
Abstract
Pancreatic cancer is a deadly disease with high mortality due to difficulties in its early diagnosis and metastasis. The tumor microenvironment induced by interactions between pancreatic epithelial/cancer cells and stromal cells is critical for pancreatic cancer progression and has been implicated in the failure of chemotherapy, radiation therapy and immunotherapy. Microenvironment formation requires interactions between pancreatic cancer cells and stromal cells. Components of the pancreatic cancer microenvironment that contribute to desmoplasia and immunosuppression are associated with poor patient prognosis. These components can facilitate desmoplasia and immunosuppression in primary and metastatic sites or can promote metastasis by stimulating angiogenesis/lymphangiogenesis, epithelial-mesenchymal transition, invasion/migration, and pre-metastatic niche formation. Some molecules participate in both microenvironment formation and metastasis. In this review, we focus on the mechanisms of pancreatic cancer microenvironment formation and discuss how the pancreatic cancer microenvironment participates in metastasis, representing a potential target for combination therapy to enhance overall survival.
Similar content being viewed by others


Background
Despite the significant advances in cancer research, pancreatic cancer is still a deadly disease. According to the latest epidemiological data, a total of 55,440 patients were newly diagnosed with pancreatic cancer, and 44,330 people died from the disease in the United States. In contrast to other cancer types with continuous increases in survival, that of pancreatic cancer decreased slightly, and the disease is commonly diagnosed at an advanced stage, leading to a 5-year survival rate of only 8% [1]. Among patients with pancreatic cancer, 90% carry a KRAS mutation, which is considered a driver gene for pancreatic cancer progression, and 50–80% have inactivating mutations in TP53, CDKN2A, and SMAD4 [2]. Pathological results have shown that the most common type of pancreatic cancer is pancreatic ductal adenocarcinoma (PDAC). Pancreatic cancer is associated with an extremely poor prognosis and high mortality because it is usually diagnosed at advanced stages with metastasized pancreatic cancer cells, requiring cellular elements that contribute to pancreatic cancer microenvironment formation.
Increasing interest has been focused on the tumor microenvironment of pancreatic cancer. The pancreatic cancer microenvironment consists of cancer cells, stromal cells and extracellular components. Stromal cells that contribute to pancreatic cancer progression are mainly pancreatic stellate cells (PSCs), regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs). These cells and cancer cells can secrete extracellular components, such as extracellular matrix (ECM), matrix metalloproteinase (MMP), growth factors, and transforming growth factor-β (TGFβ), to maintain the microenvironment. Recent studies have demonstrated that the pancreatic cancer microenvironment plays a critical role in PDAC progression [3], revealing the relationship between the microenvironment and metastasis. There are two major characteristics of the pancreatic cancer microenvironment: dense desmoplasia and extensive immunosuppression [4]. These two features can facilitate pancreatic cancer cell proliferation, the evasion of immune surveillance via the direct inhibition of anti-tumor immunity or the induction of immunosuppressive cell proliferation and metastasis. Therefore, this review discusses pancreatic cancer microenvironment formation and the mechanisms by which the microenvironment participates in metastasis to elucidate the relationship between the microenvironment and metastasis.
Characteristics of the pancreatic cancer microenvironment
Desmoplasia
In pancreatic cancer, there is extensive fibrosis at primary tumor sites, which is termed desmoplasia and documented in the pathology of PDAC. The clinical manifestations of desmoplasia are overexpression of ECM proteins and extensive transformation of fibroblastic-type cells to a myofibroblastic phenotype [5]. Desmoplasia is associated with a poor prognosis by promoting the progression of pancreatic cancer and resistance to chemotherapy [6]. These cellular components can promote desmoplasia in the pancreatic cancer microenvironment through the secretion of certain molecules, such as TGFβ, fibroblast growth factor 2 (FGF2), and connective tissue growth factor (CTGF). Among these factors, TGFβ is notable for its dual nature in cancer. It can prevent neoplastic cell growth during pancreatic intraepithelial neoplasm-1 (PanIN-1) and PanIN-2 while promoting growth during PanIN-3 due to the loss of SMAD4 and the canonical arm of the TGFβ pathway [7, 8], and it interacts at many levels with the RAS-RAF-ERK pathway [9]. Specimens from patients have shown that ECM deposition in primary tumors is associated with a poor prognosis of patients with pancreatic cancer [10]. Desmoplasia can establish a hypoxic microenvironment by enhancing the functions of antiangiogenic factors. Hypoxia, which is caused by an insufficient vasculature, is important for pancreatic cancer aggressiveness, including metabolic reprogramming, inhibition of apoptosis, sustained proliferation, treatment resistance, invasion and metastasis [11]. In contrast to other solid tumors, pancreatic cancer cells can secrete antiangiogenic factors, such as angiostatin, endostatin, and pigment epithelium-derived factors, into the hypovascular microenvironment, and ECM deposition can amplify endostatin production by cancer cells to enhance hypoxia [12,13,14]. Recent studies have reported that desmoplasia components might be potential therapeutic targets of pancreatic cancer. For instance, hyaluronan degradation by hyaluronidase PEGPH20 has been shown to increase vessel patency, drug delivery and survival in preclinical trials [15, 16], and the efficacy of hyaluronidase PEGPH20 plus gemcitabine is currently being evaluated in an ongoing phase I/II trial (NCT01453153).
Immunosuppression
The relationship between anti-tumor immunity and pancreatic cancer progression has been a hot topic in studies of pancreatic cancer. The immune system during pancreatic cancer can not only suppress tumor development or progression by destroying cells with mutations and prevent them from developing into tumor cells, but it can also promote pancreatic cancer progression by establishing favorable conditions for immunosuppression and metastasis [17, 18]. The tumor infiltrating lymphocyte (TIL) population in pancreatic cancer shows clinical correlates that higher proportions of CD4+, CD8+ and dendritic cells (DCs) of TILs can improve the prognosis of affected patients with [19]. Among these immune cells, CD8+ T cells play an essential role in killing tumor cells, and a greater number of cancer cell adjacent cytotoxic T cells significantly correlates with survival [20]. Normally, antigen-presenting cells (APCs), such as macrophages and DCs, process tumor antigens for display on major histocompatibility complex (MHC) I molecules, which activate subsequent CD8+ T cells to kill tumor cells via the granzyme, perforin, and first apoptosis signal (Fas)/FasL pathway.
Pancreatic cancer cells themselves are critical for immunosuppression by inhibiting CD8+ T cell activation and upregulating the existing regulatory immune cells. Pancreatic cancer has been shown to decrease its MHC I expression to prevent recognition by CD8+ T cells for evasion [21]. Furthermore, Fas expression is downregulated in pancreatic cancer cells, which leads to resistance to Fas-mediated apoptosis, and pancreatic cancer can induce apoptosis of CD8+ T cells by Fas/FasL counterattack [22]. Kaplan-Meier survival analysis demonstrated that high levels of Fas cytoplasmic expression in pancreatic cancer cells are significantly associated with a better outcome of pancreatic cancer [23]. Immunosuppressive cytokines such as interleukin (IL)-10 and TGFβ are also secreted during pancreatic cancer to help fibrosis, immunosuppressive phenotype formation and recruit cells involved in immune evasion to overcome the anti-tumor immunity [24,25,26]. Indoleamine 2,3-dioxygenase (IDO) catalyzes the conversion from tryptophan to kynurenine, which leads to anergy of anti-tumor T cells and enhances Treg function [27,28,29]. Moreover, high expression of IDO in pancreatic cancer cells can mediate nature killer (NK) cells dysfunction [30]. Commonly, Forkheadbox protein 3 (Foxp3), a transcription factor considered a marker of Tregs, is also expressed on cancer cells. Cancer-Foxp3 causes immunosuppression by inducing Treg accumulation via CCL5 and negatively correlates with a poor prognosis in pancreatic cancer [31]. Finally, pancreatic cancer cells express cytotoxic-T-lymphocyte associated protein 4 (CTLA-4) and the ligand for programmed cell death protein-1 (PD-1), PD-L1 [32], which is activated by the EGFR/MAPK pathway from myeloid cells [33], inhibiting T cell function. Currently, CTLA-4 and PD-1/PD-L1 have been established as therapeutic targets [34, 35]. These molecules can be blocked by monoclonal antibodies, including ipilimumab, nivolumab, and pembrolizumab. However, immune-checkpoint inhibition monotherapy may not be effective in pancreatic cancer, potentially because of the low PD-L1 expression in pancreatic cancer, highly complicated interaction between the tumor and stroma, and desmoplasia [34]. Therefore, immunotherapies combined with other therapies, such as surgery, chemotherapy, radiotherapy, targeted therapy, and other immunotherapies (Table 1), may overcome the resistance of immunotherapy
Pancreatic cancer interacts with stromal cells in the microenvironment
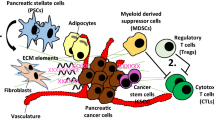
The pancreatic cancer microenvironment is characterized as dense desmoplasia and extensive immunosuppression with abundant cellular elements, mainly involving pancreatic cancer cells, Tregs, MDSCs, TAMs, and PSCs. Pancreatic cancer cells can activate or recruit other cellular elements for desmoplasia and immunosuppression and finally facilitate metastasis. Additionally, these cells can promote pancreatic cancer cell growth, proliferation, and maintenance of stemness. Compared with other types of cancer, pancreatic cancer cells only consist of approximately 10–30% of the cellular components, while dense stroma makes up 80% of the tumor mass in some patients [36]. Thus, other tumor-supporting cells are critical for the pancreatic cancer microenvironment to facilitate desmoplasia, immunosuppression, and pancreatic cancer progression.
PSCs
PSCs are considered myofibroblast-like cells that are located in exocrine regions of the pancreas [37] and share similarities with hepatic stellate cells. Quiescent PSCs store vitamin A and produce matrix metalloproteinases (MMPs) such as MMP-2, MMP-9, and MMP-13 and their inhibitors for turnover of the extracellular matrix (ECM), to maintain the modeling of normal tissue [38].
During PDAC, quiescent PSCs are activated by a variety of factors, such as IL-1, IL-6, hypoxia inducible factor 1α (HIF1α), and TGFβ, to transform them into the myofibroblast-like phenotype [39]. Activated PSCs are classified as a loss of cytoplastic lipid droplets, upregulation of MMP and ECM proteins [37], and activated PSCs acquire proliferative capacity. Additionally, activated PSCs play a vital role in the pancreatic cancer microenvironment by secreting molecules such as TGFβ, IL-6, stromal cell-derived factor-1 (SDF-1), hepatocyte growth factor (HGF) and galectin-1 to promote pancreatic cancer progression [40].
PSCs are cells that mainly contribute to desmoplasia in pancreatic cancer. Recent evidence has demonstrated that PSCs can induce desmoplasia via numerous signaling pathways, such as IL-6, paracrine sonic hedgehog (SHH) signaling, the vitamin D receptor (VDR) pathway, and the CXCL12/CXCR4 pathway [41]. Among these signaling pathways activating PSCs, SHH expression in pancreatic cancer cells is induced by KRAS by the activation of nuclear factor-κB (NFκB) [42]. GLI1, the target gene of SHH, shows a predominant signal in the stromal compartment [43]. Thus, pancreatic cancer cells express SHH to activate GLI1 in stroma to create a tumor-supportive microenvironment. In addition, PSC inhibition is a potential therapeutic target because it can inhibit desmoplasia [3, 44].
Recently, the relationship between immunosuppression and PSCs has been demonstrated. CXC chemokine ligand 12 (CXCL12) secreted by PSCs has the ability to reduce the migration of CD8+ T cells into the peritumoral stroma of pancreatic cancer [45]. Galectin-1 promotes immunosuppression in the pancreatic cancer microenvironment by inducing T cell apoptosis and Th2 cytokine secretion [46]. In addition to suppressing T cells directly, PSCs can recruit other immune cells into the tumor microenvironment to assist their immunosuppressive function. For instance, PSCs recruit Tregs via the IP-10/CXCL10 pathway [47], and IP-10 is elevated in pancreatic cancer patients, which is associated with a high stroma content and a decreased median overall survival [48]. Moreover, differentiation from peripheral blood monocytes into MDSCs can be induced by PSCs via the IL-6/signal transducer and activator of transcription 3 (STAT3) pathway to suppress T cell proliferation [49]. Although studies of the relationship between PSCs and immune evasion are still in the early stage, PSCs have potential as a target of pancreatic cancer to enhance immunotherapy in the future.
Tregs
CD4+ CD25+ Tregs (hereafter referred to as Tregs), also known as suppressor T cells, are a subtype of T cells that maintain tolerance to self-antigens and prevents autoimmune disease by suppressing or downregulating the induction and proliferation of effector T cells [50]. They are recruited by pancreatic cancer cells into the tumor microenvironment and play an important role in immunosuppression during pancreatic cancer progression. Higher proportions of Tregs in TILs are associated with progression and a poorer prognosis of patients with pancreatic ductal adenocarcinoma (PDAC) [51]. Foxp3 and CTLA-4 mRNA expression are higher in Tregs from the peripheral blood of patients with progressed and advanced pancreatic cancer, and there should further be a positive correlation between the IL-10 or TGFβ levels and the progression of pancreatic cancer [52].
Tregs can suppress tumor immunity through a variety of pathways. For instance, Tregs secrete suppressive cytokines and molecules, such as IL-10 and TGFβ, consistent with clinical findings, to inhibit effector T cell functions [53]. Another mechanism by which Tregs induce effector T cell cytolysis involves granzyme B [54, 55], the TRAIL pathway [56] and galectin-1 [57]. Moreover, Tregs can promote pancreatic cancer growth via the TRAIL pathway [58]. In addition, Tregs bind to IL-2 competitively to starve effector cells [59]. CTLA-4 expressed by Tregs can upregulate the IDO pathway in DCs [60] and effector T cells [29] and lead to their dysfunction.
MDSCs
MDSCs, which are defined as a heterogeneous population of immature myeloid cells in spleen and tumor, play a critical role in immunosuppression of pancreatic cancer. The markers of MDSCs are CD11b+ CD33+ HLA-DR− in humans. Patients with pancreatic cancer have higher MDSC and pro-MDSC cytokine levels in the peripheral blood, and MDSCs in peripheral blood may be a predictive biomarker of chemotherapy failure in pancreatic cancer patients [61]. Pancreatic cancer consistently induces the proliferation and mobility of MDSCs within the bone marrow to the tumor microenvironment [62] via cytokines, especially granulocyte macrophage colony-stimulating factor (GM-CSF), which is widely studied and produced by pancreatic cancer cells. GM-CSF is associated with differentiation from myeloid progenitor cells to MDSCs and MDSC recruitment to the pancreatic cancer microenvironment [63]. The upregulation of GM-CSF is induced by the driver gene KRASG12D [64], which is mutated in more than 90% of pancreatic cancer cases [2].
When MDSCs enter the pancreatic cancer microenvironment, they can suppress the functions of effector T cells via numerous pathways. For instance, MDSCs release reactive oxygen species (ROS), induced by cytokines such as TGFβ and IL-10 [65] released from other cells, causing oxidative stress in T cells. As a result, the CD3ζ chain in T cells cannot be translated, which inhibits antigen-dependent proliferation [66]. Polymorphonuclear cells (PMN-MDSC) possess the ability to take up, process and present antigens on MHC I proteins. These complexes can present antigen to CD8+ T cells to induce immune tolerance for immune evasion [67]. Arginase 1 (Arg1), depending on STAT3 signaling [68], and inducible nitric oxide synthase (iNOS) in MDSC are capable of depleting L-arginine from the tumor microenvironment, such that T cells are unable to multiply. STAT3 signaling pathways can be activated in MDSCs by IL-10, and STAT3 phosphorylation not only induces Arg1 expression but also upregulates PD-L1 expression on the surface of MDSCs [69], which suppresses T cell activation. In addition, MDSCs in vitro have been shown to be capable of inducing Treg development, the functions of which have been previously discussed, and targeted depletion of an MDSC subset—Gr-MDSC—leads to the accumulation of activated CD8+ T cells, apoptosis of tumor cells, and remodeling of the tumor stroma [70].
TAMs
Furthermore, macrophages derived from monocytes are phagocytic cells involved in the innate immune system. They participate in desmoplasia and immunosuppression during pancreatic cancer progression. A recent study showed that mutant KRASG12D can upregulate intercellular adhesion molecular-1 (ICAM-1) in acinar cells, serving as a chemoattractant for macrophages [71]. As a result of their plasticity, macrophages consist of a heterogeneous population of cells with different functional and phenotypic characteristics [72]. According to the activation mechanism, macrophages are classified as M1 (activated by IFN-γ and TLR ligands, the expression of higher levels of IL-12, IL-23, MHC II, and inducible nitric oxide synthase, and tumoricidal) or M2 (activated by IL-4 and IL-13, the expression of higher levels of IL-10 and TGFβ, and facilitate tumor progression) [73, 74]. Flow cytometry has demonstrated that tumor-derived factors stimulate the differentiation of macrophages, with mixed M1-like and M2-like phenotype polarization [75]. The M2-like phenotype of tumor-associated macrophages (TAMs) is immunosuppressive, and overall survival is shorter in pancreatic cancer patients with high-density M2 macrophage than low-density M2 macrophage infiltration [76]. Factors in the pancreatic cancer microenvironment, such as CSF-1, IL-4, IL-13, TGFβ and IL-10, can promote myeloid progenitor cell differentiation into monocytes and macrophages and recruit them to the tumor microenvironment [73, 77,78,79].
An immunosuppressive activity of TAMs includes the secretion of immunosuppressive cytokines, chemokines, and enzymes, such as TGFβ, IL-10, CCL17, and CCL22 [80]. Similar to MDSCs, TAMs can express more Arg1 to interfere with the metabolism of effector T cells [81], and factors such as TGFβ, IL-10, and prostaglandin E2 (PGE2) released by TAMs favor Treg recruitment and inhibit CD8+ T cells activities [82]. Moreover, NLRP3 signaling in macrophages drives the differentiation of CD4+ T cells into tumor-promoting T helper type 2 cells (Th2 cells), Th17 cells, and the Treg population, while suppressing Th1 cell polarization and cytotoxic CD8+ T cell activation [83]. TAMs may also induce apoptosis of T cells by expressing PD-L1 on their surface, which is similar to pancreatic cancer cells and MDSCs. Additionally, dectin-1 activation on macrophages via galectin-9 in the tumor microenvironment results in peritumoral immune tolerance in pancreatic cancer [84].
Furthermore, TAMs contribute to desmoplasia by facilitating PSCs. For instance, M2 macrophages can promote pancreatic fibrosis [85], and macrophages can drive PDAC fibrosis, immunosuppression and metastasis via the PI3Kγ pathway [86]. In vitro co-culture of macrophages and PSCs has demonstrated that the macrophage-stellate cell interaction is a pivotal component of desmoplasia in PDAC [87], and previous studies have demonstrated that TAMs can upregulate PSC functions. For example, TAMs can stimulate PSC proliferation and ECM secretion via TGFβ1 and PDGF, respectively [88]. In addition, hypoxia can lead to the recruitment of macrophages to activate PSCs through CCL2 secretion induced by HIF1, enhancing desmoplasia by PSCs [89]. Overall, the functions of TAMs seem to be diverse because they participate in many steps of pancreatic cancer progression.
Pancreatic cancer stem cells
Pancreatic cancer stem cells (CSCs) are considered a small subset of pancreatic cancer cells that can self-renew and generate the heterogeneous lineages of cancer cells in the tumor. They are a fundamental driving force of pancreatic cancer initiation and progression [90]. Recent studies have demonstrated that pancreatic CSCs can model the tumor microenvironment to favor their stemness maintenance, including self-renewal, tumorigenic, and metastatic potential. The main signaling pathways involved in this process are Wnt/β-catenin, hedgehog, notch, NF-κB, PI3K/Akt and PTEN, and they are dysregulated in pancreatic cancer [91,92,93,94,95].
Pancreatic CSCs can differentiate into bulk tumor cells partially in response to autocrine growth factor signaling. For example, activin and nodal, secreted by pancreatic CSCs, can cause pancreatic cancer cells to form spheres by binding to the receptors Alk4/7 [96]. Xenograft tumors from pancreatic CSCs with decreased levels of Alk4/7 can enhance sensitivity to gemcitabine and lead to longer survival times than pancreatic CSCs with high levels of Alk4/7 [96].
In addition to pancreatic CSCs themselves, stromal cells can maintain the pancreatic CSC population via paracrine signaling pathways. PSCs can secrete activin and nodal, similar to pancreatic CSCs, to promote the formation of tumor spheres in vitro and invasiveness of pancreatic CSCs in an Alk4-dependent manner [97]. HGF from PSCs can promote self-renewal of c-MetHigh pancreatic CSCs. In addition to the observations for PSCs, TAMs can also secrete factors to maintain the functions of pancreatic CSCs. A clinical study has demonstrated a positive correlation between the expression of CD44/CD133 and CD204, a marker of TAMs, and higher expression of these 3 markers was associated with shorter overall survival and disease-free survival [98]. Pancreatic CSCs can secrete IFNβ to stimulate TAM production of IFN-stimulated factor ISG15 to enhance the CSC phenotype in vitro and in vivo [99]. TAMs also produce leucine leucine (LL)-37 in the response to tumor growth factor, associated with a shorter overall survival, to increase pluripotency-associated gene expression, self-renewal, invasion and tumorigenicity of pancreatic CSCs via formyl peptide receptor 2 (FPR2)- and P2X purinoceptor 7 receptor (P2X7R)-dependent mechanisms [100]. In conclusion, the pancreatic cancer microenvironment can influence the stemness of pancreatic CSCs by multiple pathways (Fig. 1).

Development of the pancreatic cancer microenvironment. Pancreatic cancer cells secrete cytokines and chemokines to recruit or activate stromal cells for desmoplasia and immune evasion, including myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), regulatory T cells (Tregs), and pancreatic stellate cells (PSCs). Among them, PSCs are the main source of ECM deposition, and the TAM-PSC axis can facilitate desmoplasia. These cells within the pancreatic cancer microenvironment can help pancreatic cancer cells inhibit CD8+ T cells to overcome the immune surveillance by expressing or producing various factors, such as IL-10, TGFβ, PD-L1, and IDO
The pancreatic cancer microenvironment participates in metastasis
Metastasis is a major cause of morbidity and mortality in patients with pancreatic cancer, and the main pathways of metastasis are local invasion and lymphatic metastasis. Currently, only 20% of patients have resectable pancreatic cancer, with an 80% relapse rate. The majority of pancreatic cancer patients are locally advanced and unresectable disease due to vascular involvement or widespread metastasis, and the main pathways of metastasis are local invasion and lymphatic metastasis. Most patients will die with metastases to the liver, lung and/or peritoneum, the most common sites of distal metastasis [101]. Steps of successful metastasis include angiogenesis/lymphangiogenesis, epithelial-mesenchymal transition (EMT), invasion to surround tissues and migration, formation of a pre-metastatic niche, and growth at the metastatic site. Cancer cells should be depleted by anti-tumor immune cells, especially CD8+ T cells and NK cells, during each step of metastasis. Unfortunately, pancreatic cancer cells can overcome the anti-tumor immunity and metastasize to other sites due to the recruited immunosuppressive cells and their interactions.
Angiogenesis
Pancreatic cancer cells and other recruited immunosuppressive cells within the tumor microenvironment, such as TAMs and MDSCs, induce angiogenesis for the tumor blood supply and metastasis by secreting several pro-angiogenic factors, cytokines, and growth factors. Among these molecules, vascular endothelial growth factor (VEGF) plays a central role in the angiogenesis of pancreatic cancer, and the secretion of VEGF is regulated by multiple signaling pathways. For pancreatic cancer cells, STAT3 is constitutively expressed in pancreatic cancer cells, which activates the expression of VEGF for angiogenesis [102]. Mucin (MUC) 1 in pancreatic cancer cells can induce hypoxia for VEGF-A and platelet-derived growth factor (PDGF) B production, which contributes to endothelial cell tube formation [103]. NF-κB is another protein that regulates VEGF expression. Activated NF-κB in pancreatic cancer cells can upregulate VEGF [104], and xanthohumol can decrease VEGF expression and inhibit angiogenesis of pancreatic cancer by suppressing the NF-κB pathway [105]. TAMs are also involved in angiogenesis through the secretion of VEGF [106], and pharmacological targeting of tumor-infiltrating macrophages is associated with impaired angiogenesis [107]. In addition to VEGF, a recent study of xenograft models has shown that the angiogenesis of TAMs can be induced by pancreatic cancer cells, which secrete IL-35 to recruit TAMs and activate their CXCL1 and CXCL8 to stimulate angiogenesis, and the combination of a neutralizing antibody against IL-35 and gemcitabine significantly decreased monocyte infiltration and microvessel density [108].
Lymphangiogenesis and lymphatic metastasis
Similar to angiogenesis in many regards, tumor-associated lymphangiogenesis is a key step for pancreatic cancer progression, especially lymph node metastasis of pancreatic cancer. New lymphatic vessel growth can be directed by many factors derived from pancreatic cancer cells and other cells, such as M2-like TAMs [109], in the tumor microenvironment. Clinical results have shown that a high lymphatic vessel density in pancreatic ductal adenocarcinoma (PDAC) correlates with the tumor differentiation status, increased lymph node metastasis and decreased overall survival [110,111,112,113]. In comparison to normal lymph nodes, more Tregs, MDSCs, immature and tolerogenic DCs and immunosuppressive cytokines [114, 115] lie in tumor-draining lymph nodes, and the Treg density in pancreatic cancer tissue and lymph nodes correlates with pancreatic cancer lymphatic metastasis [116]. Coincidentally, a greater quantity of CD10+ PSCs in PDAC tissue indicates positive nodal metastasis and a shorter survival time [117], and M2-like TAMs can increase the lymphatic vessel density in pancreatic cancer and accelerate lymphatic metastasis [118]. These results indicate that these cells may be involved in lymphangiogenesis and lymphatic metastasis. Molecular components involved in lymphangiogenesis resemble those in angiogenesis, and VEGF-C/D appears to be important for lymphangiogenesis. Clinical studies have shown that VEGF-C/D expression increases in PDAC patients and correlates with increased lymphatic vessel invasion, lymph node metastasis, and a decreased five-year survival [119]. Knockdown of VEGF-C by anti-VEGF-C shRNA can decrease the lymphatic vessel density and inhibit tumor growth [120]. A recent study has demonstrated that microRNAs are involved in the regulation of VEGF-C production in PDAC cells. MicroRNA-206 can block K-Ras and annexin-A2 gene expression to suppress PDAC progression and downregulate VEGF-C production to inhibit lymphatic vessel formation through an NF-κB-independent pathway. In addition to VEGF, other factors within the pancreatic cancer microenvironment also participate in lymphangiogenesis and lymphatic metastasis. Inhibition of pancreatic cancer-derived SHH, an important molecule in embryonic development, reduces lymphangiogenesis and lymph node metastasis in a pancreatic cancer mouse model [121]. The proline TP53 variant stimulates lymphangiogenesis in the orthotopic pancreatic cancer mouse model [122].
Epithelial-mesenchymal transition
EMT in cancer is a process by which the cancer cells lose their cell-cell adhesion capacity and break through the basement membrane for invasion, during the initiation of metastasis. Specimens from pancreatic cancer patients indicate that EMT status is associated with portal vein invasion and lymph node metastasis [123]. A tag and track pancreatic epithelial cell experiment in premalignant lesions and pancreatic cancer mouse models has shown that pancreatic epithelial cells can invade and enter the bloodstream to become circulating epithelial cells (CECs), maintaining the mesenchymal phenotype even at the PanIN stage, and inflammation enhances EMT, invasiveness, and dissemination of pancreatic epithelial cells [124]. Another premalignant lesion, the intraductal papillary mucinous neoplasm (IPMN), which is classified as low-grade dysplasia (adenoma), intermediate-grade dysplasia (borderline), and high-grade dysplasia (carcinoma in situ) [125] and can be detected at an early stage, also undergoes EMT. Franses et al. demonstrated that CECs can be detected in mice and 88% of patients with IPMNs, and RNA-seq analysis showed that EMT of pancreatic epithelial cells in IPMNs may be driven by MUC genes [126]. Cellular elements in the tumor microenvironment, such as TAMs and PSCs, which provides the links among inflammation, premalignant lesions and cancer, can facilitate EMT. For example, M2-like TAMs can increase MMP2 and MMP9 activity in pancreatic cancer cells and decrease E-cadherin, indicating that EMT and Toll-like receptor (TLR) 4 expression and IL-10 production, are upregulated in M2-like TAMs to stimulate EMT when cocultured with pancreatic cancer cells [127]. Galectin-1, a key protein in immunosuppression and secreted by PSCs, promotes the development and metastasis of PDAC. Overexpression of galectin-1 in PSCs strongly correlates with increased expression of EMT markers in both the orthotopic xenograft tumor and in metastatic lesions of naked mice [128]. The IL-6/STAT3 pathway in PSCs, which recruits MDSCs to the pancreatic cancer microenvironment, also displays the function of promoting EMT by activating nuclear factor erythroid 2 (Nrf2) [129]. In addition to TAMs and PSCs, the relationship between pancreatic CSCs and EMT cancer cells may promote metastasis due to similar molecular characteristics, treatment resistance, and the capacity for invasion [130]. However, the exact association with EMT and pancreatic CSCs must be further investigated.
Invasion and migration
Invasion and migration are important for pancreatic cancer metastasis, especially hematological dissemination. Once pancreatic cancer cells invade capillaries in the tumor tissue, they can enter portal veins for distal metastasis, such as metastasis to the liver and lung. The pancreatic cancer microenvironment can facilitate metastasis by promoting pancreatic cancer cells invasion and migration. For instance, TAMs promote invasion by secreting matrix proteins and proteases to alter the ECM composition. Macrophage inflammatory protein-3α (MIP-3α) derived from TAMs in human pancreatic cancer tissue is considered a regulator of pancreatic cancer cell invasion [131]. MIP-3α can bind to CCR6 on pancreatic cells to upregulate their MMP9 expression, which increases pancreatic cancer cells invasion in collagen IV [132, 133]. Cancer-associated fibroblasts (CAFs) express high levels of palladin, an actin-associated protein, which promotes pancreatic cancer cells invasion by remodeling the ECM by regulating the activity of the small GTPase Cdc42 [134]. Among these cells in the microenvironment, PSCs play a more important role in tumor invasion and migration. In vitro studies have shown that CD10+ PSCs can promote the invasiveness of SUIT-2 pancreatic cancer cell lines in a murine cotransplantation model [117], and collagen-I, produced by PSCs, is the major mediator of PSC-induced haptokinesis of Panc1 and haptotaxis of UlaPaCa by activating FAK signaling via binding to integrin α2β1 [135]. Hypoxia plays a critical role in pancreatic cancer progression by inducing HIF1 to activate numerous genes involved in invasion and metastasis, such as NF-κB [136], MMP-2 [137, 138], quiescin-sulfhydryl-oxidase-1 (QSOX1) [139], CX3CR1 [140], and lysyl-oxidase (LOX) [141]. Clinical specimens have shown that SHH, induced by hypoxia via a ligand-independent mechanism [142], is overexpressed in pancreatic cancer cells and activates PSCs to secrete high levels of perineural invasion-associated molecules to promote perineural invasion in pancreatic cancer [143]. Galectin-1, which has been discussed previously, is expressed by PSCs and can induce PSC secretion of SDF-1 by NF-κB signaling and increase the migration and invasion of pancreatic cancer cells [144]. HGF, secreted by PSCs, can bind to c-Met on the surface of pancreatic cancer cells to promote their invasion and migration via the HGF/c-Met/survivin pathway [145], which is negatively regulated by the p53/p21 pathway, and HGF inhibition by AMG102 antibody can reduce pancreatic cancer metastasis dramatically in an orthotopic model of pancreatic cancer and the pancreatic cancer cell line AsPC-1 [146]. Periostin, which is a 90-kD secretory protein that was originally identified as an osteoblast-specific factor, is aberrantly upregulated in PSCs [147]. Abnormally high expression levels of periostin can increase α-SMA, periostin, collagen-1, fibronectin and TGFβ expression in PSCs and can promote growth, resistance to starvation, and invasion of pancreatic cancer cells [147]. Further studies have demonstrated that periostin creates the tumor-supportive microenvironment by binding to EGFR to trigger the Akt and Erk pathway [148].
Pre-metastatic niche
The entry of circulating tumor cells (CTCs), considered the “seed” of metastasis, into secondary or distant organ sites and growth at metastatic sites are affected by the local microenvironment encountered by CTCs. Primary tumors can prepare the supportive microenvironment, or “soil”, in metastatic sites, termed the pre-metastatic niche [149]. Molecular and cellular components, containing exosomes and factors secreted by the tumor, stroma, and bone-marrow-derived cells, can alter the pre-metastatic niche for pancreatic cancer colonization. The pre-metastatic niche can not only promote pancreatic cancer progression directly but also induce tumor dormancy at metastatic sites for recurrence at metastatic sites.
Liver is the main destination for pancreatic cancer distal metastasis, and factors derived from the pancreatic primary tumor microenvironment can induce fibrosis in liver to form a pre-metastatic niche by hepatic stellate cells (HSCs) that is similar to PSCs. For instance, a coculture study has shown that pancreatic cancer cells can directly stimulate HSC proliferation and matrix synthesis, including collagen I and c-fibronectin protein, and form an immunosuppressive microenvironment. These effects can be inhibited by antibodies against fibroblast growth factor 2 (FGF2), TGFβ1, and PDGF [150]. Similar to the occurrence of EMT at premalignant lesions, factors derived from premalignant lesions can also activate HSCs. Grunwald et al. [151] found that pancreatic tissue from patients with chronic pancreatitis, PanIN, and PDAC expresses higher levels of tissue inhibitor of metalloproteinases-1 (TIMP1) than normal pancreas. The premalignant lesions in KPC mice express TIMP1 and secrete it into the circulation to activate HSCs by binding to CD63 to trigger the PI3K pathway, but not TIMP1 protease inhibitor activity. Moreover, TIMP1 can increase the susceptibility of liver to pancreatic cancer cells. Subsequently, studies examining mutant p53 in pancreatic cancer have indicated that mutant p53 cannot inhibit pancreatic cancer progression but correlates with lymph node metastasis and upregulated PDGF receptor β signaling, which stimulate pancreatic cancer cell invasion in vitro and metastasis formation in vivo. Furthermore, the expression of PDGF receptor β correlates with poor disease-free survival in pancreatic cancer patients [152, 153]. In addition, mutant p53 together with TGFβ in pancreatic cancer cells can increase its ability to colonize the portal vein and hepatic sinusoid by secreting prometastatic mediators, including Col6A1 and Lcn2 [154].
Recent studies have demonstrated that PDAC-derived exosomes play a critical role in pre-metastatic niche formation. Costa-Silva et al. [155] demonstrated that exosomes from the pancreatic primary tumor can be taken up by Kupffer cells, upregulating TGFβ secretion and fibronectin production in HSCs to form a fibrotic microenvironment in the liver, which can recruit macrophages by macrophage migration for immunosuppression. Additionally, a greater number of exosomes are found in stage I PDAC patients who later develop liver metastasis, indicating that exosomes could promote liver metastasis and may be a diagnostic marker [155]. In addition, CD44v6 can promote pancreatic cancer growth and metastasis through the MET and VEGFR2 pathway [156], and pre-metastatic niche formation induced by exosomes requires CD44v6 [157]. In another study of exosomes derived from human breast cancer and PDAC cell lines that metastasize to the lung, liver, or both, the expression patterns of integrins in exosomes were examined, and the results indicated that the integrins α6β4 and α6β1 are associated with lung metastasis, whereas integrin αvβ5 correlates with liver metastasis [158]. Moreover, exosome integrin uptake by resident cells (lung fibroblasts and epithelial cells, liver Kupffer cells) activates Src phosphorylation and pro-inflammatory S100 gene expression, which contributes to pre-metastatic niche formation [158].
Other immune cells can also facilitate pre-metastatic niche formation. For instance, TAMs can secrete granulin to activate HSC differentiation into myofibroblasts for fibrotic microenvironment formation in liver to support metastatic PDAC growth [159]. In addition, MDSCs and neutrophils participate in liver pre-metastasis niche formation via the CXCR2 pathway. CXCR2 is a G-protein-coupled receptor for human CXC chemokines to control neutrophil and MDSC migration [160,161,162]. Steele CW et al. demonstrated that CXCR2 or Ly6G+ (a marker of neutrophils and MDSCs) cell depletion and CXCR2 inhibition can abrogate metastasis in KPC (LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx1-Cre) mice by failing to establish a metastatic niche, while CXCR2 inhibition substantially enhances sensitivity to anti-PD1 immunotherapy and prolongs survival in KPC mice [163].
Cancer dormancy at metastatic sites is responsible for the establishment of metastatic lesions and influenced by the (pre-) metastatic niche. In pancreatic cancer, downregulation of KRAS and c-Myc may be markers of dormant pancreatic cancer cells. Although KARS inhibition should be an effective strategy to treat pancreatic cancer, the rapid recurrence of primary and metastatic cancer following reactivation of KRAS supports the existence of dormant cancer cells in the animal model [164]. In addition, reactivation of c-Myc causes pancreatic cancer recurrence [165]. Lin et al. [165] demonstrated that c-Myc-negative tumor cells can express exogenous c-Myc, indicating that the dormant population is enriched by pancreatic CSCs. As mentioned before, the stemness of pancreatic CSCs is maintained by microenvironmental components such as TAMs and PSCs, indicating that TAMs and PSCs may participate in pancreatic cancer dormancy. Furthermore, Lenk et al. [166] found that liver sections of tumor-bearing KPC mice consisting of micrometastases displaying weakly proliferative and quiescent HSCs can mediate the quiescence-associated phenotype of pancreatic ductal epithelial cells, with a flattened cell morphology, Ki67-negativity and reduced proliferation, in coculture. This study showed that quiescent HSCs may induce pancreatic cancer dormancy in the liver metastatic niche, and a switch from quiescent to inflammatory activated HSC can enhance proliferation of pancreatic ductal epithelial cells (Fig. 2).

The pancreatic cancer microenvironment participates in metastasis. The pancreatic cancer microenvironment influences every step of metastasis via multiple signaling pathways. (1) The pancreatic cancer microenvironment can stimulate angiogenesis by cytokines to favor cancer cell survival and proliferation. (2) Molecules from the pancreatic cancer microenvironment can induce lymphangiogenesis to establish a pathway for lymphatic metastasis. (3) The pancreatic cancer microenvironment can facilitate the epithelial-mesenchymal transition to cause cancer cells to enter lymphatic vessel. (4) The pancreatic cancer microenvironment can play important roles in invasion and migration to facilitate metastasis. (5) Factors and exosomes derived from the pancreatic cancer microenvironment can induce pre-metastatic niche formation in liver and lung. These molecules or exosomes can activate hepatic stellate cells (HSCs) in liver for desmoplasia
Conclusions
Clinical studies have demonstrated that components within the pancreatic cancer microenvironment correlate with a poor prognosis of patients and can facilitate desmoplasia and immunosuppression or promote metastasis via numerous signaling pathways associated with the failure of immunotherapy, chemotherapy or radiation therapy. Pancreatic cancer cells can produce signaling molecules to recruit or activate stromal and immunosuppressive cells, such as Tregs, MDSCs, TAMs, and PSCs, even at a very early stage of pancreatic cancer development, to establish the microenvironment, which plays important roles in evolutionary and ecological processes in pancreatic cancer. The evolution of pancreatic cancer is divided into 3 steps: initiation, clonal expansion, and introduction to foreign microenvironments [167]. Based on the studies reviewed herein, we can infer that the selective pressures of the pancreatic cancer microenvironmental ecology can shape pancreatic cancer evolution. For example, in pancreatic cancer, TAMs can be recruited into the microenvironment to regulate the stemness of pancreatic CSCs for initiation, and pancreatic CSCs secrete factors such as IFNβ to stimulate TAMs for stemness maintenance; PSCs can secrete several factors to promote the growth of pancreatic cancer; factors/exosomes from the primary microenvironment can induce formation of the pre-metastatic niche in liver and lung; and the microenvironment in the metastatic niche can induce pancreatic cancer dormancy for recurrence.
Some factors secreted by cellular elements of the microenvironment are simultaneously involved in desmoplasia, immunosuppression, and metastasis. For example, TGFβ, IL-10 and VEGF, which are considered to be immunosuppressive cytokines secreted by pancreatic cancer cells, Tregs, and TAMs, can promote desmoplasia, angiogenesis/lymphangiogenesis, EMT, and formation of the pre-metastatic niche. STAT3, a star molecule in cancer progression, can activate other molecules involved in immunosuppression or metastasis. Thus, how are these factors regulated and what is their relationship to the 4 driver genes? Concerning the regulation of these factors, increasing knowledge of molecular pathogenesis has shown that these factors are epigenetically regulated by DNA methylation, histone modification or non-coding RNAs. For example, the hedgehog transcription factor Gli1 targets the epigenetic modifiers DNMT1 and DNMT3a, which are positive targets of oncogenic epigenetic pathways in pancreatic cancer [168]. MUC1, which participates in immunosuppression and the progression of pancreatic cancer, can be regulated by DNA methylation and histone H3 lysine 9 modification [169]. MiR-27a can activate the Ras/MAPK signaling pathway by inhibition of Sprouty2, the inhibitor of Ras/MAPK, in pancreatic cancer [170]. Moreover, exosomes, which are a hot topic in cancer research, contain non-coding RNAs and play a pivotal role in the liver metastasis of pancreatic cancer by altering the phenotype of the cells in the pre-metastatic niche. Therefore, understanding the regulation of these molecules will be important for identifying potential therapeutic targets.
A better understanding of the pathways in the tumor microenvironment during the metastasis of pancreatic cancer will facilitate a breakthrough in cancer immunotherapy studies and provide a rationale for clinical trials, which can contribute to improving the efficacy of therapy.
Abbreviations
- Akt:
-
Protein kinase B
- APC:
-
Antigen-presenting cell
- Arg1:
-
Arginase 1
- CAF:
-
Cancer-associated fibroblast
- CCL:
-
CC chemokine ligand
- Cdc42:
-
Cell division control protein 42 homolog
- CDKN2A :
-
Cyclin-dependent kinase Inhibitor 2A
- CEC:
-
Circulating epithelial cell
- c-Met:
-
Tyrosine-protein kinase Met
- CSC:
-
Cancer stem cell
- CTC:
-
Circulating tumor cell
- CTC:
-
Circulating tumor cell
- CTGF:
-
Connective tissue growth factor
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- CXCL:
-
CXC chemokine ligand
- CXCR:
-
CXC chemokine receptor
- DC:
-
Dendritic cell
- ECM:
-
Extracellular matrix
- EGFR:
-
Epidermal growth factor receptor
- EpCAM:
-
Epithelial cell adhesion molecule
- Erk:
-
Extracellular signal-regulated kinase
- FAK:
-
Focal adhesion kinase
- Fas:
-
First apoptosis signal
- FGF2:
-
Fibroblast growth factor 2
- Foxp3:
-
Forkheadbox protein 3
- FPR2:
-
Formyl peptide receptor 2
- GM-CSF:
-
Granulocyte macrophage colony-stimulating factor
- HGF:
-
Hepatocyte growth factor
- HIF:
-
Hypoxia inducible factor
- HLA:
-
Human leukocyte antigen
- HSC:
-
Hepatic stellate cell
- ICAM-1:
-
Intercellular adhesion molecular-1
- IDO:
-
Indoleamine 2,3-dioxygenase
- IFN-γ:
-
Interferon-γ
- IL:
-
Interleukin
- iNOS:
-
Inducible nitric oxide synthase
- IP-10:
-
Interferon gamma-induced protein 10
- IPMN:
-
Intraductal papillary mucinous neoplasm
- KRAS :
-
Kirsten rat sarcoma viral oncogene
- LL:
-
Leucine leucine
- MAPK:
-
Mitogen-activated protein kinase
- MDSC:
-
Myeloid-derived suppressor cell
- MHC:
-
Major histocompatibility complex
- MIP:
-
Macrophage inflammatory protein
- MMP:
-
Matrix metalloproteinase
- MUC:
-
Mucin
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NK:
-
Nature killer
- NLRP3:
-
NACHT, LRR and PYD domains-containing protein 3
- Nrf2:
-
Nuclear factor erythroid 2
- P2X7R:
-
P2X purinoceptor 7 receptor
- PD-1:
-
Programmed cell death protein-1
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PDGF:
-
Platelet-derived growth factor
- PGE2:
-
Prostaglandin E2
- PI3K:
-
Phosphoinositide 3-kinase
- PMN-MDSC:
-
Polymorphonuclear cells
- PSC:
-
Pancreatic stellate cell
- RAF:
-
Rapidly accelerated fibrosarcoma kinase
- ROS:
-
Reactive oxygen species
- SDF-1:
-
Stromal cell derived factor 1
- SHH:
-
Sonic hedgehog
- shRNA:
-
Short hairpin RNA
- SMAD4 :
-
Mothers against decapentaplegic homolog 4
- STAT3:
-
Signal transducer and activator of transcription 3
- TAM:
-
Tumor-associated macrophage
- TGFβ:
-
Transforming growth factor-β
- Th:
-
T helper
- TIL:
-
Tumor infiltrating lymphocyte
- TLR:
-
Toll-like receptor
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- Treg:
-
Regulatory T cell
- VDR:
-
Vitamin D receptor
- VEGF:
-
Vascular endothelial growth factor
- α-SMA:
-
α-Smooth muscle actin
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30.
Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6.
Chronopoulos A, Robinson B, Sarper M, Cortes E, Auernheimer V, Lachowski D, Attwood S, Garcia R, Ghassemi S, Fabry B, Del Rio HA. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat Commun. 2016;7:12630.
Neesse A, Algül H, Tuveson DA, Gress TM. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut. 2015;64:1476–84.
Ayala G, Tuxhorn JA, Wheeler TM, Frolov A, Scardino PT, Ohori M, Wheeler M, Spitler J, Rowley DR. Reactive stroma as a predictor of biochemical-free recurrence in prostate cancer. Clin Cancer Res. 2003;9:4792–801.
Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M, Ng MR, Nia HT, Grahovac J, Kao S, et al. Obesity-induced inflammation and Desmoplasia promote pancreatic Cancer progression and resistance to chemotherapy. Cancer Discov. 2016;6:852–69.
Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–21.
Whittle MC, Izeradjene K, Rani PG, Feng L, Carlson MA, DelGiorno KE, Wood LD, Goggins M, Hruban RH, Chang AE, et al. RUNX3 controls a metastatic switch in pancreatic ductal adenocarcinoma. Cell. 2015;161:1345–60.
Iglesias M, Frontelo P, Gamallo C, Quintanilla M. Blockade of Smad4 in transformed keratinocytes containing a Ras oncogene leads to hyperactivation of the Ras-dependent Erk signalling pathway associated with progression to undifferentiated carcinomas. Oncogene. 2000;19:4134–45.
Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C, Hostetter G, Shepard HM, Von Hoff DD, Han H. Desmoplasia in primary tumors and metastatic lesions of pancreatic Cancer. Clin Cancer Res. 2015;21:3561–8.
Erkan M, Kurtoglu M, Kleeff J. The role of hypoxia in pancreatic cancer: a potential therapeutic target? Expert Rev Gastroenterol Hepatol. 2016;10:301–16.
Kisker O, Onizuka S, Banyard J, Komiyama T, Becker CM, Achilles EG, Barnes CM, O'Reilly MS, Folkman J, Pirie-Shepherd SR. Generation of multiple angiogenesis inhibitors by human pancreatic cancer. Cancer Res. 2001;61:7298–304.
Erkan M, Reiser-Erkan C, Michalski CW, Deucker S, Sauliunaite D, Streit S, Esposito I, Friess H, Kleeff J. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia. 2009;11:497–508.
Samkharadze T, Erkan M, Reiser-Erkan C, Demir IE, Kong B, Ceyhan GO, Michalski CW, Esposito I, Friess H, Kleeff J. Pigment epithelium-derived factor associates with neuropathy and fibrosis in pancreatic cancer. Am J Gastroenterol. 2011;106:968–80.
Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–29.
Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112–20.
Gomez Perdiguero E, Geissmann F. Cancer immunology. Identifying the infiltrators. Science. 2014;344:801–2.
Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;15:73–86.
Fukunaga A, Miyamoto M, Cho Y, Murakami S, Kawarada Y, Oshikiri T, Kato K, Kurokawa T, Suzuoki M, Nakakubo Y, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas. 2004;28:e26–31.
Carstens JL, Correa de Sampaio P, Yang D, Barua S, Wang H, Rao A, Allison JP, VS LB, Kalluri R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun. 2017;8:15095.
Ryschich E, Notzel T, Hinz U, Autschbach F, Ferguson J, Simon I, Weitz J, Frohlich B, Klar E, Buchler MW, Schmidt J. Control of T-cell-mediated immune response by HLA class I in human pancreatic carcinoma. Clin Cancer Res. 2005;11:498–504.
von Bernstorff W, Spanjaard RA, Chan AK, Lockhart DC, Sadanaga N, Wood I, Peiper M, Goedegebuure PS, Eberlein TJ. Pancreatic cancer cells can evade immune surveillance via nonfunctional Fas (APO-1/CD95) receptors and aberrant expression of functional Fas ligand. Surgery. 1999;125:73–84.
He C, Jiang H, Geng S, Sheng H, Shen X, Zhang X, Zhu S, Chen X, Yang C, Gao H. Expression and prognostic value of c-Myc and Fas (CD95/APO1) in patients with pancreatic cancer. Int J Clin Exp Pathol. 2014;7:742–50.
Bellone G, Turletti A, Artusio E, Mareschi K, Carbone A, Tibaudi D, Robecchi A, Emanuelli G, Rodeck U. Tumor-associated transforming growth factor-beta and interleukin-10 contribute to a systemic Th2 immune phenotype in pancreatic carcinoma patients. Am J Pathol. 1999;155:537–47.
Moo-Young TA, Larson JW, Belt BA, Tan MC, Hawkins WG, Eberlein TJ, Goedegebuure PS, Linehan DC. Tumor-derived TGF-beta mediates conversion of CD4+Foxp3+ regulatory T cells in a murine model of pancreas cancer. J Immunother. 2009;32:12–21.
Principe DR, DeCant B, Mascarinas E, Wayne EA, Diaz AM, Akagi N, Hwang R, Pasche B, Dawson DW, Fang D, et al. TGFbeta signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res. 2016;76:2525–39.
Munn DH. Blocking IDO activity to enhance anti-tumor immunity. Front Biosci (Elite Ed). 2012;4:734–45.
Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561–84.
Moon YW, Hajjar J, Hwu P, Naing A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J Immunother Cancer. 2015;3:51.
Peng Y-P, Zhang J-J, Liang W-B, Tu M, Lu Z-P, Wei J-S, Jiang K-R, Gao W-T, Wu J-L, Xu Z-K, et al. Elevation of MMP-9 and IDO induced by pancreatic cancer cells mediates natural killer cell dysfunction. BMC Cancer. 2014;14:738.
Wang X, Lang M, Zhao T, Feng X, Zheng C, Huang C, Hao J, Dong J, Luo L, Li X, et al. Cancer-FOXP3 directly activated CCL5 to recruit FOXP3(+) Treg cells in pancreatic ductal adenocarcinoma. Oncogene. 2017;36:3048–58.
Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, Clendenin C, Stanger BZ, Furth EE, Wherry EJ, Vonderheide RH. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res. 2015;3:399–411.
Pergamo M, Miller G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther. 2017;24:100–5.
Kimbara S, Kondo S. Immune checkpoint and inflammation as therapeutic targets in pancreatic carcinoma. World J Gastroenterol. 2016;22:7440–52.
Feng M, Xiong G, Cao Z, Yang G, Zheng S, Song X, You L, Zheng L, Zhang T, Zhao Y. PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett. 2017;407:57–65.
Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J, Friess H. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol. 2012;9:454–67.
Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest. 2007;117:50–9.
Ferdek PE, Jakubowska MA. Biology of pancreatic stellate cells-more than just pancreatic cancer. Pflugers Arch. 2017;9:1039–50.
Zhan HX, Zhou B, Cheng YG, Xu JW, Wang L, Zhang GY, Hu SY. Crosstalk between stromal cells and cancer cells in pancreatic cancer: new insights into stromal biology. Cancer Lett. 2017;392:83–93.
Wu Q, Tian Y, Zhang J, Zhang H, Gu F, Lu Y, Zou S, Chen Y, Sun P, Xu M, et al. Functions of pancreatic stellate cell-derived soluble factors in the microenvironment of pancreatic ductal carcinoma. Oncotarget. 2017;8:102721–38.
Fu Y, Liu S, Zeng S, Shen H. The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma. Mol Cancer. 2018;17:62.
Nakashima H, Nakamura M, Yamaguchi H, Yamanaka N, Akiyoshi T, Koga K, Yamaguchi K, Tsuneyoshi M, Tanaka M, Katano M. Nuclear factor-kappaB contributes to hedgehog signaling pathway activation through sonic hedgehog induction in pancreatic cancer. Cancer Res. 2006;66:7041–9.
Lauth M, Bergstrom A, Shimokawa T, Tostar U, Jin Q, Fendrich V, Guerra C, Barbacid M, Toftgard R. DYRK1B-dependent autocrine-to-paracrine shift of hedgehog signaling by mutant RAS. Nat Struct Mol Biol. 2010;17:718–25.
Kozono S, Ohuchida K, Eguchi D, Ikenaga N, Fujiwara K, Cui L, Mizumoto K, Tanaka M. Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res. 2013;73:2345–56.
Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, Marshall JF, Chin-Aleong J, Chelala C, Gribben JG, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145:1121–32.
Tang D, Yuan Z, Xue X, Lu Z, Zhang Y, Wang H, Chen M, An Y, Wei J, Zhu Y, et al. High expression of Galectin-1 in pancreatic stellate cells plays a role in the development and maintenance of an immunosuppressive microenvironment in pancreatic cancer. Int J Cancer. 2012;130:2337–48.
Lunardi S, Lim SY, Muschel RJ, Brunner TB. IP-10/CXCL10 attracts regulatory T cells: implication for pancreatic cancer. Oncoimmunology. 2015;4:e1027473.
Lunardi S, Jamieson NB, Lim SY, Griffiths KL, Carvalho-Gaspar M, Al-Assar O, Yameen S, Carter RC, McKay CJ, Spoletini G, et al. IP-10/CXCL10 induction in human pancreatic cancer stroma influences lymphocytes recruitment and correlates with poor survival. Oncotarget. 2014;5:11064–80.
Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, Fuchs JR, Eubank TD, Frankel WL, Bekaii-Saab T, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013;73:3007–18.
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8.
Tang Y, Xu X, Guo S, Zhang C, Tang Y, Tian Y, Ni B, Lu B, Wang H. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PLoS One. 2014;9:e91551.
Wang X, Wang L, Mo Q, Dong Y, Wang G, Ji A. Changes of Th17/Treg cell and related cytokines in pancreatic cancer patients. Int J Clin Exp Pathol. 2015;8:5702–8.
Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson JT, Whiteside TL. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin Cancer Res. 2007;13:4345–54.
Grossman WJ, Verbsky JW, Tollefsen BL, Kemper C, Atkinson JP, Ley TJ. Differential expression of granzymes a and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood. 2004;104:2840–8.
Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. 2005;174:1783–6.
Ren X, Ye F, Jiang Z, Chu Y, Xiong S, Wang Y. Involvement of cellular death in TRAIL/DR5-dependent suppression induced by CD4(+) CD25(+) regulatory T cells. Cell Death Differ. 2007;14:2076–84.
Garin MI, Chu CC, Golshayan D, Cernuda-Morollon E, Wait R, Lechler RI. Galectin-1: a key effector of regulation mediated by CD4+CD25+ T cells. Blood. 2007;109:2058–65.
Beyer K, Normann L, Sendler M, Kading A, Heidecke CD, Partecke LI, von Bernstorff W. TRAIL promotes tumor growth in a syngeneic murine Orthotopic pancreatic Cancer model and affects the host immune response. Pancreas. 2016;45:401–8.
Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8:1353–62.
Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, Belladonna ML, Fioretti MC, Alegre ML, Puccetti P. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. 2003;4:1206–12.
Markowitz J, Brooks TR, Duggan MC, Paul BK, Pan X, Wei L, Abrams Z, Luedke E, Lesinski GB, Mundy-Bosse B, et al. Patients with pancreatic adenocarcinoma exhibit elevated levels of myeloid-derived suppressor cells upon progression of disease. Cancer Immunol Immunother. 2015;64:149–59.
Porembka MR, Mitchem JB, Belt BA, Hsieh CS, Lee HM, Herndon J, Gillanders WE, Linehan DC, Goedegebuure P. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol Immunother. 2012;61:1373–85.
Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–35.
Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–47.
Torroella-Kouri M, Rodriguez D, Caso R. Alterations in macrophages and monocytes from tumor-bearing mice: evidence of local and systemic immune impairment. Immunol Res. 2013;57:86–98.
Otsuji M, Kimura Y, Aoe T, Okamoto Y, Saito T. Oxidative stress by tumor-derived macrophages suppresses the expression of CD3 zeta chain of T-cell receptor complex and antigen-specific T-cell responses. Proc Natl Acad Sci U S A. 1996;93:13119–24.
Nagaraj S, Gabrilovich DI. Regulation of suppressive function of myeloid-derived suppressor cells by CD4+ T cells. Semin Cancer Biol. 2012;22:282–8.
Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, Blosser RL, Tam AJ, Bruno T, Zhang H, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest. 2013;123:1580–9.
Pinton L, Solito S, Damuzzo V, Francescato S, Pozzuoli A, Berizzi A, Mocellin S, Rossi CR, Bronte V, Mandruzzato S. Activated T cells sustain myeloid-derived suppressor cell-mediated immune suppression. Oncotarget. 2016;7:1168–84.
Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, Greenberg PD, Hingorani SR. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014;63:1769–81.
Liou GY, Doppler H, Necela B, Edenfield B, Zhang L, Dawson DW, Storz P. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 2015;5:52–63.
Gordon S, Pluddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev. 2014;262:36–55.
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–86.
Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13.
Karnevi E, Andersson R, Rosendahl AH. Tumour-educated macrophages display a mixed polarisation and enhance pancreatic cancer cell invasion. Immunol Cell Biol. 2014;92:543–52.
Hu H, Hang JJ, Han T, Zhuo M, Jiao F, Wang LW. The M2 phenotype of tumor-associated macrophages in the stroma confers a poor prognosis in pancreatic cancer. Tumour Biol. 2016;37:8657–64.
Sica A, Saccani A, Mantovani A. Tumor-associated macrophages: a molecular perspective. Int Immunopharmacol. 2002;2:1045–54.
Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–6.
Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86:1065–73.
Hao NB, Lu MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol. 2012;2012:948098.
Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J, Sotomayor EM, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–49.
Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol. 2015;36:229–39.
Daley D, Mani VR, Mohan N, Akkad N, Pandian G, Savadkar S, Lee KB, Torres-Hernandez A, Aykut B, Diskin B, et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J Exp Med. 2017;214:1711–24.
Daley D, Mani VR, Mohan N, Akkad N, Ochi A, Heindel DW, Lee KB, Zambirinis CP, Pandian GSB, Savadkar S, et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat Med. 2017;23:556–67.
Xue J, Sharma V, Hsieh MH, Chawla A, Murali R, Pandol SJ, Habtezion A. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat Commun. 2015;6:7158.
Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P, Schmid MC, Sun P, Mose E, Bouvet M, et al. Macrophage PI3Kgamma drives pancreatic ductal adenocarcinoma progression. Cancer Discov. 2016;6:870–85.
Shi C, Washington MK, Chaturvedi R, Drosos Y, Revetta FL, Weaver CJ, Buzhardt E, Yull FE, Blackwell TS, Sosa-Pineda B, et al. Fibrogenesis in pancreatic cancer is a dynamic process regulated by macrophage-stellate cell interaction. Lab Investig. 2014;94:409–21.
Schmid-Kotsas A, Gross HJ, Menke A, Weidenbach H, Adler G, Siech M, Beger H, Grunert A, Bachem MG. Lipopolysaccharide-activated macrophages stimulate the synthesis of collagen type I and C-fibronectin in cultured pancreatic stellate cells. Am J Pathol. 1999;155:1749–58.
Li N, Li Y, Li Z, Huang C, Yang Y, Lang M, Cao J, Jiang W, Xu Y, Dong J, Ren H. Hypoxia inducible factor 1 (HIF-1) recruits macrophage to activate pancreatic stellate cells in pancreatic ductal adenocarcinoma. Int J Mol Sci. 2016;17:799.
Abel EV, Simeone DM. Biology and clinical applications of pancreatic cancer stem cells. Gastroenterology. 2013;144:1241–8.
Zhang Y, Morris JP, Yan W, Schofield HK, Gurney A, Simeone DM, Millar SE, Hoey T, Hebrok M, Pasca di Magliano M. Canonical wnt signaling is required for pancreatic carcinogenesis. Cancer Res. 2013;73:4909–22.
Lee JY, Song SY, Park JY. Notch pathway activation is associated with pancreatic cancer treatment failure. Pancreatology. 2014;14:48–53.
Abel EV, Kim EJ, Wu J, Hynes M, Bednar F, Proctor E, Wang L, Dziubinski ML, Simeone DM. The notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PLoS One. 2014;9:e91983.
Onishi H, Katano M. Hedgehog signaling pathway as a new therapeutic target in pancreatic cancer. World J Gastroenterol. 2014;20:2335–42.
Chen K, Huang YH, Chen JL. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34:732–40.
Lonardo E, Hermann PC, Mueller MT, Huber S, Balic A, Miranda-Lorenzo I, Zagorac S, Alcala S, Rodriguez-Arabaolaza I, Ramirez JC, et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell. 2011;9:433–46.
Lonardo E, Frias-Aldeguer J, Hermann PC, Heeschen C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle. 2012;11:1282–90.
Hou YC, Chao YJ, Tung HL, Wang HC, Shan YS. Coexpression of CD44-positive/CD133-positive cancer stem cells and CD204-positive tumor-associated macrophages is a predictor of survival in pancreatic ductal adenocarcinoma. Cancer. 2014;120:2766–77.
Sainz B Jr, Martin B, Tatari M, Heeschen C, Guerra S. ISG15 is a critical microenvironmental factor for pancreatic cancer stem cells. Cancer Res. 2014;74:7309–20.
Sainz B Jr, Alcala S, Garcia E, Sanchez-Ripoll Y, Azevedo MM, Cioffi M, Tatari M, Miranda-Lorenzo I, Hidalgo M, Gomez-Lopez G, et al. Microenvironmental hCAP-18/LL-37 promotes pancreatic ductal adenocarcinoma by activating its cancer stem cell compartment. Gut. 2015;64:1921–35.
Yachida S, Iacobuzio-Donahue CA. The pathology and genetics of metastatic pancreatic cancer. Arch Pathol Lab Med. 2009;133:413–22.
Wei D, Le X, Zheng L, Wang L, Frey JA, Gao AC, Peng Z, Huang S, Xiong HQ, Abbruzzese JL, Xie K. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene. 2003;22:319–29.
Kitamoto S, Yokoyama S, Higashi M, Yamada N, Takao S, Yonezawa S. MUC1 enhances hypoxia-driven angiogenesis through the regulation of multiple proangiogenic factors. Oncogene. 2013;32:4614–21.
Wang L, Zhou W, Zhong Y, Huo Y, Fan P, Zhan S, Xiao J, Jin X, Gou S, Yin T, et al. Overexpression of G protein-coupled receptor GPR87 promotes pancreatic cancer aggressiveness and activates NF-kappaB signaling pathway. Mol Cancer. 2017;16:61.
Saito K, Matsuo Y, Imafuji H, Okubo T, Maeda Y, Sato T, Shamoto T, Tsuboi K, Morimoto M, Takahashi H, et al. Xanthohumol inhibits angiogenesis by suppressing nuclear factor-kappaB activation in pancreatic cancer. Cancer Sci. 2018;109:132–40.
Esposito I, Menicagli M, Funel N, Bergmann F, Boggi U, Mosca F, Bevilacqua G, Campani D. Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. J Clin Pathol. 2004;57:630–6.
Griesmann H, Drexel C, Milosevic N, Sipos B, Rosendahl J, Gress TM, Michl P. Pharmacological macrophage inhibition decreases metastasis formation in a genetic model of pancreatic cancer. Gut. 2017;66:1278–85.
Huang C, Li Z, Li N, Li Y, Chang A, Zhao T, Wang X, Wang H, Gao S, Yang S, et al. Interleukin 35 expression correlates with microvessel density in pancreatic ductal adenocarcinoma, recruits monocytes, and promotes growth and angiogenesis of xenograft tumors in mice. Gastroenterology. 2018;154:675–88.
Kurahara H, Takao S, Maemura K, Mataki Y, Kuwahata T, Maeda K, Sakoda M, Iino S, Ishigami S, Ueno S, et al. M2-polarized tumor-associated macrophage infiltration of regional lymph nodes is associated with nodal lymphangiogenesis and occult nodal involvement in pN0 pancreatic cancer. Pancreas. 2013;42:155–9.
Munn DH, Mellor AL. The tumor-draining lymph node as an immune-privileged site. Immunol Rev. 2006;213:146–58.
Kurahara H, Takao S, Shinchi H, Maemura K, Mataki Y, Sakoda M, Hayashi T, Kuwahata T, Minami K, Ueno S, Natsugoe S. Significance of lymphangiogenesis in primary tumor and draining lymph nodes during lymphatic metastasis of pancreatic head cancer. J Surg Oncol. 2010;102:809–15.
Wang Z, Wu J, Li G, Zhang X, Tong M, Wu Z, Liu Z. Lymphangiogenesis and biological behavior in pancreatic carcinoma and other pancreatic tumors. Mol Med Rep. 2012;5:959–63.
Inman KS. Complex role for the immune system in initiation and progression of pancreatic cancer. World J Gastroenterol. 2014;20:11160–23.
Podgrabinska S, Kamalu O, Mayer L, Shimaoka M, Snoeck H, Randolph GJ, Skobe M. Inflamed lymphatic endothelium suppresses dendritic cell maturation and function via mac-1/ICAM-1-dependent mechanism. J Immunol. 2009;183:1767–79.
Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science. 2010;328:749–52.
Jiang Y, Du Z, Yang F, Di Y, Li J, Zhou Z, Pillarisetty VG, Fu D. FOXP3+ lymphocyte density in pancreatic cancer correlates with lymph node metastasis. PLoS One. 2014;9:e106741.
Ikenaga N, Ohuchida K, Mizumoto K, Cui L, Kayashima T, Morimatsu K, Moriyama T, Nakata K, Fujita H, Tanaka M. CD10+ pancreatic stellate cells enhance the progression of pancreatic cancer. Gastroenterology. 2010;139:1041–51. 1051.e1041–1048
Kurahara H, Shinchi H, Mataki Y, Maemura K, Noma H, Kubo F, Sakoda M, Ueno S, Natsugoe S, Takao S. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J Surg Res. 2011;167:e211–9.
Kurahara H, Takao S, Maemura K, Shinchi H, Natsugoe S, Aikou T. Impact of vascular endothelial growth factor-C and -D expression in human pancreatic cancer: its relationship to lymph node metastasis. Clin Cancer Res. 2004;10:8413–20.
Shi Y, Tong M, Wu Y, Yang Z, Hoffman RM, Zhang Y, Tian Y, Qi M, Lin Y, Liu Y, et al. VEGF-C ShRNA inhibits pancreatic cancer growth and lymphangiogenesis in an orthotopic fluorescent nude mouse model. Anticancer Res. 2013;33:409–17.
Bailey JM, Mohr AM, Hollingsworth MA. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene. 2009;28:3513–25.
Otto N, Schulz P, Scholz A, Hauff P, Schlegelberger B, Detjen KM, Wiedenmann B. The proline TP53 variant stimulates likely lymphangiogenesis in an orthotopic mouse model of pancreatic cancer. Br J Cancer. 2012;106:348–57.
Yamada S, Fuchs BC, Fujii T, Shimoyama Y, Sugimoto H, Nomoto S, Takeda S, Tanabe KK, Kodera Y, Nakao A. Epithelial-to-mesenchymal transition predicts prognosis of pancreatic cancer. Surgery. 2013;154:946–54.
Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–61.
Castellano-Megias VM, Andres CI, Lopez-Alonso G, Colina-Ruizdelgado F. Pathological features and diagnosis of intraductal papillary mucinous neoplasm of the pancreas. World J Gastrointest Oncol. 2014;6:311–24.
Franses JW, Basar O, Kadayifci A, Yuksel O, Choz M, Kulkarni AS, Tai E, Vo KD, Arora KS, Desai N, et al. Improved detection of circulating epithelial cells in patients with Intraductal papillary mucinous neoplasms. Oncologist. 2018;23:121–7.
Liu CY, Xu JY, Shi XY, Huang W, Ruan TY, Xie P, Ding JL. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab Investig. 2013;93:844–54.
Tang D, Zhang J, Yuan Z, Zhang H, Chong Y, Huang Y, Wang J, Xiong Q, Wang S, Wu Q, et al. PSC-derived Galectin-1 inducing epithelial-mesenchymal transition of pancreatic ductal adenocarcinoma cells by activating the NF-kappaB pathway. Oncotarget. 2017;8:86488–502.
Wu YS, Chung I, Wong WF, Masamune A, Sim MS, Looi CY. Paracrine IL-6 signaling mediates the effects of pancreatic stellate cells on epithelial-mesenchymal transition via Stat3/Nrf2 pathway in pancreatic cancer cells. Biochim Biophys Acta. 1861;2017:296–306.
Li Y, Kong D, Ahmad A, Bao B, Sarkar FH. Pancreatic cancer stem cells: emerging target for designing novel therapy. Cancer Lett. 2013;338:94–100.
Kleeff J, Kusama T, Rossi DL, Ishiwata T, Maruyama H, Friess H, Buchler MW, Zlotnik A, Korc M. Detection and localization of Mip-3alpha/LARC/exodus, a macrophage proinflammatory chemokine, and its CCR6 receptor in human pancreatic cancer. Int J Cancer. 1999;81:650–7.
Kimsey TF, Campbell AS, Albo D, Wilson M, Wang TN. Co-localization of macrophage inflammatory protein-3alpha (Mip-3alpha) and its receptor, CCR6, promotes pancreatic cancer cell invasion. Cancer J. 2004;10:374–80.
Campbell AS, Albo D, Kimsey TF, White SL, Wang TN. Macrophage inflammatory protein-3alpha promotes pancreatic cancer cell invasion. J Surg Res. 2005;123:96–101.
Goicoechea SM, Garcia-Mata R, Staub J, Valdivia A, Sharek L, McCulloch CG, Hwang RF, Urrutia R, Yeh JJ, Kim HJ, Otey CA. Palladin promotes invasion of pancreatic cancer cells by enhancing invadopodia formation in cancer-associated fibroblasts. Oncogene. 2014;33:1265–73.
Lu J, Zhou S, Siech M, Habisch H, Seufferlein T, Bachem MG. Pancreatic stellate cells promote hapto-migration of cancer cells through collagen I-mediated signalling pathway. Br J Cancer. 2014;110:409–20.
Cheng ZX, Sun B, Wang SJ, Gao Y, Zhang YM, Zhou HX, Jia G, Wang YW, Kong R, Pan SH, et al. Nuclear factor-kappaB-dependent epithelial to mesenchymal transition induced by HIF-1alpha activation in pancreatic cancer cells under hypoxic conditions. PLoS One. 2011;6:e23752.
Zhu S, Zhou Y, Wang L, Zhang J, Wu H, Xiong J, Zhang J, Tian Y, Wang C, Wu H. Transcriptional upregulation of MT2-MMP in response to hypoxia is promoted by HIF-1alpha in cancer cells. Mol Carcinog. 2011;50:770–80.
Zhu SK, Zhou Y, Cheng C, Zhong S, Wu HQ, Wang B, Fan P, Xiong JX, Yang HJ, Wu HS. Overexpression of membrane-type 2 matrix metalloproteinase induced by hypoxia-inducible factor-1alpha in pancreatic cancer: implications for tumor progression and prognosis. Mol Clin Oncol. 2014;2:973–81.
Shi CY, Fan Y, Liu B, Lou WH. HIF1 contributes to hypoxia-induced pancreatic cancer cells invasion via promoting QSOX1 expression. Cell Physiol Biochem. 2013;32:561–8.
Zhao T, Gao S, Wang X, Liu J, Duan Y, Yuan Z, Sheng J, Li S, Wang F, Yu M, et al. Hypoxia-inducible factor-1alpha regulates chemotactic migration of pancreatic ductal adenocarcinoma cells through directly transactivating the CX3CR1 gene. PLoS One. 2012;7:e43399.
Miller BW, Morton JP, Pinese M, Saturno G, Jamieson NB, McGhee E, Timpson P, Leach J, McGarry L, Shanks E, et al. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol Med. 2015;7:1063–76.
Onishi H, Kai M, Odate S, Iwasaki H, Morifuji Y, Ogino T, Morisaki T, Nakashima Y, Katano M. Hypoxia activates the hedgehog signaling pathway in a ligand-independent manner by upregulation of Smo transcription in pancreatic cancer. Cancer Sci. 2011;102:1144–50.
Li X, Wang Z, Ma Q, Xu Q, Liu H, Duan W, Lei J, Ma J, Wang X, Lv S, et al. Sonic hedgehog paracrine signaling activates stromal cells to promote perineural invasion in pancreatic cancer. Clin Cancer Res. 2014;20:4326–38.
Qian D, Lu Z, Xu Q, Wu P, Tian L, Zhao L, Cai B, Yin J, Wu Y, Staveley-O’Carroll KF, et al. Galectin-1-driven upregulation of SDF-1 in pancreatic stellate cells promotes pancreatic cancer metastasis. Cancer Lett. 2017;397:43–51.
Yang XP, Liu SL, Xu JF, Cao SG, Li Y, Zhou YB. Pancreatic stellate cells increase pancreatic cancer cells invasion through the hepatocyte growth factor /c-met/survivin regulated by P53/P21. Exp Cell Res. 2017;357:79–87.
Pothula SP, Xu Z, Goldstein D, Biankin AV, Pirola RC, Wilson JS, Apte MV. Hepatocyte growth factor inhibition: a novel therapeutic approach in pancreatic cancer. Br J Cancer. 2016;114:269–80.
Erkan M, Kleeff J, Gorbachevski A, Reiser C, Mitkus T, Esposito I, Giese T, Buchler MW, Giese NA, Friess H. Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology. 2007;132:1447–64.
Liu Y, Li F, Gao F, Xing L, Qin P, Liang X, Zhang J, Qiao X, Lin L, Zhao Q, Du L. Role of microenvironmental periostin in pancreatic cancer progression. Oncotarget. 2017;8:89552–65.
Liu Y, Cao X. Characteristics and significance of the pre-metastatic niche. Cancer Cell. 2016;30:668–81.
Tien YW, Wu YM, Lin WC, Lee HS, Lee PH. Pancreatic carcinoma cells stimulate proliferation and matrix synthesis of hepatic stellate cells. J Hepatol. 2009;51:307–14.
Grunwald B, Harant V, Schaten S, Fruhschutz M, Spallek R, Hochst B, Stutzer K, Berchtold S, Erkan M, Prokopchuk O, et al. Pancreatic Premalignant Lesions Secrete Tissue Inhibitor of Metalloproteinases-1, Which Activates Hepatic Stellate Cells Via CD63 Signaling to Create a Premetastatic Niche in the Liver. Gastroenterology. 2016;151:1011–1024.e1017.
Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, Jamieson NB, Oien KA, Lowy AM, Brunton VG, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A. 2010;107:246–51.
Weissmueller S, Manchado E, Saborowski M, Morris JP, Wagenblast E, Davis CA, Moon SH, Pfister NT, Tschaharganeh DF, Kitzing T, et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell. 2014;157:382–94.
Zhong Y, Macgregor-Das A, Saunders T, Whittle MC, Makohon-Moore A, Kohutek ZA, Poling J, Herbst BT, Javier BM, Cope L, et al. Mutant p53 together with TGFbeta signaling influence organ-specific Hematogenous colonization patterns of pancreatic Cancer. Clin Cancer Res. 2017;23:1607–20.
Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, Becker A, Hoshino A, Mark MT, Molina H, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816–26.
Matzke-Ogi A, Jannasch K, Shatirishvili M, Fuchs B, Chiblak S, Morton J, Tawk B, Lindner T, Sansom O, Alves F, et al. Inhibition of Tumor Growth and Metastasis in Pancreatic Cancer Models by Interference With CD44v6 Signaling. Gastroenterology. 2016;150:513–525.e510.
Jung T, Castellana D, Klingbeil P, Cuesta Hernandez I, Vitacolonna M, Orlicky DJ, Roffler SR, Brodt P, Zoller M. CD44v6 dependence of premetastatic niche preparation by exosomes. Neoplasia. 2009;11:1093–105.
Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329–35.
Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A, Ireland L, Sakai T, Sakai K, Kim YS, et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol. 2016;18:549–60.
Cacalano G, Lee J, Kikly K, Ryan AM, Pitts-Meek S, Hultgren B, Wood WI, Moore MW. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science. 1994;265:682–4.
Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. 2010;120:2423–31.
Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, Kaplan RN, Mackall CL. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. 2014;6:237ra267.
Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, Foth M, Bryson S, McDaid K, Wilson Z, et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell. 2016;29:832–45.
Collins MA, Brisset JC, Zhang Y, Bednar F, Pierre J, Heist KA, Galban CJ, Galban S, di Magliano MP. Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PLoS One. 2012;7:e49707.
Lin WC, Rajbhandari N, Wagner KU. Cancer cell dormancy in novel mouse models for reversible pancreatic cancer: a lingering challenge in the development of targeted therapies. Cancer Res. 2014;74:2138–43.
Lenk L, Pein M, Will O, Gomez B, Viol F, Hauser C, Egberts JH, Gundlach JP, Helm O, Tiwari S, et al. The hepatic microenvironment essentially determines tumor cell dormancy and metastatic outgrowth of pancreatic ductal adenocarcinoma. Oncoimmunology. 2017;7:e1368603.
Makohon-Moore A, Iacobuzio-Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Publ Group. 2016;16:553–65.
He S, Wang F, Yang L, Guo C, Wan R, Ke A, Xu L, Hu G, Xu X, Shen J, Wang X. Expression of DNMT1 and DNMT3a are regulated by GLI1 in human pancreatic cancer. PLoS One. 2011;6:e27684.
Yamada N, Nishida Y, Tsutsumida H, Hamada T, Goto M, Higashi M, Nomoto M, Yonezawa S. MUC1 expression is regulated by DNA methylation and histone H3 lysine 9 modification in cancer cells. Cancer Res. 2008;68:2708–16.
Ma Y, Yu S, Zhao W, Lu Z, Chen J. miR-27a regulates the growth, colony formation and migration of pancreatic cancer cells by targeting Sprouty2. Cancer Lett. 2010;298:150–8.
Funding
This study was supported by the National Nature Science Foundation of China (2016, 81672960, to LY; 2016, 81672443, to YZ).
Author information
Authors and Affiliations
Contributions
Study concept and design: BR, MC, GY, HW, MF, LY, and YZ. Drafting of the manuscript: BR. Critical revision of the manuscript for important intellectual content: MC, GY, HW, MF, LY, YZ. Obtained funding: LY and YZ. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ren, B., Cui, M., Yang, G. et al. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol Cancer 17, 108 (2018). https://doi.org/10.1186/s12943-018-0858-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-018-0858-1