
Abstract
Glioblastoma (GBM), the predominant and primary malignant intracranial tumor, poses a formidable challenge due to its immunosuppressive microenvironment, thereby confounding conventional therapeutic interventions. Despite the established treatment regimen comprising surgical intervention, radiotherapy, temozolomide administration, and the exploration of emerging modalities such as immunotherapy and integration of medicine and engineering technology therapy, the efficacy of these approaches remains constrained, resulting in suboptimal prognostic outcomes. In recent years, intensive scrutiny of the inhibitory and immunosuppressive milieu within GBM has underscored the significance of cellular constituents of the GBM microenvironment and their interactions with malignant cells and neurons. Novel immune and targeted therapy strategies have emerged, offering promising avenues for advancing GBM treatment. One pivotal mechanism orchestrating immunosuppression in GBM involves the aggregation of myeloid-derived suppressor cells (MDSCs), glioma-associated macrophage/microglia (GAM), and regulatory T cells (Tregs). Among these, MDSCs, though constituting a minority (4–8%) of CD45+ cells in GBM, play a central component in fostering immune evasion and propelling tumor progression, angiogenesis, invasion, and metastasis. MDSCs deploy intricate immunosuppressive mechanisms that adapt to the dynamic tumor microenvironment (TME). Understanding the interplay between GBM and MDSCs provides a compelling basis for therapeutic interventions. This review seeks to elucidate the immune regulatory mechanisms inherent in the GBM microenvironment, explore existing therapeutic targets, and consolidate recent insights into MDSC induction and their contribution to GBM immunosuppression. Additionally, the review comprehensively surveys ongoing clinical trials and potential treatment strategies, envisioning a future where targeting MDSCs could reshape the immune landscape of GBM. Through the synergistic integration of immunotherapy with other therapeutic modalities, this approach can establish a multidisciplinary, multi-target paradigm, ultimately improving the prognosis and quality of life in patients with GBM.
Similar content being viewed by others


Introduction
Glioblastoma (GBM) is categorized as a WHO grade IV glioma [1], representing the most prevalent, primary, and malignant tumor in the brain, and is recognized for its crazy invasiveness. The median survival time of GBM cases is roughly 12.5–15 months, with 2-year and 5-year survival rates of merely 25% and 10%, respectively [2]. The standard therapeutic approach for GBM typically involves surgical intervention complemented by chemotherapy, radiotherapy (RT), or targeted therapy [3]. Nevertheless, the treatment efficacy for GBM remains suboptimal due to the considerable genetic variability and intratumoral heterogeneity inherent to GBM [4]. Recently, the impact of the tumor microenvironment (TME), particularly the immunosuppressive milieu, on the heterogeneity of GBM and its immune "cold" environment has been increasingly recognized [5, 6].
The onset of GBM can be conceptualized through the 'Swiss cheese model', which represents a culmination of successive failures in various host defense mechanisms [7]. Notably, the immune system serves as the ultimate bulwark against GBM initiation and progression. Vigilantly surveilling within the body, the immune system engages with cancer throughout its developmental stages. An imbalance in this intricate interaction underscores that cancer, beyond uncontrolled cellular proliferation, also represents a manifestation of immune dysfunction. From this vantage point forward, immunotherapy has become an inherent approach to cancer treatment [8]. Although immunotherapies targeting programmed cell death protein 1 (PD-1) or cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) have shown efficiency in certain tumors [9], their consistent failures in the case of GBM are attributed to its classification as an “immunologically cold” tumor. GBM typically manifests minimal expression of neoantigens, exacerbating the immunosuppressive milieu through numerous immune checkpoints and immune-inhibitory cytokines [10]. Moreover, owing to its significant intratumoral heterogeneity, the positive responses observed in a small cluster of patients to immunotherapies or other treatment modalities cannot be extrapolated to represent the overall treatment sensitivity of GBM. Consequently, patients’ responses to GBM treatments are frequently transient, and tumor recurrence is nearly universal. These challenges underscore the imperative necessity of enhancing existing GBM treatment strategies.
Hence, investigating the interplay between the TME, with particular emphasis on some specific components, and tumors and intervening in this interaction holds significant therapeutic promise for regulating tumor immunosuppression [11]. This review encapsulates the immunomodulatory processes and associated molecular characteristics within the immunosuppressive milieu of GBM. The latest research concentrates on delineating the component of TME within these processes, intending to selectively modulate the immunosuppressive microenvironment of GBM, thereby offering potential therapeutic avenues. Figure 1 shows the current challenges of treatment in GBM.

The current challenges of treatment in GBM. Due to its highly dynamic and complex microenvironment components and unique intratumoral heterogeneity, GBM is in urgent need of one or more combination therapies for precise target attacks. These therapies can be drugs, exogenous editing methods, new bioengineering, and so on
The immune regulation in glioblastoma
Two cell types can be simply described the central nervous system (CNS), which are glia and neurons, and glioma originate from glia, which include ependymal cells, microglia, astrocytes, and oligodendrocytes [12]. The heterogeneity of TME in GBM shows considerable variability, and the crosstalk between malignant cells and microenvironment is critical for tumor cell proliferation and migration, contributing to the suppression of infiltration and activation of T cells. The major infiltrating cells in the glioma TME are immune cell populations like tumor-associated myeloid cells (TAMCs), which include tumor-associated macrophages (TAMs) and microglias, myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs), and neutrophils [13]. Microglias are distributed throughout the CNS and play a crucial component in regulating immunity homeostasis in the brain. It is the resident TAMs of the CNS and secretes immunosuppressive factors like interleukin-10 (IL-10) and transforming growth factor-β (TGF-β) or other anti-tumor stimulating factors like IL-12 and tumor necrosis factor-α (TNF-α) based on the “heat” or “cold” status of the TME [14]. It has been shown that in GBM, TAMs lack the costimulatory molecules that are essential for the activation of lymphocytes, like CD40, CD86, and CD80, and secreting IL-6, IL-1β, and TNF-α, which are important for the response of innate immune [15]. At the same time, their ability to make the leukocyte antigen (HLA) class II molecules upregulation is impaired but showed increased expression in immunosuppressive ligands like B7-H1 and Fas ligand [16, 17]. MDSCs are heterogeneous and come from immature bone marrow cells that are recruited during tumorigenesis and then infiltrated into tumors, promoting vascularization and becoming major mechanisms of immune surveillance, including polarization of M1 macrophages, antigen presentation of DC, cytotoxicity of natural killer cells (NK cells), and activation of T cells [18]. They have substantial overlap with TAM in the GBM mouse model: They have the phenotypic characteristics of M1 and M2 macrophages and exhibit important functional and phenotypic plasticity based on their local TME [19]. Moreover, CD33+ MDSC have been discovered at higher levels in the peripheral blood (PB) of GBM patients than in healthy persons, and healthy persons-derived CD14+ monocytes (MONs) exposed to GBM cells may gain MDSC-like features, like upregulating the production of immunosuppressive factors like B7-H1, IL-10, and TGF-β, and inducing apoptosis in activated lymphocytes [20].
The blood–brain barrier (BBB) is one of the key components of the adaptive changes in TME. The BBB, which, like a semipermeable membrane, consists of endothelial cells (ECs), foot processes from astrocytes, and pericytes, separates the CNS from the peripheral immune system so that naive T cells cannot cross the BBB, but activated T cells can [21]. Thus, it rigidly regulates the lymphocytes infiltrating the CNS, and therefore, there is an overall decrease in immune surveillance in GBM compared to other tumors. As the GBM progresses, it can disrupt the BBB and induce inflammation, which leads to leakage and damage of peripheral blood vessels, resulting in inadequate oxygen delivery, and insufficient blood flow creates hypoxic regions within the tumor, which subsequently attract macrophages and further enhance tumor tumorigenicity [22].
Based on the molecular characteristics encompassing gene expression profiles, DNA methylation profiles, and transcription profiles in GBM, GBM can be classified into three distinct subtypes: mesenchymal, proneural, and classical, each marked by specific molecular features. The gene expression of the proneural subtype, including the receptor tyrosine kinase (RTK) I/LGm6 DNA methylation group, exhibiting molecular alterations such as cell cycle-dependent kinase 4 (CDK4) and platelet-derived growth factor receptor alpha (PDGFRA) amplification, predominates among younger adults. The gene expression of the classical subtype, including the RTK II DNA methylation group, is distinguished by frequent epidermal growth factor receptor (EGFR) amplification and deficiency of cyclin-dependent kinase inhibitor 2A/B (CDKN2A/B). The gene expression of the mesenchymal subtype is defined by the deficiency of neurofibromin 1 (NF1) and heightened infiltration of TAMs. While most GBM manifests these three molecular subtypes, the coexistence of multiple molecular phenotypes is commonplace, all of which are intricately linked to telomerase reverse transcriptase (TERT) promoter mutations [1, 23, 24]. Another classification method, leveraging single-cell sequencing technology, focuses on the sub-cellular subtyping of GBM. This approach categorizes internal tumor cells into distinct subclones, revealing the internal heterogeneity of GBM. The identified tumor cell subtypes include mesenchymal-like (MES-like), neural progenitor-like (NPC-like), oligodendrocyte progenitor-like (OPC-like), and astrocyte-like (AC-like) subclones. This refined classification offers a comprehensive insight into the diverse cellular composition within GBM [25]. Each subtype corresponds to a unique immunosuppressive microenvironment, with inherent heterogeneity within each subtype. The immunosuppressive processes in GBM primarily involve intricate crosstalk among genetic alterations, epigenetic changes, metabolite regulation, and various microenvironmental components. These influencing factors encompass glioma-associated macrophages/microglias (GAMs), MDSCs, and T cells. Signaling factors such as TNF-α [26], NF1 [24], and IL-33 [2] are employed, impacting pathways such as TGF-β/Smad and nuclear factor kappa-B (NF-κB) pathways [27, 28]. This intricate interplay with immune cells further fortifies the immunosuppressive microenvironment through CTLA-4, PD-1, and T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) among other targets [29,30,31,32,33]. Moreover, individuals with GBM frequently manifest systemic immunosuppression, characterized by the inhibition of activation of T cells through the IL-10-TGF-β pathway following DCs activation at the deep cervical lymph nodes [5]. This activation is triggered by tumor-associated antigens (TAAs) drained from the GBM. Additionally, peripheral components such as gut microbiota can undergo metabolic changes influenced by GBM, leading to the activation of more regulatory T cells (Tregs). These Tregs are then recruited to the GBM microenvironment, where they exert immunosuppressive effects [34]. Sometimes, the older age of onset [35] and glucocorticoids [36, 37] can also lead to systemic immunosuppression. In both the blood pool and bone marrow pool, chemokines secreted by GBM play a pivotal role in activating and recruiting MDSCs to enter the GBM microenvironment. Simultaneously, they can prohibit the activation of normal immune cells in the bone marrow pool, mediating immunosuppression [38, 39]. This process can be elucidated in more detail in subsequent sections. Notably, within the local microenvironment of GBM, the BBB undergoes modifications induced by GBM, rendering it selectively permeable for immune cells [40, 41]. This selective permeability allows TME to reject normal immune cells while facilitating the entry of immunosuppressive cells. The intricacies of immunosuppression within the GBM microenvironment will be expounded upon in the following sections. Figure 2 illustrates the systemic immune response in the presence of GBM.

Molecular mechanism of crosstalk between GBM and systemic immunity. GBM is the most common and lethal brain malignancy in adults. It not only leads to the reprogramming of local immunity in the brain but also affects peripheral immunity to some extent. The microenvironment of GBM is complex, and immune cells are heterogeneous and are mainly composed of MDSCs, microglia, astrocytes, Tregs, blood vessels, and the ECM. The secretion of numerous cytokines, chemokines, and metabolites by GBM can affect the systemic immune system through the blood, lymphatic vessels, and paracrine pathways. Similarly, these channels can also affect the occurrence and development of GBM. OPCs oligodendrocyte progenitor cells; AMPAR α-amino-3-hydroxy-5-methyl-4-isoxazole-propionica
The status of epigenetic mechanisms in glioblastoma regarding immune regulation
In GBM, immune attacks can instigate epigenetic changes in tumor cells, subsequently influencing their immune responsiveness. However, the impact of immune attacks varies among different tumor subtypes. These epigenetic alterations encompass not only histone modifications [42], chromatin remodeling [43], and DNA methylation [44], but also specific non-coding RNA molecules (such as miRNAs and lncRNAs) [45] and metabolites that exert post-transcriptional modifying effects. Current research suggests that in GBM, epigenetics pertains to the regulation of various pathways, including the Notch [46], Hedgehog [47], and WNT pathways [48].
In spontaneous GBM mouse models, activating colony-stimulating factor 1 receptor (CSF-1R) signaling can induce increased methylation in the interferon regulatory factor 8 (IRF8) promoter region. This methylation reduces GBM sensitivity to interferon-gamma (IFN-γ) and responsiveness to TAMs, ultimately promoting immune evasion [49]. The core area of GBM is characterized by extreme hypoxia, which induces the m6A demethylase alkB homolog 5 (ALKBH5). Inactivation of ALKBH5 significantly inhibits the recruitment of hypoxia-induced TAMs and immunosuppression. However, hypoxia-induced ALKBH5 also reduces m6A deposition in the lncRNA nuclear enriched abundant transcript 1 (NEAT1), promoting the repositioning of splicing factor proline and glutamine-rich (SFPQ) near the promoter of C-X-C motif chemokine ligand 8 (CXCL8). This leads to the re-expression of CXCL8/IL-8, partially restoring TAM recruitment and tumor progression [50]. Hence, this process is bidirectional, underscoring the complexity of epigenetic regulation in developing GBM and its role in intratumoral heterogeneity. In another context involving m6A-related epigenetic regulation, the YY1-CDK9 transcription complex increases the programmatic expression of m6A, subsequently downregulating MHC-related genes and interferon-related genes. Notably, the dataset in Cancer Genome Atlas (TCGA) about GBM reveals a correlation between the transcription complex and low CD8+ T cell infiltration. Targeting the YY1-CDK9 transcription complex can enhance GBM's responsiveness to PD-1 therapy [51].
Furthermore, lysine demethylase 6B (KDM6B) exhibits high expression in MDSCs within the GBM microenvironment. Specific knockdown of KDM6B in MDSCs enhances proinflammatory pathway activity and improves the prognosis of mice with GBM. KDM6B deficiency inhibits secretion of immunosuppressive mediators such as MAF BZIP transcription factor B (MAFB), suppressor of cytokine signaling 3 (SOCS3), and signal regulatory protein alpha (SIRPA), thereby enhancing the efficacy of anti-PD-1/programmed cell death 1 ligand 1 (PD-L1) therapy [52]. In humans, presence of X chromosome inactivation escape gene KDM6A [53] results in lower CD8+ T cell levels in male GBM microenvironments than in female GBM microenvironments [54]. Moreover, T cells in the male GBM microenvironment are more prone to exhaustion. Another transcription factor (TF), zinc finger protein 148 (ZNF148), promptly binds to pentraxin 3 (PTX3) promoter region and upregulates PTX3 expression. In GBM, downregulating the expression of ZNF148 could diminish PTX3 expression, consequently reducing the proliferation and migration of transformed DCs (t-DCs) and restraining the expression of costimulatory, thereby diminishing the tumor-promoting ability of t-DCs in vivo [55].
Regarding metabolic regulation, acetylation has emerged as a prevalent epigenetic modification in GBM. Fatty acids and acetate act as regulators of acetylation. Fatty acids undergo oxidation to generate acetyl-CoA, inducing the acetylation of NF-κB/RelA, which upregulates CD47 transcription, thereby enhancing the phagocytic resistance of GBM cells [56]. Acetate indirectly activates pyruvate dehydrogenase (PDH) by facilitating the conversion of pyruvate to acetyl-CoA, resulting in increased histone acetylation and modulating the stemness of glioblastoma stem cells (GSCs) [57]. Acetate salts inhibit the expression of histone deacetylase (HDAC), promote multiple miRNA expression, and hinder GBM cell proliferation, invasion, migration, and angiogenesis. Additionally, these acetate salt molecules regulate genes associated with mammalian targets of rapamycin complex 2 (mTORC2), thereby impeding GBM development [58]. At the same time, lactate is traditionally viewed as a metabolic byproduct of tumor metabolism. Recent research [59] highlights its role in enhancing chromatin accessibility and histone acetylation through aerobic metabolism and ATP-citrate lyase (ACLY) dependency. This protective mechanism shields malignant cells from death caused by nutrient deprivation [60]. Moreover, lactate accumulation induces the lactylation of histone lysine [59]. In GSCs with enhanced glycolysis, lactate induces the lactylation of H3K18, promoting the expression of the lncRNA LINC01127. This, in turn, activates the MAP4K4/JNK pathway, enabling GSCs to sustain self-renewal [61]. Palmitoylation, a post-translational modification (PTM) crucial for regulating protein transport, stability, and cellular localization, is catalyzed by palmitoyl transferases, such as Asp-His-His-Cys 9 (DHHC9). In GBM cells, DHHC9 palmitoylates glucose transporters 1 (GLUT1), enhancing its membrane localization and promoting glycolysis and tumor progression. Knocking out DHHC9 inhibits this process, offering potential improvements in patient outcomes [62].
In some specific cases, EGFR-chimeric antigen receptor T cell (CAR-T) therapy (EGFR-CAR-T) effectively prohibits the progress of GBM cells in vitro and of those derived from malignant cells and patient-derived xenografts in mice [63, 64]. However, mice quickly resist EGFR-CAR-T therapy, limiting its potential clinical application. Genomic and transcriptomic analyses of GBM cells co-cultured with EGFR-CAR-T reveal increased immunosuppressive gene activity and enhancer activity. Bromodomain-containing protein 4 (BRD4), another epigenetic factor acting on promoter and enhancer regions, is important for the activation of these immunosuppressive genes [65,66,67]. Inhibiting BRD4 with the inhibitor JQ1 disrupts the activation of these immunosuppressive genes. The treatment combining JQ1 and EGFR-CAR-T suppresses the metastasis and development of GBM cells, extending the survival time of mice [63]. The mutation of H3.3-G34R/V is common in diffuse midline gliomas (DMG) [1], whereas the mutation in G34R of pediatric high-grade gliomas (pHGGs) can lead to functional loss of DNA repair, resulting in genomic instability and the accumulation of extrachromosomal DNA. Leaked DNA can activate the cGAS/STING (cyclic GMP-AMP synthase/stimulator of interferon genes) pathway, inducing the release of immunostimulatory cytokines. Combination therapy involving DNA damage response inhibitors (DDRi) and RT in H3.3-G34R pHGG mice can significantly increase median survival [68]. Table 1 shows the epigenetic alterations associated with immune regulation in GBM [49,50,51,52, 54, 63, 66, 68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84].
Role of the transcriptome in the TME of glioblastoma
The transcriptome generally refers to the collection of all transcription products within cells under physiological conditions [85]. GBM is defined as a kind of tumor with great changes in the transcriptome that are dysregulated transcriptome. Current findings from multitranscriptomic analyses indicate that, in comparison to those in other tumors, infiltrating lymphocytes in GBM TME express various co-inhibitory immune checkpoints and demonstrate significant functional impairments, resembling a phenotype consistent with T cell exhaustion [86]. This exhaustion phenotype is characterized by the expression of HLA-DR+, TIM-3+, PD-1+, CD39+, and CD45RO+[87]. Through techniques such as spatial transcriptomics (ST) and single-cell RNA sequencing (scRNA-seq), it becomes evident that GBM cells could induce local environmental changes through signaling and structural alterations. These changes contribute to chemotherapy resistance and immune escape. Notably, the subtypes of GBM cells present in different microenvironment locations vary, and this situation may evolve due to species changes and tumor recurrence. The ability to observe and verify these changes at the single-cell level [28] explains why certain treatment strategies, effective in cell and animal models, may be less effective in patients. Moreover, the responsiveness of GBM to specific treatments may vary among patients and could be diminished by recurrence.
EZH2-92aa, encoded by the circular form of enhancer of zeste 2 (EZH2), overexpresses within GBM as well as contributing to the immune evasion of GSCs against NK cells [88]. Moreover, fibroleukin 2 (FGL2) exhibits heightened expression in GSCs and GBM cells. FGL2 suppresses CD103+ DC polarization induced by granulocyte–macrophage colony-stimulating factor (GM-CSF) by inhibiting NF-κB, p38, and signal transduction and transcription factor 1/5 (STAT1/5) activation. Low FGL2 and high GM-CSF expression correlate with CD8+ T cell infiltration and improve prognosis [89]. 67% of GBM samples highly expresse chondroitin sulfate proteoglycan 4 (CSPG4), and targeting CSPG4 by CAR-T effectively controls GBM growth in a mouse model [90]. Under normoxic conditions, GBM cells inhibit T cell proliferation by expressing indoleamine 2,3-dioxygenase-2 (IDO2). However, IDO2 is downregulated in GBM cells under hypoxic conditions, restoring T cell proliferation possibly through the reduction of kynurenine, a metabolite produced by GBM cells [91]. Moreover, GBM cells, especially those in the GBM mesenchymal subtype, highly express guanylate-binding protein 5 (GBP5). Increased GBP5 expression is positively related to poor outcomes in patients with GBM. High expression of GBP5 promotes the proliferation, migration, and invasion of GBM both in vitro and in vivo, while RNA interference-mediated silencing of GBP5 yields adverse consequences. Targeting GBP5 in GBM impedes the development of GBM and extends the mice's survival, and the Src/ERK1/2/MMP3 axis is crucial for GBP5-mediated malignant cell invasiveness [92].
STAT3 plays a crucial role in GBM development, contributing to early GSC formation and the mesenchymal transformation (MET) of GBM upon activation. As a key driver of stem cell transcription factors, STAT3 has become a significant target for GBM treatment. The STAT3 inhibitor BZA reduces the self-renewal capacity and expression of stemness markers in GSCs [93]. In the mesenchymal subtype or isocitrate dehydrogenase 1 (IDH1) wild-type (WT) subtype of GBM, elevated levels of herpes virus entry mediator (HVEM) have been observed using multiple omics technologies [94]. HVEM is implicated in various immune regulatory processes, including promoting Treg differentiation, inhibiting antigen processing, and presenting major histocompatibility complexes I (MHC I) molecules and αβT. Furthermore, the expression of PD-1, CTLA-4, TIM-3, V-domain Ig suppressor of T cell activation (VISTA), and lymphocyte activating 3 (LAG3) positively correlates with HVEM, suggesting its potential role in immune suppression within the GBM microenvironment [94, 95]. High levels of lysosomal-associated membrane protein 2A (LAMP2A) in GBM and the TME are associated with temozolomide (TMZ) resistance and tumor progression. Its elevated expression is associated with poor overall survival (OS) in patients with GBM. Highly expressed LAMP2A in GSCs facilitates their acquisition of stemness while decreasing the release of IFN-γ in the TME. Loss of LAMP2A weakens GSC-mediated tumorigenic activity [96].
Identifying various distributed genes in GBM establishes a valuable reference database for researchers, offering insights into potential therapeutic targets. Table 2 presents the current GBM genes, biological targets, and immune-related targets [17, 47, 56, 60, 69, 97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239,240]. So, characterizing the transcriptome of GBM has yielded profound insights into the highly variable transcriptomic features of GBM and its microenvironmental cell components. This has transformed our comprehension of GBM, enabling the prediction and customization of treatment strategies. Nevertheless, the functional roles of many gene changes in the GBM transcriptome remain enigmatic [241]. Therefore, the development of methods to predict GBM gene functions using multi-omics techniques and leveraging these predictions for potential targeted therapies represents an innovative predictive framework. This approach holds promise for expanding the repertoire of GBM targets and creating new opportunities for clinical translation.
One of the predominant methods for predicting targets based on the transcriptome involves utilizing databases, patient-derived samples for cell interaction and prognosis analysis, and scRNA-seq. Krishna et al. used scRNA-seq datasets from patient-derived samples [242] and identified that integrin subunit beta 2 (ITGB2) was highly enriched in immune and stromal environments, including T cells, fat cells, microglias, macrophages, and newly formed oligodendrocytes through scRNA-seq datasets from patient-derived samples. Unique genes within these cell populations include collagen type VI alpha 3 chain (COL6A3), TNF superfamily member 9 (TNFSF9), and serpin family E member 1 (SERPINE1) (microglia); thrombospondin 1 (THBS1, in newly formed oligodendrocytes); and integrin subunit alpha M (ITGAM) and THBS1 (OPC) in patients with stromal infiltration [243]. B7-H3 is upregulated in IDH1-WT gliomas within the immune checkpoint family, particularly in the mesenchymal subtype. Fusion gene analysis reveals strong positive correlations between B7-H3 and inducible T cell costimulator (ICOS), PD-1, TIM-3, LAG3, and IDO [244]. PTX3, another highly expressed protein in GBM, is also correlated with poorer survival in Zhang et al.'s list and is closely related to TIM-3, PD-1/PD-L1, and B7-H3 expression in the GBM TME [245]. According to the results of Gene Ontology (GO) analysis, Kaplan–Meier (K-M) survival analysis, and Pearson correlation analysis, CD163 expression is positively correlated with the malignancy of gliomas, especially in IDH1-WT GBM and mesenchymal subtypes. It is closely related to immune checkpoint markers (B7-H4, B7-H3, LAG3, TIM-3, and PD-1/PD-L1) and other macrophage markers arginase 1 (ARG1), TGF-β, IL-10, and IL-6 [246].
Recently, using single-cell sequencing results for classifying cell components in the GBM microenvironment and predicting patient prognosis and treatment responsiveness through immune scoring based on bioinformatics analysis has gained prominence [247]. Diverse classification results provide researchers and clinicians with a range of evaluation criteria to address the high heterogeneity of GBM treatment. In a study by Yang et al.[248], scoring small nucleolar RNA host genes (SNHGs) revealed that GBMs with high SNHG scores are connected with a poorer prognosis, a greater incidence of the mesenchymal subtype, and increased infiltration of immunosuppressive cells. Further analysis indicated that high SNHG scores correlate with a weakened reaction to anti-PD-1/PD-L1 immunotherapy. High SNHG scores were observed to be more sensitive to targeting EGFR or ERK-MAPK pathways in tumors. MyD88 is a critical adaptor protein in the Toll-like receptor (TLR)/MyD88/NF-κB pathway [249]. In GBM, especially in the mesenchymal subtype, MyD88 is most highly expressed and negatively correlated with PD-1 expression. Patients with high MyD88 expression exhibit an increased immune phenotype score (IPS) [250], and similar results are observed in subsets of PD-1+/CTLA-4− treatment and PD-1+/CTLA-4+ treatment [251]. The mRNA stemness index (mRNAsi) reflects the gene expression characteristics of cancer stem cells (CSCs) [252]. Moreover, TNF alpha-induced protein 8 like 2 (TNFAIP8L2) is an emerging immune checkpoint biomarker that may be a potential target for immunotherapy. Immune cell infiltration and stemness feature analysis showed a significant correlation between TNFAIP8L2 and the CSC index in GSCs, and high TNFAIP8L2 expression decreases macrophage and DC infiltration by promoting M2 macrophage and Treg approach [253]. The Tumor-Infiltrating Immune Cells-related lncRNA screening framework (TIIClnc), developed based on machine learning principles, can predict the response to immunotherapy by assessing immune cell infiltration levels. Moreover, TIIClnc positively relates to the expression of PD-1/PD-L1 and CD8 while providing better predictive accuracy [254]. Patients with a pathological diagnosis of GBM were exclusively considered. The results depicted in the heatmap also illustrate the heterogeneity of gene expression within GBM to a certain extent, showcasing differences in expression among different patients [255].
Indeed, while omics technologies offer a wealth of information for target prediction, the sheer volume of data can be overwhelming. It is essential to recognize that genes exhibiting differences in the transcriptome may experience altered expression in response to changes in the TME. A lack of consistency and the presence of numerous prediction scoring systems can impact the accuracy of clinical applications. Consequently, the validation of these prediction insights through multiomics technologies and fundamental experimental research becomes imperative. This ensures a full-scale comprehension of the function of genes and enhances the reliability of predictions made from transcriptomic variances in diverse contexts.
Metabolism regulates the immune response in glioblastoma
Based on existing studies on GBM, it has been demonstrated that metabolites play a crucial role in the onset and progression of GBM. Particularly, previous treatment approaches that categorize GBM based on IDH mutation status have shown promising outcomes for patients. Various types of metabolites serve as a double-edged sword in the pathogenesis of GBM. Therefore, this section will provide a brief overview of three key metabolites: glucose, fat, and proteins (amino acids). Metabolites implicated in distinct cellular processes and functions will be delineated separately in the subsequent discussion.
Classical glucose metabolism states in glioblastoma
The Warburg effect is a key metabolic aberration in cancer, including GBM [256]. The Warburg effect denotes the phenomenon wherein tumor cells predominantly depend on aerobic glycolysis for their metabolic needs in the presence of ample nutrients. This deviation from normal physiological processes assists tumor cells in acquiring a swift energy supply, facilitating their rapid proliferation and invasive capabilities [257]. There has been significant interest in the metabolic products of the glycolytic pathway, and therapeutic strategies have primarily targeted these products. However, recent research has indicated that, in addition to the glycolytic pathway, other metabolites, including fatty acids and amino acids, also play regulatory roles in the onset and progression of GBM through existing pathways [258].
In GBM, the influence of IDH1-mutant on epigenetics has gained recognition. D-2HG [259] is one of the earliest known metabolites, and its role in tumor cells is well understood. Recent findings indicate that D-2HG in the microenvironment of GBM can be absorbed by CD8+ T cells and target lactate dehydrogenase (LDH), reducing the NAD+/NADH ratio in CD8+ T cells and resulting in diminished cytotoxicity and impaired interferon-gamma signaling. These characteristics have been validated in clinical samples from IDH1-mutant glioma patients [260]. Another glycolytic metabolite, lactate, functions as an upstream regulator and can be modulated by a micropeptide called MP31, which is encoded in the 5' UTR region of protein tyrosine phosphatase (PTEN). MP31 binds to LDH in mitochondria, inhibiting the conversion of lactate to pyruvate, inducing lysosomal alkalization, inhibiting lysosomal function, and impeding the fusion of lysosomes with mitochondria [239]. Additionally, MP31 enhances GBM cell sensitivity to TMZ by inhibiting the protective mechanism of mitochondria [261].
Classical fat and amino acid metabolism states in glioblastoma
Fatty acid (FA) metabolism, primarily mediated by fatty acid oxidation (FAO), contributes to immune suppression in GBM [239]. Various FA transport proteins in Tregs are notably elevated in GBM [262]. Inhibiting FA transport or FAO processes, particularly through the FA transport protein CD36, can reduce Treg-mediated immune suppression, resulting in a significant survival benefit in tumor-bearing mice [263]. Additionally, DHHC9, a key transferase involved in S-acylation and lipidation [264], promotes GBM onset, development, and glycolysis by palmitoylating GLUT1. Elevated DHHC9 levels are connected with poor prognosis in GBM patients [62]. Amino acid metabolism, particularly tryptophan metabolism, regulated by aryl hydrocarbon receptor (AHR), influences the immunosuppressive microenvironment in GBM [265]. The tryptophan metabolite kynurenine promotes MDSCs infiltration by binding to AHR and acting as a transcription factor [266], resulting in decreased CD8+ T cell infiltration [267]. Kynurenine binding to AHR induces Treg differentiation and inhibits CD8+ T cell function in coculture with dendritic cells and naïve T cells [268]. Furthermore, kynurenine stimulates AHR in TAMs, promoting the expression levels of the chemokine receptor C–C motif chemokine receptor 2 (CCR2) and increasing MDSCs recruitment via the CCR2-CCL2 (C–C motif chemokine ligand 2) axis [121]. Consequently, kynurenine primarily modulates the functions of various immune cells through AHR signaling, inducing an immunosuppressive microenvironment and ultimately promoting GBM progression.
These findings underscore the intricate interplay of metabolic regulations in the functional reprogramming of GBM. The dynamic and complex nature of this interaction enhances our understanding of GBM's high heterogeneity and opens avenues for discovering new therapeutic targets. Indeed, it is essential to acknowledge that metabolites exert effects not only on tumor cells but also on normal tissues. Consequently, selecting appropriate metabolite targets to specifically target tumor cells while sparing normal cells is a critical consideration. This necessitates thorough deliberation to minimize potential off-target effects and maximize therapeutic efficacy.
GBM-TME crosstalk
TME of GBM encompasses elements from both the tumor niche and the tumor bioenvironment, exhibiting high dynamism and complexity. It comprises a diverse array of immune cells, primarily myeloid cells and microglias, along with blood vessels, extracellular matrix (ECM), and components of the CNS, including neurons and glial cells. This composition varies across different regions of the tumor [269, 270]. Notably, GSCs represent a prominent component with distinctive characteristics [271]. Recent ST and scRNA-seq analyses affirm the pervasive presence of GSCs [272], highlighting their status as a cellular functional state rather than a discrete cell cluster [273, 274]. GSCs exhibit a dynamic interplay with GBM cells, contributing to the development of therapeutic resistance. They secrete chemokines and pro-angiogenic factors that foster ECs proliferation and recruit immunosuppressive cells, particularly macrophages, forming immunosuppressive phenotypes [275,276,277]. Another critical feature is the GBM-associated vascular niche, which facilitates oxygen and nutrient supply to the highly vascularized tumor [278, 279]. Together with the BBB, it constitutes a protective physical microenvironment in GBM, influencing drug resistance, recurrence, and invasion [40, 41]. The collaborative actions of tumor cells, stromal cells, and proinflammatory cells act a pivotal role in formatting the new vessels in GBM, leading to vessel distortion or leakage. This phenomenon contributes to tumor cell growth, invasion, and the release of chemokines [280]. Another crucial set of microenvironmental components contributing to the formation of the microenvironment in GBM is the GBM-associated matrix microenvironment. This component encompasses GBM-associated stromal cells (GASCs), which exhibit similar phenotype and function to mesenchymal stem cells (MSCs) and cancer-associated fibroblasts (CAFs) [281]. GASCs may originate from the reverse differentiation from some brain cells (such as ECs, astrocytes, perivascular cells, or vascular smooth muscle cells) or bone marrow-derived MSCs [282]. GASCs play a component in promoting angiogenesis and tumor development within the GBM microenvironment [283], showing a negative correlation with GBM prognosis [284]. Another matrix microenvironment component is the ECM, which undergoes dynamic changes and manifests spatial heterogeneity during GBM development [285], thereby facilitating GBM invasion and influencing the plasticity of local microenvironment components [286]. Recent reports have highlighted the interaction between GBM and neurons [287]. GBM growth driven by neuronal activity can be regulated by some factors such as synaptic adhesion molecule neuroligin-3, brain-derived neurotrophic factor (BDNF) [288] or through neurotransmitter receptors like glutamatergic excitatory synapses (interacting with astrocytes) [287, 289, 290], dopaminergic receptors (D2 and D4 subtypes) [291], and γ-aminobutyric acid (GABA) receptors [292, 293]. In summary, TME is a pivotal participant and target for therapy in tumor development. A comprehensive understanding of the diverse components involved in cells and molecules in the GBM microenvironment and their crosstalk is essential for developing a more effective treatment strategy. Within the immune components, this fraction significantly contributes to the distinctive immunosuppressive milieu of GBM. Therefore, a brief description is given above, and a detailed exploration of the immune components will be provided in the subsequent discussion.
GBM is susceptible to high infiltration by immune cells in the TME [294]. Predominant among these immune populations are myeloid cells, encompassing TAMs (this section refers to GAMs), MDSCs, and neutrophils. Additionally, nonimmune-associated cells, such as neurons, assume a crucial component in GBM progression [295]. There is mounting evidence suggesting that these stromal cells infiltrating into TME foster the growth of GBM and orchestrate the immunosuppressive microenvironment, conferring resistance to immune therapies, including immune checkpoint inhibitors (ICIs) [296]. Following infiltration into the TME, tumor cells manipulate these stromal cells, promoting tumor progression, suppressing anti-tumor immunity, and instigating resistance to immunotherapy [297, 298]. In summary, these discoveries significantly enhance our comprehension of the intricate interplay between cancer cells and stromal cells in the GBM microenvironment (Fig. 3).

Interactions between GBM and cellular components of the TME. The TME, which includes cells and the ECM, is essential for the initiation and progression of GBM. The formation of a GBM immunosuppressive microenvironment mainly depends on the connection between GBM cells and multifarious stromal cells through different metabolites, cytokines, and signaling pathways, forming a huge hybrid immunosuppressive network. The mechanism of immunosuppression is extremely intricate, so eliminating tumors by a single targeted therapy is incredibly difficult. GAM Glioma-associated macrophage/microglia; AHR Aryl hydrocarbon receptor
Crosstalk between glioblastoma and myeloid lineage cells
The interaction between GAMs and GBM represents a prevalent phenomenon within the TME, given that GAMs occupy the largest proportion of all cells [299]. GAMs within GBM comprise brain-resident microglia and bone marrow-derived macrophages, originating from embryonic yolk sac and bone marrow progenitor cells, respectively [300]. Morphologically, microglia are characterized as highly branched quiescent cells with a larger size, whereas macrophages exhibit superior migratory ability, reduced branching, and smaller size [301]. The distribution of these cell types varies dynamically among different tumors. For instance, in GBM, microglia are more infiltrated and widespread, while the core of metastatic brain tumors lacks microglia and is instead populated by macrophages [294, 302]. scRNA-seq analysis provides further insights into this heterogeneity. Moreover, the composition ratio of GAM differs between primary GBM (pGBM) and recurrent GBM (rGBM), with microglia predominant in pGBM and macrophages more prevalent in rGBM [303]. Genetic mutations, such as the classical IDH1-mutant, can alter this ratio, resulting in an abundance of microglia and fewer macrophages in the early stages of IDH1-mutant GBM compared to IDH-WT tumors. However, during tumor progression, macrophage infiltration increases in the IDH-mutant mouse model compared with the IDH-WT mouse model [304]. Additionally, the functional characterization of GAM is a rapidly advancing field. The conventional classification of pro-inflammatory M1 and anti-inflammatory M2 proves overly simplistic for the intricate GBM microenvironment. Current classifications, informed by scRNA-seq, reveal that GAM may exist in a continuous or poorly differentiated state, co-expressing genes characteristic of both M1 and M2 phenotypes, exhibiting high plasticity with dynamic changes [298, 305].
GAMs can induce the transformation of GBM cells into a MES-like status. Oncostatin M (OSM), which originates from GAMs, activates STAT3 through its interaction with oncostatin M receptor (OSMR) or leukemia inhibitory factor (LIF) receptor (LIFR) subunit alpha and with GP130 on GBM cells, prompting the transformation of GBM cells into mesenchymal subtypes in vitro and in vivo [306, 307]. In recent years, in GBM, the significance of circadian locomotor output cycles kaput (CLOCK) transcriptomics has been acknowledged [308]. Elevated CLOCK expression in GBM facilitates the recruitment of GAMs, shaping an immunosuppressive TME through the up-regulation of olfactomedin-like 3 (OLFML3) [69]. CLOCK regulates the legumain (LGMN) signal by forming a complex with brain and muscle ARNT-like 1 (BMAL1), promoting immunosuppressive microglias infiltration and resulting in a poor prognosis. Inhibiting the CLOCK-OLFML3-HIF-1α-LGMN-CD162 axis demonstrates the potential to reduce microglial infiltration, enhance the infiltration, activation, and cytotoxicity of CD8+ T cells, and exhibit synergistic effects with anti-PD-1 therapy [309]. GAMs strategically position themselves close to GBM-associated ECs and participate in vascular endothelial growth factor (VEGF)-induced GAMs polarization [310, 311]. Within the microenvironment of GBM, ECs have been identified as a primary source of IL-6. Both IL-6 and CSF-1 induce elevated expression of ARG1 and selective activation of GAMs [312], mediated by hypoxia-inducible factor 2α (HIF-2α) transcription, which induced by peroxisome proliferator-activated receptor γ (PPARγ) [313]. So, targeting EC-derived IL-6 is an effective and potential treatment in GBM [310]. M2 macrophages exhibit high expression of integrin αvβ5 (ITGαvβ5), which supports their phenotypic maintenance and contributes to the immunosuppressive microenvironment. Osteopontin (OPN), secreted by GBM cells, acts as the primary ligand for ITGαvβ5. Deleting OPN reduces M2 macrophage infiltration, enhances GBM cell sensitivity to CD8+ T cell cytotoxicity, and improves survival in mouse models [147]. ITGαvβ3 drives M2 macrophage polarization and abnormal angiogenesis in GBM through the Src-PI3K-YAP signaling pathway [314]. Slit guidance ligand 2 (SLIT2) activates and promotes the chemoattraction and polarization of GAMs via the phosphoinositide-3 kinase-γ (PI3K-γ) pathway, mediating GBM immune suppression and abnormal angiogenesis [100]. EZH2 inhibition results in increased M1 marker expression and reduced M2 markers in microglia, decreasing the number of CD206+ PB MON-derived macrophages and enhancing microglial phagocytic ability [73]. TIM-3, a common co-inhibitory immune checkpoint in GBM, regulates GBM cell malignancy and induces macrophage migration and polarization toward an anti-inflammatory or pro-tumor phenotype through the IL-6 pathway [33]. In GBM metabolism associated with GAMs, the metabolite lactate from GBM can regulate GAM polarization [59], and exposure to lactate promotes an up-regulation in M2 phenotype markers and decreasing inducible nitric oxide synthase (iNOS) levels, inducing GBM immune escape. High levels of lactate in the GBM TME upregulate the sonic hedgehog (SHH) signaling pathway and facilitate the insulin-like growth factor-binding protein 6 (IGFBP6) expression in microglia, influencing microglial polarization [315]. C-X-C motif chemokine receptor 4 (CXCR4) signaling promotes MET within GBM and shortens survival. DExH-box helicase 9 (DHX9) can enhance macrophage infiltration and polarize them into M2 GAMs in GBM [316]. Silencing DHX9 reduces CSF-1 expression, restoring the inhibitory effect of targeting transcription factor 12 (TCF12) on malignant progression and TAM infiltration in GBM [317]. Overexpression of bradykinin receptor 1 (B1R) and IL-1β promotes vascular cell adhesion molecule 1 (VCAM-1) and cell adhesion molecules intercellular adhesion molecule 1 (ICAM-1) expression, enhancing migratory and adhesive abilities of GBM cells [318]. B1R also contributes to the pro-tumor chemokines and cytokines secretion, like CCL5, IL-6, CXCL11, and IL-8, in GBM, promoting MON infiltration into the TME [319].
In addition to interactions with GAMs, GBM engages with various immune cells, including neutrophils, DCs, NK cells, and MDSCs. Neutrophil infiltration in GBM begins early and persists throughout tumor progression. In vivo experiments suggest that early-infiltrating neutrophils may initially inhibit tumor progression, but this function is lost as tumors progress, leading to a pro-tumor functional phenotype, particularly in tumor protein P53 (TP53)-induced GBM [320]. Ligands of galectin 9 (LGALS9) can bind to TIM-3 receptors on DCs in the cerebrospinal fluid (CSF), inhibiting antigen recognition and presentation. This results in anti-tumor immune response failure mediated through T cells. Blocking exosomal LGALS9 allows sustained tumor antigen presentation and durable anti-tumor immune activity in GBM [321]. Annexin A1 (ANXA1) is implicated in DC maturation and is related to worse outcomes in patients with GBM [322]. Silencing cytokine-inducible SH2 (CIS) containing protein in NK cells increases production levels of IFN-γ and TNF-α, enhancing cancer cells apoptosis mediated by allogeneic NK cells and improving overall survival in mice with GBM [323]. GBM cells can secrete LDH5, which induces natural-killer group 2 member D (NKG2D) ligands upregulation, leading to NKG2D downregulation in NK cells [196]. Leukocyte immunoglobulin-like receptor subfamily B member 2 (LILRB2) promotes MDSCs formation and expansion, prohibiting CD8+ T cells from normal function through exosomes, creating an immunosuppressive TME [324]. CXCL1/2/3 secreted by GBM cells and CXCR2 expressed by polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) create an axis that regulates PMN-MDSCs output from the bone marrow, resulting in a significant up-regulation in PMN-MDSCs in GBM-draining lymph nodes and spleen [122, 325]. Further details about these interactions are available in Table 2 for the involved cell types.
Interaction between glioblastoma cells and T cells
Exhaustion of CD8+ T cells and Tregs infiltration act as key components in the immunosuppressive TME within GBM [326]. Transcriptome changes, epigenetic alterations, and the inhibition of certain stromal cells in GBM often contribute to functional impairments in CD8+ T cells, leading to a decline in their anti-tumor capabilities. Within the tumor immunosuppressive microenvironment of GBM, T cell function is adversely affected by cytokines and metabolites and is directly inhibited by tumor cells, Tregs, GAMs, and MDSCs. These inhibitory effects are primarily mediated through the surface receptors of these immune cells [327].
scRNA-seq results have highlighted that S100A4 is important in regulating Tregs and bone marrow-derived cells in GBM. Increased expression of S100A4 in Treg cells is related to worse outcomes in patients with GBM [328]. GPNMB is predominantly expressed on macrophages in GBM. Macrophages with high levels of GPNMB induce MET in tumor cells and inhibit T-cell activation, fostering an immunosuppressive microenvironment. Targeting glycoprotein nonmetastatic melanoma protein B (GPNMB) could enhance tumor sensitivity to molecularly targeted therapies and create a more favorable environment for immune responses from T cells [329]. Moreover, the immune checkpoint TIM-3 has been identified as an inhibitor of microglia and CD8+ T cell function, playing a critical role in GBM cell proliferation and tumorigenesis. Targeting TIM-3 upregulates the presence of NK cells, DCs, CD8+ T cells, and microglias characterized by proliferative and active phenotypes. An upregulation of the secretion of immune-stimulating factors such as IFN-γ, CLL2, IL-1β, CCL5, and CXCL10 into the TME of GBM accompanies this. Ultimately, TIM-3 blockade could induce profound pro-inflammatory changes in the TME, inducing T-cell activation and generating immune memory, thereby inhibiting the recurrence of tumor [32]. The overexpression of common immune checkpoint molecules in the GBM microenvironment can also impact T cell function (Table 2 and Fig. 3).
Interaction between glioblastoma cells and neurons
Recent research has underscored the growing recognition of the nervous system as a crucial regulator of cancer, as it plays a role in various stages, from tumorigenesis to malignant growth and metastatic spread. In the context of GBM, this relationship is bidirectional. Not only does the nervous system regulate GBM progression, but GBM also can remodel and hijack the nervous system, affecting its structure and function [330]. Interactions between the nervous system and GBM extend beyond the local TME, influencing systemic processes. Neurons and glial cells, which support the CNS, impact the function and infiltration of immune cells by releasing paracrine factors. This intricate interplay between the nervous system and GBM adds an extra aspect of complicity to understanding the TME and its impact on cancer progression [331].
The relationship between sensory stimuli and the development or progression of brain tumors, including GBM, is an intriguing area of research [332]. Reports suggest that sensory signals, such as visual or olfactory stimulation, may influence the development and behavior of brain tumors, potentially through signaling pathways such as mammalian target of rapamycin (mTOR) signal [333]. For instance, visual stimulation has been linked to the development of optic nerve gliomas in mice with specific gene mutations. Similarly, olfactory stimulation has been associated with promoting GBM, and this effect has been attributed to mTOR signal. The mTOR signal is a crucial regulator of cellular processes, including cell growth and proliferation. The mTOR signal in the context of GBM may also impact the immune microenvironment. Activation of mTOR signal promotes the immunosuppressive microglial formation by regulating the activity of the transcription factors STAT3 and NF-κB. This, in turn, hinders the T-cell proliferation and immune response, allowing GBM cells to escape from the anti-tumor immunity as well as facilitating the growth of tumors in experimental models [334]. Susan et al. [335] explored the potential therapeutic implications of targeting mTOR in the context of GBM. Inhibition of the mTOR pathway, such as rapamycin (RAPA), has been investigated to reinduce anti-tumor immune activity. Using RAPA in a training method related to taste-immune association learning demonstrated the ability to reinstate a proinflammatory, anti-tumor TME. This approach has shown promising outcomes in animal models, suggesting that modulating mTOR signal is a potential method to enhance anti-tumor immunity in GBM.
The intricate interplay of GBM and its microenvironment adds another layer of complexity to understanding and treating this aggressive brain tumor. The high degree of intratumor heterogeneity in GBM, coupled with rapid lineage switching, is rooted in its permissive epigenetic and transcriptomic landscape. One fascinating aspect is GBM's ability to mimic the transcriptomic state of normal neuronal populations, a strategy employed to evade immune attacks by imitating the developmental trajectory of normal neurons [25, 336, 337]. Efforts to limit GBM plasticity within these neural-like pathways are advanced to enhance the validity in targeting tumor heterogeneity [338]. Despite genetic mutations, the transcriptional signature of GBM cells tends to converge on similar neural-like states. However, significant differences exist between the core and edge of GBM, highlighting distinct biological properties. Notably, immune infiltration-related injury programs dominate this phenomenon, leading to the generation of hyperproliferative injured neural progenitor cells (iNPCs). iNPCs constitute a substantial proportion of resting GBM cells and can be activated by interferon within the T cell niche [339]. The microenvironment at the immuno-cold edge of the tumor appears to influence GBM's trajectory, resembling normal neuronal development. This environment prompts the differentiation of tumor cells into aggressive AC-like cells [340]. These findings underscore the crucial role of local components within the TME in shaping the fate of GBM cells. Understanding and potentially manipulating these interactions could offer new avenues for therapeutic interventions aimed at targeting specific cellular states and enhancing treatment outcomes in GBM patients.
The complex interactions among CNS, GBM, and the immune system highlight the complex nature of this disease. The regulatory crosstalk between these systems influences the delicate balance between pro-tumor inflammation and anticancer immunity. Understanding these interactions necessitates an interdisciplinary approach, bringing together expertise from neuroscience, developmental biology, immunology, and cancer biology. Collaboration across these diverse fields is crucial for unraveling the complexities of GBM and developing targeted therapeutic strategies. Insights gained from this interdisciplinary collaboration could pave the way for innovative approaches that disrupt the regulatory pathways exploited by GBM. By leveraging knowledge from multiple disciplines, researchers and clinicians may identify new therapeutic targets, enhance treatment efficacy, and ultimately improve outcomes for individuals affected by GBM.
The role of MDSCs in the initiation and development of glioblastoma
In this section, we focus exclusively on MDSCs, as their relatively limited representation belies their essential component in initiating and progressing the comprehensive immunosuppressive microenvironment in GBM. This significance extends beyond their direct immunosuppressive functions, encompassing intricate interactions with other stromal cells. Specifically, MDSCs are involved in priming or modulating the functions of additional immunosuppressive cells while concurrently impeding the functions of normal immune components.
MDSCs constitute the significant role in the immunosuppressive TME of GBM and cancer cells' response to immunity. In the GBM microenvironment, GAM emerges as the predominant immunosuppressive component, accounting for up to 50% of all living cells in GBM [341]. However, it is noteworthy that MDSCs (accounting for 4%-8% of all CD45+ cells in GBM) [342] primarily mediate the formation of GAMs, and their inhibitory effect surpasses that of GAMs and Tregs. Within the TME, enhanced infiltration of B cells, cytotoxic T cells (CTLs), T cells, and NK cells correlates with a more favorable prognosis. Conversely, heightened infiltration of MDSCs is associated with a poorer prognosis [343,344,345]. Under pathological conditions, MDSCs function as immunosuppressive regulatory cells originating from the bone marrow [346]. For instance, following infection or in the context of tumors, they accumulate in the PB and tissues [344, 345, 347], a phenomenon not observed under physiological conditions [342]. This accumulation signifies the pathological activation of neutrophils and MONs. MDSCs exert their immunosuppressive effects by inhibiting the release of inflammatory factors and activating immunosuppressive cells, thereby mediating the suppression of the body's anti-tumor immunity [348]. They can be categorized into two types: monocytic myeloid-derived suppressor cells (M-MDSCs) and PMN-MDSCs. These subtypes exhibit distinct phenotypes with unique gene expression profiles yet share certain similarities. PMN-MDSCs, resembling the morphology of neutrophils, predominantly induce long-term immune tolerance. Conversely, M-MDSCs, resembling MONs, tend to polarize into GAMs, playing a rapid immunosuppressive role thereafter [348]. MDSCs are recognized as pivotal components implicated in the immune evasion of tumors. Escalation during the induction and activation of MDSCs can enhance tumor immunosuppression, thereby contributing to tumor progression, encompassing angiogenesis, invasion, and metastasis [349]. Therefore, in the following section, we will elaborate on how MDSCs mediate these processes in GBM and the possible mechanisms.
Regulatory mechanisms of MDSC origin
MDSCs predominantly originate from the bone marrow, although their presence is not limited to this site, and they can extend to peripheral lymphoid organs like the liver, spleen, and other tissues [350]. The prevailing theory supporting MDSC genesis is the double signal theory. This theory involves the orchestration of signals through GM-CSF, granulocyte colony-stimulating factor (G-CSF), and CSF-1. These signals activate transcription factors such as STAT3, IRF8, and CCAAT/enhancer binding protein β (C/EBPβ), thereby promoting proliferation within the BM. Pathologically, a downregulation of IRF8 signaling occurs, resulting in immature myeloid cells (IMCs) accumulating in spleen and bone marrow. These IMCs subsequently differentiate into PMN-MDSCs or M-MDSCs upon peripheral activation. Under physiological conditions, PMN-MDSCs or M-MDSCs can further differentiate into DCs, polymorphonuclear neutrophils (PMNs), and MONs [351,352,353,354,355]. Notably, this differentiation lasts longer than normal and exhibits specific expression profiles and characteristics that support tissue angiogenesis and immune cell suppression under pathological conditions [356]. Physiologically, various signals, including endoplasmic reticulum stress (ERS), VEGF, IL-6, macrophage colony-stimulating factor (M-CSF), IL-3, IFN-γ, thrombopoietin (TPO), GM-CSF, receptor tyrosine kinase (c-Kit) ligands, lipopolysaccharide (LPS), FMS-like tyrosine kinase 3 ligands (FLT3L), and IL-1β, with GM-CSF upregulate and mediate the differentiation of MDSCs [299, 346]. A pivotal role in the generation of PMN-MDSCs is ascribed to the downregulation of IRF8 in hematopoietic progenitor cells, as it induces PMN-MDSC generation and participates in STAT3/STAT5-mediated anti-tumor processes (Fig. 4) [299, 357,358,359,360,361,362].

Mechanisms of MDSC generation, recruitment, and activation. HSCs in the BM proliferate and differentiate into IMCs under the stimulation of various signaling pathways, such as the IRF8 signaling pathway. Subsequently, IMCs are recruited and differentiated into MDSCs, including M-MDSCs and PMN-MDSCs, via a variety of chemokines in the PB. Then, MDSCs are activated by a variety of cellular mediators released by tumor cells, thereby exerting immunosuppressive effects and maintaining an immunosuppressive microenvironment. c-Kit Receptor tyrosine kinase; C/EBPβ CCAAT/enhancer binding protein β; CSF-1/M-CSF Macrophage colony-stimulating factor-1; e-MDSCs early-stage myeloid-derived suppressor cells; FATP2 Fatty acid transport protein 2; FCN1 Ficolin 1; FLT3L Fms-related tyrosine kinase 3 ligand; FN1 Fibronectin 1; G-CSF Granulocyte colony-stimulating factor; GM-CSF Granulocyte–macrophage colony-stimulating factor; HSC Hematopoietic stem cell; IL Interleukin; IMC Immature myeloid cells; IRF Interferon regulatory factor; LPS Lipopolysaccharide; M-MDSCs Monocytic myeloid-derived suppressor cells; miRNA Micro RNA; PMN-MDSCs Polymorphonuclear myeloid-derived suppressor cells; Rb Retinoblastoma; RORC1 Receptor-related orphan receptor γ; SOCS3 Suppressor of cytokine signaling 3; STAT Signal transduction and transcription factor; TPO Thrombopoietin; VEGF Vascular endothelial growth factor
In non-IRF8-regulated cell populations, granulocyte-monocyte progenitors (MLPGs) can undergo differentiation into PMN-MDSCs through the downregulation of the retinoblastoma gene (Rb) [363]. The crucial transcription factors C/EBPα and C/EBPβ, generated by bone marrow cells, play opposing roles in MDSC generation, where C/EBPβ promotes MDSC generation and C/EBPα inhibits MDSC generation [364], C/EBPβ regulates MDSC generation by controlling GM-CSF and G-CSF, and it also modulates the expression of iNOS, NADPH oxidase 2 (NOX2), and ARG1, influencing the essential functions of MDSCs, particularly M-MDSCs. Additionally, retinoic acid receptor-related orphan receptor γ (RORC1) enhances the expression of C/EBP-β through the SOCS3 and B cell lymphoma 3 (Bcl3), promoting MDSC generation. Furthermore, C/EBPβ can facilitate the differentiation of MDSCs into TAMs [365]. CD33+ MDSC-like cells and CD14+ PMN-MDSCs promote the aggregation and differentiation of PMN-MDSCs in peripheral blood mononuclear cells (PBMCs) [347, 359, 366].
MDSCs infiltrate the TME under the influence of cytokines or some signaling molecules, promoting the growth and progression of tumors through suppressing the normal anti-tumor immunity [367]. M-CSF, GM-CSF, G-CSF, and other cytokines are important in maintaining metabolic reprogramming, proliferation, and epigenetic modifications in MDSCs. Soluble cell factors, including IL-6, TNF, IL-4, IL-1 family cytokines, and IL-13 [367], not only facilitate the metastasis and invasion of cancer cells but also control MDSCs accumulating and activating in the TME [368, 369]. Consequently, a strong correlation has been established between the aggregation of MDSCs and the invasion of tumor cells in the TME. Among the earliest transcription factors implicated in MDSC generation is the STAT family, including STAT3, STAT5, and STAT6. Notably, STAT3 and its downstream pathways, involving the upregulation of c-Myc, Bcl-xL, Cyclin D, S100A8/A9, and NOX2, along with cooperation with cytokines like IL-6, GM-CSF, and G-CSF, are implicated in MDSC accumulation and immunosuppressive mechanisms [347, 370,371,372]. Specifically, S100A8/A9 can directly bind to membrane receptors, promoting MDSC migration. Moreover, STAT3 is able to bind with the promoter of ARG1, participating in immunosuppression [373].
Recently, microRNAs (miRNAs) have garnered increasing attention in MDSC development; these molecules play pivotal roles in regulating MDSC proliferation, maturation, and immunosuppressive functions. For instance, miR155-5p, which is induced by TGF-β, inhibits phosphatidylinositol-3,4,5-trisphosphate 5-phosphatase 1 (SHIP-1) and promotes STAT3 activation, thereby supporting MDSC proliferation and differentiation [374]. Similarly, miR-30a-5p facilitates the activation of MDSC by targeting SOCS3 downstream of the JAK/STAT3 pathway, encouraging the production of IL-10, ARG1, and reactive oxygen species (ROS) [375]. Furthermore, miR-494 downregulates the expression of PTEN, promotes the PI3K/Akt signal, and modulates the accumulation of MDSC [376]. Additionally, miR-21-5p, miR-223-3p, and others have been implicated in MDSC development [347, 377].
The classification of MDSCs in glioblastoma
The general classification of MDSCs
As previously mentioned, MDSCs are broadly categorized as two main clusters: PMN-MDSCs and M-MDSCs. PMN-MDSCs emerge early in the PB or peripheral lymphoid organs of individuals with tumors, potentially representing an early stage of MDSC development. Notably, they possess migratory capabilities and constitute over 75% of MDSCs, playing a crucial role in the expansion of MDSC populations and their migration to and residence within tumor tissues [352, 361]. Conversely, TAMs can differentiate from M-MDSCs within the microenvironment and exhibit more pronounced immunosuppressive effects than PMN-MDSCs [359, 378].
Early-stage MDSCs (e-MDSCs) represent a newly recognized third subtype of suppressive MDSCs. These cells have been identified as bone marrow cells lacking markers for mature MONs or neutrophils in both the PB and the TME. Classified as immature MDSCs due to the absence of mature lineage markers, it remains to be established whether they serve as precursors for other MDSC subsets [379]. In vitro experiments have indicated that e-MDSCs may exhibit the lowest suppressive capacity in the TME [380], demonstrating the weakest ability to restrain T cell proliferation. Unlike other MDSCs, the accumulation of e-MDSCs does not appear to be correlated with overall survival in cancer patients [380, 381]. Ongoing research explores markers associated with eosinophilic granulocytes, such as the high expression of CD123, as potential identifiers for e-MDSCs [382]. Recent findings from scRNA-seq suggest that markers such as CD14, CD15, and CD16 may also be useful for identifying e-MDSCs [383]. For GBM, e-MDSC is a unique subset of MDSCs present in it, which is hardly observed in other grades of glioma [383].
The molecular classification of MDSC
In the early stages of molecular studies in mice, CD11b and Gr1 were utilized for labeling MDSCs, with different Ly6G and Ly6C expressions used to classify PMN-MDSCs and M-MDSCs: Ly6G+/Ly6Clo/CD11b+ for PMN-MDSCs and Ly6G−/Ly6Chi/CD11b+ for M-MDSCs [343]. Currently, CD49d is considered a specific marker for M-MDSCs [384], while lectin-like oxidized low-density lipoprotein receptor 1 (LOX1) is becoming a novel specific marker for PMN-MDSCs [299]. In humans, PMN-MDSCs are markered with CD14−/CD11b+/CD66b+/CD15+, while M-MDSCs are markered with HLA−/DR−/low/CD11b+/CD15−/CD14+ (Fig. 4) [343]. In the context of GBM, vascular noninflammatory molecule 2 (VNN2+) may serve as a unique marker for MDSCs [385].
In recent scRNA-seq studies of GBM, the role of e-MDSCs has gained gradual recognition. e-MDSCs interact with GSCs and contribute significantly to the transformation of tumors into more malignant mesenchymal types, correlating with a poor prognosis [383]. scRNA-seq has identified two distinct types of GBM: e-MDSCs and M-MDSCs. e-MDSCs primarily participate in the immunosuppression process in GBM. Simultaneously, M-MDSCs primarily function as recruits, attracting PMN-MDSCs, TAMs, and Tregs in GBM. Additionally, M-MDSCs are capable of transforming into each other. Under the influence of the extracellular matrix and inflammatory factors (FN1, FLNA, VCAN, CD44, FCN1, CXCL2, S100, CXCL3, etc.), e-MDSCs can transform into M-MDSCs. This transformation leads to an increase in glycolysis-related genes and antioxidant and stress processes associated genes downregulating [383].
The mechanism of MDSC recruitment in glioblastoma
MDSCs in tumors play a key component in the development of tumors, and tumors can secrete specific chemokines to facilitate the MDSCs’ recruitment. Chemokines such as CXCR4-CXCL12, CXCR2-CXCL5/8, and CCR2-CCL2 [386], with CXCR2-CXCL5, are particularly significant in primarily regulating M-MDSCs’ recruitment [387]. In human colorectal cancer, the expression of CCL2 increases with cancer progression, and CCL2 deficiency has been associated with reduced infiltration of intratumoral MDSCs and smaller tumor sizes in spontaneous mouse models of colon cancer [388]. Similarly, the upregulation of the expression of CCL15 in colorectal cancer can enhance M-MDSCs’ recruitment [389]. PMN-MDSCs’ recruitment is mediated mainly by chemokines such as CXCR1-CXCL8, CXCR2-CXCL8, CCR5-CCL5, CXCL6, and CXCL12 [388, 390, 391]. Additionally, CCL2, CCL3, and hypoxia have been identified as factors contributing to the recruitment of PMN-MDSCs. IL-8 is also considered one of the inducers of MDSC mobilization [367]. In brain metastasis, CXCL10 emerges as a crucial mediator that establishes the premetastatic niche and contributes to immune suppression in brain tumors [392].
Observations from PB and intratumoral studies in glioma patients reveal a notable proliferation of PMN-MDSCs and M-MDSCs in patients with GBM compared to that in healthy individuals’ PB. GBMs are among the tumors exhibiting the highest levels of MDSCs in PB [22]. Within the PB in patients with GBM, PMN-MDSCs emerge as the dominant subset, with M-MDSCs constituting almost the entirety of MDSC subpopulations [393]. In high-grade gliomas (HGGs) with IDH-mutant, intratumoral studies indicate that PMN-MDSCs are the predominant subset [394]. Moreover, the increased percentage of PMN-MDSCs within the tumor may suggest BBB disruption [395], highlighting the heterogeneity of MDSCs and the TME in GBM. Elevated MDSC levels in the PB and increased infiltration of MDSCs in GBM are indicative of a poorer prognosis [342, 396], with the degree of M-MDSC infiltration correlating with glioma grade [396, 397]. Radiomics development has further confirmed the robust correlation between high MDSC infiltration and poor prognosis in gliomas [398]. Notably, in patients with rGBM, the MDSC population in the TME does not significantly differ from that observed before treatment. This indicates that the persistence of MDSCs is essential in the rGBM [399, 400].
In the GBM TME, numerous constituents contribute to tumor progression, particularly influencing MDSCs. For instance, GBM cells can secrete IL-8, resulting in the upregulation of CCR2 [401]. CCR2 has dual functions, not only facilitating the recruitment of MDSCs but also activating MDSCs within the TME of GBM [396, 402]. GSCs are proficient in secreting substantial amounts of macrophage migration inhibitory factor (MIF) [112], thereby augmenting the production of ARG1 through a CXCR2-dependent pathway, consequently impeding immune function [403]. Notably, while inhibiting MIF does not directly impede tumor progression, it diminishes the infiltration of MDSCs, underscoring its specificity in targeting MDSCs in GBM [403]. Furthermore, GBM cells secrete galectin-1, eliciting stimulation of tumor angiogenesis. Recent investigations have demonstrated that inhibiting galectin-1 significantly diminishes MDSCs’ amount in the microenvironment and improves the mice with GBM in prognosis [404], a phenomenon potentially linked to the regulation of LGALS1 [405]. The histone methyltransferase G9a is pivotal in the GSC-mediated tumor immune microenvironment (TIME). It upregulates the Notch pathway by binding to the H3K9me2 modification on the promoter of F-box and WD-40 domain protein 7 (Fbxw7), which can suppress Notch signal, thereby fostering the recruitment of MDSCs in GBM [406]. FGL2 in GBM exhibits a positive correlation with the increase of MDSCs, notwithstanding its lack of association with the conventional upregulation of PD-1 or CD39 [407]. Notably, activation of the Notch pathway in GBM induces upregulation of CCL2, thereby promoting the recruitment of MDSCs [408]. In addition to the IDO mechanism, the upregulation of complement factor H (CFH) or FH-like protein 1 (FHL-1) can similarly facilitate the infiltration of intratumoral MDSCs in GBM [136].
LOX1 is recognized as a distinctive marker for PMN-MDSCs, playing a vital component in suppressing T-cell proliferation within GBM and contributing to early recurrence and progression [409]. Recent investigations specifically focusing on GBM with epidermal growth factor receptor variant III (EGFRvIII) mutations have uncovered an increasing abundance of PMN-MDSCs, correlating with resistance to PD-1 and CTLA-4 inhibitors. Subsequent studies have elucidated the regulatory axis involving CXCL1/2/3 and the CXCR2 receptor expressed by PMN-MDSCs, influencing PMN-MDSCs’ production and recruitment in bone marrow [325]. These findings underscore an intricate interplay among genetic mutations, TME heterogeneity, and resistance to ICIs in GBM. In contrast to PMN-MDSCs, M-MDSCs in GBM manifest heightened expression of integrin β1 and dipeptidyl peptidase-4 (DPP-4). Inhibiting DPP-4 has been shown to diminish tumor migration mediated by the pERK signaling pathway, while targeting integrin β1 eradicates the immunosuppressive phenotype of MDSCs. Notably, the concurrent inhibition of these targets has been shown to enhance survival outcomes in mice bearing GBM [72].
Hence, MDSCs recruited to tumors are influenced by many factors that vary across different cancers, resulting in high variability. Consequently, therapeutic interventions aimed at blocking MDSC recruitment to tumors by targeting a specific chemokine or cytokine may have limited impact. Nonetheless, a potentially more effective approach could involve targeting specific chemokine receptors, as certain receptors can interact with multiple chemokines.
Immunosuppressive effect of MDSC in glioblastoma
The signaling molecular involved in immunosuppression in MDSC
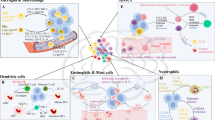
MDSCs exhibit weaker immunosuppressive abilities than normal bone marrow-derived suppressor cells, yet they exert a prolonged inhibitory effect, leading to sustained immune suppression. The immunosuppressive mechanisms of MDSCs encompass various pathways, including Toll-like receptor signaling [410], certain proinflammatory cytokines (like IL-13, IL-4, PGE2, IFN-γ, and IL-1β) [411], and exosome secretion [412]. Activation of NF-κB signal facilitates iNOS2 expression [358], an essential player inhibiting M-MDSCs’ function. Additionally, ERS is another crucial factor activated by tumor hypoxia, low pH, and proinflammatory cytokines. This activation leads to increased expression of ERS-related proteins (CHOP, LOX1, DR5), IL-6, C/EBPβ, and pSTAT3, further enhancing MDSC recognition and targeting of immune cells in the TME [357, 413, 414]. Notably, ERS has distinct impacts on PMN-MDSCs and M-MDSCs, with inositol-requiring enzyme 1α (IRE1α) and human-activating transcription factor 6 (ATF6) playing critical components in the immunosuppressive activity of PMN-MDSCs. In contrast, M-MDSCs are less dependent on ERS and rely predominantly on IL-6-mediated immunosuppression [358]. Different cytokines exert diverse effects on MDSCs [343]. TNF-α and IFN-γ can promote the formation of a proinflammatory phenotype in the GBM microenvironment by reducing MDSC numbers [415]. This process is activated by JAK/STAT signal, inducing IRF1 downregulation, promoting the secretion of PD-L1, and altering the immunoescape microenvironment [416]. However, the upregulated expression of FAT atypical cadherin 1 (FAT1) enhances IL-1β, IL-10, PD-L1, IL-6, and HIF-1α secretion through AP-1 signal. This promotes the function of MDSCs and establishes a TIME within GBM [417]. Table 3 [72, 112, 122, 324, 354, 388, 401, 406, 409, 418,419,420,421,422,423,424,425,426,427,428,429,430,431,432,433,434,435,436,437,438,439,440] and Fig. 5 provide a comprehensive summary of the main immunosuppressive pathways targeting the TME [441].

Immunosuppressive role of MDSC in the TME. Once infiltrated into the tumor, MDSCs can promote tumor progression and exert immunosuppressive effects in a variety of ways. Among them, the most important is the release of multiple cytokines to directly inhibit the activity of CTLs and activate and enhance the function of Tregs, directly inhibiting anti-tumor immunity to create a tumor immunosuppression microenvironment. In addition, it can also inhibit the antigen presentation function of DCs and the tumor-killing function of NK cells and enhance autoimmune suppression through the exosome pathway. Arg1 Arginase 1; COX2 Cyclooxygenase 2; CTL Cytotoxic T cells; DC Dendritic cells; IDO Indoleamine 2,3-dioxygenase 1; IL Interleukin; MDSC Myeloid-derived suppressor cell; miRNA microRNA; MPO Myeloperoxidase; NK cell Natural killer cell; PGE2 Prostaglandin E2; PNT Peroxynitrite; ROS Reactive oxygen species; SLC7A11 Solute carrier family 7 member 11; TGF Transforming growth factor; Treg T regulatory cells; VEGF Vascular endothelial growth factor
Exosomes, which are double-membrane extracellular vesicles (EVs), play a pivotal role in regulating MDSC function by secreting proteins and miRNAs [442]. Their inhibitory effect on the myeloid cell differentiation is facilitated by TGF-β secretion [443]. Moreover, EVs induce the accumulation of ARG1, cyclooxygenase 2 (COX2), IL-6, and VEGF, thereby enhancing the function of MDSCs [320]. By utilizing heat shock protein 72 (HSP72), EVs activate the TLR2/MyD88 pathway, synergizing with IL-6 to improve the immune inhibitory function of MDSCs [412]. Furthermore, EVs interact with IL-10 and IL-16, participating in microenvironmental regulation, promoting angiogenesis, and activating STAT1/3 to enhance the immunosuppressive function of MDSCs [444, 445]. In GBM, EVs can initiate MDSC differentiation under low-oxygen conditions through retinoic acid related-orphan receptor α (RORα) and PTEN via miR-10a and miR-21, respectively, to promote immune suppression [442]. Another class of miRNAs, miR-92a and miR-29a, can activate MDSCs by targeting high mobility group protein B1 (HMGB1) and cAMP-dependent protein kinase regulatory type I-α (Prkar1a) [446]. Additionally, miR-155, miR-27b, miR-1260a, miR-126-3p, miR-494-3p, miR-320, and miR-494-3p may also be associated with the activation of MDSCs [447, 448].
In GBM, the secretion of EVs involves a unique mechanism in which these vesicles interact with heparan sulfate proteoglycans (HSPGs) and MDSCs, inducing the transformation of MDSCs [441]. This process can be inhibited by heparin, leading to a reduction in the number of MDSCs in GBM [449]. EVs derived from GBM cells can reprogram normal MONs, promoting their differentiation into MDSCs and subsequent suppression of T cell function [449]. Again, heparin can inhibit this reprogramming process and restore T cell function. A recently discovered factor, LILRB2, has been found to propagate between GBM cells through vesicles, inducing the formation, expansion, and shaping of the TIME by promoting MDSCs [324]. EVs secreted under hypoxic conditions have shown an enhanced ability to induce or facilitate the generation and transformation of MDSCs, leading to increased infiltration into the TME and subsequent suppression of immune cell function [450, 451]. Additionally, MDSCs can interact with tumor-associated B cells or regulatory B cell (Breg) cells through EVs, transmitting PD-L1 to regulate B cell function and consequently inhibiting the typical immune function of CD8+ T cells, thereby suppressing immune function in GBM [452]. The inhibitory effect of EVs on T cell function is also indirectly mediated through MDSCs [453].
Metabolism regulation of immunosuppression through the MDSC in glioblastoma
Immunosuppressive factors such as nitric oxide (NO), ROS, and peroxynitrite (PNT) play crucial roles in the immunosuppressive functional mechanism of MDSCs [454]. NO is a key molecule mediating immunosuppression in MDSCs, especially M-MDSCs, and is primarily metabolized by iNOS in the TME, induced by IL-1β, IFN-γ, and TNF-α, which is included in Th1 cytokines, participating in the inhibition of the IL-2-associated receptor [455]. In PMN-MDSCs, the ROS pathway plays a pivotal role, and ROS is mainly produced by NOX2. Phosphorylation of STAT3 can directly regulate NOX3 and increase ROS production [456]. PMN-MDSCs can generate a substantial amount of ROS by mediating TGF-β, GM-CSF, IL-6, and IL-10, inducing T-cell death [457]. Reactive nitrogen species (RNS) also play a complementary role [458]. PNT serves as another mediator, with MDSCs nitrating amino acids through PNT to form TCR-CD8 nitrate complexes [459]. This interferes with the antigen–antibody recognition process, inhibiting antigen-specific immune activation. PNT can also reduce the efficiency of MHC I binding with peptides on the membrane of cancer cells, nitrating CCL2, STAT1, and Lymphocyte cell-specific protein-tyrosine kinase (LCK) to inhibit anti-tumor immunity [356]. Nitration of CCL2 cannot induce T cell migration but does not affect the migration of MDSCs, thereby exacerbating the TIME to some extent.
In the context of oxidation, polyunsaturated fatty acids (PUFAs) play a crucial role in free radical-mediated peroxidation. The accumulation of oxidized lipids, such as prostaglandin E2 (PGE2), fatty acid transport protein 2 (FATP2), and arachidonic acid, also contributes to MDSC-mediated immune suppression through oxidative stress [460]. PGE2 can engage in NF-κB signaling to mediate immune suppression; it can activate the Ras/Erk pathway, elevate TGF-β levels, and mediate NK cell inhibition [461]. Recent studies have indicated that lipid peroxidation combined with ferroptosis plays a specific role in the immunosuppression mediated by PMN-MDSCs. Ferroptosis induces the production of lipid peroxidation products in PMN-MDSCs, inhibiting the normal function of T cells [462, 463]. In GBM, MDSCs can take up and utilize lactate produced by tumor cells. Estrogen is also crucial in the immunosuppressive mechanism of MDSCs in GBM [464]. The forkhead box protein P3 (FOXP3) promoter region contains estrogen receptors, and estrogen can inhibit its expression, thereby suppressing the function of Tregs. Progesterone can enhance this process, while androgens can increase FOXP3 expression, inhibiting the immunosuppressive function of MDSCs [464].
Other critical mechanisms include the upregulation of ARG1 via Th2-mediated signaling to deplete arginine [465], the upregulation of solute carrier family 7 members 11 (SLC7A11) to limit cysteine utilization [466], the increased activity of IDO to decrease local tryptophan levels [467], and the increased activity of IDO to decrease local tryptophan levels [466, 468]. PMN-MDSCs can also suppress the antigen-presenting capacity of DCs by upregulating myeloperoxidase (MPO) expression. Significant improvements in the cross-presentation of TAAs by DCs were observed in tumor-bearing mice lacking MDSCs or MPO [469, 470]. Furthermore, MPO can catalyze the generation of peroxidized lipids via PMN-MDSCs, contributing to immune suppression [469]. In addition, PI3K-γ has been shown to contribute to the upregulation of iNOS and ARG1 in MDSCs to mediate immunosuppression [471]. PMN-MDSCs can also facilitate tumor angiogenesis by releasing proangiogenic cytokines like basic fibroblast growth factor (bFGF) and VEGF, facilitate metastasis of tumor by releasing matrix metalloproteinases, and contribute to the progression of epithelial-to-mesenchymal transition (EMT) [472].
MDSCs can produce immunosuppressive factors like IL-10 and TGF-β, inducing Treg activation and affecting NK cell function [473]. PMN-MDSCs can directly inhibit NK cell activity by upregulating PD-L1. Most studies suggest that MDSC-mediated immunosuppression of T cell function in lymphoid organs or PB via the ROS pathway requires closer intercellular contact, as the ROS pathway is sensitive, and only closer intercellular contact allows ROS to act quickly for maximum efficiency [474]. However, not all of the above mechanisms operate synchronously, and the specific mechanism depends on the subtype of MDSCs produced in various cancers. The proportion of PMN-MDSCs to M-MDSCs is also crucial for immune suppression, as they have different immunosuppressive mechanisms. PMN-MDSCs are more inclined to induce immunosuppression through PGE2, ROS, ARG1, and PNT, while M-MDSCs rely more on IL-10, TGF-β, PD-L1, and NO [353, 475]. It is noteworthy that male mice have more M-MDSCs, while female mice have more PMN-MDSCs in PB [430]. Therefore, the ROS pathway in PMN-MDSCs requires closer intercellular contact, while M-MDSCs rely on producing large quantities of NO, ARG1, and other immunosuppressive cytokines for immune suppression. The half-life of these molecules is much longer than that of ROS, so M-MDSCs do not need closer attachment with T cells. Therefore, M-MDSCs can effectively inhibit nonspecific responses of T cells, and their suppressive activity is greater than that of PMN-MDSCs on a per-cell basis [476,477,478]. However, compared to peripheral MDSCs, intratumoral MDSCs exhibit stronger suppressive activity [479, 480]. Different TMEs can explain the distinct ratio of PMN-MDSCs to M-MDSCs or changes in MDSCs function in various tissues.
There is a higher infiltration of PMN-MDSCs in IDH-mutant GBM compared to IDH-WT. However, while M-MDSCs infiltrate less, their immunosuppressive effect is more pronounced in GBM. In addition to the previously mentioned inhibitory mechanisms, hypoxia-inducible heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) promotes exosome packaging miRNA [341, 481]. MDSCs can take up exosomes, activating MDSCs through dual-specificity phosphatase-3 (DUSP3)/ERK signal and inhibiting T cells through PD-L1 through a HIF-1α-dependent pathway [353]. Current research suggests that the NF-κB-related pathway is crucial in mediating TIME development in GBM and determining the anti-inflammatory or proinflammatory phenotype of MDSCs [482]. The NF-κB pathway, along with the JAK pathway, is associated with the anti-inflammatory pathways linked to MDSCs [482]. It can increase IDO levels through the STAT3 pathway, thereby enhancing the significant immunosuppressive function of MDSCs [482, 483]. The use of NF-κB inhibitors in combination with standard GBM treatment regimens, such as TMZ, can enhance anti-tumor immunity in GBM mouse models [482].
Heterogeneity of MDSC regulated by the TME
Expression profile of MDSC in different tumors
MDSCs exhibit distinct gene expression profiles and characteristics depending on their infiltration into different organs. Recent studies have analyzed individual subtypes of MDSCs, and the results indicate that the TME may enhance the function of MDSCs by altering their properties. PMN-MDSCs exhibit higher generation of inflammatory cytokines and activation of downstream targets in the NF-κB signaling pathway [353, 484], including IL-6, M-CSF, IFN-γ, ERS regulatory factors, and mitogen-activated protein kinase (MAPK) signal [353]. While M-MDSCs upregulate other factors, like IL-6, TGF-β, and PI3K [480]. MDSCs within prostate or lung cancer have higher expression levels of ARG1, ARG2, NOS2, NOS3, and S100A9 than splenic MDSCs, with ARG1 being the highest. This effect is associated with the significantly enhanced inhibitory activity of MDSCs in the TME [485,486,487,488]. As for myeloma, NF-κB pathway-related genes, IRF1, COX2/PTGS2, CSF-1, IL-4R, STAT1, STAT3, STAT6, and IL-8 is high expression, promoting MDSC maturation and infiltration, thereby enhancing the TIME [489]. HIF-1α plays a crucial component in differentiating M-MDSCs into TAMs [479]. It facilitates the immune inhibitory activity of MDSCs by upregulating iNOS and ARG1 and acting in conjunction with PD-L1 [479]. HIF-1α also regulates glycolysis in MDSCs [490]. Under hypoxic conditions, the tyrosine phosphatase activity of CD45 increases in M-MDSCs, selectively reducing the activity of STAT3 and promoting the transformation of MDSCs into TAMs [491]. The upregulation of sialylation of CD45 protein dimers induces increased expression of the CD45 phosphatase. Thus, treatment with sialidase can eliminate the impact of hypoxia on the excitation and differentiation of STAT3 in MDSCs.
Immunosuppressive function of TME-driven MDSC
The TME serves as a critical component in the activation and immunosuppressive function of MDSCs, and emerging evidence suggests that hypoxic conditions within the tumor, particularly through the HIF-1α-associated pathway, play a significant component in this process. As mentioned earlier, MDSCs can hinder the priming of nonspecific antigen-T cells in hypoxic environments [479], thereby reshaping the TME. HIF-1α promotes TAMs differentiating from some MDSCs, inhibiting anti-tumor immunity by downregulating STAT3. In a lung metastasis model, MDSCs differentiate into fibroblasts with the participation of Kruppel-like factor 4 (KLF4) and ferroptosis suppressor protein 1 (Fsp1), contributing to the establishment of the lung metastatic TIME [492]. Moreover, the process of MDSC differentiation into TAMs may involve the regulation of ARG1 and iNOS. MDSCs lacking HIF-1α cannot differentiate into TAMs but acquire the characteristics of DCs [493]. HIF-1α binds to the proximal promoter of PD-1/PD-L1, increasing PD-L1 expression in MDSC membranes and leading to more potent immunosuppressive activity, especially in M-MDSCs [481, 494,495,496]. In addition, M-MDSCs can be regulated by various factors to differentiate into macrophages. In a breast tumor model, TLR7/8 agonists induce splenic MDSCs to differentiate into macrophages [497]. In an ovarian tumor model, thrombin stimulation can cause peritoneal MONs to differentiate into TAMs [498]. High expression of IL-6 and LIF in ovarian cancer ascites promotes the differentiation of MONs into TAMs [499]. Furthermore, in the spleen, M-MDSCs can differentiate into DCs upon STAT3 inhibition. In vitro, MDSCs can differentiate into Tregs under the induction of IL-10 and IFN-γ [343]. However, the transformation between MDSCs and TAMs has not been observed in GBM.
Variations in glycolysis and oxidative phosphorylation in tumors significantly influence MDSCs’ function in immunosuppression. To sustain the pathologically rapid proliferation in cancer cells, most cancer cells predominantly utilize aerobic glycolysis, which is known as the Warburg effect. In mice, the augmentation of glycolysis is concomitant with the increased activity of ARG1 in MDSCs. The resultant activation of AMP-activated protein kinase (AMPK) enhances ATP synthesis, maintaining the energy supply for MDSCs [500]. Simultaneously, tumor-associated MDSCs elevate FA uptake and engage in FAO, a metabolic shift controlled by lactate and hypoxia. However, the specific regulatory mechanisms of this process and its potential implications for targeted therapy remain to be precisely elucidated. MDSCs’ heightened activity in tumor immunosuppression is closely related to the increased FAO-related gene expression. This effect can be mitigated by FAO inhibitors [480]. Spleen-derived MDSCs restrain immune reactions by antigens in T cells through the ROS-dependent pathway. Similarly, tumor-derived MDSCs exhibit more potent antigen-specific suppression activity, primarily suppressing responses to anti-CD3/28 stimulation through the production of NO and secretion of ARG1 [479].
Certainly, GBM exhibits dynamic changes, and it is imperative to scrutinize the interactions among immune components from the perspective of spatiotemporal dynamic evolution. In recent years, the fusion of scRNA-seq with lineage tracing has facilitated researchers in gaining insights into the dynamic evolution within the GBM TME. As previously discussed, the early phases of GBM development are characterized by pro-inflammatory microglia and innate immunity [395]. However, these microglia are swiftly modified by tumor cells to foster tumor growth [501]. Simultaneously, bone marrow-derived MONs are recruited in the initial stages, expediting disease progression [502]. Conversely, the later stages of GBM predominantly consist of anti-inflammatory macrophages and MDSCs [270]. Recent studies, however, reveal that this macrophage population is more akin to microglia in terms of single-cell typing [395]. e-MDSC components in GBM, known as e-MDSC, may evolve into M-MDSC during GBM development, engaging in interactions with GSCs to sustain GSC growth and facilitate GBM infiltration into the pseudopalisading region [383]. Lineage tracing results further indicate that the early stages of GBM prompt the urgent mobilization of bone marrow to generate MDSCs [395]. Neutrophils are observed to infiltrate the early stages of mesenchymal subtypes GBM [320], initially exerting a tumor-suppressive role through their cytotoxic and immuno-activating activities [503]. However, they transition to a pro-tumor phenotype during tumor development, expediting tumor growth. Similar to PMN-MDSCs, neutrophils are present only in the early and late stages of GBM, a process potentially associated with BBB disruption [395]. Regarding T cells, they exhibit a "rejection" effect in GBM TME, resulting in minimal internal effector T cell infiltration [38]. Only in the early stages of GBM development do CD8+ T cells exhibit normal function; however, due to insufficient stimulation, they enter a non-responsive state. T cells are likely to elicit a response, but subsequently, GBM antigens inhibit T cell activity [504]. Consequently, most T cells comprise immunosuppressive Tregs, persistently circulating throughout GBM development [505]. For B cells, their recruitment to the GBM microenvironment occurs early on, exerting inhibitory effects. Furthermore, MDSCs undergo conversion to Bregs mediated by PD-L1, intensifying inhibitory effects [452].
Current treatment strategies and progress of glioblastoma
The conventional treatment paradigm for GBM involves gross total resection (GTR) whenever feasible, followed by adjuvant RT and chemotherapy, typically utilizing TMZ [1]. The STUPP therapy (postoperative RT combined with TMZ) proposed by Stupp et al. in 2005 was previously considered the gold standard for GBM treatment, and it is still a kind of chief treatment in most GBM cases today [144]. This approach, established in an era with limited genetic mutation testing, has demonstrated effectiveness. Despite the emergence of alternative treatments, it remains the primary therapeutic strategy for GBM in cases where specific target sites are not well defined. According to diagnosed tumor position and magnitude, patients commonly receive tumor resection first, following the combination of chemotherapy and RT, incorporating emerging therapies as deemed appropriate. Clinical studies have consistently indicated that aggressive surgical tumor resection correlates with favorable outcomes for GBM patients [327]. However, owing to the diverse locations of brain tumor growth, the surgical approach and prognosis can vary. GTR may not be achievable for all GBMs, especially those in functional areas or proximity to the brainstem, where subtotal resection (STR) might be the chosen course. Given the recurrence tendencies of GBM and the limitations of surgical resection, reliance solely on conventional RT and chemotherapy often proves inadequate. Consequently, various innovative treatment approaches have recently been developed for GBM. Figure 6 provides an overview of the existing treatment strategies for GBM.

Existing therapeutic strategies against GBM. Currently, there are various therapeutic strategies for GBM, but single-targeted therapy has poor efficacy, and combining multiple treatments is necessary to achieve therapeutic efficacy. The current view is that the initial treatment consists of surgery, RT, and chemotherapy, followed by a variety of other targeted therapies, including immunotherapy, tumor-related vaccine therapy, virus-killing therapy, engineering-based adjuvant therapy, and TTFields. CAR chimeric antigen receptor; BiTe bispecific T-cell engager; DC dendritic cell; ADC antibody–drug conjugate; TTField tumor treatment field
ICI therapy in glioblastoma
ICIs represent an extensively researched class of immunotherapy drugs for GBM, demonstrating efficacy in clinical trials across various malignancies [28]. Prominent targets in ICI therapy, like PD-L1 and CTLA-4, have exhibited promising outcomes in numerous tumors [506]. Data from multiple omics studies and clinical samples underscore elevated PD-L1 expression in GBM, positioning it as a potential and promising immunotherapeutic target [507]. Preclinical findings suggest that anti-PD-1/PD-L1 therapy can shift macrophage polarization from M2 to M1, transforming the immunosuppressive microenvironment into a pro-inflammatory state and ultimately prolonging survival in GBM-afflicted mice [508]. In the GBM mouse model, CTLA-4 blockage can recover CD4+ T-cell proliferation, producing stronger anti-tumor ability, while T cells are conferred resistance to Treg suppression in tumors to significantly prolong the survival of mice without causing experimental allergic encephalomyelitis (EAE) [509]. LAG3, also known as CD223, is a marker of exhaustion in T cells expressed on various T-cell surfaces and significantly reduces their ability to produce IFN-γ [510], which is expressed in tumor-associated perivascular lymphocytes and tumor-infiltrating lymphocytes (TILs) in human GBM [511]. Preclinical models have shown that early blocking of LAG3 significantly promotes prognosis in mice with GBM and is highly effective in eradicating tumors along with anti-PD-1/PD-L1 therapy. T-cell immune receptor with Ig and ITIM domains (TIGIT) is another nonclassical checkpoint expressed in various immune cells, like activated T cells, Tregs, and NK cells [512]. Its high expression has been shown to have an overall inhibitory phenotype in various tumor models, which is associated with reduced production of tumor-killing related cytokines and poor survival. Combined anti-TIGIT with anti-PD-1/PD-L1 significantly improved the survival in GBM mouse models compared with only anti-PD-1/PD-L1 therapy, which was attributed to enhancing the function of T cell and downregulating PMN-MDSCs and DCs amount [235, 513]. TIM-3, a membrane protein, is selectively expressed in immune cells, which acts as an immune checkpoint to regulate innate and adaptive immunity [514]. Studies have shown that it is one of the most up-regulated co-inhibitory immune checkpoints and is closely related to the poor prognosis of GBM [31]. Blocking TIM-3 not only inhibited its induction of macrophage migration and transition to a pro-tumor phenotype but also inhibited the tumorigenicity of GBM in vivo, thereby extending mouse survival. Furthermore, TIM3’s expression upregulates in cancer cells, microglias, and macrophages within TME in diffuse intrinsic pontine glioma (DIPG) patients. Blocking TIM-3 can directly inhibit tumor growth and strengthen CD8+ T-cell and microglia's function, resulting in durable anti-tumor immune memory, thereby eliminating tumors and preventing their recurrence [32]. Despite these encouraging preclinical results, clinical trials involving PD-1, CTLA-4, and other immunotherapies for GBM have, regrettably, not yielded substantial success. Even though combinations of ICIs with various adjuvant therapies have shown promise in preclinical models, translating these results into successful clinical outcomes remains a significant challenge [515,516,517]. Ongoing clinical trials investigating immunotherapy in GBM are outlined in Table 4.
Presently, the latest preclinical trials involve combining ICIs with other treatment strategies to achieve effective progress in terms of survival benefits. Notably, the simultaneous blockade of PD-1, VEGF, and angiopoietin 2 (Ang-2/ANGPT2) has shown significant promise in prolonging the survival time of GBM mice. This triple therapy demonstrated improvements in the number of CTLs and reduced the infiltration of MDSCs and Tregs. Transcriptome analysis of the GBM microvasculature indicated that triple therapy could promote tumor vascular normalization, potentially limiting or preventing cancer progression and metastasis [515]. Despite these promising preclinical results, translating such findings into successful clinical outcomes has proven challenging. Clinical trials involving ICIs in combination therapy have been disappointing, partly due to the BBB, which hinders these agents from reaching effective therapeutic concentrations within the intracranial space [518]. Consequently, researchers are exploring small molecule immunotargeted drugs, particularly immunomodulatory cytokines, as a research hotspot in immunomodulatory therapy. Immunomodulatory cytokines like TNF-α and IFN-α can traverse the BBB and have been demonstrated effective at reversing GBM-induced immunosuppression. Therapeutic regimens employing IFN-α and TNF-α to counteract the immunosuppressive microenvironment of GBM have shown promise in preclinical models and early clinical trials [519,520,521]. IFN-α can facilitate the differentiation of DCs, strengthen NK-cell, T-cell, and macrophage’s anti-tumor ability, as well as increase TAA expression. Additionally, IFN-α has exhibited the ability to prohibit tumor angiogenesis through disrupting ECs growth and promoting the synthesis of angiosuppressive chemokines such as CXCL1, CXCL9, and CXCL10 [522]. TNF-α has also been demonstrated to induce DCs’ maturation and enhance the infiltration of T cells within GBM mice models [523].
ICI therapy represents a prominent and promising field in cancer treatment and has demonstrated benefits in various tumors. However, there are no ICIs for GBM that get permission from the Food and Drug Administration (FDA), although a few are in clinical trials. ICIs remain ineffective against GBM as monotherapy, indicating there are limitations and deficiencies in our current preclinical model. Current preclinical models have been established by orthotopic injection of murine glioma cell, patient-derived xenograft model, or genetically engineered mouse model, which cannot recapitulate the complexity and heterogeneity of the patient’s GBM microenvironment [524]. Therefore, mouse models for evaluating immunotherapies in preclinical settings must be carefully considered. In addition, there is a BBB in the brain, which strictly regulates the barrier between the CNS and the PB, allowing small-molecule, lipid-soluble drugs to be passively diffused across, but water-soluble drugs and large-molecule to be largely inaccessible since tightknit connectivity networks [525]. Thus, many drugs, such as monoclonal antibodies, have poor anti-tumor effects due to the insufficient delivery of the BBB. It is necessary to consider improving the delivery system to increase drug delivery to cancer. However, with GBM progression, the integrity of the BBB is gradually lost, followed by the increase of tight junction permeability [526]. Its disruption allows for the delivery of drugs, which can recruit immune cells from the peripheral; however, it strengthens tumorigenicity through facilitating pro-tumorigenic-cell infiltration, like immunosuppressive macrophages [527]. In addition, the BBB is kept perfectly in some areas of the tumor [279]. Thus, systemic treatment of GBM has to conquer these difficult limits to become valid. In addition, ICI can lead to treatment-related toxicity. The expression levels of CTLA-4 and PD-1 strike a subtle balance in self-immunotolerance and autoimmunity [528, 529]. The direct toxicity of anti-CTLA-4 and anti-PD-1/PD-L1 is little, and their vital toxicity is an autoimmune disease known as delayed immune-related side effects (irSEs), which can influence all organs, especially skin, kidney, endocrine system, and gastrointestinal tract [530]. It is well known that the unique heterogeneity of GBM leads to its resistance to most treatments. It has a unique TME consisting of 20% to 40% immune cells, mostly from bone marrow, with various proportions in bone marrow-derived circulating macrophages and tissue-resident microglia [531]. The MON-derived macrophage and lymphocyte infiltration are higher in IDH-WT GBM. However, the immune pool in IDH-mutant GBM is almost microglia [294]. Currently, the standard therapy for adult GBM is RT and TMZ chemotherapy, followed by maintenance TMZ chemotherapy after surgical resection [10]. However, in preclinical models and GBM patients, systemic chemotherapy, including TMZ, has an inherent immunosuppressive effect, which allows the already minimal number of T cells in TME to rapidly deplete or develop tolerance to tumor antigens, leading to a possible failure of immunotherapy to promote TILs effector function [532]. Since there is little T cell infiltration in GBM, neoadjuvant immunotherapy provides drug therapy before chemotherapy, RT, and surgical resection can help address complications associated with its immunosuppressive environment [30]. It has been shown that PD-1 blockade for neoadjuvant therapy leads to the upregulation of T cells and IFN genes within the tumor and the reduction of the cell cycle in rGBM, thereby promoting anti-tumor responses [533]. Therefore, combination therapy and neoadjuvant therapy are necessary to address the unique immune microenvironment of GBM, such as multi-factor immunosuppressive TME and heterogeneity in cancer. Additionally, TMZ can reduce the number of anti-inflammatory MDSCs, although their number significantly increases at the late stage of the tumor, which is the focus of current ICI therapy [294, 395]. In GBM, the origin and function of MDSCs also vary depending on the gender of the patient [430]. Notably, therapies targeting MDSCs will be discussed later, showcasing significant benefits in improving the immunosuppressive microenvironment of GBM.
Molecular-based therapy in glioblastoma
Targeted therapy in cancer treatment focuses on addressing proteins that regulate the growth, division, and spread of tumor cells while minimizing the impact on normal cells. This approach aligns with the principles of precision medicine, tailoring treatments based on the specific characteristics of the individual and their cancer [534]. As our understanding of the genetic and protein changes underlying tumors deepens, researchers can design treatments targeting these aberrations. The two main targeted therapies are small-molecule drugs and monoclonal antibodies. Monoclonal antibodies, or therapeutic antibodies, are laboratory-produced proteins designed to bind to specific targets on tumor cells. They can mark cancer cells, making them more visible to the immune system for detection and destruction. Some monoclonal antibodies directly inhibit tumor cell growth or trigger self-destructive mechanisms in these cells. Additionally, certain antibodies are engineered to carry toxins that can selectively destroy tumor cells. Small-molecule drugs, compact enough to traverse the BBB, can bind to specific targets on tumor cells, impeding their growth or inducing cell death. This makes them particularly relevant for brain cancers such as GBM. In addition to targeting tumor proto-oncogenes or mutated genes, emerging targeted therapies encompass tumor epigenetics and metabolism. This diversification allows for a more comprehensive and personalized approach to cancer treatment. Table 5 provides an overview of ongoing clinical trials focused on targeted therapies for GBM.
Extensive transcriptomic and proteomic analyses have identified numerous potential therapeutic targets in GBM, with a particular emphasis on angiogenesis as a pivotal process in GBM initiation and progression. Noteworthy interventions targeting VEGF or EGFRvIII, such as bevacizumab and cetuximab, have been extensively investigated in clinical monotherapy, showcasing variable efficacy. VEGF, a key angiogenic factor and regulator of the innate immune response, significantly influences GBM pathology [535,536,537]. Elevated VEGF levels contribute to a threefold increase in tumor volume, marked vascular architecture remodeling, and an approximately 50% reduction in GAMs infiltration. Bevacizumab, a VEGF inhibitor, promotes tumor vascular normalization, mitigates GBM-related edema, and significantly enhances patient symptoms [538]. EGFRvIII, the predominant mutant form of EGFR in GBM, plays an important component in the progression of tumors. Studies indicate EGFRvIII expressing with a substantial proportion of GBM patients (40% ~ 60%), establishing its significance in regulating angiogenesis, growth, and metastasis [539]. Preclinical studies validate cetuximab's efficacy in suppressing GBM cell growth and enhancing the effectiveness of therapeutic modalities, including radiation therapy [540]. Aquaporin 4 (AQP4), a prominent aquaporin in the CNS, emerges as a crucial determinant of glioma cell fate and an ideal biomarker for precise diagnosis and treatment [246]. TMZ suppresses AQP4 expression through MAPK signaling pathway activation, suggesting the therapeutic potential of targeting the AQP4-MAPK pathway [541]. Inhibition of AQP4 enhances GBM sensitivity to TMZ, influencing TMZ efficacy by regulating sodium pump α3 subunit protein (ATP1A3) [542]. AQP4's role in maintaining BBB integrity positions selective inhibition as a promising avenue for innovative therapies. PDGFRA amplification characterizes proneural subtypes, emphasizing its pivotal role [543,544,545]. Analysis of the database of TCGA and clinical samples reveals that elevated EPH receptor A2 (EPHA2) expression correlates with PDGF signaling pathway upregulation [151]. Prohibiting EPHA2 and PDGFRA simultaneously shows synergistic results in malignant cells in GBM.
The circadian rhythm, a conserved phenomenon, is a crucial regulatory system maintaining normal cell and tissue homeostasis. It plays a pivotal role in regulating various tumor-related processes, including tumor cell proliferation, survival, metabolism, DNA repair, and inflammation [546]. The transcription factors CLOCK and BMAL1 [308], key components of the circadian rhythm mechanism, form a heterodimeric complex with either pro-tumor or anti-tumor effects depending on the TME and cancer type [69]. In GBM, the CLOCK-BMAL1 complex is identified as an oncogenic factor fostering proliferation and migration in tumor cells [547] through enabing NF-κB signal [548]. Targeting CLOCK or its heterodimeric partner BMAL1 induces cell cycle arrest and apoptosis by attenuating mitochondrial metabolic function and inhibiting key enzymes in the tricarboxylic acid (TCA) cycle [548]. Furthermore, the CLOCK-BMAL1 complex suppresses anti-tumor immunity by upregulating chemokines, leading to immunosuppressive microglial infiltration into the GBM TME [309]. Additionally, it contributes to angiogenesis and cancer progression, associated with adverse clinical outcomes in GBM through activating TANK binding kinase 1 (TBK1) signaling pathway in ECs [549]. Inhibiting the CLOCK-BMAL1 complex counteracts its tumor-promoting effects on GBM and enhances BBB permeability [550, 551], increasing the effective concentration of therapeutic drugs in the brain. This underscores the potential of CLOCK-BMAL1 as an important treatment target in GBM [552, 553].
The CNS is pivotal for development and oncology, exerting regulatory control over stem and precursor cell populations and influencing tumor growth and metastasis. This recognition has given rise to an emerging field known as cancer neuroscience. Increasingly, studies underscore the critical involvement of the nervous system in cancer initiation and metastasis, forming the basis for figuring out the relation of neurological processes and tumorigenesis [554]. For GBM, infiltration into the brain often follows organized anatomical structures, such as blood vessels and white matter tracts containing neuronal axons. This observation suggests the active participation of neuronal populations in GBM progression. Recent investigations into GBM pathobiology reveal a bidirectional signaling relationship between cancers and neurons, establishing a feedback loop characterized by heightened brain activity, increased proliferation, and synaptic integration. This suggests the intriguing possibility that neuronal activity itself contributes to tumor invasion and progression. Specifically, callosal projection neurons in the hemisphere opposite primary GBM drive tumor progression and widespread infiltration, with Ssemaphorin 4F (SEMA4F) emerging as a key regulator dependent on neuronal activity [555]. This finding unveils a novel mechanism in GBM progression regulated by neuronal activity. In the intricate interplay between neurons and GBM, the physical interaction between potassium voltage-gated channel subfamily a regulatory beta subunit 2 (KCNAB2 or Kvβ2) and Ether-a-go-go 2 (EAG2) forms a potassium channel complex, regulating intracellular Ca2+ concentration in tumor cells, promoting growth, invasion, and chemoresistance in GBM. Inhibition of the EAG2-Kvβ2 complex mitigates cancer aggressiveness, extending survival time in mice with GBM, even in GBM resistant to TMZ [288, 556, 557]. These findings highlight the potential of targeting the EAG2-Kvβ2 complex as a therapeutic strategy for GBM, particularly in cases where resistance to conventional treatment poses a challenge [558, 559].
Epigenetic modifications, pervasive in tumors, play pivotal roles in establishing and maintaining heterogeneity in GBM. Aberrant epigenetic regulation is a primary driver for GBM initiation, with dysregulation of epigenetic regulators contributing to tumor formation. DNA methylation, orchestrated by DNA methyltransferases (DNMTs), represents a reversible process converting 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC). Transformations in 5mC to 5hmC patterning are documented in various human cancers, with lower 5hmC levels correlating negatively with glioma grade [560]. Hypermethylation in the promoter of O6-methylguanine DNA methyltransferase (MGMT) occurs in approximately 40% of GBM cases [561], serving as a key marker for evaluating GBM sensitivity to TMZ treatment and prognostic outcomes [562]. Gliomas, including GBM, exhibit overall hypermethylation in CpG islands, which is glioma CpG island methylator phenotype (G-CIMP) [563], which is recognized as a prognostic indicator for glioma patient survival. Most GBMs are characterized as G-CIMP negative [564]. Consequently, drugs designed to suppress DNMTs are anticipated to induce DNA hypomethylation, potentially activating tumor-suppressing genes. DNMT inhibitors, such as 5-azacytidine and decitabine [565], demonstrate anti-tumor effects in preclinical GBM models and FDA approval as Class I epigenetic drugs for treating various tumors [566, 567]. Histone modification, a multifaceted process, involves diverse mechanisms such as lactylation, methylation, ubiquitination, acetylation, phosphorylation, and adenosine diphosphate (ADP) ribosylation [568], facilitated by various enzymes. Aberrations in histone modification contribute significantly to glioma progression, particularly histone acetylation and methylation in GBM [569]. EZH2, known as histone methyltransferase in polycomb repressive complex 2 (PRC2), modulates gene expression [570, 571] by inhibiting PTEN and activating the NF-κB pathway in GBM [572, 573]. Conflicting opinions exist regarding the efficacy of the EZH2 inhibitor tazemetostat in GBM clinical trials, emphasizing the need for cautious consideration until its specific benefits are delineated for GBM patients [574, 575], especially tumors with H3K27M mutation [576,577,578]. Histone deacetylation, mediated by HDACs, promotes a closed chromatin conformation, inhibiting tumor suppressors [579]. Class I HDACs (HDAC1, 2, 3, and 8) mainly operate within nucleus and primarily inhibit gene transcription, while Class II HDACs (HDAC4, 5, 6, 7, 9, and 10) shuttle between the nucleus and cytoplasm. Class III comprises NAD+-dependent protein deacetylases involved in various cellular processes, and Class IV contains HDAC11, whose sequence is homologous to catalytic core regions of Class I and II HDACs. The multifaceted functions of Class III and IV HDACs in GBM pathogenesis have yet to be fully elucidated [580, 581]. Notably, researchers have observed a downregulation in the mRNA levels in Class II and IV HDACs in GBM compared to low-grade astrocytoma [582]. HDAC inhibitors (HDACIs) have become potential treatments in GBM, impacting oncogene transcription, cell cycle regulation, apoptosis, and differentiation [583]. Recent advancements highlight lactate-derived histone lactylation as a novel modification implicated in GBM progression [59]. This modification induced by the Warburg effect upregulates LINC01127 expression via NF-κB signaling pathway, promoting cancer cell self-renewal through the MAP4K4/JNK/NF-κB axis [61]. Inhibiting lactate production, the substrate for histone lactylation, suppresses GBM progression, making lactylation a potential target for GBM treatment. Elevating lactate production within the TME plays a pivotal role in shaping an acidic microenvironment conducive to tumor promotion, supporting tumor growth, and serving as a cellular substrate for lactylation within the microenvironment. Targeting lactylation emerges as a potentially effective treatment strategy for GBM.
Metabolic reprogramming is a prominent hallmark of tumors, with tumor cells autonomously modulating adaptations through diverse metabolic pathways to meet heightened bioenergetic and biosynthetic demands, which are crucial for proliferation and survival while alleviating oxidative stress. In the local microenvironment, poor vascular differentiation leads to inefficient delivery of oxygen, nutrients, and metabolic waste removal, creating conditions where cancer cells, by rapid proliferation, outcompete anti-tumor immune cells for limited nutrients [584]. Consequently, cancer cells establish a unique anti-immune metabolic microenvironment. Immune cells undergo metabolic adaptations associated with a tolerance phenotype, such as T cells relying on aerobic glycolysis and glutamine catabolism [585]. GBM, with heightened metabolic demands, presents an opportunity for treatment by targeting tumor metabolism [569, 586]. The sodium/hydrogen exchanger 1 (NHE1), from SLC9A1, plays a pivotal role in keeping the microenvironment alkaline within the tumor, supporting aerobic glycolysis crucial for tumor progression [587]. High SLC9A1 expression is observed in the classical and mesenchymal subtypes, indicating a positive correlation with GBM malignancy. NHE1 promotes GBM cell migration and invasion, impacting cell adhesion and ECM rearrangement [588]. Inhibition of NHE1 reduces tumor volume, invasion, angiogenesis, TAM infiltration, and cytokine secretion, enhances the immune system to resist tumors, as well as improves sensitivity to anti-PD-1/PD-L1 immunotherapy and TMZ in mice with GBM [589]. GBM's metabolic shift toward glucose oxidation results in elevated ROS production, requiring upregulation of redox pathways, such as glutathione synthesis [590]. BPM31510 and valerenic acid show promise in inducing oxidative stress and inhibiting GBM progression [591, 592]. Lactate, once considered a glycolysis byproduct [593], now plays a crucial role in metabolic coupling, immune responses, and intercellular communication in the TME [594]. Targeting lactate metabolism, specifically monocarboxylate transporters (MCTs) and LDH, presents therapeutic potential [595, 596]. MCT inhibitors and LDHA inhibitors like NHI-1 and NHI-2 show effectiveness against GBM [60, 597, 598], affecting cell viability and inducing apoptosis and differentiation [171]. GBM utilizes an internal immune escape mechanism through LDH5 secretion, suppressing NK cell recognition [196]. Targeting LDH5 may enhance tumor recognition [599]. The IDO pathway [136], perilipin-2 (PLIN2) [600], ketone oxidation [601], and amino acid metabolism [602] are additional GBM treatment targets, often combined with other therapies to enhance efficacy, impacting immune or epigenetic pathways for improved patient survival.
Targeted CAR modification in glioblastoma
The CAR represents a synthetic modular protein characterized by division into three distinct domains: intracellular, transmembrane, and extracellular domain. The extracellular domain is capable of recognizing target antigens independently of MHC presentation. The transmembrane domain serves the crucial function of integrating the extracellular and intracellular domains, playing a pivotal component in information transmission. The intracellular domain assumes responsibility for T cell stimulation, facilitating proliferation of T cells and inducing cytotoxicity, thereby contributing to the anti-tumor effect [603, 604]. The modification of CAR significantly augments the anti-tumor activity of immune cells [605, 606]. Notably, Table 6 provides a list of CAR immune cells targeting GBM.
The advancement of immunotherapy has broadened the therapeutic landscape for GBM patients. Immunotherapy employing CAR-T technology, commonly known as CAR-T therapy, represents an innovative approach to targeting tumors. This method contains extracting T cells from the patient's blood, modifying them with genetic engineering to give specific antigen recognition domains to T cells, and subsequently reintroducing the modified T cells into the patient to eliminate the tumor [607, 608]. CAR-T can specifically recognize cancer cells, thereby enabling targeted cytotoxicity [609]. To mitigate the risk of CAR-T cells targeting normal cells, TAAs must remain either undetectable or minimally expressed in normal tissues [610]. This strategy is a potential therapy for leukemia as well as certain solid tumors. CAR-T therapy, leveraging specific tumor antigens, has been applied to GBM treatment. IL-13 plays a regulatory component in the responses to inflammation and immunity within the TME by binding to IL13Rα1, and it also interacts with the high-affinity decoy receptor IL13Rα2 [611, 612]. Notably, research has demonstrated abundant expression of the IL-13 receptor in GBM patients, with minimal binding sites in normal individuals, rendering it a potential target for CAR-T therapy in GBM [613]. In GBM, IL13Rα2 is related to aggressiveness and worse outcomes. Studies have indicated that CAR-T cells, transfected with human anti-IL13Rα2 CAR and mouse anti-IL13Rα2 CAR, exhibit enhanced expansion capabilities in T cells and more effective inhibitory in GBM growth [614]. Noteworthy clinical outcomes have been observed, such as increased immune cells and cytokines in CSF of patients with rGBM following IL13Rα2-CAR-T therapy, leading to subsidence of cancer cells in the spinal canal and spine [615]. Additionally, investigations have demonstrated favorable tolerance and anti-tumor responses in patients with rGBM treated with intracranial infusion of IL13Ra2 CAR-T [616]. Approximately 40% of newly diagnosed GBM cases exhibit EGFR expression and amplification. Notably, about 50% of GBM patients with EGFR amplification harbor the constitutively active EGFRvIII oncogenic variant, which is characteristically low or absent in normal tissues. This unique expression profile renders EGFRvIII a practical, feasible, and safe therapeutic target for GBM [617, 618]. In a research conducted by Rourke et al. in 2017, CAR-T targeting EGFRvIII in ten patients with EGFRvIII+ rGBM was found nonsignificant in prognosis [619]. However, post-surgery observations in seven patients revealed increased CAR-T cells in tumor-infiltrating area. Concurrently, elevations in Tregs were noted, accompanied by heightened expression of inhibitory molecules like PD-L1, IL-10, IDO, and TGF-β. Upregulation of these immunosuppressive factors in the TME led to continuous loss of the EGFRvIII antigen, resulting in diminished CAR-T efficacy. Furthermore, adoptive transfer of CAR-T cells in 18 patients, previously subjected to MDSC and Treg depletion through chemotherapy and IL-2 infusion to support CAR-T cell expansion demonstrated prolonged durability of CAR-T cells but lacked objective responses [620, 621]. These findings suggest that CAR-T targeting EGFRvIII induces a supplementary immune response in the TME. Consequently, it is implied that EGFRvIII CAR-T therapy may exhibit enhanced effectiveness when utilized in conjunction with other immunotherapies to potentiate the anti-tumor immune response or reprogram the TIME. Additionally, EPHA2 is frequently overexpressed within GBM and is correlated with prognosis [622]. Targeting EPHA2 with CAR-T therapy can facilitate IL-2 and IFN-γ secretion, exhibiting significant cytotoxicity against malignant cells as well as extending mice outcomes [623, 624].
Given limited success observed with CAR-T-related therapies, the exploration of other immune cells within the GBM microenvironment holds significant promise for advancing effective immunotherapy strategies [625]. Macrophages, an integral role in innate immune system, can proficiently infiltrate tumors, engulf and eliminate abnormal cells, uptake antigens, and present them to T cells [626]. These distinctive attributes underscore the potential of CAR expression on macrophages to enhance targeting and represent a viable avenue for immunotherapeutic interventions. A noteworthy study has reported the successful generation of CAR macrophages (CAR-MΦ) through the utilization of a cavity-injectable nanoporter-hydrogel system, demonstrating efficacy in preventing GBM recurrence [627]. These engineered CAR-MΦ exhibit a remarkable ability to locate phagocytic GSCs, impeding their residual presence. This mechanism stimulates an adaptive anti-tumor immune response within the TME. Significantly, these CAR-MΦ have demonstrated the capacity to lead to enduring anti-tumor immunity, effectively preventing the recurrence of GBM post-surgery.
NK cells can be subject to genetic engineering to express CAR, resulting in increased protein levels in GBM, a condition associated with a worse prognosis. Following the stereotactic injection of Erb-B2 receptor tyrosine kinase 2 (ErbB2)-specific CAR NK cells into the tumor, a notable extension of asymptomatic survival time was observed, extending from 73 days to 200.5 days. CAR-NK therapy exhibited curative effects in immunocompetent mice, curing a significant proportion of subcutaneous tumor-bearing and GBM-bearing mice while enhancing the innate immune system to resist tumors. This, in turn, led to the acquisition of enduring anti-tumor immune responses [628]. Moreover, CD155/CD112, through interaction with DNAX accessory molecule-1 (DNAM-1) and TIGIT on NK cells, exerts immunomodulatory effects and enhances their expression in GBM. These findings position CAR-NK as a potential therapy in GBM [234]. Although neutrophils possess an efficient ability to traverse physiological barriers in response to pathogens, their short lifespan and resistance to genome editing have constrained their broader application in immunotherapy. Chang et al. employed gene editing technology to induce CAR neutrophils, incorporating specific gamma signaling domains produced by human pluripotent stem cells, demonstrating a favorable anti-GBM effect. CAR-neutrophils, thus engineered, can deliver and release nano drugs that influence the TME without inducing additional inflammation, providing a more targeted approach to GBM treatment [629]. While exploring CAR non-T cell treatment in GBM is still in the early stage, preclinical findings indicate an unlimited potential for applying this strategy, making it a promising avenue for future research. As with various other treatments, combination therapy involving CAR appears to be a prevailing trend in GBM treatment.
A parallel therapeutic strategy is the application of bispecific T cell engagers (BiTE), which involves linking an agonist antibody fragment targeting the TCR complex CD3ε to a tumor antigen [630] and a gene-fusion antibody fragment promoting the crosstalks in T cells and target cells, like cancer cells. This design establishes an artificial immune synapse to enhance killing target cells by T cells [631, 632]. The current landscape of BiTE therapy in GBM is an emerging area of investigation. The promising outcomes observed in BiTE therapy targeting IL13Rα2 [633, 634], EGFRvIII [635], EGFR [633], and Fn14 [636], along with notable efficacy in GBM animal models, provide a robust foundation for subsequent clinical translation studies.
Application of glioblastoma vaccine therapy
Vaccine therapy, as the earliest developed form of immunotherapy, has emerged as a crucial approach for researchers to modulate the immune system, enhancing local immune responses to achieve therapeutic effects [637]. It holds longstanding promise for instigating potent anti-tumor immunity, directing cytotoxicity toward tumors while preserving normal tissue, and establishing durable immune memory capable of monitoring tumor recurrence [638, 639]. In the spectrum of immunotherapy strategies for GBM, vaccine therapy stands out as a method to target tumor antigens, surmount the internal immunosuppressive milieu within the tumor, and augment the immune response against the tumor. Multiple TAAs have been identified in GBM, some of which present as promising candidates for vaccine-directed immunotherapy [640]. These cancer vaccines are meticulously crafted to instigate the development of long-term memory in tumor-specific effector T cells, aiming to eradicate cancer cells and forestall tumor recurrence [640, 641]. Table 7 provides an overview of ongoing clinical trials exploring various vaccine therapies for GBM, encompassing peptide vaccines, cell vaccines, mRNA vaccines, and more.
GBM-associated TAAs identified thus far encompass but are not confined to IDH1, HSP, Wilms tumor protein (WT1), survivin, IL13Ra2, EGFRvIII, and IL-4 [642,643,644]. EGFRvIII, expressed heterogeneously in approximately one-third of GBM patients, is absent in normal tissues and serves as an independent adverse prognostic marker, presenting a crucial target for antitumor immunotherapy [645]. Investigations have demonstrated that the introduction of Rindopepimut, a 14-amino acid peptide vaccine targeting EGFRvIII, significantly extended patients with GBM prognosis, particularly combined with TMZ, showcasing the vaccine's remarkable efficacy [646]. Combining Rindopepimut with the VEGFR inhibitor bevacizumab has demonstrated prolonged progression-free survival (PFS) in rGBM [619]. Survivin, an anti-apoptotic protein prevalent in brain tumors, is associated with a poorer prognosis and is scarcely found in normal tissues, rendering it an appealing vaccine target. SurVaxM, a survivin-targeted peptide vaccine, received orphan drug designation from the FDA owing to its capacity to stimulate T cell immunity and inhibit the survivin pathway. Clinical research has indicated that SurVaxM can enhance the PFS of patients with survivin-positive rGBM [647]. DCs, as the most critical type of antigen-presenting cell (APC), are essential for stimulating primary T-cell proliferation. As for brain tumor immunotherapy, a significant focus is placed on DC vaccines, involving the in vitro production of autologous DCs pretreated with tumor antigens, which are reintroduced into patients as immunotherapy [648]. While autologous cell vaccines, particularly DC vaccines, are intricate and costly, they have demonstrated the capacity to elicit robust immune responses [649]. DCVax-L, an autologous cell vaccine comprising DCs pulsed with autologous tumor lysate to stimulate the immune response, has exhibited promising outcomes. Patients with MES gene expression characteristics treated with DCVax-L displayed higher CD8+ T cell infiltration to TME, significantly extending outcomes compared to patients with other gene expression profiles in GBM [650]. Cytomegalovirus (CMV), a double-stranded DNA virus, has been detected in various tumor types, including GBM [651]. Persistent chronic inflammation and immunosuppression in GBM can reactivate CMV, offering a potential therapeutic avenue [652]. The CMV phosphoprotein 65 (pp65) RNA, expressed in over 90% GBM but not in the normal tissue, serves as a novel target [651]. Targeting CMV pp65 mRNA-pulsed DC vaccines has induced robust anti-tumor immunity by upregulating CCL3. Deposing with some antigen, like tetanus/diphtheria (Td) toxoid, enhances tumor-antigen-specific DC infiltration into draining lymph nodes, related to a notable improvement in the OS of GBM patients [653]. Vaccine therapy for GBM holds promise in preclinical and early clinical assessments. Combined strategies, including immune checkpoint blockade (ICB), Treg depletion, and enhanced DC migration, may synergize with tumor-specific vaccines to enhance patient outcomes. The future of GBM vaccine therapy may involve combinatorial approaches that integrate the identification of tumor-specific antigens with vaccines and block immunosuppressive pathways, thereby mitigating the strength and duration of antitumor immunity in GBM patients [654].
Oncolytic viruses, immunotoxins, and antibody-coupled drug therapy
Immunotherapy, encompassing strategies such as ICI, cytokine-based therapies, vaccine therapies, T cell therapies, and viral therapies designed to specifically target tumors, has emerged as a focal point in anti-tumor research [655]. Oncolytic viruses (OVs) operate primarily through two mechanisms: some infect and selectively replicate within tumor cells, while others involve the introduction of transgenes promoting anti-tumor effects into non-replicating viruses [656]. Current research aims to express novel transgenes in viruses, preserving their replication and lytic capabilities to enhance tumor clearance and patient survival. Several oncolytic viruses are undergoing clinical development, like herpes simplex virus (HSV), adenovirus (ADV), vaccinia virus, coxsackievirus, measles virus (MV), poliovirus (PV), reovirus, and Newcastle disease virus (NDV), with many in early clinical trials [657,658,659]. However, like other treatments, oncolytic virus therapy encounters challenges in patient selection. Identifying patients likely to respond to oncolytic virus treatment remains challenging, and reliable biomarkers and predictive factors for OV therapy response are yet to be fully elucidated [660]. Table 8 provides an overview of current clinical trials related to virotherapy strategies for GBM, encompassing OVs, immunotoxins (ITs), and antibody–drug conjugates (ADCs), among others.
CCL5, an inflammatory chemokine that facilitates immune cell chemotaxis through interaction with CCR1/CCR5, undergoes methylation-induced silencing in the progression of solid tumors [661]. Consequently, restoring or augmenting CCL5 expression is a prospective therapeutic strategy for overcoming the TIME in GBM. However, the inherent challenges of its short half-life, delivery to the TME, and potential off-target toxic effects limit its efficacy in tumor therapy. GBM cells infected with oncolytic HSV, targeting both EGFR and CCL5 receptors, exhibit elevated and sustained levels of CCL5 in the TME. This elevation enhances adaptive and innate immune cell infiltration. Furthermore, acting as an IgG1 anti-EGFR monoclonal antibody, it activates macrophage antibody-dependent cellular phagocytosis (ADCP) and NK cells through antibody-dependent cellular cytotoxicity (ADCC), thereby reducing EGFR signaling in cancer cells [662, 663]. This comprehensive strategy significantly prohibits cancer growth and prolongs the mice's prognosis. The ECM contributes to tumor progression by interacting with cancer cells and stromal components within the TME [664]. In GBM, the tumor ECM, consisting of proteins like collagen, fibronectin, and laminin, along with non-proteins such as hyaluronan (HA), plays a critical role [665, 666]. HA regulates cancer cell proliferation and invasion and affects chemotherapy activity by binding to CD44 and receptor for hyaluronic acid-mediated motility (RHAMM) [665]. ICOVIR17, an ADV expressing hyaluronidase, is employed to treat GBM-bearing mice [667]. This virus degrades HA, disrupting the immunosuppressive microenvironment by inhibiting the NF-κB signaling pathway. Consequently, this approach increases CD8+ T cells and macrophages infiltrating into tumors, ultimately extending mice prognosis [666]. Moreover, oncolytic HSV-1 G207 demonstrates significant efficacy in prolonging the median OS of GBM patients. As a neurophilic virus, G207 is well-suited for targeting GBM. Its ability to bypass the BBB through intratumoral inoculation enables direct infection and lysis of tumor cells. This, in turn, reverses tumor immune escape, enhances the cross-presentation of tumor antigens, and enhances the immune system resisting tumors [662, 668, 669].
ITs represent a class of therapeutic agents comprising targeted peptides, typically antibodies or antibody fragments, coupled with peptide toxins sourced from plants or bacteria [670]. Some toxins possess potent cytotoxic properties, inducing apoptosis and inhibiting protein synthesis in the cytoplasm. Consequently, ITs are recognized as crucial agents in cancer treatment and infection prevention [671]. Several pseudomonas exotoxins (PE) based ITs have undergone exploration and evaluation [672]. After recognition and binding to the target antigen, ITs undergo internalization through endocytosis mediated by receptors. The functional domain in PE then catalyzes elongation factor-2 (EF2) with ADP-ribosylation in cytoplasm. This process induces the arrest in protein synthesis, ultimately inducing cell death [673]. In the context of GBM, IL-13R has been identified by the majority of GBM cells and samples obtained from surgically resected patients [674]. Particularly, the IL13Rα2 chain, a principal binding and internalization component of IL-13, is expressed in approximately 80% of GBM tumor specimens but is minimally expressed in normal brain tissues [675]. IL13-PE38QQR is IL-13 with a truncated form of Pseudomonas aeruginosa exotoxin A (PE38QQR). This compound induces cytotoxicity through inhibiting protein synthesis, causing cell apoptosis and death [676,677,678]. Convection-enhanced delivery (CED) in CNS of IL13-PE38QQR has demonstrated significant efficacy in extending the median OS of patients with rGBM [678]. This targeted therapeutic approach capitalizes on the specific expression of IL13Rα2 in GBM tumor specimens, underscoring its potential as a promising treatment strategy for this aggressive form of brain cancer. Furthermore, when combined with concurrent 5 Gy irradiation, the cytotoxicity to GBM cells was significantly enhanced [679]. This suggests that IT-targeted IL-13R, in combination with other modalities such as RT, holds promise for enhancing treatment outcomes in GBM patients. Additionally, intratumoral injection of EGFRvIII IT has demonstrated the eradication of tumors in a GBM mouse model. The down-regulation of MGMT mediated by IT further sensitizes tumor cells to TMZ [680]. D2C7-IT (D2C7) represents a recombinant antibody fragment-based IT targeting EGFR and EGFRvIII, two predominant driver oncogenes in GBM [135]. Delivery of D2C7 via CED leads to direct tumor cell death and facilitates CD4+ and CD8+ T cells, triggering secondary immune responses [681]. While D2C7 monotherapy has demonstrated prolonged survival and promoted disease control in some patients, its efficacy is constrained by the potent immunosuppressive microenvironment in GBM [682]. Combined therapy with targeted CD40 has shown the potential to enhance the response of GBM to D2C7 treatment. CD40, a costimulatory factor in TNF receptor superfamily, is highly expressed in GBM [683]. The combination of D2C7 and anti-CD40 cytotoxic immunotherapy activates microglia and TAMs, creates a pro-inflammatory TME, inhibits exhaustion of CD8+ TILs, and increases tumor antigen-specific CD8+ TILs. This comprehensive approach has demonstrated prolonged survival and development of a long-term anti-tumor immune response in mice bearing GBM. Phase I clinical trials for this combination therapy have been initiated [684].
ADCs represent an advancing anti-cancer drug, combined with targeting precision of monoclonal antibodies and the anti-tumor effects in cytotoxic drugs [630, 685]. Currently, more than 40 ADCs have entered clinical trials, including FDA-approved examples like Adcetris and Kadcyla, used in treating CD30-overexpressing Hodgkin lymphoma and human epidermal growth factor receptor 2 (HER2)-overexpressing breast cancer, respectively [686, 687]. Application in ADCs is also gaining prominence in the treatment of GBM. AMG595 combines the highly selective anti-EGFRvIII antibody with mertansine (DM1), an anti-tubulin agent, through a non-cleavable linker. This ADC combines with the membrane and gets into the endo-lysosomal pathway of EGFRvIII+ cells, inducing mitotic arrest in tumor cells and resulting in regression of GBM [688, 689]. Galectin 3 binding protein (LGALS3BP) is vital in regulating stroma-tumor interactions and is among the most abundant surface components in tumor-derived extracellular vesicles [690]. Plasma vesicle LGALS3BP levels are related to the grade and progression of glioma [691]. Targeting LGALS3BP with an ADC has proven effective in inhibiting GBM cell growth in vivo, inducing a noticeable improvement in the survival time of mice [692]. CD97 is expressed in various immune system lineages. It is vital in inflammatory responses in a range of liquid (leukemia) and solid (ovarian, esophageal, breast, stomach, colon, pancreatic, thyroid, prostate, hepatocellular) malignancies, including GBM [693,694,695]. CD97 is associated with cell proliferation, brain invasion, and tumor metabolism in GBM [696]. It promotes Warburg metabolism through signaling mechanisms, including receptor cytoplasmic C-terminal phosphorylation, β-arrestin recruitment, and activating MAPK/ERK signal, thereby contributing to tumorigenesis in GBM [697]. The ADC targeting CD97 has demonstrated selective killing of patient-derived GBM cultures while sparing neural stem cells and non-neoplastic human astrocytes. This suggests that a CD97-targeting ADC is a potential treatment in GBM [698].
The integration of medicine and engineering technology shines brightly in glioblastoma
The convergence of medicine and engineering constitutes an emerging interdisciplinary field that embodies a collaborative and innovative approach, amalgamating medical sciences with engineering technologies [699]. In the context of cancer treatment, this fusion entails the application of biotechnology in tandem with engineering methods to optimize drug delivery and treatment targeting. Table 9 provides an overview of ongoing clinical trials focused on engineering-based treatments for GBM.
Zinc ion carriers, known for their tissue specificity, have found extensive applications in this field. They are employed to modify engineering carriers with CpG oligonucleotide nanoparticles (CpG NPs) and AMD-Zn (Zn(II)2-AMD3100), creating an injectable hydrogel system (imGEL) that, among them, the tissue-specific affinity of zinc nanoparticles and the unique tissue diffusion and resident properties of hydrogels can increase the drug efficacy [700]. When delivered into the surgical cavity, it effectively inhibits persistent GAMs activation and stimulates CTLs. The results indicate that imGEL can modulate the TIME, suppress the recurrence of GBM, and provide precious time for follow-up clinical adjuvant therapy [701]. Hydrogels have also recently been extensively used due to their tissue-specific dispersion properties. Leveraging their diffusion characteristics, Chen et al. [627] combined a special hydrogel composite structure with GSC-specific CAR-MΦ to be injected into the tumor cavity following GBM resection in mice. This approach conferred powerful tumor-immune cytotoxicity in the surgical cavity, inhibiting GBM recurrence. Moreover, direct intratumoral administration is an emerging and highly effective approach in current cancer treatments, and ultrasound (US) possesses strong tissue-penetrating capabilities and has widespread clinical applications. So, sonodynamic therapy (SDT) is a novel approach that utilizes the principles of ultrasound to activate photosensitizers previously injected into tumor tissue, generating ROS and cavitation bubbles, thus eradicating GBM cells [702]. Several GBM combination therapy approaches based on SDT have been studied, like SDT-thermotherapy, SDT-autophagy inhibition, photodynamic therapy (PDT) with SDT, and SDT-chemotherapy. The above combinatorial methods synergize tumor ablation, significantly strengthening the effectiveness of GBM treatment [703]. Another adjuvant strategy based on CED can facilitate the improved delivery of drugs to the interior of GBM [704,705,706].
However, the applications of medicine and engineering go beyond that. They can be combined with other treatment approaches, such as OVs, engineered using engineering techniques to enhance their tissue specificity for tumor tissue. Moreover, bacteria-mediated tumor therapy can stimulate the immune system and carry various drugs with genetic engineering [707, 708]. Zhu et al. [709] used C-novyi-spores with melittin-RADA32 nanofiber hybrid peptide. It armed them with metformin, inducing the infiltration of CD8+ T cells, regulating immune-active factors secretion, and promoting the polarization of M1 macrophages, thus reactivating anti-tumor immunity in the GBM microenvironment. The integration of medicine and engineering can also be combined with therapies that target tumor metabolism. Both glioma cells and TAMs overexpress α7 nicotinic acetylcholine receptors (nAChRs) [710]. A lipid complex, CDX-LIPO, has been developed to target these receptors. It can co-target tumor cells, tumor vasculatures, and TAMs to restrain aerobic glycolysis through the mTOR pathway, thereby inducing tumor autophagy, suppressing M2 macrophages, and MDSCs while activating the function of CTL, M1 macrophages, and NK cells in GBM [711]. Applying engineering techniques can also improve the effectiveness of ICIs [712].
The integration of medical and engineering technologies has recently become a prominent strategy in cancer therapy. Various treatments, including immunotherapy, cell therapy, and metabolic therapy, are being modified using engineering technologies to achieve better targeting and improved tumor specificity. Furthermore, engineering modifications can enhance the efficacy of existing treatment modalities, ultimately strengthening their tumor-killing effects. This fusion of medicine and engineering represents a powerful tool in cancer treatment, providing innovative strategies to combat the complexity of cancer and improve patient outcomes.
Tumor treating fields therapy in glioblastoma
Tumor treating fields (TTFs) represents a physical therapy approach in cancer treatment that involves applying low intensity, intermediate frequency, alternating electric fields (1–3 V/cm and 100 kHz to 300 kHz). This disrupts the processes of the mitotic spindle in rapidly dividing tumor cells, leading to chromosome missegregation, incomplete cytoplasmic separation, mitotic catastrophe, and p53-dependent and independent apoptosis [713]. TTFs have shown efficacy in extending patients with GBM prognosis, leading to FDA approval for treating GBM and rGBM after surgery and RT with adjuvant TMZ (combined with TMZ to extend median PFS to 6 months) [714,715,716,717]. TTFs have been observed to cause cell cycle arrest at the G2/S phase or disrupt G1/synthesis, along with enhancing ROS production to augment radiation-induced apoptosis [718,719,720]. Additionally, TTFs can delay DNA damage repair and enhance radiation-mediated DNA damage. The combination of TTFs with radiation treatment has been shown to promote caspase-3 and poly ADP-ribose polymerase (PARP) cleavage, contributing to a more effective killing of GBM cells [721]. Moreover, TTFs activate autophagy by inducing miR-29b, which inhibits the Akt2/mTOR/p70S6K/4EBP1 axis signaling, thereby inhibiting GBM progression in vitro [722]. TTFs have demonstrated multifaceted effects in GBM treatment. TTFs not only impact the cell cycle and apoptosis but also exhibit potential in modulating various signaling pathways associated with GBM proliferation and progression. For instance, TTFs have been found to reduce eukaryotic translation initiation factor 4A3 (EIF4A3)-mediated circMMD biosynthesis, which is elevated in GBM. The circMMD expressed highly is related to worse outcomes in GBM cases. By inhibiting Wnt/β-catenin pathway activation, TTFs contribute to the suppression of GBM proliferation [723]. Moreover, TTFs have been shown to induce anti-tumor immunity, potentially enhancing immunotherapy. TTFs promote the infiltration of tumor-infiltrating leukocytes in the TME. This results in increased PD-L1 expression in macrophages and DCs, as well as elevated release of IFN-γ by CTLs [724]. Combining TTFs and anti-PD-1/PD-L1 significantly reduces tumor volume, enhances anti-tumor immunity, and achieves a more potent anti-tumor effect. It's noteworthy that TTFs do not seem to adversely affect crucial functions of T cells involved in anti-tumor immunity. The secretion of IFN-γ, cytotoxic degranulation, and antigen-directed cytotoxic function in T cells exposed to TTFs remain unaffected. Although TTFs inhibit the T-cell activity in proliferation, the viability of non-proliferative T cells is not compromised [725, 726]. Interestingly, TTFs have been related to a significant upregulation in tumor antigen-specific infiltration of T cells in patients who received TTFs combined with standard chemoradiotherapy in GBM, with no apparent alteration in their proliferative capacity [725].
The recent study highlights the potential of TTFs in triggering immunogenic responses in GBM. TTFs-induced mitotic catastrophe leads to the local disruption of the nuclear envelope, resulting in the release of micronucleus within the cell. This, in turn, activates DNA sensing pathways such as cGAS/STING and is absent in melanoma 2 (AIM2), eliciting various inflammatory mediators, such as IL-6, CXLC10, IL-8, type 1 interferon, IL-1, and type 1 interferon-responsive genes production [727]. In addition to the in vitro findings, TTFs have been shown to stimulate STING/AIM2-mediated anti-tumor immunity in mice with GBM. This stimulation promotes T cell activation in the microenvironment and the generation of durable memory T cells. As a result, mice treated with TTFs were protected from re-challenge by the same GBM cell line [728]. These findings suggest that TTFs may have an immunomodulatory effect by promoting anti-tumor immunity. This highlights the potential of TTFs as a therapeutic strategy not only for directly targeting GBM but also for mounting an effective anti-tumor reaction. The implications of this research extend beyond GBM, hinting at the possibility of using TTFs as cancer immunotherapy for other solid tumors.
In the contemporary landscape, therapies for GBM have transitioned into an epoch characterized by comprehensive interventions. The profound intratumoral heterogeneity inherent to GBM renders singular therapeutic modalities susceptible to heightened drug resistance and recurrent manifestations. Moreover, propelled by advancements in scRNA-seq, the discernment of various drug-sensitive and drug-resistant cellular clusters within GBM has become feasible. Consequently, the amalgamation of diverse treatment modalities emerges as a strategic imperative for surmounting the constraints precipitated by the heterogeneity intrinsic to singular treatment modalities. This strategic amalgamation is oriented towards realizing a comprehensive therapeutic impact, delineating a departure from unilaterally oriented approaches.
Potential prospects for targeting MDSC in glioblastoma
The heightened infiltration of MDSCs within the TME intricately correlates with tumor invasiveness, compromised efficacy of immunotherapy, and a more unfavorable prognosis. Elevated MDSC levels are discernible in the peripheral circulation of GBM patients, a phenomenon mediated by arginase activity and G-CSF, with ensuing reversible dysfunction observed in T cells [19]. Consequently, targeting MDSCs stands out as a promising therapeutic avenue in the GBM treatment landscape. Four primary therapeutic strategies have evolved for MDSC targeting: the inhibition of MDSC generation, depletion of MDSC populations, curbing MDSC recruitment to the TME, and interference with the immunosuppressive functionality of MDSC. Refer to Table 10 and Fig. 7 for a comprehensive summary of available MDSC-targeting strategies in tumors.

Therapeutic strategies targeting MDSC. Current therapeutic strategies targeting MDSCs can include four steps: suppressing the generation or expansion of MDSCs, depleting the existing MDSCs, restraining the recruitment of MDSCs, and regulating the immunosuppressive function of MDSCs. Akt Protein kinase B; ApoE Apolipoprotein E; ATRA All-trans-retinoicacid; BTK Bruton’s tyrosine kinase; C/EBPβ CCAAT/enhancer binding protein β; CAR-T Chimeric antigen receptor T-Cell immunotherapy; CHK1 Checkpoint kinase 1; CK2 Casein kinase 2; CSF-1 Macrophage colony-stimulating factor-1; Erk Extracellular regulated protein kinases; Fbxw7 F-box and WD-40 domain protein 7; GCN2 General control nonderepressible 2 kinase; IDO Indoleamine2,3-dioxygenase1; IFN-γ Interferon γ; IL Interleukin; iNOS inducible nitric oxide synthase; IRF Interferon regulatory factor; iRGD internalizing RGD; JAK Janus Kinase; LRP8 Low-density lipoprotein receptor-related protein 8; LXRβ Liver X receptor β; MDSC Myeloid-derived suppressor cells; MIF Macrophage migration inhibitory factor; NLRP3 NOD-like receptor thermal protein domain associated protein 3; PGE2 Prostaglandin E2; PI3K Phosphoinositide-3 kinase; STAT Signal transduction and transcription factor; TLR2 Toll-like receptor 2; TMZ Temozolomide; TNF Tumor necrosis factor; TRAIL-R Tumor necrosis factor-related apoptosis-inducing ligand receptor; VEGF Vascular endothelial growth factor
Suppression of MDSC generation
In recent years, ICIs have emerged as pivotal components of cancer therapy. Sen et al. demonstrated that combining oral checkpoint kinase 1 (CHK1) inhibitor SRA737 with gemcitabine significantly augmented the amount of CD8+ T cells, DCs, and M1 macrophages in small cell Lung cancer (SCLC) models [729]. This therapy concomitantly induced a marked reduction in M2 macrophages and MDSCs. The resultant attenuation of the immunosuppressive microenvironment holds promise for strengthening anti-tumor results combined with anti-PD-L1/anti-PD-1 [730]. Targeting CD33, a standard marker for human MDSCs, is applied to treat acute myeloid leukemia [731]. Recent studies have revealed that metformin, belonging to a class of drugs capable of activating the AMPK pathway and inhibiting the mTOR pathway, can diminish the levels of S100A8/A9 and ARG1. This reduction, coupled with an upregulation in CD8+ T cells, collectively inhibits the population of PMN-MDSCs when combined with ICIs [732]. Additionally, all-trans retinoic acid (ATRA) can impede retinoic acid signaling, prompting the conversion of MDSCs into MONs and DCs [356]. This process involves the activation of extracellular regulated protein kinases 1/2 (ERK1/2) and generating glutathione, which has anti-angiogenic effects in breast cancer [733]. ATRA-based therapies are presently undergoing evaluation in melanoma, renal cell carcinoma (RCC), and lung cancer, showcasing significant reductions in MDSC and improved prognoses. Casein kinase 2 (CK2) inhibitors represent an additional strategy for impeding MDSC differentiation, particularly targeting PMN-MDSCs differentiation by regulating the Notch phosphorylation pathway [734,735,736]. When combined with anti-CTLA-4, CK2 inhibitors can inhibit bone marrow cell differentiation and diminish PMN-MDSC generation [734]. While MDSCs are traditionally considered to originate from the bone marrow, recent studies have illuminated the spleen as an additional reservoir of MDSCs [737]. In lung adenocarcinoma, researchers have identified substantial migration of MDSC precursors from the spleen to the TME. These cells promote CCR2 signaling, which is crucial for recruiting spleen-derived MDSCs in vivo [738, 739]. Notably, splenectomy, either before or after tumor development, significantly attenuates MDSC responsiveness and retards tumor progression. Liver X Receptors (LXRs) activate genes about glucose metabolism, cholesterol, and FA regulation transcription [740]. Agonists of LXR, such as GW3965 and RGX-104, currently undergoing Phase I clinical trials, have demonstrated potent anti-tumor effects in immune-competent mice, inhibiting tumorigenesis, including GBM [741, 742]. These agonists induce the up-regulation of apolipoprotein E (ApoE), a transcriptional target of LXR, which acts on the LRP8 receptor on MDSCs. This action reduces the abundance of tumor-infiltrating and systemic MDSCs, concurrently increasing CD8+ and CD4+ T cells infiltrating into the microenvironment. This modulation aims to reverse tumor immune evasion and promote anti-tumor immunity [743].
Depletion of MDSC
MDSCs, highly heterogeneous cells originating from BM, impose limitations on the efficacy of immunotherapy in tumors. The elimination of MDSCs within the TIME has demonstrated a substantial enhancement in the anti-tumor effects of immunotherapy, leading to a noteworthy extension in the mice’s prognosis in tumors. MDSCs in both mice and tumor-afflicted patients exhibit a significantly heightened ERS response compared to their counterparts in normal mice and healthy individuals. Multiple factors can induce ERS in MDSCs, among which an elevation in ROS within MDSCs is noteworthy [744, 745]. Induction of DR5 expression in mouse MDSCs through ERS inducers has been observed. Targeting DR5 effectively eliminates MDSCs via caspase-8-mediated apoptosis, facilitating the expansion and augmenting the cytotoxic activity of CD8+ T cells. This, in turn, significantly amplifies the anti-tumor efficacy of anti-CTLA-4, particularly in weakly immunogenic tumors [403]. Resiquimod, a TLR7/8 agonist, exerts anti-viral and anti-tumor immunomodulatory effects by stimulating various cytokines secretion [746, 747]. In a breast cancer mouse model, resiquimod induces F4/80+ macrophages and CD11c+/I-A+ DCs, differentiating from MDSCs. These differentiated cells exhibit heightened proliferation-inducing activity on antigen-primed T cells and robustly stimulate the proliferation of CD4+ and CD8+ T cells, reinforcing anti-cancer immunity [748]. Furthermore, the loss of the serine-threonine kinase general control nonderepressible 2 (GCN2), a key driver in the polarization of MDSCs, leads to the transition of immunosuppressive MDSCs to an antitumor-responsive phenotype in the TME. This transition is achieved by promoting the transcription of cyclic-AMP response binding protein 2/ATF4 (CREB2/ATF4), strengthening proinflammatory responses, and enhancing IFN-γ secreted by CD8+ T cells [749]. Notably, patients with pre-existing or newly diagnosed systemic autoimmune conditions have been reported to exhibit a significantly increased likelihood of developing tumors, particularly melanoma [750]. Excessive immunosuppressive therapy in cancer patients can induce elevated IFN-γ, potentially triggering de novo autoinflammation and exacerbating pre-existing autoimmune conditions [751]. The expansion of MDSCs derived from systemic lupus erythematosus (SLE) in the context of melanoma has been implicated in driving systemic macrophage polarization. Notably, SLE-derived MDSCs interact with autoimmune macrophages to suppress CD40 expression and IL-27 production on the cell surface. This inhibition of CD40/IL-27 signaling in tumors is associated with increased TAM infiltration and resistance to ICB. In GBM, the selective depletion of MDSCs using low doses of 5-Fluorouracil (5-FU) has demonstrated increased activated-T-cell amount and extended mice prognosis [403]. Oral administration of the 5-FU prodrug capecitabine in rGBM patients activated anti-tumor immunity, including CD8+ T cells and NK cells. This treatment also led to reduced circulating MDSCs, which is related to a more favorable prognosis [752]. Conversely, dexamethasone, used to treat peritumoral edema in GBM patients, promotes abnormal myeloid lineage cell proliferation in the bone marrow. This increased proportion of MDSCs contributes to the immunosuppressive microenvironment in GBM. This effect is associated with the immunosuppressive response to corticosteroids and is considered reversible [752]. Consequently, the management of peritumoral edema during the perioperative period in GBM warrants reevaluation.
GBM necessitates a comprehensive treatment approach, emphasizing maximal surgical resection followed by a combination of RT, chemotherapy, and immunotherapy or targeted therapy. Maximal surgical resection not only aims at reducing the tumor burden but has also been observed to decrease MDSCs: tumor debulking significantly diminishes MDSCs. It facilitates CD4+ and CD8+ T-cell recruitment. This synergistic approach, especially when combined with immunotherapy, strengthens anti-tumor efficacy [753]. Elevated TIGIT expression on TIL has been associated with reduced CTL cytokine production and poorer survival outcomes [754]. In a murine GBM model, TIGIT blocking stimulated anti-tumor CTL responses and concurrently reduced the number of immunosuppressive PMN-MDSCs [235]. Within the GBM microenvironment, pro-angiogenic cytokines such as VEGF and Ang-2 are highly expressed. These cytokines drive tumor angiogenesis and vascular permeability while negatively regulating T cells and the innate immune response [755, 756]. Targeted VEGF therapy has shown promise in alleviating immunosuppression, allowing T cells to enter the TME and function effectively. Combined with ICIs, anti-VEGF/Ang-2 treatment has demonstrated enhanced infiltration of CD8+ T cells, reduced immunosuppressive MDSCs, and diminished FOXP3+ Tregs, thereby improving the efficiency of immunotherapy [515]. The TIME poses a significant obstacle to CAR-T therapy in GBM. Notably, GBM patient TME cells, including MDSCs, exhibit significantly elevated levels of IL15Rα [757]. IL15Rα-targeted CAR-T (CAR-IL15-T) effectively depletes MDSCs within the TME, inhibits the secretion of immunosuppressive molecules by MDSCs, and extends the survival of GBM mouse models. Moreover, combining B7-H3-targeted CAR-T and OVs with chemokine CXCL11 (oAd-CXCL11) achieves superior anti-tumor effects in GBM. oAd-CXCL11 contributes to TIME reprogramming by facilitating M1 macrophage, CD8+ T cell, and NK cell infiltration while concurrently depleting MDSCs, Tregs, and M2 macrophages [758].
Restraining of MDSC recruitment to the TME
Two distinct sets of signals govern the recruitment of MDSCs. Firstly, there is the induction of emergency myelopoiesis and the modulation of myeloid cell differentiation, primarily mediated through G-CSF and GM-CSF. The second signal involves the activation of MDSCs, predominantly mediated by pro-inflammatory cytokines, like IL-6, IL-1β, IFN-γ, and IL-4 [759, 760]. Research has demonstrated that mitogen-activated protein kinase (MEK) inhibitors can reduce GM-CSF and IL-6 production, thereby restraining the recruitment of MDSCs while concurrently promoting CD8+ T-cell recruitment. This microenvironment reprogramming aims to restore the sensitivity of Kirsten rat sarcoma viral oncogene (KRAS)-mutant tumors to PARP inhibitors and anti-PD-1/PD-L1 therapy [761]. The synergistic combination with MEK inhibitor, PARP inhibitor, and anti-PD-1/PD-L1 therapy has shown potential for achieving a more sustained anti-tumor response [761, 762]. Inhibition of the CXCL12/CXCR4 signaling pathway has been identified as another strategy to modulate MDSC recruitment and enhance anti-tumor responses. Targeting this pathway not only inhibits tumor cell proliferation but also restrains the recruitment of CXCR4+ M-MDSCs to the TME. Additionally, it contributes to restoring BBB integrity and induces immunogenic cell death (ICD), thereby sensitizing tumors to complementary therapies such as RT and fostering an anti-GBM immune response [763]. Within specific cancer types like oral and lung cancers, PMN-MDSCs constitute the predominant myeloid cell subpopulation. SX-682, an oral small-molecule CXCR1/CXCR2 inhibitor currently undergoing clinical evaluation, demonstrates significant efficacy in inhibiting the recruitment of CXCR1+ PMN-MDSCs. This inhibition is accompanied by an enhancement in the accumulation of endogenous or adoptively transferred T cells, thus facilitating the effectiveness of T cell-based immunotherapies, including ICBs and adoptive T cell transfer. Importantly, this occurs without altering the expression of CXCR2 ligands and the trafficking of CXCR1+ macrophages [764, 765]. In patients with head and neck squamous cell carcinoma (HNSCC), CD14+ M-MDSCs and CXCR1/2+/CD15+ PMN-MDSCs evident infiltration is observed both in the circulation and at tumor sites [765]. Notably, MDSCs within tumors exhibit a more pronounced immunosuppressive effect than those present in the circulation. The small-molecule inhibitor SX-682 has demonstrated efficacy in mitigating MDSCs accumulating within tumors through blocking CXCR1/2, thereby inhibiting PMN-MDSCs recruiting [766,767,768]. This intervention enhances the anti-tumor efficiency in NK cells. Importantly, SX-682 does not directly alter the proliferation, survival, or sensitivity of tumor cells to NK cells, and it does not affect the immunosuppressive function of PMN-MDSC. TAMs play multifaceted roles in tumor development, making them an attractive target for therapeutic intervention [769]. However, targeting TAMs with CSF-1R inhibitors has shown limited antitumor efficacy. Tumor cells producing CSF-1 can down-regulate granulocyte-specific chemokine in CAFs through HDAC2-mediated pathways, inhibiting myeloid cells recruited into tumor. Paradoxically, blocking CSF-1R can result in CAFs secreting numerous cytokines, recruiting PMN-MDSCs into the tumor. The use of CXCR2 inhibitors can counteract the adverse effects of CSF-1R blockade. As most chemokines bind to CXCR2, up-regulation of CXCR2 induced by CSF-1R blockade can be mitigated by CXCR2 inhibitors, preventing the chemokines secreted by CAFs from functioning. This inhibition of CXCR2 enhances the antitumor effect of CSF-1R inhibitors by restraining the recruitment of PMN-MDSCs. In the breast cancer models, the PARP inhibitor inhibits the recruitment of MDSCs mediated by CXCR4. This inhibition is achieved by reducing stromal cell-derived factor 1 alpha (SDF1α) released by CAFs, thereby augmenting the anti-tumor effect of EGFRvIII targeted CAR-T therapy [770].
RT has been a longstanding and integral component of GBM treatment, contributing to enhanced local control rates and extended survival. Despite its importance, RT can induce local inflammatory responses, including generating complement C5a, a classical inducer of MDSCs [771,772,773]. Consequently, there is an induction of MDSC recruitment. Resistance to tumor RT arises from mechanisms such as STING signal activated through RT. This activation induces IFN-β secretion within tumor cells, inducing the secretion of chemokines like CCL12, CCL2, and CCL7. These chemokines attract CCR2+ M-MDSCs to the TME [774,775,776]. However, it's noteworthy that RT, particularly at high doses, can also decrease MDSC levels. Ablative hypofractionated radiotherapy (AHFRT), instead of conventionally fractionated radiotherapy (CFRT), has been observed to downregulate the amount and immunosuppressive function in MDSCs. This effect is attributed to reduced intratumor hypoxia and VEGF [777]. Combining a single dose of AHFRT with anti-PD-1/PD-L1 treatment activates CD8+ T cells and reduces MDSC levels. This strategy induces the generation in T cells and DCs, further leading to the elimination of MDSCs in GBM-bearing mice [749]. In the GBM microenvironment, chemokines CCL2 and CCL7, secreted by both tumor and non-tumor cells, redundantly contribute to the migration of CCR2+/CX3CR1+ M-MDSCs into the TME. This population of MDSCs can directly impede CD4+ and CD8+ T-cell proliferation and activation, exacerbating the TIME in GBM [778]. Furthermore, CCL2 expression has been verified to negatively correlate with the survival time of GBM patients, with patients with low expression of CCL2 surviving longer than those with high expression of CCL2 [421]. Disruption of the CCL2/CCR2 axis inhibited intratumoral MDSCs’ recruitment and led to the related accumulation of these cells in the BM but had no effect on the intratumoral T cell population [401]. Additionally, studies have shown that gram-negative bacteria/LPS can induce the production of TLR4-dependent CXCL1 in hepatocytes, which induces CXCR2+ PMN-MDSCs infiltrating in TME, thereby regulating the formation of an immunosuppressive microenvironment in hepatocytes and promoting liver tumor growth [736]. Neomycin treatment can block CXCL1 and PMN-MDSC accumulating and inhibit tumor growth. Sunitinib, a tyrosine kinase inhibitor, is the oral compound permitted by the FDA for first-line treatment of various cancers [779]. In the mouse glioma model, CD4+ T cells increased, and MDSCs recruitment decreased after sunitinib treatment, and the reduced amount of MDSCs was consistent with the increased CD4+ T cell quantity and higher proliferation ability, resulting in tumor reduction and significantly prolonged mouse survival [780]. The CXCR4/CXCL12 signaling pathway is crucial in the homing and migration of immune cells [781]. CXCR4 is commonly expressed in hematopoietic cells like MDSCs, T cells, microglia, and B cells, overexpressing in various tumors, including GBM. It contributes to tumor treatment resistance by recruiting immunosuppressive bone marrow cells and promoting abnormal tumor angiogenesis [782]. Anti-CXCR4 therapy can reduce the amount of immunosuppressive tumor-infiltrating leukocytes, like MDSCs and intracranial microglial cells. Targeting MDSC with anti-CXCR4 promotes anti-PD-1 anti-tumor immune responses and improves GBM mouse survival through modulation of the myeloid and T cell TME and the underlying tumor bed vasculature [783,784,785]. Therefore, targeting MDSC to reprogram the immunosuppressive microenvironment is promising to enhance the efficacy of other anti-tumor immunotherapies in GBM.
Regulation of MDSC’s immunosuppressive function
The success of immune checkpoint therapy has instilled optimism regarding the potential cure for cancer. However, a substantial proportion of patients remain unresponsive, and many experience relapse due to immune escape. Among the critical elements contributing to resistance to ICIs, the presence of MDSCs within cancers stands out. MDSCs drive T-cell exhaustion and dysfunction, ultimately leading to immunosuppression. Therefore, the strategic targeting of MDSCs to convert GBM from a "cold" tumor, refractory to immune response, to a "hot" tumor that responds favorably to immunotherapy holds significant therapeutic promise (Fig. 7).
In the pursuit of developing targeted therapies against MDSCs to counteract immunosuppression, MIF has emerged as a notable candidate. MIF exhibits expression in different tumors, including GBM, lung cancer, and breast cancer. Several immune cells, like neutrophils, T cells, MONs, and macrophages, can produce MIF [786]. Particularly noteworthy is the induction of MIF expression by glucocorticoids, commonly used for edema in GBM patients. The levels of MIF increase with glioma grade and upregulation of MIF is related to worse outcomes [787]. Investigations have revealed that M-MDSCs express elevated CD74, a MIF cognate receptor, and are expressed within the TME of GBM [112]. Ibudilast, a brain-permeable inhibitor, can effectively restrain the MIF/CD74 signaling pathway, diminish the immunosuppressive functions in MDSCs, and enhance the activity of CD8+ T cells in the microenvironment. Furthermore, clinically approved MIF inhibitors have been developed, showcasing the potential for repurposing in treating GBM [788,789,790]. The IRF8 has been identified as a crucial player in normal bone marrow formation and the secretion of certain pro-inflammatory type 1 cytokines, like IL-12p40 and CCL5 [791]. Notably, a robust negative correlation exists between the expression of IRF8 and the presence of MDSCs in tumors. Increased expression of IRF8 has been shown to mitigate the pro-tumorigenic capabilities of cancer-induced MDSCs [792]. As MDSCs emerge in response to cancer-derived factors [759], several transcription factors are implicated in STAT3 or STAT5 signaling pathways, with the activation of STAT3 or STAT5 playing various roles in MDSC biology [418, 793,794,795,796,797]. Research indicates that MDSC-inducing factors like GM-CSF and G-CSF in TME promote IRF8 downregulating through STAT3 and STAT5-dependent signals. The reduction in IRF8 is correlated with an increase in MDSC frequency [798, 799]. Downregulation of IRF8 in MDSCs can also influence the expression of Bax and Bcl-xL, suppressing FAS-mediated spontaneous apoptosis and facilitating evasion from elimination by CTLs [800]. Elevated levels of IRF8 have been demonstrated to alleviate the immunosuppressive characteristics of MDSCs, thereby enhancing the efficacy of immunotherapy. MDSCs, known as major producers of IL-6, exhibit significantly higher IL-6 production compared to tumor cells in tumor-bearing mice [797]. IL-6, generated by MDSCs, serves a dual role by safeguarding these cells from TNF-α-mediated necroptosis and sustaining their immunosuppressive functions within the TME. This is achieved through up-regulation of DNMT1 and DNMT3b via STAT3 activation in an autocrine pathway. Additionally, IL-6 can enhance the immunosuppressive abilities of MDSCs by increasing ARG1 activity and ROS production through STAT3 signaling [368]. The STAT3 plays a pivotal role in MDSC functions, and its inhibition has been shown to disrupt MDSC-mediated immunosuppression [801]. Blocking STAT3 induces apoptosis in MDSCs and reduces the expression of immunosuppressive factors [802, 803]. IDO is associated with tumor invasiveness and advanced metastasis [804]. IDO-positive cancer patients often exhibit high expression of inhibitory MDSCs, which inhibit T-cell activation and facilitate FOXP3+ Tregs’ differentiation and activation through the production of kynurenine [467, 805, 806]. Inhibiting IDO with a selective inhibitor has been shown to reverse the (TIME by reducing the infiltration of MDSCs and Tregs and eliminating their suppressive functions in vivo. Cysteine, crucial for mammalian protein synthesis and cell proliferation, is required by T cells for antigen presentation and activation [807]. MDSCs, lacking the neutral amino acid transporters, acquire cysteine from the environment without exporting it. This consumption of cysteine limits its availability in the extracellular environment, suppressing the T-cell activation and anti-tumor immunity [808,809,810]. Targeting amino acid metabolism to inhibit MDSC function and restore the antitumor effect of T cells represents a potential strategy [466, 811, 812]. Entinostat, an HDAC inhibitor, has been shown to reduce MDSC infiltration and its inhibitory functions through STAT3-mediated down-regulation of ARG1. When combined with ICIs, entinostat significantly alters innate immune cells' infiltration and activity, leading to a more effective adaptive immune reation [429, 803, 813].
Conclusions
The intricate and highly heterogeneous TME is essential in the initiation and advancement in GBM. GBM is characterized by pronounced intratumor heterogeneity and a variable immunosuppressive milieu, contributing to drug resistance, frequent recurrence, and rapid disease progression. Among the significant contributors to the TME of GBM, MDSCs emerge as pivotal players, showcasing their essential role in shaping the immune landscape of aggressive brain tumors. The occurrence, recruitment, and dynamic functional alterations of MDSCs exhibit remarkable diversity across distinct stages of glioma development, orchestrated by various regulatory mechanisms. This diversity is further complicated by the profound influence of the heterogeneous microenvironment within gliomas on the function and differentiation of MDSCs. Figure 8 illustrates the timeline of key events in the establishment of targeting MDSCs as a novel therapeutic approach.

Timeline depicting the history of targeted MDSC anti-tumor therapy strategy. ARG1 Arginase 1; ATRA All-trans-retinoic acid; Cox2 Cyclooxygenase 2; GBM Glioblastoma; HDAC Histone deacetylase; IDO Indoleamine 2,3-dioxygenase; M-MDSCs Monocytic myeloid-derived suppressor cells; MDSCs Myeloid-derived suppressor cells; miRNA MicroRNA; NOS Nitric oxide synthase; PMN-MDSCs Polymorphonuclear myeloid-derived suppressor cells; STAT Signal transduction, and transcription factor
As indicated earlier, compelling evidence underscores the significance of the intricate interactions between tumor cells and stromal cells in developing GBM and resistance to immunotherapy. Cancer cells actively recruit and instruct stromal cells, including MDSCs and T cells, during their evolution. Conversely, infiltrating stromal cells are vital to enhance the aggressiveness of cancer cells, leading to resistance against immunotherapy. These observations highlight the potential of targeting the interaction in the tumor and the microenvironment as a promising therapeutic strategy for GBM. Recently, ICI has profoundly transformed the tumor treatment landscape, gaining FDA approval for its safety and feasibility in various malignancies. However, its efficacy in clinical trials for GBM remains under investigation. Presently, the standard treatment for GBM involves post-tumor resection RT combined with TMZ, constituting the primary therapeutic approach. It's important to note that both RT and TMZ have immunosuppressive effects. Additionally, the GBM microenvironment poses a challenging barrier to anti-tumor immune responses, emphasizing the need for a nuanced understanding of this complexity in developing immunotherapeutic strategies. Hence, there is an urgent imperative for combination therapies aimed at transforming these "cold" tumors into "hot," thereby augmenting existing immunotherapy approaches. MDSCs, by inhibiting host immune responses to tumors, play a pivotal role in immunotherapy resistance.
In Tables 4, 5, 6, 7, 8, a comprehensive summary revealed that a substantial portion of clinical studies across immune checkpoint therapy, targeted therapy, CAR-T, tumor vaccine therapy, OVs, ADCs, ITs, and integration of medicine and engineering technology encountered early-stage treatment failures and excessive complications, leading to premature trial termination. Upon systematic categorization of these clinical trials, it was observed that targeted therapy for GBM boasts the highest number of ongoing trials (69 in Active and recruiting), positioning it as the most actively pursued modality. Cancer-related vaccines, recognized as a burgeoning treatment avenue, also exhibit a noteworthy count of ongoing trials in the "Active" status. However, an assessment of the maturity of extant treatment methods, particularly those in phase II and more advanced, indicates that immune checkpoint therapy, tumor vaccine therapy, and targeted therapy lead the landscape. This underscores the relative maturity and safety of immunotherapy and targeted therapy within the contemporary spectrum of novel treatment approaches for GBM. While the field of tumor vaccines is steadily advancing, the anticipated progress in treatment strategies across these three domains is a promising prospect for the future. Despite the multitude of ongoing clinical trials, the impact on the prognosis of GBM remains limited, emphasizing the urgent need for innovative and effective treatment modalities for patients.
Consequently, combining alternative strategies that target MDSCs with active or passive immunotherapy holds the promise of synergistic effects. Most of the existing therapeutic strategies for MDSC are in the early stages of clinical trials. However, existing MDSC-targeting treatments face challenges due to the unclear phenotype, significant heterogeneity, and complex origin and functional networks of MDSCs [814]. To address these challenges, it is essential to employ high-throughput proteomics and genomics technologies to investigate the phenotype and characteristics of MDSCs in various tumor types. This will pave the way for precise methods to eliminate MDSCs. Moreover, the complexity of MDSC binding to tumor cells makes isolating MDSCs challenging, leading researchers to focus primarily on the overall MDSC population rather than tumor-infiltrating MDSCs. As different MDSC subtypes exhibit distinct regulatory mechanisms, identifying and understanding their unique functions is crucial for accurately targeting specific subtypes. Notably, MDSCs share similar phenotypes with normal bone marrow cells, posing a challenge for selective targeting. Therefore, targeting MDSCs in tumor patients must consider tumor site, stage, molecular type, and others. Various drugs have been demonstrated to inhibit the effects of MDSCs in tumors, with some receiving FDA approval, others undergoing clinical trials, and some being studied in preclinical models [815]. However, the intricate mechanisms involved in the generation, recruitment, activation, and immune suppression of MDSCs make it seemingly impossible to induce potent antitumor effects through a single approach. Consequently, combining MDSC-targeted therapy with other immunotherapies emerges as the preferred strategy.
Availability of data and materials
Not applicable.
Abbreviations
- 5-FU:
-
5-Fluorouracil
- 5hmC:
-
5-Hydroxymethylcytosine
- 5mC:
-
5-Methylcytosine
- AC-like:
-
Astrocyte-like
- ACLY:
-
ATP-citrate lyase
- ADC:
-
Antibody–drug conjugate
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- ADCP:
-
Antibody-dependent cellular phagocytosis
- ADP:
-
Adenosine diphosphate
- ADV:
-
Adenovirus
- AHFRT:
-
Ablative hypofractionated radiotherapy
- AHR:
-
Aryl hydrocarbon receptor
- AIM2:
-
Absent in melanoma 2
- Akt:
-
Protein kinase B
- ALKBH5:
-
AlkB homolog 5
- AMPK:
-
AMP-activated protein kinase
- Ang-2/ANGPT2:
-
Angiopoietin 2
- ANXA1:
-
Annexin A1
- AP-1:
-
Activator protein-1
- APC:
-
Antigen-presenting cell
- ApoE:
-
Apolipoprotein E
- AQP4:
-
Aquaporin 4
- ARG1:
-
Arginase 1
- ATF:
-
Activating transcription factor
- ATP1A3:
-
Sodium pump α3 subunit protein
- ATRA:
-
All-trans-retinoicacid
- B1R:
-
Bradykinin receptor 1
- BBB:
-
Blood–brain barrier
- Bcl3:
-
B cell lymphoma 3
- BDNF:
-
Brain-derived neurotrophic factor
- bFGF:
-
Basic fibroblast growth factor
- BiTE:
-
Bispecific T cell engagers
- BMAL1:
-
Brain and muscle ARNT-like 1
- BRD4:
-
Bromodomain-containing protein 4
- Breg:
-
Regulatory B cell
- BTK:
-
Bruton’s tyrosine kinase
- c-Kit:
-
Receptor tyrosine kinase
- C/EBPβ:
-
CCAAT/enhancer binding protein β
- CAF:
-
Cancer-associated fibroblast
- CAR-MΦ:
-
Chimeric antigen receptor macrophages
- CAR-T:
-
Chimeric antigen receptor T-cell immunotherapy
- CCR2:
-
C–C motif chemokine receptor 2
- CCL2:
-
C–C motif chemokine receptor 2
- CDK 4:
-
Cycle-dependent kinase 4
- CDKN:
-
Cyclin-dependent kinase inhibitor
- CED:
-
Convection-enhanced delivery
- CFH:
-
Complement factor H
- CFRT:
-
Conventionally fractionated radiotherapy
- cGAS/STING:
-
Cyclic GMP-AMP synthase/stimulator of interferon genes
- CHK1:
-
Checkpoint kinase 1
- CHOP:
-
C/EBP homologous protein
- CIS:
-
Cytokine-inducible SH2-containing protein
- CK2:
-
Casein kinase 2
- CLOCK:
-
Circadian locomotor output cycles kaput
- CMV:
-
Cytomegalovirus
- CNS:
-
Central nervous system
- COL6A3:
-
Collagen type VI alpha 3 chain
- COX2/PTGS2:
-
Cyclooxygenase 2
- CpG NP:
-
CpG oligonucleotide nanoparticle
- CPT1:
-
Carnitine palmitoyl transferase I
- CREB:
-
Cyclic-AMP response binding protein
- CSC:
-
Cancer stem cell
- CSF:
-
Cerebrospinal fluid
- CSF-1/M-CSF:
-
Macrophage colony-stimulating factor-1
- CSF-1R:
-
Colony-stimulating factor 1 receptor
- CSPG4:
-
Chondroitin sulfate proteoglycan 4
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- CTLs:
-
Cytotoxic T cells
- CXCL:
-
C-X-C motif chemokine ligand
- CXCR:
-
C-X-C motif chemokine receptor
- DCs:
-
Dendritic cells
- DDRi:
-
DNA damage response inhibitors
- DHHC9:
-
Asp-His-His-Cys 9
- DHX9:
-
DExH-box helicase 9
- DIPG:
-
Diffuse intrinsic pontine glioma
- DM1:
-
Mertansine
- DMG:
-
Diffuse midline gliomas
- DNAM-1:
-
DNAX accessory molecule-1
- DNMT:
-
DNA methyltransferase
- DPP-4:
-
Dipeptidyl peptidase-4
- DUSP3:
-
Dual-specificity phosphatase 3
- EAE:
-
Experimental allergic encephalomyelitis
- EAG2:
-
Ether-a-go-go 2
- EC:
-
Endothelial cell
- ECM:
-
Extracellular matrix
- e-MDSCs:
-
Early-stage myeloid-derived suppressor cells
- EF2:
-
Elongation factor-2
- EGFR:
-
Epidermal growth factor receptor
- EGFRvIII:
-
Epidermal growth factor receptor variant III
- EIF4A3:
-
Eukaryotic translation initiation factor 4A3
- EMT:
-
Epithelial to mesenchymal transition
- ENTPD2:
-
Ectonucleoside triphosphate diphosphohydrolase 2
- EPHA2:
-
EPH receptor A2
- ErbB2:
-
Erb-B2 receptor tyrosine kinase 2
- ERK:
-
Extracellular regulated protein kinases
- ERS:
-
Endoplasmic reticulum stress
- EVs:
-
Extracellular vehicles
- EZH2:
-
Enhancer of zeste 2
- FA:
-
Fatty acid
- FAO:
-
Fatty acid oxidation
- FAT1:
-
FAT atypical cadherin 1
- FATP2:
-
Fatty acid transport protein 2
- Fbxw7:
-
F-box and WD-40 domain protein 7
- FCN1:
-
Ficolin 1
- FDA:
-
Food and Drug Administration
- FGL2:
-
Fibroleukin 2
- FHL-1:
-
FH-like protein 1
- FLNA:
-
Filamin A
- FLT3L:
-
Fms-related tyrosine kinase 3 ligand
- FN1:
-
Fibronectin 1
- FOXP3:
-
Forkhead box protein P3
- Fsp1:
-
Ferroptosis suppressor protein 1
- G-CIMP:
-
Glioma CpG island methylator phenotype
- G-CSF:
-
Granulocyte colony-stimulating factor
- GABA:
-
γ-Aminobutyric acid
- GAM:
-
Glioma-associated macrophages/microglia
- GASC:
-
GBM-associated stromal cell
- GBM:
-
Glioblastoma
- GBP5:
-
Guanylate binding protein 5
- GCN2:
-
General control nonderepressible 2 kinase
- GLUT1:
-
Glucose transporters 1
- GM-CSF:
-
Granulocyte–macrophage colony-stimulating factor
- GO:
-
Gene ontology
- GPNMB:
-
Glycoprotein nonmetastatic melanoma protein B
- GSCs:
-
Glioblastoma stem cells
- GTR:
-
Gross total resection
- HA:
-
Hyaluronan
- HDAC:
-
Histone deacetylase
- HDACI:
-
Histone deacetylase inhibitors
- HER2:
-
Human epidermal growth factor receptor 2
- HGG:
-
High-grade gliomas
- HIF:
-
Hypoxia-inducible factor
- HLA:
-
Human leukocyte antigen
- HMGB1:
-
High mobility group protein B1
- hnRNPA1:
-
Hypoxia-inducible heterogeneous nuclear ribonucleoprotein A1
- HNSCC:
-
Head and neck squamous cell carcinoma
- HSC:
-
Hematopoietic stem cell
- HSP72:
-
Heat shock protein 72
- HSPGs:
-
Heparan sulfate proteoglycans
- HSV:
-
Herpes simplex virus
- HVEM:
-
Herpes virus entry mediator
- ICAM:
-
Intercellular adhesion molecule
- ICBs:
-
Immune checkpoint blockade
- ICD:
-
Immunogenic cell death
- ICIs:
-
Immune checkpoint inhibitors
- ICOS:
-
Inducible T cell costimulator
- IDH1:
-
Isocitrate dehydrogenase 1
- IDO:
-
Indoleamine2,3-dioxygenase
- IFN-γ:
-
Interferon γ
- IGFBP6:
-
Insulin-like growth factor-binding protein 6
- IL:
-
Interleukin
- IMCs:
-
Immature myeloid cells
- iNOS:
-
Inducible nitric oxide synthase
- iNPC:
-
Injured neural progenitor cells
- IPS:
-
Immune phenotype score
- IRE1α:
-
Inositol-requiring enzyme 1α
- irSEs:
-
Immune-related side effects
- IRF:
-
Interferon regulatory factor
- iRGD:
-
Internalizing RGD
- IT:
-
Immunotoxins
- ITGαvβ5:
-
Integrin αvβ5
- ITGAM:
-
Integrin subunit alpha M
- ITGB2:
-
Integrin subunit beta 2
- JAK:
-
Janus Kinase
- K–M:
-
Kaplan–Meier
- KCNAB2/Kvβ2:
-
Potassium voltage-gated channel subfamily a regulatory beta subunit 2
- KDM6B:
-
Lysine demethylase 6B
- KLF4:
-
Kruppel-like factor 4
- KRAS:
-
Kirsten rat sarcoma viral oncogene
- LAG3:
-
Lymphocyte activating 3
- LAMP2A:
-
Lysosomal-associated membrane protein 2A
- LCK:
-
Lymphocyte cell-specific protein-tyrosine kinase
- LDH:
-
Lactate dehydrogenase
- LGALS1:
-
Galectin-1
- LGALS3BP:
-
Galectin 3 binding protein
- LGMN:
-
Legumain
- LIF:
-
Leukemia inhibitory factor
- LIFR:
-
LIF receptor subunit alpha
- LILRB2:
-
Leukocyte immunoglobulin-like receptor subfamily B member 2
- LOX1:
-
Lectin-like oxidized low-density lipoprotein receptor 1
- LPS:
-
Lipopolysaccharide
- LRP8:
-
Low-density lipoprotein receptor-related protein 8
- LXR:
-
Liver X receptor
- LXRβ:
-
Liver X receptor β
- M-MDSCs:
-
Monocytic myeloid-derived suppressor cells
- MAFB:
-
MAF BZIP transcription factor B
- MAPK:
-
Mitogen-activated protein kinase
- MCT:
-
Monocarboxylate transporter
- MEK:
-
Mitogen-activated protein kinase
- MET:
-
Mesenchymal transformation
- MDSCs:
-
Myeloid-derived suppressor cells
- MES-like:
-
Mesenchymal-like
- MGMT:
-
Major histocompatibility complexes I
- MHC I:
-
Major histocompatibility complexes I
- MIF:
-
Macrophage migration inhibitory factor
- miRNA:
-
Micro RNA
- MLPGs:
-
Granulocyte-monocyte progenitors
- MON:
-
Monocytes
- MPO:
-
Myeloperoxidase
- mRNAsi:
-
MRNA stemness index
- MSC:
-
Mesenchymal stem cell
- mTOR:
-
Mammalian target of rapamycin
- mTORC2:
-
Mammalian target of rapamycin complex 2
- MV:
-
Measles virus
- nAChR:
-
Nicotinic acetylcholine receptor
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NDV:
-
Newcastle disease virus
- NEAT1:
-
Nuclear enriched abundant transcript 1
- NF1:
-
Neurofibromin 1
- NF-κB:
-
Nuclear factor kappa-B
- NHE1:
-
Sodium/hydrogen exchanger 1
- NK cells:
-
Natural killer cells
- NKG2D:
-
Natural-killer group 2 member D
- NLRP3:
-
NOD-like receptor thermal protein domain associated protein 3
- NOX2:
-
NADPH oxidase 2
- NPC-like:
-
Neural progenitor-like
- NSCL:
-
Non-small-cell lung cancer
- OLFML3:
-
Olfactomedin-like 3
- OPC-like:
-
Oligodendrocyte progenitor-like
- OPN:
-
Osteopontin
- OS:
-
Overall survival
- OSM:
-
Oncostatin M
- OSMR:
-
Oncostatin M receptor
- OV:
-
Oncolytic virus
- PARP:
-
Poly ADP-ribose polymerase
- PB:
-
Peripheral blood
- PBMCs:
-
Peripheral blood mononuclear cells
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed cell death 1 ligand 1
- PDE5:
-
Phosphodiesterase 5
- PDGF:
-
Platelet-derived growth factor
- PDGFRA:
-
Platelet-derived growth factor receptor alpha
- PDH:
-
Pyruvate dehydrogenase
- PDT:
-
Photodynamic therapy
- PE38QQR:
-
Pseudomonas aeruginosa exotoxin A
- PET:
-
Positron emission tomography
- PFS:
-
Progression-free survival
- pGBM:
-
Primary GBM
- PGE2:
-
Prostaglandin E2
- pHGG:
-
Pediatric high-grade glioma
- PI3K:
-
Phosphoinositide-3 kinase
- PLIN2:
-
Perilipin-2
- PMN:
-
Morphology of neutrophils
- PMN-MDSCs:
-
Polymorphonuclear myeloid-derived suppressor cells
- PNT:
-
Peroxynitrite
- pp65:
-
Phosphoprotein 65
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- PRC2:
-
Polycomb repressive complex 2
- Prkar1a:
-
CAMP-dependent protein kinase regulatory type I-α
- PTEN:
-
Protein tyrosine phosphatase
- PTM:
-
Post-translational modification
- PTX3:
-
Pentraxin 3
- PUFAs:
-
Polyunsaturated fatty acids
- PV:
-
Poliovirus
- RAPA:
-
Rapamycin
- Rb:
-
Retinoblastoma
- RCC:
-
Renal cell carcinoma
- rGBM:
-
Recurrent GBM
- RHAMM:
-
Receptor for hyaluronic acid-mediated motility
- RNS:
-
Reactive nitrogen species
- RORC1:
-
Receptor-related orphan receptor γ
- RORα:
-
Retinoic acid related-orphan receptor α
- ROS:
-
Reactive oxygen species
- RT:
-
Radiotherapy
- RTK:
-
Receptor tyrosine kinase
- SCLC:
-
Small cell Lung cancer
- scRNA-seq:
-
Single-cell RNA sequencing
- SDF1α:
-
Stromal cell-derived factor 1 alpha
- SDT:
-
Sonodynamic therapy
- SEMA4F:
-
Ssemaphorin 4F
- SERPINE1:
-
Serpin family E member 1
- SFPQ:
-
Splicing factor proline and glutamine-rich
- SHH:
-
Sonic hedgehog
- SHIP-1:
-
Phosphatidylinositol-3,4,5-trisphosphate 5-phosphatase 1
- SIRPA:
-
Signal regulatory protein alpha
- SLC7A11:
-
Solute carrier family 7 members 11
- SLE:
-
Systemic lupus erythematosus
- SLIT2:
-
Slit guidance ligand 2
- SNHG:
-
Small nucleolar RNA host genes
- SOCS3:
-
Suppressor of cytokine signaling 3
- SSAO:
-
Semi carbazide-sensitive amine oxidase
- ST:
-
Spatial transcriptomics
- STAT:
-
Signal transduction and transcription factor
- STR:
-
Subtotal resection
- TAMs:
-
Tumor-associated macrophages
- TAMCs:
-
Tumor-associated myeloid cells
- TBK1:
-
TANK binding kinase 1
- TCA:
-
Tricarboxylic acid
- TCF:
-
Transcription factor
- TCGA:
-
The Cancer Genome Atlas
- Td:
-
Tetanus/diphtheria
- TF:
-
Transcription factor
- TGF:
-
Transforming growth factor
- THBS1:
-
Thrombospondin 1
- TIGIT:
-
T cell immune receptor with Ig and ITIM domains
- TIIClnc:
-
Tumor-Infiltrating Immune Cells-related lncRNA screening framework
- TIL:
-
Tumor-infiltrating lymphocyte
- TIM-3:
-
T-cell immunoglobulin and mucin-domain containing 3
- TIME:
-
Tumor immune microenvironment
- TLR:
-
Toll-like receptor
- TME:
-
Tumor microenvironment
- TMZ:
-
Temozolomide
- TNF:
-
Tumor necrosis factor
- TNFAIP8L2:
-
TNF alpha-induced protein 8 like 2
- TNFSF9:
-
TNF superfamily member 9
- TP53:
-
Tumor protein P53
- TPO:
-
Thrombopoietin
- TRAIL-R:
-
Tumor necrosis factor-related apoptosis-inducing ligand receptor.
- TRET:
-
Telomerase reverse transcriptase
- Tregs:
-
Regulatory T cells
- TTF:
-
Tumor treating field
- US:
-
Ultrasound
- VCAM:
-
Vascular cell adhesion molecule
- VCAN:
-
Versican core protein
- VEGF:
-
Vascular endothelial growth factor
- VISTA:
-
V-domain Ig suppressor of T cell activation
- VNN2:
-
Vascular non-inflammatory molecule 2
- WT:
-
Wild-type
- WT1:
-
Wilms tumor protein
- ZNF148:
-
Zinc finger protein 148
References
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23(8):1231–51.
De Boeck A, Ahn BY, D’Mello C, Lun X, Menon SV, Alshehri MM, et al. Glioma-derived IL-33 orchestrates an inflammatory brain tumor microenvironment that accelerates glioma progression. Nat Commun. 2020;11(1):4997.
Weller M, van den Bent M, Preusser M, Le Rhun E, Tonn JC, Minniti G, et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol. 2021;18(3):170–86.
Qazi MA, Vora P, Venugopal C, Sidhu SS, Moffat J, Swanton C, et al. Intratumoral heterogeneity: pathways to treatment resistance and relapse in human glioblastoma. Ann Oncol. 2017;28(7):1448–56.
Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer. 2020;20(1):12–25.
Broekman ML, Maas SLN, Abels ER, Mempel TR, Krichevsky AM, Breakefield XO. Multidimensional communication in the microenvirons of glioblastoma. Nat Rev Neurol. 2018;14(8):482–95.
Teran Pumar OY, Lathia JD, Watson DC, Bayik D. 'Slicing' glioblastoma drivers with the Swiss cheese model. Trends Cancer. 2023.
Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol. 2019;20(9):1100–9.
Gong J, Chehrazi-Raffle A, Reddi S, Salgia R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6(1):8.
Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15(7):422–42.
Liu K, Cui JJ, Zhan Y, Ouyang QY, Lu QS, Yang DH, et al. Reprogramming the tumor microenvironment by genome editing for precision cancer therapy. Mol Cancer. 2022;21(1):98.
Linares CA, Varghese A, Ghose A, Shinde SD, Adeleke S, Sanchez E, et al. Hallmarks of the Tumour Microenvironment of Gliomas and Its Interaction with Emerging Immunotherapy Modalities. Int J Mol Sci. 2023;24(17).
Dapash M, Hou D, Castro B, Lee-Chang C, Lesniak MS. The Interplay between Glioblastoma and Its Microenvironment. Cells. 2021;10(9).
Price G, Bouras A, Hambardzumyan D, Hadjipanayis CG. Current knowledge on the immune microenvironment and emerging immunotherapies in diffuse midline glioma. EBioMedicine. 2021;69: 103453.
Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006;8(3):261–79.
Schartner JM, Hagar AR, Van Handel M, Zhang L, Nadkarni N, Badie B. Impaired capacity for upregulation of MHC class II in tumor-associated microglia. Glia. 2005;51(4):279–85.
Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7–H1 expression in tumor-associated macrophages. Clin Cancer Res. 2013;19(12):3165–75.
Domingues P, González-Tablas M, Otero Á, Pascual D, Miranda D, Ruiz L, et al. Tumor infiltrating immune cells in gliomas and meningiomas. Brain Behav Immun. 2016;53:1–15.
Raychaudhuri B, Rayman P, Ireland J, Ko J, Rini B, Borden EC, et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol. 2011;13(6):591–9.
Rodrigues JC, Gonzalez GC, Zhang L, Ibrahim G, Kelly JJ, Gustafson MP, et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro Oncol. 2010;12(4):351–65.
Brown NF, Carter TJ, Ottaviani D, Mulholland P. Harnessing the immune system in glioblastoma. Br J Cancer. 2018;119(10):1171–81.
Grabowski MM, Sankey EW, Ryan KJ, Chongsathidkiet P, Lorrey SJ, Wilkinson DS, et al. Immune suppression in gliomas. J Neurooncol. 2021;151(1):3–12.
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell. 2016;164(3):550–63.
Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell. 2017;32(1):42-56.e6.
Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 2019;178(4):835-49.e21.
Kaffes I, Szulzewsky F, Chen Z, Herting CJ, Gabanic B, Velázquez Vega JE, et al. Human Mesenchymal glioblastomas are characterized by an increased immune cell presence compared to Proneural and Classical tumors. Oncoimmunology. 2019;8(11): e1655360.
Metelli A, Salem M, Wallace CH, Wu BX, Li A, Li X, et al. Immunoregulatory functions and the therapeutic implications of GARP-TGF-β in inflammation and cancer. J Hematol Oncol. 2018;11(1):24.
Kreatsoulas D, Bolyard C, Wu BX, Cam H, Giglio P, Li Z. Translational landscape of glioblastoma immunotherapy for physicians: guiding clinical practice with basic scientific evidence. J Hematol Oncol. 2022;15(1):80.
Geraldo LHM, Garcia C, da Fonseca ACC, Dubois LGF, de Sampaio ESTCL, Matias D, et al. Glioblastoma Therapy in the Age of Molecular Medicine. Trends Cancer. 2019;5(1):46–65.
Preddy I, Nandoliya K, Miska J, Ahmed AU. Checkpoint: Inspecting the barriers in glioblastoma immunotherapies. Semin Cancer Biol. 2022;86(Pt 3):473–81.
Guo Q, Shen S, Guan G, Zhu C, Zou C, Cao J, et al. Cancer cell intrinsic TIM-3 induces glioblastoma progression. iScience. 2022;25(11):105329.
Ausejo-Mauleon I, Labiano S, de la Nava D, Laspidea V, Zalacain M, Marrodán L, et al. TIM-3 blockade in diffuse intrinsic pontine glioma models promotes tumor regression and antitumor immune memory. Cancer Cell. 2023.
Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin Cancer Res. 2017;23(1):124–36.
Lyu Y, Yang H, Chen L. Metabolic regulation on the immune environment of glioma through gut microbiota. Semin Cancer Biol. 2022;86(Pt 2):990–7.
Montecino-Rodriguez E, Berent-Maoz B, Dorshkind K. Causes, consequences, and reversal of immune system aging. J Clin Invest. 2013;123(3):958–65.
Gustafson MP, Lin Y, New KC, Bulur PA, O’Neill BP, Gastineau DA, et al. Systemic immune suppression in glioblastoma: the interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol. 2010;12(7):631–44.
Otvos B, Alban TJ, Grabowski MM, Bayik D, Mulkearns-Hubert EE, Radivoyevitch T, et al. Preclinical Modeling of Surgery and Steroid Therapy for Glioblastoma Reveals Changes in Immunophenotype that are Associated with Tumor Growth and Outcome. Clin Cancer Res. 2021;27(7):2038–49.
Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med. 2018;24(9):1459–68.
Chae M, Peterson TE, Balgeman A, Chen S, Zhang L, Renner DN, et al. Increasing glioma-associated monocytes leads to increased intratumoral and systemic myeloid-derived suppressor cells in a murine model. Neuro Oncol. 2015;17(7):978–91.
Steeg PS. The blood-tumour barrier in cancer biology and therapy. Nat Rev Clin Oncol. 2021;18(11):696–714.
van Tellingen O, Yetkin-Arik B, de Gooijer MC, Wesseling P, Wurdinger T, de Vries HE. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat. 2015;19:1–12.
Zang LL, Wang XJ, Li XB, Wang SQ, Xu WR, Xie XB, et al. SAHA-based novel HDAC inhibitor design by core hopping method. J Mol Graph Model. 2014;54:10–8.
Liau BB, Sievers C, Donohue LK, Gillespie SM, Flavahan WA, Miller TE, et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell. 2017;20(2):233-46.e7.
de Souza CF, Sabedot TS, Malta TM, Stetson L, Morozova O, Sokolov A, et al. A Distinct DNA Methylation Shift in a Subset of Glioma CpG Island Methylator Phenotypes during Tumor Recurrence. Cell Rep. 2018;23(2):637–51.
Uddin MS, Mamun AA, Alghamdi BS, Tewari D, Jeandet P, Sarwar MS, et al. Epigenetics of glioblastoma multiforme: From molecular mechanisms to therapeutic approaches. Semin Cancer Biol. 2022;83:100–20.
Bazzoni R, Bentivegna A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers (Basel). 2019;11(3).
Rimkus TK, Carpenter RL, Sirkisoon S, Zhu D, Pasche BC, Chan MD, et al. Truncated Glioma-Associated Oncogene Homolog 1 (tGLI1) Mediates Mesenchymal Glioblastoma via Transcriptional Activation of CD44. Cancer Res. 2018;78(10):2589–600.
Wang G, Shen J, Sun J, Jiang Z, Fan J, Wang H, et al. Cyclophilin A Maintains Glioma-Initiating Cell Stemness by Regulating Wnt/β-Catenin Signaling. Clin Cancer Res. 2017;23(21):6640–9.
Gangoso E, Southgate B, Bradley L, Rus S, Galvez-Cancino F, McGivern N, et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell. 2021;184(9):2454-70.e26.
Dong F, Qin X, Wang B, Li Q, Hu J, Cheng X, et al. ALKBH5 Facilitates Hypoxia-Induced Paraspeckle Assembly and IL8 Secretion to Generate an Immunosuppressive Tumor Microenvironment. Cancer Res. 2021;81(23):5876–88.
Qiu Z, Zhao L, Shen JZ, Liang Z, Wu Q, Yang K, et al. Transcription Elongation Machinery Is a Druggable Dependency and Potentiates Immunotherapy in Glioblastoma Stem Cells. Cancer Discov. 2022;12(2):502–21.
Goswami S, Raychaudhuri D, Singh P, Natarajan SM, Chen Y, Poon C, et al. Myeloid-specific KDM6B inhibition sensitizes glioblastoma to PD1 blockade. Nat Cancer. 2023.
Balaton BP, Brown CJ. Escape Artists of the X Chromosome. Trends Genet. 2016;32(6):348–59.
Lee J, Nicosia M, Hong ES, Silver DJ, Li C, Bayik D, et al. Sex-Biased T-cell Exhaustion Drives Differential Immune Responses in Glioblastoma. Cancer Discov. 2023;13(9):2090–105.
Cheng S, Liu L, Wang D, Li Y, Li S, Yuan J, et al. Upregulation of the ZNF148/PTX3 axis promotes malignant transformation of dendritic cells in glioma stem-like cells microenvironment. CNS Neurosci Ther. 2023;29(9):2690–704.
Jiang N, Xie B, Xiao W, Fan M, Xu S, Duan Y, et al. Fatty acid oxidation fuels glioblastoma radioresistance with CD47-mediated immune evasion. Nat Commun. 2022;13(1):1511.
Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2(31):31ra4.
Mekala JR, Kurappalli RK, Ramalingam P, Moparthi NR. N-acetyl l-aspartate and Triacetin modulate tumor suppressor MicroRNA and class I and II HDAC gene expression induce apoptosis in Glioblastoma cancer cells in vitro. Life Sci. 2021;286: 120024.
Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575–80.
Torrini C, Nguyen TTT, Shu C, Mela A, Humala N, Mahajan A, et al. Lactate is an epigenetic metabolite that drives survival in model systems of glioblastoma. Mol Cell. 2022;82(16):3061-76.e6.
Li L, Li Z, Meng X, Wang X, Song D, Liu Y, et al. Histone lactylation-derived LINC01127 promotes the self-renewal of glioblastoma stem cells via the cis-regulating the MAP4K4 to activate JNK pathway. Cancer Lett. 2023;579: 216467.
Zhang Z, Li X, Yang F, Chen C, Liu P, Ren Y, et al. DHHC9-mediated GLUT1 S-palmitoylation promotes glioblastoma glycolysis and tumorigenesis. Nat Commun. 2021;12(1):5872.
Xia L, Liu JY, Zheng ZZ, Chen YJ, Ding JC, Hu YH, et al. BRD4 inhibition boosts the therapeutic effects of epidermal growth factor receptor-targeted chimeric antigen receptor T cells in glioblastoma. Mol Ther. 2021;29(10):3011–26.
Dong Z, Liu Y, Wang C, Hao Y, Fan Q, Yang Z, et al. Tumor Microenvironment Modulating CaCO(3)-based Colloidosomal Microreactors Can Generally Reinforce Cancer Immunotherapy. Adv Mater. 2023:e2308254.
Donati B, Lorenzini E, Ciarrocchi A. BRD4 and Cancer: going beyond transcriptional regulation. Mol Cancer. 2018;17(1):164.
Gusyatiner O, Bady P, Pham MDT, Lei Y, Park J, Daniel RT, et al. BET inhibitors repress expression of interferon-stimulated genes and synergize with HDAC inhibitors in glioblastoma. Neuro Oncol. 2021;23(10):1680–92.
Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell. 2018;33(3):401-16.e8.
Haase S, Banerjee K, Mujeeb AA, Hartlage CS, Núñez FM, Núñez FJ, et al. H3.3-G34 mutations impair DNA repair and promote cGAS/STING-mediated immune responses in pediatric high-grade glioma models. J Clin Invest. 2022;132(22).
Chen P, Hsu WH, Chang A, Tan Z, Lan Z, Zhou A, et al. Circadian Regulator CLOCK Recruits Immune-Suppressive Microglia into the GBM Tumor Microenvironment. Cancer Discov. 2020;10(3):371–81.
Wang Z, Hu P, Tang F, Lian H, Chen X, Zhang Y, et al. HDAC6 promotes cell proliferation and confers resistance to temozolomide in glioblastoma. Cancer Lett. 2016;379(1):134–42.
Dejaegher J, Solie L, Hunin Z, Sciot R, Capper D, Siewert C, et al. DNA methylation based glioblastoma subclassification is related to tumoral T-cell infiltration and patient survival. Neuro Oncol. 2021;23(2):240–50.
Bayik D, Bartels CF, Lovrenert K, Watson DC, Zhang D, Kay K, et al. Distinct Cell Adhesion Signature Defines Glioblastoma Myeloid-Derived Suppressor Cell Subsets. Cancer Res. 2022;82(22):4274–87.
Yin Y, Qiu S, Li X, Huang B, Xu Y, Peng Y. EZH2 suppression in glioblastoma shifts microglia toward M1 phenotype in tumor microenvironment. J Neuroinflammation. 2017;14(1):220.
Xiong W, Li C, Wan B, Zheng Z, Zhang Y, Wang S, et al. N6-Methyladenosine Regulator-Mediated Immue Patterns and Tumor Microenvironment Infiltration Characterization in Glioblastoma. Front Immunol. 2022;13: 819080.
Tompa M, Kraboth Z, Galik B, Kajtar B, Gyenesei A, Kalman B. Epigenetic Suppression of the IL-7 Pathway in Progressive Glioblastoma. Biomedicines. 2022;10(9).
Ratnam NM, Sonnemann HM, Frederico SC, Chen H, Hutchinson MND, Dowdy T, et al. Reversing Epigenetic Gene Silencing to Overcome Immune Evasion in CNS Malignancies. Front Oncol. 2021;11: 719091.
Ferreira WAS, Vitiello GAF, da Silva MT, de Oliveira EHC. Comprehensive analysis of epigenetics regulation, prognostic and the correlation with immune infiltrates of GPX7 in adult gliomas. Sci Rep. 2022;12(1):6442.
Hansen LJ, Yang R, Roso K, Wang W, Chen L, Yang Q, et al. MTAP loss correlates with an immunosuppressive profile in GBM and its substrate MTA stimulates alternative macrophage polarization. Sci Rep. 2022;12(1):4183.
Wu X, Wan Q, Wang J, Hou P, Zhang Q, Wang Q, et al. Epigenetic Activation of lncRNA MIR155HG Mediated by Promoter Hypomethylation and SP1 is Correlated with Immune Infiltration in Glioma. Onco Targets Ther. 2022;15:219–35.
Wiencke JK, Accomando WP, Zheng S, Patoka J, Dou X, Phillips JJ, et al. Epigenetic biomarkers of T-cells in human glioma. Epigenetics. 2012;7(12):1391–402.
Bam M, Chintala S, Fetcko K, Williamsen BC, Siraj S, Liu S, et al. Genome wide DNA methylation landscape reveals glioblastoma’s influence on epigenetic changes in tumor infiltrating CD4+ T cells. Oncotarget. 2021;12(10):967–81.
Yi L, Cui Y, Xu Q, Jiang Y. Stabilization of LSD1 by deubiquitinating enzyme USP7 promotes glioblastoma cell tumorigenesis and metastasis through suppression of the p53 signaling pathway. Oncol Rep. 2016;36(5):2935–45.
Yoon S, Wu X, Armstrong B, Habib N, Rossi JJ. An RNA Aptamer Targeting the Receptor Tyrosine Kinase PDGFRα Induces Anti-tumor Effects through STAT3 and p53 in Glioblastoma. Mol Ther Nucleic Acids. 2019;14:131–41.
Saidi D, Cheray M, Osman AM, Stratoulias V, Lindberg OR, Shen X, et al. Glioma-induced SIRT1-dependent activation of hMOF histone H4 lysine 16 acetyltransferase in microglia promotes a tumor supporting phenotype. Oncoimmunology. 2018;7(2): e1382790.
Rao A, Barkley D, França GS, Yanai I. Exploring tissue architecture using spatial transcriptomics. Nature. 2021;596(7871):211–20.
Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin Cancer Res. 2018;24(17):4175–86.
Mohme M, Schliffke S, Maire CL, Rünger A, Glau L, Mende KC, et al. Immunophenotyping of Newly Diagnosed and Recurrent Glioblastoma Defines Distinct Immune Exhaustion Profiles in Peripheral and Tumor-infiltrating Lymphocytes. Clin Cancer Res. 2018;24(17):4187–200.
Zhong J, Yang X, Chen J, He K, Gao X, Wu X, et al. Circular EZH2-encoded EZH2-92aa mediates immune evasion in glioblastoma via inhibition of surface NKG2D ligands. Nat Commun. 2022;13(1):4795.
Yan J, Zhao Q, Gabrusiewicz K, Kong LY, Xia X, Wang J, et al. FGL2 promotes tumor progression in the CNS by suppressing CD103(+) dendritic cell differentiation. Nat Commun. 2019;10(1):448.
Pellegatta S, Savoldo B, Di Ianni N, Corbetta C, Chen Y, Patané M, et al. Constitutive and TNFα-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci Transl Med. 2018;10(430).
Mohapatra SR, Sadik A, Tykocinski LO, Dietze J, Poschet G, Heiland I, et al. Hypoxia Inducible Factor 1α Inhibits the Expression of Immunosuppressive Tryptophan-2,3-Dioxygenase in Glioblastoma. Front Immunol. 2019;10:2762.
Yu X, Jin J, Zheng Y, Zhu H, Xu H, Ma J, et al. GBP5 drives malignancy of glioblastoma via the Src/ERK1/2/MMP3 pathway. Cell Death Dis. 2021;12(2):203.
Wightman SM, Alban TJ, Chen X, Lathia JD, Wang Y, Stark GR. Bazedoxifene inhibits sustained STAT3 activation and increases survival in GBM. Transl Oncol. 2021;14(11): 101192.
Han MZ, Wang S, Zhao WB, Ni SL, Yang N, Kong Y, et al. Immune checkpoint molecule herpes virus entry mediator is overexpressed and associated with poor prognosis in human glioblastoma. EBioMedicine. 2019;43:159–70.
Sordo-Bahamonde C, Lorenzo-Herrero S, Granda-Díaz R, Martínez-Pérez A, Aguilar-García C, Rodrigo JP, et al. Beyond the anti-PD-1/PD-L1 era: promising role of the BTLA/HVEM axis as a future target for cancer immunotherapy. Mol Cancer. 2023;22(1):142.
Auzmendi-Iriarte J, Otaegi-Ugartemendia M, Carrasco-Garcia E, Azkargorta M, Diaz A, Saenz-Antoñanzas A, et al. Chaperone-Mediated Autophagy Controls Proteomic and Transcriptomic Pathways to Maintain Glioma Stem Cell Activity. Cancer Res. 2022;82(7):1283–97.
Li X, Su W, Wu H, Xu J, Tang H, Chen X, et al. FOXM1 maintains fatty acid homoeostasis through the SET7-H3K4me1-FASN axis. Cell Death Discov. 2023;9(1):310.
De Martino M, Daviaud C, Minns HE, Lazarian A, Wacker A, Costa AP, et al. Radiation therapy promotes unsaturated fatty acids to maintain survival of glioblastoma. Cancer Lett. 2023;570: 216329.
Minami JK, Morrow D, Bayley NA, Fernandez EG, Salinas JJ, Tse C, et al. CDKN2A deletion remodels lipid metabolism to prime glioblastoma for ferroptosis. Cancer Cell. 2023;41(6):1048-60.e9.
Geraldo LH, Xu Y, Jacob L, Pibouin-Fragner L, Rao R, Maissa N, et al. SLIT2/ROBO signaling in tumor-associated microglia and macrophages drives glioblastoma immunosuppression and vascular dysmorphia. J Clin Invest. 2021;131(16).
Ham SW, Jeon HY, Jin X, Kim EJ, Kim JK, Shin YJ, et al. TP53 gain-of-function mutation promotes inflammation in glioblastoma. Cell Death Differ. 2019;26(3):409–25.
Sielska M, Przanowski P, Pasierbińska M, Wojnicki K, Poleszak K, Wojtas B, et al. Tumour-derived CSF2/granulocyte macrophage colony stimulating factor controls myeloid cell accumulation and progression of gliomas. Br J Cancer. 2020;123(3):438–48.
Palumbo P, Lombardi F, Augello FR, Giusti I, Dolo V, Leocata P, et al. Biological effects of selective COX-2 inhibitor NS398 on human glioblastoma cell lines. Cancer Cell Int. 2020;20:167.
Chen P, Zhao D, Li J, Liang X, Li J, Chang A, et al. Symbiotic Macrophage-Glioma Cell Interactions Reveal Synthetic Lethality in PTEN-Null Glioma. Cancer Cell. 2019;35(6):868-84.e6.
Yee PP, Wei Y, Kim SY, Lu T, Chih SY, Lawson C, et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat Commun. 2020;11(1):5424.
Yoon J, Grinchuk OV, Kannan S, Ang MJY, Li Z, Tay EXY, et al. A chemical biology approach reveals a dependency of glioblastoma on biotin distribution. Sci Adv. 2021;7(36):eabf6033.
Bhat KPL, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell. 2013;24(3):331–46.
Cheng X, Geng F, Pan M, Wu X, Zhong Y, Wang C, et al. Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress. Cell Metab. 2020;32(2):229-42.e8.
Choi SH, Tamura K, Khajuria RK, Bhere D, Nesterenko I, Lawler J, et al. Antiangiogenic variant of TSP-1 targets tumor cells in glioblastomas. Mol Ther. 2015;23(2):235–43.
Zhang XN, Yang KD, Chen C, He ZC, Wang QH, Feng H, et al. Pericytes augment glioblastoma cell resistance to temozolomide through CCL5-CCR5 paracrine signaling. Cell Res. 2021;31(10):1072–87.
Rodero M, Marie Y, Coudert M, Blondet E, Mokhtari K, Rousseau A, et al. Polymorphism in the microglial cell-mobilizing CX3CR1 gene is associated with survival in patients with glioblastoma. J Clin Oncol. 2008;26(36):5957–64.
Alban TJ, Bayik D, Otvos B, Rabljenovic A, Leng L, Jia-Shiun L, et al. Glioblastoma Myeloid-Derived Suppressor Cell Subsets Express Differential Macrophage Migration Inhibitory Factor Receptor Profiles That Can Be Targeted to Reduce Immune Suppression. Front Immunol. 2020;11:1191.
Parik S, Fernández-García J, Lodi F, De Vlaminck K, Derweduwe M, De Vleeschouwer S, et al. GBM tumors are heterogeneous in their fatty acid metabolism and modulating fatty acid metabolism sensitizes cancer cells derived from recurring GBM tumors to temozolomide. Front Oncol. 2022;12: 988872.
Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011;1(5):442–56.
Villa GR, Hulce JJ, Zanca C, Bi J, Ikegami S, Cahill GL, et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell. 2016;30(5):683–93.
Chen A, Jiang Y, Li Z, Wu L, Santiago U, Zou H, et al. Chitinase-3-like 1 protein complexes modulate macrophage-mediated immune suppression in glioblastoma. J Clin Invest. 2021;131(16).
Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–72.
Yeini E, Ofek P, Pozzi S, Albeck N, Ben-Shushan D, Tiram G, et al. P-selectin axis plays a key role in microglia immunophenotype and glioblastoma progression. Nat Commun. 2021;12(1):1912.
Iwata R, Hyoung Lee J, Hayashi M, Dianzani U, Ofune K, Maruyama M, et al. ICOSLG-mediated regulatory T-cell expansion and IL-10 production promote progression of glioblastoma. Neuro Oncol. 2020;22(3):333–44.
Yin J, Kim SS, Choi E, Oh YT, Lin W, Kim TH, et al. ARS2/MAGL signaling in glioblastoma stem cells promotes self-renewal and M2-like polarization of tumor-associated macrophages. Nat Commun. 2020;11(1):2978.
Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao CC, et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci. 2019;22(5):729–40.
Hu J, Zhao Q, Kong LY, Wang J, Yan J, Xia X, et al. Regulation of tumor immune suppression and cancer cell survival by CXCL1/2 elevation in glioblastoma multiforme. Sci Adv. 2021;7(5).
Pang L, Guo S, Khan F, Dunterman M, Ali H, Liu Y, et al. Hypoxia-driven protease legumain promotes immunosuppression in glioblastoma. Cell Rep Med. 2023:101238.
Yuan W, Zhang Q, Gu D, Lu C, Dixit D, Gimple RC, et al. Dual Role of CXCL8 in Maintaining the Mesenchymal State of Glioblastoma Stem Cells and M2-Like Tumor-Associated Macrophages. Clin Cancer Res. 2023;29(18):3779–92.
Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25(3):462–9.
Chen CH, Wei KC, Liao WC, Lin YY, Chen HC, Feng LY, et al. Prognostic value of an APOBEC3 deletion polymorphism for glioma patients in Taiwan. J Neurosurg. 2023;138(5):1325–37.
Johnson A, Severson E, Gay L, Vergilio JA, Elvin J, Suh J, et al. Comprehensive Genomic Profiling of 282 Pediatric Low- and High-Grade Gliomas Reveals Genomic Drivers, Tumor Mutational Burden, and Hypermutation Signatures. Oncologist. 2017;22(12):1478–90.
Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7–H1 expression and immunoresistance in glioma. Nat Med. 2007;13(1):84–8.
Guo X, Qiu W, Li B, Qi Y, Wang S, Zhao R, et al. Hypoxia-induced neuronal activity in glioma patients polarizes microglia by potentiating RNA m6A demethylation. Clin Cancer Res. 2023.
Di Tomaso T, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A, et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res. 2010;16(3):800–13.
Waight JD, Chand D, Dietrich S, Gombos R, Horn T, Gonzalez AM, et al. Selective FcγR Co-engagement on APCs Modulates the Activity of Therapeutic Antibodies Targeting T Cell Antigens. Cancer Cell. 2018;33(6):1033-47.e5.
Ni X, Wu W, Sun X, Ma J, Yu Z, He X, et al. Interrogating glioma-M2 macrophage interactions identifies Gal-9/Tim-3 as a viable target against PTEN-null glioblastoma. Sci Adv. 2022;8(27):eabl5165.
Newman JP, Wang GY, Arima K, Guan SP, Waters MR, Cavenee WK, et al. Interleukin-13 receptor alpha 2 cooperates with EGFRvIII signaling to promote glioblastoma multiforme. Nat Commun. 2017;8(1):1913.
Steven A, Seliger B. Control of CREB expression in tumors: from molecular mechanisms and signal transduction pathways to therapeutic target. Oncotarget. 2016;7(23):35454–65.
Chandramohan V, Bao X, Keir ST, Pegram CN, Szafranski SE, Piao H, et al. Construction of an immunotoxin, D2C7-(scdsFv)-PE38KDEL, targeting EGFRwt and EGFRvIII for brain tumor therapy. Clin Cancer Res. 2013;19(17):4717–27.
Zhai L, Bell A, Ladomersky E, Lauing KL, Bollu L, Nguyen B, et al. Tumor Cell IDO Enhances Immune Suppression and Decreases Survival Independent of Tryptophan Metabolism in Glioblastoma. Clin Cancer Res. 2021;27(23):6514–28.
Pilanc P, Wojnicki K, Roura AJ, Cyranowski S, Ellert-Miklaszewska A, Ochocka N, et al. A Novel Oral Arginase 1/2 Inhibitor Enhances the Antitumor Effect of PD-1 Inhibition in Murine Experimental Gliomas by Altering the Immunosuppressive Environment. Front Oncol. 2021;11: 703465.
Ye L, Park JJ, Dong MB, Yang Q, Chow RD, Peng L, et al. In vivo CRISPR screening in CD8 T cells with AAV-Sleeping Beauty hybrid vectors identifies membrane targets for improving immunotherapy for glioblastoma. Nat Biotechnol. 2019;37(11):1302–13.
Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012;109(17):6662–7.
Wang M, Jia J, Cui Y, Peng Y, Jiang Y. CD73-positive extracellular vesicles promote glioblastoma immunosuppression by inhibiting T-cell clonal expansion. Cell Death Dis. 2021;12(11):1065.
Liu G, Ying H, Zeng G, Wheeler CJ, Black KL, Yu JS. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. 2004;64(14):4980–6.
Liu G, Yu JS, Zeng G, Yin D, Xie D, Black KL, et al. AIM-2: a novel tumor antigen is expressed and presented by human glioma cells. J Immunother. 2004;27(3):220–6.
Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. 2010;17(4):362–75.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96.
Bleeker FE, Atai NA, Lamba S, Jonker A, Rijkeboer D, Bosch KS, et al. The prognostic IDH1( R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010;119(4):487–94.
Sigmond J, Honeywell RJ, Postma TJ, Dirven CM, de Lange SM, van der Born K, et al. Gemcitabine uptake in glioblastoma multiforme: potential as a radiosensitizer. Ann Oncol. 2009;20(1):182–7.
Wei J, Marisetty A, Schrand B, Gabrusiewicz K, Hashimoto Y, Ott M, et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J Clin Invest. 2019;129(1):137–49.
Sa JK, Chang N, Lee HW, Cho HJ, Ceccarelli M, Cerulo L, et al. Transcriptional regulatory networks of tumor-associated macrophages that drive malignancy in mesenchymal glioblastoma. Genome Biol. 2020;21(1):216.
Minami N, Hong D, Stevers N, Barger CJ, Radoul M, Hong C, et al. Imaging biomarkers of TERT or GABPB1 silencing in TERT-positive glioblastoma. Neuro Oncol. 2022;24(11):1898–910.
Goffart N, Lombard A, Lallemand F, Kroonen J, Nassen J, Di Valentin E, et al. CXCL12 mediates glioblastoma resistance to radiotherapy in the subventricular zone. Neuro Oncol. 2017;19(1):66–77.
Gai QJ, Fu Z, He J, Mao M, Yao XX, Qin Y, et al. EPHA2 mediates PDGFA activity and functions together with PDGFRA as prognostic marker and therapeutic target in glioblastoma. Signal Transduct Target Ther. 2022;7(1):33.
Kim Y, Kim E, Wu Q, Guryanova O, Hitomi M, Lathia JD, et al. Platelet-derived growth factor receptors differentially inform intertumoral and intratumoral heterogeneity. Genes Dev. 2012;26(11):1247–62.
Suvà ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell. 2014;157(3):580–94.
Koschmann C, Calinescu AA, Nunez FJ, Mackay A, Fazal-Salom J, Thomas D, et al. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci Transl Med. 2016;8(328):328ra28.
Sun X, Klingbeil O, Lu B, Wu C, Ballon C, Ouyang M, et al. BRD8 maintains glioblastoma by epigenetic reprogramming of the p53 network. Nature. 2023;613(7942):195–202.
Al-Mayhani TF, Heywood RM, Vemireddy V, Lathia JD, Piccirillo SGM, Watts C. A non-hierarchical organization of tumorigenic NG2 cells in glioblastoma promoted by EGFR. Neuro Oncol. 2019;21(6):719–29.
Cho HJ, Zhao J, Jung SW, Ladewig E, Kong DS, Suh YL, et al. Distinct genomic profile and specific targeted drug responses in adult cerebellar glioblastoma. Neuro Oncol. 2019;21(1):47–58.
El Hindy N, Keyvani K, Pagenstecher A, Dammann P, Sandalcioglu IE, Sure U, et al. Implications of Dll4-Notch signaling activation in primary glioblastoma multiforme. Neuro Oncol. 2013;15(10):1366–78.
Park NI, Guilhamon P, Desai K, McAdam RF, Langille E, O’Connor M, et al. ASCL1 Reorganizes Chromatin to Direct Neuronal Fate and Suppress Tumorigenicity of Glioblastoma Stem Cells. Cell Stem Cell. 2017;21(2):209-24.e7.
Xia X, Li X, Li F, Wu X, Zhang M, Zhou H, et al. A novel tumor suppressor protein encoded by circular AKT3 RNA inhibits glioblastoma tumorigenicity by competing with active phosphoinositide-dependent Kinase-1. Mol Cancer. 2019;18(1):131.
Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell. 2012;22(1):21–35.
Zhou Y, Xiao D, Jiang X, Nie C. EREG is the core onco-immunological biomarker of cuproptosis and mediates the cross-talk between VEGF and CD99 signaling in glioblastoma. J Transl Med. 2023;21(1):28.
Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, et al. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med. 2011;208(2):313–26.
Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455(7216):1129–33.
Maus A, Peters GJ. Glutamate and α-ketoglutarate: key players in glioma metabolism. Amino Acids. 2017;49(1):21–32.
Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Cell Biol. 2015;17(12):1556–68.
Marin-Valencia I, Yang C, Mashimo T, Cho S, Baek H, Yang XL, et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012;15(6):827–37.
Yuan F, Sun Q, Zhang S, Ye L, Xu Y, Deng G, et al. The dual role of p62 in ferroptosis of glioblastoma according to p53 status. Cell Biosci. 2022;12(1):20.
Phillips RE, Soshnev AA, Allis CD. Epigenomic Reprogramming as a Driver of Malignant Glioma. Cancer Cell. 2020;38(5):647–60.
Wu W, Yu T, Wu Y, Tian W, Zhang J, Wang Y. The miR155HG/miR-185/ANXA2 loop contributes to glioblastoma growth and progression. J Exp Clin Cancer Res. 2019;38(1):133.
Verdugo E, Puerto I, Medina M. An update on the molecular biology of glioblastoma, with clinical implications and progress in its treatment. Cancer Commun (Lond). 2022;42(11):1083–111.
Libby CJ, Gc S, Benavides GA, Fisher JL, Williford SE, Zhang S, et al. A role for GLUT3 in glioblastoma cell invasion that is not recapitulated by GLUT1. Cell Adh Migr. 2021;15(1):101–15.
Sanborn RE, Pishvaian MJ, Callahan MK, Weise A, Sikic BI, Rahma O, et al. Safety, tolerability and efficacy of agonist anti-CD27 antibody (varlilumab) administered in combination with anti-PD-1 (nivolumab) in advanced solid tumors. J Immunother Cancer. 2022;10(8).
Oji Y, Hashimoto N, Tsuboi A, Murakami Y, Iwai M, Kagawa N, et al. Association of WT1 IgG antibody against WT1 peptide with prolonged survival in glioblastoma multiforme patients vaccinated with WT1 peptide. Int J Cancer. 2016;139(6):1391–401.
Cen L, Carlson BL, Schroeder MA, Ostrem JL, Kitange GJ, Mladek AC, et al. p16-Cdk4-Rb axis controls sensitivity to a cyclin-dependent kinase inhibitor PD0332991 in glioblastoma xenograft cells. Neuro Oncol. 2012;14(7):870–81.
Verreault M, Schmitt C, Goldwirt L, Pelton K, Haidar S, Levasseur C, et al. Preclinical Efficacy of the MDM2 Inhibitor RG7112 in MDM2-Amplified and TP53 Wild-type Glioblastomas. Clin Cancer Res. 2016;22(5):1185–96.
Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337(6099):1231–5.
Hu LS, Ning S, Eschbacher JM, Baxter LC, Gaw N, Ranjbar S, et al. Radiogenomics to characterize regional genetic heterogeneity in glioblastoma. Neuro Oncol. 2017;19(1):128–37.
Chakraborty S, Li L, Tang H, Xie Y, Puliyappadamba VT, Raisanen J, et al. Cytoplasmic TRADD confers a worse prognosis in glioblastoma. Neoplasia. 2013;15(8):888–97.
Peng G, Yuan X, Yuan J, Liu Q, Dai M, Shen C, et al. miR-25 promotes glioblastoma cell proliferation and invasion by directly targeting NEFL. Mol Cell Biochem. 2015;409(1–2):103–11.
Belotti Y, Tolomeo S, Yu R, Lim WT, Lim CT. Prognostic Neurotransmitter Receptors Genes Are Associated with Immune Response, Inflammation and Cancer Hallmarks in Brain Tumors. Cancers (Basel). 2022;14(10).
Chen J, Wang H, Deng C, Fei M. SLC12A5 as a novel potential biomarker of glioblastoma multiforme. Mol Biol Rep. 2023;50(5):4285–99.
Yang J, Yang Q. Identification of Core Genes and Screening of Potential Targets in Glioblastoma Multiforme by Integrated Bioinformatic Analysis. Front Oncol. 2020;10: 615976.
Lu WC, Xie H, Yuan C, Li JJ, Li ZY, Wu AH. Identification of potential biomarkers and candidate small molecule drugs in glioblastoma. Cancer Cell Int. 2020;20:419.
Méndez O, Zavadil J, Esencay M, Lukyanov Y, Santovasi D, Wang SC, et al. Knock down of HIF-1alpha in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol Cancer. 2010;9:133.
Wu T, Li Y, Chen B. B4GALT3 promotes cell proliferation and invasion in glioblastoma. Neurol Res. 2020;42(6):463–70.
Hormigo A, Gu B, Karimi S, Riedel E, Panageas KS, Edgar MA, et al. YKL-40 and matrix metalloproteinase-9 as potential serum biomarkers for patients with high-grade gliomas. Clin Cancer Res. 2006;12(19):5698–704.
Yu S, Yu X, Sun L, Zheng Y, Chen L, Xu H, et al. GBP2 enhances glioblastoma invasion through Stat3/fibronectin pathway. Oncogene. 2020;39(27):5042–55.
Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23(6):839–52.
Hu C, Leche CA 2nd, Kiyatkin A, Yu Z, Stayrook SE, Ferguson KM, et al. Glioblastoma mutations alter EGFR dimer structure to prevent ligand bias. Nature. 2022;602(7897):518–22.
Zottel A, Šamec N, Kump A, Raspor Dall'Olio LR, Pužar Dominkuš P, Romih R, et al. Analysis of miR-9–5p, miR-124–3p, miR-21–5p, miR-138–5p, and miR-1–3p in Glioblastoma Cell Lines and Extracellular Vesicles. Int J Mol Sci. 2020;21(22).
Ma S, Guo Z, Wang B, Yang M, Yuan X, Ji B, et al. A Computational Framework to Identify Biomarkers for Glioma Recurrence and Potential Drugs Targeting Them. Front Genet. 2021;12: 832627.
Patil SS, Gokulnath P, Bashir M, Shwetha SD, Jaiswal J, Shastry AH, et al. Insulin-like growth factor binding protein-2 regulates β-catenin signaling pathway in glioma cells and contributes to poor patient prognosis. Neuro Oncol. 2016;18(11):1487–97.
Shiina S, Ohno M, Ohka F, Kuramitsu S, Yamamichi A, Kato A, et al. CAR T Cells Targeting Podoplanin Reduce Orthotopic Glioblastomas in Mouse Brains. Cancer Immunol Res. 2016;4(3):259–68.
Yang Y, Yan R, Zhang L, Meng X, Sun W. Primary glioblastoma transcriptome data analysis for screening survival-related genes. J Cell Biochem. 2020;121(2):1901–10.
Crane CA, Austgen K, Haberthur K, Hofmann C, Moyes KW, Avanesyan L, et al. Immune evasion mediated by tumor-derived lactate dehydrogenase induction of NKG2D ligands on myeloid cells in glioblastoma patients. Proc Natl Acad Sci U S A. 2014;111(35):12823–8.
Festuccia C, Mancini A, Colapietro A, Gravina GL, Vitale F, Marampon F, et al. The first-in-class alkylating deacetylase inhibitor molecule tinostamustine shows antitumor effects and is synergistic with radiotherapy in preclinical models of glioblastoma. J Hematol Oncol. 2018;11(1):32.
Tu Z, Ouyang Q, Long X, Wu L, Li J, Zhu X, et al. Protein Disulfide-Isomerase A3 Is a Robust Prognostic Biomarker for Cancers and Predicts the Immunotherapy Response Effectively. Front Immunol. 2022;13: 837512.
Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–31.
Lin Z, Xu H, Yang R, Li Z, Zheng H, Zhang Z, et al. Effective treatment of a BRAF V600E-mutant epithelioid glioblastoma patient by vemurafenib: a case report. Anticancer Drugs. 2022;33(1):100–4.
Garcia I, Aldaregia J, Marjanovic Vicentic J, Aldaz P, Moreno-Cugnon L, Torres-Bayona S, et al. Oncogenic activity of SOX1 in glioblastoma. Sci Rep. 2017;7:46575.
Sang J, Li X, Lv L, Zhang C, Zhang X, Li G. Circ‑TOP2A acts as a ceRNA for miR‑346 and contributes to glioma progression via the modulation of sushi domain‑containing 2. Mol Med Rep. 2021;23(4).
Yu K, Lin CJ, Hatcher A, Lozzi B, Kong K, Huang-Hobbs E, et al. PIK3CA variants selectively initiate brain hyperactivity during gliomagenesis. Nature. 2020;578(7793):166–71.
Mokgautsi N, Kuo YC, Tang SL, Liu FC, Chen SJ, Wu ATH, et al. Anticancer Activities of 9-chloro-6-(piperazin-1-yl)-11H-indeno[1,2-c] quinolin-11-one (SJ10) in Glioblastoma Multiforme (GBM) Chemoradioresistant Cell Cycle-Related Oncogenic Signatures. Cancers (Basel). 2022;14(1).
Zhao H, Zhou X, Yuan G, Hou Z, Sun H, Zhai N, et al. CDC6 is up-regulated and a poor prognostic signature in glioblastoma multiforme. Clin Transl Oncol. 2021;23(3):565–71.
Cui K, Chen JH, Zou YF, Zhang SY, Wu B, Jing K, et al. Hub biomarkers for the diagnosis and treatment of glioblastoma based on microarray technology. Technol Cancer Res Treat. 2021;20:1533033821990368.
Drexler R, Schüller U, Eckhardt A, Filipski K, Hartung TI, Harter PN, et al. DNA methylation subclasses predict the benefit from gross total tumor resection in IDH-wildtype glioblastoma patients. Neuro Oncol. 2023;25(2):315–25.
Wang Z, Gao L, Guo X, Lian W, Deng K, Xing B. Development and Validation of a Novel DNA Methylation-Driven Gene Based Molecular Classification and Predictive Model for Overall Survival and Immunotherapy Response in Patients With Glioblastoma: A Multiomic Analysis. Front Cell Dev Biol. 2020;8: 576996.
Guo C, Liu S, Greenaway F, Sun MZ. Potential role of annexin A7 in cancers. Clin Chim Acta. 2013;423:83–9.
Naqvi AAT, Jairajpuri DS, Hussain A, Hasan GM, Alajmi MF, Hassan MI. Impact of glioblastoma multiforme associated mutations on the structure and function of MAP/microtubule affinity regulating kinase 4. J Biomol Struct Dyn. 2021;39(5):1781–94.
Babic I, Anderson ES, Tanaka K, Guo D, Masui K, Li B, et al. EGFR mutation-induced alternative splicing of Max contributes to growth of glycolytic tumors in brain cancer. Cell Metab. 2013;17(6):1000–8.
Li G, Yang T, Chen Y, Bao J, Wu D, Hu X, et al. USP5 Sustains the Proliferation of Glioblastoma Through Stabilization of CyclinD1. Front Pharmacol. 2021;12: 720307.
Izaguirre DI, Zhu W, Hai T, Cheung HC, Krahe R, Cote GJ. PTBP1-dependent regulation of USP5 alternative RNA splicing plays a role in glioblastoma tumorigenesis. Mol Carcinog. 2012;51(11):895–906.
Kałuzińska-Kołat Ż, Kośla K, Kołat D, Płuciennik E, Bednarek AK. Antineoplastic Nature of WWOX in Glioblastoma Is Mainly a Consequence of Reduced Cell Viability and Invasion. Biology (Basel). 2023;12(3).
Lefave CV, Squatrito M, Vorlova S, Rocco GL, Brennan CW, Holland EC, et al. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. Embo j. 2011;30(19):4084–97.
Sun T, Xu YJ, Jiang SY, Xu Z, Cao BY, Sethi G, et al. Suppression of the USP10/CCND1 axis induces glioblastoma cell apoptosis. Acta Pharmacol Sin. 2021;42(8):1338–46.
Turchi L, Sakakini N, Saviane G, Polo B, Saurty-Seerunghen MS, Gabut M, et al. CELF2 Sustains a Proliferating/OLIG2+ Glioblastoma Cell Phenotype via the Epigenetic Repression of SOX3. Cancers (Basel). 2023;15(20).
Ohno M, Matsuzaki J, Kawauchi J, Aoki Y, Miura J, Takizawa S, et al. Assessment of the Diagnostic Utility of Serum MicroRNA Classification in Patients With Diffuse Glioma. JAMA Netw Open. 2019;2(12): e1916953.
Lu C, Wei Y, Wang X, Zhang Z, Yin J, Li W, et al. DNA-methylation-mediated activating of lncRNA SNHG12 promotes temozolomide resistance in glioblastoma. Mol Cancer. 2020;19(1):28.
Ito H, Watari K, Shibata T, Miyamoto T, Murakami Y, Nakahara Y, et al. Bidirectional Regulation between NDRG1 and GSK3β Controls Tumor Growth and Is Targeted by Differentiation Inducing Factor-1 in Glioblastoma. Cancer Res. 2020;80(2):234–48.
Evans SM, Putt M, Yang XY, Lustig RA, Martinez-Lage M, Williams D, et al. Initial evidence that blood-borne microvesicles are biomarkers for recurrence and survival in newly diagnosed glioblastoma patients. J Neurooncol. 2016;127(2):391–400.
Lu-Emerson C, Duda DG, Emblem KE, Taylor JW, Gerstner ER, Loeffler JS, et al. Lessons from anti-vascular endothelial growth factor and anti-vascular endothelial growth factor receptor trials in patients with glioblastoma. J Clin Oncol. 2015;33(10):1197–213.
Dong Y, Han Q, Zou Y, Deng Z, Lu X, Wang X, et al. Long-term exposure to imatinib reduced cancer stem cell ability through induction of cell differentiation via activation of MAPK signaling in glioblastoma cells. Mol Cell Biochem. 2012;370(1–2):89–102.
Lerner RG, Grossauer S, Kadkhodaei B, Meyers I, Sidorov M, Koeck K, et al. Targeting a Plk1-Controlled Polarity Checkpoint in Therapy-Resistant Glioblastoma-Propagating Cells. Cancer Res. 2015;75(24):5355–66.
Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17(2):170–82.
Zhu Z, Mesci P, Bernatchez JA, Gimple RC, Wang X, Schafer ST, et al. Zika Virus Targets Glioblastoma Stem Cells through a SOX2-Integrin α(v)β(5) Axis. Cell Stem Cell. 2020;26(2):187-204.e10.
Braun CJ, Stanciu M, Boutz PL, Patterson JC, Calligaris D, Higuchi F, et al. Coordinated Splicing of Regulatory Detained Introns within Oncogenic Transcripts Creates an Exploitable Vulnerability in Malignant Glioma. Cancer Cell. 2017;32(4):411-26.e11.
Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. 2016;352(6288):aad3018.
Fan Q, Aksoy O, Wong RA, Ilkhanizadeh S, Novotny CJ, Gustafson WC, et al. A Kinase Inhibitor Targeted to mTORC1 Drives Regression in Glioblastoma. Cancer Cell. 2017;31(3):424–35.
Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25(1):55–7.
Bernhart E, Damm S, Heffeter P, Wintersperger A, Asslaber M, Frank S, et al. Silencing of protein kinase D2 induces glioma cell senescence via p53-dependent and -independent pathways. Neuro Oncol. 2014;16(7):933–45.
Rodón L, Gonzàlez-Juncà A, Inda Mdel M, Sala-Hojman A, Martínez-Sáez E, Seoane J. Active CREB1 promotes a malignant TGFβ2 autocrine loop in glioblastoma. Cancer Discov. 2014;4(10):1230–41.
Lin W, Niu R, Park SM, Zou Y, Kim SS, Xia X, et al. IGFBP5 is an ROR1 ligand promoting glioblastoma invasion via ROR1/HER2-CREB signaling axis. Nat Commun. 2023;14(1):1578.
Lupo KB, Matosevic S. CD155 immunoregulation as a target for natural killer cell immunotherapy in glioblastoma. J Hematol Oncol. 2020;13(1):76.
Raphael I, Kumar R, McCarl LH, Shoger K, Wang L, Sandlesh P, et al. TIGIT and PD-1 Immune Checkpoint Pathways Are Associated With Patient Outcome and Anti-Tumor Immunity in Glioblastoma. Front Immunol. 2021;12: 637146.
Zhao C, Gomez GA, Zhao Y, Yang Y, Cao D, Lu J, et al. ETV2 mediates endothelial transdifferentiation of glioblastoma. Signal Transduct Target Ther. 2018;3:4.
Franceschi S, Corsinovi D, Lessi F, Tantillo E, Aretini P, Menicagli M, et al. Mitochondrial enzyme GLUD2 plays a critical role in glioblastoma progression. EBioMedicine. 2018;37:56–67.
Jin L, Ge H, Long Y, Yang C, Chang YE, Mu L, et al. CD70, a novel target of CAR T-cell therapy for gliomas. Neuro Oncol. 2018;20(1):55–65.
Huang N, Li F, Zhang M, Zhou H, Chen Z, Ma X, et al. An Upstream Open Reading Frame in Phosphatase and Tensin Homolog Encodes a Circuit Breaker of Lactate Metabolism. Cell Metab. 2021;33(1):128-44.e9.
Shan L, Zhu X, Qiu HZ, Zuo ED, Cheng X. Prognostic significance of TMEM131L in glioma and establishment of oxidative stress prognostic model. Front Neurol. 2023;14:1162394.
Hahn WC, Bader JS, Braun TP, Califano A, Clemons PA, Druker BJ, et al. An expanded universe of cancer targets. Cell. 2021;184(5):1142–55.
Sai Krishna AVS, Ramu A, Hariharan S, Sinha S, Donakonda S. Characterization of tumor microenvironment in glioblastoma multiforme identifies ITGB2 as a key immune and stromal related regulator in glial cell types. Comput Biol Med. 2023;165: 107433.
Wang S, Xu X. An Immune-Related Gene Pairs Signature for Predicting Survival in Glioblastoma. Front Oncol. 2021;11: 564960.
Zhang C, Zhang Z, Li F, Shen Z, Qiao Y, Li L, et al. Large-scale analysis reveals the specific clinical and immune features of B7–H3 in glioma. Oncoimmunology. 2018;7(11): e1461304.
Zhang H, Wang Y, Zhao Y, Liu T, Wang Z, Zhang N, et al. PTX3 mediates the infiltration, migration, and inflammation-resolving-polarization of macrophages in glioblastoma. CNS Neurosci Ther. 2022;28(11):1748–66.
Liu S, Zhang C, Maimela NR, Yang L, Zhang Z, Ping Y, et al. Molecular and clinical characterization of CD163 expression via large-scale analysis in glioma. Oncoimmunology. 2019;8(7):1601478.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–7.
Fan Y, Gao Z, Xu J, Wang H, Guo Q, Xue H, et al. Identification and validation of SNHG gene signature to predict malignant behaviors and therapeutic responses in glioblastoma. Front Immunol. 2022;13: 986615.
Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer. 2009;9(1):57–63.
Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, et al. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017;18(1):248–62.
Feng S, Liang X, Li J, Wang Z, Zhang H, Dai Z, et al. Immunogenic cell death related risk model to delineate ferroptosis pathway and predict immunotherapy response of patients with GBM. Front Immunol. 2022;13: 992855.
Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN, et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell. 2018;173(2):338-54.e15.
Bai KH, Zhang YY, Li XP, Tian XP, Pan MM, Wang DW, et al. Comprehensive analysis of tumor necrosis factor-α-inducible protein 8-like 2 (TIPE2): A potential novel pan-cancer immune checkpoint. Comput Struct Biotechnol J. 2022;20:5226–34.
Zhang H, Zhang N, Wu W, Zhou R, Li S, Wang Z, et al. Machine learning-based tumor-infiltrating immune cell-associated lncRNAs for predicting prognosis and immunotherapy response in patients with glioblastoma. Brief Bioinform. 2022;23(6).
Zhao Z, Zhang K-N, Wang Q, Li G, Zeng F, Zhang Y, et al. Chinese Glioma Genome Atlas (CGGA): A Comprehensive Resource with Functional Genomic Data from Chinese Glioma Patients. Genomics Proteomics Bioinformatics. 2021;19(1):1–12.
King JL, Benhabbour SR. Glioblastoma Multiforme-A Look at the Past and a Glance at the Future. Pharmaceutics. 2021;13(7).
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science. 2009;324(5930):1029–33.
Caniglia JL, Jalasutram A, Asuthkar S, Sahagun J, Park S, Ravindra A, et al. Beyond glucose: alternative sources of energy in glioblastoma. Theranostics. 2021;11(5):2048–57.
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30.
Notarangelo G, Spinelli JB, Perez EM, Baker GJ, Kurmi K, Elia I, et al. Oncometabolite d-2HG alters T cell metabolism to impair CD8(+) T cell function. Science. 2022;377(6614):1519–29.
Huang N, Chen Z, Yang X, Gao Y, Zhong J, Li Y, et al. Upstream Open Reading Frame-encoded MP31 Disrupts the Mitochondrial Quality Control Process and Inhibits Tumorigenesis in Glioblastoma. Neuro Oncol. 2023.
Miska J, Lee-Chang C, Rashidi A, Muroski ME, Chang AL, Lopez-Rosas A, et al. HIF-1α Is a Metabolic Switch between Glycolytic-Driven Migration and Oxidative Phosphorylation-Driven Immunosuppression of Tregs in Glioblastoma. Cell Rep. 2019;27(1):226-37.e4.
Wang H, Franco F, Tsui YC, Xie X, Trefny MP, Zappasodi R, et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol. 2020;21(3):298–308.
Gu M, Jiang H, Tan M, Yu L, Xu N, Li Y, et al. Palmitoyltransferase DHHC9 and acyl protein thioesterase APT1 modulate renal fibrosis through regulating β-catenin palmitoylation. Nat Commun. 2023;14(1):6682.
Gabriely G, Quintana FJ. Role of AHR in the control of GBM-associated myeloid cells. Semin Cancer Biol. 2020;64:13–8.
Perng P, Lim M. Immunosuppressive Mechanisms of Malignant Gliomas: Parallels at Non-CNS Sites. Front Oncol. 2015;5:153.
Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478(7368):197–203.
Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A. 2010;107(46):19961–6.
Perus LJM, Walsh LA. Microenvironmental Heterogeneity in Brain Malignancies. Front Immunol. 2019;10:2294.
Bikfalvi A, da Costa CA, Avril T, Barnier JV, Bauchet L, Brisson L, et al. Challenges in glioblastoma research: focus on the tumor microenvironment. Trends Cancer. 2023;9(1):9–27.
Almairac F, Turchi L, Sakakini N, Debruyne DN, Elkeurti S, Gjernes E, et al. ERK-Mediated Loss of miR-199a-3p and Induction of EGR1 Act as a “Toggle Switch” of GBM Cell Dedifferentiation into NANOG- and OCT4-Positive Cells. Can Res. 2020;80(16):3236–50.
Ren Y, Huang Z, Zhou L, Xiao P, Song J, He P, et al. Spatial transcriptomics reveals niche-specific enrichment and vulnerabilities of radial glial stem-like cells in malignant gliomas. Nat Commun. 2023;14(1):1028.
Natsume A, Ito M, Katsushima K, Ohka F, Hatanaka A, Shinjo K, et al. Chromatin regulator PRC2 is a key regulator of epigenetic plasticity in glioblastoma. Cancer Res. 2013;73(14):4559–70.
Jung J, Kim LJ, Wang X, Wu Q, Sanvoranart T, Hubert CG, et al. Nicotinamide metabolism regulates glioblastoma stem cell maintenance. JCI Insight. 2017;2(10).
Seano G. Targeting the perivascular niche in brain tumors. Curr Opin Oncol. 2018;30(1):54–60.
Huang Z, Cheng L, Guryanova OA, Wu Q, Bao S. Cancer stem cells in glioblastoma–molecular signaling and therapeutic targeting. Protein Cell. 2010;1(7):638–55.
Prager BC, Bhargava S, Mahadev V, Hubert CG, Rich JN. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer. 2020;6(3):223–35.
Dréan A, Goldwirt L, Verreault M, Canney M, Schmitt C, Guehennec J, et al. Blood-brain barrier, cytotoxic chemotherapies and glioblastoma. Expert Rev Neurother. 2016;16(11):1285–300.
Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. 2018;20(2):184–91.
Red-Horse K, Crawford Y, Shojaei F, Ferrara N. Endothelium-microenvironment interactions in the developing embryo and in the adult. Dev Cell. 2007;12(2):181–94.
Clavreul A, Menei P. Mesenchymal Stromal-Like Cells in the Glioma Microenvironment: What Are These Cells? Cancers (Basel). 2020;12(9).
Taghipour M, Omidvar A, Razmkhah M, Ghaderi A, Mojtahedi Z. Comparative Proteomic Analysis of Tumor Mesenchymal-Like Stem Cells Derived from High Grade versus Low Grade Gliomas. Cell J. 2017;19(2):250–8.
Clavreul A, Etcheverry A, Tétaud C, Rousseau A, Avril T, Henry C, et al. Identification of two glioblastoma-associated stromal cell subtypes with different carcinogenic properties in histologically normal surgical margins. J Neurooncol. 2015;122(1):1–10.
Shahar T, Rozovski U, Hess KR, Hossain A, Gumin J, Gao F, et al. Percentage of mesenchymal stem cells in high-grade glioma tumor samples correlates with patient survival. Neuro Oncol. 2017;19(5):660–8.
Miroshnikova YA, Mouw JK, Barnes JM, Pickup MW, Lakins JN, Kim Y, et al. Tissue mechanics promote IDH1-dependent HIF1α-tenascin C feedback to regulate glioblastoma aggression. Nat Cell Biol. 2016;18(12):1336–45.
Perrin SL, Samuel MS, Koszyca B, Brown MP, Ebert LM, Oksdath M, et al. Glioblastoma heterogeneity and the tumour microenvironment: implications for preclinical research and development of new treatments. Biochem Soc Trans. 2019;47(2):625–38.
Venkataramani V, Tanev DI, Strahle C, Studier-Fischer A, Fankhauser L, Kessler T, et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature. 2019;573(7775):532–8.
Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell. 2015;161(4):803–16.
Marcus HJ, Carpenter KL, Price SJ, Hutchinson PJ. In vivo assessment of high-grade glioma biochemistry using microdialysis: a study of energy-related molecules, growth factors and cytokines. J Neurooncol. 2010;97(1):11–23.
Robert SM, Buckingham SC, Campbell SL, Robel S, Holt KT, Ogunrinu-Babarinde T, et al. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci Transl Med. 2015;7(289):289ra86.
Dolma S, Selvadurai HJ, Lan X, Lee L, Kushida M, Voisin V, et al. Inhibition of Dopamine Receptor D4 Impedes Autophagic Flux, Proliferation, and Survival of Glioblastoma Stem Cells. Cancer Cell. 2016;29(6):859–73.
Young SZ, Bordey A. GABA’s control of stem and cancer cell proliferation in adult neural and peripheral niches. Physiology (Bethesda). 2009;24:171–85.
Habela CW, Ernest NJ, Swindall AF, Sontheimer H. Chloride accumulation drives volume dynamics underlying cell proliferation and migration. J Neurophysiol. 2009;101(2):750–7.
Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell. 2020;181(7):1643-60.e17.
Aslan K, Turco V, Blobner J, Sonner JK, Liuzzi AR, Núñez NG, et al. Heterogeneity of response to immune checkpoint blockade in hypermutated experimental gliomas. Nat Commun. 2020;11(1):931.
Pombo Antunes AR, Scheyltjens I, Duerinck J, Neyns B, Movahedi K, Van Ginderachter JA. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. Elife. 2020;9.
Xuan W, Khan F, James CD, Heimberger AB, Lesniak MS, Chen P. Circadian regulation of cancer cell and tumor microenvironment crosstalk. Trends Cell Biol. 2021;31(11):940–50.
Xuan W, Lesniak MS, James CD, Heimberger AB, Chen P. Context-Dependent Glioblastoma-Macrophage/Microglia Symbiosis and Associated Mechanisms. Trends Immunol. 2021;42(4):280–92.
van Vlerken-Ysla L, Tyurina YY, Kagan VE, Gabrilovich DI. Functional states of myeloid cells in cancer. Cancer Cell. 2023;41(3):490–504.
Chen P, Hsu WH, Han J, Xia Y, DePinho RA. Cancer Stemness Meets Immunity: From Mechanism to Therapy. Cell Rep. 2021;34(1): 108597.
Chen Z, Ross JL, Hambardzumyan D. Intravital 2-photon imaging reveals distinct morphology and infiltrative properties of glioblastoma-associated macrophages. Proc Natl Acad Sci U S A. 2019;116(28):14254–9.
Friebel E, Kapolou K, Unger S, Núñez NG, Utz S, Rushing EJ, et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell. 2020;181(7):1626-42.e20.
Pombo Antunes AR, Scheyltjens I, Lodi F, Messiaen J, Antoranz A, Duerinck J, et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat Neurosci. 2021;24(4):595–610.
Friedrich M, Sankowski R, Bunse L, Kilian M, Green E, Ramallo Guevara C, et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat Cancer. 2021;2(7):723–40.
Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017;18(1):234.
Hara T, Chanoch-Myers R, Mathewson ND, Myskiw C, Atta L, Bussema L, et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell. 2021;39(6):779-92.e11.
Chen M, Ren R, Lin W, Xiang L, Zhao Z, Shao B. Exploring the oncostatin M (OSM) feed-forward signaling of glioblastoma via STAT3 in pan-cancer analysis. Cancer Cell Int. 2021;21(1):565.
Petković M, Henis M, Heese O, Relógio A. Chronotherapy in Glioblastoma: state of the art and future perspectives. EBioMedicine. 2023;89: 104470.
Xuan W, Hsu WH, Khan F, Dunterman M, Pang L, Wainwright DA, et al. Circadian Regulator CLOCK Drives Immunosuppression in Glioblastoma. Cancer Immunol Res. 2022;10(6):770–84.
Wang Q, He Z, Huang M, Liu T, Wang Y, Xu H, et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2α. Nat Commun. 2018;9(1):559.
Sattiraju A, Kang S, Giotti B, Chen Z, Marallano VJ, Brusco C, et al. Hypoxic niches attract and sequester tumor-associated macrophages and cytotoxic T cells and reprogram them for immunosuppression. Immunity. 2023;56(8):1825-43.e6.
Casati G, Giunti L, Iorio AL, Marturano A, Galli L, Sardi I. Hippo Pathway in Regulating Drug Resistance of Glioblastoma. Int J Mol Sci. 2021;22(24).
Zhang Y, Zhang B, Lv C, Zhang N, Xing K, Wang Z, et al. Single-cell RNA sequencing identifies critical transcription factors of tumor cell invasion induced by hypoxia microenvironment in glioblastoma. Theranostics. 2023;13(11):3744–60.
Cui X, Morales RT, Qian W, Wang H, Gagner JP, Dolgalev I, et al. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials. 2018;161:164–78.
Longhitano L, Vicario N, Forte S, Giallongo C, Broggi G, Caltabiano R, et al. Lactate modulates microglia polarization via IGFBP6 expression and remodels tumor microenvironment in glioblastoma. Cancer Immunol Immunother. 2023;72(1):1–20.
Khan AB, Lee S, Harmanci AS, Patel R, Latha K, Yang Y, et al. CXCR4 expression is associated with proneural-to-mesenchymal transition in glioblastoma. Int J Cancer. 2023;152(4):713–24.
Liu L, Zhou X, Cheng S, Ge Y, Chen B, Shi J, et al. RNA-binding protein DHX9 promotes glioma growth and tumor-associated macrophages infiltration via TCF12. CNS Neurosci Ther. 2023;29(4):988–99.
Shen CK, Huang BR, Yeh WL, Chen CW, Liu YS, Lai SW, et al. Regulatory effects of IL-1β in the interaction of GBM and tumor-associated monocyte through VCAM-1 and ICAM-1. Eur J Pharmacol. 2021;905: 174216.
Shen CK, Huang BR, Charoensaensuk V, Yang LY, Tsai CF, Liu YS, et al. Bradykinin B1 Receptor Affects Tumor-Associated Macrophage Activity and Glioblastoma Progression. Antioxidants (Basel). 2023;12(8).
Magod P, Mastandrea I, Rousso-Noori L, Agemy L, Shapira G, Shomron N, et al. Exploring the longitudinal glioma microenvironment landscape uncovers reprogrammed pro-tumorigenic neutrophils in the bone marrow. Cell Rep. 2021;36(5): 109480.
Wang M, Cai Y, Peng Y, Xu B, Hui W, Jiang Y. Exosomal LGALS9 in the cerebrospinal fluid of glioblastoma patients suppressed dendritic cell antigen presentation and cytotoxic T-cell immunity. Cell Death Dis. 2020;11(10):896.
Chen R, Chen C, Han N, Guo W, Deng H, Wang Y, et al. Annexin-1 is an oncogene in glioblastoma and causes tumour immune escape through the indirect upregulation of interleukin-8. J Cell Mol Med. 2022;26(15):4343–56.
Nakazawa T, Morimoto T, Maeoka R, Matsuda R, Nakamura M, Nishimura F, et al. CIS deletion by CRISPR/Cas9 enhances human primary natural killer cell functions against allogeneic glioblastoma. J Exp Clin Cancer Res. 2023;42(1):205.
Wu P, Guo Y, Xiao L, Yuan J, Tang C, Dong J, et al. LILRB2-containing small extracellular vesicles from glioblastoma promote tumor progression by promoting the formation and expansion of myeloid-derived suppressor cells. Cancer Immunol Immunother. 2023;72(7):2179–93.
Yeo AT, Shah R, Aliazis K, Pal R, Xu T, Zhang P, et al. Driver mutations dictate the immunological landscape and response to checkpoint immunotherapy of glioblastoma. Cancer Immunol Res. 2023.
Kamran N, Chandran M, Lowenstein PR, Castro MG. Immature myeloid cells in the tumor microenvironment: Implications for immunotherapy. Clin Immunol. 2018;189:34–42.
Janjua TI, Rewatkar P, Ahmed-Cox A, Saeed I, Mansfeld FM, Kulshreshtha R, et al. Frontiers in the treatment of glioblastoma: Past, present and emerging. Adv Drug Deliv Rev. 2021;171:108–38.
Abdelfattah N, Kumar P, Wang C, Leu JS, Flynn WF, Gao R, et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat Commun. 2022;13(1):767.
Xiong A, Zhang J, Chen Y, Zhang Y, Yang F. Integrated single-cell transcriptomic analyses reveal that GPNMB-high macrophages promote PN-MES transition and impede T cell activation in GBM. EBioMedicine. 2022;83: 104239.
Venkataramani V, Yang Y, Schubert MC, Reyhan E, Tetzlaff SK, Wißmann N, et al. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell. 2022;185(16):2899-917.e31.
Mancusi R, Monje M. The neuroscience of cancer. Nature. 2023;618(7965):467–79.
Pan Y, Hysinger JD, Barron T, Schindler NF, Cobb O, Guo X, et al. NF1 mutation drives neuronal activity-dependent initiation of optic glioma. Nature. 2021;594(7862):277–82.
Chen P, Wang W, Liu R, Lyu J, Zhang L, Li B, et al. Olfactory sensory experience regulates gliomagenesis via neuronal IGF1. Nature. 2022;606(7914):550–6.
Dumas AA, Pomella N, Rosser G, Guglielmi L, Vinel C, Millner TO, et al. Microglia promote glioblastoma via mTOR-mediated immunosuppression of the tumour microenvironment. Embo j. 2020;39(15): e103790.
Hetze S, Barthel L, Lückemann L, Günther HS, Wülfing C, Salem Y, et al. Taste-immune associative learning amplifies immunopharmacological effects and attenuates disease progression in a rat glioblastoma model. Brain Behav Immun. 2022;106:270–9.
Tejero R, Huang Y, Katsyv I, Kluge M, Lin JY, Tome-Garcia J, et al. Gene signatures of quiescent glioblastoma cells reveal mesenchymal shift and interactions with niche microenvironment. EBioMedicine. 2019;42:252–69.
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–401.
Richards LM, Whitley OKN, MacLeod G, Cavalli FMG, Coutinho FJ, Jaramillo JE, et al. Gradient of Developmental and Injury Response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nat Cancer. 2021;2(2):157–73.
Garcia-Diaz C, Pöysti A, Mereu E, Clements MP, Brooks LJ, Galvez-Cancino F, et al. Glioblastoma cell fate is differentially regulated by the microenvironments of the tumor bulk and infiltrative margin. Cell Rep. 2023;42(5): 112472.
De Silva MI, Stringer BW, Bardy C. Neuronal and tumourigenic boundaries of glioblastoma plasticity. Trends Cancer. 2023;9(3):223–36.
Pang L, Khan F, Heimberger AB, Chen P. Mechanism and therapeutic potential of tumor-immune symbiosis in glioblastoma. Trends Cancer. 2022;8(10):839–54.
González-Tablas Pimenta M, Otero Á, Arandia Guzman DA, Pascual-Argente D, Ruíz Martín L, Sousa-Casasnovas P, et al. Tumor cell and immune cell profiles in primary human glioblastoma: Impact on patient outcome. Brain Pathol. 2021;31(2):365–80.
Wu Y, Yi M, Niu M, Mei Q, Wu K. Myeloid-derived suppressor cells: an emerging target for anticancer immunotherapy. Mol Cancer. 2022;21(1):184.
Valencia JC, Erwin-Cohen RA, Clavijo PE, Allen C, Sanford ME, Day CP, et al. Myeloid-Derived Suppressive Cell Expansion Promotes Melanoma Growth and Autoimmunity by Inhibiting CD40/IL27 Regulation in Macrophages. Cancer Res. 2021;81(23):5977–90.
Yu S, Ren X, Li L. Myeloid-derived suppressor cells in hematologic malignancies: two sides of the same coin. Exp Hematol Oncol. 2022;11(1):43.
Aarts CEM, Kuijpers TW. Neutrophils as myeloid-derived suppressor cells. Eur J Clin Invest. 2018;48(S2): e12989.
Tumino N, Fiore PF, Pelosi A, Moretta L, Vacca P. Myeloid derived suppressor cells in tumor microenvironment: Interaction with innate lymphoid cells. Semin Immunol. 2022;61–64: 101668.
Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016;37(3):208–20.
Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer. 2020;20(11):662–80.
Wu C, Hua Q, Zheng L. Generation of Myeloid Cells in Cancer: The Spleen Matters. Front Immunol. 2020;11:1126.
Tcyganov E, Mastio J, Chen E, Gabrilovich DI. Plasticity of myeloid-derived suppressor cells in cancer. Curr Opin Immunol. 2018;51:76–82.
Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol. 2015;98(6):913–22.
Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;21(8):485–98.
Wang Y, Johnson KCC, Gatti-Mays ME, Li Z. Emerging strategies in targeting tumor-resident myeloid cells for cancer immunotherapy. J Hematol Oncol. 2022;15(1):118.
Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res. 2017;5(1):3–8.
Grover A, Sanseviero E, Timosenko E, Gabrilovich DI. Myeloid-Derived Suppressor Cells: A Propitious Road to Clinic. Cancer Discov. 2021;11(11):2693–706.
Alicea-Torres K, Sanseviero E, Gui J, Chen J, Veglia F, Yu Q, et al. Immune suppressive activity of myeloid-derived suppressor cells in cancer requires inactivation of the type I interferon pathway. Nat Commun. 2021;12(1):1717.
Tcyganov EN, Hanabuchi S, Hashimoto A, Campbell D, Kar G, Slidel TW, et al. Distinct mechanisms govern populations of myeloid-derived suppressor cells in chronic viral infection and cancer. J Clin Invest. 2021;131(16).
Veglia F, Hashimoto A, Dweep H, Sanseviero E, De Leo A, Tcyganov E, et al. Analysis of classical neutrophils and polymorphonuclear myeloid-derived suppressor cells in cancer patients and tumor-bearing mice. J Exp Med. 2021;218(4).
Netherby CS, Messmer MN, Burkard-Mandel L, Colligan S, Miller A, Cortes Gomez E, et al. The Granulocyte Progenitor Stage Is a Key Target of IRF8-Mediated Regulation of Myeloid-Derived Suppressor Cell Production. J Immunol. 2017;198(10):4129–39.
Patel S, Fu S, Mastio J, Dominguez GA, Purohit A, Kossenkov A, et al. Unique pattern of neutrophil migration and function during tumor progression. Nat Immunol. 2018;19(11):1236–47.
Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest. 2013;123(4):1580–9.
Mastio J, Condamine T, Dominguez G, Kossenkov AV, Donthireddy L, Veglia F, et al. Identification of monocyte-like precursors of granulocytes in cancer as a mechanism for accumulation of PMN-MDSCs. J Exp Med. 2019;216(9):2150–69.
Muench DE, Olsson A, Ferchen K, Pham G, Serafin RA, Chutipongtanate S, et al. Mouse models of neutropenia reveal progenitor-stage-specific defects. Nature. 2020;582(7810):109–14.
Strauss L, Sangaletti S, Consonni FM, Szebeni G, Morlacchi S, Totaro MG, et al. RORC1 Regulates Tumor-Promoting “Emergency” Granulo-Monocytopoiesis. Cancer Cell. 2015;28(2):253–69.
Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. 2019;569(7754):73–8.
Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19(2):108–19.
Weber R, Groth C, Lasser S, Arkhypov I, Petrova V, Altevogt P, et al. IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cell Immunol. 2021;359: 104254.
Dai H, Xu H, Wang S, Ma J. Connections between Metabolism and Epigenetic Modification in MDSCs. Int J Mol Sci. 2020;21(19):7356.
Kumar V, Cheng P, Condamine T, Mony S, Languino L, McCaffrey J, et al. CD45 phosphatase regulates the fate of myeloid cells in tumor microenvironment by inhibiting STAT3 activity. The Journal of Immunology. 2016;196(1_Supplement):211.4-.4.
Kwak T, Wang F, Deng H, Condamine T, Kumar V, Perego M, et al. Distinct Populations of Immune-Suppressive Macrophages Differentiate from Monocytic Myeloid-Derived Suppressor Cells in Cancer. Cell Rep. 2020;33(13): 108571.
Su YL, Banerjee S, White SV, Kortylewski M. STAT3 in Tumor-Associated Myeloid Cells: Multitasking to Disrupt Immunity. Int J Mol Sci. 2018;19(6).
Ravi A, Hellmann MD, Arniella MB, Holton M, Freeman SS, Naranbhai V, et al. Genomic and transcriptomic analysis of checkpoint blockade response in advanced non-small cell lung cancer. Nat Genet. 2023.
Li L, Zhang J, Diao W, Wang D, Wei Y, Zhang CY, et al. MicroRNA-155 and MicroRNA-21 promote the expansion of functional myeloid-derived suppressor cells. J Immunol. 2014;192(3):1034–43.
Yu H, Liu Y, McFarland BC, Deshane JS, Hurst DR, Ponnazhagan S, et al. SOCS3 Deficiency in Myeloid Cells Promotes Tumor Development: Involvement of STAT3 Activation and Myeloid-Derived Suppressor Cells. Cancer Immunol Res. 2015;3(7):727–40.
Liu Y, Lai L, Chen Q, Song Y, Xu S, Ma F, et al. MicroRNA-494 is required for the accumulation and functions of tumor-expanded myeloid-derived suppressor cells via targeting of PTEN. J Immunol. 2012;188(11):5500–10.
McClure C, Brudecki L, Ferguson DA, Yao ZQ, Moorman JP, McCall CE, et al. MicroRNA 21 (miR-21) and miR-181b couple with NFI-A to generate myeloid-derived suppressor cells and promote immunosuppression in late sepsis. Infect Immun. 2014;82(9):3816–25.
Ballesteros I, Rubio-Ponce A, Genua M, Lusito E, Kwok I, Fernández-Calvo G, et al. Co-option of Neutrophil Fates by Tissue Environments. Cell. 2020;183(5):1282-97.e18.
Glover A, Zhang Z, Shannon-Lowe C. Deciphering the roles of myeloid derived suppressor cells in viral oncogenesis. Front Immunol. 2023;14:1161848.
Lang S, Bruderek K, Kaspar C, Höing B, Kanaan O, Dominas N, et al. Clinical Relevance and Suppressive Capacity of Human Myeloid-Derived Suppressor Cell Subsets. Clin Cancer Res. 2018;24(19):4834–44.
Okła K, Czerwonka A, Wawruszak A, Bobiński M, Bilska M, Tarkowski R, et al. Clinical Relevance and Immunosuppressive Pattern of Circulating and Infiltrating Subsets of Myeloid-Derived Suppressor Cells (MDSCs) in Epithelial Ovarian Cancer. Front Immunol. 2019;10:691.
Khan ANH, Emmons TR, Wong JT, Alqassim E, Singel KL, Mark J, et al. Quantification of Early-Stage Myeloid-Derived Suppressor Cells in Cancer Requires Excluding Basophils. Cancer Immunol Res. 2020;8(6):819–28.
Jackson C, Cherry C, Bom S, Dykema AG, Thompson E, Zheng M, et al. Distinct Myeloid Derived Suppressor Cell Populations Promote Tumor Aggression in Glioblastoma. bioRxiv. 2023.
Haile LA, Gamrekelashvili J, Manns MP, Korangy F, Greten TF. CD49d is a new marker for distinct myeloid-derived suppressor cell subpopulations in mice. J Immunol. 2010;185(1):203–10.
Soler DC, Young AB, Cooper KD, Kerstetter-Fogle A, Barnholtz-Sloan JS, Gittleman H, et al. The ratio of HLA-DR and VNN2(+) expression on CD14(+) myeloid derived suppressor cells can distinguish glioblastoma from radiation necrosis patients. J Neurooncol. 2017;134(1):189–96.
Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–5.
Ozga AJ, Chow MT, Luster AD. Chemokines and the immune response to cancer. Immunity. 2021;54(5):859–74.
Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, et al. CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep. 2015;12(2):244–57.
Inamoto S, Itatani Y, Yamamoto T, Minamiguchi S, Hirai H, Iwamoto M, et al. Loss of SMAD4 Promotes Colorectal Cancer Progression by Accumulation of Myeloid-Derived Suppressor Cells through the CCL15-CCR1 Chemokine Axis. Clin Cancer Res. 2016;22(2):492–501.
Sceneay J, Chow MT, Chen A, Halse HM, Wong CS, Andrews DM, et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. 2012;72(16):3906–11.
Reichel CA, Puhr-Westerheide D, Zuchtriegel G, Uhl B, Berberich N, Zahler S, et al. C-C motif chemokine CCL3 and canonical neutrophil attractants promote neutrophil extravasation through common and distinct mechanisms. Blood. 2012;120(4):880–90.
Guldner IH, Wang Q, Yang L, Golomb SM, Zhao Z, Lopez JA, et al. CNS-Native Myeloid Cells Drive Immune Suppression in the Brain Metastatic Niche through Cxcl10. Cell. 2020;183(5):1234-48.e25.
Gielen PR, Schulte BM, Kers-Rebel ED, Verrijp K, Petersen-Baltussen HM, ter Laan M, et al. Increase in both CD14-positive and CD15-positive myeloid-derived suppressor cell subpopulations in the blood of patients with glioma but predominance of CD15-positive myeloid-derived suppressor cells in glioma tissue. J Neuropathol Exp Neurol. 2015;74(5):390–400.
Raghavan JV, Ganesh RA, Sonpatki P, Naik D, John AE, Arunachalam P, et al. Immuno-phenotyping of IDH-mutant grade 3 astrocytoma and IDH-wildtype glioblastoma reveals specific differences in cells of myeloid origin. Oncoimmunology. 2021;10(1):1957215.
Yeo AT, Rawal S, Delcuze B, Christofides A, Atayde A, Strauss L, et al. Single-cell RNA sequencing reveals evolution of immune landscape during glioblastoma progression. Nat Immunol. 2022;23(6):971–84.
Richard SA. Explicating the Pivotal Pathogenic, Diagnostic, and Therapeutic Biomarker Potentials of Myeloid-Derived Suppressor Cells in Glioblastoma. Dis Markers. 2020;2020:8844313.
De Leo A, Ugolini A, Veglia F. Myeloid Cells in Glioblastoma Microenvironment. Cells. 2020;10(1).
Hsu JB, Lee GA, Chang TH, Huang SW, Le NQK, Chen YC, et al. Radiomic Immunophenotyping of GSEA-Assessed Immunophenotypes of Glioblastoma and Its Implications for Prognosis: A Feasibility Study. Cancers (Basel). 2020;12(10).
Magri S, Musca B, Bonaudo C, Tushe A, Russo MG, Masetto E, et al. Sustained Accumulation of Blood-Derived Macrophages in the Immune Microenvironment of Patients with Recurrent Glioblastoma after Therapy. Cancers (Basel). 2021;13(24).
Alban TJ, Alvarado AG, Sorensen MD, Bayik D, Volovetz J, Serbinowski E, et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight. 2018;3(21).
Flores-Toro JA, Luo D, Gopinath A, Sarkisian MR, Campbell JJ, Charo IF, et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc Natl Acad Sci U S A. 2020;117(2):1129–38.
Kumar R, de Mooij T, Peterson TE, Kaptzan T, Johnson AJ, Daniels DJ, et al. Modulating glioma-mediated myeloid-derived suppressor cell development with sulforaphane. PLoS ONE. 2017;12(6): e0179012.
Otvos B, Silver DJ, Mulkearns-Hubert EE, Alvarado AG, Turaga SM, Sorensen MD, et al. Cancer Stem Cell-Secreted Macrophage Migration Inhibitory Factor Stimulates Myeloid Derived Suppressor Cell Function and Facilitates Glioblastoma Immune Evasion. Stem Cells. 2016;34(8):2026–39.
Verschuere T, Toelen J, Maes W, Poirier F, Boon L, Tousseyn T, et al. Glioma-derived galectin-1 regulates innate and adaptive antitumor immunity. Int J Cancer. 2014;134(4):873–84.
Chen Q, Han B, Meng X, Duan C, Yang C, Wu Z, et al. Immunogenomic analysis reveals LGALS1 contributes to the immune heterogeneity and immunosuppression in glioma. Int J Cancer. 2019;145(2):517–30.
Cao Y, Liu B, Cai L, Li Y, Huang Y, Zhou Y, et al. G9a promotes immune suppression by targeting the Fbxw7/Notch pathway in glioma stem cells. CNS Neurosci Ther. 2023.
Yan J, Kong LY, Hu J, Gabrusiewicz K, Dibra D, Xia X, et al. FGL2 as a Multimodality Regulator of Tumor-Mediated Immune Suppression and Therapeutic Target in Gliomas. J Natl Cancer Inst. 2015;107(8).
Otani Y, Yoo JY, Lewis CT, Chao S, Swanner J, Shimizu T, et al. NOTCH-Induced MDSC Recruitment after oHSV Virotherapy in CNS Cancer Models Modulates Antitumor Immunotherapy. Clin Cancer Res. 2022;28(7):1460–73.
Chai E, Zhang L, Li C. LOX-1+ PMN-MDSC enhances immune suppression which promotes glioblastoma multiforme progression. Cancer Manag Res. 2019;11:7307–15.
Zhou H, Jiang M, Yuan H, Ni W, Tai G. Dual roles of myeloid-derived suppressor cells induced by Toll-like receptor signaling in cancer. Oncol Lett. 2021;21(2):149.
Hu X, Li B, Li X, Zhao X, Wan L, Lin G, et al. Transmembrane TNF-α promotes suppressive activities of myeloid-derived suppressor cells via TNFR2. J Immunol. 2014;192(3):1320–31.
Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest. 2010;120(2):457–71.
Condamine T, Kumar V, Ramachandran IR, Youn JI, Celis E, Finnberg N, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest. 2014;124(6):2626–39.
Mohamed E, Sierra RA, Trillo-Tinoco J, Cao Y, Innamarato P, Payne KK, et al. The Unfolded Protein Response Mediator PERK Governs Myeloid Cell-Driven Immunosuppression in Tumors through Inhibition of STING Signaling. Immunity. 2020;52(4):668-82.e7.
Pituch KC, Miska J, Krenciute G, Panek WK, Li G, Rodriguez-Cruz T, et al. Adoptive Transfer of IL13Rα2-Specific Chimeric Antigen Receptor T Cells Creates a Pro-inflammatory Environment in Glioblastoma. Mol Ther. 2018;26(4):986–95.
Qian J, Wang C, Wang B, Yang J, Wang Y, Luo F, et al. The IFN-γ/PD-L1 axis between T cells and tumor microenvironment: hints for glioma anti-PD-1/PD-L1 therapy. J Neuroinflammation. 2018;15(1):290.
Irshad K, Srivastava C, Malik N, Arora M, Gupta Y, Goswami S, et al. Upregulation of Atypical Cadherin FAT1 Promotes an Immunosuppressive Tumor Microenvironment via TGF-β. Front Immunol. 2022;13: 813888.
Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13(1):23–35.
Chiu DK, Xu IM, Lai RK, Tse AP, Wei LL, Koh HY, et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology. 2016;64(3):797–813.
Li B, Zhang S, Huang N, Chen H, Wang P, Yang J, et al. CCL9/CCR1 induces myeloid-derived suppressor cell recruitment to the spleen in a murine H22 orthotopic hepatoma model. Oncol Rep. 2019;41(1):608–18.
Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016;76(19):5671–82.
Sun H, Yang W, Tian Y, Zeng X, Zhou J, Mok MTS, et al. An inflammatory-CCRK circuitry drives mTORC1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular carcinoma. Nat Commun. 2018;9(1):5214.
Toh B, Wang X, Keeble J, Sim WJ, Khoo K, Wong WC, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011;9(9): e1001162.
OuYang LY, Wu XJ, Ye SB, Zhang RX, Li ZL, Liao W, et al. Tumor-induced myeloid-derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J Transl Med. 2015;13:47.
David JM, Dominguez C, Hamilton DH, Palena C. The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines (Basel). 2016;4(3).
Allard D, Turcotte M, Stagg J. Targeting A2 adenosine receptors in cancer. Immunol Cell Biol. 2017;95(4):333–9.
Chiu DK, Tse AP, Xu IM, Di Cui J, Lai RK, Li LL, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. 2017;8(1):517.
Alghamri MS, McClellan BL, Avvari RP, Thalla R, Carney S, Hartlage CS, et al. G-CSF secreted by mutant IDH1 glioma stem cells abolishes myeloid cell immunosuppression and enhances the efficacy of immunotherapy. Sci Adv. 2021;7(40):eabh3243.
Christmas BJ, Rafie CI, Hopkins AC, Scott BA, Ma HS, Cruz KA, et al. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunol Res. 2018;6(12):1561–77.
Bayik D, Zhou Y, Park C, Hong C, Vail D, Silver DJ, et al. Myeloid-Derived Suppressor Cell Subsets Drive Glioblastoma Growth in a Sex-Specific Manner. Cancer Discov. 2020;10(8):1210–25.
Xu M, Zhao Z, Song J, Lan X, Lu S, Chen M, et al. Interactions between interleukin-6 and myeloid-derived suppressor cells drive the chemoresistant phenotype of hepatocellular cancer. Exp Cell Res. 2017;351(2):142–9.
Oft M. IL-10: master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol Res. 2014;2(3):194–9.
Bourgeois-Daigneault MC, Roy DG, Falls T, Twumasi-Boateng K, St-Germain LE, Marguerie M, et al. Oncolytic vesicular stomatitis virus expressing interferon-γ has enhanced therapeutic activity. Mol Ther Oncolytics. 2016;3:16001.
Li S, Sun R, Chen Y, Wei H, Tian Z. TLR2 limits development of hepatocellular carcinoma by reducing IL18-mediated immunosuppression. Cancer Res. 2015;75(6):986–95.
Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. 2013;190(7):3783–97.
Qiu W, Guo X, Li B, Wang J, Qi Y, Chen Z, et al. Exosomal miR-1246 from glioma patient body fluids drives the differentiation and activation of myeloid-derived suppressor cells. Mol Ther. 2021;29(12):3449–64.
Iwata T, Kondo Y, Kimura O, Morosawa T, Fujisaka Y, Umetsu T, et al. PD-L1(+)MDSCs are increased in HCC patients and induced by soluble factor in the tumor microenvironment. Sci Rep. 2016;6:39296.
Li R, Li H, Luo HJ, Lin ZX, Jiang ZW, Luo WH. SSAO inhibitors suppress hepatocellular tumor growth in mice. Cell Immunol. 2013;283(1–2):61–9.
Guha P, Gardell J, Darpolor J, Cunetta M, Lima M, Miller G, et al. STAT3 inhibition induces Bax-dependent apoptosis in liver tumor myeloid-derived suppressor cells. Oncogene. 2019;38(4):533–48.
Huang CY, Chung CL, Hu TH, Chen JJ, Liu PF, Chen CL. Recent progress in TGF-β inhibitors for cancer therapy. Biomed Pharmacother. 2021;134: 111046.
Jung MY, Aibaidula A, Brown DA, Himes BT, Cumba Garcia LM, Parney IF. Superinduction of immunosuppressive glioblastoma extracellular vesicles by IFN-γ through PD-L1 and IDO1. Neurooncol Adv. 2022;4(1):vdac017.
Tian X, Shen H, Li Z, Wang T, Wang S. Tumor-derived exosomes, myeloid-derived suppressor cells, and tumor microenvironment. J Hematol Oncol. 2019;12(1):84.
Valenti R, Huber V, Filipazzi P, Pilla L, Sovena G, Villa A, et al. Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor-beta-mediated suppressive activity on T lymphocytes. Cancer Res. 2006;66(18):9290–8.
Wang J, De Veirman K, De Beule N, Maes K, De Bruyne E, Van Valckenborgh E, et al. The bone marrow microenvironment enhances multiple myeloma progression by exosome-mediated activation of myeloid-derived suppressor cells. Oncotarget. 2015;6(41):43992–4004.
Wang J, De Veirman K, Faict S, Frassanito MA, Ribatti D, Vacca A, et al. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J Pathol. 2016;239(2):162–73.
Guo X, Qiu W, Wang J, Liu Q, Qian M, Wang S, et al. Glioma exosomes mediate the expansion and function of myeloid-derived suppressor cells through microRNA-29a/Hbp1 and microRNA-92a/Prkar1a pathways. Int J Cancer. 2019;144(12):3111–26.
Ridder K, Sevko A, Heide J, Dams M, Rupp AK, Macas J, et al. Extracellular vesicle-mediated transfer of functional RNA in the tumor microenvironment. Oncoimmunology. 2015;4(6): e1008371.
Bruns H, Böttcher M, Qorraj M, Fabri M, Jitschin S, Dindorf J, et al. CLL-cell-mediated MDSC induction by exosomal miR-155 transfer is disrupted by vitamin D. Leukemia. 2017;31(4):985–8.
Himes BT, Fain CE, Tritz ZP, Nesvick CL, Jin-Lee HJ, Geiger PA, et al. Use of heparin to rescue immunosuppressive monocyte reprogramming by glioblastoma-derived extracellular vesicles. J Neurosurg. 2022:1–11.
Guo X, Qiu W, Liu Q, Qian M, Wang S, Zhang Z, et al. Immunosuppressive effects of hypoxia-induced glioma exosomes through myeloid-derived suppressor cells via the miR-10a/Rora and miR-21/Pten Pathways. Oncogene. 2018;37(31):4239–59.
Musatova OE, Rubtsov YP. Effects of glioblastoma-derived extracellular vesicles on the functions of immune cells. Front Cell Dev Biol. 2023;11:1060000.
Lee-Chang C, Rashidi A, Miska J, Zhang P, Pituch KC, Hou D, et al. Myeloid-Derived Suppressive Cells Promote B cell-Mediated Immunosuppression via Transfer of PD-L1 in Glioblastoma. Cancer Immunol Res. 2019;7(12):1928–43.
Himes BT, Peterson TE, de Mooij T, Garcia LMC, Jung MY, Uhm S, et al. The role of extracellular vesicles and PD-L1 in glioblastoma-mediated immunosuppressive monocyte induction. Neuro Oncol. 2020;22(7):967–78.
Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer. 2014;134(12):2853–64.
García-Ortiz A, Serrador JM. Nitric Oxide Signaling in T Cell-Mediated Immunity. Trends Mol Med. 2018;24(4):412–27.
Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182(9):5693–701.
Wang L, Kuang Z, Zhang D, Gao Y, Ying M, Wang T. Reactive oxygen species in immune cells: A new antitumor target. Biomed Pharmacother. 2021;133: 110978.
Barry ST, Gabrilovich DI, Sansom OJ, Campbell AD, Morton JP. Therapeutic targeting of tumour myeloid cells. Nat Rev Cancer. 2023;23(4):216–37.
Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007;13(7):828–35.
Take Y, Koizumi S, Nagahisa A. Prostaglandin E Receptor 4 Antagonist in Cancer Immunotherapy: Mechanisms of Action. Front Immunol. 2020;11:324.
Porta C, Consonni FM, Morlacchi S, Sangaletti S, Bleve A, Totaro MG, et al. Tumor-Derived Prostaglandin E2 Promotes p50 NF-κB-Dependent Differentiation of Monocytic MDSCs. Cancer Res. 2020;80(13):2874–88.
Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol. 2020;16(3):278–90.
Kim R, Hashimoto A, Markosyan N, Tyurin VA, Tyurina YY, Kar G, et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature. 2022;612(7939):338–46.
Sharpe MA, Baskin DS, Jenson AV, Baskin AM. Hijacking Sexual Immuno-Privilege in GBM-An Immuno-Evasion Strategy. Int J Mol Sci. 2021;22(20).
Timosenko E, Hadjinicolaou AV, Cerundolo V. Modulation of cancer-specific immune responses by amino acid degrading enzymes. Immunotherapy. 2017;9(1):83–97.
Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70(1):68–77.
Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep. 2015;13(2):412–24.
Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. 2011;208(10):1949–62.
Ugolini A, Tyurin VA, Tyurina YY, Tcyganov EN, Donthireddy L, Kagan VE, et al. Polymorphonuclear myeloid-derived suppressor cells limit antigen cross-presentation by dendritic cells in cancer. JCI Insight. 2020;5(15).
Zhou J, Nefedova Y, Lei A, Gabrilovich D. Neutrophils and PMN-MDSC: Their biological role and interaction with stromal cells. Semin Immunol. 2018;35:19–28.
Davis RJ, Moore EC, Clavijo PE, Friedman J, Cash H, Chen Z, et al. Anti-PD-L1 Efficacy Can Be Enhanced by Inhibition of Myeloid-Derived Suppressor Cells with a Selective Inhibitor of PI3Kδ/γ. Cancer Res. 2017;77(10):2607–19.
Taki M, Abiko K, Ukita M, Murakami R, Yamanoi K, Yamaguchi K, et al. Tumor Immune Microenvironment during Epithelial-Mesenchymal Transition. Clin Cancer Res. 2021;27(17):4669–79.
Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010;70(1):99–108.
Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol. 2010;184(6):3106–16.
Solito S, Marigo I, Pinton L, Damuzzo V, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann N Y Acad Sci. 2014;1319:47–65.
Haverkamp JM, Smith AM, Weinlich R, Dillon CP, Qualls JE, Neale G, et al. Myeloid-derived suppressor activity is mediated by monocytic lineages maintained by continuous inhibition of extrinsic and intrinsic death pathways. Immunity. 2014;41(6):947–59.
Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol. 2010;40(1):22–35.
Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moreno C, Scumpia PO, Laface DM, et al. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol Med. 2011;17(3–4):281–92.
Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207(11):2439–53.
Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol Res. 2015;3(11):1236–47.
Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211(5):781–90.
Achyut BR, Angara K, Jain M, Borin TF, Rashid MH, Iskander ASM, et al. Canonical NFκB signaling in myeloid cells is required for the glioblastoma growth. Sci Rep. 2017;7(1):13754.
Yu J, Wang Y, Yan F, Zhang P, Li H, Zhao H, et al. Noncanonical NF-κB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J Immunol. 2014;193(5):2574–86.
Youn JI, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol. 2012;91(1):167–81.
Haverkamp JM, Crist SA, Elzey BD, Cimen C, Ratliff TL. In vivo suppressive function of myeloid-derived suppressor cells is limited to the inflammatory site. Eur J Immunol. 2011;41(3):749–59.
Sceneay J, Griessinger CM, Hoffmann SHL, Wen SW, Wong CSF, Krumeich S, et al. Tracking the fate of adoptively transferred myeloid-derived suppressor cells in the primary breast tumor microenvironment. PLoS ONE. 2018;13(4): e0196040.
Guha P, Gardell J, Rabinowitz B, Lopes M, DaSilva NA, Rowley D, et al. Monocytic and granulocytic myeloid-derived suppressor cell plasticity and differentiation are organ-specific. Oncogene. 2021;40(3):693–704.
Alshetaiwi H, Pervolarakis N, McIntyre LL, Ma D, Nguyen Q, Rath JA, et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci Immunol. 2020;5(44).
Perez C, Botta C, Zabaleta A, Puig N, Cedena MT, Goicoechea I, et al. Immunogenomic identification and characterization of granulocytic myeloid-derived suppressor cells in multiple myeloma. Blood. 2020;136(2):199–209.
Salminen A, Kauppinen A, Kaarniranta K. AMPK activation inhibits the functions of myeloid-derived suppressor cells (MDSC): impact on cancer and aging. J Mol Med (Berl). 2019;97(8):1049–64.
Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity. 2016;44(2):303–15.
Shi Y, Ou L, Han S, Li M, Pena MM, Pena EA, et al. Deficiency of Kruppel-like factor KLF4 in myeloid-derived suppressor cells inhibits tumor pulmonary metastasis in mice accompanied by decreased fibrocytes. Oncogenesis. 2014;3(11): e129.
Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344(6186):921–5.
Liu G, Bi Y, Shen B, Yang H, Zhang Y, Wang X, et al. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1α-dependent glycolysis. Cancer Res. 2014;74(3):727–37.
Palazón A, Martínez-Forero I, Teijeira A, Morales-Kastresana A, Alfaro C, Sanmamed MF, et al. The HIF-1α hypoxia response in tumor-infiltrating T lymphocytes induces functional CD137 (4–1BB) for immunotherapy. Cancer Discov. 2012;2(7):608–23.
Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci U S A. 2017;114(5):1117–22.
Trellakis S, Bruderek K, Hütte J, Elian M, Hoffmann TK, Lang S, et al. Granulocytic myeloid-derived suppressor cells are cryosensitive and their frequency does not correlate with serum concentrations of colony-stimulating factors in head and neck cancer. Innate Immun. 2013;19(3):328–36.
Mishalian I, Bayuh R, Levy L, Zolotarov L, Michaeli J, Fridlender ZG. Tumor-associated neutrophils (TAN) develop pro-tumorigenic properties during tumor progression. Cancer Immunol Immunother. 2013;62(11):1745–56.
Duluc D, Delneste Y, Tan F, Moles MP, Grimaud L, Lenoir J, et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood. 2007;110(13):4319–30.
Hammami I, Chen J, Murschel F, Bronte V, De Crescenzo G, Jolicoeur M. Immunosuppressive activity enhances central carbon metabolism and bioenergetics in myeloid-derived suppressor cells in vitro models. BMC Cell Biol. 2012;13:18.
Sevenich L. Brain-Resident Microglia and Blood-Borne Macrophages Orchestrate Central Nervous System Inflammation in Neurodegenerative Disorders and Brain Cancer. Front Immunol. 2018;9:697.
Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017;77(9):2266–78.
Hedrick CC, Malanchi I. Neutrophils in cancer: heterogeneous and multifaceted. Nat Rev Immunol. 2022;22(3):173–87.
Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA, Fecci PE. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin Cancer Res. 2018;24(16):3792–802.
DiDomenico J, Lamano JB, Oyon D, Li Y, Veliceasa D, Kaur G, et al. The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. Oncoimmunology. 2018;7(7): e1448329.
Amaria RN, Reddy SM, Tawbi HA, Davies MA, Ross MI, Glitza IC, et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat Med. 2018;24(11):1649–54.
Jang BS, Kim IA. A Radiosensitivity Gene Signature and PD-L1 Status Predict Clinical Outcome of Patients with Glioblastoma Multiforme in The Cancer Genome Atlas Dataset. Cancer Res Treat. 2020;52(2):530–42.
Rao G, Latha K, Ott M, Sabbagh A, Marisetty A, Ling X, et al. Anti-PD-1 Induces M1 Polarization in the Glioma Microenvironment and Exerts Therapeutic Efficacy in the Absence of CD8 Cytotoxic T Cells. Clin Cancer Res. 2020;26(17):4699–712.
Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, Archer GE, et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res. 2007;13(7):2158–67.
Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat Rev Immunol. 2015;15(1):45–56.
Harris-Bookman S, Mathios D, Martin AM, Xia Y, Kim E, Xu H, et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int J Cancer. 2018;143(12):3201–8.
Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10(1):48–57.
Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. 2018;7(8): e1466769.
Zhang H, Song Y, Yang H, Liu Z, Gao L, Liang X, et al. Tumor cell-intrinsic Tim-3 promotes liver cancer via NF-κB/IL-6/STAT3 axis. Oncogene. 2018;37(18):2456–68.
Di Tacchio M, Macas J, Weissenberger J, Sommer K, Bähr O, Steinbach JP, et al. Tumor Vessel Normalization, Immunostimulatory Reprogramming, and Improved Survival in Glioblastoma with Combined Inhibition of PD-1, Angiopoietin-2, and VEGF. Cancer Immunol Res. 2019;7(12):1910–27.
Zhang B, Xie L, Liu J, Liu A, He M. Construction and validation of a cuproptosis-related prognostic model for glioblastoma. Front Immunol. 2023;14:1082974.
Lee AH, Sun L, Mochizuki AY, Reynoso JG, Orpilla J, Chow F, et al. Neoadjuvant PD-1 blockade induces T cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat Commun. 2021;12(1):6938.
Caccese M, Indraccolo S, Zagonel V, Lombardi G. PD-1/PD-L1 immune-checkpoint inhibitors in glioblastoma: A concise review. Crit Rev Oncol Hematol. 2019;135:128–34.
Weiss T, Puca E, Silginer M, Hemmerle T, Pazahr S, Bink A, et al. Immunocytokines are a promising immunotherapeutic approach against glioblastoma. Sci Transl Med. 2020;12(564).
Kristó K, Szekeres M, Makai Z, Márki Á, Kelemen A, Bali L, et al. Preparation and investigation of core-shell nanoparticles containing human interferon-α. Int J Pharm. 2020;573: 118825.
Zhao P, Wang Y, Kang X, Wu A, Yin W, Tang Y, et al. Dual-targeting biomimetic delivery for anti-glioma activity via remodeling the tumor microenvironment and directing macrophage-mediated immunotherapy. Chem Sci. 2018;9(10):2674–89.
Budhwani M, Mazzieri R, Dolcetti R. Plasticity of Type I Interferon-Mediated Responses in Cancer Therapy: From Anti-tumor Immunity to Resistance. Front Oncol. 2018;8:322.
DeCordova S, Shastri A, Tsolaki AG, Yasmin H, Klein L, Singh SK, et al. Molecular Heterogeneity and Immunosuppressive Microenvironment in Glioblastoma. Front Immunol. 2020;11:1402.
Khalsa JK, Cheng N, Keegan J, Chaudry A, Driver J, Bi WL, et al. Immune phenotyping of diverse syngeneic murine brain tumors identifies immunologically distinct types. Nat Commun. 2020;11(1):3912.
Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32(11):1959–72.
Larsson HB, Stubgaard M, Frederiksen JL, Jensen M, Henriksen O, Paulson OB. Quantitation of blood-brain barrier defect by magnetic resonance imaging and gadolinium-DTPA in patients with multiple sclerosis and brain tumors. Magn Reson Med. 1990;16(1):117–31.
Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016;17(9):2445–59.
Baruch K, Deczkowska A, Rosenzweig N, Tsitsou-Kampeli A, Sharif AM, Matcovitch-Natan O, et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease. Nat Med. 2016;22(2):135–7.
Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139–48.
Huang J, Liu F, Liu Z, Tang H, Wu H, Gong Q, et al. Immune Checkpoint in Glioblastoma: Promising and Challenging. Front Pharmacol. 2017;8:242.
Arrieta VA, Najem H, Petrosyan E, Lee-Chang C, Chen P, Sonabend AM, et al. The Eclectic Nature of Glioma-Infiltrating Macrophages and Microglia. Int J Mol Sci. 2021;22(24).
Galluzzi L, Humeau J, Buqué A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol. 2020;17(12):725–41.
Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25(3):477–86.
Shergalis A, Bankhead A 3rd, Luesakul U, Muangsin N, Neamati N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol Rev. 2018;70(3):412–45.
Reardon DA, Groves MD, Wen PY, Nabors L, Mikkelsen T, Rosenfeld S, et al. A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clin Cancer Res. 2013;19(4):900–8.
Brown N, McBain C, Nash S, Hopkins K, Sanghera P, Saran F, et al. Multi-Center Randomized Phase II Study Comparing Cediranib plus Gefitinib with Cediranib plus Placebo in Subjects with Recurrent/Progressive Glioblastoma. PLoS ONE. 2016;11(5): e0156369.
Van Den Bent M, Eoli M, Sepulveda JM, Smits M, Walenkamp A, Frenel JS, et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro Oncol. 2020;22(5):684–93.
Turkowski K, Brandenburg S, Mueller A, Kremenetskaia I, Bungert AD, Blank A, et al. VEGF as a modulator of the innate immune response in glioblastoma. Glia. 2018;66(1):161–74.
Chuntova P, Hou Y, Naka R, Yamamichi A, Chen T, Goretsky Y, et al. Novel EGFRvIII-CAR transgenic mice for rigorous preclinical studies in syngeneic mice. Neuro Oncol. 2022;24(2):259–72.
Porret E, Kereselidze D, Dauba A, Schweitzer-Chaput A, Jegot B, Selingue E, et al. Refining the delivery and therapeutic efficacy of cetuximab using focused ultrasound in a mouse model of glioblastoma: An (89)Zr-cetuximab immunoPET study. Eur J Pharm Biopharm. 2023;182:141–51.
Chen Y, Gao F, Jiang R, Liu H, Hou J, Yi Y, et al. Down-Regulation of AQP4 Expression via p38 MAPK Signaling in Temozolomide-Induced Glioma Cells Growth Inhibition and Invasion Impairment. J Cell Biochem. 2017;118(12):4905–13.
Lan YL, Chen C, Wang X, Lou JC, Xing JS, Zou S, et al. Gamabufotalin induces a negative feedback loop connecting ATP1A3 expression and the AQP4 pathway to promote temozolomide sensitivity in glioblastoma cells by targeting the amino acid Thr794. Cell Prolif. 2020;53(1): e12732.
du Chatinier A, Meel MH, Das AI, Metselaar DS, Waranecki P, Bugiani M, et al. Generation of immunocompetent syngeneic allograft mouse models for pediatric diffuse midline glioma. Neurooncol Adv. 2022;4(1):vdac079.
Pearson JRD, Regad T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct Target Ther. 2017;2:17040.
Cui X, Zhao J, Li G, Yang C, Yang S, Zhan Q, et al. Blockage of EGFR/AKT and mevalonate pathways synergize the antitumor effect of temozolomide by reprogramming energy metabolism in glioblastoma. Cancer Commun (Lond). 2023.
Sulli G, Lam MTY, Panda S. Interplay between Circadian Clock and Cancer: New Frontiers for Cancer Treatment. Trends Cancer. 2019;5(8):475–94.
Liu T, Wang Z, Ye L, Duan Y, Jiang H, He H, et al. Nucleus-exported CLOCK acetylates PRPS to promote de novo nucleotide synthesis and liver tumour growth. Nat Cell Biol. 2023;25(2):273–84.
Wang Z, Su G, Dai Z, Meng M, Zhang H, Fan F, et al. Circadian clock genes promote glioma progression by affecting tumour immune infiltration and tumour cell proliferation. Cell Prolif. 2021;54(3): e12988.
Pang L, Dunterman M, Xuan W, Gonzalez A, Lin Y, Hsu WH, et al. Circadian regulator CLOCK promotes tumor angiogenesis in glioblastoma. Cell Rep. 2023;42(2): 112127.
Solt LA, Wang Y, Banerjee S, Hughes T, Kojetin DJ, Lundasen T, et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature. 2012;485(7396):62–8.
De A, Beligala DH, Birkholz TM, Geusz ME. Anticancer Properties of Curcumin and Interactions With the Circadian Timing System. Integr Cancer Ther. 2019;18:1534735419889154.
Wu S, Calero-Pérez P, Villamañan L, Arias-Ramos N, Pumarola M, Ortega-Martorell S, et al. Anti-tumour immune response in GL261 glioblastoma generated by Temozolomide Immune-Enhancing Metronomic Schedule monitored with MRSI-based nosological images. NMR Biomed. 2020;33(4): e4229.
Zhang SL, Lahens NF, Yue Z, Arnold DM, Pakstis PP, Schwarz JE, et al. A circadian clock regulates efflux by the blood-brain barrier in mice and human cells. Nat Commun. 2021;12(1):617.
Winkler F, Venkatesh HS, Amit M, Batchelor T, Demir IE, Deneen B, et al. Cancer neuroscience: State of the field, emerging directions. Cell. 2023;186(8):1689–707.
Huang-Hobbs E, Cheng YT, Ko Y, Luna-Figueroa E, Lozzi B, Taylor KR, et al. Remote neuronal activity drives glioma progression through SEMA4F. Nature. 2023;619(7971):844–50.
Huang X, Dubuc AM, Hashizume R, Berg J, He Y, Wang J, et al. Voltage-gated potassium channel EAG2 controls mitotic entry and tumor growth in medulloblastoma via regulating cell volume dynamics. Genes Dev. 2012;26(16):1780–96.
Francisco MA, Wanggou S, Fan JJ, Dong W, Chen X, Momin A, et al. Chloride intracellular channel 1 cooperates with potassium channel EAG2 to promote medulloblastoma growth. Journal of Experimental Medicine. 2020;217(5).
Venkataramani V, Schneider M, Giordano FA, Kuner T, Wick W, Herrlinger U, et al. Disconnecting multicellular networks in brain tumours. Nat Rev Cancer. 2022;22(8):481–91.
Dong W, Fekete A, Chen X, Liu H, Beilhartz GL, Chen X, et al. A designer peptide against the EAG2-Kvβ2 potassium channel targets the interaction of cancer cells and neurons to treat glioblastoma. Nat Cancer. 2023;4(10):1418–36.
Kraus TF, Globisch D, Wagner M, Eigenbrod S, Widmann D, Münzel M, et al. Low values of 5-hydroxymethylcytosine (5hmC), the “sixth base,” are associated with anaplasia in human brain tumors. Int J Cancer. 2012;131(7):1577–90.
Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–4.
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003.
Malta TM, de Souza CF, Sabedot TS, Silva TC, Mosella MS, Kalkanis SN, et al. Glioma CpG island methylator phenotype (G-CIMP): biological and clinical implications. Neuro Oncol. 2018;20(5):608–20.
Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stütz AM, et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. 2014;506(7489):445–50.
Akasaki Y, Kikuchi T, Homma S, Koido S, Ohkusa T, Tasaki T, et al. Phase I/II trial of combination of temozolomide chemotherapy and immunotherapy with fusions of dendritic and glioma cells in patients with glioblastoma. Cancer Immunol Immunother. 2016;65(12):1499–509.
Gallitto M, Cheng He R, Inocencio JF, Wang H, Zhang Y, Deikus G, et al. Epigenetic preconditioning with decitabine sensitizes glioblastoma to temozolomide via induction of MLH1. J Neurooncol. 2020;147(3):557–66.
Ma R, Rei M, Woodhouse I, Ferris K, Kirschner S, Chandran A, et al. Decitabine increases neoantigen and cancer testis antigen expression to enhance T-cell-mediated toxicity against glioblastoma. Neuro Oncol. 2022;24(12):2093–106.
Dai X, Lv X, Thompson EW, Ostrikov KK. Histone lactylation: epigenetic mark of glycolytic switch. Trends Genet. 2022;38(2):124–7.
Dewdney B, Jenkins MR, Best SA, Freytag S, Prasad K, Holst J, et al. From signalling pathways to targeted therapies: unravelling glioblastoma’s secrets and harnessing two decades of progress. Signal Transduct Target Ther. 2023;8(1):400.
Duan R, Du W, Guo W. EZH2: a novel target for cancer treatment. J Hematol Oncol. 2020;13(1):104.
Rasras S, Zibara K, Vosoughi T, Deris ZZ. Genetics and Epigenetics of Glioblastoma: Therapeutic Challenges. Clinical Cancer Investigation Journal. 2018;7:43.
Yang R, Wang M, Zhang G, Bao Y, Wu Y, Li X, et al. E2F7-EZH2 axis regulates PTEN/AKT/mTOR signalling and glioblastoma progression. Br J Cancer. 2020;123(9):1445–55.
Liu H, Sun Y, Qi X, Gordon RE, O’Brien JA, Yuan H, et al. EZH2 Phosphorylation Promotes Self-Renewal of Glioma Stem-Like Cells Through NF-κB Methylation. Front Oncol. 2019;9:641.
Chen X, Ma H, Wang Z, Zhang S, Yang H, Fang Z. EZH2 Palmitoylation Mediated by ZDHHC5 in p53-Mutant Glioma Drives Malignant Development and Progression. Cancer Res. 2017;77(18):4998–5010.
Gounder M, Schöffski P, Jones RL, Agulnik M, Cote GM, Villalobos VM, et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: an international, open-label, phase 2 basket study. Lancet Oncol. 2020;21(11):1423–32.
Dhar S, Gadd S, Patel P, Vaynshteyn J, Raju GP, Hashizume R, et al. A tumor suppressor role for EZH2 in diffuse midline glioma pathogenesis. Acta Neuropathol Commun. 2022;10(1):47.
Wiese M, Schill F, Sturm D, Pfister S, Hulleman E, Johnsen SA, et al. No Significant Cytotoxic Effect of the EZH2 Inhibitor Tazemetostat (EPZ-6438) on Pediatric Glioma Cells with Wildtype Histone 3 or Mutated Histone 3.3. Klin Padiatr. 2016;228(3):113–7.
Mohammad F, Weissmann S, Leblanc B, Pandey DP, Højfeldt JW, Comet I, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med. 2017;23(4):483–92.
Pei Y, Liu KW, Wang J, Garancher A, Tao R, Esparza LA, et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell. 2016;29(3):311–23.
Li Y, Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med. 2016;6(10).
Pathania R, Ramachandran S, Mariappan G, Thakur P, Shi H, Choi JH, et al. Combined Inhibition of DNMT and HDAC Blocks the Tumorigenicity of Cancer Stem-like Cells and Attenuates Mammary Tumor Growth. Cancer Res. 2016;76(11):3224–35.
Lucio-Eterovic AK, Cortez MA, Valera ET, Motta FJ, Queiroz RG, Machado HR, et al. Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: class II and IV are hypoexpressed in glioblastomas. BMC Cancer. 2008;8:243.
Hazane-Puch F, Arnaud J, Trocmé C, Faure P, Laporte F, Champelovier P. Sodium Selenite Decreased HDAC Activity, Cell Proliferation and Induced Apoptosis in Three Human Glioblastoma Cells. Anticancer Agents Med Chem. 2016;16(4):490–500.
Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23(1):27–47.
Bader JE, Voss K, Rathmell JC. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol Cell. 2020;78(6):1019–33.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Parks SK, Chiche J, Pouysségur J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat Rev Cancer. 2013;13(9):611–23.
Guan X, Luo L, Begum G, Kohanbash G, Song Q, Rao A, et al. Elevated Na/H exchanger 1 (SLC9A1) emerges as a marker for tumorigenesis and prognosis in gliomas. J Exp Clin Cancer Res. 2018;37(1):255.
Hasan MN, Luo L, Ding D, Song S, Bhuiyan MIH, Liu R, et al. Blocking NHE1 stimulates glioma tumor immunity by restoring OXPHOS function of myeloid cells. Theranostics. 2021;11(3):1295–309.
Polewski MD, Reveron-Thornton RF, Cherryholmes GA, Marinov GK, Cassady K, Aboody KS. Increased Expression of System xc- in Glioblastoma Confers an Altered Metabolic State and Temozolomide Resistance. Mol Cancer Res. 2016;14(12):1229–42.
Lu Q, Ding Y, Li Y, Lu Q. 5-HT receptor agonist Valerenic Acid enhances the innate immunity signal and suppresses glioblastoma cell growth and invasion. Int J Biol Sci. 2020;16(12):2104–15.
Sun J, Patel CB, Jang T, Merchant M, Chen C, Kazerounian S, et al. High levels of ubidecarenone (oxidized CoQ(10)) delivered using a drug-lipid conjugate nanodispersion (BPM31510) differentially affect redox status and growth in malignant glioma versus non-tumor cells. Sci Rep. 2020;10(1):13899.
Ferguson BS, Rogatzki MJ, Goodwin ML, Kane DA, Rightmire Z, Gladden LB. Lactate metabolism: historical context, prior misinterpretations, and current understanding. Eur J Appl Physiol. 2018;118(4):691–728.
Ippolito L, Morandi A, Giannoni E, Chiarugi P. Lactate: A Metabolic Driver in the Tumour Landscape. Trends Biochem Sci. 2019;44(2):153–66.
Caruso JP, Koch BJ, Benson PD, Varughese E, Monterey MD, Lee AE, et al. pH, Lactate, and Hypoxia: Reciprocity in Regulating High-Affinity Monocarboxylate Transporter Expression in Glioblastoma. Neoplasia. 2017;19(2):121–34.
Park SJ, Smith CP, Wilbur RR, Cain CP, Kallu SR, Valasapalli S, et al. An overview of MCT1 and MCT4 in GBM: small molecule transporters with large implications. Am J Cancer Res. 2018;8(10):1967–76.
Ferreira NN, Granja S, Boni FI, Ferreira LMB, Reis RM, Baltazar F, et al. A novel strategy for glioblastoma treatment combining alpha-cyano-4-hydroxycinnamic acid with cetuximab using nanotechnology-based delivery systems. Drug Deliv Transl Res. 2020;10(3):594–609.
Dorneburg C, Fischer M, Barth TFE, Mueller-Klieser W, Hero B, Gecht J, et al. LDHA in Neuroblastoma Is Associated with Poor Outcome and Its Depletion Decreases Neuroblastoma Growth Independent of Aerobic Glycolysis. Clin Cancer Res. 2018;24(22):5772–83.
Daniele S, Giacomelli C, Zappelli E, Granchi C, Trincavelli ML, Minutolo F, et al. Lactate dehydrogenase-A inhibition induces human glioblastoma multiforme stem cell differentiation and death. Sci Rep. 2015;5:15556.
Yi K, Zhan Q, Wang Q, Tan Y, Fang C, Wang Y, et al. PTRF/cavin-1 remodels phospholipid metabolism to promote tumor proliferation and suppress immune responses in glioblastoma by stabilizing cPLA2. Neuro Oncol. 2021;23(3):387–99.
Maurer GD, Brucker DP, Bähr O, Harter PN, Hattingen E, Walenta S, et al. Differential utilization of ketone bodies by neurons and glioma cell lines: a rationale for ketogenic diet as experimental glioma therapy. BMC Cancer. 2011;11:315.
Hitosugi T, Fan J, Chung TW, Lythgoe K, Wang X, Xie J, et al. Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol Cell. 2011;44(6):864–77.
Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov. 2022;21(11):799–820.
Nehama D, Woodell AS, Maingi SM, Hingtgen SD, Dotti G. Cell-based therapies for glioblastoma: Promising tools against tumor heterogeneity. Neuro Oncol. 2023;25(9):1551–62.
Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell. 2018;23(2):181-92.e5.
Kim GB, Aragon-Sanabria V, Randolph L, Jiang H, Reynolds JA, Webb BS, et al. High-affinity mutant Interleukin-13 targeted CAR T cells enhance delivery of clickable biodegradable fluorescent nanoparticles to glioblastoma. Bioact Mater. 2020;5(3):624–35.
Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–28.
Ma W, Wang Y, Zhang R, Yang F, Zhang D, Huang M, et al. Targeting PAK4 to reprogram the vascular microenvironment and improve CAR-T immunotherapy for glioblastoma. Nat Cancer. 2021;2(1):83–97.
Bagley SJ, Desai AS, Linette GP, June CH, O’Rourke DM. CAR T-cell therapy for glioblastoma: recent clinical advances and future challenges. Neuro Oncol. 2018;20(11):1429–38.
Lin YJ, Mashouf LA, Lim M. CAR T Cell Therapy in Primary Brain Tumors: Current Investigations and the Future. Front Immunol. 2022;13: 817296.
Thaci B, Brown CE, Binello E, Werbaneth K, Sampath P, Sengupta S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol. 2014;16(10):1304–12.
Zhang D, Li AM, Hu G, Huang M, Yang F, Zhang L, et al. PHGDH-mediated endothelial metabolism drives glioblastoma resistance to chimeric antigen receptor T cell immunotherapy. Cell Metab. 2023;35(3):517-34.e8.
Debinski W, Gibo DM, Hulet SW, Connor JR, Gillespie GY. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin Cancer Res. 1999;5(5):985–90.
Xu C, Bai Y, An Z, Hu Y, Zhang C, Zhong X. IL-13Rα2 humanized scFv-based CAR-T cells exhibit therapeutic activity against glioblastoma. Mol Ther Oncolytics. 2022;24:443–51.
Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med. 2016;375(26):2561–9.
Alizadeh D, Wong RA, Gholamin S, Maker M, Aftabizadeh M, Yang X, et al. IFNγ Is Critical for CAR T Cell-Mediated Myeloid Activation and Induction of Endogenous Immunity. Cancer Discov. 2021;11(9):2248–65.
Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci U S A. 1992;89(10):4309–13.
Dmello C, Zhao J, Chen L, Gould A, Castro B, Arrieta VA, et al. Checkpoint kinase 1/2 inhibition potentiates anti-tumoral immune response and sensitizes gliomas to immune checkpoint blockade. Nat Commun. 2023;14(1):1566.
O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399).
Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J Immunother. 2019;42(4):126–35.
Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38(8):947–53.
Miao H, Gale NW, Guo H, Qian J, Petty A, Kaspar J, et al. EphA2 promotes infiltrative invasion of glioma stem cells in vivo through cross-talk with Akt and regulates stem cell properties. Oncogene. 2015;34(5):558–67.
Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013;21(3):629–37.
Wang D, Prager BC, Gimple RC, Aguilar B, Alizadeh D, Tang H, et al. CRISPR Screening of CAR T Cells and Cancer Stem Cells Reveals Critical Dependencies for Cell-Based Therapies. Cancer Discov. 2021;11(5):1192–211.
Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. 2021;13(591).
Gatto L, Nunno VD, Franceschi E, Brandes AA. Chimeric antigen receptor macrophage for glioblastoma immunotherapy: the way forward. Immunotherapy. 2021;13(11):879–83.
Chen C, Jing W, Chen Y, Wang G, Abdalla M, Gao L, et al. Intracavity generation of glioma stem cell-specific CAR macrophages primes locoregional immunity for postoperative glioblastoma therapy. Sci Transl Med. 2022;14(656):eabn1128.
Zhang C, Burger MC, Jennewein L, Genßler S, Schönfeld K, Zeiner P, et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J Natl Cancer Inst. 2016;108(5).
Chang Y, Cai X, Syahirah R, Yao Y, Xu Y, Jin G, et al. CAR-neutrophil mediated delivery of tumor-microenvironment responsive nanodrugs for glioblastoma chemo-immunotherapy. Nat Commun. 2023;14(1):2266.
Zinn S, Vazquez-Lombardi R, Zimmermann C, Sapra P, Jermutus L, Christ D. Advances in antibody-based therapy in oncology. Nat Cancer. 2023;4(2):165–80.
Zhou S, Liu M, Ren F, Meng X, Yu J. The landscape of bispecific T cell engager in cancer treatment. Biomark Res. 2021;9(1):38.
Nagorsen D, Kufer P, Baeuerle PA, Bargou R. Blinatumomab: a historical perspective. Pharmacol Ther. 2012;136(3):334–42.
Yin Y, Rodriguez JL, Li N, Thokala R, Nasrallah MP, Hu L, et al. Locally secreted BiTEs complement CAR T cells by enhancing killing of antigen heterogeneous solid tumors. Mol Ther. 2022;30(7):2537–53.
Pituch KC, Zannikou M, Ilut L, Xiao T, Chastkofsky M, Sukhanova M, et al. Neural stem cells secreting bispecific T cell engager to induce selective antiglioma activity. Proc Natl Acad Sci U S A. 2021;118(9).
Gardell JL, Matsumoto LR, Chinn H, DeGolier KR, Kreuser SA, Prieskorn B, et al. Human macrophages engineered to secrete a bispecific T cell engager support antigen-dependent T cell responses to glioblastoma. J Immunother Cancer. 2020;8(2).
Li G, Zhang Z, Cai L, Tang X, Huang J, Yu L, et al. Fn14-targeted BiTE and CAR-T cells demonstrate potent preclinical activity against glioblastoma. Oncoimmunology. 2021;10(1):1983306.
Delgado-Martín B, Medina M. Advances in the Knowledge of the Molecular Biology of Glioblastoma and Its Impact in Patient Diagnosis, Stratification, and Treatment. Adv Sci (Weinh). 2020;7(9):1902971.
Sampson JH, Mitchell DA. Vaccination strategies for neuro-oncology. Neuro Oncol. 2015;17 Suppl 7(Suppl 7):vii15-vii25.
Swartz AM, Batich KA, Fecci PE, Sampson JH. Peptide vaccines for the treatment of glioblastoma. J Neurooncol. 2015;123(3):433–40.
Srinivasan VM, Ferguson SD, Lee S, Weathers SP, Kerrigan BCP, Heimberger AB. Tumor Vaccines for Malignant Gliomas. Neurotherapeutics. 2017;14(2):345–57.
Naghavian R, Faigle W, Oldrati P, Wang J, Toussaint NC, Qiu Y, et al. Microbial peptides activate tumour-infiltrating lymphocytes in glioblastoma. Nature. 2023;617(7962):807–17.
Lin H, Wang K, Xiong Y, Zhou L, Yang Y, Chen S, et al. Identification of Tumor Antigens and Immune Subtypes of Glioblastoma for mRNA Vaccine Development. Front Immunol. 2022;13: 773264.
Kozielski KL, Ruiz-Valls A, Tzeng SY, Guerrero-Cázares H, Rui Y, Li Y, et al. Cancer-selective nanoparticles for combinatorial siRNA delivery to primary human GBM in vitro and in vivo. Biomaterials. 2019;209:79–87.
Wang QT, Nie Y, Sun SN, Lin T, Han RJ, Jiang J, et al. Tumor-associated antigen-based personalized dendritic cell vaccine in solid tumor patients. Cancer Immunol Immunother. 2020;69(7):1375–87.
Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28(31):4722–9.
Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol. 2015;17(6):854–61.
Fenstermaker RA, Ciesielski MJ, Qiu J, Yang N, Frank CL, Lee KP, et al. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol Immunother. 2016;65(11):1339–52.
Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011;17(6):1603–15.
Prins RM, Cloughesy TF, Liau LM. Cytomegalovirus immunity after vaccination with autologous glioblastoma lysate. N Engl J Med. 2008;359(5):539–41.
Liau LM, Ashkan K, Brem S, Campian JL, Trusheim JE, Iwamoto FM, et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination With Extension of Survival Among Patients With Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023;9(1):112–21.
Lawler SE. Cytomegalovirus and glioblastoma; controversies and opportunities. J Neurooncol. 2015;123(3):465–71.
Streblow DN, Nelson JA. Models of HCMV latency and reactivation. Trends Microbiol. 2003;11(7):293–5.
Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519(7543):366–9.
Wang X, Hu M, Xing F, Wang M, Wang B, Qian D. Human cytomegalovirus infection promotes the stemness of U251 glioma cells. J Med Virol. 2017;89(5):878–86.
Sener U, Ruff MW, Campian JL. Immunotherapy in Glioblastoma: Current Approaches and Future Perspectives. Int J Mol Sci. 2022;23(13).
Santos Apolonio J, Lima de Souza Gonçalves V, Cordeiro Santos ML, Silva Luz M, Silva Souza JV, Rocha Pinheiro SL, et al. Oncolytic virus therapy in cancer: A current review. World J Virol. 2021;10(5):229–55.
Farrera-Sal M, Moya-Borrego L, Bazan-Peregrino M, Alemany R. Evolving Status of Clinical Immunotherapy with Oncolytic Adenovirus. Clin Cancer Res. 2021;27(11):2979–88.
Zhu S, Zhang T, Zheng L, Liu H, Song W, Liu D, et al. Combination strategies to maximize the benefits of cancer immunotherapy. J Hematol Oncol. 2021;14(1):156.
Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642–62.
Stavrakaki E, Dirven CMF, Lamfers MLM. Personalizing Oncolytic Virotherapy for Glioblastoma: In Search of Biomarkers for Response. Cancers (Basel). 2021;13(4).
Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell. 2019;35(6):885-900.e10.
Friedman GK, Johnston JM, Bag AK, Bernstock JD, Li R, Aban I, et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N Engl J Med. 2021;384(17):1613–22.
Tian L, Xu B, Chen Y, Li Z, Wang J, Zhang J, et al. Specific targeting of glioblastoma with an oncolytic virus expressing a cetuximab-CCL5 fusion protein via innate and adaptive immunity. Nat Cancer. 2022;3(11):1318–35.
Silver DJ, Siebzehnrubl FA, Schildts MJ, Yachnis AT, Smith GM, Smith AA, et al. Chondroitin sulfate proteoglycans potently inhibit invasion and serve as a central organizer of the brain tumor microenvironment. J Neurosci. 2013;33(39):15603–17.
Whatcott CJ, Han H, Posner RG, Hostetter G, Von Hoff DD. Targeting the tumor microenvironment in cancer: why hyaluronidase deserves a second look. Cancer Discov. 2011;1(4):291–6.
Kiyokawa J, Kawamura Y, Ghouse SM, Acar S, Barçın E, Martínez-Quintanilla J, et al. Modification of Extracellular Matrix Enhances Oncolytic Adenovirus Immunotherapy in Glioblastoma. Clin Cancer Res. 2021;27(3):889–902.
Park JB, Kwak HJ, Lee SH. Role of hyaluronan in glioma invasion. Cell Adh Migr. 2008;2(3):202–7.
Chen X, Liu J, Li Y, Zeng Y, Wang F, Cheng Z, et al. IDH1 mutation impairs antiviral response and potentiates oncolytic virotherapy in glioma. Nat Commun. 2023;14(1):6781.
Ling AL, Solomon IH, Landivar AM, Nakashima H, Woods JK, Santos A, et al. Clinical trial links oncolytic immunoactivation to survival in glioblastoma. Nature. 2023;623(7985):157–66.
Hjortland GO, Garman-Vik SS, Juell S, Olsen OE, Hirschberg H, Fodstad O, et al. Immunotoxin treatment targeted to the high-molecular-weight melanoma-associated antigen prolonging the survival of immunodeficient rats with invasive intracranial human glioblastoma multiforme. J Neurosurg. 2004;100(2):320–7.
Carter P. Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer. 2001;1(2):118–29.
Havaei SM, Aucoin MG, Jahanian-Najafabadi A. Pseudomonas Exotoxin-Based Immunotoxins: Over Three Decades of Efforts on Targeting Cancer Cells With the Toxin. Front Oncol. 2021;11: 781800.
Wei H, Xiang L, Wayne AS, Chertov O, FitzGerald DJ, Bera TK, et al. Immunotoxin resistance via reversible methylation of the DPH4 promoter is a unique survival strategy. Proc Natl Acad Sci U S A. 2012;109(18):6898–903.
Joshi BH, Plautz GE, Puri RK. Interleukin-13 receptor alpha chain: a novel tumor-associated transmembrane protein in primary explants of human malignant gliomas. Cancer Res. 2000;60(5):1168–72.
Rahaman SO, Sharma P, Harbor PC, Aman MJ, Vogelbaum MA, Haque SJ. IL-13R(alpha)2, a decoy receptor for IL-13 acts as an inhibitor of IL-4-dependent signal transduction in glioblastoma cells. Cancer Res. 2002;62(4):1103–9.
Kunwar S. Convection enhanced delivery of IL13-PE38QQR for treatment of recurrent malignant glioma: presentation of interim findings from ongoing phase 1 studies. Acta Neurochir Suppl. 2003;88:105–11.
Vogelbaum MA, Sampson JH, Kunwar S, Chang SM, Shaffrey M, Asher AL, et al. Convection-enhanced delivery of cintredekin besudotox (interleukin-13-PE38QQR) followed by radiation therapy with and without temozolomide in newly diagnosed malignant gliomas: phase 1 study of final safety results. Neurosurgery. 2007;61(5):1031–7; discussion 7–8.
Kunwar S, Prados MD, Chang SM, Berger MS, Lang FF, Piepmeier JM, et al. Direct intracerebral delivery of cintredekin besudotox (IL13-PE38QQR) in recurrent malignant glioma: a report by the Cintredekin Besudotox Intraparenchymal Study Group. J Clin Oncol. 2007;25(7):837–44.
Kawakami K, Kawakami M, Liu Q, Puri RK. Combined effects of radiation and interleukin-13 receptor-targeted cytotoxin on glioblastoma cell lines. Int J Radiat Oncol Biol Phys. 2005;63(1):230–7.
Huang L, He H, Wang K, Ma X, Chen X, Chen W, et al. EGFRvIII-targeted immunotoxin combined with temozolomide and bispecific antibody for the eradication of established glioblastoma. Biomed Pharmacother. 2022;155: 113659.
Chandramohan V, Bao X, Yu X, Parker S, McDowall C, Yu YR, et al. Improved efficacy against malignant brain tumors with EGFRwt/EGFRvIII targeting immunotoxin and checkpoint inhibitor combinations. J Immunother Cancer. 2019;7(1):142.
Desjardins A, Randazzo D, Chandramohan V, Peters K, Johnson M, Landi D, et al. CTIM-23. A PHASE 1 TRIAL OF D2C7-IT IN COMBINATION WITH ATEZOLIZUMAB IN RECURRENT WHO GRADE IV MALIGNANT GLIOMA (MG). Neuro-Oncology. 2020;22(Supplement_2):ii38-ii.
Chonan M, Saito R, Shoji T, Shibahara I, Kanamori M, Sonoda Y, et al. CD40/CD40L expression correlates with the survival of patients with glioblastomas and an augmentation in CD40 signaling enhances the efficacy of vaccinations against glioma models. Neuro Oncol. 2015;17(11):1453–62.
Parker S, McDowall C, Sanchez-Perez L, Osorio C, Duncker PC, Briley A, et al. Immunotoxin-αCD40 therapy activates innate and adaptive immunity and generates a durable antitumor response in glioblastoma models. Sci Transl Med. 2023;15(682):eabn5649.
Leal M, Sapra P, Hurvitz SA, Senter P, Wahl A, Schutten M, et al. Antibody-drug conjugates: an emerging modality for the treatment of cancer. Ann N Y Acad Sci. 2014;1321:41–54.
Okeley NM, Alley SC, Senter PD. Advancing antibody drug conjugation: from the laboratory to a clinically approved anticancer drug. Hematol Oncol Clin North Am. 2014;28(1):13–25.
Baron JM, Boster BL, Barnett CM. Ado-trastuzumab emtansine (T-DM1): a novel antibody-drug conjugate for the treatment of HER2-positive metastatic breast cancer. J Oncol Pharm Pract. 2015;21(2):132–42.
Hamblett KJ, Kozlosky CJ, Siu S, Chang WS, Liu H, Foltz IN, et al. AMG 595, an Anti-EGFRvIII Antibody-Drug Conjugate, Induces Potent Antitumor Activity against EGFRvIII-Expressing Glioblastoma. Mol Cancer Ther. 2015;14(7):1614–24.
Marin BM, Porath KA, Jain S, Kim M, Conage-Pough JE, Oh JH, et al. Heterogeneous delivery across the blood-brain barrier limits the efficacy of an EGFR-targeting antibody drug conjugate in glioblastoma. Neuro Oncol. 2021;23(12):2042–53.
Capone E, Iacobelli S, Sala G. Role of galectin 3 binding protein in cancer progression: a potential novel therapeutic target. J Transl Med. 2021;19(1):405.
Rana R, Chauhan K, Gautam P, Kulkarni M, Banarjee R, Chugh P, et al. Plasma-Derived Extracellular Vesicles Reveal Galectin-3 Binding Protein as Potential Biomarker for Early Detection of Glioma. Front Oncol. 2021;11: 778754.
Dufrusine B, Capone E, Ponziani S, Lattanzio R, Lanuti P, Giansanti F, et al. Extracellular LGALS3BP: a potential disease marker and actionable target for antibody-drug conjugate therapy in glioblastoma. Mol Oncol. 2023;17(8):1460–73.
Aust G, Steinert M, Schütz A, Boltze C, Wahlbuhl M, Hamann J, et al. CD97, but not its closely related EGF-TM7 family member EMR2, is expressed on gastric, pancreatic, and esophageal carcinomas. Am J Clin Pathol. 2002;118(5):699–707.
Ward Y, Lake R, Yin JJ, Heger CD, Raffeld M, Goldsmith PK, et al. LPA receptor heterodimerizes with CD97 to amplify LPA-initiated RHO-dependent signaling and invasion in prostate cancer cells. Cancer Res. 2011;71(23):7301–11.
Aust G, Zheng L, Quaas M. To Detach, Migrate, Adhere, and Metastasize: CD97/ADGRE5 in Cancer. Cells. 2022;11(9).
Chidambaram A, Fillmore HL, Van Meter TE, Dumur CI, Broaddus WC. Novel report of expression and function of CD97 in malignant gliomas: correlation with Wilms tumor 1 expression and glioma cell invasiveness. J Neurosurg. 2012;116(4):843–53.
Ravn-Boess N, Roy N, Hattori T, Bready D, Donaldson H, Lawson C, et al. The expression profile and tumorigenic mechanisms of CD97 (ADGRE5) in glioblastoma render it a targetable vulnerability. Cell Rep. 2023;42(11): 113374.
Parakh S, Nicolazzo J, Scott AM, Gan HK. Antibody Drug Conjugates in Glioblastoma—Is There a Future for Them? Front Oncol. 2021;11: 718590.
Tran PHL, Xiang D, Tran TTD, Yin W, Zhang Y, Kong L, et al. Exosomes and Nanoengineering: A Match Made for Precision Therapeutics. Adv Mater. 2020;32(18): e1904040.
Correa S, Grosskopf AK, Lopez Hernandez H, Chan D, Yu AC, Stapleton LM, et al. Translational Applications of Hydrogels. Chem Rev. 2021;121(18):11385–457.
Zhang Y, You H, Wang Y, Chen Q, Guo Q, Chu Y, et al. A Micro-Environment Regulator for Filling the Clinical Treatment Gap after a Glioblastoma Operation. Adv Healthc Mater. 2022;11(3): e2101578.
Lin X, Song J, Chen X, Yang H. Ultrasound-Activated Sensitizers and Applications. Angew Chem Int Ed Engl. 2020;59(34):14212–33.
Guo QL, Dai XL, Yin MY, Cheng HW, Qian HS, Wang H, et al. Nanosensitizers for sonodynamic therapy for glioblastoma multiforme: current progress and future perspectives. Mil Med Res. 2022;9(1):26.
Young JS, Aghi MK. Chronic convection-enhanced intratumoural delivery of chemotherapy for glioblastoma. Lancet Oncol. 2022;23(11):1347–8.
van Vuurden DG. Convection-enhanced delivery: chemosurgery in diffuse intrinsic pontine glioma. Lancet Oncol. 2018;19(8):1001–3.
Vogelbaum MA, Aghi MK. Convection-enhanced delivery for the treatment of glioblastoma. Neuro Oncol. 2015;17 Suppl 2(Suppl 2):ii3-ii8.
Cheng Z, Li M, Dey R, Chen Y. Nanomaterials for cancer therapy: current progress and perspectives. J Hematol Oncol. 2021;14(1):85.
Janku F, Zhang HH, Pezeshki A, Goel S, Murthy R, Wang-Gillam A, et al. Intratumoral Injection of Clostridium novyi-NT Spores in Patients with Treatment-refractory Advanced Solid Tumors. Clin Cancer Res. 2021;27(1):96–106.
Zhu L, Liu J, Qiu M, Chen J, Liang Q, Peng G, et al. Bacteria-mediated metformin-loaded peptide hydrogel reprograms the tumor immune microenvironment in glioblastoma. Biomaterials. 2022;288: 121711.
Pucci S, Bolchi C, Bavo F, Pallavicini M, De Palma C, Renzi M, et al. Evidence of a dual mechanism of action underlying the anti-proliferative and cytotoxic effects of ammonium-alkyloxy-stilbene-based α7- and α9-nicotinic ligands on glioblastoma cells. Pharmacol Res. 2022;175: 105959.
Zheng Z, Zhang J, Jiang J, He Y, Zhang W, Mo X, et al. Remodeling tumor immune microenvironment (TIME) for glioma therapy using multi-targeting liposomal codelivery. J Immunother Cancer. 2020;8(2).
Zhang P, Miska J, Lee-Chang C, Rashidi A, Panek WK, An S, et al. Therapeutic targeting of tumor-associated myeloid cells synergizes with radiation therapy for glioblastoma. Proc Natl Acad Sci U S A. 2019;116(47):23714–23.
Mittal S, Klinger NV, Michelhaugh SK, Barger GR, Pannullo SC, Juhász C. Alternating electric tumor treating fields for treatment of glioblastoma: rationale, preclinical, and clinical studies. J Neurosurg. 2018;128(2):414–21.
Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA. 2017;318(23):2306–16.
Killock D. CNS cancer: TTFields improve survival. Nat Rev Clin Oncol. 2018;15(3):136.
Tan AC, Ashley DM, López GY, Malinzak M, Friedman HS, Khasraw M. Management of glioblastoma: State of the art and future directions. CA Cancer J Clin. 2020;70(4):299–312.
Moser JC, Salvador E, Deniz K, Swanson K, Tuszynski J, Carlson KW, et al. The Mechanisms of Action of Tumor Treating Fields. Cancer Res. 2022;82(20):3650–8.
Kim EH, Kim YH, Song HS, Jeong YK, Lee JY, Sung J, et al. Biological effect of an alternating electric field on cell proliferation and synergistic antimitotic effect in combination with ionizing radiation. Oncotarget. 2016;7(38):62267–79.
Clark PA, Gaal JT, Strebe JK, Pasch CA, Deming DA, Kuo JS, et al. The effects of tumor treating fields and temozolomide in MGMT expressing and non-expressing patient-derived glioblastoma cells. J Clin Neurosci. 2017;36:120–4.
Tanzhu G, Chen L, Xiao G, Shi W, Peng H, Chen D, et al. The schemes, mechanisms and molecular pathway changes of Tumor Treating Fields (TTFields) alone or in combination with radiotherapy and chemotherapy. Cell Death Discov. 2022;8(1):416.
Kessler AF, Frömbling GE, Gross F, Hahn M, Dzokou W, Ernestus RI, et al. Effects of tumor treating fields (TTFields) on glioblastoma cells are augmented by mitotic checkpoint inhibition. Cell Death Discov. 2018;4:12.
Kim EH, Jo Y, Sai S, Park MJ, Kim JY, Kim JS, et al. Tumor-treating fields induce autophagy by blocking the Akt2/miR29b axis in glioblastoma cells. Oncogene. 2019;38(39):6630–46.
Xu S, Luo C, Chen D, Tang L, Cheng Q, Chen L, et al. circMMD reduction following tumor treating fields inhibits glioblastoma progression through FUBP1/FIR/DVL1 and miR-15b-5p/FZD6 signaling. J Exp Clin Cancer Res. 2023;42(1):64.
Voloshin T, Kaynan N, Davidi S, Porat Y, Shteingauz A, Schneiderman RS, et al. Tumor-treating fields (TTFields) induce immunogenic cell death resulting in enhanced antitumor efficacy when combined with anti-PD-1 therapy. Cancer Immunol Immunother. 2020;69(7):1191–204.
Diamant G, Simchony Goldman H, Gasri Plotnitsky L, Roitman M, Shiloach T, Globerson-Levin A, et al. T Cells Retain Pivotal Antitumoral Functions under Tumor-Treating Electric Fields. J Immunol. 2021;207(2):709–19.
Lee YJ, Seo HW, Baek JH, Lim SH, Hwang SG, Kim EH. Gene expression profiling of glioblastoma cell lines depending on TP53 status after tumor-treating fields (TTFields) treatment. Sci Rep. 2020;10(1):12272.
Chen D, Le SB, Hutchinson TE, Calinescu AA, Sebastian M, Jin D, et al. Tumor Treating Fields dually activate STING and AIM2 inflammasomes to induce adjuvant immunity in glioblastoma. J Clin Invest. 2022;132(8).
Wan D, Jiang W, Hao J. Research Advances in How the cGAS-STING Pathway Controls the Cellular Inflammatory Response. Front Immunol. 2020;11:615.
Sen T, Della Corte CM, Milutinovic S, Cardnell RJ, Diao L, Ramkumar K, et al. Combination Treatment of the Oral CHK1 Inhibitor, SRA737, and Low-Dose Gemcitabine Enhances the Effect of Programmed Death Ligand 1 Blockade by Modulating the Immune Microenvironment in SCLC. J Thorac Oncol. 2019;14(12):2152–63.
Tie Y, Tang F, Wei YQ, Wei XW. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol. 2022;15(1):61.
Fultang L, Panetti S, Ng M, Collins P, Graef S, Rizkalla N, et al. MDSC targeting with Gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine. 2019;47:235–46.
Kim SH, Li M, Trousil S, Zhang Y, Pasca di Magliano M, Swanson KD, et al. Phenformin Inhibits Myeloid-Derived Suppressor Cells and Enhances the Anti-Tumor Activity of PD-1 Blockade in Melanoma. J Invest Dermatol. 2017;137(8):1740–8.
Bauer R, Udonta F, Wroblewski M, Ben-Batalla I, Santos IM, Taverna F, et al. Blockade of Myeloid-Derived Suppressor Cell Expansion with All-Trans Retinoic Acid Increases the Efficacy of Antiangiogenic Therapy. Cancer Res. 2018;78(12):3220–32.
Hashimoto A, Gao C, Mastio J, Kossenkov A, Abrams SI, Purandare AV, et al. Inhibition of Casein Kinase 2 Disrupts Differentiation of Myeloid Cells in Cancer and Enhances the Efficacy of Immunotherapy in Mice. Cancer Res. 2018;78(19):5644–55.
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97–103.
Zhang Q, Ma C, Duan Y, Heinrich B, Rosato U, Diggs LP, et al. Gut Microbiome Directs Hepatocytes to Recruit MDSCs and Promote Cholangiocarcinoma. Cancer Discov. 2021;11(5):1248–67.
Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325(5940):612–6.
Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A. 2012;109(7):2491–6.
Lee M, Park CS, Lee YR, Im SA, Song S, Lee CK. Resiquimod, a TLR7/8 agonist, promotes differentiation of myeloid-derived suppressor cells into macrophages and dendritic cells. Arch Pharm Res. 2014;37(9):1234–40.
Pencheva N, Buss CG, Posada J, Merghoub T, Tavazoie SF. Broad-spectrum therapeutic suppression of metastatic melanoma through nuclear hormone receptor activation. Cell. 2014;156(5):986–1001.
Tavazoie MF, Pollack I, Tanqueco R, Ostendorf BN, Reis BS, Gonsalves FC, et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell. 2018;172(4):825-40.e18.
LXR Agonism Depletes MDSCs to Promote Antitumor Immunity. Cancer Discov. 2018;8(3):263.
Halaby MJ, Hezaveh K, Lamorte S, Ciudad MT, Kloetgen A, MacLeod BL, et al. GCN2 drives macrophage and MDSC function and immunosuppression in the tumor microenvironment. Sci Immunol. 2019;4(42).
Santos CX, Tanaka LY, Wosniak J, Laurindo FR. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid Redox Signal. 2009;11(10):2409–27.
Ranjan A, Srivastava SK. Penfluridol suppresses glioblastoma tumor growth by Akt-mediated inhibition of GLI1. Oncotarget. 2017;8(20):32960–76.
Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166(1):678–89.
Yuen KC, Liu LF, Gupta V, Madireddi S, Keerthivasan S, Li C, et al. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat Med. 2020;26(5):693–8.
Peereboom DM, Alban TJ, Grabowski MM, Alvarado AG, Otvos B, Bayik D, et al. Metronomic capecitabine as an immune modulator in glioblastoma patients reduces myeloid-derived suppressor cells. JCI Insight. 2019;4(22).
Ostrand-Rosenberg S, Horn LA, Ciavattone NG. Radiotherapy Both Promotes and Inhibits Myeloid-Derived Suppressor Cell Function: Novel Strategies for Preventing the Tumor-Protective Effects of Radiotherapy. Front Oncol. 2019;9:215.
Ma Q, Shilkrut M, Zhao Z, Li M, Batty N, Barber B. Autoimmune comorbidities in patients with metastatic melanoma: a retrospective analysis of us claims data. BMC Cancer. 2018;18(1):145.
Tocut M, Brenner R, Zandman-Goddard G. Autoimmune phenomena and disease in cancer patients treated with immune checkpoint inhibitors. Autoimmun Rev. 2018;17(6):610–6.
Moyes KW, Davis A, Hoglund V, Haberthur K, Lieberman NA, Kreuser SA, et al. Effects of tumor grade and dexamethasone on myeloid cells in patients with glioma. Oncoimmunology. 2018;7(11): e1507668.
Choi SH, Stuckey DW, Pignatta S, Reinshagen C, Khalsa JK, Roozendaal N, et al. Tumor Resection Recruits Effector T Cells and Boosts Therapeutic Efficacy of Encapsulated Stem Cells Expressing IFNβ in Glioblastomas. Clin Cancer Res. 2017;23(22):7047–58.
Chu X, Tian W, Wang Z, Zhang J, Zhou R. Co-inhibition of TIGIT and PD-1/PD-L1 in Cancer Immunotherapy: Mechanisms and Clinical Trials. Mol Cancer. 2023;22(1):93.
Scholz A, Harter PN, Cremer S, Yalcin BH, Gurnik S, Yamaji M, et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol Med. 2016;8(1):39–57.
Kloepper J, Riedemann L, Amoozgar Z, Seano G, Susek K, Yu V, et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc Natl Acad Sci U S A. 2016;113(16):4476–81.
Zannikou M, Duffy JT, Levine RN, Seblani M, Liu Q, Presser A, et al. IL15 modification enables CAR T cells to act as a dual targeting agent against tumor cells and myeloid-derived suppressor cells in GBM. J Immunother Cancer. 2023;11(2).
Wang G, Zhang Z, Zhong K, Wang Z, Yang N, Tang X, et al. CXCL11-armed oncolytic adenoviruses enhance CAR-T cell therapeutic efficacy and reprogram tumor microenvironment in glioblastoma. Mol Ther. 2023;31(1):134–53.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–68.
Sonda N, Chioda M, Zilio S, Simonato F, Bronte V. Transcription factors in myeloid-derived suppressor cell recruitment and function. Curr Opin Immunol. 2011;23(2):279–85.
Yang B, Li X, Fu Y, Guo E, Ye Y, Li F, et al. MEK Inhibition Remodels the Immune Landscape of Mutant KRAS Tumors to Overcome Resistance to PARP and Immune Checkpoint Inhibitors. Cancer Res. 2021;81(10):2714–29.
Kinoshita R, Sato H, Yamauchi A, Takahashi Y, Inoue Y, Sumardika IW, et al. Newly developed anti-S100A8/A9 monoclonal antibody efficiently prevents lung tropic cancer metastasis. Int J Cancer. 2019;145(2):569–75.
Alghamri MS, Banerjee K, Mujeeb AA, Mauser A, Taher A, Thalla R, et al. Systemic Delivery of an Adjuvant CXCR4-CXCL12 Signaling Inhibitor Encapsulated in Synthetic Protein Nanoparticles for Glioma Immunotherapy. ACS Nano. 2022;16(6):8729–50.
Sun L, Clavijo PE, Robbins Y, Patel P, Friedman J, Greene S, et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight. 2019;4(7).
Greene S, Robbins Y, Mydlarz WK, Huynh AP, Schmitt NC, Friedman J, et al. Inhibition of MDSC Trafficking with SX-682, a CXCR1/2 Inhibitor, Enhances NK-Cell Immunotherapy in Head and Neck Cancer Models. Clin Cancer Res. 2020;26(6):1420–31.
Alfaro C, Sanmamed MF, Rodríguez-Ruiz ME, Teijeira Á, Oñate C, González Á, et al. Interleukin-8 in cancer pathogenesis, treatment and follow-up. Cancer Treat Rev. 2017;60:24–31.
Schott AF, Goldstein LJ, Cristofanilli M, Ruffini PA, McCanna S, Reuben JM, et al. Phase Ib Pilot Study to Evaluate Reparixin in Combination with Weekly Paclitaxel in Patients with HER-2-Negative Metastatic Breast Cancer. Clin Cancer Res. 2017;23(18):5358–65.
Lu Z, Zou J, Li S, Topper MJ, Tao Y, Zhang H, et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature. 2020;579(7798):284–90.
Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell. 2017;32(5):654-68.e5.
Sun R, Luo H, Su J, Di S, Zhou M, Shi B, et al. Olaparib Suppresses MDSC Recruitment via SDF1α/CXCR4 Axis to Improve the Anti-tumor Efficacy of CAR-T Cells on Breast Cancer in Mice. Mol Ther. 2021;29(1):60–74.
Surace L, Lysenko V, Fontana AO, Cecconi V, Janssen H, Bicvic A, et al. Complement is a central mediator of radiotherapy-induced tumor-specific immunity and clinical response. Immunity. 2015;42(4):767–77.
Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9(11):1225–35.
Shastri A, Choudhary G, Teixeira M, Gordon-Mitchell S, Ramachandra N, Bernard L, et al. Antisense STAT3 inhibitor decreases viability of myelodysplastic and leukemic stem cells. J Clin Invest. 2018;128(12):5479–88.
Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun. 2017;8(1):1736.
Proia TA, Singh M, Woessner R, Carnevalli L, Bommakanti G, Magiera L, et al. STAT3 Antisense Oligonucleotide Remodels the Suppressive Tumor Microenvironment to Enhance Immune Activation in Combination with Anti-PD-L1. Clin Cancer Res. 2020;26(23):6335–49.
Chen HM, Ma G, Gildener-Leapman N, Eisenstein S, Coakley BA, Ozao J, et al. Myeloid-Derived Suppressor Cells as an Immune Parameter in Patients with Concurrent Sunitinib and Stereotactic Body Radiotherapy. Clin Cancer Res. 2015;21(18):4073–85.
Lan J, Li R, Yin LM, Deng L, Gui J, Chen BQ, et al. Targeting Myeloid-derived Suppressor Cells and Programmed Death Ligand 1 Confers Therapeutic Advantage of Ablative Hypofractionated Radiation Therapy Compared With Conventional Fractionated Radiation Therapy. Int J Radiat Oncol Biol Phys. 2018;101(1):74–87.
Takacs GP, Kreiger CJ, Luo D, Tian G, Garcia JS, Deleyrolle LP, et al. Glioma-derived CCL2 and CCL7 mediate migration of immune suppressive CCR2(+)/CX3CR1(+) M-MDSCs into the tumor microenvironment in a redundant manner. Front Immunol. 2022;13: 993444.
Ashman LK, Griffith R. Therapeutic targeting of c-KIT in cancer. Expert Opin Investig Drugs. 2013;22(1):103–15.
Raychaudhuri B, Rayman P, Huang P, Grabowski M, Hambardzumyan D, Finke JH, et al. Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J Neurooncol. 2015;122(2):293–301.
Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res. 2014;124:31–82.
Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120(3):694–705.
Wu A, Maxwell R, Xia Y, Cardarelli P, Oyasu M, Belcaid Z, et al. Combination anti-CXCR4 and anti-PD-1 immunotherapy provides survival benefit in glioblastoma through immune cell modulation of tumor microenvironment. J Neurooncol. 2019;143(2):241–9.
Ban Y, Mai J, Li X, Mitchell-Flack M, Zhang T, Zhang L, et al. Targeting Autocrine CCL5-CCR5 Axis Reprograms Immunosuppressive Myeloid Cells and Reinvigorates Antitumor Immunity. Cancer Res. 2017;77(11):2857–68.
Blattner C, Fleming V, Weber R, Himmelhan B, Altevogt P, Gebhardt C, et al. CCR5(+) Myeloid-Derived Suppressor Cells Are Enriched and Activated in Melanoma Lesions. Cancer Res. 2018;78(1):157–67.
Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3(10):791–800.
Mitchell RA, Metz CN, Peng T, Bucala R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem. 1999;274(25):18100–6.
O’Reilly C, Doroudian M, Mawhinney L, Donnelly SC. Targeting MIF in Cancer: Therapeutic Strategies, Current Developments, and Future Opportunities. Med Res Rev. 2016;36(3):440–60.
Hassel JC, Jiang H, Bender C, Winkler J, Sevko A, Shevchenko I, et al. Tadalafil has biologic activity in human melanoma. Results of a pilot trial with Tadalafil in patients with metastatic Melanoma (TaMe). Oncoimmunology. 2017;6(9):e1326440.
Yu SJ, Ma C, Heinrich B, Brown ZJ, Sandhu M, Zhang Q, et al. Targeting the crosstalk between cytokine-induced killer cells and myeloid-derived suppressor cells in hepatocellular carcinoma. J Hepatol. 2019;70(3):449–57.
Tamura T, Ozato K. ICSBP/IRF-8: its regulatory roles in the development of myeloid cells. J Interferon Cytokine Res. 2002;22(1):145–52.
Stewart TJ, Liewehr DJ, Steinberg SM, Greeneltch KM, Abrams SI. Modulating the expression of IFN regulatory factor 8 alters the protumorigenic behavior of CD11b+Gr-1+ myeloid cells. J Immunol. 2009;183(1):117–28.
Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–35.
Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest. 2012;122(11):4094–104.
Reilley MJ, McCoon P, Cook C, Lyne P, Kurzrock R, Kim Y, et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: results of a phase 1b trial. J Immunother Cancer. 2018;6(1):119.
Hossain DM, Pal SK, Moreira D, Duttagupta P, Zhang Q, Won H, et al. TLR9-Targeted STAT3 Silencing Abrogates Immunosuppressive Activity of Myeloid-Derived Suppressor Cells from Prostate Cancer Patients. Clin Cancer Res. 2015;21(16):3771–82.
Smith AD, Lu C, Payne D, Paschall AV, Klement JD, Redd PS, et al. Autocrine IL6-Mediated Activation of the STAT3-DNMT Axis Silences the TNFα-RIP1 Necroptosis Pathway to Sustain Survival and Accumulation of Myeloid-Derived Suppressor Cells. Cancer Res. 2020;80(15):3145–56.
Waight JD, Netherby C, Hensen ML, Miller A, Hu Q, Liu S, et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest. 2013;123(10):4464–78.
Hashimoto A, Sarker D, Reebye V, Jarvis S, Sodergren MH, Kossenkov A, et al. Upregulation of C/EBPα inhibits suppressive activity of myeloid cells and potentiates antitumor response in mice and patients with cancer. Clin Cancer Res. 2021;27(21):5961–78.
Hu X, Bardhan K, Paschall AV, Yang D, Waller JL, Park MA, et al. Deregulation of apoptotic factors Bcl-xL and Bax confers apoptotic resistance to myeloid-derived suppressor cells and contributes to their persistence in cancer. J Biol Chem. 2013;288(26):19103–15.
Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32(1):19–25.
Bitsch R, Kurzay A, Özbay Kurt F, De La Torre C, Lasser S, Lepper A, et al. STAT3 inhibitor Napabucasin abrogates MDSC immunosuppressive capacity and prolongs survival of melanoma-bearing mice. J Immunother Cancer. 2022;10(3).
Li X, Su X, Liu R, Pan Y, Fang J, Cao L, et al. HDAC inhibition potentiates anti-tumor activity of macrophages and enhances anti-PD-L1-mediated tumor suppression. Oncogene. 2021;40(10):1836–50.
Munn DH. Blocking IDO activity to enhance anti-tumor immunity. Front Biosci (Elite Ed). 2012;4(2):734–45.
Xiang H, Ramil CP, Hai J, Zhang C, Wang H, Watkins AA, et al. Cancer-Associated Fibroblasts Promote Immunosuppression by Inducing ROS-Generating Monocytic MDSCs in Lung Squamous Cell Carcinoma. Cancer Immunol Res. 2020;8(4):436–50.
Feng S, Cheng X, Zhang L, Lu X, Chaudhary S, Teng R, et al. Myeloid-derived suppressor cells inhibit T cell activation through nitrating LCK in mouse cancers. Proc Natl Acad Sci U S A. 2018;115(40):10094–9.
Li T, Li X, Zamani A, Wang W, Lee CN, Li M, et al. c-Rel Is a Myeloid Checkpoint for Cancer Immunotherapy. Nat Cancer. 2020;1(5):507–17.
Sato H, Watanabe H, Ishii T, Bannai S. Neutral amino acid transport in mouse peritoneal macrophages. J Biol Chem. 1987;262(27):13015–9.
Xu P, Yin K, Tang X, Tian J, Zhang Y, Ma J, et al. Metformin inhibits the function of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Biomed Pharmacother. 2019;120: 109458.
Qin G, Lian J, Huang L, Zhao Q, Liu S, Zhang Z, et al. Metformin blocks myeloid-derived suppressor cell accumulation through AMPK-DACH1-CXCL1 axis. Oncoimmunology. 2018;7(7): e1442167.
Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. 2011;118(20):5498–505.
Dávila-González D, Choi DS, Rosato RR, Granados-Principal SM, Kuhn JG, Li WF, et al. Pharmacological Inhibition of NOS Activates ASK1/JNK Pathway Augmenting Docetaxel-Mediated Apoptosis in Triple-Negative Breast Cancer. Clin Cancer Res. 2018;24(5):1152–62.
Nigam S, McCarl L, Kumar R, Edinger RS, Kurland BF, Anderson CJ, et al. Preclinical ImmunoPET Imaging of Glioblastoma-Infiltrating Myeloid Cells Using Zirconium-89 Labeled Anti-CD11b Antibody. Mol Imaging Biol. 2020;22(3):685–94.
Li MO, Wolf N, Raulet DH, Akkari L, Pittet MJ, Rodriguez PC, et al. Innate immune cells in the tumor microenvironment. Cancer Cell. 2021;39(6):725–9.
Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther. 2021;6(1):362.
Acknowledgements
This study was supported by the National Key Research and Development Program of China (2020YFA0804200, 2023YFC3404800), the National Natural Science Foundation of China (82073166, 82273203). H.Y. is supported by the Program for Professors of Special Appointment (Eastern Scholar) at the Shanghai Institutions of Higher Learning (SSF151005).
Author information
Authors and Affiliations
Contributions
LH, LCX, DWZ, HY, and YM designed and wrote the manuscript. LCX, AKH, and DWZ revised the manuscript preparation. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lin, H., Liu, C., Hu, A. et al. Understanding the immunosuppressive microenvironment of glioma: mechanistic insights and clinical perspectives. J Hematol Oncol 17, 31 (2024). https://doi.org/10.1186/s13045-024-01544-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-024-01544-7