
Abstract
The production of isobutanol in microorganisms has recently been achieved by harnessing the highly active 2-keto acid pathways. Since these 2-keto acids are precursors of amino acids, we aimed to construct an isobutanol production platform in Corynebacterium glutamicum, a well-known amino-acid-producing microorganism. Analysis of this host’s sensitivity to isobutanol toxicity revealed that C. glutamicum shows an increased tolerance to isobutanol relative to Escherichia coli. Overexpression of alsS of Bacillus subtilis, ilvC and ilvD of C. glutamicum, kivd of Lactococcus lactis, and a native alcohol dehydrogenase, adhA, led to the production of 2.6 g/L isobutanol and 0.4 g/L 3-methyl-1-butanol in 48 h. In addition, other higher chain alcohols such as 1-propanol, 2-methyl-1-butanol, 1-butanol, and 2-phenylethanol were also detected as byproducts. Using longer-term batch cultures, isobutanol titers reached 4.0 g/L after 96 h with wild-type C. glutamicum as a host. Upon the inactivation of several genes to direct more carbon through the isobutanol pathway, we increased production by ∼25% to 4.9 g/L isobutanol in a ∆pyc∆ldh background. These results show promise in engineering C. glutamicum for higher chain alcohol production using the 2-keto acid pathways.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The production of fuels from renewable materials has received increased attention due to the limited supply of fossil fuels and increasing environmental concerns. The annual production of ethanol in the USA reached 9.2 billion gallons in 2008 (Seiferlein 2009). Although ethanol production by fermentation has a long history and plays an important role for transition to bio-based fuels, it is not an ideal gasoline replacement due to its low energy density (∼70% of gasoline), high vapor pressure, and high hygroscopicity, making ethanol incompatible with the current infrastructure. In contrast, higher chain alcohols possess elevated energy densities and are compatible with the existing infrastructure. 1-Butanol production by fermentation was first accomplished with Clostridium acetobutylicum as a host about a century ago and has recently received significant attention (Borden and Papoutsakis 2007; Ezeji et al. 2004). In the past few years, the production of this alcohol and several other higher chain alcohols such as 1-propanol, isobutanol, 2-methyl-1-butanol, and 3-methyl-1-butanol have been demonstrated in Escherichia coli (Atsumi et al. 2008a, b; Atsumi and Liao 2008; Cann and Liao 2008; Connor and Liao 2008; Shen and Liao 2008). Isobutyraldehyde and isobutanol production by photosynthetic CO2 recycling has also been accomplished in Synechococcus elongatus (Atsumi et al. 2009b).
Corynebacterium glutamicum is a rapidly growing, gram-positive soil bacterium and has been the workhorse of industrial amino acid production (Leuchtenberger et al. 2005). The discovery of this organism’s ability to produce glutamate with high efficiency led to the development of amino acid production by fermentation. In 2005, C. glutamicum was used to produce 1.5 million tons of glutamate as monosodium glutamate per year as well as several thousand tons of other amino acids such as lysine, isoleucine, tryptophan, and threonine. These processes have been engineered extensively, with valine and lysine production titers reaching ∼100 g/L (Leuchtenberger et al. 2005). Because these 2-keto acid pathways for higher chain alcohol production share common precursors with amino acids, C. glutamicum shows potential for the production of higher chain alcohols. To our knowledge, this organism has never been used to synthesize any alcohols beyond ethanol (Inui et al. 2004).
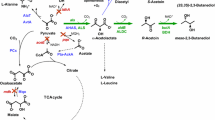
Valine, which shares the 2-keto acid precursor, 2-ketoisovalerate (KIV), with isobutanol, has been produced in C. glutamicum from glucose by overexpression of the valine biosynthesis pathway (ilvBNCDE) and elimination of carbon competing pathways (Blombach et al. 2008; Radmacher et al. 2002). To produce isobutanol in C. glutamicum, we applied a similar strategy by overexpression of the 2-keto acid synthesis pathway (Fig. 1). This pathway includes alsS (Bacillus subtilis) and ilvCD (C. glutamicum) along with downstream genes for the subsequent decarboxylation (kivd from L. lactis) and reduction (adhA from C. glutamicum) of 2-ketoisovalerate to isobutanol.

2-Keto acid based higher alcohol production pathways from glucose in C. glutamicum. KIV 2-ketoisovalerate, KIC 2-ketoisocaproate, KV 2-ketovalerate, KB 2-ketobutyrate, KMV 2-keto-3-methyl-valerate, PEP phosphoenolpyruvate, OAA oxaloacetate
Materials and methods
Reagents
All restriction enzymes and antarctic phosphatase were purchased from New England Biolabs (Ipswich, MA, USA). The DNA ligation kit was supplied by Roche (Manheim, Germany). KOD DNA polymerase was purchased from EMD Chemicals (San Diego, CA, USA). Oligonucleotides were ordered from Integrated DNA Technologies (Coralville, IA, USA).
Strains and plasmids
All strains, plasmids, and oligonucleotides used are listed in Table 1. E. coli XL1-Blue (Stratagene) was used to propagate all plasmids. All corynebacteria strains used were derived from C. glutamicum ATCC 13032, which was used as wild type (WT). Gene knockout in C. glutamicum was achieved using pK19mobsacB as previously described (Schäfer et al. 1994). To begin construction of the isobutanol production plasmid, a multiple cloning site and a transcriptional terminator from the ilvBNC operon of C. glutamicum was made by primer elongation using oligonucleotides (K673–K676). The PCR product was digested with MfeI and PstI and ligated into pEP2 digested with EcoRI and PstI to obtain pKS122. The cat (encoding chloramphenicol acetyltransferase) gene was cloned from pZA31 (Lutz and Bujard 1997) with primers K709 and K710, digested with SalI and NotI, and ligated into pKS122 cut with the same enzymes to obtain pKS133. The eftu promoter, which transcribes the eftu gene (Cg0587), was amplified from genomic DNA (ATCC 13032) with primers K717 and K718, digested with PstI and SalI, and ligated into pKS133 cut with the same restriction enzymes to obtain pKS140. To clone alsS of B. subtilis, the gene was amplified from pSA69 (Atsumi et al. 2008b) with primers K753 and A124, digested with XhoI and SalI, and ligated into pKS140 cut with SalI to obtain pKS149. IlvC and ilvD of C. glutamicum were amplified by PCR from genomic DNA (ATCC 13032) with primers K764 and K765 and K766 and K767, respectively. The PCR products were fused together by sequential overlap extension, digested with XhoI and NotI, and ligated into pKS149 cut with SalI and NotI to give pKS152. pKS154 was constructed by amplifying kivd of L. lactis from pSA55 (Atsumi et al. 2008b) with K852 and K853, digesting the PCR product with NotI and SpeI and ligating into pKS152 cut with the same restriction enzymes. adhA (Cg3107) of C. glutamicum was amplified from genomic DNA using primers K887 and K888, and the PCR product was digested with SpeI and ligated into pKS154 cut with the same enzyme to obtain pKS167. For construction of pKS160, the eftu promoter was amplified with primers K717 and K718, digested with PstI and SalI, and ligated into pKS122 cut with the same enzymes. Inactivation of chromosomal genes was done as previously described (Schäfer et al. 1994). Plasmid p∆pyc was constructed using primers K976 + K977 and K978 + K979 to amplify the terminal regions of pyc from genomic DNA for construction of the pyc fragment. Likewise, plasmid p∆ppc was constructed using K972 + K973 and K974 + K975, plasmid p∆aceE was constructed using K960 + K961 and K962 + K963, and plasmid p∆pgi with primers K950 + K951 and K952 + K953 and plasmid p∆ilvE was constructed using K21 + K22 and K23 + K24. The two PCR products were fused together by sequential overlap extension, digested with MfeI and HindIII (pyc, aceE, and pgi fragments) or with HindIII and EcoRI (ppc and ilvE fragments), and ligated into pK19mobsacB cut with EcoRI and HindIII. All knockouts were confirmed by PCR (Table 2).
Medium and cultivation
E. coli cultures for plasmid construction and propagation were grown in Luria–Bertani (LB) media at 37°C and shaken at 250 rpm. For stepwise construction of the isobutanol pathway, the cultures were grown in 20-mL media CGIII (Menkel et al. 1989), but with 40 g/L glucose in a 250-mL screw-cap shake flask. Longer-term production cultures were done in 20 mL CGIII media but with 70 g/L glucose in a 250-mL screw-cap flask. All strains with an aceE knockout were cultured in the same production media but with 4.5 g/L acetate to allow for adequate cell growth. All C. glutamicum production cultures were inoculated to an initial OD600 nm of 0.1 from a 3-mL overnight culture grown in CGIII at 30°C in a rotary shaker (250 rpm). After inoculation, production cultures were shaken at 250 rpm and 30°C. The pH values of long-term production cultures were checked at every sampling time, and 10 M NaOH was added as needed to adjust to pH 6.8. Antibiotics were added appropriately (kanamycin 25 μg/mL).
Tolerance assays
Cultures of E. coli BW25113 were grown at 30°C and 37°C in LB with 2% glucose (LBG) in a 500-mL baffled shake flask at 250 rpm. C. glutamicum was grown in LBG and CGIII media at 30°C in a 500-mL baffled shake flask at 250 rpm. After an OD of 1.0 was reached, 5-mL aliquots were moved into test tubes, isobutanol was added at varied concentrations (0%, 1.0%, 1.25%, 1.5%, 1.75%, and 2.0%), and the cultures were incubated at 30°C and shaken at 250 rpm. After 4 h, each culture was diluted and spread onto LB plates for colony counting after 24 h of incubation at 30°C. Percent viability was calculated as the number of cells remaining viable after isobutanol exposure normalized with the cell number obtained with no isobutanol exposure.
Enzymatic assays
For crude extract enzyme assays, 5-mL CGIII cultures of C. glutamicum harboring the relevant plasmids were grown overnight at 30°C in 5 mL CGIII media in test tubes. Crude extracts were prepared by concentrating the cultures by 5-fold in 0.1 M phosphate buffer (pH 7.1) and lysing them with 0.1 mM glass beads. The acetolactate synthase (ALS) assay was performed as previously reported (Holtzclaw and Chapman 1975). KDC activity was assayed by measuring the generation of isobutyraldehyde from 2-ketoisovalerate as previously described (Atsumi et al. 2009a). Reductive ADH activity was detected by measuring NADH oxidation at 340 nm in 1 mL of 50 mM phosphate buffer (pH 7.1), 100 mM isobutyraldehyde, 250 μM NADH, and 100 μL crude extract. Oxidative ADH activity was detected by measuring NAD+ reduction at 340 nm in 1 mL of 50 mM phosphate buffer (pH 7.1), 500 mM ethanol or isobutanol, 250 μM NAD+, and 100 μL crude extract. Total protein concentrations were measured by Bradford assay.
Metabolite detection
The alcohol compounds produced were quantified using gas chromatography equipped with flame ionization as previously described (Atsumi et al. 2008a). All other compounds were measured by applying filtered fermentation broth to an Agilent 1100 high-performance liquid chromatograph equipped with an autosampler (Agilent Technologies) and a Bio-Rad Aminex HPX87 column (5 mM H2SO4; 0.3 mL/min; column temperature, 35°C; Bio-Rad Laboratories, Hercules, CA, USA).
Results
C. glutamicum exhibits better isobutanol tolerance than E. coli
The capability of C. glutamicum to tolerate isobutanol toxicity was evaluated by exposing the organism to isobutanol at different concentrations and determining the percentage of cells that remained viable relative to no alcohol exposure. In addition to C. glutamicum, we also performed the same experiment with E. coli, which is a successful isobutanol-producing organism (Atsumi et al. 2008b). Initially, both organisms were cultured in LB with 2% glucose (LBG) at 30°C to compare their tolerance levels in identical growth conditions (Fig. 2a). However, because LBG is not an optimal growth medium for C. glutamicum, the isobutanol tolerance of this organism was assayed in CGIII medium as well (Fig. 2b). Likewise, we investigated the ability of E. coli to tolerate isobutanol toxicity at its optimal growth temperature, 37°C (Fig. 2b). We found that C. glutamicum is more tolerant of isobutanol toxicity than E. coli for all concentrations and conditions tested (Fig. 2). No significant difference in the viability of E. coli at 30°C or 37°C was found; however, the viability of C. glutamicum was improved in CGIII media in comparison with LBG media at 10 and 12.5 g/L isobutanol. In LBG media with 10 g/L isobutanol, 51% of C. glutamicum cells remained viable after exposure, whereas 30% and 24% of E. coli cells remained viable when incubated at 30°C and 37°C, respectively. For all concentrations at or below 12.5 g/L isobutanol in both mediums tested, C. glutamicum displayed 2-fold better viability in comparison with E. coli. At 20 g/L isobutanol, C. glutamicum showed 18% cell viability in LBG, which is 4-fold higher than the 4% cell viability obtained with E. coli incubated at 37°C. This beneficial characteristic of C. glutamicum further increased our motivation to pursue higher chain alcohol production with this host.

Comparison of isobutanol tolerance of C. glutamicum and E. coli by exposure to isobutanol and calculation of% viable cells remaining. a E. coli and C. glutamicum cultured in LBG medium at 30°C, b E. coli and C. glutamicum cultured in LBG medium at 37°C and CGIII medium at 30°C, respectively. Data shown are the result of three independent experiments
Constructing the isobutanol production pathway
We aimed to produce isobutanol as a primary fermentation product from glucose using C. glutamicum. First, a plasmid for expression of the isobutanol pathway was constructed. The promoter of eftu (P eftu ), encoding elongation factor TU, was chosen for transcription of the entire isobutanol operon. Second, we selected ALS (acetolactate synthase) of B. subtilis (Holtzclaw and Chapman 1975) to catalyze the first reaction from pyruvate. To ensure that both P eftu and ALS have functional activity in C. glutamicum, crude extract ALS enzyme assays (Holtzclaw and Chapman 1975) were performed. The activity of the heterologous ALS in wild-type C. glutamicum was found to be 6,700 U mg−1, whereas an activity of 100 U mg−1 was detected with no ALS expression (Table 3), confirming that ALS and the P eftu have good activity in C. glutamicum. Following ALS, the native enzymes, IlvC and IlvD, were overexpressed to convert the 2-acetolactate produced from pyruvate by ALS to 2-ketoisovalerate. The promiscuous KDC of L. lactis, encoded by kivd, was employed to decarboxylate KIV to isobutyraldehyde, which is then reduced by an alcohol dehydrogenase to produce isobutanol (Fig. 1). Because expression of KDC in C. glutamicum has never been accomplished, crude extract enzyme assays were performed to confirm it has activity for KIV in this host. Indeed, we verified the activity of KDC to be 2.3 ± 0.6 U mg−1 from C. glutamicum harboring pKS167, whereas no KDC activity was detected with an empty plasmid (pKS160). For our initial attempt at isobutanol production, we selected adhA, a native alcohol dehydrogenase previously identified to be NADH specific (Arndt and Eikmanns 2007) and determined its activity for isobutyraldehyde reduction to be 0.62 ± 0.06 U mg−1, whereas an activity of 0.09 U mg−1 was detected with no adhA overexpression (Table 3). In C. glutamicum, adhA has been demonstrated to be required for growth on ethanol, generating acetaldehyde and NADH (Arndt and Eikmanns 2007); however, we did not detect any oxidative activity with isobutanol as substrate with this enzyme.
Isobutanol production with C. glutamicum
The effects of overexpression of the three steps from pyruvate to KIV (alsS-ilvCD) followed by sequential overexpression of kivd and a native alcohol dehydrogenase, encoded by adhA, on isobutanol production were determined (Fig. 3). Overexpression of alsS-ilvCD without kivd did not produce any isobutanol, demonstrating that C. glutamicum does not have an endogenous KDC with activity toward KIV. The added overexpression of kivd resulted in the production of 2.2 g/L isobutanol in 48 h (Fig. 3) and, in addition, 0.4 g/L of 3-methyl-1-butanol (Table 4), confirming that one or more of the five putative and confirmed ADHs of C. glutamicum (Arndt and Eikmanns 2007) are capable of reducing isobutyraldehyde to isobutanol. Isobutyraldehyde was detected (350 mg/L), indicating the need for overexpression of an alcohol dehydrogenase. By overexpression of adhA, the final isobutanol titer was increased to 2.6 g/L, and isobutyraldehyde accumulation was reduced to 50 mg/L (Fig. 3). Also, 1-propanol, 1-butanol, 2-methyl-1-butanol, and 2-phenylethanol were detected as byproducts (Table 4), demonstrating this organism’s ability to produce several higher alcohols.

Effects of the systematic overexpression of the synthetic isobutanol pathway on isobutanol production in C. glutamicum. Plus symbols denote overexpression of the indicated gene(s) or the addition of 40 g/L glucose to the media. Data obtained are the result of three independent fermentations
Strain improvement by gene knockout
Batch fermentations of C. glutamicum harboring pKS167 (P eftu ::alsS-ilvCD-kivd-adhA) were performed to characterize long-term isobutanol production in different backgrounds (Fig. 4). WT produced 4.0 g/L isobutanol in 96 h (Fig. 4a) and the overall product yield was found to be 0.08 g/g (isobutanol/glucose), which is 19% of the theoretical maximum. HPLC analysis led to the detection of only 0.9 g/L acetate in the fermentation medium (Fig. 4d). We attempted to increase the isobutanol titer by inactivating several enzymes that may consume precursors of the isobutanol pathway. First, we implemented single knockouts of ppc (encoding phosphoenolpyruvate carboxylase), pyc (encoding pyruvate carboxylase), and ilvE (encoding valine transaminase) to eliminate the consumption of phosphoenolpyruvate, pyruvate, and KIV, respectively. The ∆ppc and ∆ilvE strains were found to be ineffective at increasing the isobutanol titer and performed as WT (data not shown), and the ∆pyc strain slightly improved the isobutanol titer to 4.3 g/L (Fig. 4a). Furthermore, inactivation of PYC led to the accumulation of 2.6 g/L lactate (Fig. 4c), whereas acetate production was relatively unaffected (Fig. 4d). To direct the accumulated lactate in the ∆pyc strain toward isobutanol synthesis, we disrupted ldh (encoding lactate dehydrogenase (LDH)) and found that it increased the final isobutanol titer by ∼25% to 4.9 g/L isobutanol (Fig. 4a) as well as increased cell growth (Fig. 4e).

Long-term isobutanol production in C. glutamicum harboring pKS167 (P eftu ::alsS-ilvCD-kivd-adhA). a Isobutanol production, b glucose consumption, c lactate production, d acetate production, and e cell growth. WT (open circles), ∆pyc (open diamonds), ∆pyc∆ldh (closed circles), ∆aceE (open squares), ∆aceE∆ldh (closed squares), and ∆aceE∆ldh∆pgi (open triangles). Data obtained are the result of three independent fermentations
Because we did not detect significant organic acid accumulation in WT, we hypothesized that the pyruvate dehydrogenase complex (PDHc) is a major consumer of pyruvate and constructed a ∆aceE strain deficient in PDHc activity. As shown in Fig. 4a, isobutanol production in the ∆aceE background was severely inhibited and only accumulated to 1.4 g/L isobutanol, while cell growth was similar to the other strains tested (Fig. 4e). Moreover, inactivation of the PDHc resulted in a significant accumulation of 8.3 g/L lactate (Fig. 4c), indicating that it is the major consumer of pyruvate. Next, we aimed to direct this accumulated lactate through the isobutanol pathway with the additional inactivation of LDH by constructing a ∆aceE∆ldh strain. The deletion of ldh in the ∆aceE host did direct the carbon flux away toward isobutanol during the first 24 h. Ultimately, however, this strain did not increase isobutanol production over WT (Fig. 4a) and the strain excreted 2.1 g/L acetate (Fig. 4d). This is perhaps due to the activity of pyruvate:quinone oxidoreductase that has been shown to compete for pyruvate to generate acetate (Blombach et al. 2008). At this point, we predicted that isobutanol production in ∆aceE strains might be inhibited due to an insufficient supply of NADPH that may be generated from the TCA cycle. To increase NADPH availability, pgi (encoding phosphoglucose isomerase, PGI) was knocked out to create a ∆aceE∆ldh∆pgi strain. However, isobutanol production with this strain was eliminated (Fig. 4a). Also, the glucose consumption rate of the ∆aceE∆ldh∆pgi strain was strongly inhibited (Fig. 4b), suggesting that PGI is an essential path for glucose metabolism toward isobutanol. Similar to previous studies regarding valine production in C. glutamicum (Blombach et al. 2008) and isobutanol production in E. coli (Atsumi et al. 2008b), we found that all strains produced isobutanol primarily in the stationary phase (Fig. 4e), and valine was never detected above concentrations present in fresh medium (data not shown).
Discussion
We engineered C. glutamicum for isobutanol production and produced several other higher chain alcohols as byproducts (1-propanol, 1-butanol, 2-methyl-1-butanol, 3-methyl-1-butanol, and 2-phenylethanol), highlighting the capacity of this host for the construction of several different higher chain alcohol production platforms.
Despite the promising use of isobutanol as a fuel, its toxic effects on microbes may be an important factor limiting its production (Brynildsen and Liao 2009). Here, we found that C. glutamicum has an increased ability to tolerate isobutanol toxicity in comparison with E. coli at all concentrations and conditions tested (0–20 g/L isobutanol), showing that C. glutamicum has potential to be a higher alcohol producer as the tolerance of E. coli is similar to that of C. acetobutylicum (Atsumi et al. 2008a; Lin and Blaschek 1983), a natural 1-butanol producer. Corynebacteria possess a thick cell wall that helps protect the cell against external stresses such as freeze damage, pressure, and hyperosmotic shock (Marienfeld et al. 1997). Similar to mycobacteria, the cell wall of corynebacteria contains mycolic acids linked to polysaccharides which, in mycobacteria, form a second bilayer surrounding the cell wall and limiting its permeability (Eggeling and Sahm 2001). The higher isobutanol tolerance of C. glutamicum may be the result of these protective characteristics, and this trait may prove to be advantageous for higher chain alcohol production once alcohol titers reach toxic levels.
To begin construction of the isobutanol pathway, the promoter of eftu was chosen for expression of the isobutanol operon. This promoter has been shown to be effective for strong gene expression in C. glutamicum for the production of lysine (Becker et al. 2005). With this promoter, we were able to successfully express ALS of B. subtilis and KDC of L. lactis in C. glutamicum and obtain good activities. C. glutamicum also possesses native acetohydroxyacid synthase (AHAS) enzymes capable of generating 2-acetolactate, one of which has been used for valine production (Radmacher et al. 2002); however, ALS has been shown to be beneficial for isobutanol production in E. coli, and, unlike AHAS (encoded by ilvBN), ALS is not feedback-inhibited by valine (Elisakova et al. 2005; Holtzclaw and Chapman 1975), and it is more specific for acetolactate synthase activity.
Overexpression of alsS-ilvCD without kivd did not produce any isobutanol, indicating that C. glutamicum does not have an endogenous KDC with activity toward KIV. The additional overexpression of kivd and adhA resulted in improved production of isobutanol, and we also detected 1-propanol, 1-butanol, 2-phenylethanol, 2-methyl-1-butanol, and 3-methyl-1-butanol as byproducts. We therefore conclude that the overexpessed KDC is active in C. glutamicum and that this organism may be engineered to produce a wide array of higher alcohols.
Long-term fermentations with WT harboring pKS167 (P eftu ::alsS-ilvCD-kivd-adhA) produced 4.0 g/L isobutanol in 96 h and gave a yield that is 19% of the theoretical maximum (isobutanol/glucose). WT produced isobutanol slowly for the first 24 h when the cells are growing, whereas 85% of the isobutanol was produced after the cells reach stationary phase. In an effort to improve isobutanol productivity, we targeted pathways that were potentially consuming several isobutanol precursors: phosphoenolpyruvate, pyruvate, and KIV. The inactivations of PPC and IlvE did not increase isobutanol production, and disruption of PYC only slightly improved the final titer. However, the absence of PYC resulted in the accumulation of 2.6 g/L lactic acid, indicating that PYC actively consumes pyruvate, which might explain why WT does not produce lactate. Our results also indicate that the pyruvate made available in the ∆pyc strain is more effectively consumed by LDH, outcompeting ALS of the isobutanol pathway. This is plausible, as the K m value of ALS for pyruvate is 13.6 mM (Atsumi et al. 2009a), whereas LDH has a lower K m of 7.4 mM (Dietrich et al. 2009), which may explain the increased efficacy of LDH for pyruvate consumption. Our results are in agreement with this argument, as the additional inactivation of the competitive LDH (∆pyc∆ldh) improved isobutanol production by ∼25% to 4.9 g/L isobutanol and increased the yield to 23% of the theoretical maximum.
Previous studies concerning valine production in C. glutamicum have shown that inactivation of the PDHc resulted in significant improvement of valine production and high product yield (Blombach et al. 2008; Blombach et al. 2007). In our system, we also found that inactivation of the PDHc increases pyruvate availability; however, LDH becomes the more dominant enzyme, and lactic acid becomes the primary product, similar to our results in the ∆pyc strain. It seemed that by constructing a ∆aceE∆ldh strain, we may convert this “available” pyruvate to isobutanol. Initially, this double knockout strain produced isobutanol well in comparison with WT but productivity slowed drastically after the cells entered the stationary phase. We hypothesized that TCA cycle activity might be important for isobutanol production in C. glutamicum, as this metabolic pathway supplies ATP and NAD(P)H. More specifically, NADPH has been shown to be a critical cofactor for the production of amino acids in C. glutamicum, including lysine and valine (Kabus et al. 2007; Marx et al. 2003), and in fact, the introduction of a pgi knockout in a strain deficient in PDHc activity has been shown to increase the valine yield from glucose (Blombach et al. 2008). Unfortunately, redirecting the carbon flow through the pentose phosphate pathway by disruption of PGI (∆ldh∆aceE∆pgi) eliminated isobutanol production and severely stunted glucose consumption. This is in contrast with results obtained for valine production (Blombach et al. 2008); however, unlike the valine pathway that ultimately consumes 2 mole of NADPH per mole of valine produced, here, the isobutanol pathway consumes 1 mole of NADPH and 1 mole of NADH per mole of isobutanol produced. This difference in redox cofactor requirement may be one reason for the ineffectiveness of a pgi knockout on isobutanol production. Also, several studies have shown that PGI mutants dramatically disturb NADPH metabolism and retard growth and glucose consumption in C. glutamicum (Marx et al. 2003).
Our initial attempts to produce isobutanol in C. glutamicum resulted in the maximum production of 4.9 g/L in a ∆pyc∆ldh background. This success emphasizes the efficacy of the 2-keto acid based pathways for higher alcohol production, and future studies should be aimed to increase the low isobutanol yield. There have been several cases of generating mutants of C. glutamicum that overproduce valine, as well as several other pertinent amino acids for higher alcohol production such as isoleucine and threonine, by applying random mutagenesis coupled with a selection for growth in the presence of an amino acid analogue (Shimura 1972a, b; Uemura et al. 1972). In addition, other metabolic engineering tools and experiences (Barkovich and Liao 2001; Liao and Delgado 1993) might prove to be useful to help elucidate the effects of the mutations introduced and to direct the metabolic flux to the desirable pathway(s). Perhaps this would be an effective method to develop isobutanol overproducers as valine and isobutanol share the same 2-keto acid precursor, KIV. As a first attempt at isobutanol production in C. glutamicum, our primary challenge was the effective expression of heterologous enzymes (KDC and ALS) that are important for isobutanol production. The success we have had with this more-tolerant host supports future studies to engineer several higher chain alcohol production platforms in C. glutamicum using the 2-keto acid pathways.
References
Arndt A, Eikmanns BJ (2007) The alcohol dehydrogenase gene adhA in Corynebacterium glutamicum is subject to carbon catabolite repression. J Bacteriol 189:7408–7416
Atsumi S, Liao JC (2008) Directed evolution of Methanococcus jannaschii citramalate synthase for biosynthesis of 1-propanol and 1-butanol by Escherichia coli. Appl Environ Microbiol 74:7802–7808
Atsumi S, Cann AF, Connor MR, Shen CR, Smith KM, Brynildsen MP, Chou KJ, Hanai T, Liao JC (2008a) Metabolic engineering of Escherichia coli for 1-butanol production. Metab Eng 10:305–311
Atsumi S, Hanai T, Liao JC (2008b) Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 451:86–89
Atsumi S, Li Z, Liao JC (2009a) Acetolactate synthase from Bacillus subtilis serves as a 2-ketoisovalerate decarboxylase for isobutanol biosynthesis in Escherichia coli. Appl Environ Microbiol 75:6306–6311
Atsumi S, Higashide W, Liao JC (2009b) Direct photosynthetic recycling of carbon dioxide to isobutyraldehyde. Nat Biotechnol 27:1177–1180
Barkovich R, Liao JC (2001) Metabolic engineering of isoprenoids. Metab Eng 3:27–39
Becker J, Klopprogge C, Zelder O, Heinzle E, Wittmann C (2005) Amplified expression of fructose 1, 6-bisphosphatase in Corynebacterium glutamicum increases in vivo flux through the pentose phosphate pathway and lysine production on different carbon sources. Appl Environ Microbiol 71:8587–8596
Blombach B, Schreiner ME, Holatko J, Bartek T, Oldiges M, Eikmanns BJ (2007) L-valine production with pyruvate dehydrogenase complex-deficient Corynebacterium glutamicum. Appl Environ Microbiol 73:2079–2084
Blombach B, Schreiner ME, Bartek T, Oldiges M, Eikmanns BJ (2008) Corynebacterium glutamicum tailored for high-yield L-valine production. Appl Microbiol Biotechnol 79:471–479
Borden JR, Papoutsakis ET (2007) Dynamics of genomic-library enrichment and identification of solvent tolerance genes for Clostridium acetobutylicum. Appl Environ Microbiol 73:3061–3068
Brynildsen MP, Liao JC (2009) An integrated network approach identifies the isobutanol response network of Escherichia coli. Mol Syst Biol 5:277
Cann AF, Liao JC (2008) Production of 2-methyl-1-butanol in engineered Escherichia coli. Appl Microbiol Biotechnol 81:89–98
Connor MR, Liao JC (2008) Engineering of an Escherichia coli strain for the production of 3-methyl-1-butanol. Appl Environ Microbiol 74:5769–5775
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645
Dietrich C, Nato A, Bost B, Le Marechal P, Guyonvarch A (2009) Regulation of ldh expression during biotin-limited growth of Corynebacterium glutamicum. Microbiology 155:1360–1375
Eggeling L, Sahm H (2001) The cell wall barrier of Corynebacterium glutamicum and amino acid efflux. J Biosci Bioeng 92:201–213
Elisakova V, Patek M, Holatko J, Nesvera J, Leyval D, Goergen JL, Delaunay S (2005) Feedback-resistant acetohydroxy acid synthase increases valine production in Corynebacterium glutamicum. Appl Environ Microbiol 71:207–213
Ezeji TC, Qureshi N, Blaschek HP (2004) Acetone butanol ethanol (ABE) production from concentrated substrate: reduction in substrate inhibition by fed-batch technique and product inhibition by gas stripping. Appl Microbiol Biotechnol 63:653–658
Holtzclaw WD, Chapman LF (1975) Degradative acetolactate synthase of Bacillus subtilis: purification and properties. J Bacteriol 121:917–922
Inui M, Kawaguchi H, Murakami S, Vertés AA, Yukawa H (2004) Metabolic engineering of Corynebacterium glutamicum for fuel ethanol production under oxygen-deprivation conditions. J Mol Microbiol Biotechnol 8:243–254
Kabus A, Georgi T, Wendisch VF, Bott M (2007) Expression of the Escherichia coli pntAB genes encoding a membrane-bound transhydrogenase in Corynebacterium glutamicum improves L-lysine formation. Appl Microbiol Biotechnol 75:47–53
Leuchtenberger W, Huthmacher K, Drauz K (2005) Biotechnological production of amino acids and derivatives: current status and prospects. Appl Microbiol Biotechnol 69:1–8
Liao JC, Delgado J (1993) Advances in metabolic control analysis. Biotechnol Progr 9:221–223
Lin YL, Blaschek HP (1983) Butanol production by a butanol-tolerant strain of Clostridium acetobutylicum in extruded corn broth. Appl Environ Microbiol 45:966–973
Lutz R, Bujard H (1997) Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res 25:1203–1210
Marienfeld S, Uhlemann EM, Schmid R, Kramer R, Burkovski A (1997) Ultrastructure of the Corynebacterium glutamicum cell wall. Antonie Van Leeuwenhoek 72:291–297
Marx A, Hans S, Mockel B, Bathe B, de Graaf AA, McCormack AC, Stapleton C, Burke K, O’Donohue M, Dunican LK (2003) Metabolic phenotype of phosphoglucose isomerase mutants of Corynebacterium glutamicum. J Biotechnol 104:185–197
Menkel E, Thierbach G, Eggeling L, Sahm H (1989) Influence of increased aspartate availability on lysine formation by a recombinant strain of Corynebacterium glutamicum and utilization of fumarate. Appl Environ Microbiol 55:684–688
Messerotti LJ, Radford AJ, Hodgson AL (1990) Nucleotide sequence of the replication region from the Mycobacterium–Escherichia coli shuttle vector pEP2. Gene 96:147–148
Radmacher E, Vaitsikova A, Burger U, Krumbach K, Sahm H, Eggeling L (2002) Linking central metabolism with increased pathway flux: L-valine accumulation by Corynebacterium glutamicum. Appl Environ Microbiol 68:2246–2250
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73
Seiferlein KE (2009) Annual energy review 2008. In: Annual Energy Review. Energy Information Administration. http://www.eia.doe.gv/emeu/aer/contents.html. Accessed October 6, 2009
Shen CR, Liao JC (2008) Metabolic engineering of Escherichia coli for 1-butanol and 1-propanol production via the keto-acid pathways. Metab Eng 10:312–320
Shimura K (1972a) Isoleucine. In: Yamada K, Kinoshita S, Tsunoda T, Aida K (eds) The microbial production of amino acids. Halsted, New York, pp 491–513
Shimura K (1972b) Threonine. In: Yamada K, Kinoshita S, Tsunoda T, Aida K (eds) The microbial production of amino acids. Halsted, New York, pp 453–472
Uemura T, Sugisaki Z, Takamura Y (1972) Valine. In: Yamada K, Kinoshita S, Tsunoda T, Aida K (eds) The microbial production of amino acids. Halsted, New York, pp 339–368
Acknowledgments
We are grateful to Margaret Chen for her experimental assistance and to Dr. Sinskey and Dr. Lessard (MIT–Department of Biology) for the generous donation of pEP2.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Electronic supplementary figure
(PDF 33 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Smith, K.M., Cho, KM. & Liao, J.C. Engineering Corynebacterium glutamicum for isobutanol production. Appl Microbiol Biotechnol 87, 1045–1055 (2010). https://doi.org/10.1007/s00253-010-2522-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2522-6