fifty years of inorganic biochemistry: developments, trends, highlights, impact and citations. J Inorg Biochem 212:111230. https://doi.org/10.1016/j.jinorgbio.2020.111230
Marchesini A, Kroneck PMH (1979) Ascorbate oxidase from Cucurbita pepo medullosa. New method of purification and reinvestigation of properties. Eur J Biochem 101:65–76. https://doi.org/10.1111/j.1432-1033.1979.tb04217.x
Marchesini A, Minelli M, Merkle H, Kroneck PMH (1979) Mavicyanin, a blue copper protein from Cucurbita pepo medullosa. Purification and characterization. Eur J Biochem 101:77–84. https://doi.org/10.1111/j.1432-1033.1979.tb04218.x
Kroneck PMH, Armstrong FA, Merkle H, Marchesini A (1982) Ascorbate oxidase: molecular properties and catalytic activity. In: Seib PA, Tolbert BM (eds) Ascorbic acid: chemistry, metabolism, and uses. Advances in chemistry, vol 200. American Chemical Society, pp 223–248. .https://doi.org/10.1021/ba-1982-0200.ch010
Gray HB, Banci L, Luchinat C, Turano P (2012) Ivano Bertini 1940–2012. Nat Struct Mol Biol 19:868–869. https://doi.org/10.1038/nsmb.2369
Santos H (2012) António Xavier and his contribution to the development of bioinorganic chemistry. FEBS Lett 586:476–478. https://doi.org/10.1016/j.febslet.2012.02.007
Ambler RP, Bruschi M, Le Gall J (1969) The structure of cytochrome c’3 from Desulfovibrio gigas (NCIB 9332). FEBS Lett 5:115–117. https://doi.org/10.1016/0014-5793(69)80308-X
Haser R, Pierrot M, Frey M, Payan F, Astier JP, Bruschi M, Le Gall J (1979) Structure and sequence of the multihaem cytochrome c3. Nature 282:806–810. https://doi.org/10.1038/282806a0
Matias PM, Morais J, Coelho R, Carrondo MA, Wilson K, Dauter Z, Sieker L (1996) Cytochrome c3 from Desulfovibrio gigas: crystal structure at 1.8 Å resolution and evidence for a specific calcium-binding site. Prot Sci 5:1342–1354. https://doi.org/10.1002/pro.5560050713
Coutinho IB, Turner DL, Le Gall J, Xavier AV (1992) Revision of the haem-core architecture in the tetraheam cytochrome c3 from Desulvovibrio baculatus by two-dimensional 1H NMR. Eur J Biochem 209:329–333. https://doi.org/10.1111/j.1432-1033.1992.tb17293.x
Bruschi M, Leroy G, Bonicel J, Campese D, Dolla A (1996) The cytochrome c3 superfamily : amino acid sequence of a dimeric octahaem cytochrome c3 (Mr 26000) isolated from Desulfovibrio gigas. Biochem J 320:933–938. https://doi.org/10.1042/bj3200933
Coutinho IB, Turner DL, Liu M-Y, Le Gall J, Xavier AV (1996) Structure of the three-haem core of cytochrome c551.5 determined by 1H NMR. J Biol Inorg Chem 1:305–311. https://doi.org/10.1007/s007750050058
Fonseca BM, Paquete CM, Salgueiro CA, Louro RO (2012) The role of intramolecular interactions in the functional control of multiheme cytochromes c. FEBS Lett 586:504–509. https://doi.org/10.1016/j.febslet.2011.08.019
Steuber J, Cypionka H, Kroneck PMH (1994) Mechanism of dissimilatory sulfite reduction by Desulfovibrio desulfuricans: purification of a membrane-bound sulfite reductase and coupling with cytochrome c3 and hydrogenase. Arch Microbiol 162:255–260. https://doi.org/10.1007/BF00301847
Einsle O, Foerster S, Mann K, Fritz G, Messerschmidt A, Kroneck PMH (2001) Spectroscopic investigation and determination of reactivity and structure of the tetraheme cytochrome c3 from Desulfovibrio desulfuricans Essex 6. Eur J Biochem 268:3028–3035. https://doi.org/10.1046/j.1432-1327.2001.02195.x
Umhau S, Fritz G, Diederichs K, Breed J, Welte W, Kroneck PMH (2001) Three-dimensional structure of the nonaheme cytochrome c from Desulfovibrio desulfuricans Essex in the Fe(III) State at 1.89 Å resolution. Biochemistry 40:1308–1316. https://doi.org/10.1021/bi001479a
Fritz G, Griesshaber D, Seth O, Kroneck PMH (2001) Nonaheme cytochrome c, a new physiological electron acceptor for [Ni, Fe] hydrogenase in the sulfate-reducing bacterium Desulfovibrio desulfuricans Essex: primary sequence, molecular parameters, and redox properties. Biochemistry 40:1317–1324. https://doi.org/10.1021/bi001480+
Ma J-G, Zhang J, Franco Jia S-L, Moura I, Moura JJG, Kroneck PMH, Shelnutt JA (1998) The structural origin of nonplanar heme distortions in tetraheme ferricytochromes c3. Biochemistry 37:12431–12442. https://doi.org/10.1021/bi981189i
https://www.bmb.uga.edu/history. Accessed 30 Nov 2021
Mccormack F (1998) The RED & BLACK. https://www.redandblack.com/news/professor-retired-but-still-experimenting-with-success/article_a36f617e-f7b4-539c-a10d-b7ad39f5f05d.html. Accessed 30 Nov 2021
Peck HD Jr, Le Gall J (1994) Inorganic microbial sulfur metabolism. Methods in enzymology, vol 243. Academic Press, San Diego
Sigal N, Senez JC, Le Gall J, Sebald M (1963) Base composition of the deoxyribonucleic acid of sulfate reducing bacteria. J Bacteriol 85:1315–1318. https://doi.org/10.1128/JB.85.6.1315-1318.1963
Probst I, Bruschi M, Pfennig N, Le Gall J (1977) Cytochrome c-551.5 (c7) from Desulfuromonas acetoxidans. Biochim Biophys Acta 460:58–64. https://doi.org/10.1016/0005-2728(77)90151-7
Cookson DJ, Moore GR, Pitt RC, Williams RJP, Campbell ID, Ambler RP, Bruschi M, Le Gall J (1978) Structural homology of cytochromes c. Eur J Biochem 83:261–275. https://doi.org/10.1111/j.1432-1033.1978.tb12091.x
Fauque G, Herve D, Le Gall J (1979) Structure-function relationship in hemoproteins: the role of cytochrome c3 in the reduction of colloidal sulfur by sulfate-reducing bacteria. Arch Microbiol 121:261–264. https://doi.org/10.1007/BF00425065
Le Gall J, Peck HD Jr (1987) Amino-terminal amino acid sequences of electron transfer proteins from Gram-negative bacteria as indicators of their cellular localization: the sulfate-reducing bacteria. FEMS Microbiol Rev 46:35–40. https://doi.org/10.1016/0378-1097(87)90184-4
Saint-Martin P, Lespinat PA, Fauque G, Berlier Y, Le Gall J, Moura I, Teixeira M, Xavier AV, Moura JJG (1988) Hydrogen production and deuterium-proton exchange reactions catalyzed by Desulfovibrio nickel(II)-substituted rubredoxin. Proc Natl Acad Sci USA 85:9378–9380. https://doi.org/10.1073/pnas.85.24.9378
Denariaz G, Ketchum PA, Payne WJ, Liu M-Y, Le Gall J, Moura I, Moura JJG (1994) An unusual hemoprotein capable of reversible binding of nitric oxide from the gram-positive Bacillus halodenitrificans. Arch Microbiol 162:316–322. https://doi.org/10.1007/BF00263778
Le Gall J, Xavier AV (1996) Anaerobes response to oxygen: the sulfate-reducing bacteria. Anaerobe 2:1–9. https://doi.org/10.1006/anae.1996.0001
Pereira IAC, Pachecho I, Liu M-Y, Le Gall J, Xavier AV, Teixeira M (1997) Multiheme cytochromes from the sulfur-reducing bacterium Desulfuromonas acetoxidans. Eur J Biochem 248:323–328. https://doi.org/10.1111/j.1432-1033.1997.00323.x
Matias PM, Morais J, Coelho AV, Meijers R, Gonzalez A, Thompson AW, Sieker L, Le Gall J, Carrondo MA (1998) A preliminary analysis of the three-dimensional structure of dimeric di-haem split-Soret cytochrome c from Desulfovibrio desulfuricans ATCC 27774 at 2.5-Å resolution using the MAD phasing method: a novel cytochrome fold with a stacked-haem arrangement. J Biol Inorg Chem 2:507–514. https://doi.org/10.1007/s007750050162
Matias PM, Coelho R, Pereira IAC, Coelho AV, Thompson AW, Sieker LC, Le Gall J, Carrondo MA (1999) The primary and three-dimensional structures of a nine-haem cytochrome c from Desulfovibrio desulfuricans ATCC 27774 reveal a new member of the Hmc family. Structure 7:119–130. https://doi.org/10.1016/S0969-2126(99)80019-7
Matias PM, Coelho AV, Valente FMA, Placido D, Le Gall J, Xavier AV, Pereira IAC, Carrondo MA (2002) Sulfate respiration in Desulfovibrio vulgaris Hildenborough. Structure of the 16-heme cytochrome c HmcA at 2.5 Å resolution and a view of its role in transmembrane electron transfer. J Biol Chem 277:47907–47916. https://doi.org/10.1074/jbc.M207465200
Fareleira P, Santos BS, António C, Moradas-Ferreira P, Le Gall J, Xavier AV, Santos H (2003) Response of a strict anaerobe to oxygen: survival strategies in Desulfovibrio gigas. Microbiology 149:1513–1522. https://doi.org/10.1099/mic.0.26155-0
Zöphel A, Kennedy MC, Beinert H, Kroneck PMH (1988) Investigations on microbial sulfur respiration. 1. Activation and reduction of elemental sulfur in several strains of eubacteria. Arch Microbiol 150:72–77. https://doi.org/10.1007/BF00409720
Zöphel A, Kennedy MC, Beinert H, Kroneck PMH (1991) Investigations on microbial sulfur respiration. Isolation, purification, and characterization of cellular components from Spirillum 5175. Eur J Biochem 195:849–856. https://doi.org/10.1111/j.1432-1033.1991.tb15774.x
Schumacher W, Kroneck PMH, Pfennig N (1992) Comparative systematic study on “Spirillum” 5175, Campylobacter and Wolinella species. Description of “Spirillum” 5175 as Sulfurospirillum deleyianum gen. nov., spec. nov.*. Arch Microbiol 158:287–293. https://doi.org/10.1007/BF00245247
Sosa Torres ME, Rito Morales A, Solano Peralta A, Kroneck PMH (2020) Sulfur, the versatile non-metal. Met Ions Life Sci 20:19–49. https://doi.org/10.1515/9783110589757-002
Sosa Torres ME, Kroneck PMH (2021) Introduction: from rocks to living cells. Met Ions Life Sci 21:1–32. https://doi.org/10.1515/9783110589771-001
Lehnert N, Coruzzi G, Hegg E, Seefeldt L, Stein L (2016) NSF workshop report: feeding the world in the 21st century: grand challenges in the nitrogen cycle, National Science Foundation: Arlington, VA
Louro RO, Catarino T, Salgueiro CA, Le Gall J, Turner DL, Xavier AV (1998) Molecular basis for energy transduction: mechanisms of cooperativity in multihaem cytochromes. In: Canters GW, Vijgenboom E (eds) Biological electron transfer chains: genetics, composition and mode of operation. NATO ASI Series (Series C: Mathematical and Physical Sciences), vol 512. Springer, Dordrecht, pp 209–223. https://doi.org/10.1007/978-94-011-5133-7_15
Hartshorne RS, Jepson BN, Clarke TA, Field SJ, Fredrickson J, Zachara J, Shi L, Butt JN, Richardson DJ (2007) Characterization of Shewanella oneidensis MtrC: a cell-surface decaheme cytochrome involved in respiratory electron transport to extracellular electron acceptors. J Biol Inorg Chem 12:1083–1094. https://doi.org/10.1007/s00775-007-0278-y
Sharma S, Cavallaro G, Rosato A (2010) A systematic investigation of multiheme c-type cytochromes in prokaryotes. J Biol Inorg Chem 15(15):559–571. https://doi.org/10.1007/s00775-010-0623-4
Clarke TA, Edwards MJ, Gates AJ, Hall A, White GF, Bradley J, Reardon CL, Shi L, Beliaev AS, Marshall MJ, Wang Z, Watmough NJ, Fredrickson JK, Zachara JM, Butt JN, Richardson DJ (2011) Structure of a bacterial cell surface decaheme electron conduit. Proc Natl Acad Sci USA 108:9384–9389. https://doi.org/10.1073/pnas.1017200108
Breuer M, Rosso KM, Blumberger J (2014) Electron flow in multiheme bacterial cytochromes is a balancing act between heme electronic interaction and redox potentials. Proc Natl Acad Sci USA 111:611–616. https://doi.org/10.1073/pnas.1316156111
Breuer M, Rosso KM, Blumberger J, Butt JN (2015) Multi-haem cytochromes in Shewanella oneidensis MR-1: structures, functions and opportunities. J R Soc Interface 12:20141117. https://doi.org/10.1098/rsif.2014.1117
Edwards MJ, Richardson DJ, Paquete CM, Clarke TA (2019) Role of multiheme cytochromes involved in extracellular anaerobic respiration in bacteria. Prot Sci 29:830–842. https://doi.org/10.1002/pro.3787
Futera Z, Ide I, Kayser B, Garg K, Jiang X, van Wonderen JH, Butt JN, Ishii H, Pecht I, Sheves M, Cahen D, Blumberger J (2020) Coherent electron transport across a 3 nm bioelectronic junction made of multi-heme proteins. J Phys Chem Lett 11:9766–9774. https://doi.org/10.1021/acs.jpclett.0c02686
Trindade IB, Paquete CM, Louro RO (2021) Extracellular redox chemistry. Met Ions Life Sci 21:229–269. https://doi.org/10.1515/9783110589771-008
Igarashi N, Moriyama H, Fujiwara T, Fukumori Y, Tanaka N (1997) The 2.8 Å structure of hydroxylamine oxidoreductase from a nitrifying chemoautotrophic bacterium, Nitrosomonas europaea. Nat Struct Mol Biol 4:276–284. https://doi.org/10.1038/nsb0497-276
Kurnikov IV, Ratner MA, Pacheco AA (2005) Redox equilibria in hydroxylamine oxidoreductase. electrostatic control of electron redistribution in multielectron oxidative processes. Biochemistry 44:1856–1863. https://doi.org/10.1021/bi048060v
Parey K, Fielding AJ, Sörgel M, Rachel R, Huber H, Ziegler C, Rajendran C (2016) In meso crystal structure of a novel membrane-associated octaheme cytochrome c from the Crenarchaeon Ignicoccus hospitalis. FEBS J 283:3807–3820. https://doi.org/10.1111/febs.13870
Caranto JD, Vilbert AC, Lancaster KM (2016) Nitrosomonas europaea cytochrome P460 is a direct link between nitrification and nitrous oxide emission. Proc Natl Acad Sci USA 113:14704–14709. https://doi.org/10.1073/pnas.1611051113
Caranto JD, Lancaster KM (2017) Nitric oxide is an obligate bacterial nitrification intermediate produced by hydroxylamine oxidoreductase. Proc Natl Acad Sci USA 114:8217–8222. https://doi.org/10.1073/pnas.1704504114
Smith MA, Majer SH, Vilbert AC, Lancaster KM (2019) Controlling a burn: outer-sphere gating of hydroxylamine oxidation by a distal base in cytochrome P460. Chem Sci 10:3756–3764. https://doi.org/10.1039/C9SC00195F
Coleman RE, Lancaster KM (2020) Heme P460: a (cross) link to nitric oxide. Acc Chem Res 53:2925–2935. https://doi.org/10.1021/acs.accounts.0c00573
Roldán MD, Sears HJ, Cheesman MR, Ferguson SJ, Thomson AJ, Berks BC, Richardson DJ (1998) Spectroscopic characterization of a novel multiheme c-type cytochrome widely implicated in bacterial electron transport. J Biol Chem 273:28785–28790. https://doi.org/10.1074/jbc.273.44.28785
Bruschi M, Bertrand P, More C, Leroy G, Bonicel J, Haladjian J, Chottard G, Pollock WBR, Voordouw G (1992) Biochemical and spectroscopic characterization of the high molecular weight cytochrome c from Desulfovibrio vulgaris Hildenborough expressed in Desulfovibrio desulfuricans G200. Biochemistry 12:3281–3288. https://doi.org/10.1021/bi00127a033
Florens L, Bruschi M (1994) Recent advances in the characterization of the hexadecahemic cytochrome c from Desulfovibrio. Biochimie 76:561–568. https://doi.org/10.1016/0300-9084(94)90180-5
Mowat CG, Rothery E, Miles CS, McIver L, Doherty MK, Drewette K, Taylor P, Walkinshaw MD, Chapman SK, Reid GA (2004) Octaheme tetrathionate reductase is a respiratory enzyme with novel heme ligation. Nature Struct Mol Biol 11:1023–1024. https://doi.org/10.1038/nsmb827
Atkinson SJ, Mowat CG, Reid GA, Chapman SK (2007) An octaheme c-type cytochrome from Shewanella oneidensis can reduce nitrite and hydroxylamine. FEBS Lett 581:3805–3808. https://doi.org/10.1016/j.febslet.2007.07.005
Reid GA, Miles CS, Moysey RK, Pankhurst KL, Chapman SK (2000) Catalysis in fumarate reductase. Biochim Biophys Acta 1459:310–315. https://doi.org/10.1016/S0005-2728(00)00166-3
Page CC, Moser CC, Chen X, Dutton PL (1999) Natural engineering principles of electron tunnelling in biological oxidation–reduction. Nature 402:47–52. https://doi.org/10.1038/46972
Mulder A, van de Graaf AA, Robertson LA, Kuenen JG (1995) Anaerobic ammonium oxidation discovered in a denitrifying fluidized bed reactor. FEMS Microbiol Ecol 16:177–183. https://doi.org/10.1016/0168-6496(94)00081-7
Kuenen JG (2020) Anammox and beyond. Environ Microbiol 22:525–536. https://doi.org/10.1111/1462-2920.14904
Kartal B, van Niftrik L, Keltjens JT, Op den Camp HJM, Jetten MSM (2012) Anammox: growth physiology, cell biology, and metabolism. Adv Microb Physiol 60:211–262. https://doi.org/10.1016/B978-0-12-398264-3.00003-6
Kartal B, Maalcke WJ, de Almeida NM, Cirpus I, Gloerich J, Geerts W, Op den Camp HJM, Harhangi HR, Janssen-Megens EM, Francoijs K-J, Stunnenberg HG, Keltjens JT, Jetten MSM, Strous M (2011) Molecular mechanism of anaerobic ammonium oxidation. Nature 479:127–130. https://doi.org/10.1038/nature10453
Reimann J, Jetten MSM, Keltjens JT (2015) Metal enzymes in “impossible” microorganisms catalyzing the anaerobic oxidation of ammonium and methane. Met Ions Life Sci 15:257–313. https://doi.org/10.1007/978-3-319-12415-5_7
Kartal B, Keltjens JT (2016) Anammox biochemistry: a tale of heme c proteins. Trends Biochem Sci 41:998–1011. https://doi.org/10.1016/j.tibs.2016.08.015
Maalcke WJ, Dietl A, Marritt SJ, Butt JN, Jetten MSM, Keltjens JT, Barends TRM, Kartal B (2014) Structural basis of biological NO generation by octaheme oxidoreductases. J Biol Chem 289:1228–1242. https://doi.org/10.1074/jbc.M113.525147
Maalcke WJ, Reimann J, de Vries S, Butt JN, Dietl A, Kip N, Mersdorf U, Barends TRM, Jetten MSM, Keltjens JT, Kartal B (2016) Characterization of anammox hydrazine dehydrogenase, a key N2-producing enzyme in the global nitrogen cycle. J Biol Chem 291:17077–17092. https://doi.org/10.1074/jbc.M116.735530
Akram M, Dietl A, Mersdorf U, Prinz S, Maalcke WJ, Keltjens JT, Ferousi C, de Almeida NM, Reimann J, Kartal B, Jetten MSM, Parey K, Barends TRM (2019) A 192-heme electron transfer network in the hydrazine dehydrogenase complex. Sci Adv 5:4310. https://doi.org/10.1126/sciadv.aav4310
Soler-Jofra A, Laureni M, Warmerdam M, Pérez J, van Loosdrecht M (2020) Hydroxylamine metabolism of Ca Kuenenia stuttgartiensis. Water Res 184:116188. https://doi.org/10.1016/j.watres.2020.116188
Bache R, Kroneck PMH, Merkle H, Beinert H (1983) A survey of EPR-detectable components in sulfur-reducing bacteria. Biochim Biophys Acta 722:417–426. https://doi.org/10.1007/BF00409720
Wolfe RS, Pfennig N (1977) Reduction of sulfur by spirillum 5175 and syntrophism with Chlorobium. Appl Environ Microbiol 33:427–433. https://doi.org/10.1128/AEM.33.2.427-433.1977
Schumacher W, Kroneck PMH (1992) Anaerobic energy metabolism of the sulfur-reducing bacterium “Spirillum” 5175 during dissimilatory nitrate reduction to ammonia. Arch Microbiol 157:464–470. https://doi.org/10.1007/BF00249106
Eisenmann E, Beuerle J, Sulger K, Kroneck PMH, Schumacher W (1995) Lithotrophic growth of Sulfurospirillum deleyianum with sulfide as electron donor coupled to respiratory reduction of nitrate to ammonia. Arch Microbiol 164:180–185. https://doi.org/10.1007/BF02529969
Schumacher W, Kroneck PMH (1991) Dissimilatory hexaheme c nitrite reductase of “Spirillum” strain 5175: purification and properties. Arch Microbiol 156:70–74. https://doi.org/10.1007/BF00418190
Schumacher W, Hole U, Kroneck PMH (1994) Ammonia-forming cytochrome c nitrite reductase from Sulfurospirillum deleyianum is a tetraheme protein: new aspects of the molecular composition and spectroscopic properties. Biochem Biophys Res Commun 205:911–916. https://doi.org/10.1006/bbrc.1994.2751
Strehlitz B, Gründig B, Schumacher W, Kroneck PMH, Vorlop K-D, Kotte H (1996) A nitrite sensor based on a highly sensitive nitrite reductase mediator-coupled amperometric detection. Anal Chem 68:807–816. https://doi.org/10.1021/ac950692n
Einsle O, Schumacher W, Kurun E, Nath U, Kroneck PMH (1998) Cytochrome c nitrite reductase from Sulfurospirillum deleyianum and Wolinella succinogenes. In: Canters GW, Vijgenboom E (eds) Biological electron transfer chains: genetics, composition and mode of operation. NATO ASI Series (Series C: Mathematical and Physical Sciences), vol 512. Springer, Dordrecht, pp 197–208. https://doi.org/10.1007/978-94-011-5133-7_14
Rudolf M, Kroneck PMH (2005) The nitrogen cycle: its biology. Met Ions Biol Systems 43:76–103 (PMID: 16370115)
Stach P, Einsle O, Schumacher W, Kurun E, Kroneck PMH (2000) Bacterial cytochrome c nitrite reductase: new structural and functional aspects. J Inorg Biochem 79:381–385. https://doi.org/10.1016/S0162-0134(99)00248-2
Rudolf M, Einsle O, Neese F, Kroneck PMH (2002) Pentahaem cytochrome c nitrite reductase: reaction with hydroxylamine, a potential reaction intermediate and substrate. Biochem Soc Trans 30:649–653. https://doi.org/10.1042/bst0300649
Brittain T, Blackmore R, Greenwood RC, Thomson AJ (1992) Bacterial nitrite-reducing enzymes. Eur J Biochem 209:793–802. https://doi.org/10.1111/j.1432-033.1992.tb17350.x
Darwin A, Hussain H, Griffiths L, Grove J, Sambongi Y, Busby S, Cole JA (1993) Regulation and sequence of the structural gene for cytochrome c552 from Escherichia coli: not a hexahaem but a 50kDa tetrahaem nitrite reductase. Mol Microbiol 9:1255–1265. https://doi.org/10.1111/j.1365-2958.1993.tb01255.x
Hussain H, Grove J, Griffiths L, Busby S, Cole JA (1994) A seven-gene operon essential for formate-dependent nitrite reduction to ammonia by enteric bacteria. Mol Microbiol 12:153–163. https://doi.org/10.1111/j.1365-2958.1994.tb01004.xd
Eaves DJ, Grove J, Staudenmann W, James P, Poole RK, White SA, Griffiths L, Cole JA (1998) Involvement of products of the nrfEFG genes in the covalent attachment of haem c to a novel cysteine–lysine motif in the cytochrome c552 nitrite reductase from Escherichia coli. Mol Microbiol 28:205–216. https://doi.org/10.1046/j.1365-2958.1998.00792.x
Einsle O, Messerschmidt A, Stach P, Bourenkov GP, Bartunik HD, Huber R, Kroneck PMH (1999) Structure of cytochrome c nitrite reductase. Nature 400:476–479. https://doi.org/10.1038/22802
Einsle O, Stach P, Messerschmidt A, Simon J, Kröger A, Huber R, Kroneck PMH (2000) Cytochrome c nitrite reductase from Wolinella succinogenes. Structure at 1.6 Å resolution, inhibitor binding, and heme-packing motifs. J Biol Chem 275:39608–39618. https://doi.org/10.1074/jbc.M006188200
Einsle O (2011) Structure and function of formate-dependent cytochrome c nitrite reductase, NrfA. Meth Enzymol 496:399–420. https://doi.org/10.1016/B978-0-12-386489-5.00016-6
Bamford VA, Angove HC, Seward HE, Thomson AJ, Cole JA, Butt JN, Hemmings AM, Richardson DJ (2002) Structure and spectroscopy of the periplasmic cytochrome c nitrite reductase from Escherichia coli. Biochemistry 41:2921–2931. https://doi.org/10.1021/bi015765d
Lockwood CWJ, Burlat B, Cheesman MR, Kern M, Simon J, Clarke TA, Richardson DJ, Butt JN (2015) Resolution of key roles for the distal pocket histidine in cytochrome c nitrite reductases. J Am Chem Soc 137:3059–3068. https://doi.org/10.1021/ja512941j
Almeida MG, Macieira S, Goncalves LL, Huber R, Cunha CA, Romão MJ, Costa C, Lampreia J, Moura JJG, Moura I (2003) The isolation and characterization of cytochrome c nitrite reductase subunits (NrfA and NrfH) from Desulfovibrio desulfuricans ATCC 27774. Re-evaluation of the spectroscopic data and redox properties. Eur J Biochem 270:3904–3915. https://doi.org/10.1046/j.1432-1033.2003.03772.x
Cunha CA, Macieira S, Dias JM, Almeida G, Goncalves LL, Costa C, Lampreia J, Huber R, Moura JJG, Moura I, Romão MJ (2003) Cytochrome c nitrite reductase from Desulfovibrio desulfuricans ATCC 27774. The relevance of the two calcium sites in the structure of the catalytic subunit (NrfA). J Biol Chem 278:17455–17465. https://doi.org/10.1074/jbc.M211777200
Youngblut M, Judd ET, Srajer V, Sayyed B, Goelzer T, Elliott SJ, Schmidt M, Pacheco AA (2012) Laue crystal structure of Shewanella oneidensis cytochrome c nitrite reductase from a high-yield expression system. J Biol Inorg Chem 17:647–662. https://doi.org/10.1007/s00775-012-0885-0
Rodrigues ML, Oliveira TF, Pereira IAC, Archer M (2006) X-ray structure of the membrane-bound cytochrome c quinol dehydrogenase NrfH reveals novel haem coordination. EMBO J 25:5951–5960. https://doi.org/10.1038/sj.emboj.7601439
Todorovic S, Rodrigues ML, Matos D, Pereira IAC (2012) Redox properties of lysine- and methionine-coordinated hemes ensure downhill electron transfer in NrfH2A4 nitrite reductase. J Phys Chem B 116:5637–5643. https://doi.org/10.1021/jp301356m
Campeciño J, Lagishetty S, Wawrzak Z, Sosa Alfaro V, Lehnert N, Reguera G, Hu J, Hegg EL (2020) Cytochrome c nitrite reductase from the bacterium Geobacter lovleyi represents a new NrfA subclass. J Biol Chem 295:11455–11465. https://doi.org/10.1074/jbc.RA120.013981
Sosa Alfaro V, Campeciño J, Tracy M, Elliott SJ, Hegg EL, Lehnert N (2021) Elucidating electron storage and distribution within the pentaheme scaffold of cytochrome c nitrite reductase (NrfA). Biochemistry 60:1853–1867. https://doi.org/10.1021/acs.biochem.0c00977
Welsh A, Chee-Sanford JC, Connor LM, Löffler FE, Sanford RA (2014) Refined NrfA phylogeny improves PCR-based nrfA gene detection. Appl Environ Microbiol 80:2110–2119. https://doi.org/10.1128/AEM.03443-13
Cannon J, Sanford RA, Connor L, Yang WH, Chee-Sanford J (2019) Sequence alignments and validation of PCR primers used to detect phylogenetically diverse nrfA genes associated with dissimilatory nitrate reduction to ammonium (DNRA). Data Brief 25:104016. https://doi.org/10.1016/j.dib.2019.104016
Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, Basham D, Chillingworth T, Davies RM, Feltwell T, Holroyd S, Jagels K, Karlyshev AV, Moule S, Pallen MJ, Penn CW, Quail MA, Rajandream MA, Rutherford KM, van Vliet AH, Whitehead S, Barrell BG (2000) The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403:665–668. https://doi.org/10.1038/35001088
Iverson TM, Arciero DM, Hsu BT, Logan MSP, Hooper AB, Rees DC (1998) Heme packing motifs revealed by the crystal structure of the tetra-heme cytochrome c554 from Nitrosomonas europaea. Nat Struct Biol 5:1005–1012. https://doi.org/10.1038/2975
Simon J, Gross R, Einsle O, Kroneck PMH, Kröger A, Klimmek O (2000) A NapC/NirT-type cytochrome c (NrfH) is the mediator between the quinone pool and the cytochrome c nitrite reductase of Wolinella succinogenes. Mol Microbiol 35:686–696. https://doi.org/10.1046/j.1365-2958.2000.01742.x
Simon J, Pisa R, Stein T, Eichler R, Klimmek O, Gross R (2001) The tetraheme cytochrome c NrfH is required to anchor the cytochrome c nitrite reductase (NrfA) in the membrane of Wolinella succinogenes. Eur J Biochem 268:5776–5782. https://doi.org/10.1046/j.0014-2956.2001.02520.x
Einsle O, Stach P, Messerschmidt A, Klimmek O, Simon J, Kröger A, Kroneck PMH (2002) Crystallization and preliminary X-ray analysis of the membrane-bound cytochrome c nitrite reductase complex (NrfHA) from Wolinella succinogenes. Acta Crystallogr D 58:341–342. https://doi.org/10.1107/S090744490102039X
Gwyer JD, Richardson DJ, Butt JN (2004) Resolving complexity in the interactions of redox enzymes and their inhibitors: contrasting mechanisms for the inhibition of a cytochrome c nitrite reductase revealed by protein film voltammetry. Biochemistry 43:15086–15094. https://doi.org/10.1021/bi049085x
Stein N, Love D, Judd ET, Elliott SJ, Bennett B, Pacheco A (2015) Correlations between the electronic properties of Shewanella oneidensis cytochrome c nitrite reductase (ccNiR) and its structure: effects of heme oxidation state and active site ligation. Biochemistry 54:3749–3758. https://doi.org/10.1021/acs.biochem.5b00330
Jafferji A, Allen JWA, Ferguson SJ, Fülöp V (2000) X-ray crystallographic study of cyanide binding provides insights into the structure-function relationship for cytochrome cd1 nitrite reductase from Paracoccus pantotrophus. J Biol Chem 275:25089–25094. https://doi.org/10.1074/jbc.M001377200
Pisa R, Stein T, Eichler R, Gross R, Simon J (2002) The nrfI gene is essential for the attachment of the active site haem group of Wolinella succinogenes cytochrome c nitrite reductase. Mol Microbiol 43:763–770. https://doi.org/10.1046/j.1365-2958.2002.02784.x
Howes BD, Feis A, Raimondi L, Indiani C, Smulevich G (2001) The critical role of the proximal calcium ion in the structural properties of horseradish peroxidase. J Biol Chem 276:40704–40711. https://doi.org/10.1074/jbc.M107489200
Murphy MJ, Siegel LM, Tove SR, Kamin H (1974) Siroheme: a new prosthetic group participating in six-electron reduction reactions catalyzed by both sulfite and nitrite reductases. Proc Nat Acad Sci USA 71:612–616. https://doi.org/10.1073/pnas.71.3.612
Young LJ, Siegel LM (1988) On the reaction of ferric heme proteins with nitrite and sulfite. Biochemistry 27:2790–2800. https://doi.org/10.1021/bi00408a020
Crane BR, Getzoff ED (1996) The relationship between structure and function for the sulfite reductases. Curr Opt Struct Biol 6:44–756. https://doi.org/10.1016/S0959-440X(96)80003-0
Crane BR, Siegel LM, Getzoff ED (1997) Structures of the siroheme- and Fe4S4-containing active center of sulfite reductase in different states of oxidation: heme activation via reduction-gated exogenous ligand exchange. Biochemistry 36:12101–12119. https://doi.org/10.1021/bi971065q
Crane BR, Siegel LM, Getzoff ED (1997) Probing the catalytic mechanism of sulfite reductase by X-ray crystallography: structures of the Escherichia coli hemoprotein in complex with substrates, inhibitors, intermediates, and products. Biochemistry 36:12120–12137. https://doi.org/10.1021/bi971066i
Fauque GD, Barton LL (2012) Hemoproteins in dissimilatory sulfate- and sulfur-reducing prokaryotes. Adv Microb Physiol 60:1–90. https://doi.org/10.1016/B978-0-12-398264-3.00001-2
Askenasy I, Stroupe ME (2020) The Siroheme-[4Fe-4S] Coupled Center. Met Ions Life Sci 20:343–380. https://doi.org/10.1515/9783110589757-016
Einsle O, Messerschmidt A, Huber R, Kroneck PMH, Neese F (2002) Mechanism of the six-electron reduction of nitrite to ammonia by cytochrome c nitrite reductase. J Am Chem Soc 124:11737–11745. https://doi.org/10.1021/ja0206487
Bykov D, Neese F (2011) Substrate binding and activation in the active site of cytochrome c nitrite reductase: a density functional study. J Biol Inorg Chem 16:417–430. https://doi.org/10.1007/s00775-010-0739-6
Bykov D, Neese F (2012) Reductive activation of the heme iron–nitrosyl intermediate in the reaction mechanism of cytochrome c nitrite reductase: a theoretical study. J Biol Inorg Chem 17:741–760. https://doi.org/10.1007/s00775-012-0893-0
Bykov D, Plog M, Neese F (2014) Heme-bound nitroxyl, hydroxylamine, and ammonia ligands as intermediates in the reaction cycle of cytochrome c nitrite reductase: a theoretical study. J Biol Inorg Chem 19:97–112. https://doi.org/10.1007/s00775-013-1065-6
Bykov D, Neese F (2015) Six-electron reduction of nitrite to ammonia by cytochrome c nitrite reductase: insights from density functional theory studies. Inorg Chem 54:9303–9316. https://doi.org/10.1021/acs.inorgchem.5b01506
Martins G, Rodrigues L, Cunha FM, Matos D, Hildebrandt P, Murgida DH, Pereira IAC, Todorovic S (2010) Substrate binding to a nitrite reductase induces a spin transition. J Phys Chem B 114:5563–5566. https://doi.org/10.1021/jp9118502
Milton RD, Minteer SD (2017) Enzymatic bioelectrosynthetic ammonia production: recent electrochemistry of nitrogenase, nitrate reductase, and nitrite reductase. ChemPlusChem 82:513–521. https://doi.org/10.1002/cplu.201600442
Poock SR, Leach ER, Moir JWB, Cole JA, Richardson DJ (2002) Respiratory detoxification of nitric oxide by the cytochrome c nitrite reductase of Escherichia coli. J Biol Chem 277:23664–23669. https://doi.org/10.1074/jbc.M200731200
Youngblut M, Pauly DJ, Stein N, Walters D, Conrad JA, Moran GR, Bennett B, Pacheco AA (2014) Shewanella oneidensis cytochrome c nitrite reductase (ccNiR) does not disproportionate hydroxylamine to ammonia and nitrite, despite a strongly favorable driving force. Biochemistry 53:2136–2144. https://doi.org/10.1021/bi401705d
Angove HC, Cole JA, Richardson DJ, Butt JN (2002) Protein film voltammetry reveals distinctive fingerprints of nitrite and hydroxylamine reduction by a cytochrome c nitrite reductase. J Biol Chem 277:23374–23381. https://doi.org/10.1074/jbc.M200495200
Ali M, Stein N, Mao Y, Shahid S, Schmidt M, Bennett B, Pacheco AA (2019) Trapping of a putative intermediate in the cytochrome c nitrite reductase (ccNiR)-catalyzed reduction of nitrite: implications for the ccNiR reaction mechanism. J Am Chem Soc 141:13358–13371. https://doi.org/10.1021/jacs.9b03036
Da Silva S, Cosnier S, Almeida MG, Moura JJG (2004) An efficient poly(pyrrole-viologen)-nitrite reductase biosensor for the mediated detection of nitrite. Electrochem Commun 6:404–408. https://doi.org/10.1016/j.elecom.2004.02.007
Almeida MG, Silveira CM, Guigliarelli B, Bertrand P, Moura JJG, Moura I, Léger C (2007) A needle in a haystack: the active site of the membrane-bound complex cytochrome c nitrite reductase. FEBS Lett 581:284–288. https://doi.org/10.1016/j.febslet.2006.12.023
Chen H, Mousty C, Cosnier S, Silveira C, Moura JJG, Almeida MG (2007) Highly sensitive nitrite biosensor based on the electrical wiring of nitrite reductase by [ZnCr-AQS] LDH. Electrochem Commun 9:2240–2245. https://doi.org/10.1016/j.elecom.2007.05.030
Silveira CM, Besson S, Moura I, Moura JJG, Almeida MG (2010) Measuring the cytochrome c nitrite reductase activity—practical considerations on the enzyme assays. Bioinorg Chem Appl. https://doi.org/10.1155/2010/634597
Almeida MG, Serra A, Silveira CM, Moura JJG (2010) Nitrite biosensing via selective enzymes—a long but promising route. Sensors 10:11530–11555. https://doi.org/10.3390/s101211530
Paes de Sousa PM, Pauleta SR, Simões Gonçalves ML, Pettigrew GW, Moura I, Moura JJG, Correia dos Santos MM (2011) Artefacts induced on c-type haem proteins by electrode surfaces. J Biol Inorg Chem 16:209–215. https://doi.org/10.1007/s00775-010-0717-z
Correia C, Rodrigues M, Silveira CM, Moura JJG, Ochoteco E, Jubete E, Almeida MG (2013) Nitrite biosensing using cytochrome c nitrite reductase: towards a disposable strip electrode. In: Gabriel J, Schier J, Van Huffel S, Conchon E, Correia C, Fred A, Gamboa H (eds) Biomedical engineering systems and technologies, communications in computer and information science, vol 357. Springer, Berlin, pp 41–50. https://doi.org/10.1007/978-3-642-38256-7_3
Gomes FO, Maia LB, Cordas C, Delerue-Matos C, Moura I, Moura JJG, Morais S (2018) Nitric oxide detection using electrochemical third-generation biosensors-based on heme proteins and porphyrins. Electroanalysis 30:2485–2503. https://doi.org/10.1002/elan.201800421
Gomes FO, Maia LB, Loureiro JA, Carmo Pereira M, Delerue-Matos C, Moura I, Moura JJG, Morais S (2019) Biosensor for direct bioelectrocatalysis detection of nitric oxide using nitric oxide reductase incorporated in carboxylated single-walled carbon nanotubes/lipidic 3 bilayer nanocomposite. Bioelectrochemistry 127:76–86. https://doi.org/10.1016/j.bioelechem.2019.01.010
Paoli M, Marles-Wright J, Smith A (2002) Structure-function relationships in heme-proteins. DNA Cell Biol 21:271–280. https://doi.org/10.1089/104454902753759690
Voigt P, Knapp E-W (2003) Tuning heme redox potentials in the cytochrome c subunit of photosynthetic reaction centers. J Biol Chem 278:51993–52001. https://doi.org/10.1074/jbc.M307560200
Reedy CJ, Elvekrog MM, Gibney BR (2008) Development of a heme protein structure–electrochemical function database. Nucleic Acids Res 36:D307–D313. https://doi.org/10.1093/nar/gkm814
Timmons AJ, Symes MD (2015) Converting between the oxides of nitrogen using metal–ligand coordination complexes. Chem Soc Rev 44:6708–6722. https://doi.org/10.1039/C5CS00269A
Hosseinzadeh P, Lu Y (2016) Design and fine-tuning redox potentials of metalloproteins involved in electron transfer in bioenergetics. Biochim Biophys Acta 1857:557–581. https://doi.org/10.1016/j.bbabio.2015.08.006
Huang J, Zarzycki J, Gunner MR, Parson WW, Kern JF, Junko J, Ducat DC, Kramer DM (2020) Mesoscopic to macroscopic electron transfer by hopping in a crystal network of cytochromes. J Am Chem Soc 142:10459–10467. https://doi.org/10.1021/jacs.0c02729
Teixeira LR, Cordas CM, Fonseca MP, Duke NEC, Pokkuluri PR, Salgueiro CA (2020) Modulation of the redox potential and electron/proton transfer mechanisms in the outer membrane cytochrome OmcF from Geobacter sulfurreducens. Front Microbiol 10:29412020. https://doi.org/10.3389/fmicb.2019.02941
Stroka JR, Kandemir B, Matson EM, Bren KL (2020) Electrocatalytic multielectron nitrite reduction in water by an iron complex. ACS Catal 10:13968–13972. https://doi.org/10.1021/acscatal.0c03600
Ferrer JC, Guillemette JG, Bogumil R, Inglis SC, Smith M, Mauk AG (1993) Identification of Lys79 as an iron ligand in one form of alkaline yeast Iso-1-ferricytochrome c. J Am Chem Soc 115(1993):7507–7508. https://doi.org/10.1021/ja00069a062
Rossell FI, Ferrer JC, Mauk AG (1998) Proton-linked protein conformational switching: definition of the alkaline conformational transition of yeast iso-1-ferricytochrome c. J Am Chem Soc 120:11234–11245. https://doi.org/10.1021/ja971756
Johnson EA, Rice SL, Preimesberger MR, Nye DB, Gilevicius L, Wenke BB, Brown JM, Witman GB, Lecomte JTJ (2014) Characterization of THB1, a Chlamydomonas reinhardtii truncated hemoglobin: linkage to nitrogen metabolism and identification of lysine as the distal heme ligand. Biochemistry 53:4573–4589. https://doi.org/10.1021/bi5005206
Ubbink M, Campos AP, Teixeira M, Hunt NI, Hill HAO, Canters GW (1994) Characterization of mutant Met100Lys of cytochrome c-550 from Thiobacillus versutus with lysine-histidine heme ligation. Biochemistry 33:10051–10059. https://doi.org/10.1021/bi00199a032
Louro RO, de Waal EC, Ubbink M, Turner DL (2002) Replacement of the methionine axial ligand in cytochrome c(550) by a lysine: effects on the haem electronic structure. FEBS Lett 510:185–188. https://doi.org/10.1016/S0014-5793(01)03272-0
Du J, Perera R, Dawson JH (2011) Alkylamine-ligated H93G myoglobin cavity mutant: a model system for endogenous lysine and terminal amine ligation in heme proteins such as nitrite reductase and cytochrome f. Inorg Chem 50:1242–1249. https://doi.org/10.1021/ic101644u
Du J, Sono M, Dawson JH (2011) The H93G myoglobin cavity mutant as a versatile scaffold for modeling heme iron coordination structures in protein active sites and their characterization with magnetic circular dichroism spectroscopy. Coord Chem Rev 255:700–716. https://doi.org/10.1016/j.ccr.2011.01.029
Tse W, Whitmore N, Cheesman MR, Watmough NJ (2021) Influence of the heme distal pocket on nitrite binding orientation and reactivity in Sperm Whale myoglobin. Biochem J 478:927–942. https://doi.org/10.1042/BCJ20200596
Copeland DM, Soares AS, West AH, Richter-Addo GB (2006) Crystal structures of the nitrite and nitric oxide complexes of horse heart myoglobin. J Inorg Biochem 100:1413–1425. https://doi.org/10.1016/j.jinorgbio.2006.04.011
Yi J, Safo MK, Richter-Addo GB (2008) The nitrite anion binds to human hemoglobin via the uncommon O-nitrito mode. Biochemistry 47:8247–8249. https://doi.org/10.1021/bi801015c
Sundararajan M, Neese F (2015) Distal histidine modulates the unusual O-binding of nitrite to myoglobin: evidence from the quantum chemical analysis of EPR parameters. Inorg Chem 54(15):7209–7217. https://doi.org/10.1021/acs.inorgchem.5b00557
Tikhonova TV, Slutsky A, Antipov AN, Boyko KM, Polyakov KM, Sorokin DY, Zvyagilskaya RA, Popov VO (2006) Molecular and catalytic properties of a novel cytochrome c nitrite reductase from nitrate-reducing haloalkaliphilic sulfur-oxidizing bacterium Thioalkalivibrio nitratireducens. Biochim Biophys Acta 1764:715–723. https://doi.org/10.1016/j.bbapap.2005.12.021
Tikhonova TV, Trofimov AA, Popov VO (2012) Octaheme nitrite reductases: structure and properties. Biochem Mosc 77:1129–1138. https://doi.org/10.1134/S0006297912100057
Tikhonova T, Tikhonov A, Trofimov AA, Polyakov K, Boyko K, Cherkashin E, Rakitina T, Sorokin D, Popov V (2012) Comparative structural and functional analysis of two octaheme nitrite reductases from closely related Thioalkalivibrio species. FEBS J. 279:4052–4061. https://doi.org/10.1111/j.1742-4658.2012.08811.x
Andoralov V, Shleev S, Dergousova N, Kulikova O, Popov V, Tikhonov T (2021) Octaheme nitrite reductase: The mechanism of intramolecular electron transfer and kinetics of nitrite bioelectroreduction. Bioelectrochemistry 138:107699. https://doi.org/10.1016/j.bioelechem.2020.107699
Dong H, Yu B (2007) Geomicrobiological processes in extreme environments: a review. Episodes 300:202–216. https://doi.org/10.18814/epiiugs/2007/v30i3/003
Ferreira MR, Fernandes TM, Salgueiro CA (2020) Thermodynamic properties of triheme cytochrome PpcF from Geobacter metallireducens reveal unprecedented functional mechanism. Biochim Biophys Acta 1861:148271. https://doi.org/10.1016/j.bbabio.2020.148271
Acknowledgements
My sincere thanks go to R. Bache, H. Beinert, O. Einsle, G. Fritz, R. Huber, M. C. Kennedy, A. Kröger, A. Messerschmidt, F. Neese, M. Rudolf, N. Pfennig, W. Schumacher, J. Simon, P. Stach, J. Steuber, and A. Zöphel for the exciting excursion into the world of microbes and pentaheme nitrite reductase, and all the students and co-workers cited in the references for their valuable contributions. Work in the laboratory was supported by the Deutsche Forschungsgemeinschaft, the Volkswagen-Stiftung, the German–Israel Foundation, and the University of Konstanz.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no competing financial interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kroneck, P.M.H. Nature's nitrite-to-ammonia expressway, with no stop at dinitrogen. J Biol Inorg Chem 27, 1–21 (2022). https://doi.org/10.1007/s00775-021-01921-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-021-01921-4