
Abstract
Background
2-Phenylethanol is a specific aromatic alcohol with a rose-like smell, which has been widely used in the cosmetic and food industries. At present, 2-phenylethanol is mainly produced by chemical synthesis. The preference of consumers for “natural” products and the demand for environmental-friendly processes have promoted biotechnological processes for 2-phenylethanol production. Yet, high 2-phenylethanol cytotoxicity remains an issue during the bioproduction process.
Results
Corynebacterium glutamicum with inherent tolerance to aromatic compounds was modified for the production of 2-phenylethanol from glucose and xylose. The sensitivity of C. glutamicum to 2-phenylethanol toxicity revealed that this host was more tolerant than Escherichia coli. Introduction of a heterologous Ehrlich pathway into the evolved phenylalanine-producing C. glutamicum CALE1 achieved 2-phenylethanol production, while combined expression of the aro10. Encoding 2-ketoisovalerate decarboxylase originating from Saccharomyces cerevisiae and the yahK encoding alcohol dehydrogenase originating from E. coli was shown to be the most efficient. Furthermore, overexpression of key genes (aroGfbr, pheAfbr, aroA, ppsA and tkt) involved in the phenylpyruvate pathway increased 2-phenylethanol titer to 3.23 g/L with a yield of 0.05 g/g glucose. After introducing a xylose assimilation pathway from Xanthomonas campestris and a xylose transporter from E. coli, 3.55 g/L 2-phenylethanol was produced by the engineered strain CGPE15 with a yield of 0.06 g/g xylose, which was 10% higher than that with glucose. This engineered strain CGPE15 also accumulated 3.28 g/L 2-phenylethanol from stalk hydrolysate.
Conclusions
In this study, we established and validated an efficient C. glutamicum strain for the de novo production of 2-phenylethanol from corn stalk hydrolysate. This work supplied a promising route for commodity 2-phenylethanol bioproduction from nonfood lignocellulosic feedstock.
Graphical Abstract

Similar content being viewed by others


Introduction
Aromatic compounds are a very important class of fine chemicals with a broad market prospect. For example, 2-phenylethanol represents a specific aromatic alcohol with a rose-like smell utilized as a flavor ingredient in the cosmetic and food industries [1]. Natural 2-phenylethanol (about US$ 1000 per kilo) is traditionally extracted from the flower essential oils, such as rose essential oil, lily essential oil, and jasmine essential oil [2]. However, the low concentration of 2-phenylethanol in these plants makes the extraction process costly, thereby restricting the availability. At present, 2-phenylethanol is also produced by chemical synthesis with advantages in yield and cost (about US$ 5 per kilo) [2]; yet, this method requires high temperature since some toxic by-products seriously lower the product quality [3]. The US and European food agencies stipulate that flavors obtained from the biotechnological process are regarded as “natural” products if the used feedstocks are of natural origin [1, 4]. Consumers' preference for “natural” products and the demand for environment-friendly processes have promoted a biotechnological process for 2-phenylethanol production, which is at present considered the most commercially viable route.
2-Phenylethanol can be transformed from L-phenylalanine via the Ehrlich pathway and the styrene-derived pathway using two main types of microbial cell factories (including Saccharomyces cerevisiae and Escherichia coli) [5,6,7]. Ehrlich pathway is the most important pathway in which L-phenylalanine is converted to 2-phenylethanol through a three-step enzymatic reaction including transamination, decarboxylation, and reduction. After introducing a self-sufficient cofactor system, the maximum conversion ratios of 95% and 88% were achieved for S. cerevisiae and E. coli, respectively [7, 8]. Moreover, Machas et al. found that the styrene-derived pathway for producing 2-phenylethanol from L-phenylalanine through deamination, decarboxylation, epoxidation, isomerization, and reduction is more efficient and exhibits higher productivity compared to the Ehrlich pathway [9].
Several microorganisms could naturally produce 2-phenylethanol from cheaper carbon substrate such as glucose but at relatively low concentration [10, 11]. Thus, metabolic engineering strategies were made to de novo synthesize 2-phenylethanol from cheaper substrate, which represented a more economic approach. The engineered S. cerevisiae with connection of central carbon metabolism and aromatic amino acids metabolism produced 13 mM 2-phenylethanol with a yield of 0.113 mol/mol [5]. To increase 2-phenylethanol productivity, the fast-growing E. coli was also engineered as host for de novo synthesis. Kang et al. constructed the Ehrlich pathway and overexpressed the phenylpyruvate pathway in E. coli, and the engineered strain could produce 0.29 g/L of 2-phenylethanol from glucose [12]. In comparison, another engineered L-phenylalanine producing E. coli with the overexpression of styrene-derived artificial cascades produced up to 1.82 g/L of 2-phenylethanol from glucose [9]. Although various strategies were developed, the concentration is still insufficient for industrial production. One of the bottlenecks for bio-production of 2-phenylethanol is cytotoxicity. The hydrophobic nature of 2-phenylethanol led to collapse of the transmembrane gradient and disruption of the cell membrane, which, in turn, decreased cell viability [1, 13]. In order to reduce the cytotoxicity and increase yield, 2-phenylethanol was extracted from the fermentation broth by in situ product removal (ISPR) technologies [1]. With biodiesel as the extractant, high-level 2-phenylethanol production of 9.1 g/L was achieved from glycerol or glucose [6]. Alternatively, biosynthesis of 2-phenylethanol from nonfood carbohydrate feedstocks with robust and tolerant microorganisms without additional extractants could be a more economical strategy.
Corynebacterium glutamicum is a rapidly growing facultatively anaerobic, Gram-positive bacterium that has been utilized for large-scale industrial production of a variety of amino acids. The development of metabolic engineering for C. glutamicum has increased the spectrum of products produced by this host strain [14]. For the past years, the product range of C. glutamicum has been extended to a wide variety of alcohols due to its pronounced resistance [15, 16]. C. glutamicum was found to possess better tolerance to alcohols with longer alkyl chains than E. coli, such as isobutanol and pentanol isomers [16, 17]. Besides alcohols, C. glutamicum was innately tolerant to aromatic compounds and toxic inhibitors derived from lignocellulose biomass hydrolysates, thus these hydrolysates could be directly utilized as feedstock without detoxification [18,19,20]. Recently, C. glutamicum was evolved into a promising host strain for the bioproduction of aromatic chemicals involved in the shikimate pathway, including hydroxybenzoic acids and protocatechuic acid, by inactivating its natural catabolic pathways for aromatic compounds [21, 22]. In particular, C. glutamicum exhibited a high tolerance to phenol, catechol and benzoic acid, thus it was engineered for high-level production of muconic acid from lignin [23].
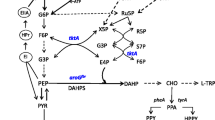
In this study, the C. glutamicum ATCC 21420 was rationally engineered to produce 2-phenylethanol (Fig. 1). C. glutamicum was shown to be a suitable host strain for 2-phenylethanol production through analysis of this host’s sensitivity, and two more tolerant strains were obtained by adaptive laboratory evolution. The mutations in the evolved strains were identified by whole-genome resequencing. Several 2-keto acid decarboxylases (KDC) and alcohol dehydrogenases (ADH) were introduced and screened for efficient conversion of phenylpyruvate to 2-phenylethanol. Then, the key genes of the phenylpyruvate pathway were overexpressed to drive more carbon flux to phenylpyruvate. Finally, the feasibility of 2-phenylethanol production from lignocellulosic feedstock with engineered C. glutamicum was evaluated.

The engineering strategies for de novo production of 2-phenylethanol by C. glutamicum. The red arrows indicated that pathways were overexpressed. Metabolites: G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; GAP, glyceraldehyde-3-phosphate;PEP, phosphoenolpyruvate; PYR, pyruvate; OAA, oxaloacetate; CIT, citrate; AcCoA, Acetyl-CoA; Ru5P, ribulose-5P; X5P, xylulose-5P; R5P, ribose-5P; S7P, sedoheptulose‑7‑phosphate; E4P, erythrose‑4‑phosphate; DAHP, 3-deoxy-D-arabinoheptulosonate 7-phosphate; DHQ, 3-dehydroquinate; DHS, 3-dehydroshikimate; SA, shikimate; S3P, Shikimate-3-phosphate; EPSP, 5-enolpyruvyl shikimate 3-phosphate; CHA, chorismate; PPA, prephenate; PPY, phenylpyruvate; PPAL, phenylacetaldehyde. Genes/enzymes: pts, phosphoenolpyruvate phosphotransferase system; xylE, xylose transporter; xylA xylose isomerase; xylB, xylulokinase; tkt, transketolase; tal, transaldolase; aroG, DAHP synthase; ppsA, phosphoenolpyruvate synthase; aroL, shikimate kinase II; aroA, 5-enolpyruvylshikimate-3-phosphate synthetase; pheA, chorismate mutase-prephenate dehydratase; aro10, 2-ketoisovalerate decarboxylase; yahK, alcohol dehydrogenase
Materials and methods
Bacterial strains, plasmids, and culture media
All bacterial strains, their relevant characteristics, and sources are shown in Table 1. During the process of plasmid construction, E. coli JM110 and DH5α used for the gene cloning were cultured with LB medium containing appropriate concentrations of antibiotics (25 mg/L chloramphenicol; 50 mg/L kanamycin). Transferring plasmid DNA into C. glutamicum strain was performed by electroporation, and the recombinant clones were screened on brain heart infusion sorbitol (BHIS) agar supplemented with appropriate concentrations of antibiotics (10 mg/L chloramphenicol; 25 mg/L kanamycin). Shanghai Jiuqian Chemical Co., China, provided the detoxified corn stalk hydrolysate, which was prepared by enzymatic hydrolysis. The sterilized corn stalk hydrolysate (115 °C/15 min) contained 148.7 g/L glucose, 67.3 g/L xylose, 0.06 g/L furfural and 0.18 g/L phenolics.
Construction of plasmids and strains
The genes of aro10, pmkdc, ipdc, yahK, yjgB, aroH, aroGfbr, pheAfbr, aroL, ppsA and xylE were synthesized by Genewiz Biotech (Suzhou, China) according to codon preference of C. glutamicum. The genes adhA, aroA and tkt, and the Phom promoter were amplified from the genomic DNA of C. glutamicum ATCC 13032 [24]. The Phom promoter was fused to the xylE gene by overlap extension PCR. The xylAB genes under Ptac promoter was amplified from the plasmid pX‑xcbAB [25]. DNA oligonucleotides used in this study were listed in Additional file 1: Table S1. The PCR products with suitable Shine Dalgarno sequence and restriction sites were inserted into pEC-XK99E and pXMJ19, yielding a series of pEC-XK99E-based and pXMJ19-based expression plasmids.
Tolerance test and adaptive laboratory evolution
To investigate 2-phenylethanol tolerance, C. glutamicum was cultured with LB medium at 30 °C, while E. coli was cultured with LB medium at 37 °C. Briefly, 1 mL of preculture was inoculated into 100 mL LB medium supplemented with different concentrations of 2-phenylethanol (from 1 g/L to 5 g/L). Bacteria were grown with shaking at 200 rpm for 48 h. To further enhance 2-phenylethanol tolerance, C. glutamicum ATCC 21420 cells were repeatedly cultivated with LB medium supplemented with 2-phenylethanol using two independent cultures. As the growth of C. glutamicum ATCC 21420 was significantly reduced with LB medium containing 3 g/L 2-phenylethanol, this concentration was used for adaptive laboratory evolution. The culture was serially passed every 24 h for 60 times.
Whole genome sequencing
De novo whole-genome sequencing of C. glutamicum ATCC 21420 was performed on the illumine PE150 platform and PacBio Sequel system by Genewiz Biotech (Suzhou, China). The coding genes were annotated with NCBI nr database by Diamond. Then the functions of genes were annotated by GO database, and the pathways were annotated using KEGG database. DNA re-sequencing of CALE1 and CALE2 was performed on the illumina HiseqXten/Novaseq/MGI2000 system by Genewiz Biotech (Suzhou, China). Pipeline of Sentieon (V202112.02) was used to map clean data to reference genome (C. glutamicum ATCC 21420), remove duplication and call SNV/InDel. Annotation for SNV/InDel was performed by Annovar (V21 Apr 2018). Breakdancer and CNVnator were used to analyze genomic structure variation.
Cultivation conditions for 2-phenylethanol production
Single clones of C. glutamicum were grown in 10 mL of brain heart infusion (BHI) medium supplemented with appropriate concentrations of antibiotics at 30 °C and 200 rpm. After overnight incubation, cells of the preculture were transferred into 50 mL modified AY medium containing 60 g/L glucose, xylose, or sugar mixture. The modified AY medium contained 7 g/L (NH4)2SO4, 2 g/L urea, 0.5 g/L KH2PO4, 0.5 g/L MgSO4 ∙7 H2O, 0.5 g/L K2HPO4, 6 mg/L FeSO4 ∙7 H2O, 0.4 mg/L biotin, 6 mg/L MnSO4 ∙H2O, 0.4 mg/L thiamine, 5 g/L yeast extract, 1 g/L casamino acids, and 21 g/L 3-morpholinopropanesulfonic acid (MOPS) (pH 7.5) [26]. To induce expression, 1.0 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to the cultivation medium. For cultivation with corn stalk hydrolysate as feedstock, the total sugar concentration was adjusted to 60 g/L, which contained 41.3 g/L glucose and 18.7 g/L xylose.
Analytical methods
The concentrations of L-phenylalanine and 2-phenylethanol were measured by HPLC (HP 1200, Agilent, USA) equipped with a reversed phase column of Poroshell 120 SB-C18 (4.6 × 150 mm, 2.7 micron, Agilent, USA) and a UV absorbance detector (Agilent, USA) at 258 nm with the mobile phase of 70% water and 30% acetonitrile containing 0.1% trifluoroacetic acid at the flow rate of 0.6 mL/min. Organic acids and sugars were measured by HPLC (HP 1200, Agilent, USA) equipped with a cation-exchange column of Aminex HPX-87H (7.8 × 300 mm, BioRad, USA), a refractive index (RI) detector (Agilent, USA), and a UV absorbance detector (Agilent, USA) with the mobile phase of 3 mM sulfuric acid solution at the flow rate of 0.5 mL/min. Acetate, lactate, and shikimate were detected by the UV detector at 210 nm, while the RI detector was used to measure glucose and xylose. Cell growth was monitored by measuring the optical density of cultures at 600 nm (OD600), and one unit of optical density corresponded to 0.31 dry cell weight (DCW) (g/L) [27]. The determination of reductive activity of AdhA was performed as described previously [16].
Results and discussion
Cytotoxic effect of 2-phenylethanol on C. glutamicum
The cytotoxicity of higher alcohols is generally far greater than that of alcohols with a shorter alkyl chain such as ethanol [17]. 2-Phenylethanol could severely affect the cellular metabolism of S. cerevisiae during its bioproduction, including the reduction of respiratory capacity and restriction of nutrient uptake [28]. In this study, the toxicity of 2-phenylethanol against C. glutamicum was examined, and the most widely used chassis E. coli was also tested as a control. For the direct comparison, E. coli MG1655 and C. glutamicum ATCC 21,420 were cultivated in LB medium supplemented with 2-phenylethanol (from 1 g/L to 5 g/L) (Fig. 2A, B). The tolerance of the two hosts to 2-phenylethanol was measured by the relative growth rate after 2-phenylethanol addition. With the addition of 2 g/L 2-phenylethanol, the relative growth rate of C. glutamicum was reduced to 61%, while the growth of E. coli cells was almost stopped. After adding 3 g/L 2-phenylethanol, the relative growth rate of C. glutamicum was 53%; while the 2-phenylethanol was higher than 4 g/L, cell growth was severely inhibited. The results showed that C. glutamicum exhibited a better tolerance towards 2-phenylethanol than E. coli.
The tolerance mechanisms of C. glutamicum to aromatic compounds have been elucidated e.g., complex structures of cell envelope, and effective damage repair and defense mechanisms [29, 30]. These protective mechanisms might be the reason why C. glutamicum could stand against alcohols [17, 31]. However, further investigation is required. To further enhance 2-phenylethanol tolerance, adaptive laboratory evolution of C. glutamicum ATCC 21420 was performed. The cells were repeatedly cultivated containing 3 g/L 2-phenylethanol using two independent cultures. Two evolved strains, CALE1 and CALE2, were obtained after this selection process, displaying normal growth with 3 g/L 2-phenylethanol (Fig. 2C). The evolved strain CALE1 showed a higher biomass at 20 h and exhibited a higher growth rate than strain CALE2 in the presence of 2-phenylethanol. Analysis of the production performance showed that the phenylpyruvate pathway of the evolved strain was almost unaffected after evolution, as these evolved strains produced comparable concentrations of phenylalanine to C. glutamicum ATCC 21420 (Additional file 1: Fig. S1).

Growth profiles of E. coli A and C. glutamicum ATCC 21420 B strains in the presence of different concentrations of 2-phenylethanol, and evolved C. glutamicum C strains in 3 g/L 2-phenylethanol. E. coli was cultured in LB medium at 37 °C, while C. glutamicum was cultured in LB medium at 30 °C
Identification of mutations in the evolved strains by whole-genome resequencing
The de novo whole genome sequence of C. glutamicum ATCC 21420 spans 3303123 bp, with a G + C content of 54.3%, containing 3047 predicted coding sequences (GenBank accession No. CP121344). The mutations in CALE1 and CALE2 relative to the parent strain were identified by resequencing their genomes. CALE1 possessed 10 mutations including 9 single nucleotides variants (SNVs) and 1 insertion/deletion (InDel), while CALE2 only possessed 4 SNVs and 2 InDels (Table 2). Of the 10 mutations in CALE1, 8 mutations were found in intragenic regions, while 2 mutations occurred in intergenic regions. The CG_2122 gene, annotated as a mycothiol system anti-sigma-R factor (100% amino acid identity with RshA of C. glutamicum ATCC 13032), could not be expressed properly in CALE1 caused by a mutation in the translational start codon of this gene (from ATG to AGG). The rshA gene coding for an anti-sigma factor controls the function of the stress-responsive sigma factor SigH in C. glutamicum ATCC 13032, and genes related to mycothiol synthesis and recycling, protein degradation, SOS-response to DNA damage and heat stress were upregulated in the rshA deletion mutant [32]. Mycothiol was found to contribute significantly to resistance to various poisonous chemicals in C. glutamicum [33]. Accordingly, the evolved strain CALE1 might release the repression of SigH and improve the resistance to 2-phenylethanol. A nonsynonymous mutation was found in CG_2149 gene of CALE1, which annotated as a cell wall biosynthesis protein (97.5% amino acid identity with LcpA of C. glutamicum ATCC 13032). LcpA is responsible for the transfer of arabinogalactan onto peptidoglycan involved in cell wall biosynthesis [34]. C. glutamicum has a complex structured cell wall, which gives it inherent ability to withstand adverse external environments [35]. The amino acid change I136F occurred in the LCP-domain of LcpA, near the position that binding the pyrophosphate head group of the substrate (R138), might affect its catalytic efficiency [34].
The evolved strain CALE2 had a nonsynonymous mutation in CG_1170 gene (amino acid change H339Y), which annotated as a primary RNA polymerase sigma factor SigA. This mutation located in the promoter-recognizing 3.0 domain of SigA [36]. Mutations in sigma factors were identified in adaptive evolution and their importance in natural evolution was illustrated [37]. Specially, ethanol tolerance of E. coli could be greatly improved by overexpression of randomly mutated main sigma factor RpoD (σ70) [38]. In general, mutations in sigma factors could cause global transcription-level alterations [37, 39]. Considering that both evolved strains CALE1 and CALE2 possessed mutations associated with the regulation of sigma factors, the phenotype of improved 2-phenylethanol tolerance might be complex and controlled by a multitude of genes.
Construction of 2-phenylethanol synthesis pathway
Metabolic engineered E. coli could produce aromatic alcohols from glucose by expanding the shikimate pathway with the connection of the Ehrlich pathway [40]. The Ehrlich pathway was also constructed in C. glutamicum ATCC 13032 for isobutanol production by introducing a heterologous 2-keto acid decarboxylase; then, 2-phenylethanol was detected as a by-product due to the promiscuous activity of this enzyme [16]. Considering the complex feedback regulation in branched metabolic pathways, the phenylalanine-producing CALE1 strain derived from C. glutamicum ATCC21420 was selected as starting strain as it possessed an improved flux in the phenylpyruvate pathway and relatively strong 2-phenylethanol tolerance. Three 2-keto acid decarboxylases (IpdC from Azospirillum brasilense, PmKDC from Proteus mirabilis, and Aro10 from S. cerevisiae) and two alcohol dehydrogenases (YjgB and YahK from E. coli) were evaluated in C. glutamicum for 2-phenylethanol synthesis, as these enzymes were already successfully used for the production of alcohols [7, 40, 41]. The native NAD-dependent alcohol dehydrogenase AdhA, which appeared to be functional for converting phenylacetaldehyde, was also tested [16].
As shown in Fig. 3, all nine combinations of decarboxylase and dehydrogenase could achieve 2-phenylethanol production, while the choice had a huge impact on the 2-phenylethanol production (from 0.19 to 1.02 g/L). As for decarboxylase, the maximum concentration of 2-phenylethanol with an expression of Aro10 was about 1.8-fold higher than that of PmKDC and IpdC, while there was no difference between PmKDC and IpdC, which could be because Aro10 has a relatively higher affinity to phenylpyruvate (Km of 0.10 mM) compared to IpdC (Km of 1.08 mM) and PmKDC (Km of 0.62 mM) [42, 43]. The Aro10 with excellent property has been used for efficient production of 2-phenylethanol in engineered yeast and E. coli [7, 44], and it also performed well in C. glutamicum. On the other hand, 2-phenylethanol production via introduction of AdhA was substantially lower in comparison to YahK and YjgB, and there was no significant difference between YahK and YjgB. This might be that AdhA preferred alcohols as substrates, such as ethanol, when C. glutamicum was growing on the substrate, despite adequately contributing to producing isobutanol and pentanol [16, 17]. The crude extract enzyme assays were performed to confirm whether AdhA was successfully expressed. For strain CGPE4, the specific activity toward phenylacetaldehyde was 0.02 U/mg, and there was no significant difference compared to the CALE1 strain with only introduction of Aro10 (Additional file 1: Table S2). However, the specific activity toward isobutyraldehyde was detected to be 0.46 U/mg, whereas an activity of 0.05 U/mg was detected without introduction of AdhA (Additional file 1: Table S2). These results demonstrated the efficient expression of AdhA, however, it could not efficiently convert phenylacetaldehyde to 2-phenylethanol. Moreover, the CALE1 strain with only introduction of Aro10 also accumulated 0.26 g/L 2-phenylethanol (Additional file 1: Fig. S2), indicating that endogenous alcohol dehydrogenases of C. glutamicum were capable of reducing phenylacetaldehyde to 2-phenylethanol (Additional file 1: Table S2). In the engineered strain CGPE4 with the expression of Aro10 and YahK, maximal 2-phenylethanol production reached 1.02 g/L after 60 h cultivation.

Comparison of 2-phenylethanol production by C. glutamicum CALE1 strain with various 2-keto acid decarboxylases and alcohol dehydrogenases. 2-Phenylethanol production was determined after 60 h fermentation in modified AY medium supplemented with 60 g/L glucose. The values were given as the averages and standard deviations of three independent cultures
Driving carbon flux into the phenylpyruvate biosynthesis pathway
The enzymes involved in the synthesis of phenylalanine in C. glutamicum are tightly regulated by feedback inhibition. Overexpression of aroGS180F encoding a mutated feedback-resistant 3-deoxy-D-arabino-heptulosonate 7-phosphate (DAHP) synthase originating from E. coli cumulatively improved carbon flux towards the shikimate pathway [21, 22]. Additionally, co-expression of aroH encoding a wild-type DAHP synthase and pheAfbr encoding a mutant prephenate dehydratase/chorismite mutase originating from E. coli significantly increased phenylalanine production and decreased shikimate production [45]. As 2-phenylethanol shares the precursor with phenylalanine, combinations of aroGfbr/aroH and pheAfbr were introduced to evaluate their effects on 2-phenylethanol production. As expected, co-expression of the committed enzymes led to a substantial increase in 2-phenylethanol production, while the combination of aroGfbr and pheAfbr (CGPE10) exhibited better performance with a production titer of 2.05 g/L compared with the combination of aroH and pheAfbr (CGPE11) with a production titer of 1.88 g/L (Table 2). Even so, the intermediate shikimate was incidentally accumulated, indicating bottlenecks in the downstream shikimate pathway. A previous study indicated that high-level expression of the downstream genes of shikimate and moderate expression of the upstream genes of shikimate could balance the carbon flux to phenylalanine [27]. The two steps catalyzed by shikimate kinase (aroL) and 5-enolpyruvylshikimate-3-phosphate (EPSP) synthase (aroA) in E. coli [46, 47] and C. glutamicum [27] were limiting steps in the synthesis of aromatic amino acids, whose enhanced expression could increase phenylalanine and tyrosine production and reduce intermediates accumulation. Consequently, these genes were individually overexpressed in the CGPE10 strain. As shown in Table 3, overexpression of the native aroA (CGPE13) increased 2-phenylethanol production to 2.48 g/L with simultaneously reduced intermediate shikimate accumulation (1.49 g/L). However, enhanced expression of aroL from E. coli did not obviously affect 2-phenylethanol production (CGPE12), indicating that expression of shikimate kinase gene might not be a rate-limiting factor in our strain background. It might also be the result of the inhibition of shikimate kinase activity by shikimate [46, 48]. The accumulation of shikimate indicated that the downstream pathway of shikimate should be further optimized. In previous reports on 4-hydroxybenzoic acid production with C. glutamicum ATCC 13032 and R, simultaneous overexpression of genes aroCKB, aroD, aroE and aroA in the shikimate pathway could effectively increase product titers and reduce the formation of shikimate and 3-dehydroshikimate [49, 50]. In particular, introduction of a gene aroK encoding shikimate-resistant shikimate kinase from Methanocaldococcus jannaschii led to a great decrease in the formation of shikimate and 3-dehydroshikimatae. We thus expected to overcome the accumulation of intermediates during 2-phenylethanol production through enhanced expression of all genes in the shikimate pathway based on the chromosomal integration in future work.
Adequate supply of phosphoenolpyruvate (PEP) and erythrose‑4‑phosphate (E4P) is a crucial factor for achieving the maximum carbon flux toward shikimate pathway [27]. Utilization of a non-PTS alternative glucose uptake route and enhanced expression of pentose phosphate pathway genes are effective strategies for the overproduction of shikimate and other aromatics [21, 27, 51, 52]. To increase PEP availability for 2-phenylethanol synthesis, we attempted to block the PTS glucose uptake route by deleting ptsH gene; however, the engineering failed due to the infeasibility of chromosome manipulation in this strain background. Alternatively, a PEP synthase from E. coli that could covert pyruvate to PEP was introduced into CGPE13 [53,54,55]. Secondly, the native tkt gene was overexpressed to increase the E4P availability. The resulting strain CGPE14 produced 3.23 g/L 2-phenylethanol, increasing by 30% compared to the CGPE13 strain (Table 3).
2-Phenylethanol production from glucose and xylose mixtures
Biomass-derived xylose is an economical feedstock for the biotechnological production of value-added chemicals. Usually, C. glutamicum is not able to use xylose as the sole carbon source; however, after introducing xylose isomerase (xylA) and xylulokinase (xylB) from Xanthomonas campestris, the engineered strain was able to grow with xylose, showing fast growth rate [56, 57]. This strain simultaneously consumed glucose and xylose mixture with weak carbon catabolite repression. Accordingly, the two genes xylAB were heterologously expressed in CGPE14 strain. To facilitate xylose transportation, a pentose transporter gene xylE from E. coli was further expressed under the weak constitutive Phom promoter [24]. The xylose utilization ability and the production performance of the resulting strain CGPE15 were evaluated. As shown in Fig. 4, the CGPE15 strain could use xylose as sole carbon and produced 3.55 g/L 2-phenylethanol with a yield of 0.06 g/g xylose, which was 10% higher than that with glucose. Specifically, the accumulation of by-products lactate and acetate derived from pyruvate dramatically decreased when xylose was utilized, indicating a lower pyruvate availability. This might be attributed to the fact that XylE was used for xylose transportation but not the PTS system, which phosphorylated its substrate with phosphoenolpyruvate. This observation was consistent with the previous study in which the by-product lactate accumulation was strongly reduced with xylose fermentation [58]. Notably, the biomass obtained by CGPE15 strain with xylose was 17% lower than that with glucose. This reflected a significant change in the distribution of carbon metabolism in C. glutamicum, which was strongly influenced by the substrate. The increased TCA flux accompanied by high CO2 formation when utilizing xylose was induced by the PPP limitation to supply NADPH, leading to lower biomass yield on xylose [59]. Furthermore, the production of 2-phenylethanol from sugar mixtures of glucose/xylose with different proportions was carried out. The 2-phenylethanol titer and biomass formation obtained for sugar mixture were in between the values obtained for a single substrate. Also, the 2-phenylethanol production increased with the ratio of xylose, along with reduced by-products accumulation. These results suggested that xylose was a better feedstock for 2-phenylethanol production in the CGPE15 strain background.

De novo production of 2-phenylethanol by C. glutamicum CGPE15 strain with glucose A, xylose B and different combinations of sugar C. a, 60 g/L glucose; b, 40 g/L glucose and 20 g/L xylose; c, 30 g/L glucose and 30 g/L xylose; d, 20 g/L glucose and 40 g/L xylose; e, 60 g/L xylose
2-Phenylethanol production from plant biomass hydrolysates
Lignocellulose-treated sugars are usually mixed with microbial growth inhibitors, e.g., furans and phenolics [60, 61]. Suitable microbial hosts for bio-based chemicals production with these sugars should not only tolerate the toxicity of the final product, but also growth inhibitors from culture medium to achieve high titer and yield [19]. Therefore, the production of 2-phenylethanol and sugars utilization with a real hydrolysate were examined to evaluate its potential for practical application. The corn stalk hydrolysate treated with commercial Cellic CTec2 cellulase was used as feedstock. A total of 60 g/L sugar mixture containing 41.3 g/L glucose and 18.7 g/L xylose from corn stalk hydrolysate was adjusted to correspond to the cultivation with the single substrate. The analytical reagent sugar mixture was also used as the substrate for comparison. As shown in Fig. 5A, the engineered strain CGPE15 could effectively utilize the hydrolysate for 2-phenylethanol production, consuming glucose and xylose within 60 h. In addition, both sugars were simultaneously utilized at the start of the cultivation without a typical carbon catabolite repression. A similar result was observed for a glutamic acid-producing C. glutamicum expressing xylAB and araE when co-utilizing glucose and xylose [62]. The 2-phenylethanol titer of 3.28 g/L with corn stalk hydrolysate was obtained, which was comparable to that with analytical pure glucose and xylose of 3.33 g/L (Fig. 5B). There was no obvious difference in the carbon uptake manner between the cultivation with sugar mixture and corn stalk hydrolysate, although the growth with hydrolysate was slightly inhibited due to the presence of low contents of inhibitors. Even so, the selection of the robust strain C. glutamicum for 2-phenylethanol production showed an obvious advantage when lignocellulosic wastes were used as the feedstock considering its ability to withstand growth inhibitors and 2-phenylethanol.

De novo production of 2-phenylethanol by C. glutamicum CGPE15 strain with mixture of glucose and xylose A and corn stalk hydrolysate B
Conclusions
In this study, we established and validated an efficient C. glutamicum strain for the de novo production of 2-phenylethanol. The tolerance test of C. glutamicum indicated that the strain has high potential as a 2-phenylethanol production host. Mutations associated with the regulation of sigma factors were identified in both more tolerant evolved strains CALE1 and CALE2. Several KDCs and ADHs enzymes were screened to construct an Ehrlich pathway for the biosynthesis of 2-phenylethanol, and Aro10 originating from S. cerevisiae and YahK originating from E. coli were found to be the most efficient. 2-Phenylethanol production further increased after expressing key genes involved in the phenylpyruvate biosynthesis pathway. After engineering a xylose assimilation pathway, the final strain CGPE15 could produce 3.28 g/L 2-phenylethanol from a real and practical corn stalk hydrolysate. It is expected that 2-phenylethanol production could be further improved by reducing the by-products accumulation and optimizing the fermentation process. This study provides a promising route for commodity 2-phenylethanol bioproduction from lignocellulosic biomass.
References
Wang Y, Zhang H, Lu X, Zong H, Zhuge B. Advances in 2-phenylethanol production from engineered microorganisms. Biotechnol Adv. 2019;37:403–9.
Hua D, Xu P. Recent advances in biotechnological production of 2-phenylethanol. Biotechnol Adv. 2011;29:654–60.
Etschmann MM, Bluemke W, Sell D, Schrader J. Biotechnological production of 2-phenylethanol. Appl Microbiol Biotechnol. 2002;59:1–8.
Serra S, Fuganti C, Brenna E. Biocatalytic preparation of natural flavours and fragrances. Trends Biotechnol. 2005;23:193–8.
Hassing E-J, de Groot PA, Marquenie VR, Pronk JT. Daran J-MG: Connecting central carbon and aromatic amino acid metabolisms to improve de novo 2-phenylethanol production in Saccharomyces cerevisiae. Metab Eng. 2019;56:165–80.
Sekar BS, Lukito BR, Li Z. Production of natural 2-phenylethanol from glucose or glycerol with coupled Escherichia coli strains expressing l-phenylalanine biosynthesis pathway and artificial biocascades. ACS Sustainable Chemistry & Engineering. 2019;7:12231–9.
Wang P, Yang X, Lin B, Huang J, Tao Y. Cofactor self-sufficient whole-cell biocatalysts for the production of 2-phenylethanol. Metab Eng. 2017;44:143–9.
Wang Z, Jiang M, Guo X, Liu Z, He X. Reconstruction of metabolic module with improved promoter strength increases the productivity of 2-phenylethanol in Saccharomyces cerevisiae. Microb Cell Fact. 2018;17:60.
Machas MS, McKenna R, Nielsen DR. Expanding upon styrene biosynthesis to engineer a novel route to 2-phenylethanol. Biotechnol J. 2017;12:1700310.
Liu C, Zhang K, Cao W, Zhang G, Chen G, Yang H, Wang Q, Liu H, Xian M, Zhang H: Genome mining of 2-phenylethanol biosynthetic genes from Enterobacter sp. CGMCC 5087 and heterologous overproduction in Escherichia coli. Biotechnology for Biofuels 2018, 11:305.
Kim T-Y, Lee S-W, Oh M-K. Biosynthesis of 2-phenylethanol from glucose with genetically engineered Kluyveromyces marxianus. Enzyme Microb Technol. 2014;61–62:44–7.
Kang Z, Zhang C, Du G, Chen J. Metabolic Engineering of Escherichia coli for Production of 2-phenylethanol from Renewable Glucose. Appl Biochem Biotechnol. 2014;172:2012–21.
Seward R, Willetts JC, Dinsdale MG, Lloyd D. The effects of ethanol, hexan-1-ol, and 2-phenylethanol on cider yeast growth, viability, and energy status; synergistic inhibition. J Inst Brew. 1996;102:439–43.
Lee J-H, Wendisch VF. Production of amino acids – Genetic and metabolic engineering approaches. Biores Technol. 2017;245:1575–87.
Sasaki Y, Eng T, Herbert RA, Trinh J, Chen Y, Rodriguez A, Gladden J, Simmons BA, Petzold CJ, Mukhopadhyay A. Engineering Corynebacterium glutamicum to produce the biogasoline isopentenol from plant biomass hydrolysates. Biotechnol Biofuels. 2019;12:41.
Smith KM, Cho K-M, Liao JC. Engineering Corynebacterium glutamicum for isobutanol production. Appl Microbiol Biotechnol. 2010;87:1045–55.
Vogt M, Brüsseler C. Ooyen Jv, Bott M, Marienhagen J: Production of 2-methyl-1-butanol and 3-methyl-1-butanol in engineered Corynebacterium glutamicum. Metab Eng. 2016;38:436–45.
Lange J, Müller F, Takors R, Blombach B. Harnessing Novel chromosomal integration loci to utilize an organosolv-derived hemicellulose fraction for isobutanol production with engineered Corynebacterium glutamicum. Microb Biotechnol. 2018;11:257–63.
Fischer CR, Klein-Marcuschamer D, Stephanopoulos G. Selection and optimization of microbial hosts for biofuels production. Metab Eng. 2008;10:295–304.
Shen X-H, Zhou N-Y, Liu S-J. Degradation and assimilation of aromatic compounds by Corynebacterium glutamicum: another potential for applications for this bacterium? Appl Microbiol Biotechnol. 2012;95:77–89.
Kogure T, Suda M, Hiraga K, Inui M. Protocatechuate overproduction by Corynebacterium glutamicum via simultaneous engineering of native and heterologous biosynthetic pathways. Metab Eng. 2021;65:232–42.
Kallscheuer N, Marienhagen J. Corynebacterium glutamicum as platform for the production of hydroxybenzoic acids. Microb Cell Fact. 2018;17:70.
Becker J, Kuhl M, Kohlstedt M, Starck S, Wittmann C. Metabolic engineering of Corynebacterium glutamicum for the production of cis, cis-muconic acid from lignin. Microb Cell Fact. 2018;17:115.
Zhang Y, Shang X, Lai S, Zhang G, Liang Y, Wen T. Development and Application of an Arabinose-Inducible Expression System by Facilitating Inducer Uptake in Corynebacterium glutamicum. Appl Environ Microbiol. 2012;78:5831–8.
Mao Y, Li G, Chang Z, Tao R, Cui Z, Wang Z. Tang Y-j, Chen T, Zhao X: Metabolic engineering of Corynebacterium glutamicum for efficient production of succinate from lignocellulosic hydrolysate. Biotechnol Biofuels. 2018;11:95.
Okai N, Miyoshi T, Takeshima Y, Kuwahara H, Ogino C, Kondo A. Production of protocatechuic acid by Corynebacterium glutamicum expressing chorismate-pyruvate lyase from Escherichia coli. Appl Microbiol Biotechnol. 2016;100:135–45.
Zhang C, Zhang J, Kang Z, Du G, Chen J. Rational engineering of multiple module pathways for the production of L-phenylalanine in Corynebacterium glutamicum. J Ind Microbiol Biotechnol. 2015;42:787–97.
Stark D, Zala D, Münch T, Sonnleitner B, Marison IW, von Stockar U. Inhibition aspects of the bioconversion of l-phenylalanine to 2-phenylethanol by Saccharomyces cerevisiae. Enzyme Microb Technol. 2003;32:212–23.
Chen C, Pan J, Yang X, Xiao H, Zhang Y, Si M, Shen X, Wang Y. Global transcriptomic analysis of the response of Corynebacterium glutamicum to ferulic acid. Arch Microbiol. 2017;199:325–34.
Chen C, Pan J, Yang X, Guo C, Ding W, Si M, Zhang Y, Shen X, Wang Y. Global transcriptomic analysis of the response of Corynebacterium glutamicum to Vanillin. PLoS ONE. 2016;11:e0164955–e0164955.
Tsuge Y, Hori Y, Kudou M, Ishii J, Hasunuma T, Kondo A. Detoxification of furfural in Corynebacterium glutamicum under aerobic and anaerobic conditions. Appl Microbiol Biotechnol. 2014;98:8675–83.
Busche T, Šilar R, Pičmanová M, Pátek M, Kalinowski J. Transcriptional regulation of the operon encoding stress-responsive ECF sigma factor SigH and its anti-sigma factor RshA, and control of its regulatory network in Corynebacterium glutamicum. BMC Genomics. 2012;13:445.
Liu Y-B, Long M-X, Yin Y-J, Si M-R, Zhang L, Lu Z-Q, Wang Y, Shen X-H. Physiological roles of mycothiol in detoxification and tolerance to multiple poisonous chemicals in Corynebacterium glutamicum. Arch Microbiol. 2013;195:419–29.
Baumgart M, Schubert K, Bramkamp M, Frunzke J. Impact of LytR-CpsA-Psr proteins on cell wall biosynthesis in Corynebacterium glutamicum. J Bacteriol. 2016;198:3045–59.
Barry CE, Mdluli K. Drug sensitivity and environmental adaptation of myocobacterial cell wall components. Trends Microbiol. 1996;4:275–81.
Pátek M, Nešvera J. Sigma factors and promoters in Corynebacterium glutamicum. J Biotechnol. 2011;154:101–13.
King T, Ishihama A, Kori A, Ferenci T. A regulatory trade-off as a source of strain variation in the species Escherichia coli. J Bacteriol. 2004;186:5614–20.
Alper H, Stephanopoulos G. Global transcription machinery engineering: a new approach for improving cellular phenotype. Metab Eng. 2007;9:258–67.
Lanza AM, Alper HS. Global Strain Engineering by Mutant Transcription Factors. In: Williams JA, editor. Strain Engineering: Methods and Protocols. Totowa, NJ: Humana Press; 2011. p. 253–74.
Koma D, Yamanaka H, Moriyoshi K, Ohmoto T, Sakai K. Production of aromatic compounds by metabolically engineered Escherichia coli with an expanded Shikimate pathway. Appl Environ Microb. 2012;78:6203–16.
Liu J, Jiang J, Bai Y. Fan T-p, Zhao Y, Zheng X, Cai Y: Mimicking a new 2-phenylethanol production pathway from Proteus mirabilis JN458 in Escherichia coli. J Agric Food Chem. 2018;66:3498–504.
Spaepen S, Versées W, Gocke D, Pohl M, Steyaert J, Vanderleyden J. Characterization of phenylpyruvate decarboxylase, involved in auxin production of Azospirillum brasilense. J Bacteriol. 2007;189:7626–33.
Kneen MM, Stan R, Yep A, Tyler RP, Saehuan C, McLeish MJ. Characterization of a thiamin diphosphate-dependent phenylpyruvate decarboxylase from Saccharomyces cerevisiae. FEBS J. 2011;278:1842–53.
Shen L, Nishimura Y, Matsuda F, Ishii J, Kondo A. Overexpressing enzymes of the Ehrlich pathway and deleting genes of the competing pathway in Saccharomyces cerevisiae for increasing 2-phenylethanol production from glucose. J Biosci Bioeng. 2016;122:34–9.
Zhang C, Zhang J, Kang Z, Du G, Yu X, Wang T, Chen J. Enhanced production of l-phenylalanine in Corynebacterium glutamicum due to the introduction of Escherichia coli wild-type gene aroH. J Ind Microbiol Biotechnol. 2013;40:643–51.
Ding D, Liu Y, Xu Y, Zheng P, Li H, Zhang D, Sun J. Improving the Production of L-phenylalanine by identifying key enzymes through multi-enzyme reaction system in vitro. Sci Rep. 2016;6:32208.
Juminaga D, Baidoo EE, Redding-Johanson AM, Batth TS, Burd H, Mukhopadhyay A, Petzold CJ, Keasling JD. Modular engineering of L-tyrosine production in Escherichia coli. Appl Environ Microbiol. 2012;78:89–98.
Millar G, Lewendon A, Hunter MG, Coggins JR: The cloning and expression of the aroL gene from Escherichia coli K12. Purification and complete amino acid sequence of shikimate kinase II, the aroL-gene product. Biochemical Journal 1986, 237:427–437.
Syukur Purwanto H, Kang M-S, Ferrer L, Han S-S, Lee J-Y, Kim H-S, Lee J-H. Rational engineering of the shikimate and related pathways in Corynebacterium glutamicum for 4-hydroxybenzoate production. J Biotechnol. 2018;282:92–100.
Kitade Y, Hashimoto R, Suda M, Hiraga K, Inui M. Production of 4-hydroxybenzoic acid by an aerobic growth-arrested bioprocess using metabolically engineered Corynebacterium glutamicum. Appl Environ Microbiol. 2018;84:e02587-e12517.
Vargas-Tah A, Gosset G: Production of Cinnamic and p-Hydroxycinnamic Acids in Engineered Microbes. Frontiers in Bioengineering and Biotechnology 2015, 3.
Chen X, Li M, Zhou L, Shen W, Algasan G, Fan Y, Wang Z. Metabolic engineering of Escherichia coli for improving shikimate synthesis from glucose. Biores Technol. 2014;166:64–71.
Shen T, Liu Q, Xie X, Xu Q, Chen N. Improved production of tryptophan in genetically engineered Escherichia coli with TktA and PpsA overexpression. J Biomed Biotechnol. 2012;2012: 605219.
Padilla L, Agosin E. Heterologous expression of Escherichia coli ppsA (phosphoenolpyruvate synthetase) and galU (UDP-glucose pyrophosphorylase) genes in Corynebacterium glutamicum, and its impact on trehalose synthesis. Metab Eng. 2005;7:260–8.
Nakagawa A, Minami H, Kim J-S, Koyanagi T, Katayama T, Sato F, Kumagai H. A bacterial platform for fermentative production of plant alkaloids. Nat Commun. 2011;2:326.
Mao Y, Li G, Chang Z, Tao R, Cui Z, Wang Z, Tang YJ, Chen T, Zhao X. Metabolic engineering of Corynebacterium glutamicum for efficient production of succinate from lignocellulosic hydrolysate. Biotechnol Biofuels. 2018;11:95.
Meiswinkel TM, Gopinath V, Lindner SN, Nampoothiri KM, Wendisch VF. Accelerated pentose utilization by Corynebacterium glutamicum for accelerated production of lysine, glutamate, ornithine and putrescine. Microb Biotechnol. 2013;6:131–40.
Buschke N, Schröder H, Wittmann C. Metabolic engineering of Corynebacterium glutamicum for production of 1,5-diaminopentane from hemicellulose. Biotechnol J. 2011;6:306–17.
Buschke N, Becker J, Schäfer R, Kiefer P, Biedendieck R, Wittmann C. Systems metabolic engineering of xylose-utilizing Corynebacterium glutamicum for production of 1,5-diaminopentane. Biotechnol J. 2013;8:557–70.
Wang X, Khushk I, Xiao Y, Gao Q, Bao J. Tolerance improvement of Corynebacterium glutamicum on lignocellulose derived inhibitors by adaptive evolution. Appl Microbiol Biotechnol. 2018;102:377–88.
Palmqvist E, Hahn-Hägerdal B: Fermentation of lignocellulosic hydrolysates. II: inhibitors and mechanisms of inhibition. Bioresource Technology 2000, 74:25–33.
Jin C, Huang Z, Bao J. High-titer glutamic acid production from lignocellulose using an engineered Corynebacterium glutamicum with simultaneous co-utilization of xylose and glucose. ACS Sustainable Chemistry & Engineering. 2020;8:6315–22.
Funding
This research was supported by National Natural Science Foundation of China (22078227), Natural Science Foundation of the Higher Education Institutions of Jiangsu Province (19KJB180029), Natural Science Foundation of Jiangsu Province (BK20210144) and Taizhou Science and Technology Support Plan (Agriculture) Project (TN202221).
Author information
Authors and Affiliations
Contributions
NZ and WX conceived and designed the experiments. NZ, GW, YS, JL and XG carried out the experiments. YW prepared figures. NZ, WX, GW, YS and JL analyzed the data. NZ and WX wrote the paper. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Primers used in this study. Table S2. Specific activity of the AdhA for phenylacetaldehyde and isobutyraldehyde. Figure S1. L-Phenylalanine production by C. glutamicum. Figure S2. Comparison of 2-phenylethanol production by C. glutamicum CGPE7 and CALE1(pEC-aro10).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, N., Xia, W., Wang, G. et al. Engineering Corynebacterium glutamicum for de novo production of 2-phenylethanol from lignocellulosic biomass hydrolysate. Biotechnol Biofuels 16, 75 (2023). https://doi.org/10.1186/s13068-023-02327-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-023-02327-x