
Abstract
Background
Global trend of the introduction of high levels of relatively cheap carbohydrates to reduce the amount of costly protein in the aquatic animal feed production has affected the aquaculture of an economically important cyprinid fish, blunt snout bream (Megalobrama amblycephala). This dietary shift has resulted in increased prevalence of metabolic disorders, often causing economic losses. High dietary intake of carbohydrates, associated with obesity, is one of the major causes of non-alcoholic fatty liver disease (NAFLD) in humans.
Results
We have conducted an eight-week feeding trial to better understand how a high-carbohydrate diet (HCBD) affects the liver health in this fish. Hepatosomatic index and lipid content were significantly (P < 0.05) higher in the HCBD group. Histology results also suggested pathological changes in the livers of HCBD group, with excessive lipid accumulation and indication of liver damage. Metabolomics and serum biochemistry analyses showed that a number of metabolites indicative of liver damage were increased in the HCBD group. This group also exhibited low levels of betaine, which is a metabolite crucial for maintaining the healthy liver functions. Transcriptomic and qPCR analyses indicated that HCBD had a strong impact on the expression of a large number of genes associated with the NAFLD and insulin signalling pathways, which may lead to the development of insulin resistance in hepatocytes, pathological liver changes, and eventually the NAFLD.
Conclusions
Transcriptomics, metabolomics and histology results all indicate early symptoms of liver damage. However whether these would actually lead to the development of NAFLD after a longer period of time, remains inconclusive. Additionally, a very high number of upregulated genes in the HCBD group associated with several neurodegenerative diseases is a strong indication of neurodegenerative changes caused by the high-carbohydrate diet in blunt snout bream. This suggests that fish might present a good model to study neurodegenerative changes associated with high-carbohydrate diet in humans.
Similar content being viewed by others


Background
Dietary carbohydrates are the main source of energy for herbivorous animals. Glucose and other monosaccharides obtained from the digestion of dietary carbohydrates are metabolized in the liver, the major site of carbohydrate metabolism and triglyceride synthesis, to provide precursors (such as acetyl-CoA) for the synthesis of ATP and fatty acids. Carbohydrates are usually the cheapest dietary source of energy, and their appropriate incorporation reduces the catabolism of more costly dietary proteins and lipids for energy. Hence the inclusion of high levels of carbohydrates is favored by the aquaculture industry, as it usually leads to a reduction in the cost of formulated diets [1,2,3]. However, the ability of fish to use dietary carbohydrates varies widely among and within species. Although the inclusion of high percentage of dietary carbohydrates appears to induce positive effects on growth and digestibility in several fish species [4,5,6], in comparison to mammals, fishes are generally considered to be poor utilizers of carbohydrates, with high dietary protein requirements and low tolerance to carbohydrates [7,8,9,10]. In herbivores (partial or predominant), though, if the appropriate level of carbohydrates is not included in the diet, there may be negative effects on the nutrient utilization, growth, metabolism, and health of the fish [11, 12]. In comparison to mammals, the source and the type of dietary carbohydrate have much higher impacts on the utilization of carbohydrates in fish. For example, dietary glucose is known to result in reduced growth performance and feed utilization in comparison to dietary starch in several fish species [2, 13, 14]. In mammals, over-ingestion of simple carbohydrates, such as sucrose and fructose, is a major cause of nonalcoholic fatty liver disease (NAFLD) [15]. Although high dietary intake of carbohydrates has been associated with increased risks of obesity in fish, and zebrafish and medaka were used as models to investigate whether fish livers are affected by excessive dietary carbohydrates, including changes in liver size, cellular architecture, gene expression patterns, and lipid accumulation, the correlation between high carbohydrate diet and NAFLD in fish remains inconclusive [16,17,18].
Blunt snout bream (Megalobrama amblycephala), a predominantly herbivorous cyprinid fish native to China, has rapidly joined the ranks of the handful of economically most important fishes in Chinese freshwater aquaculture during the last two decades [19, 20]. As it was domesticated barely more than half a century ago from a very small founding population [21], with the rapid development of the aquaculture of this species, founder effect (reduction in genetic diversity) has contributed to the increasing appearance of growth depression, early maturation [22] and low disease resistance in cultured populations [23]. The global trend of introduction of high levels of relatively cheap carbohydrates and fat in order to reduce the amount of costly protein in aquatic animal feed production has affected the aquaculture of this species as well [3, 24,25,26]. This dietary shift has resulted in increased prevalence of metabolic disorders, such as high deposition of body lipids and weakened immune system, in many farmed fish species, including the blunt snout bream. These disorders often result in high mortality and low market quality of the farmed fish, causing economic losses to the industry [11, 26, 27].
Few studies that attempted to examine the impacts of HCBD on blunt snout bream have produced inconclusive results: Li et al. [26] found that this fish utilizes dietary lipids better than carbohydrates; but Li et al. [28] found that high dietary lipids cause metabolic stress, whereas HCBD did not produce any adverse results; Zhou et al. [27], however, found that HCBD causes the swelling of hepatocytes. The latter is in agreement with the results of our previous study, where we found that high-carbohydrate diets caused signs of liver damage [29].
Therefore, to contribute to a better understanding of the NAFLD in the blunt snout bream (essential for the aquaculture of this species) and fish in general, we have investigated how a high carbohydrate diet impacts the gene expression in blunt snout bream by sequencing the entire transcriptome. To provide additional focus on the NAFLD indices, we have also studied the liver histology, levels of some metabolites and expression of specific genes associated with the NAFLD.
Methods
Diet preparation and proximate analysis
Two diets (Table 1) were formulated to contain approximately 30% proteins and 8% lipids. The control diet was formulated according to the current blunt snout bream aquaculture industry standards (25–30% carbohydrates) [30, 31] to contain 26.6% carbohydrates. As Zhou et al. have proposed that diets containing 31–47% carbohydrates cause a stress response in blunt snout bream [27, 31], the HCBD was formulated to contain over 34% of carbohydrates (Table 1). As a result, the total energy was higher in the HCBD diet (23.5 vs. 19.1 MJ/kg). The ingredients (Table 1) were powdered, mixed, pelleted, air-dried, then oven-dried (105 °C) overnight, sealed in airtight bags, and stored at −20 °C until use. The crude protein, crude lipid, moisture, and ash in the feeds were determined using standard methods [32]: moisture by oven-drying at 105 °C, ash using a muffle furnace at 550 °C, crude protein (N × 6.25) using the Kjeldahl method after acid digestion with the Kjeltec system (Kjeltec 2300 Analyzer, Foss Tecator, Hoganas, Sweden), crude lipid using the ether extraction method with the Soxtec System HT (Soxtec System HT6, Tecator). Energy contents of the diets were measured with a bomb calorimeter Parr 6200 equipped with a Parr 1108 Oxygen Bomb and a Parr 6510 water handling system (Parr Instrument Company, Moline, IL, USA).
Animal rearing, experimental procedures and sample collection
Healthy one-year-old blunt snout breams (n = 270; average weight = 48.13 ± 0.02 g) were obtained from the experimental base of the Huazhong Agricultural University, Wuhan, China, distributed into six aerated fiberglass tanks (45 individuals per half-filled 1 m3 tank) and fed the control diet during the acclimation period (14 days). At the onset of the experiment, three tanks were randomly assigned to two different experimental diet groups: control and HCBD (each n = 135). Three tanks in each group (3 × 45 = 135) ensured three replicates per diet. The period of the rearing trial was the standard [33] 8 weeks, during which the water temperature was 25 ± 0.5 °C and the photoperiod was natural. Between 25 and 50% of the water was replaced in each tank daily. The fish were fed by hand twice daily. At the end of the experiment, after a 12-h fast, all of the surviving fish (control = 128, HCBD = 132) were tranquilized in 20–30 mg/L MS-222 buffered to pH 7.0–7.5, and weighed pooled together. Five individuals from each tank were then randomly selected and euthanized in buffered MMS at 300 mg/L concentration. After weighing, blood was collected from the caudal vein using 1 mL syringes and plasma samples prepared according to the standard protocol for clinical chemistry measurements, using sodium heparin as anticoagulant. All blood samples were centrifuged immediately after collection and supernatant (plasma) stored at −80 °C. After dissection, entire livers were collected and weighed in order to calculate the hepatosomatic index (HSI). A small portion of each liver was immediately fixed by submerging into 1 mL of paraformaldehyde in a 2 mL Eppendorf tube for histopathological examination by H&E and Oil red O staining [34], whereas samples for transcriptomic and metabolomic analyses were placed in 2 mL plastic tubes, immediately flash-frozen in liquid nitrogen for 6 h, and stored at −80 °C. Liver lipid content was calculated from the Oil red O staining results using Image-pro plus 6.0 (Media Cybernetics, Inc., Rockville, MD, USA). For details regarding determining the growth performance, please see our previous publication [29]. Data were analyzed statistically with SPSS 20 (IBM, Chicago, IL, USA), where statistical differences between the two groups were determined using the unpaired two-tailed analysis (t test). Thresholds for statistical significance were set at P < 0.05 (significant) and P < 0.01 (highly significant). Results were expressed as means ± SD.
Metabolomic analysis
As we have studied the metabolome and serum biochemistry parameters in detail in our previous study [29], here we have focused exclusively on the parameters indicative of liver damage [28]. Among the 30 collected serum samples, ten samples were chosen randomly from each group for clinical biochemistry and metabolomic analyses. Hence, 1H NMR metabolomic analysis was conducted only for betaine in liver samples (not published before), whereas for relevant metabolites in serum samples (α-glucose, β-glucose, succinate and tyrosine), as well as the serum biochemistry parameters (alanine transaminase - ALT, aspartate transaminase - AST, triglycerides - TG, high-density lipoprotein - HDL and low-density lipoprotein - LDL) we referred to the appropriate data (HCBD group) from the previous study.
Clinical biochemistry
Serum biochemistry parameters were measured with commercial kits produced by Jiancheng Bioengineering Institute (Nanjing, China) with a Tecan analyzer (Tecan Ltd) as described before [29].
Nuclear magnetic resonance (1H–NMR) spectroscopy
Conducted as described before [29]. Briefly: liver tissues (about 50 mg) were homogenized in cold methanol and water (v/v = 2:1) using a Qiagen TissueLyser (Retsch GmBH, Germany). All plasma NMR spectra were acquired at 298 K on a Bruker Avance III 600 MHz NMR spectrometer (600.13 MHz for 1H frequency) equipped with a cryogenic probe (Bruker Biospin, Germany). One-dimensional 1H NMR spectra were acquired with the Carr–Purcell–Meiboom–Gill pulse train [35]. The NMR spectroscopic analysis, NMR data processing, and multivariate data analysis were performed as previously described [36]. The cutoff value (|r| > 0.602) was based on the significance threshold value of P < 0.05, which was determined according to the test for significance of the Pearson’s product-moment correlation coefficient (n = 10, P < 0.05).
Transcriptome analysis
Among the 30 collected liver samples, one sample from each tank (3 control +3 HCBD) was randomly selected for the transcriptome sequencing, and sent (in liquid nitrogen) to the Biomarker Biotechnology Corporation (Beijing, China). The total RNA was extracted, cDNA libraries constructed and sequenced, and transcriptome assembled and annotated, as described before [37], so only a brief overview is given here:
-
RNA extraction: the total RNA was extracted using RNAiso Plus Reagent (Takara Bio Inc., Dalian, China) according to the manufacturer’s instructions. The quality of the total RNA from the individual tissue samples was evaluated using electrophoresis in 1% agarose gels. The RNA was quantified spectrophotometrically with NanoDrop 2000 spectrophotometer (Thermo Scientific, Delaware, USA) and Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA).
-
cDNA library preparation and Illumina sequencing: high-quality RNA (5 μg, 100 ng/μL) was used for the preparation of RNA-seq libraries using the Illumina HiSeq™ 2500 platform according to the manufacturer’s manual [38] (Biomarker Technologies Co., Ltd., Beijing, China). All raw transcriptome data have been deposited in the NCBI Short Read Archive (SRA) with the accession numbers SRR5763110, SRR5763111, SRR5763112, SRR5763116, SRR5763117 and SRR5763107.
-
De novo assembly of sequencing reads: raw reads of the transcriptome datasets (control and HCBD) were cleaned by filtering out adaptor-only reads (nt length of the recognized adaptor ≤13 and the remaining adaptor-excluded nt length ≤ 35) and low-quality reads: reads in which more than 80.01% of the bases had a Q value of ≤ 30 were filtered out with the Fastq_filter software (Biomarker Technologies Co., Ltd). The clean reads were then assembled with Trinity platform [39] using three implemented modules, Inchworm, Chrysalis, and Butterfly, with the following parameters: ‘K-mer = 25, and group pairs distance = 300. Redundant sequences were eliminated, and the longest transcripts were recognized as unigenes, which were grouped together for the final assembly and subsequent annotation.
-
Annotation: Open reading frames (ORFs) of transcripts and unigene sequences were predicted by the TransDecoder package integrated into Trinity, with the minimum ORF length set at 100 bp. Unigene reads were queried in a number of databases: NCBI’s BLASTn and BLASTx (Ref-Seq nr); as well as UniProtKB/Swiss-Prot (manually annotated and reviewed) and UniProtKB/TrEMBL (automatically annotated and not reviewed) [40]. Each unigene sequence was allocated a gene name according to the BLAST hit with the highest score. Getorf program implemented in EMBOSS software package (version 1.20.0) [41] was employed to predict the open reading frames (ORFs) and to select the longest ORF for each unigene using default parameters.
-
Ontology analysis: these unigenes were used to query a series of databases, using BLASTx with the E-value cutoff = 1e−5. Blast2GO program [42] was used to produce Gene Ontology (GO) annotations. COG [43] and KEGG [44] databases were also queried. KEGG Orthology (KO) assignment was obtained using the KAAS server (bi-directional best hit method) [45]. KEGG enrichment analysis was conducted using KOBAS software [46] to statistically test the enrichment of differentially expressed genes in KEGG pathways.
-
Gene expression analysis: The numbers of reads produced by the RNA-Seq analyses were normalized to reads per kilobase of transcripts per million (RPKM) to compute the gene expression levels [47]. Detection of differentially expressed genes was performed by EBseq software [48] using pairwise comparison. Benjamini-Hochberg procedure [49] (α < 0.01) was used to control for the false discovery rate (FDR). Genes were defined as differentially expressed (DEG) when they exhibited the following parameters: FDR < 0.001 and |log2 ratio| > 1 (the RPKM value of a gene in one sample is at least two-fold higher than in another sample).
qPCR analysis
This method was used to further investigate the expression of 13 genes associated with the NAFLD pathway (Additional file 1) (http://www.kegg.jp/kegg/pathway.html) that were significantly differentially expressed in response to HCBD in the transcriptomic results. The total RNA was extracted and cDNA prepared, from the same six liver samples that were used for the transcriptome sequencing, as described in the previous section. The cDNA libraries were serially diluted 10-fold and used as templates for qPCR with the primers listed in Additional file 2. Primers were designed using Primer Premier 5 software (Premier Biosoft, USA) [50] on the basis of the sequences of these 13 genes obtained from the transcriptome data, and synthesized by the Tsingke Biological Technology company (Co. Ltd., Wuhan). qPCR was performed with LightCycler®480 II (Roche Diagnostics GmbH, Germany) and SYBR® Premix Ex Taq™ (Takara Bio Inc.) according to the manufacturer’s instructions. In brief: the total qPCR mixture reaction volume of 10 mL contained 5 mL Light-Cycler480 SYBR Green I Master, 3.6 mL ddH2O, 0.4 mL of forward and reverse primers and 1 mL of cDNA template. PCR procedure was as follows: pre-incubation at 95 °C for 5 min, followed by 45 cycles of 10 s at 95 °C, 10 s at the temperature adjusted to the specific primer pair (Additional file 2), and 10 s at 72 °C. Melt curve analysis was performed at the end. Reactions were performed in triplicate for each sample. Expression levels of the target genes were normalized to the expression levels of two reference genes: 18S rRNA and Rpl13a [51], and expressed as fold changes relative to the target gene expression in the control group using the 2–ΔΔCt method [52]. The qPCR data were analyzed statistically using Microsoft Excel and SPSS. Student-T test, implemented in SPSS, was applied to test the statistical significance of the differences between the two groups, where significance thresholds were set at P < 0.05 (significant) and P < 0.01 (highly significant).
Results
Hepatosomatic index and liver histology
After the 60-day feeding period, survival rates of both groups were very high. Although the difference was not statistically significant (P = 0.530), survival rate of the HCBD group was higher: 97.78 ± 2.22 vs. 94.82 ± 7.14 (of the control group). On the contrary, HSI values were significantly (P = 0.020) higher in the HCBD group: 1.52 ± 0.13 vs. 1.40 ± 0.14. Liver lipid content and histology analyses have revealed the cause for the significant difference in the two HSI values: the control group had regular hepatocytes with large and spherical nuclei centrally located in a moderate cytoplasm and a small number of lipid droplets (Fig. 1a, c); in contrast, the HCBD group exhibited swollen hepatocytes with large diffused lipid vacuoles, abnormal endothelial cells in the central liver vein, inflammatory infiltrate and some hypertrophy of the hepatocytes (Fig. 1b, d). Moreover, significant fat accumulation in livers of the HCBD group was observed using the oil red O staining method (Fig. 1e, f): the lipid content in livers of the HCBD group was significantly (P = 0.006) higher than in the control group: 19.14 ± 2.30% vs. 3.95 ± 0.37% respectively.

Photomicrographs of representative hematoxylin- and eosin-stained (a-d) and Oil red O-stained (E and F) histological liver sections of blunt snout bream fed control diet (a, c and e) and high-carbohydrate diet (b, d and f). Arrows indicate examples of a: normal hepatocytes with regular, round nuclei; b: swollen hepatocytes with large diffused lipid vacuoles, c: absent nucleus and d: abnormal nucleus. CV and IF labels indicate examples of central liver vein with abnormal endothelial cells and inflammatory infiltrate respectively. In the Oil red O-staining section, to evaluate lipid content in livers (e and f), lipids are stained red. Magnification ×400 (a, b, e and f) and ×1000 (c and d)
Serum biochemistry
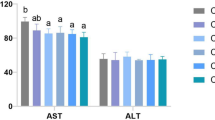
The HCBD has induced changes in all five studied serum biochemistry parameters: apart from TG, which was significantly reduced (P < 0.05), the remaining four parameters (AST, ALT, LDL, and HDL) were all significantly (LDL highly significantly P < 0.01) elevated in the HCBD group in comparison to the control group (Fig. 2).

Serum biochemistry parameters associated with liver damage. * above bar indicates significant difference (P < 0.05) and ** indicates highly significant difference (P < 0.01). Results were analyzed by t-test
The metabolomics of serum and liver
The studied metabolites, indicative of liver damage, with significantly different abundance in the blood sera and livers of the control and HCBD groups, inferred from 1H–NMR metabolite profiles, are shown in Fig. 3. Metabolites belonging to two different metabolic pathways were (highly) significantly more abundant in the HCBD group. In glycolysis pathway, it was α-glucose (P < 0.01) and β-glucose (P < 0.05), and in tricarboxylic acid (TCA) cycle pathway it was succinate (P < 0.05) and tyrosine (P < 0.01) (Fig. 3a, Additional file 3). In liver, betaine was highly significantly reduced in the HCBD group (P < 0.01; Fig. 3b , Additional files 3, 4 and 5).

Metabolites in plasma and liver extracts significantly different between the control and HCBD diet groups. a Plasma metabolomics and b Liver metabolomics. Results were analyzed by t-test. * above bars indicates significant difference (P < 0.05) and ** indicates highly significant difference (P < 0.01)
Impacts of high-carbohydrate diet on the transcriptome
Non-normalized cDNA libraries were constructed from six tissue samples (3 control +3 HCBD) and Solexa/Illumina paired-end sequencing yielded 157,063 transcripts and 96,427 unigenes. After stringent quality assessment and data filtering, 22,415,280 high-quality reads from the control group and 19,837,027 from the HCBD group were selected for further analysis. GC contents of the control and HCBD samples were 47.76 and 47.80%, respectively. Using the GO annotation to assign all 96,427 unigenes to the three main categories, those assigned to the ‘molecular function’ (14,901, 36.56%) and ‘biological processes’ (14,583, 35.78%) were the most abundant, with ‘cellular components’ (11,271, 27.66%) lagging behind (Fig. 4a). Within the ‘biological processes’ category, the most frequent GO terms were ‘cellular process’, ‘metabolic process’, and ‘single-organism process’; among the ‘cellular components’, genes involved in ‘cell part’, ‘organelle’ and ‘macromolecular complex’ functions were the most abundant; whereas in the category of molecular function, the term ‘binding’ accounted for the highest proportion of annotations, followed by ‘catalytic activity’ and ‘molecular transducer activity’.

Transcriptome profiles of the HCBD and control groups. a GO annotation of all unigenes and DEG (differentially expressed) unigenes; b MA plot of differential gene expression levels in the two groups, where expression intensity is on the x-axis (log2 FPKM) and differences in the gene expression levels (fold change) are on the y-axis (log2 FC), each dot represents one gene, the log2 (FC) is plotted against the mean expression level for each gene, red dots represent genes whose abundance is significantly up-regulated, green dots - down-regulated, and black dots unchanged (or non-significantly changed) regulation; c COG annotation of and DEGs; d KEGG annotation of and DEGs
Among the 96,427 unigenes, 5556 significantly differentially expressed genes (DEGs) were identified by pairwise comparison of the HCBD with the control group; 1318 of them were upregulated and 4238 were downregulated in the HCBD group (Fig. 4b). Among these, 4440 were successfully annotated: 1935 using the COG database, 1686 using the GO database, 1895 using the KEGG database, 3973 using the Pfam database, 2294 using the Swiss-Prot database, and 3265 using the NR database.
Distribution of the 1686 genes successfully annotated using GO database in the three GO categories generally followed the pattern observed among all annotated unigenes (Fig. 4a), with some notable exceptions: in the ‘biological process’ category, genes associated with metabolic processes were more abundant (second, vs third most abundant), as were genes associated with multi-organism processes, signaling and rhythmic process, whereas biological adhesion and reproductive process categories exhibited reduced abundance. In the ‘cellular component’ category, the most notable was the complete absence of collagen trimer-associated DEG unigenes. In the ‘molecular function’ category, most notable was a strong increase in genes associated with structural molecule and channel regulator activities, whereas guanyl-nucleotide exchange factor activity-associated unigenes were notably reduced.
Among the 1935 DEGs successfully annotated using the COG database (Fig. 4c), the major group was ‘General function prediction only’ (412 unigenes; 15.92%), followed by ‘Translation, ribosomal structure, and biogenesis’ (348 unigenes; 13.45%), ‘Posttranslational modification, protein turnover, chaperones’ (227 unigenes; 8.77%), and ‘Energy production and conversion’ (202 unigenes, 7.81%). In ‘cell mobility’ category, only three unigenes were differentially expressed, and none in ‘extracellular structures’ and ‘nuclear structure’ categories.
To further clarify the biological pathways affected by the HCBD, the 1895 DEGs were mapped with the KEGG Pathway tools, and assigned to a total of 319 pathways, divided into six KEGG categories (Fig. 4d): the most strongly represented functional category was ‘Human Diseases’, containing 709 unigenes, the highest number of which (145) were associated with Alzheimer’s disease. Importantly, a large number (96) of DEGs in this category were also associated with NAFLD. Expectedly, a large number (592) of DEGs were associated with six different metabolic processes in ‘Metabolism’ category, with OXPHOS and Carbon metabolism at the top. By far the highest number (269) of DEGs was associated with Ribosome in the ‘Genetic Information Processing’ category (Fig. 4d, Additional file 6).
qPCR
Among the 13 DEGs associated with the NAFLD pathway and Insulin signaling pathway (Additional files 1 and 7) (http://www.genome.jp/kegg/) studied by qPCR, ten were (highly) significantly upregulated in the HCBD group (in comparison to the control group), whereas three were (highly) significantly downregulated (Fig. 5).

Expression of 13 DEGs from the transcriptome analysis associated with NAFLD, studied by qPCR. Data were normalized to 18 s rRNA and Rpl13a as reference genes and presented as a fold change between the control and HCBD groups (mean ± SE). HCBD is the high-carbohydrate diet group. Results were analyzed by t-test. (*) above bar indicates significant (P < 0.05) and (**) highly significant differences (P < 0.01)
Discussion
Hepatosomatic index and liver histology
The observed lipid vacuolization of hepatocytes and increased HSI indicate that the HCBD has resulted in de novo lipid synthesis [9, 53] and lipid accumulation in livers. Abnormal hepatocytes and lipid accumulation have been observed previously in association with high-carbohydrate diet in blunt snout bream [27], as well as in a number of other fish species, e.g., Nile tilapia (Oreochromis niloticus), gilthead sea bream [54], juvenile yellow catfish (Pelteobagrus fulvidraco) [55], juvenile Labeo rohita [56], and L. rohita fry [57]. High lipid accumulation was also the most likely cause for the high HSI value observed in the HCBD group. Increased HSI in association with high-carbohydrate diet was also observed in a number of other fish species, including golden pompano (Trachinotus ovatus) [58] and gilthead sea bream (Sparus aurata) [59, 60]. Therefore, high carbohydrate intake has caused fat accumulation and inflammatory infiltration in the liver, both of which are indicative of impaired liver health and precursors of the fatty liver disease [61,62,63].
Serum biochemistry
High ALT and AST levels are indicators of liver damage and are associated with NAFLD [64, 65]. We hypothesize that an explanation for the much higher LDL levels observed here might be increased cholesterol synthesis in the liver in association with high dietary carbohydrates, resulting in an increase in the amount of LDL required to transport this cholesterol to other tissues. High levels of LDL are associated with obesity in mammals [66], and are generally a predictor of several chronic diseases [67]. LDL is also one of the causes of hepatic inflammation, and most importantly plays a critical role in the development of NAFLD [68]. Increased HDL levels in the HCBD group are somewhat confusing in this context, as these two metabolites are generally in negative correlation [69, 70]. The observed increase in both LDL and HDL levels may therefore indicate a general disorder in lipid metabolism caused by high de novo lipid synthesis. Also somewhat surprising was the lower level of triglycerides in the serum of the HCBD group, despite the high lipid accumulation in liver and marginally higher body weight [29]. However, TG levels in serum are driven by a large number of factors, and thus not particularly informative [71]. Liver dysfunction, attributable to liver damage associated with fatty liver, may be a part of the explanation for this phenomenon, insofar as the capacity of a damaged liver to pick up the extra TGs from the serum might be somewhat impaired. Additionally, low serum TGs have been associated with low betaine levels in mice [72], which was observed in the metabolomic analysis in our study (discussed in more detail in the following section). All these results are in agreement with previous observations that HCBD causes a metabolic stress response in blunt snout bream [31].
The metabolomics of serum and liver
A part of the increase in HSI in the fish fed the HCBD may hypothetically be attributed to increased glycogen deposition in liver [1, 59, 73]. A previous study has also shown that blunt snout breams fed a high-carbohydrate diet had significantly higher hepatic glycogen content than those fed a control diet [27]. Our metabolomics results show that the high dietary carbohydrate intake induced significant increases in plasma α/β-glucose, succinate and tyrosine (Fig. 2), all of which can also be converted to acetyl-CoA in the TCA cycle pathway, which connects the glucose metabolism with the fatty acid metabolism. Therefore, although it is possible that the excessive carbohydrates available in the diet may have resulted in increased storage of α/β-glucose as glycogen, a large increase in the lipid content in the livers of the HCBD group suggests that it is much more likely that the majority of the superfluous carbohydrates were redirected to de novo lipid synthesis in the liver.
It is known that a decline in betaine is correlated with lower serum TGs in a diabetic mouse model [72]. Therefore, lower serum TG levels in the HCBD group are also consistent with the liver metabolomics results, where betaine was significantly reduced in the HCBD group. Betaine is the product of the irreversible oxidation of choline in liver and kidney. It can effectively prevent fructose-induced NAFLD and improve liver function by inhibiting inflammatory factors, reducing lipid peroxidation, reducing endoplasmic reticulum stress, and preventing apoptosis [74,75,76]. This indicates that a decline in liver betaine may be a consequence of the pathological hepatic changes associated with the high carbohydrate diet.
Impacts of high-carbohydrate diet on gene expression
Thirteen DEGs were directly associated with the NAFLD pathway and insulin signaling pathway (Additional file 1), and their expression was further studied by qPCR (Fig. 5). The expression of the suppressor of cytokine signaling 3 (SOCS3) gene, which plays an important role in the pathogenesis of obesity in animal models [77], was downregulated (P < 0.05). This gene correlates negatively with the insulin receptor (INSR) and the insulin receptor substrate (IRS) [78], both of which were accordingly upregulated (P < 0.01). INSR is a member of the receptor tyrosine kinase family, which binds insulin and other ligands that regulate glucose uptake and release, as well as the synthesis and storage of carbohydrates, lipids, and proteins [79]. Insulin can also regulate its own expression by binding to cell-surface INSR and stimulating phosphorylation-dependent signaling cascade [80]. IRS phosphorylates the P13 kinase, which was significantly upregulated in the HCBD group (Fig. 5), by binding to its regulatory subunit p85 [81] and thereby generates Phosphatidylinositol-(3,4,5)-triphosphate (PIP3), which in turn leads to the activation of several PIP3-dependent serine/threonine kinases, including PDPK1 and subsequently AKT/PKB [79, 82].
Upregulation in the expression of INSR and IRS genes has further led to upregulation of the downstream elements in this signaling pathway (Fig. 5, Additional file 1): phosphoinositide-3-kinase (PI3K), phosphoinositide-dependent protein kinase 1 (PDPK1), and RAC-gamma serine/threonine-protein kinase (AKT) in the HCBD group (although the transcriptomic data show downregulation of PI3K, see Table 2, qPCR analysis of the same sample has repeatedly shown upregulation of this gene). Normally, AKT is phosphorylated by PDPK1, which is a master kinase activated by several growth factors and hormones that plays an important role in various signaling pathways, including insulin signaling, and is believed to play an important role in regulating the organism size [83]. The increase in AKT/PKB expression appears to have inhibited Glycogen synthase kinase 3 beta (GSK3β) expression in this study (significantly downregulated, Fig. 5). GSK3β was probably inactivated because its expression correlates negatively with AKT [84, 85], and glycogen (starch) synthase (GYS). As GYS is an enzyme that catalyzes the final step of glycogen synthesis [86], the inactivation of GSK3β by AKT promotes glucose storage as glycogen. Thus these results indicate that the HCBD has resulted in increased glycogen synthesis. By promoting glucose storage, insulin inhibited the production and release of glucose in the liver by blocking gluconeogenesis and glycogenolysis [87]. This mechanism is likely to lead to insulin resistance (Fig. 6 and Additional file 1), which is an important component of the pathogenesis of NAFLD in humans, where insulin resistance in adipose tissue leads to increases in circulating glucose and the availability of lipid substrates for the accumulation of hepatic lipids [88].

A hypothetical mechanism through which the observed changes in transcriptome, serum biochemistry, and serum and liver metabolomics caused by high-carbohydrate diet can lead to fatty liver disease
The expression of acetyl-CoA carboxylase (ACC) was significantly upregulated as a result of the downregulation of its inhibitor: AMP-activated protein kinase (AMPK) (Figs. 5, 6 and Additional file 1). AMPK regulates insulin sensitivity: a continuous reduction in AMPK activity is accompanied by insulin resistance, whereas AMPK activation increases insulin sensitivity. AMPK also controls a number of key players in various metabolic pathways and is therefore a major regulator of glucose and lipid metabolisms [89]. This downregulation of AMPK and upregulation of ACC, which is crucial for hepatic de novo fatty acid synthesis, are strongly indicative of heightened de novo lipogenesis [61, 90,91,92,93]. Thus these results indicate that prolonged exposure of hepatocytes to high glucose concentrations associated with the HCBD has resulted in insulin resistance and increased lipogenesis in liver [94], which is also in agreement with our histological observations. These early symptoms of hepatic metabolic disorders, when prolonged, can lead to a pathological accumulation of TGs in liver and eventually contribute to the onset of fatty liver disease [63, 95, 96].
Annotation results for the fructose-1,6-bisphosphatase 1 (FBP) gene indicate the existence of several isoforms, some of which were up- and some down-regulated (Table 2). In mammals, this gene partakes in carbohydrate transport and metabolism via the insulin signaling pathway, which is associated with NAFLD. It plays a role in regulating glucose sensing and insulin secretion of pancreatic beta-cells, and is an important negative regulator of appetite [97]. Some previous studies found that high carbohydrate diets result in increased FBP expression [2, 98,99,100]. However, the existence of several differently regulated paralogs in our results indicates that its regulation mechanism and functions in fish might be more complex than in mammals.
Hepatic IL6, which is an important proinflammatory adipocytokine that is almost always significantly elevated in the adipose cells of obese and insulin-resistant individuals and NAFLD patients, was also upregulated in the HCBD group (Fig. 5). Increased lipid accumulation in liver is usually observed in association with elevated expression of this gene [101,102,103], which is in agreement with the liver histology results in this study. Another gene associated with de novo fatty acid synthesis, MAX-like protein X (MLX-ChREBP), was also upregulated in the HCBD group. Based on many reports that found that ChREBP-MLX is a liver transcription factor always responsive to the consumption of high levels of carbohydrates and required for the induction of the expression of lipogenic genes in response to high dietary glucose [104,105,106,107], it has been proposed that the activation of ChREBP-MLX may be the glucose-dependent mechanism responsible for the synergistic induction of fatty acid synthesis by glucose and insulin [108]. Increased transcription of the Transcription factor activator protein 1 (AP1) gene, which was also upregulated in the HCBD group (Fig. 5), has been linked to obesity, hepatic lipid metabolism, and NAFLD [109]. The expression of interleukin 6 (IL6), transcription factor AP-1 (AP1), phosphoenolpyruvate carboxykinase (PEPCK) gene was also upregulated in the HCBD group (Table 2, Additional file 1). This is a key enzyme in the synthesis of glucose in liver and kidney, which also plays a role in hepatic energy metabolism [110, 111]. It is not clear why would a gene associated with increased gluconeogenesis be upregulated in association with HCBD. However, glucose level in blood is maintained within well-defined limits, in part due to the precise regulation of PEPCK gene expression. For example, overexpression of this enzyme in mice results in symptoms of the type II diabetes mellitus [112]. Because of the importance of the blood glucose homeostasis, a number of hormones regulate a set of genes (including PEPCK) in liver, which modulate the rate of glucose synthesis. Thus the upregulation of this gene may be a reflection of hormonal disbalance caused by high glucose levels in blood [113,114,115,116,117].
Correlation between transcriptomic and qPCR results was relatively low: Pearson correlation coefficient (R2 = 0.3226), significance analysis (P = 0.0428), and linear regression (red line) are shown (Additional file 7). The underlying cause might be different data normalization methods used for qPCR and RNA-seq data analyses. In qPCR, reference genes are used to normalize gene expression, where the expression of target gene is calculated relative to the expression of reference gene. This method assumes that the expression of reference gene(s) is constant for all samples. However in transcriptomic data analysis (RNA-seq), in most cases we assume that each sample has the same total expressed mRNA, which can be expressed as read count per million mapped reads (RPKM). Thus, both individual differences between samples and variation in reference gene expression can cause discrepancies between the results obtained by these two methods. Also, for subtle differences, it is not unlikely that only one method will detect a significant difference in expression, as qPCR and RNA-seq use different scales. Furthermore, in cases where qPCR analysis indicates upregulation, whereas the transcriptomics indicates downregulation, we should not exclude a possibility that the primers designed for qPCR may have non-specifically amplified both (or more) paralogs present in the genome. As a result of a relatively recent (≈8 MYA) genome duplication, teleost fish possess a large number of duplicated genes [118]. Many paralogs have retained their functions [119, 120], and their expression regulation is often very complex [121,122,123]. Such circumstances may easily result in inconsistent expression analysis results.
Conclusions
All of our results, which includes HSI values, liver histology, serum biochemistry, serum and liver metabolomics, liver transcriptomics and specific gene expression analyses, show signs of high accumulation of lipids in the liver of blunt snout bream in association with high-carbohydrate diet. Some of the results even indicate pathological liver changes usually associated with NAFLD. Specifically, high levels of LDL, HDL, AST and ALT observed in the serum are all consistent with our hepatic histology results, which indicate excessive lipid accumulation and liver damage. The decline in the liver betaine may be a consequence of pathological hepatic changes associated with the high-carbohydrate diet. Transcriptomic profiles showed that the HCBD had a strong impact on the expression of genes involved in the NAFLD and insulin signaling pathways, which may lead to the development of insulin resistance in hepatocytes and eventually NAFLD. An example is IL6, upregulation of which is strongly associated with insulin-resistance and NAFLD. These results are in agreement with previous observations that HCBD causes stress in blunt snout bream [31]. However, although high HSI and hepatic lipid accumulation correlate negatively with hepatic health in fish, further research will be needed to fully assess whether these pathological changes associated with high-carbohydrate diet would actually lead to the development of NAFLD after a longer period of time. Based on our results, we propose that the duration of these future studies should be more than 60 days and that blood serum parameters should be regularly monitored during the feeding trial.
Finally, an additional interesting finding in our study, not directly discussed here, is a very high number of upregulated genes in the HCBD group associated with a number of neurodegenerative diseases (Fig. 4d): Alzheimer’s (145 genes), Huntington’s (131 genes) and Parkinson’s (127 genes). This is a strong indication of neurodegenerative changes associated with high-carbohydrate diet in blunt snout bream, which has been observed in humans as well [124,125,126]. Therefore, fish might present a good model to study neurodegenerative changes associated with high-carbohydrate diet in humans.
Abbreviations
- ACC:
-
Acetyl-CoA carboxylase
- AKT:
-
RAC-gamma serine/threonine-protein kinase
- ALT:
-
Alanine transaminase
- AMPK:
-
AMP-activated protein kinase
- AP1:
-
Transcription factor AP-1
- AST:
-
Aspartate transaminase
- Bp:
-
Basepair
- BSB:
-
Blunt snout bream
- cDNA:
-
Complementary DNA
- ChREBP:
-
MAX-like protein X
- DEGs:
-
Differentially expressed genes
- FBP:
-
Fructose-1,6-bisphosphatase 1
- FDR:
-
False discovery rate
- FPKM:
-
Fragments per kilobase million
- GO:
-
Gene ontology
- GSK3β:
-
Glycogen synthase kinase 3 beta
- GYS:
-
Glycogen (starch) synthase
- HCBD:
-
High-carbohydrate diet
- HDL:
-
High-density lipoprotein
- HSI:
-
Hepatosomatic index
- IL6:
-
Interleukin 6
- INSR:
-
Insulin receptor
- IRS:
-
Insulin receptor substrate
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- KO:
-
KEGG orthology
- KOG:
-
Eukaryotic Orthologous Groups
- LDL:
-
Low-density lipoprotein
- mRNA:
-
Messenger RNA
- MYA:
-
Million years ago
- NAFLD:
-
Nonalcoholic fatty liver disease
- NCBI:
-
National Center for Biotechnology Information
- Nr database:
-
Non-redundant database
- ORF:
-
Open reading frame
- PCR:
-
Polymerase chain reaction
- PDK1:
-
Phosphoinositide-dependent protein kinase1
- PEPCK:
-
Phosphoenolpyruvate carboxykinase
- PI3K:
-
Phosphoinositide-3-kinase
- RPKM:
-
Reads per kilobase of exon model per million mapped reads
- SNP:
-
Single nucleotide polymorphism
- SOCS3:
-
Suppressor of cytokine signaling 3
- SSR:
-
Simple sequence repeats or microsatellites
- TCA:
-
Tricarboxylic acid
- TG:
-
Triglyceride
References
Wilson RP. Utilization of dietary carbohydrate by fish. Aquaculture. 1994;124:67–80.
Enes P, Peres H, Couto A, Oliva-Teles A. Growth performance and metabolic utilization of diets including starch, dextrin, maltose or glucose as carbohydrate source by gilthead sea bream (Sparus Aurata) juveniles. Fish Physiol Biochem. 2010;36:903–10.
Ren M, Habte-Tsion HM, Xie J, Liu B, Zhou Q, Ge X, et al. Effects of dietary carbohydrate source on growth performance, diet digestibility and liver glucose enzyme activity in blunt snout bream, Megalobrama Amblycephala. Aquaculture. 2015;438:75–81.
Rippe JM, Angelopoulos TJ. Sucrose, high-fructose corn syrup, and fructose, their metabolism and potential health effects: what do we really know? Adv Nutr. 2013;4:236–45.
Macdonald IA. Dietary strategies for the management of cardiovascular risk: role of dietary carbohydrates. Proc Nutr Soc. 2014;73:167–71.
Postic C, Dentin R, Girard J. Role of the liver in the control of carbohydrate and lipid homeostasis. Diabetes Metab. 2004;30:398–408.
Enes P, Panserat S, Kaushik S, Oliva-Teles A. Nutritional regulation of hepatic glucose metabolism in fish. Fish Physiol Biochem. 2009;35:519–39.
Goldsmith JR, Jobin C. Think small: zebrafish as a model system of human pathology. J Biomed Biotechnol. 2012;2012:817341.
Hemre GI, Mommsen TP, Krogdahl Å. Carbohydrates in fish nutrition: effects on growth, glucose metabolism and hepatic enzymes. Aquac Nutr. 2002;8:175–94.
Moon TW. Glucose intolerance in teleost fish: fact or fiction? Comp Biochem Physiol Part B. 2001;129:243–9.
Li X, Jiang Y, Liu W, Ge X. Protein-sparing effect of dietary lipid in practical diets for blunt snout bream (Megalobrama Amblycephala) fingerlings: effects on digestive and metabolic responses. Fish Physiol Biochem. 2012;38:529–41.
Polakof S, Panserat S, Soengas JL, Moon TW. Glucose metabolism in fish: a review. J Comp Physiol B. 2012;182:1015–45.
White JS. Challenging the fructose hypothesis: new perspectives on fructose consumption and metabolism. Adv Nutr. 2013;4:246–56.
Jafri A. Effect of dietary carbohydrate-to-lipid ratio on growth and body composition of walking catfish (Clarias Batrachus). Aquaculture. 1998;161:159–68.
Lee SM, Kim KD, Lall SP. Utilization of glucose, maltose, dextrin and cellulose by juvenile flounder (Paralichthys Olivaceus). Aquaculture. 2003;221:427–38.
Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nat Rev Genet. 2007;8:353–67.
Goessling W, Sadler KC. Zebrafish: an important tool for liver disease research. Gastroenterology. 2015;149:1361–77.
Matsumoto T, Terai S, Oishi T, Kuwashiro S, Fujisawa K, Yamamoto N, et al. Medaka as a model for human nonalcoholic steatohepatitis. Dis Model Mech. 2010;3:431–40.
Bureau CF. China fisheries yearbook. Beijing: Chinese Ag; 2013.
FAO. Fishery and aquaculture statistics. 2014.
Ke H. The artificial reproduction and culture experiment of Megalobrama Amblycephala. Acta Hydrobial Sin. 1965;5:282–3. (In China)
Li S, Cai W. Effects of two-way selection on biochemical genetics of blunt snout bream (Megalobrama Amblycephala). J Fish China. 1996;3:1–5. (In China)
Liang T, Wang DD, Zhang GR, Wei KJ, Wang WM, Zou GW. Molecular cloning and expression analysis of two B-defensin genes in the blunt snout bream (Megalobrama Amblycephala). Comp Biochem Physiol Part B. 2013;166:91–8.
Gao W, Liu YJ, Tian LX, Mai KS, Liang GY, Yang HJ, et al. Effect of dietary carbohydrate-to-lipid ratios on growth performance, body composition, nutrient utilization and hepatic enzymes activities of herbivorous grass carp (Ctenopharyngodon Idella). Aquac Nutr. 2009;16:327–33.
Li XF, Lu KL, Liu WB, Jiang GZ, Xu WN. Effects of dietary lipid and carbohydrate and their interaction on growth performance and body composition of juvenile blunt snout bream, Megalobrama Amblycephala. Bamidgeh. 2014;66:931.
Li XF, Wang Y, Liu WB, Jiang GZ, Zhu J. Effects of dietary carbohydrate/lipid ratios on growth performance, body composition and glucose metabolism of fingerling blunt snout bream Megalobrama Amblycephala. Aquac Nutr. 2013;19:701–8.
Zhou CP, Ge XP, Liu B, Xie J, Miao LH. Effect of high dietary carbohydrate on the growth performance and physiological responses of juvenile wuchang bream, Megalobrama Amblycephala. Asian Australas J Anim Sci. 2013;26:1598–608.
Li XF, Liu WB, Lu KL, Xu WN, Wang Y. Dietary carbohydrate/lipid ratios affect stress, oxidative status and non-specific immune responses of fingerling blunt snout bream, Megalobrama Amblycephala. Fish Shellfish Immunol. 2012;33:316–23.
Prathomya P, Prisingkorn W, Jakovlić I, Deng F-Y, Zhao Y-H, Wang W-M. 1H NMR-based metabolomics approach reveals metabolic alterations in response to dietary imbalances in Megalobrama Amblycephala. Metabolomics. 2017;13:17.
Zhang Y, Liu B, Zhou C, Ge X, Xie J, Xu P. The research progress of nutritional requirement for Megalobrama Amblycephala Yih. J Anhui Agric. 2010;32:18239–41. (In China)
Zhou C, Liu B, Ge X, Xie J, Xu P, Liu B, et al. Effect of dietary carbohydrate on the growth performance, immune response, hepatic antioxidant abilities and heat shock protein 70 expression of Wuchang bream, Megalobrama Amblycephala. J Appl Ichthyol. 2013;29:1348–56.
AOAC International A. Official methods of analysis of AOAC international. Agric Chem Contam drugs. 1995:I.
Tan Q, Xie S, Zhu X, Lei W, Yang Y. Effect of dietary carbohydrate sources on growth performance and utilization for gibel carp (Carassius Auratus Gibelio) and Chinese longsnout catfish (Leiocassis Longirostris Gunther). Aquac Nutr. 2006;12:61–70.
Churukian CJ. Lillie’s oil red O method for neutral lipids. J Histotechnol. 1999;22:309–11.
Stejskal EO, Tanner JE. Spin diffusion measurements: spin echoes in the presence of a time-dependent field gradient. J Chem Phys. 1965;42:288.
An Y, Xu W, Li H, Lei H, Zhang L, Hao F, et al. High-fat diet induces dynamic metabolic alterations in multiple biological matrices of rats. J. Proteome Res American Chemical Society. 2013;12:3755–68.
Liu X, Xu X, Li B, Wang X, Wang G, Li M. RNA-seq transcriptome analysis of maize inbred carrying nicosulfuron-tolerant and nicosulfuron-susceptible alleles. Int J Mol Sci. 2015;16:5975–89.
HiSeq ® 2500 System Guide. 2015;
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52.
Consortium U. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–69.
Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16:276–7.
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6.
Galperin MY, Makarova KS, Wolf YI, Koonin EV. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015;43:D261–9.
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27:29–34.
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:182–5.
Mao X, Cai T, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21:3787–93.
Mortazavi A, Brian AW, Kenneth M, Lorian S, Barbara W. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8.
Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BMG, et al. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013;29:1035–43.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300.
Rychlik W, Rhoads RE. A computer program for choosing optimal oligonudeotides for filter hybridization, sequencing and in vitro amplification of DNA. Nucleic Acids Res. 1989;17:8543–51.
Luo W, Zhang J, Wen JF, Liu H, Wang WM, Gao ZX. Molecular cloning and expression analysis of major histocompatibility complex class I, IIA and IIB genes of blunt snout bream (Megalobrama Amblycephala). Dev Comp Immunol. 2014;42:169–73.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the2-DDCT method. Methods. 2001;25:402–8.
Shimeno S, Ming D-C, Takeda M. Metabolic response to dietary carbohydrate to lipid ratios in Oreochromis Niloticus. Nippon Suisan Gakkaishi. 1993;59:827–33.
Bou M, Todorčević M, Fontanillas R, Capilla E, Gutiérrez J, Navarro I. Adipose tissue and liver metabolic responses to different levels of dietary carbohydrates in gilthead sea bream (Sparus Aurata). Comp Biochem Physiol A Mol Integr Physiol. 2014;175:72–81.
Wang LN, Bin LW, Le LK, Xu WN, Sen CD, Zhang CN, et al. Effects of dietary carbohydrate/lipid ratios on non-specific immune responses, oxidative status and liver histology of juvenile yellow catfish Pelteobagrus Fulvidraco. Aquaculture. 2014;426–427:41–8.
Kumar S, Sahu NP, Pal AK, Choudhury D, Yengkokpam S, Mukherjee SC. Effect of dietary carbohydrate on haematology, respiratory burst activity and histological changes in L. Rohita juveniles. Fish Shellfish Immunol. 2005;19:331–44.
Mohapatra M, Sahu NP, Chaudhari A. Utilization of gelatinized carbohydrate in diets of Labeo Rohita fry. Aquac Nutr. 2003;9:189–96.
Zhou C, Ge X, Niu J, Lin H, Huang Z, Tan X. Effect of dietary carbohydrate levels on growth performance, body composition, intestinal and hepatic enzyme activities, and growth hormone gene expression of juvenile golden pompano, Trachinotus Ovatus. Aquaculture. 2015;437:390–7.
Moreira IS, Peres H, Couto A, Enes P, Oliva-Teles A. Temperature and dietary carbohydrate level effects on performance and metabolic utilisation of diets in European sea bass (Dicentrarchus Labrax) juveniles. Aquaculture. 2008;274:153–60.
Enes P, Panserat S, Kaushik S, Oliva-teles A. Growth performance and metabolic utilization of diets with native and waxy maize starch by gilthead sea bream ( Sparus Aurata ) juveniles. Aquaculture. 2008;274:101–8.
Song Z, Deaciuc I, Zhou Z, Song M, Chen T, Hill D, et al. Involvement of AMP-activated protein kinase in beneficial effects of betaine on high-sucrose diet-induced hepatic steatosis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G894–902.
Duvnjak M, Lerotić I, Baršić N, Tomašić V, Jukić LV, Velagić V. Pathogenesis and management issues for non-alcoholic fatty liver disease. World J Gastroenterol. 2007;13:4539–50.
Bergheim I, Weber S, Vos M, Krämer S, Volynets V, Kaserouni S, et al. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: role of endotoxin. J Hepatol. 2008;48:983–92.
Clark JM, Van Natta M, Kleiner D, Chalasani NP, McCullogh AJ, Neuschwander-Tetri BA, et al. Association of AST and ALT with liver histology in adults with NAFLD. Hepatology. 2007;738A:46.
Mahtab MA, Rahman S, Karim F, Hossain MF, Khan M. Serum ALT is a better predictor of fibrosis than serum AST in patients with NAFLD: experience from a tertiary centre in Bangladesh. J Gastroenterol Hepatol. 2008;23(Suppl.5):A26–7.
Waldram A, Holmes E, Wang Y, Rantalainen M, Wilson ID, Tuohy KM, et al. Top-down systems biology modeling of host metabotype-microbiome associations in obese rodents. J Proteome Res American Chemical Society. 2009;8:2361–75.
Pedersen BK, Saltin B. Evidence for prescribing exercise as therapy in chronic disease. Scand J Med Sci Sports. 2006;16(Suppl 1):3–63.
Lai YS, Yang TC, Chang PY, Chang SF, Ho SL, Chen HL, et al. Electronegative LDL is linked to high-fat, high-cholesterol diet-induced nonalcoholic steatohepatitis in hamsters. J Nutr Biochem. 2016;30:44–52.
Kjaer MA, Vegusdal A, Berge GM, Galloway TF, Hillestad M, Krogdahl S, et al. Characterisation of lipid transport in Atlantic cod (Gadus Morhua) when fasted and fed high or low fat diets. Aquaculture. 2009;288:325–36.
Tocher DR, Bendiksen EÅ, Campbell PJ, Bell JG. The role of phospholipids in nutrition and metabolism of teleost fish. Aquaculture. 2008;280:21–34.
Macdonald I. Diet and triglyceride metabolism. J Clin Pathol. 1973;26:22–5.
Jung GY, Won SB, Kim J, Jeon S, Han A, Kwon YH. Betaine alleviates hypertriglycemia and tau hyperphosphorylation in db/db mice. Toxicol Res. 2013;29:7–14.
Zhou C, Ge X, Liu B, Xie J, Chen R, Ren M. Effect of high dietary carbohydrate on the growth performance, blood chemistry, hepatic enzyme activities and growth hormone gene expression of Wuchang bream (Megalobrama Amblycephala) at two temperatures. Asian-Australas J Anim Sci. 2015;28:207–14.
Ji C, Shinohara M, Vance D, Than TA, Ookhtens M, Chan C, et al. Effect of transgenic extrahepatic expression of betaine-homocysteine methyltransferase on alcohol or homocysteine-induced fatty liver. Alcohol Clin Exp Res. 2008;32:1049–58.
Kharbanda KK, Mailliard ME, Baldwin CR, Sorrell MF, Tuma DJ. Accumulation of proteins bearing atypical isoaspartyl residues in livers of alcohol-fed rats is prevented by betaine administration: effects on protein-L-isoaspartyl methyltransferase activity. J Hepatol. 2007;46:1119–25.
Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–99.
Tang W, Zou J, Chen X, Zheng J, Zeng H, Liu Z, et al. Association of two polymorphisms within and near SOCS3 gene with obesity in three nationalities in Xinjiang province of China. Acta Pharmacol Sin CPS and SIMM. 2011;32:1381–6.
Ueki K, Kondo T, Kahn CR, Ueki K, Kondo T, Kahn CR. Suppressor of cytokine signaling 1 ( SOCS-1 ) and SOCS-3 cause insulin resistance through inhibition of tyrosine Phosphorylation of insulin receptor substrate proteins by discrete mechanisms suppressor of cytokine signaling 1 ( SOCS-1 ) and SOCS-3 cause I. Mol Cell Biol. 2004;24:5434–46.
INSR_GeneCards [Internet]. [cited 2016 May 8]. Available from: http://www.genecards.org/cgi-bin/carddisp.pl?gene=INSR. Accessed 5 Aug 2017.
Lynn FC. Meta-regulation: microRNA regulation of glucose and lipid metabolism. Trends Endocrinol Metab. 2009;20:452–9.
White M. The insulin signalling system and the IRS proteins. Diabetologia. 1997;40:S2–17.
Farese RV, Sajan MP, Standaert ML. Insulin-sensitive protein Kinases (atypical protein Kinase C and protein Kinase B/Akt): actions and defects in obesity and type II diabetes. Soc. Exp Biol Med. 2005;230:593–605.
Bayascas JR, Wullschleger S, Sakamoto K, Garcia-Martinez JM, Clacher C, Komander D, et al. Mutation of the PDK1 PH domain inhibits protein kinase B/Akt, leading to small size and insulin resistance. Mol. Cell Biol. 2008;28:3258–72.
Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1.
Kitagishi Y, Kobayashi M, Kikuta K, Matsuda S. Roles of PI3K/AKT/GSK3/mTOR pathway in cell signaling of mental illnesses. Depress Res Treat. 2012(2012):8. http://dx.doi.org/10.1155/2012/752563.
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9.
Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806.
Hazlehurst JM, Woods C, Marjot T, Cobbold JF, Tomlinson JW. Non-alcoholic fatty liver disease and diabetes. Metabolism. 2016;65:1096–108.
Godlewski J, Nowicki MO, Bronisz A, Nuovo G, Palatini J, De Lay M, et al. MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in Glioma cells. Mol Cell. 2010;37:620–32.
Park H, Kaushik VK, Constant S, Prentki M, Przybytkowski E, Ruderman N, et al. Coordinate regulation of malonyl-CoA decarboxylase, sn-glycerol-3-phosphate acyltransferase, and acetyl-CoA carboxylase by AMP-activated protein kinase in rat tissues in response to exercise. J Biol Chem. 2002;277:32571–7.
Saha AK, Ruderman NB. Malonyl-CoA and AMP-activated protein kinase: an expanding partnership. Mol Cell Biochem. 2003;253:65–70.
Diraison F, Moulin P, Beylot M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab. 2003;29:478–85.
Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest American Society for Clinical Investigation. 2005;115:1343–51.
Zang M, Zuccollo A, Hou X, Nagata D, Walsh K, Herscovitz H, et al. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J Biol Chem. 2004;279:47898–905.
Diraison F, Dusserre E, Vidal H, Sothier M, Beylot M. Increased hepatic lipogenesis but decreased expression of lipogenic gene in adipose tissue in human obesity. Am J Physiol-Endoc Metab. 2002;282:E46–51.
Koo H-Y, Wallig MA, Chung BH, Nara TY, Cho BHS, Nakamura MT. Dietary fructose induces a wide range of genes with distinct shift in carbohydrate and lipid metabolism in fed and fasted rat liver. Biochim Biophys Acta. 2008;1782:341–8.
FBP1_GeneCards [Internet]. [cited 2017 Jun 12]. Available from: http://www.genecards.org/cgi-bin/carddisp.pl?gene=FBP1&keywords=FBP1. Accessed 6 Dec 2017.
Rocha F, Dias J, Engrola S, Gavaia P, Geurden I, Dinis MT, et al. Glucose metabolism and gene expression in juvenile zebrafish (Danio Rerio) challenged with a high carbohydrate diet: effects of an acute glucose stimulus during late embryonic life. Br J Nutr. 2015;113:403–13.
Enes P, Panserat S, Kaushik S, Oliva-Teles A. Rapid metabolic adaptation in European sea bass (Dicentrarchus Labrax) juveniles fed different carbohydrate sources after heat shock stress. Comp Biochem Physiol - A Mol Integr Physiol. 2006;145:73–81.
Shikata T, Iwanaga S, Shimeno S. Effects of dietary glucose, fructose, and galactose on hepatopancreatic enzyme activities and body composition in carp*. Fish Sci. 1994;60:613–7.
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91.
Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R, Simsolo RB. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest. 1995;95:2111–9.
Haukeland JW, Damås JK, Konopski Z, Løberg EM, Haaland T, Goverud I, et al. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J Hepatol. 2006;44:1167–74.
Dentin R, Pégorier J-P, Benhamed F, Foufelle F, Ferré P, Fauveau V, et al. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem. 2004;279:20314–26.
Ishii S, Iizuka K, Miller BC, Uyeda K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc Natl Acad Sci U S A. 2004;101:15597–602.
Ma L, Tsatsos NG, Towle HC. Direct role of ChREBP.Mlx in regulating hepatic glucose-responsive genes. J Biol Chem. 2005;280:12019–27.
Koo HY, Miyashita M, Simon Cho BH, Nakamura MT. Replacing dietary glucose with fructose increases ChREBP activity and SREBP-1 protein in rat liver nucleus. Biochem Biophys Res Commun. 2009;390:285–9.
Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A. 2004;101:7281–6.
Hasenfuss SC, Bakiri L, Thomsen MK, Williams EG, Auwerx J, Wagner EF. Regulation of steatohepatitis and PPAR?? Signaling by distinct AP-1 dimers. Cell Metab. 2014;19:84–95.
She P, Shiota M, Shelton KD, Chalkley R, Postic C, Magnuson MA. Phosphoenolpyruvate carboxykinase is necessary for the integration of hepatic energy metabolism. Mol Cell Biol. 2000;20:6508–17.
Hanson RW, Reshef L. Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression. Annu Rev Biochem. 1997;66:581–611.
Magnusson I, Rothman DL, Katz LD, Shulman RG, Shulman GI. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J Clin Invest. 1992;90:1323–7.
Méndez-Lucas A, Duarte JAG, Sunny NE, Satapati S, He T, Fu X, et al. PEPCK-M expression in mouse liver potentiates, not replaces, PEPCK-C mediated gluconeogenesis. J Hepatol. 2013;59:105–13.
Liu K, Ba X, Yu J, Li J, Wei Q, Han G, et al. The phosphoenolpyruvate carboxykinase of mycobacterium tuberculosis induces strong cell-mediated immune responses in mice. Mol Cell Biochem. 2006;288:65–71.
Holyoak T, Sullivan SM, Nowak T. Structural insights into the mechanism of PEPCK catalysis. Biochemistry. 2006;45:8254–63.
Chakravarty K, Cassuto H, Reshef L, Hanson RW. Factors that control the tissue-specific transcription of the gene for Phosphoenolpyruvate Carboxykinase-C. Crit Rev Biochem Mol Biol. 2005;40:129–54.
Nash JT, Szabo DT, Carey GB. Polybrominated Diphenyl ethers Alter hepatic Phosphoenolpyruvate Carboxykinase enzyme kinetics in male Wistar rats: implications for lipid and glucose metabolism. J Toxicol Environ Heal Part A. 2013;76:142–56.
Taylor JS, Van de Peer Y, Braasch I, Meyer A. Comparative genomics provides evidence for an ancient genome duplication event in fish. Philos Trans R Soc L B Biol Sci 2001;356:1661–1679.
Pasquier J, Cabau C, Nguyen T, Jouanno E, Severac D, Braasch I, et al. Gene evolution and gene expression after whole genome duplication in fish: the PhyloFish database. BMC Genomics. 2016;17:368.
Crollius HR, Weissenbach J. Fish genomics and biology. Genome Res. 2015;15:1675–82. doi:10.1101/gr.3735805. http://genome.cshlp.org/content/15/12/1675.
Jakovlić I, Liu H, Wang W-M. Identification, origin and evidence for retained functionality of two IκBα paralogs in Megalobrama Amblycephala. Dev Comp Immunol. 2016;62:89–96.
Jakovlić I, Wang W-M. Expression of Hox paralog group 13 genes in adult and developing Megalobrama Amblycephala. Gene Expr Patterns. 2016;21:63–8.
Johnston IA, Bower NI, Macqueen DJ. Growth and the regulation of myotomal muscle mass in teleost fish. J Exp Biol. 2011;214:1617–28.
Henderson ST. High carbohydrate diets and Alzheimer’s disease. Med Hypotheses. 2004;62:689–700.
Seneff S, Wainwright G, Mascitelli L. Nutrition and Alzheimer’s disease: the detrimental role of a high carbohydrate diet. Eur J Intern Med European Federation of Internal Med. 2011;22:134–40.
Correia SC, Santos RX, Carvalho C, Cardoso S, Candeias E, Santos MS, et al. Insulin signaling, glucose metabolism and mitochondria: major players in Alzheimer’s disease and diabetes interrelation. Brain Res. 2012;1441:64–78.
Acknowledgments
We would like to thank the members of College of Fisheries Huazhong Agricultural University, Key Lab of Freshwater Animal Breeding, and Ministry of Agriculture for technical help. We also wish to thank Professor George Britton and colleague Shao-Kui Yi and Fang-Yu Deng for helpful discussions.
Funding
This study was supported by Natural Science Foundation of China (No. 31401976) and the earmarked fund for the Modern Agro-industry Technology Research System entitled “Staple Freshwater Fishery Industry Technology System” (No. CARS-46-05). The funders had no role in the design of the study, collection, analysis, and interpretation of data, nor in writing the manuscript.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional files, as well as in the NCBI Short Read Archive (SRA), under accession numbers SRR5763110, SRR5763111, SRR5763112, SRR5763116, SRR5763117 and SRR5763107.
Author information
Authors and Affiliations
Contributions
The authors’ responsibilities were as follows: WW and YZ conceptualized the study; WP, WW and YZ designed the study; WP, HL and YZ conducted the research; WP, PP and YZ provided the materials; WP, PP and YZ collected the data; WP, PP and HL analyzed the data; WP, IJ and YZ interpreted the data; WP and IJ wrote the manuscript; and WW, IJ and YZ revised the manuscript critically for important intellectual content. All authors read the final manuscript, and provided comments and suggestions for improvements.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Efforts were made to minimize suffering as much as possible, and all animals were handled and experimental procedures conducted in accordance with the guidelines for the care and use of animals for scientific purposes set by the Ministry of Science and Technology, Beijing, China (No. 398, 2006). The study was approved by the Institutional Animal Care and Use Ethics Committee of Huazhong Agricultural University. The permit number for conducting animal experiments of the Huazhong Agricultural University is SCXK(Hubei)2015–0019.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Genes differentially expressed between HCBD and control groups in two KEGG pathways. (A) Non-alcoholic fatty liver disease pathway (P = 0.0564, Enrichment factor = 1.42) and (B) Insulin signaling pathway (P = 1, Enrichment factor = 1.13). (DOCX 397 kb)
Additional file 2:
Primers designed for qPCR. (DOCX 15 kb)
Additional file 3:
1H NMR signal assignment of metabolites in serum and liver extracts. s - singlet; d - doublet; t - triplet; q - quartet; m - multiplet; b - broad peak; dd - doublet of doublets; S - serum; L - liver. (DOCX 13 kb)
Additional file 4:
OPLS-DA scores plots and corresponding loading plots. OPLS-DA scores plots (left) and corresponding loading plots (right), derived from 1H NMR data, reveal a significant (r = 0.602) decrease in the amount of betaine in livers of HCBD group (blue triangles) in comparison to the Control diet group (black squares). The colour code corresponds to the absolute value of the OPLS-DA correlation coefficient |r|, which indicates the contribution of the corresponding variable to the group separation. In these loading plots, the hot-colored (the red end of the spectrum) metabolites contributed more significantly to the intergroup differences than the cold-colored (blue) ones. (DOCX 98 kb)
Additional file 5:
The 600 MHz 1H NMR spectra of plasma (P) and liver (L) from control (A) and HCBD (B) groups. The dotted regions were vertically expanded 16 times in the spectra of plasma and liver extracts. The keys for metabolites are given in Additional file 3. (DOCX 3909 kb)
Additional file 6:
Top 20 KEGG pathway. (DOCX 14 kb)
Additional file 7:
Correlation analysis between transcriptome and qPCR data. Analysis was conducted for the 13 genes associated with NAFLD and insulin signaling pathways (Fig. 5). Transcriptome is log2FC (x-axis) and qPCR is 2-ΔΔCT (y-axis). Pearson correlation coefficient (R2 = 0.3226), significance analysis (P = 0.0428), and linear regression (red line) are shown. (DOCX 29 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Prisingkorn, W., Prathomya, P., Jakovlić, I. et al. Transcriptomics, metabolomics and histology indicate that high-carbohydrate diet negatively affects the liver health of blunt snout bream (Megalobrama amblycephala). BMC Genomics 18, 856 (2017). https://doi.org/10.1186/s12864-017-4246-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-4246-9