
Abstract
Pancreatic cancer is currently one of the most lethal diseases. In recent years, increasing evidence has shown that reprogrammed metabolism may play a critical role in the carcinogenesis, progression, treatment and prognosis of pancreatic cancer. Affected by internal or external factors, pancreatic cancer cells adopt extensively distinct metabolic processes to meet their demand for growth. Rewired glucose, amino acid and lipid metabolism and metabolic crosstalk within the tumor microenvironment contribute to unlimited pancreatic tumor progression. In addition, the metabolic reprogramming involved in pancreatic cancer resistance is also closely related to chemotherapy, radiotherapy and immunotherapy, and results in a poor prognosis. Reflective of the key role of metabolism, the number of preclinical and clinical trials about metabolism-targeted therapies for pancreatic cancer is increasing. The poor prognosis of pancreatic cancer patients might be largely improved after employing therapies that regulate metabolism. Thus, investigations of metabolism not only benefit the understanding of carcinogenesis and cancer progression but also provide new insights for treatments against pancreatic cancer.
Similar content being viewed by others


Background
Pancreatic cancer is one of the most aggressive diseases; it has a poor prognosis, and its five-year survival rate remains lower than 9% despite decades of continuous efforts. According to recent data, pancreatic cancer is the fourth and sixth leading cause of cancer-related mortality in the U.S. and China, respectively [1, 2]. Furthermore, it is predicted to be the second leading cause of cancer-related deaths in the U.S. in 2030 [3]. Surgery remains the only way to cure pancreatic cancer. However, because most patients are diagnosed with a nonresectable disease due to the lack of symptoms in the early stage, only up to 20% of patients have the opportunity to receive initial surgical resection [4]. Even for patients who undergo a successful operation, over 80% of them still eventually develop local recurrence or metastases [5]. Therefore, in addition to surgery, comprehensive treatment following multidisciplinary management should be given more attention. Indeed, there remain many challenging problems. Chemotherapy is still recommended as the primary treatment for patients who have nonresectable pancreatic cancer and patients who undergo resection. Depending on different patient statuses, there are several distinct recommended chemotherapy regimens, which mainly include gemcitabine, FOLFIRINOX, and albumin-bound paclitaxel. However, the rapid and common development of chemoresistance usually leads to poor prognosis [6]. Although radiation is another relatively well-established anticancer method, it is currently regarded as a palliative way to relieve pain caused by advanced pancreatic cancer [7]. In addition to chemotherapy and radiotherapy, immunotherapy and targeted therapy are emerging as remarkable anticancer strategies [8,9,10]. Nevertheless, many successful immunotherapies against other cancer types are not as effective in pancreatic cancer treatment [11, 12], and most clinical trials focusing on targeted therapy fail to show satisfying outcomes [13]. Therefore, breakthroughs in pancreatic cancer treatment are needed.
In pancreatic cancer cells, several genetic alterations are considered to be the basis for pancreatic cancer progression and its dismal prognosis; these alterations include oncogenic KRAS mutations, which occur in over 90% of cases, and inactivating mutations in suppressor genes such as TP53, SMAD4, and CDKN2A [14]. Moreover, the aforementioned dilemma in comprehensive treatments is also largely determined by other biological features, such as extensive dense desmoplasia, hypoperfusion and an immunosuppressive microenvironment [15]. Additionally, many recent reports have indicated that distinct cancer metabolism is important for restricting the therapeutic effect.
Reprogrammed cellular energy metabolism, one of the emerging hallmarks of cancer [16], has been refocused over the past decade [17]. Cancer cells rewire many metabolic pathways to facilitate their survival, unlimited cell growth, and division. In addition, they also rely on extensive metabolic interactions with other nonmalignant cells and with the extracellular matrix (ECM) within the tumor microenvironment [18, 19]. Beyond the tissue level, the local tumor can affect host metabolism via cachexia, impairing antitumor immunity [20]. Interestingly, several recent studies also demonstrated that metabolic alterations can promote pancreatic tumorigenesis and metastasis through epigenetic regulation [21, 22], emphasizing the vital role of metabolism in pancreatic cancer development. Furthermore, many studies clearly showed that pancreatic tumor metabolism is closely associated with chemoresistance [23], radioresistance [24] and immunosuppression [25]. Recently, pancreatic cancer was also stratified into different metabolic subgroups (quiescent, glycolytic, cholesterogenic and mixed), which could predict different prognoses and responses to therapy [26, 27]. Therefore, the metabolic features of pancreatic cancer provide attractive therapeutic opportunities for novel and personalized treatments [27, 28].
Metabolic features of pancreatic cancer
Although reprogrammed metabolism is a general characteristic of cancer, different cancers show distinct metabolic addictions, which are mainly determined by their specific genetic mutations, tissue of origin or tumor microenvironment [29, 30]. Even in the same pancreatic cancer patient, the primary tumor and metastatic lesions exhibit relatively different metabolic gene expression [31]. Therefore, metabolic alteration of pancreatic cancer is a collective scenario mediated by multiple factors. In addition to the genomic characterization of pancreatic cancer cells [32], there is a complex and harsh microenvironment within the pancreatic tumor. The dense stroma results in elevated solid stress and interstitial fluid pressure that compress the vasculature, leading to hypoperfusion [33]. However, cancer cells exhibit extraordinary growth advantages in relatively hypoxic and nutrient-poor niches. They survive and thrive mainly in three ways: (1) Reprogramming intracellular energy metabolism of nutrients, including glucose, amino acids, and lipids. (2) Improving nutrient acquisition by scavenging and recycling. (3) Conducting metabolic crosstalk with other components within the microenvironment [34].
Intracellular metabolism
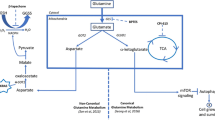
In the 1920s, Otto Warburg’s pioneering work demonstrated that tumor cells consume more glucose than normal cells. They subsequently turn most glucose-derived carbon into lactate even in the presence of sufficient oxygen. This process is named aerobic glycolysis or the Warburg effect [35]. It indeed provides some tangible advantages to cancer cells. First, compared with oxidative phosphorylation (OXPHOS), ample glycolytic flux achieves a higher rate of ATP production [36]. Second, it provides tumor cells with plenty of intermediates required for rapid and vast biosynthesis with a proper ATP/ADP ratio. Third, it plays an important role in maintaining redox balance and modulating chromatin state. Fourth, it creates a low immunity microenvironment and enhances cancer cell invasion [37]. Since Warburg’s initial publications, many studies have been conducted to uncover the metabolism of tumors. Cancers with different tissue origins exhibit distinct metabolic changes, even driven by the same oncogenes [38]. For pancreatic cancer cells, genetic mutations and stromal cues are thought to drive heterogeneous metabolic phenotypes [39,40,41,42,43], which mainly include the Warburg, reverse Warburg, lipid-dependence, and glutaminolysis phenotypes [44]. Therefore, pancreatic cancer cells exhibit complex and heterogeneous reprogramming of glucose, amino acid and lipid metabolism (Fig. 1).

The landscape of metabolic pathways in pancreatic cancer cells. The metabolism of glucose, amino acids and lipids is largely reprogrammed, which is mainly due to changes in key enzymes and transporters. Furthermore, some of them are closely regulated by oncogenic KRAS. Additionally, micropinocytosis and autophagy are also promoted by mutant KRAS, but they are controlled by other regulatory mechanisms within pancreatic cancer cells as well. Long solid arrows imply shifts or bioconversions. The dotted arrow means positive regulation, whereas the blunt end means negative regulation. Red arrowheads following the enzymes, transporters, and processes represent the effects induced by mutant KRAS: upward means upregulation; downward means downregulation. The black symbols represent the changes induced by other or unknown reasons. In addition, those following tildes indicate that they are dually regulated under different conditions. ACLY, ATP citrate lyase; ASNS, asparagine synthetase; CARM1, coactivator-associated arginine methyltransferase 1; CS, citrate synthetase; F-6P, fructose 6-phosphate; F-1,6BP, fructose 1,6-bisphosphate; F-2,6BP, fructose 2,6-bisphosphate; GFPT1, glutamine:fructose 6-phosphate amidotransferase 1; G-6P, glucose 6-phosphate; HK1/2, hexokinase 1/2; HMGCR, 3-hydroxy-3-methylglutaryl coenzyme A reductase; HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A; ME1, malic enzyme; MUFA, monounsaturated fatty acid; PFK1, phosphofructokinase 1; PRODH1, proline oxidase; PUFA, polyunsaturated fatty acid; RPE, ribulose-5-phosphate epimerase; SCD1, stearoyl-CoA desaturase; SFA, saturated fatty acid; TCA, tricarboxylic acid
Glucose
Glucose metabolism is relatively well documented in the rewired metabolic network. In the process of elevated aerobic glycolysis in pancreatic cancer cells, the expression of glucose transporter 1 (GLUT1) and its translocation to the cancer cell membrane are promoted, increasing glucose uptake [39, 45]. In addition to absorption, pancreatic cancer also shows upregulated expression of many genes encoding rate-limiting glycolytic enzymes, such as hexokinase 1/2, phosphofructokinase 1 and lactate dehydrogenase A (LDHA, the subunit of LDH), enhancing the Warburg effect and glycolytic flux to lactate [39]. In contrast to glycolysis, phosphorylated pyruvate dehydrogenase kinase 1 (PDHK1) inhibits the pyruvate dehydrogenase (PDH) complex, suppressing mitochondrial OXPHOS in pancreatic cancer cells [46]. To address the excess of acid products, such as lactate, from aerobic glycolysis, pancreatic cancer cells robustly express monocarboxylate transporter 1 (MCT1), MCT4 and CD147 on the plasma membrane to accelerate lactate flux [47,48,49]. The nonoxidative pentose phosphate pathway (PPP) originating from glycolysis offers materials for anabolism, including DNA synthesis. In this enhanced metabolic pathway, pancreatic cancer cells display increased ribulose 5-phosphate isomerase (RPIA) and ribulose-5-phosphate-3-epimerase (RPE) expression [50, 51]. The hexosamine biosynthesis pathway (HBP), another branch of glucose metabolism, provides the substrate for protein and lipid glycosylation, which is considered to be critical for tumor progression [52]. In addition, the rate-limiting enzyme of HBP, glutamine:fructose-6-phosphate amidotransferase-1 (GFPT1), is upregulated in pancreatic cancer cells [39]. In addition to those enhanced glycolytic enzymes, pancreatic cancer cells also overexpress more Nampt than adjacent normal tissues. Nampt is an essential enzyme that recycles nicotinamide adenine dinucleotide (NAD), a vital cofactor in many redox reactions, to sustain the high level of glycolytic flux within cancer cells [53]
Such distinct metabolic features are regulated by various factors. KRAS, a kind of small GTPase, is persistently activated upon mutation in pancreatic cancer and continuously stimulates downstream effectors (e.g., PI3K and RAF) [54]. As a result, the expression of GLUT1 and the key enzymes in glucose metabolism mentioned before are promoted (Fig. 1). Regarding the underlying mechanisms, the KRAS-driven MAPK pathway and transcription factor MYC might be prominent mediators, but the refined regulation of the respective enzymes remains to be further studied [39, 54]. In addition to the regulation of glycolysis, mutant KRAS signaling stimulates mitochondrial translocation of phosphoglycerate kinase 1 (PGK1), leading to phosphorylated PDHK1 and restricted OXPHOS in pancreatic cancer cells [46]. Glucose deprivation also promotes KRAS mutations in turn, suggesting the complex interaction between metabolism and the oncogene [55]. Furthermore, some KRAS-driven overexpressed enzymes, such as RPIA, are still preserved in some pancreatic cancer cell lines with KRAS ablation, sustaining nonoxidative PPP and cancer cell survival in a KRAS-independent manner [50]. Therefore, the relationship between KRAS and reprogrammed metabolism needs further study. Additionally, mutant TP53 is a key player in enhancing the Warburg effect and reducing mitochondrial activity [45, 56]. In pancreatic cancer cells, TP53 can increase glucose uptake by increasing paraoxonase 2 expression and impairing the expression of TP53-induced glycolysis and apoptosis regulator (TIGAR), which degrades fructose-2,6-bisphosphate, an effective enhancer of glycolysis, to fructose-6-phosphate [40, 57, 58]. Moreover, mutant TP53 in pancreatic cancer cells also maintains the cytoplasmic stabilization of glyceraldehyde-3-phosphate dehydrogenase, a key enzyme in glycolysis, to support the Warburg effect and confer sensitivity to glycolysis inhibitors (2-deoxyglucose, also known as 2-DG) [59]. However, pancreatic cancer cells with normal TP53 status are resistant to LDHA inhibition due to decreased dependence on glycolysis [57].In addition to KRAS and TP53 signaling in pancreatic cancer cells, LDHA could also be comprehensively regulated by deacetylation modification and other oncogenic transcription factors, such as Forkhead box protein M1 (FOXM1) [60, 61]. Additionally, there are many other mechanisms regulating glucose metabolism in pancreatic cancer cells. For example, hypoxia-inducible factor-1 (HIF-1) mainly induced by hypoxia contributes to strengthened glycolysis and the upregulated expression of HBP-related enzyme (GFPT2, the isoform of GFPT1) [62]. HIF-1 also inhibits PDH, leading to compromised mitochondrial oxidation [63]. Pyruvate kinase muscle isozyme 2 (PKM2) expression in pancreatic cancer cells is largely determined by nutrient conditions, and PKM2 is overexpressed under normal conditions. However, low glucose levels decrease PKM2 expression, which maintains cell survival by promoting autophagy and biomacromolecule accumulation and reducing oxidative stress [64].
Amino acids
Amino acid metabolism is also widely rewired in pancreatic cancer cells. Several amino acid transporters are highly expressed in pancreatic cancer cells to satisfy the increased need [65, 66]. Among various amino acids, glutamine (Gln) metabolism is critical for cancer cell survival as the main source of nitrogen and carbon, contributing to macromolecular synthesis and redox balance [67, 68]. Initially, Gln entering mitochondria is deaminated to glutamate (Glu) by glutaminase 1 (GLS1). In many cancer cell lines, aminotransferase or glutamate dehydrogenase (GDH) usually catalyzes the conversion from Gln-derived Glu to α-ketoglutarate (α-KG), which depends on different situations [67]. However, in pancreatic cancer cells, GDH is repressed, but the expression of cytoplasmic aspartate transaminase (GOT1) is promoted [69]. In this process, mitochondrial aspartate transaminase (GOT2) converts Gln-derived Glu and oxaloacetate (OAA) to aspartate (Asp) and α-KG in mitochondria. After that, Asp enters the cytoplasm and is turned into OAA by upregulated GOT1. Then, cytoplasmic OAA is converted to malate through malate dehydrogenase 1 (MDH1) and subsequently oxidized to pyruvate by malic enzyme. In addition, sufficient reducing power is generated to resist reactive oxygen species (ROS) and achieve redox control in pancreatic cancer cells [69]. Consistently, pancreatic cancer cells upregulate the expression of GOT1 in the acidic microenvironment to deal with increased ROS generation and support cancer cell survival [70].
This pathway in pancreatic cancer cells is primarily dominated by mutant KRAS, which results in GDH repression and GOT1 promotion. Therefore, it is named KRAS-driven noncanonical Gln metabolism [69]. Additionally, the regulation of some other processes is also involved in this process. For example, arginine methylation of MDH1 induced by coactivator-associated arginine methyltransferase 1 (CARM1) suppresses tumor growth, but KRAS activation and oxidative stress relieve such inhibition [71]. Furthermore, a recent report revealed that pancreatic cancer cells have compensatory metabolic networks of Gln, exhibiting recovered Gln-derived metabolic intermediates and tumor growth after long-term GLS1 inhibition [72]. In particular, Gln metabolism is vital for the viability and proliferation of hypoxic pancreatic cancer cells, which is mainly mediated by upregulated GLS2, the isoform of GLS1 [62]. Thus, the shift of Gln metabolism in pancreatic cancer is worth further study.
In addition to Gln, other amino acids are also key players in pancreatic cancer progression. Pancreatic cancer cells show overexpressed proline (Pro) oxidase (PRODH1), which contributes to Pro-derived Glu and promotes the survival and proliferation of pancreatic cancer cells, especially under glucose- or Gln-limited conditions [19]. Additionally, many pancreatic tumors from clinical cases display negative or low asparagine (Asn) synthetase expression, indicating the dependence of exogenous Asn. Therefore, plasma Asn depletion mediated by erythrocyte-entrapped L-asparaginase (ERY-ASP) might be a novel therapeutic strategy [73]. Moreover, increased circulating and intracellular branched-chain amino acids are also related to pancreatic cancer progression, which might be the result of enhanced tissue protein breakdown and decreased tumor use mediated by mutant TP53 [38, 56, 74].
Pancreatic cancer also has an extraordinary amino acid degradation ability, possessing urea cycle pathways comparable to those in the liver, which are critical for cancer as well. Obesity or constitutively active AKT, a kinase known to accelerate cancer growth, can induce high expression of arginase 2 that catabolizes arginine into urea and ornithine within the mitochondria of pancreatic cancer cells [75].
Lipids
Lipid metabolism is also essential for cancer progression [76]. It not only provides ample building blocks for rapid membrane formation but also produces signaling molecules and substrates for the posttranslational modification of proteins. Lipids can be acquired via biosynthesis and diet. In contrast to normal cells relying on dietary fat, approximately 93% of triacylglycerol fatty acids in tumor cells are de novo synthesized from mitochondrial citrate [77], which is the intermediate between mitochondria and cytosolic acetyl coenzyme A (CoA) [78]. In pancreatic cancer, many enzymes participating in de novo fatty acids and cholesterol synthesis are obviously upregulated, including citrate synthase (CS), ATP citrate lyase (ACLY), fatty acid synthase (FASN), stearoyl-CoA desaturase (SCD1) and 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) [78, 79] (Fig. 1). Additionally, hypoxia or oncogenic KRAS could also induce the uptake of monounsaturated fatty acids from extracellular lysophospholipids [80]. In de novo cholesterol synthesis, in addition to the elevated canonical pathway, overexpressed aldo-keto reductase 1B10 (AKR1B10) could metabolize farnesal and geranylgeranyl in pancreatic cancer, providing intermediates for cholesterol synthesis [81]. Additionally, those intermediates are also significant for protein prenylation, which activates KRAS and its downstream carcinogenic signaling pathways [81, 82]. Cholesterol acquisition also highly relies on enhanced extracellular uptake in pancreatic cancer cells. Compared with the modestly increased cholesterol synthesis pathway, overactive low-density lipoprotein receptor (LDLR)-mediated uptake of cholesterol-rich lipoproteins is predominant in murine pancreatic cancer cells [79]. After that, excessive free cholesterol is stored as cholesteryl ester within pancreatic cancer cells after esterification, which is mediated by highly expressed acyl-CoA cholesterol acyl-transferase-1 (ACAT-1) [83].
Among different fatty acids, saturated and monounsaturated fatty acids are considered to promote the growth of pancreatic cancer cells [84]. Polyunsaturated fatty acids, mainly containing the ω3 and ω6 families, dually affect pancreatic cancer. ω3 fatty acids inhibit cancer cell proliferation via reducing AKT phosphorylation, but ω6 fatty acids increase AKT phosphorylation [85]. However, a transcriptomics and metabolomics study revealed that lipase and a panel of fatty acids are significantly decreased in pancreatic tumors, and two saturated fatty acids, palmitate, and stearate, showed an obvious ability to inhibit the proliferation of pancreatic cancer cells [86]. Therefore, the role of fatty acids in pancreatic cancer is complicated and still not very clear. Cholesterol also participates in pancreatic cancer progression. Statins, inhibitors of cholesterol de novo synthesis, contribute to improved survival in pancreatic cancer patients in some clinical studies, but the underlying mechanism is still under debate [87]. However, a finished phase II clinical trial combining simvastatin with gemcitabine in advanced pancreatic cancer treatment failed to show clinical benefit (NCT00944463) [88]. Nevertheless, several clinical trials combining statins with other agents in pancreatic cancer treatments are still ongoing (NCT03889795) (NCT03889795). In contrast to the positive role of cholesterol in pancreatic cancer progression, high-level free cholesterol with ACAT-1 inhibition results in severe endoplasmic reticulum (ER) stress and cancer cell apoptosis [83].
Improving nutrient acquisition by scavenging and recycling
In addition to reprogramming the metabolism of glucose, amino acids and lipids within cells, pancreatic cancer cells have multiple other mechanisms by which they acquire enough fuels for survival and growth.
Macropinocytosis is a process located in the cell membrane that represents bulk extracellular fluid uptake through large endocytic vacuoles, which is crucial for maintaining the amino acid supply of pancreatic cancer cells after subsequent intracellular digestion and degeneration [89, 90]. Oncogenic KRAS plays a key role in promoting macropinocytosis with the help of αvβ3 and galectin-3 on the surface of tumor cells. Moreover, the mutant KRAS/galectin-3/αvβ3 complex also maintains the redox balance of pancreatic cancer cells [91, 92]. Autophagy is another critical cellular process that degrades cellular macromolecules and organelles, affording the recycling of intracellular bioenergetic components. Therefore, it plays a key role in maintaining energy homeostasis and metabolic fuel sources in the tumor [93]. Furthermore, autophagy also enables pancreatic cancer progression via controlling ROS production and sustaining OXPHOS [94]. In turn, elevated ROS promotes autophagy in pancreatic cancer cells [94]. This process is also under the regulation of KRAS. Surprisingly, recent significant studies suggested that the inhibition of KRAS and/or downstream RAF-MEK-ERK signaling pathway could obviously upregulate autophagic flux, which might be the metabolic adaption of compromised glycolysis and mitochondrial activity. Consistent with these findings, combining MEK and autophagy inhibition showed exciting results in both preclinical and clinical studies [95, 96]. TP53 also participates in the regulation of autophagy in pancreatic cancer, but the mechanism is unclear and merits further study [97, 98].
Both macropinocytosis and autophagy undergo degradation in lysosomes to regenerate nutrients. In this process, SLC38A9, an arginine-regulated transporter, facilitates the release of amino acids from lysosomes and activates mechanistic target of rapamycin complex 1 (mTORC1), both of which support pancreatic tumor growth [99]. mTORC1 is a homodimer composed of four mTOR units and a regulatory-associated protein. It is mainly activated by high levels of intracellular amino acids and growth factor signaling and phosphorylates multiple downstream targets and regulates metabolism and tumor progression [100]. Under nutrient-sufficient conditions, activated mTORC1 suppresses autophagy and inhibits the utilization of extracellular proteins through macropinocytosis [101,102,103]. However, under nutrient-poor circumstances, inactive mTORC1 contributes to the increase in autophagy and macropinocytosis [102,103,104] to sustain pancreatic tumor growth.
There are several other regulatory relationships involved. For example, MiT/TFE proteins promote the expression of autophagy-lysosome genes with the help of importin 8, regardless of the inhibition from active mTORC1 [105]. Recently, a study suggested that deprivation of amino acids could also induce protein scavenging independently of mTORC1 and that mTOR inhibition could restrict protein synthesis and preserve the intracellular amino acid pool, sustaining the growth of murine pancreatic tumor cells under amino acid deprivation [106].
Metabolic crosstalk within the microenvironment
The pancreatic cancer microenvironment is highly heterogeneous. In addition to cancer cells, ECM and stromal cells are also present. The interest in their interactions in metabolism has been increasing recently [107, 108] (Fig. 2).

Metabolic crosstalk within the microenvironment. There is extensive and heterogeneous metabolic crosstalk within pancreatic tumors. Pancreatic cancer cells can adopt relatively distinct metabolic patterns under different oxygen and nutrition conditions. Black arrows imply shift, positive regulation or fueling, whereas blunt ends indicate inhibition. Ala, alanine; HGF, hepatocyte growth factor
Pancreatic cancer cells are surrounded by tight desmoplasia composed of a collagen meshwork, resulting in hypoxic and nutrient-poor conditions, especially in the tumor core. This collagen provides Pro to fuel cancer cells [19]. Additionally, irregular shear stress caused by dense desmoplasia leads to PI3K/AKT signaling upregulation and ROS production, both of which can enhance glycolysis in cancer cells [109]. Furthermore, there are massive amounts of lactate in the tumor, which is distinctly treated by different pancreatic cancer cells under normoxia and hypoxia. Connexin-43 channels, a kind of gap junction, are important for transporting excess lactate from glycolytic pancreatic cancer cells in the tumor core to the periphery, supplying substrates for OXPHOS in better-perfused normoxic cancer cells and producing a suitable chemical milieu for pancreatic tumor growth [110]. Additionally, lactate within the microenvironment can also be sensed by GPR81, a Gi-coupled receptor on the pancreatic cancer membrane, promoting the expression of MCTs and CD147. In addition, activated GPR81 upregulates peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) and increases mitochondrial biogenesis and respiration. Therefore, both lactate absorption and use are improved, which is especially significant for the growth of pancreatic cancer cells in low glucose conditions [111]. A recent study also revealed that circulating lactate contributes to the TCA cycle of pancreatic tumors as a primary substrate during the fasted state [112]. Additionally, lactate produced by glycolytic cancer cells has some nonmetabolic roles in tumors, such as improving invasiveness, decreasing antitumor immunity and facilitating angiogenesis [37, 113].
Cancer-associated fibroblasts (CAFs), the major type of stromal cells, can be stimulated by neighboring cancer cells, to exhibit aerobic glycolysis and secrete high-energy metabolites, such as pyruvate and lactate. Adjacent cancer cells, particularly in normoxic regions, uptake and use the metabolites in OXPHOS. Such a pattern between CAFs and cancer cells is named the reverse Warburg effect or two-compartment metabolic coupling model, which has attracted considerable attention [44, 114, 115]. Therefore, more recent studies have refocused the effects of OXPHOS in pancreatic cancer cells [116, 117]. CAFs also enrich the microenvironment by releasing exosomes, which contain TCA cycle intermediates, amino acids, and lipids. They are key to pancreatic tumor growth, especially under nutrient-deprived conditions. However, pancreatic cancer cells display suppressed mitochondrial OXPHOS and increased glycolysis upon absorbing these exosomes [118]. The majority of CAFs are derived from pancreatic stellate cells (PSCs), which are activated during carcinogenesis. They can secrete autophagy-derived alanine to support pancreatic cancer cell metabolism, especially after being stimulated by cancer cells [119]. In addition to direct metabolite supply, activated PSCs can be particularly stimulated by oncogenic KRAS in adjacent pancreatic cancer cells, reciprocally enhancing downstream pathways of oncogenic KRAS signaling in cancer cells, including metabolic regulation [120]. Furthermore, enhanced glycolytic metabolism in pancreatic cancer cells can also be induced by paracrine hepatocyte growth factor (HGF) from PSCs [121]. Another recent study indicated that PSCs promote pancreatic cancer progression in a particular manner depending on PKM2 in either cancer cells or PSCs, but the mechanism is unclear and remains to be further studied [122]. In addition, saturated and monounsaturated fatty acids seem to be opposite players in fibrosis and activation of PSCs [123], but their particular roles in activated PSCs of pancreatic cancer are still unknown. Nevertheless, LipidemTM, an ω3 fatty acid-rich emulsion, shows the ability to decrease PSC proliferation and inhibit the invasive capacity of pancreatic cancer cells, especially in combination with gemcitabine [124].
Tumor-associated macrophages (TAMs), another essential type of stromal cell with immune functions, also participate in metabolic crosstalk within the microenvironment. Compared with steady-state macrophages in the normal pancreas, TAMs exhibit an elevated glycolytic signature, promoting pancreatic cancer vascularization and metastasis [125]. Another study showed that TAMs promote aerobic glycolysis in neighboring pancreatic cancer cells via paracrine signaling. Subsequently, lactate in the microenvironment promotes the procancer M2-like polarization of TAMs, which leads to low immunity [126]. Such a relationship was also revealed in preclinical experiments on novel molecules, providing hits for further research. Metavert, an inhibitor of glycogen synthase kinase 3β and histone deacetylases, could normalize the glucose metabolism of pancreatic cancer cells and transform M2-like TAMs to the anticancer M1 phenotype in mouse models [127].
Adipocytes also have extensive metabolic interactions with pancreatic cancer cells. After coculture with pancreatic cancer cells, adipocytes exhibited smaller size, mesenchymal phenotypes, decreased lipid content and multiple altered metabolic pathways. Such tumor-associated adipocytes could also promote the aggressiveness of pancreatic cancer cells [128]. In addition, a brief study in murine cell lines suggested that pancreatic cancer cells could inhibit Gln degeneration in cocultured adipocytes and then predispose them to Gln secretion. In turn, Gln derived from adipocytes facilitates cancer cell proliferation [129]. In addition to the direct interaction between pancreatic cancer cells and adipocytes, adipocyte accumulation within the microenvironment could interact with PSCs and tumor-associated neutrophils as well, enhancing tumor progression, particularly in obese patients [130]. There are many other stromal components besides the abovementioned components, and the metabolic crosstalk within the microenvironment of pancreatic cancer remains largely unclear.
Chemoresistance and metabolism
Chemotherapy is still the most fundamental systemic treatment against the majority of cancers. According to the NCCN guidelines, there are several distinct chemotherapy regimens against pancreatic cancer with different statuses. Among them, gemcitabine (also known as 2,2-difluorodeoxycytidin, dFdC), the nucleoside analog of deoxycytidine, currently remains the cornerstone of chemotherapy in pancreatic cancer treatments [131]. As a type of prodrug, gemcitabine enters pancreatic cancer cells and undergoes a series of phosphorylation events with precise regulation. After that, its derivatives can interfere with DNA synthesis and block cancer cell cycle progression [132, 133] (Fig. 3). Nano albumin-bound (nab) paclitaxel delivers a high concentration of paclitaxel within pancreatic tumors, resulting in the inhibition of microtubule depolymerization and cancer cell division [134, 135]. Benefiting from the synergistic effects [136, 137], the clinical application of nab paclitaxel often occurs in combination with gemcitabine. In addition to gemcitabine, 5-fluorouracil (5-FU), an analogue of uracil, also exerts anticancer effects by damaging DNA and RNA and inhibiting thymidylate synthase (TS) [138]. Although the clinical benefits produced by 5-FU are lower than those of gemcitabine, 5-FU is still widely applied in treating pancreatic cancer partly due to its lower toxicity [139]. In recent years, increasing evidence has shown that the FOLFIRINOX regimen (5-FU, leucovorin, irinotecan and oxaliplatin) can achieve longer overall survival than gemcitabine-based therapy, especially in patients with good status [140,141,142,143,144]. Compared with other chemotherapy regimens, the underlying mechanism of gemcitabine resistance is relatively well documented [131]. At present, there is increasing evidence that gemcitabine resistance is related to the metabolism of glucose, amino acids, and lipids (Fig. 4). Moreover, metabolic profiling revealed that there is an obvious difference in the metabolome between gemcitabine-sensitive and gemcitabine-resistant pancreatic cancer cell lines [145].

The metabolism and mechanisms of gemcitabine action. Gemcitabine plays an anticancer role after a series of phosphorylations in pancreatic cancer cells. Asterisks indicate that dFdCTP is the most active metabolite of gemcitabine that produces anticancer effects. Intermediates in gray ovals have anticancer functions. Long solid arrows represent shifts or bioconversions. Dotted arrows mean positive regulations, while dotted blunt ends mean negative regulations. dFdC, 2’,2’-difluorodeoxycytidine (gemcitabine); dFdCMP, 2’,2’-difluorodeoxycytidine 5’-monophosphate; dFdCDP, 2’,2’-difluorodeoxycytidine 5’-diphosphate; dFdCTP, 2’,2’-difluorodeoxycytidine 5’-triphosphate; dFdU, 2’,2’-difluorodeoxyuridine; dFdUMP, 2’,2’-difluorodeoxyuridine 5’-monophosphate; dFdUDP, 2’,2’-difluorodeoxyuridine 5’-diphosphate; dFdUTP, 2’,2’-difluorodeoxyuridine 5’-triphosphate; dUMP, deoxyuridine monophosphate; dTMP, deoxythymidine monophosphate; dTTP, deoxythymidine triphosphate; CDP, cytidine diphosphate; dCDP, deoxycytidine diphosphate; dCTP, deoxycytidine triphosphate; CDA, cytidine deaminase; dCK, deoxycytidine kinase; DCTD, deoxycytidylate deaminase; hNTs, human nucleosides transporters; NDPK, nucleoside diphosphate kinase; NMPK, nucleoside monophosphate kinase; RR, ribonucleotide reductase; TS, thymidylate synthase; 5’-NT, 5’-nucleotidase

Gemcitabine resistance and metabolism. Gemcitabine and some intrinsic characteristics of pancreatic cancer cells produce chemoresistance. Ovals are biological substances, rectangles are processes, and hexagons are statuses. Red names indicate corresponding inhibitors. Arrows represent positive regulations, and lines with blunt ends represent negative regulations. CR, chemoresistance; EMT, epithelial-mesenchymal transition; FBP1, fructose-1,6-bisphosphatase 1; IDH1, isocitrate dehydrogenase 1; LAT2, L-type amino acid transporter 2; noPPP, nonoxidative pentose phosphate pathway; RRM2, ribonucleotide reductase subunit-M2
Chemoresistant pancreatic cancer cell lines induced by long-term moderate gemcitabine treatment exhibit increased aerobic glycolysis and lower ROS levels than their parental cells. The increased glycolysis maintains low ROS levels that induce cancer stem cell (CSC) and epithelial-mesenchymal transition (EMT) phenotypes, contributing to chemoresistance [146]. Such enhanced glycolysis is partly mediated by increased HIF-1α. In addition to hypoxia, increased expression of MUC1, a transmembrane protein, also activates and stabilizes HIF-1α, enhancing glycolysis, nonoxidative PPP and pyrimidine biosynthesis [147,148,149]. All of these factors lead to gemcitabine resistance in pancreatic cancer cells. Owing to this mechanism, HIF-1α inhibitors (digoxin or YC1) and pyrimidine biosynthesis inhibitors (leflunomide) showed the ability to improve gemcitabine efficacy in animal studies [149]. Furthermore, MUC1 inhibition also sensitizes pancreatic cancer cell lines to 5-FU [150]. In addition, F-box and WD repeat domain-containing 7 (FBW7), a pancreatic tumor suppressor inhibited by oncogenic KRAS mutation, inhibited glycolysis in pancreatic cancer cells and enhanced the efficacy of gemcitabine in xenograft models as well [151]. In our previous research, we found that L-type amino acid transporter 2 (LAT2), an oncogenic protein in pancreatic cancer cells, could Gln-dependently activate mTOR to inhibit apoptosis and promote glycolysis. Both of them give rise to the gemcitabine-resistant phenotype, whereas mTOR inhibitor (RAD001) could solve such gemcitabine resistance [152]. Overexpressed Nampt provides massive NAD, sustaining enhanced glycolytic activity and contributing to gemcitabine resistance as well. Nampt inhibitor (FK866) reversed this resistance to sensitivity [53]. Additionally, gemcitabine also promotes the expression of the ribonucleotide reductase M2 (RRM2) subunit in pancreatic cancer cells through the ERK/E2F1 pathway, promoting deoxyribonucleotide biosynthesis and inhibiting gemcitabine-induced DNA damage [153, 154]. CG-5, a glucose transporter inhibitor, inhibits E2F1 expression and enhances gemcitabine efficacy in pancreatic cancer cells [153]. Gambogic acid and fructose-1,6-bisphosphatase 1 inhibits the ERK signaling pathway and bypasses gemcitabine resistance in mouse models with xenograft tumors [154, 155]. Increased thymidylate synthase expression in gemcitabine-treated pancreatic cancer cells might also adopt the same E2F1-dependent pathway, but this effect is not very clear yet [153]. In addition to pyrimidine biosynthesis, enhanced HBP in pancreatic cancer cells also gives rise to gemcitabine resistance by increasing the glycosylation of many proteins in several chemoresistant signaling pathways [156].
Compared with the relatively well-documented role of glucose metabolism, the role of amino acid metabolism in chemoresistance remains unclear. Enhanced Gln metabolism fuels elevated HBP and glycosylation. Moreover, Gln addiction is also significant for controlling ROS generation and activating mTOR, both of which contribute to chemoresistance [152, 156]. ROS production in pancreatic cancer cells triggered by gemcitabine treatment is presumed to be related to the dose. Low to moderately elevated ROS levels are considered to activate the nuclear factor-kappa B (NF-κB)/signal transducer and activator of transcription 3 (STAT3) signaling cascade, maintaining the CSC phenotype and inducing chemoresistance [157]. In addition to gemcitabine, low nutrient conditions also contribute to moderate ROS generation that activates HuR, an RNA binding protein. Activated HuR rapidly upregulates isocitrate dehydrogenase 1 (IDH1) to enhance NADPH recycling, which maintains redox balance and results in chemoresistance [158]. Therefore, compared with pancreatic cancer patients with high serum glucose, patients with low or normal serum glucose exhibit more severe initial gemcitabine resistance [158].
In lipid metabolism, FASN expression is upregulated, which is also crucial for gemcitabine resistance. Overexpressed FASN in pancreatic cancer cells upregulates PKM2 expression, promoting glycolysis and gemcitabine resistance [159]. PKM2 also plays a nonmetabolic role in chemoresistance by inhibiting gemcitabine-induced TP53 signaling and subsequent apoptosis [160]. In addition to PKM2, high FASN levels relieve ER stress, maintain the CSC phenotype and inhibit gemcitabine-induced apoptosis. Orlistat, a FASN inhibitor, induces ER stress and increases gemcitabine sensitivity in mouse models with orthotopic pancreatic cancer implantation [161]. ω3 fatty acids repress NF-kβ and STAT3 activation and improve the anticancer role of gemcitabine as well [162]. Furthermore, in a completed phase II clinical trial, after treatment with gemcitabine and intravenous ω3 fatty acid-rich emulsion, patients with metastatic or locally advanced pancreatic cancer exhibited reduced proinflammatory circulating growth factors and cytokines, which might contribute to the improved outcome (NCT01019382) [163]. In addition to fatty acids, cholesterol uptake disruption mediated by LDLR silencing also enhances gemcitabine-induced regression of murine pancreatic cancer cells [79]. Cholesterol also supports the function of caveolin-1 (cav-1) [164], which is the primary structural protein of caveolae, contributing to nab-paclitaxel uptake and chemosensitivity [165]. However, another study suggested that both cholesterol and cav-1 could maintain ABC transporters in caveolae, leading to drug efflux and chemoresistance to nab-paclitaxel in pancreatic tumor initiating cells with high CD133 expression [166]. In general, chemoresistance has intricate relations with the metabolism of pancreatic cancer cells, and further research is needed to achieve better chemotherapy responses.
In addition to the metabolism of glucose, amino acids, and lipids, autophagy in pancreatic cancer cells also plays a role in chemoresistance. Both nutrient limitation and gemcitabine induce autophagy in pancreatic cancer cells, which inhibits apoptosis and contributes to chemoresistance [167, 168]. Gemcitabine-induced autophagy might be mediated by a deubiquitinating protease, ubiquitin-specific peptidase 9X (USP9X). However, WP1130, a deubiquitinating enzyme inhibitor, inhibits USP9X and attenuates chemoresistance in mouse models bearing tumor xenografts [168]. Additionally, chloroquine also increases gemcitabine and 5-FU sensitivity as an autophagy inhibitor [168, 169]. In a completed phase I study, chloroquine showed promising effects in patients with unresectable or metastatic pancreatic cancer when combined with gemcitabine (NCT01777477) [170].
Although there is no evidence showing the direct relationship between metabolic crosstalk within the microenvironment and chemoresistance, the role of the microenvironment in chemoresistance has become more significant. For example, a recent study showed that CAFs in pancreatic cancer could scavenge gemcitabine and contribute to chemoresistance in murine pancreatic cancer [171]. Another study suggested that vitamin D receptors highly expressed on PSCs restrict the tumor-supportive role of PSCs and improve the delivery and efficacy of gemcitabine upon binding ligands [172]. Moreover, an OXPHOS inhibitor (metformin) could overcome CAF-induced chemotherapy resistance and enhance the efficacy of oxaliplatin in pancreatic cancer organoids [173]. In addition to CAFs, nab-paclitaxel internalization of TAMs via macropinocytosis could drive macrophage M1 polarization, restoring immune recognition in pancreatic cancer [174]. Given the extensive and vital metabolic crosstalk within the microenvironment, there might be many potential opportunities to promote the anticancer effects of current chemotherapy.
Radioresistance and metabolism
In contrast to chemotherapy, there are several controversies regarding the survival benefits of radiotherapy in pancreatic cancer [175,176,177]. However, it is still an efficient player in controlling the local progression of pancreatic cancer and other solid tumors [178]. According to the National Comprehensive Cancer Network guidelines on pancreatic cancer, radiotherapy is recommended as a neoadjuvant therapy for resectable or borderline disease, an adjuvant therapy for resected disease, a definitive treatment for locally advanced disease, and a palliative care strategy for terminal disease (relieving pain, bleeding and local obstructive symptoms). Additionally, radiotherapy is also recommended for local recurrent pancreatic cancer, but there are limited supporting data. Among many molecular and cellular pathways related to radiotherapy, some clues suggest that metabolic changes in pancreatic cancer are important factors that give rise to radioresistance [179].
Clinical investigations have shown that patients with high baseline metabolism in pancreatic cancer have a poor therapeutic response after receiving chemoradiotherapy [180, 181]. Increased glycolysis-nucleotide metabolism mediated by overexpressed MUC1 in pancreatic cancer also plays a key role in facilitating radioresistance [24]. 2-DG can increase metabolic oxidative stress and cause the radiosensitization of pancreatic cancer by inhibiting glucose metabolism [182]. Ketogenic diets represented by high fat and low carbohydrate intake increased radiotherapy sensitivity in mouse models with xenograft pancreatic cancer. However, a relevant phase I clinical trial in pancreatic cancer patients was not successful, mainly due to poor compliance (NCT01419483) [183]. In addition to glucose metabolism, upregulated FASN likely leads to radioresistance [184, 185]. Several genes involved in the cholesterol synthesis pathway are also associated with radioresistance in pancreatic cancer. Among them, overexpressed farnesyl diphosphate synthase can be inhibited by zoledronic acid (ZOL), which partly attenuated the radioresistance of pancreatic cancer cells [185]. A phase II clinical study combining ZOL with chemoradiotherapy followed by surgery in pancreatic cancer patients is ongoing (NCT03073785). The role of metabolism in pancreatic cancer radioresistance remains to be further researched.
Immunity and metabolism
Pancreatic cancer is a kind of low immunogenicity tumor that has a highly immunosuppressive microenvironment dominated by three main leukocyte subtypes: TAMs (mainly predominated by M2-type macrophages), regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) [186, 187]. Emerging evidence suggests that there are close relationships between metabolism and pancreatic cancer immunity, including immunosuppression and immunotherapy resistance.
Immunosuppression and metabolism
Glucose-dependent metabolism, especially aerobic glycolysis, is critical to fulfilling the immune functions of CD8+ effector T cells and IFN-γ production of CD4+ T cells [188, 189]. In contrast to CD4+ effector T cells (Th1, Th2, and Th17) and M1-type macrophages, Tregs and M2-type macrophages are mainly fueled by lipid oxidation and rely less on glycolysis [190, 191]. Tumor-associated MDSCs also undergo metabolic reprogramming, resulting in both enhanced fatty acid oxidation and increased glycolysis, which sustain their survival and contribute to their immunosuppressive functions [192, 193]. A study in sarcoma revealed that enhancing the glycolysis of tumor cells restricts glucose supply to nearby T cells, thereby leading to dysfunctional T cells and an immunosuppressive tumor microenvironment [25]. In addition to direct nutrition competition, tumor-derived lactate is key to remodeling immunity within the microenvironment, inducing the M2-like phenotype of TAMs and reducing CD8+ cytotoxic T cell functions [194, 195]. More significantly, lactate also upregulates MDSCs and inhibits natural killer cell activity in pancreatic cancer, resulting in an immunosuppressive microenvironment [196]. Additionally, tumor-induced interleukin 6 compromises host metabolism during caloric deficiency, giving rise to suppressed antitumor immunity [20]. In conclusion, metabolism provides novel directions for addressing immunosuppression, which limits the effects of many immunotherapies.
Immunotherapies and metabolism
Because of the low proportion of resectable cases and obvious resistance to chemotherapy and radiotherapy, immunotherapy has been rising as a novel strategy to treat pancreatic cancer. Many kinds of immunotherapies for pancreatic cancer have entered clinical trial stages, including immune checkpoint inhibitors [11], therapeutic vaccines and adoptive T cell transfers [197, 198]. However, most results are disappointing. Recently, accumulated studies have suggested that T cell-mediated immunotherapy could be optimized by modulating cell metabolism [199]. Moreover, immune checkpoint inhibitors also show the ability to support the metabolism of lymphocytes in the tumor and improve their antitumor effects [25, 200]. Given the relationship between metabolism and immunotherapy, it is appropriate to improve conventional immunotherapies through metabolic regulation.
In addition to immunotherapies focusing on improving the anticancer abilities of lymphocytes, the immunosuppressive microenvironment is emerging as a novel therapeutic target [201]. In addition to leukocyte subtypes, some cytokines also participate in immunosuppression of pancreatic cancer. For example, indoleamine 2,3-dioxygenase (IDO), a metabolic enzyme expressed in many carcinomas, including pancreatic cancer cells, degrades tryptophan within the tumor microenvironment and inhibits immune cell responses [202, 203]. Furthermore, combining IDO depletion and tumor desmoplasia inhibition showed successful antitumor effects in mouse models with pancreatic cancer [204]. Recently, a phase I/II trial combining IDO inhibitor (indoximod) and chemotherapy in patients with metastatic pancreatic cancer was completed (NCT02077881), while another phase II clinical study employing another IDO inhibitor (epacadostat) and immunotherapy or cyclophosphamide in pancreatic cancer patients is recruiting (NCT03006302). In general, given the significant role of metabolism in immunity, metabolic regulation has the potential to improve the clinical results of immunotherapies.
Clinical perspectives and conclusion
Metabolism targeted therapy is not yet recommended as regular treatment in most guidelines for treating variety of cancers. In addition to the wide use of aromatase inhibitors in treating breast cancer [205], most metabolism-targeted therapies against a variety of cancers remain in experimental and clinical trial phases. However, some of them have shown promising results. For instance, ivosidenib, an inhibitor of IDH1, improves the complete remission rate of IDH1-mutated acute myeloid leukemia with a low frequency of treatment-related adverse events in a phase I clinical trial that enrolled 258 patients [206]. In a phase IV clinical trial that enrolled 28 patients, the mechanism of diclofenac in effectively relieving actinic keratosis (a premalignant skin lesion) was well demonstrated, and the effect was largely dependent on modulating the metabolism of local lesions [207]. However, given the heterogeneity of different cancers, successful clinical applications in other cancers cannot be directly and simply applied to treating pancreatic tumors. In pancreatic cancer, many metabolic regulators have been employed in preclinical studies [208] and even in clinical trials [28, 34]. Compared with some old drugs, such as metformin [209], aspirin [210, 211], and statins, which play complex roles in metabolic regulation and have been debated over a dozen years, there are higher expectations for some new properties of other drugs, novel drug combinations and new metabolic regulators. In addition to the clinical trials mentioned above, many other clinical studies have also focused on metabolism in pancreatic cancer treatments (Table 1).
Therapies targeting altered glycolysis pathways are gaining momentum. In a phase I clinical trial in patients with solid tumors, including advanced pancreatic cancer, 2-DG produced tolerable adverse effects but tangible clinical benefits in combination with docetaxel (NCT00096707) [212]. Although PDH is inhibited by mutant KRAS [46], cancer cell survival still requires the persistence of PDH activity and mitochondrial metabolism. CPI-613, a lipoate analog, selectively inhibits tumor PDH activity and could disrupt pancreatic cancer growth in xenograft models [213]. An ongoing phase I study combining CPI-613 with modified FOLFIRINOX for treating metastatic pancreatic cancer showed a small increase in side effects and toxicity compared to that with FOLFIRINOX alone but an encouraging response rate (NCT01835041) [214].
For the noncanonical Gln metabolism of pancreatic cancer, GLS1 inhibition decreases antioxidant pools, whereas β-lapachone induces excess ROS generation specifically in pancreatic cancer cells as an NAD(P)H: quinone oxidoreductase 1-bioactivatable drug. Combining β-lapachone (ARQ761) with GLS1 inhibitors selectively leads to pancreatic cancer cell death in preclinical mouse models [215]. At present, a phase I clinical study that combined ARQ761 with gemcitabine/nab-paclitaxel in patients with advanced pancreatic cancer is ongoing (NCT02514031). For asparagine metabolism, ERY-ASP showed good tolerance in patients with metastatic pancreatic cancer in a phase I clinical study (NCT01523808) [216], and a further phase II clinical trial combining ERY-ASP with chemotherapy was completed recently (NCT02195180).
Given the comprehensive role of mTOR in metabolism, some clinical trials have employed mTOR inhibitors to address gemcitabine resistance in pancreatic cancer. However, RAD001 (everolimus) showed minimal clinical activity as a single agent in patients with metastatic and gemcitabine-resistant pancreatic cancer (NCT00409292) [217], whereas another phase II study combining everolimus with capecitabine showed moderately positive results and an acceptable toxicity profile (NCT01079702) [218]. Therefore, further clinical trials are anticipated.
The rewired glucose, amino acid, and lipid metabolism in pancreatic tumors from the cell to the microenvironment or even at the whole-body level deeply affects cancer progression. Furthermore, pancreatic cancer metabolism is also associated with anticancer treatments. Currently, more clinical trials in pancreatic cancer patients are beginning to involve metabolic regulation. Some of the completed trials even showed promising and exciting results. Given the side effects of some metabolic regulators [219], the role of metabolism targeted therapy in nonmalignant tissues should also be emphasized in future research, which largely restricts the transformation from basic study to successful clinical application. Moreover, pancreatic cancer displayed highly plastic metabolism, suggesting that cancer cells can adapt to use other metabolic pathways to bypass a certain metabolic inhibition [220]. In addition, pancreatic cancer cell lines also showed heterogeneous metabolic addictions [43]. Therefore, identification of the real metabolic hub of pancreatic cancer or combining distinct metabolism-targeted therapies in clinical trials is in high demand. In conclusion, a better understanding of pancreatic cancer metabolism and its role in treatments will benefit novel strategies, improving the prognosis of patients with pancreatic cancer.
Availability of data and materials
Not applicable.
Abbreviations
- 2-DG:
-
2-deoxyglucose
- 5-FU:
-
5-fluorouracil
- ACAT-1:
-
Acyl-CoA cholesterol acyl-transferase-1
- ACLY:
-
ATP citrate lyase
- AKR1B10:
-
Aldo-keto reductase 1B10
- Asn:
-
Asparagine
- Asp:
-
Aspartate
- CAFs:
-
Cancer-associated fibroblasts
- CARM1:
-
Coactivator-associated arginine methyltransferase 1
- cav-1:
-
caveolin-1
- CS:
-
citrate synthase
- CSC:
-
Cancer stem cell
- ECM:
-
Extracellular matrix
- EMT:
-
Epithelial-mesenchymal transition
- ER:
-
Endoplasmic reticulum
- ERY-ASP:
-
Erythrocyte-entrapped L-asparaginase
- FASN:
-
Fatty acid synthase
- FBW7:
-
F-box and WD repeat domain-containing 7
- FOXM1:
-
Forkhead box protein M1
- GDH:
-
Glutamate dehydrogenase
- GFPT1:
-
Glutamine:fructose-6-phosphate amidotransferase-1
- Gln:
-
Glutamine
- GLS1:
-
Glutaminase 1
- Glu:
-
Glutamate
- GLUT1:
-
Glucose transporter 1
- GOT1:
-
Cytoplasmic aspartate transaminase
- GOT2:
-
Mitochondrial aspartate transaminase
- HBP:
-
Hexosamine biosynthesis pathway
- HGF:
-
Hepatocyte growth factor
- HIF-1:
-
Hypoxia-inducible factor-1
- HMGCR:
-
3-hydroxy-3-methylglutaryl coenzyme A reductase
- IDH1:
-
Isocitrate dehydrogenase 1
- IDO:
-
Indoleamine 2,3-dioxygenase
- LAT2:
-
L-type amino acid transporter 2
- LDH:
-
Lactate dehydrogenase
- LDLR:
-
Low-density lipoprotein receptor
- MCT1:
-
Monocarboxylate transporter 1
- MDH1:
-
Malate dehydrogenase 1
- MDSCs:
-
Myeloid-derived suppressor cells
- mTORC1:
-
mechanistic target of rapamycin complex 1
- MUC1:
-
Mucin1
- NAD:
-
Nicotinamide adenine dinucleotide
- NF-κB:
-
Nuclear factor-kappa B
- OAA:
-
Oxaloacetate
- OXPHOS:
-
Oxidative phosphorylation
- PDH:
-
Pyruvate dehydrogenase
- PDHK1:
-
Pyruvate dehydrogenase kinase 1
- PGC-1α:
-
Peroxisome proliferator-activated receptor gamma coactivator-1α
- PGK1:
-
Phosphoglycerate kinase 1
- PKM2:
-
Pyruvate kinase muscle isozyme 2
- PPP:
-
Pentose phosphate pathway
- Pro:
-
Proline
- PRODH1:
-
Proline oxidase
- PSCs:
-
Pancreatic stellate cells
- ROS:
-
Reactive oxygen species
- RPE:
-
Ribulose-5-phosphate-3-epimerase
- RPIA:
-
Ribulose 5-phosphate isomerase
- RRM2:
-
Ribonucleotide reductase M2
- SCD1:
-
Stearoyl-CoA desaturase
- STAT3:
-
Signal transducer and activator of transcription 3
- TAMs:
-
Tumor-associated macrophages
- TIGAR:
-
TP53-induced glycolysis and apoptosis regulator
- Tregs:
-
Regulatory T cells
- TS:
-
Thymidylate synthase
- USP9X:
-
Ubiquitin-specific peptidase 9X
- ZOL:
-
Zoledronic acid
- α-KG:
-
α-ketoglutarate
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34.
Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–32.
Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–21.
Huang L, Jansen L, Balavarca Y, Molina-Montes E, Babaei M, van der Geest L, et al. Resection of pancreatic cancer in Europe and USA: an international large-scale study highlighting large variations. Gut. 2019;68:130–9.
Kleeff J, Reiser C, Hinz U, Bachmann J, Debus J, Jaeger D, et al. Surgery for recurrent pancreatic ductal adenocarcinoma. Ann Surg. 2007;245:566–72.
Kim MP, Gallick GE. Gemcitabine resistance in pancreatic cancer: picking the key players. Clin Cancer Res. 2008;14:1284–5.
Buwenge M, Macchia G, Arcelli A, Frakulli R, Fuccio L, Guerri S, et al. Stereotactic radiotherapy of pancreatic cancer: a systematic review on pain relief. J Pain Res. 2018;11:2169–78.
Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell. 2018;175:313–26.
Lee YT, Tan YJ, Oon CE. Molecular targeted therapy: treating cancer with specificity. Eur J Pharmacol. 2018;834:188–96.
Colli LM, Machiela MJ, Zhang H, Myers TA, Jessop L, Delattre O, et al. Landscape of combination immunotherapy and targeted therapy to improve cancer management. Cancer Res. 2017;77:3666–71.
Feng M, Xiong G, Cao Z, Yang G, Zheng S, Song X, et al. PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett. 2017;407:57–65.
Akce M, Zaidi MY, Waller EK, El-Rayes BF, Lesinski GB. The potential of CAR T cell therapy in pancreatic cancer. Front Immunol. 2018;9:2166.
Mosquera C, Maglic D, Zervos EE. Molecular targeted therapy for pancreatic adenocarcinoma: a review of completed and ongoing late phase clinical trials. Cancer Gene Ther. 2016;209:567–81.
Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6.
Neesse A, Algul H, Tuveson DA, Gress TM. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut. 2015;64:1476–84.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95.
Lyssiotis CA, Kimmelman AC. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 2017;27:863–75.
Olivares O, Mayers JR, Gouirand V, Torrence ME, Gicquel T, Borge L, et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat Commun. 2017;8:16031.
Flint TR, Janowitz T, Connell CM, Roberts EW, Denton AE, Coll AP, et al. Tumor-induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab. 2016;24:672–84.
McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet. 2017;49:367–76.
Carrer A, Trefely S, Zhao S, Campbell SL, Norgard RJ, Schultz KC, et al. Acetyl-CoA metabolism supports multistep pancreatic tumorigenesis. Cancer Discov. 2019;9:416–35.
Grasso C, Jansen G, Giovannetti E. Drug resistance in pancreatic cancer: impact of altered energy metabolism. Crit Rev Oncol Hematol. 2017;114:139–52.
Gunda V, Souchek J, Abrego J, Shukla SK, Goode GD, Vernucci E, et al. MUC1-mediated metabolic alterations regulate response to radiotherapy in pancreatic cancer. Clin Cancer Res. 2017;23:5881–91.
Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229–41.
Karasinska JM, Topham JT, Kalloger SE, Jang GH, Denroche RE, Culibrk L, et al. Altered gene expression along the glycolysis-cholesterol synthesis Axis is associated with outcome in pancreatic cancer. Clin Cancer Res. 2020;26:135–46.
Mehla K, Singh PK. Metabolic subtyping for novel personalized therapies against pancreatic cancer. Clin Cancer Res. 2020;26:6–8.
Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14:11–31.
Yuneva MO, Fan TW, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012;15:157–70.
Mayers JR, Vander Heiden MG. Nature and nurture: what determines tumor metabolic phenotypes? Cancer Res. 2017;77:3131–4.
Chaika NV, Yu F, Purohit V, Mehla K, Lazenby AJ, DiMaio D, et al. Differential expression of metabolic genes in tumor and stromal components of primary and metastatic loci in pancreatic adenocarcinoma. PLoS One. 2012;7:e32996.
Cancer Genome Atlas Research Network. Electronic address aadhe, cancer genome atlas research N: integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32:185–203 e113.
Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci U S A. 2012;109:15101–8.
Halbrook CJ, Lyssiotis CA. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell. 2017;31:5–19.
Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–37.
Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;292:504–7.
Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41:211–8.
Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science. 2016;353:1161–5.
Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–70.
Nagarajan A, Dogra SK, Sun L, Gandotra N, Ho T, Cai G, et al. Paraoxonase 2 facilitates pancreatic cancer growth and metastasis by stimulating GLUT1-mediated glucose transport. Mol Cell. 2017;67:685–701 e686.
Sherman MH, Yu RT, Tseng TW, Sousa CM, Liu S, Truitt ML, et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc Natl Acad Sci U S A. 2017;114:1129–34.
Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, et al. MYC/PGC-1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015;22:590–605.
Daemen A, Peterson D, Sahu N, McCord R, Du X, Liu B, et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc Natl Acad Sci U S A. 2015;112:E4410–7.
Liang C, Qin Y, Zhang B, Ji S, Shi S, Xu W, et al. Energy sources identify metabolic phenotypes in pancreatic cancer. Acta Biochim Biophys Sin Shanghai. 2016;48:969–79.
Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4:2935.
Li X, Jiang Y, Meisenhelder J, Yang W, Hawke DH, Zheng Y, et al. Mitochondria-translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol Cell. 2016;61:705–19.
Wu DH, Liang H, Lu SN, Wang H, Su ZL, Zhang L, et al. Yu S: miR-124 suppresses pancreatic ductal adenocarcinoma growth by regulating Monocarboxylate transporter 1-mediated cancer lactate metabolism. Cell Physiol Biochem. 2018;50:924–35.
Kong SC, Nohr-Nielsen A, Zeeberg K, Reshkin SJ, Hoffmann EK, Novak I, et al. Monocarboxylate transporters MCT1 and MCT4 regulate migration and invasion of pancreatic ductal adenocarcinoma cells. Pancreas. 2016;45:1036–47.
Schneiderhan W, Scheler M, Holzmann KH, Marx M, Gschwend JE, Bucholz M, et al. CD147 silencing inhibits lactate transport and reduces malignant potential of pancreatic cancer cells in in vivo and in vitro models. Gut. 2009;58:1391–8.
Santana-Codina N, Roeth AA, Zhang Y, Yang A, Mashadova O, Asara JM, et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat Commun. 2018;9:4945.
Kawada K, Toda K, Sakai Y. Targeting metabolic reprogramming in KRAS-driven cancers. Int J Clin Oncol. 2017;22:651–9.
Slawson C, Hart GW. O-GlcNAc signalling: implications for cancer cell biology. Nat Rev Cancer. 2011;11:678–84.
Ju HQ, Zhuang ZN, Li H, Tian T, Lu YX, Fan XQ, et al. Regulation of the Nampt-mediated NAD salvage pathway and its therapeutic implications in pancreatic cancer. Cancer Lett. 2016;379:1–11.
Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91–100.
Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–9.
Schofield HK, Zeller J, Espinoza C, Halbrook CJ, Del Vecchio A, Magnuson B, et al. Mutant p53R270H drives altered metabolism and increased invasion in pancreatic ductal adenocarcinoma. JCI Insight. 2018;3:97422.
Rajeshkumar NV, Dutta P, Yabuuchi S, de Wilde RF, Martinez GV, Le A, et al. Therapeutic targeting of the Warburg effect in pancreatic cancer relies on an absence of p53 function. Cancer Res. 2015;75:3355–64.
Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–20.
Butera G, Pacchiana R, Mullappilly N, Margiotta M, Bruno S, Conti P, et al. Mutant p53 prevents GAPDH nuclear translocation in pancreatic cancer cells favoring glycolysis and 2-deoxyglucose sensitivity. Biochim Biophys Acta, Mol Cell Res. 1865;2018:1914–23.
Cui J, Shi M, Xie D, Wei D, Jia Z, Zheng S, et al. FOXM1 promotes the Warburg effect and pancreatic cancer progression via transactivation of LDHA expression. Clin Cancer Res. 2014;20:2595–606.
Zhao D, Zou SW, Liu Y, Zhou X, Mo Y, Wang P, et al. Lysine-5 acetylation negatively regulates lactate dehydrogenase a and is decreased in pancreatic cancer. Cancer Cell. 2013;23:464–76.
Guillaumond F, Leca J, Olivares O, Lavaut MN, Vidal N, Berthezene P, et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A. 2013;110:3919–24.
Golias T, Papandreou I, Sun R, Kumar B, Brown NV, Swanson BJ, et al. Hypoxic repression of pyruvate dehydrogenase activity is necessary for metabolic reprogramming and growth of model tumours. Sci Rep. 2016;6:31146.
Li X, Deng S, Liu M, Jin Y, Zhu S, Deng S, et al. The responsively decreased PKM2 facilitates the survival of pancreatic cancer cells in hypoglucose. Cell Death Dis. 2018;9:133.
Kaira K, Sunose Y, Arakawa K, Ogawa T, Sunaga N, Shimizu K, et al. Prognostic significance of L-type amino-acid transporter 1 expression in surgically resected pancreatic cancer. Br J Cancer. 2012;107:632–8.
Coothankandaswamy V, Cao S, Xu Y, Prasad PD, Singh PK, Reynolds CP, et al. Amino acid transporter SLC6A14 is a novel and effective drug target for pancreatic cancer. Br J Pharmacol. 2016;173:3292–306.
Shanware NP, Mullen AR, DeBerardinis RJ, Abraham RT. Glutamine: pleiotropic roles in tumor growth and stress resistance. J Mol Med (Berl). 2011;89:229–36.
Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35:427–33.
Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–5.
Abrego J, Gunda V, Vernucci E, Shukla SK, King RJ, Dasgupta A, et al. GOT1-mediated anaplerotic glutamine metabolism regulates chronic acidosis stress in pancreatic cancer cells. Cancer Lett. 2017;400:37–46.
Wang YP, Zhou W, Wang J, Huang X, Zuo Y, Wang TS, et al. Arginine methylation of MDH1 by CARM1 inhibits glutamine metabolism and suppresses pancreatic cancer. Mol Cell. 2016;64:673–87.
Biancur DE, Paulo JA, Malachowska B, Quiles Del Rey M, Sousa CM, Wang X, et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun. 2017;8:15965.
Dufour E, Gay F, Aguera K, Scoazec JY, Horand F, Lorenzi PL, et al. Pancreatic tumor sensitivity to plasma L-asparagine starvation. Pancreas. 2012;41:940–8.
Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med. 2014;20:1193–8.
Zaytouni T, Tsai PY, Hitchcock DS, DuBois CD, Freinkman E, Lin L, et al. Critical role for arginase 2 in obesity-associated pancreatic cancer. Nat Commun. 2017;8:242.
Sunami Y, Rebelo A, Kleeff J. Lipid metabolism and lipid droplets in pancreatic cancer and stellate cells. Cancers (Basel). 2017;10:E3.
Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–77.
Swierczynski J, Hebanowska A, Sledzinski T. Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World J Gastroenterol. 2014;20:2279–303.
Guillaumond F, Bidaut G, Ouaissi M, Servais S, Gouirand V, Olivares O, et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc Natl Acad Sci U S A. 2015;112:2473–8.
Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci U S A. 2013;110:8882–7.
Chung YT, Matkowskyj KA, Li H, Bai H, Zhang W, Tsao MS, et al. Overexpression and oncogenic function of aldo-keto reductase family 1B10 (AKR1B10) in pancreatic carcinoma. Mod Pathol. 2012;25:758–66.
Zhang W, Li H, Yang Y, Liao J, Yang GY. Knockdown or inhibition of aldo-keto reductase 1B10 inhibits pancreatic carcinoma growth via modulating Kras-E-cadherin pathway. Cancer Lett. 2014;355:273–80.
Li J, Gu D, Lee SS, Song B, Bandyopadhyay S, Chen S, et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene. 2016;35:6378–88.
Yu M, Liu H, Duan Y, Zhang D, Li S, Wang F. Four types of fatty acids exert differential impact on pancreatic cancer growth. Cancer Lett. 2015;360:187–94.
Ding Y, Mullapudi B, Torres C, Mascarinas E, Mancinelli G, Diaz AM, et al. Omega-3 fatty acids prevent early pancreatic carcinogenesis via repression of the AKT pathway. Nutrients. 2018;10:1289.
Zhang G, He P, Tan H, Budhu A, Gaedcke J, Ghadimi BM, et al. Integration of metabolomics and transcriptomics revealed a fatty acid network exerting growth inhibitory effects in human pancreatic cancer. Clin Cancer Res. 2013;19:4983–93.
Huang BZ, Chang JI, Li E, Xiang AH, Wu BU. Influence of statins and cholesterol on mortality among patients with pancreatic cancer. J Natl Cancer Inst. 2017;109:2.
Hong JY, Nam EM, Lee J, Park JO, Lee SC, Song SY, et al. Randomized double-blinded, placebo-controlled phase II trial of simvastatin and gemcitabine in advanced pancreatic cancer patients. Cancer Chemother Pharmacol. 2014;73:125–30.
Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015;75:544–53.
Davidson SM, Jonas O, Keibler MA, Hou HW, Luengo A, Mayers JR, et al. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat Med. 2017;23:235–41.
Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497:633–7.
Seguin L, Camargo MF, Wettersten HI, Kato S, Desgrosellier JS, von Schalscha T, et al. Galectin-3, a Druggable vulnerability for KRAS-addicted cancers. Cancer Discov. 2017;7:1464–79.
Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab. 2017;25:1037–43.
Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–29.
Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, et al. Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med. 2019;25:620–7.
Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25:628–40.
Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300.
Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014;4:905–13.
Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, et al. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell. 2017;171:642–54 e612.
Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40:310–22.
He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93.
Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–9.
Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, Thompson CB. The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell. 2015;162:259–70.
Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–74.
Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361–5.
Nofal M, Zhang K, Han S, Rabinowitz JD. mTOR inhibition restores amino acid balance in cells dependent on catabolism of extracellular protein. Mol Cell. 2017;67:936–46 e935.
Dougan SK. The pancreatic cancer microenvironment. Cancer J. 2017;23:321–5.
Ren B, Cui M, Yang G, Wang H, Feng M, You L, et al. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol Cancer. 2018;17:108.
Tung JC, Barnes JM, Desai SR, Sistrunk C, Conklin MW, Schedin P, et al. Tumor mechanics and metabolic dysfunction. Free Radic Biol Med. 2015;79:269–80.
Dovmark TH, Saccomano M, Hulikova A, Alves F, Swietach P. Connexin-43 channels are a pathway for discharging lactate from glycolytic pancreatic ductal adenocarcinoma cells. Oncogene. 2017;36:4538–50.
Roland CL, Arumugam T, Deng D, Liu SH, Philip B, Gomez S, et al. Cell surface lactate receptor GPR81 is crucial for cancer cell survival. Cancer Res. 2014;74:5301–10.
Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, et al. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551:115–8.
San-Millan I, Brooks GA. Reexamining cancer metabolism: lactate production for carcinogenesis could be the purpose and explanation of the Warburg effect. Carcinogenesis. 2017;38:119–33.
Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001.
Chen S, Chen X, Shan T, Ma J, Lin W, Li W, et al. MiR-21-mediated metabolic alteration of cancer-associated fibroblasts and its effect on pancreatic cancer cell behavior. Int J Biol Sci. 2018;14:100–10.
Maertin S, Elperin JM, Lotshaw E, Sendler M, Speakman SD, Takakura K, et al. Roles of autophagy and metabolism in pancreatic cancer cell adaptation to environmental challenges. Am J Physiol Gastrointest Liver Physiol. 2017;313:G524–36.
Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res. 2018;24:2482–90.
Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife. 2016;5:e10250.
Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536:479–83.
Tape CJ, Ling S, Dimitriadi M, McMahon KM, Worboys JD, Leong HS, et al. Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation. Cell. 2016;165:1818.
Yan B, Jiang Z, Cheng L, Chen K, Zhou C, Sun L, et al. Paracrine HGF/c-MET enhances the stem cell-like potential and glycolysis of pancreatic cancer cells via activation of YAP/HIF-1alpha. Exp Cell Res. 2018;371:63–71.
Masamune A, Hamada S, Yoshida N, Nabeshima T, Shimosegawa T. Pyruvate kinase isozyme M2 plays a critical role in the interactions between pancreatic stellate cells and cancer cells. Dig Dis Sci. 2018;63:1868–77.
Ben-Harosh Y, Anosov M, Salem H, Yatchenko Y, Birk R. Pancreatic stellate cell activation is regulated by fatty acids and ER stress. Exp Cell Res. 2017;359:76–85.
Haqq J, Howells LM, Garcea G, Dennison AR. Targeting pancreatic cancer using a combination of gemcitabine with the omega-3 polyunsaturated fatty acid emulsion, Lipidem. Mol Nutr Food Res. 2016;60:1437–47.
Penny HL, Sieow JL, Adriani G, Yeap WH, See Chi Ee P, San Luis B, et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology. 2016;5:e1191731.
Ye H, Zhou Q, Zheng S, Li G, Lin Q, Wei L, et al. Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018;9:453.
Edderkaoui M, Chheda C, Soufi B, Zayou F, Hu RW, Ramanujan VK, et al. An inhibitor of GSK3B and HDACs kills pancreatic cancer cells and slows pancreatic tumor growth and metastasis in mice. Gastroenterology. 2018;155:1985–98 e1985.
Cai Z, Liang Y, Xing C, Wang H, Hu P, Li J, et al. Cancerassociated adipocytes exhibit distinct phenotypes and facilitate tumor progression in pancreatic cancer. Oncol Rep. 2019;42:2537–49.
Meyer KA, Neeley CK, Baker NA, Washabaugh AR, Flesher CG, Nelson BS, et al. Adipocytes promote pancreatic cancer cell proliferation via glutamine transfer. Biochem Biophys Rep. 2016;7:144–9.
Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M, et al. Obesity-induced inflammation and Desmoplasia promote pancreatic cancer progression and resistance to chemotherapy. Cancer Discov. 2016;6:852–69.
Amrutkar M, Gladhaug IP. Pancreatic cancer Chemoresistance to gemcitabine. Cancers (Basel). 2017;9:157.
Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W. Action of 2’, 2’-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991;51:6110–7.
Saif MW, Lee Y, Kim R. Harnessing gemcitabine metabolism: a step towards personalized medicine for pancreatic cancer. Ther Adv Med Oncol. 2012;4:341–6.
Yardley DA. Nab-paclitaxel mechanisms of action and delivery. J Control Release. 2013;170:365–72.
Giordano G, Pancione M, Olivieri N, Parcesepe P, Velocci M, Di Raimo T, et al. Nano albumin bound-paclitaxel in pancreatic cancer: current evidences and future directions. World J Gastroenterol. 2017;23:5875–86.
Frese KK, Neesse A, Cook N, Bapiro TE, Lolkema MP, Jodrell DI. Tuveson DA: nab-paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012;2:260–9.
Borsoi C, Leonard F, Lee Y, Zaid M, Elganainy D, Alexander JF, et al. Gemcitabine enhances the transport of nanovector-albumin-bound paclitaxel in gemcitabine-resistant pancreatic ductal adenocarcinoma. Cancer Lett. 2017;403:296–304.
Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–8.
Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–13.
Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med. 2018;379:2395–406.
Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–25.
Suker M, Beumer BR, Sadot E, Marthey L, Faris JE, Mellon EA, et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol. 2016;17:801–10.
Dhir M, Zenati MS, Hamad A, Singhi AD, Bahary N, Hogg ME, et al. FOLFIRINOX versus gemcitabine/nab-paclitaxel for neoadjuvant treatment of Resectable and borderline Resectable pancreatic head adenocarcinoma. Ann Surg Oncol. 2018;25:1896–903.
Chan KKW, Guo H, Cheng S, Beca JM, Redmond-Misner R, Isaranuwatchai W, et al. Real-world outcomes of FOLFIRINOX vs gemcitabine and nab-paclitaxel in advanced pancreatic cancer: a population-based propensity score-weighted analysis. Cancer Med. 2020;9:160–9.
Fujimura Y, Ikenaga N, Ohuchida K, Setoyama D, Irie M, Miura D, et al. Mass spectrometry-based metabolic profiling of gemcitabine-sensitive and gemcitabine-resistant pancreatic cancer cells. Pancreas. 2014;43:311–8.
Zhao H, Duan Q, Zhang Z, Li H, Wu H, Shen Q, et al. Up-regulation of glycolysis promotes the stemness and EMT phenotypes in gemcitabine-resistant pancreatic cancer cells. J Cell Mol Med. 2017;21:2055–67.
Chaika NV, Gebregiworgis T, Lewallen ME, Purohit V, Radhakrishnan P, Liu X, et al. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci U S A. 2012;109:13787–92.
Mehla K, Singh PK. MUC1: a novel metabolic master regulator. Biochim Biophys Acta. 1845;2014:126–35.
Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, et al. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell. 2017;32:71–87 e77.
Trehoux S, Duchene B, Jonckheere N, Van Seuningen I. The MUC1 oncomucin regulates pancreatic cancer cell biological properties and chemoresistance. Implication of p42-44 MAPK, Akt, Bcl-2 and MMP13 pathways. Biochem Biophys Res Commun. 2015;456:757–62.
Ji S, Qin Y, Liang C, Huang R, Shi S, Liu J, et al. FBW7 (F-box and WD repeat domain-containing 7) negatively regulates glucose metabolism by targeting the c-Myc/TXNIP (Thioredoxin-binding protein) Axis in pancreatic cancer. Clin Cancer Res. 2016;22:3950–60.
Feng M, Xiong G, Cao Z, Yang G, Zheng S, Qiu J, et al. LAT2 regulates glutamine-dependent mTOR activation to promote glycolysis and chemoresistance in pancreatic cancer. J Exp Clin Cancer Res. 2018;37:274.
Lai IL, Chou CC, Lai PT, Fang CS, Shirley LA, Yan R, et al. Targeting the Warburg effect with a novel glucose transporter inhibitor to overcome gemcitabine resistance in pancreatic cancer cells. Carcinogenesis. 2014;35:2203–13.
Xia G, Wang H, Song Z, Meng Q, Huang X, Huang X. Gambogic acid sensitizes gemcitabine efficacy in pancreatic cancer by reducing the expression of ribonucleotide reductase subunit-M2 (RRM2). J Exp Clin Cancer Res. 2017;36:107.
Jin X, Pan Y, Wang L, Ma T, Zhang L, Tang AH, et al. Fructose-1,6-bisphosphatase inhibits ERK activation and bypasses gemcitabine resistance in pancreatic cancer by blocking IQGAP1-MAPK interaction. Cancer Res. 2017;77:4328–41.
Chen R, Lai LA, Sullivan Y, Wong M, Wang L, Riddell J, et al. Disrupting glutamine metabolic pathways to sensitize gemcitabine-resistant pancreatic cancer. Sci Rep. 2017;7:7950.
Zhang Z, Duan Q, Zhao H, Liu T, Wu H, Shen Q, et al. Gemcitabine treatment promotes pancreatic cancer stemness through the Nox/ROS/NF-kappaB/STAT3 signaling cascade. Cancer Lett. 2016;382:53–63.
Zarei M, Lal S, Parker SJ, Nevler A, Vaziri-Gohar A, Dukleska K, et al. Posttranscriptional upregulation of IDH1 by HuR establishes a powerful survival phenotype in pancreatic cancer cells. Cancer Res. 2017;77:4460–71.
Tian S, Li P, Sheng S, Jin X. Upregulation of pyruvate kinase M2 expression by fatty acid synthase contributes to gemcitabine resistance in pancreatic cancer. Oncol Lett. 2018;15:2211–7.
Kim DJ, Park YS, Kang MG, You YM, Jung Y, Koo H, et al. Pyruvate kinase isoenzyme M2 is a therapeutic target of gemcitabine-resistant pancreatic cancer cells. Exp Cell Res. 2015;336:119–29.
Tadros S, Shukla SK, King RJ, Gunda V, Vernucci E, Abrego J, et al. De novo lipid synthesis facilitates gemcitabine resistance through endoplasmic reticulum stress in pancreatic cancer. Cancer Res. 2017;77:5503–17.
Hering J, Garrean S, Dekoj TR, Razzak A, Saied A, Trevino J, et al. Inhibition of proliferation by omega-3 fatty acids in chemoresistant pancreatic cancer cells. Ann Surg Oncol. 2007;14:3620–8.
Arshad A, Chung WY, Steward W, Metcalfe MS, Dennison AR. Reduction in circulating pro-angiogenic and pro-inflammatory factors is related to improved outcomes in patients with advanced pancreatic cancer treated with gemcitabine and intravenous omega-3 fish oil. HPB (Oxford). 2013;15:428–32.
Cordes N, Frick S, Brunner TB, Pilarsky C, Grutzmann R, Sipos B, et al. Human pancreatic tumor cells are sensitized to ionizing radiation by knockdown of caveolin-1. Oncogene. 2007;26:6851–62.
Chatterjee M, Ben-Josef E, Robb R, Vedaie M, Seum S, Thirumoorthy K, et al. Caveolae-mediated endocytosis is critical for albumin cellular uptake and response to albumin-bound chemotherapy. Cancer Res. 2017;77:5925–37.
Gupta VK, Sharma NS, Kesh K, Dauer P, Nomura A, Giri B, et al. Metastasis and chemoresistance in CD133 expressing pancreatic cancer cells are dependent on their lipid raft integrity. Cancer Lett. 2018;439:101–12.
Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell. 2014;159:1263–76.
Ma T, Chen W, Zhi X, Liu H, Zhou Y, Chen BW, et al. USP9X inhibition improves gemcitabine sensitivity in pancreatic cancer by inhibiting autophagy. Cancer Lett. 2018;436:129–38.
Hashimoto D, Blauer M, Hirota M, Ikonen NH, Sand J, Laukkarinen J. Autophagy is needed for the growth of pancreatic adenocarcinoma and has a cytoprotective effect against anticancer drugs. Eur J Cancer. 2014;50:1382–90.
Samaras P, Tusup M, Nguyen-Kim TDL, Seifert B, Bachmann H, von Moos R, et al. Phase I study of a chloroquine-gemcitabine combination in patients with metastatic or unresectable pancreatic cancer. Cancer Chemother Pharmacol. 2017;80:1005–12.
Hessmann E, Patzak MS, Klein L, Chen N, Kari V, Ramu I, et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut. 2018;67:497–507.
Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80–93.
Broekgaarden M, Anbil S, Bulin AL, Obaid G, Mai Z, Baglo Y, et al. Modulation of redox metabolism negates cancer-associated fibroblasts-induced treatment resistance in a heterotypic 3D culture platform of pancreatic cancer. Biomaterials. 2019;222:119421.
Cullis J, Siolas D, Avanzi A, Barui S, Maitra A, Bar-Sagi D. Macropinocytosis of nab-paclitaxel drives macrophage activation in pancreatic cancer. Cancer Immunol Res. 2017;5:182–90.
Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, Hickey H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. 2004;350:1200–10.
Loehrer PJ Sr, Feng Y, Cardenes H, Wagner L, Brell JM, Cella D, et al. Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: an eastern cooperative oncology group trial. J Clin Oncol. 2011;29:4105–12.
Rutter CE, Park HS, Corso CD, Lester-Coll NH, Mancini BR, Yeboa DN, et al. Addition of radiotherapy to adjuvant chemotherapy is associated with improved overall survival in resected pancreatic adenocarcinoma: an analysis of the National Cancer Data Base. Cancer. 2015;121:4141–9.
Badiyan SN, Molitoris JK, Chuong MD, Regine WF, Kaiser A. The role of radiation therapy for pancreatic cancer in the adjuvant and neoadjuvant settings. Surg Oncol Clin N Am. 2017;26:431–53.
Seshacharyulu P, Baine MJ, Souchek JJ, Menning M, Kaur S, Yan Y, et al. Biological determinants of radioresistance and their remediation in pancreatic cancer. Biochim Biophys Acta Rev Cancer. 1868;2017:69–92.
Dholakia AS, Chaudhry M, Leal JP, Chang DT, Raman SP, Hacker-Prietz A, et al. Baseline metabolic tumor volume and total lesion glycolysis are associated with survival outcomes in patients with locally advanced pancreatic cancer receiving stereotactic body radiation therapy. Int J Radiat Oncol Biol Phys. 2014;89:539–46.
Kurahara H, Maemura K, Mataki Y, Sakoda M, Iino S, Kawasaki Y, et al. Significance of (18) F-Fluorodeoxyglucose (FDG) uptake in response to Chemoradiotherapy for pancreatic cancer. Ann Surg Oncol. 2019;26:644–51.
Coleman MC, Asbury CR, Daniels D, Du J, Aykin-Burns N, Smith BJ, et al. 2-deoxy-D-glucose causes cytotoxicity, oxidative stress, and radiosensitization in pancreatic cancer. Free Radic Biol Med. 2008;44:322–31.
Zahra A, Fath MA, Opat E, Mapuskar KA, Bhatia SK, Ma DC, et al. Consuming a ketogenic diet while receiving radiation and chemotherapy for locally advanced lung cancer and pancreatic cancer: the University of Iowa experience of two phase 1 clinical trials. Radiat Res. 2017;187:743–54.
Yang Y, Liu H, Li Z, Zhao Z, Yip-Schneider M, Fan Q, et al. Role of fatty acid synthase in gemcitabine and radiation resistance of pancreatic cancers. Int J Biochem Mol Biol. 2011;2:89–98.
Souchek JJ, Baine MJ, Lin C, Rachagani S, Gupta S, Kaur S, et al. Unbiased analysis of pancreatic cancer radiation resistance reveals cholesterol biosynthesis as a novel target for radiosensitisation. Br J Cancer. 2014;111:1139–49.
Clark CE, Beatty GL, Vonderheide RH. Immunosurveillance of pancreatic adenocarcinoma: insights from genetically engineered mouse models of cancer. Cancer Lett. 2009;279:1–7.
Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer. 2013;108:914–23.
Cham CM, Driessens G, O'Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol. 2008;38:2438–50.
Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–51.
Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–303.
Huang SC, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15:846–55.
Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. 2015;3:1236–47.
Jian SL, Chen WW, Su YC, Su YW, Chuang TH, Hsu SC, et al. Glycolysis regulates the expansion of myeloid-derived suppressor cells in tumor-bearing hosts through prevention of ROS-mediated apoptosis. Cell Death Dis. 2017;8:e2779.
Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–63.
Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–9.
Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. 2013;191:1486–95.
Ducreux M, Seufferlein T, Van Laethem JL, Laurent-Puig P, Smolenschi C, Malka D, et al. Systemic treatment of pancreatic cancer revisited. Semin Oncol. 2019;46:28–38.
Foley K, Kim V, Jaffee E, Zheng L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016;381:244–51.
Kishton RJ, Sukumar M, Restifo NP. Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab. 2017;26:94–109.
Franchina DG, He F, Brenner D. Survival of the fittest: cancer challenges T cell metabolism. Cancer Lett. 2018;412:216–23.
Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-Centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17:651–62.
Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–74.
Wang D, Saga Y, Mizukami H, Sato N, Nonaka H, Fujiwara H, et al. Indoleamine-2,3-dioxygenase, an immunosuppressive enzyme that inhibits natural killer cell function, as a useful target for ovarian cancer therapy. Int J Oncol. 2012;40:929–34.
Manuel ER, Chen J, D'Apuzzo M, Lampa MG, Kaltcheva TI, Thompson CB, et al. Salmonella-based therapy targeting Indoleamine 2,3-dioxygenase coupled with enzymatic depletion of tumor Hyaluronan induces complete regression of aggressive pancreatic tumors. Cancer Immunol Res. 2015;3:1096–107.
Early Breast Cancer Trialists’ Collaborative G. Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet. 2015;386:1341–52.
DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with Ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378:2386–98.
Singer K, Dettmer K, Unger P, Schonhammer G, Renner K, Peter K, et al. Topical diclofenac reprograms metabolism and immune cell infiltration in actinic keratosis. Front Oncol. 2019;9:605.
Rajeshkumar NV, Yabuuchi S, Pai SG, De Oliveira E, Kamphorst JJ, Rabinowitz JD, et al. Treatment of pancreatic cancer patient-derived xenograft panel with metabolic inhibitors reveals efficacy of Phenformin. Clin Cancer Res. 2017;23:5639–47.
Bhaw-Luximon A, Jhurry D. Metformin in pancreatic cancer treatment: from clinical trials through basic research to biomarker quantification. J Cancer Res Clin Oncol. 2016;142:2159–71.
Yue W, Yang CS, DiPaola RS, Tan XL. Repurposing of metformin and aspirin by targeting AMPK-mTOR and inflammation for pancreatic cancer prevention and treatment. Cancer Prev Res (Phila). 2014;7:388–97.
Mormile R. Aspirin and pancreatic cancer-letter. Cancer Epidemiol Biomark Prev. 2017;26:978.
Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;71:523–30.
Zachar Z, Marecek J, Maturo C, Gupta S, Stuart SD, Howell K, et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J Mol Med (Berl). 2011;89:1137–48.
Alistar A, Morris BB, Desnoyer R, Klepin HD, Hosseinzadeh K, Clark C, et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: a single-Centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017;18:770–8.
Chakrabarti G, Moore ZR, Luo X, Ilcheva M, Ali A, Padanad M, et al. Targeting glutamine metabolism sensitizes pancreatic cancer to PARP-driven metabolic catastrophe induced by ss-lapachone. Cancer Metab. 2015;3:12.
Bachet JB, Gay F, Marechal R, Galais MP, Adenis A, Ms CD, et al. Asparagine Synthetase expression and phase I study with L-Asparaginase encapsulated in red blood cells in patients with pancreatic adenocarcinoma. Pancreas. 2015;44:1141–7.
Wolpin BM, Hezel AF, Abrams T, Blaszkowsky LS, Meyerhardt JA, Chan JA, et al. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J Clin Oncol. 2009;27:193–8.
Kordes S, Klumpen HJ, Weterman MJ, Schellens JH, Richel DJ, Wilmink JW. Phase II study of capecitabine and the oral mTOR inhibitor everolimus in patients with advanced pancreatic cancer. Cancer Chemother Pharmacol. 2015;75:1135–41.
Voss M, Lorenz NI, Luger AL, Steinbach JP, Rieger J, Ronellenfitsch MW. Rescue of 2-Deoxyglucose side effects by ketogenic diet. Int J Mol Sci. 2018;19:2462.
Biancur DE, Kimmelman AC. The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. Biochim Biophys Acta Rev Cancer. 1870;2018:67–75.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China Grants (81974376 to Mr. Yupei Zhao; 81972321 to Ms. Lei You; 81773215 to Mr. Weibin Wang), and the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2018PT32014).
Author information
Authors and Affiliations
Contributions
YZ, WW and LY directed and guided this study. CQ collected related literature and drafted this manuscript. GY, JY, BR, HW, GC, FZ, LY, YZ made critical revisions to this manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qin, C., Yang, G., Yang, J. et al. Metabolism of pancreatic cancer: paving the way to better anticancer strategies. Mol Cancer 19, 50 (2020). https://doi.org/10.1186/s12943-020-01169-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-020-01169-7