
Abstract
The tumor microenvironment (TME) is comprised of many different cell populations, such as cancer-associated fibroblasts and various infiltrating immune cells, and non-cell components of extracellular matrix. These crucial parts of the surrounding stroma can function as both positive and negative regulators of all hallmarks of cancer development, including evasion of apoptosis, induction of angiogenesis, deregulation of the energy metabolism, resistance to the immune detection and destruction, and activation of invasion and metastasis. This review represents a summary of recent studies focusing on describing these effects of microenvironment on initiation and progression of the head and neck squamous cell carcinoma, focusing on oral squamous cell carcinoma, since it is becoming clear that an investigation of differences in stromal composition of the head and neck squamous cell carcinoma microenvironment and their impact on cancer development and progression may help better understand the mechanisms behind different responses to therapy and help define possible targets for clinical intervention.
Similar content being viewed by others

Introduction
The head and neck cancer (HNC) is considered one of the malignities with the most severe impact on quality of life of patients, caused mainly by relatively low responsiveness to treatment and severe drug resistance [1,2,3]. HNC is a heterogeneous group of tumors arising from the mucosal surfaces of the nasal and oral cavity, oropharynx, larynx and hypopharynx. Up to 90% of these tumors are head and neck squamous cell carcinomas (HNSCCs) [4], which represent the sixth most prevalent cancer worldwide [5]. The survival rate still remains very low, since up to 25% of patients develop second cancer within 5 years after diagnosis [6]. The most important prognostic determinant of HNSCC tumors is considered the presence of lymph node metastases, since lymphatic metastatic spread correlates with a significant decrease in the survival rate of patients [7]. While primary risk factors are tobacco use and alcohol consumption [8], the role of the oncogenic human papillomaviruses (HPVs) has been implicated in HNSCC as well and many studies have suggested HPV infection as a risk factor of the HNSCC development [9,10,11].
In recent years, the outlook on cancer has changed dramatically and the tumor is no longer viewed as a bulk of malignant cancer cells, but rather as a complex tumor microenvironment (TME) that other subpopulations of cells corrupted by cancer cells get recruited into to form a self-sufficient biological structure. The stromal component of the tumor microenvironment is composed of multiple different cell types, such as cancer-associated fibroblasts, neutrophils, macrophages, regulatory T cells, myeloid-derived suppressor cells, natural killer cells, platelets and mast cells. These subpopulations of cells interact with each other as well as cancer cells via complex communication networks through various secreted cytokines, chemokines, growth factors and proteins of the extracellular matrix (ECM). This review will focus on describing these major subpopulations of cells and other factors influencing the TME and will discuss their function in the development of cancer, in particular HNSCC.
Tumor microenvironment
Cancer-associated fibroblasts
Cancer-associated fibroblasts (CAFs) are the predominant cell type within the tumor stroma and their main function is to maintain a favorable microenvironment for tumor cell growth and proliferation. CAFs modulate the microenvironment primarily via secretion of large variety of autocrine and paracrine cytokines and other tumor-promoting factors critical for tumor cell proliferation, angiogenesis, invasion, inflammation, metastasis and drug resistance. These factors include various growth factors, cytokines and chemokines, such as epidermal growth factor (EGF), hepatocyte growth factor (HGF), vascular endothelial growth factor (VEGF), C-X-C motif chemokine ligands (CXCCLs) CXCL12 and CXCL14, C-C motif chemokine ligands (CCLs) CCL5 and CCL7, and interleukins (ILs) IL-6 and IL-17A [12,13,14,15,16,17,18,19]. CAFs are also crucial producers of matrix-metalloproteinases (MMPs) and therefore play an important role in modulating the microenvironment by remodelation and degradation of ECM, which ultimately results in promotion of the invasive phenotype of cancer cells [20,21,22].
The morphology of CAFs is characterized by their elongated spindle-like shape, sharing many similarities with mesenchymal and smooth muscle cells [23]. CAFs have distinctly different morphological and biological characteristics compared with normal fibroblasts; they also differ from normal fibroblasts by their constitutively activated state. Several molecules, such as α-smooth muscle actin (α-SMA), fibroblast activation protein (FAP), fibroblast-specific protein-1 (FSP-1), platelet-derived growth factor receptor α/β (PDGFR α/β) and vimentin are considered some of the markers of activated CAFs [12, 24,25,26].
CAFs can be derived from various types of progenitor cells, such as resting resident fibroblasts or pericytes through mesothelial-mesenchymal transition (MMT) [27], endothelial cells through endothelial-mesenchymal transition (EdMT) [28], epithelial cells through epithelial-mesenchymal transition (EMT) [29], adipocytes [30] and bone marrow-derived mesenchymal cells (BDMCs) [31]. The most common marker used to detect CAFs in the tumor stroma is α-SMA, a specific marker of myofibroblasts [32]. This myofibroblast phenotype of CAFs is frequently observed in HNSCC and the upregulation of α-SMA has been correlated to poor prognosis in oral carcinoma [33]. Another marker of myofibroblasts widely used for detection of CAFs is FAP [34, 35]. FAP is overexpressed in sites of fibrosis and in the tumor stroma of various carcinomas, including HNSCC. CAFs can also be characterized by the absence of epithelial and endothelial markers, such as cluster of differentiation (CD) CD31 and cytokeratin [36, 37].
Macrophages
Macrophages are mononuclear phagocytes considered one of the most important immune cells, mainly for their prominent active role in tissue homeostasis and both innate and acquired immune response against pathogens [38]. Macrophages display a great plasticity, M1 and M2 representing the extreme activation states. However, the re-polarization of fully polarized macrophages in vitro towards the other phenotype by various cytokines has been observed [39]. These two distinct phenotypes are characterized by different receptor expression, function and cytokine and chemokine production [40,41,42,43]. The “pro-inflammatory” classically activated M1 macrophages are characterized by their activation by the T helper type 1 (Th1) cytokine interferon-γ (IFN-γ) and/or bacterial lipopolysaccharide (LPS). They produce pro-inflammatory cytokines, such as IL-12, IL-23 and tumor necrosis factor-α (TNF-α), and chemokines (CCL-5, CXCL9, CXCL10 and CXCL5). They participate in anti-tumor immunity by contributing to the Th1 response to infection, by inhibiting proliferation and by exerting cytotoxic activity [44,45,46]. The “anti-inflammatory” alternatively activated M2 macrophages play an immunoregulatory role and are involved in the tissue remodeling, wound healing, angiogenesis and tumor progression [47,48,49,50]. The M2 phenotype is induced by various Th cytokines (IL-4, IL-10, IL-13) and is characterized by increased secretion of anti-inflammatory cytokines, such as IL-1 receptor antagonist (IL-1ra), IL-10 and TGF-β [51,52,53].
Tumor-associated macrophages (TAMs) represent a major component of the macrophage population largely contributing to proliferation, invasion and metastasis of tumor cells, promotion of tumor progression, angiogenesis and suppression of T cell antitumor immune response. Recent studies suggested the correlation between the level of infiltration of TAMs and a poor outcome in HNSCC, which could be used as a potential prognostic marker [54,55,56]. In the past years, TAMs have been considered a large subpopulation of macrophages within the M2 phenotype, however it has become clear TAMs are able to adopt a wide range of different activation states between M1 and M2, expressing both M2 and M1 markers, such as upregulated IL-10 (M2) [57], arginase-1 (M2) [58], peroxisome proliferator-activated receptor γ (PPARγ) (M2) [59], TNF-α (M1) [60], MMP-9 (M1) [61] and increased levels of interferon-(INF)-inducible chemokines CCL2, CCL5, CXCL9, CXCL10 and CXCL16 (M1) [62].
Neutrophils
Neutrophils, also known as polymorphonuclear leukocytes (PMNs), are essential effector cells of the innate immune system and the most predominant leukocyte population present in the circulation [63]. Neutrophils, along with macrophages, represent the first line of defense against pathogens and first responders at the site of infection and injury [64], they are also directly involved in adaptive immunity responses, playing an important role in mediation of T cell independent antibody responses [65], as well as antigen presentation and T cell activation [66, 67]. Until recently, neutrophils were thought to act only as phagocytic cells by producing lytic enzymes and reactive oxygen species (ROS). However, neutrophils are able to form neutrophil extracellular traps (NETs) by releasing their cytotoxic cytosolic and granule proteins on a scaffold of decondensed chromatin [68] in a cell death process called NETosis [69, 70]. It has been reported NETs activate platelets and promote thrombosis [71, 72], and indeed an increased risk of cancer-associated venous thromboembolism (VTE) has been reported in many types of cancer, including the HNSCC [73].
The identification and characterization of the neutrophil population based on the expression of specific surface markers remains difficult since these specific markers have yet to be identified. For the identification of pure human neutrophil subpopulations, many studies use various markers individually or in combination, such as CD11b, CD14, CD15, CD16, CD62L and CD66b [74,75,76].
The contribution of tumor-associated neutrophils (TANs) to the cancer progression remains unclear, the main reason being TANs show both pro- and anti-tumor properties. In TANs, in an analogy to TAMs, a phenotypic duplicity in a form of polarization states has been observed [77]. These anti-tumor and pro-tumor phenotypes within the neutrophil population have been termed N1 and N2, respectively. The pro-tumor N2 phenotype is characterized by increased expression of angiogenesis and invasion promoting factors CXCR4, VEGF and MMP-9 with absent IFN-β [78] and is acquired by neutrophils following the TGF-β treatment [77]. However, neutrophils can revert back to the cytotoxic N1 phenotype upon the TGF-β blockade or in the presence of the IFN-β [79], while expressing high levels of intercellular adhesion molecule 1 (ICAM1) and TNF-α as well as increasing NETs formation.
Myeloid-derived suppressor cells
Myeloid-derived suppressor cells (MDSCs) comprise a heterogeneous population of immature inhibitory immune cells in various stages of myelopoiesis [80]. This cell population plays a crucial role in negative regulation of the immune response in many pathological conditions, such as cancer and inflammation, by inhibiting both the adaptive and innate immunity. MDSCs are induced by various tumor-derived factors in the microenvironment, mainly granulocyte-macrophage colony-stimulating factor (GM-CSF), VEGF and IL-6 [81], and modulate the inflammatory microenvironment via depletion of many amino acids (such as L-arginin, L-tryptophan and L-cystein) [82,83,84], via increased production of nitric oxid (NO), ROS, inducible NO synthase (iNOS) and arginase-1 [85,86,87], and via expression of programmed death receptor ligand 1 (PD-L1), which ultimately inhibits T cell activation and proliferation and causes T cell apoptosis [88]. MDSCs also regulate the activity of natural killer (NK) cells and the induction of immunosuppressive regulatory T cells (Tregs) [89, 90].
MDSCs were originally described in peripheral blood of HNSCC patients as immature CD34+ cells exhibiting the ability to suppress the activity of T cells [91,92,93]. The identification of MDSCs based on the expression of surface markers is challenging mainly because of the phenotypic diversity of the MDSCs population, since different subpopulations within the MDSCs express combinations of various myeloid markers, including CD11b, CD33, CD14, CD15 and CD16 but lack the expression of HLA-DR. Although MDSCs have been first discovered for their immune-suppressive function in cancer, recently the presence of MDSCs has also been linked to other processes within the TME, such as promotion of tumor angiogenesis via production of pro-angiogenic factors [94, 95], degradation of ECM via production of significant levels of MMPs, especially MMP-9, and most importantly the formation of premetastatic niches.
Regulatory T-cells (Tregs)
Regulatory T cells comprise a unique subset of T cells responsible for suppression of excessive immune response, for maintaining self-tolerance and homeostasis, and for regulation of other immune cells, including CD4 and CD8 T-cells, B cells, NK cells, macrophages and dendritic cells; and the loss of these cells ultimately results in various autoimmune diseases [96]. Tregs are characterized by their expression of markers CD4, CD25 and transcription factor forkhead box P3 (FOXP3) [97]. However, the markers CD4 and CD25 are also expressed by effector T cells, thereby making it difficult to distinguish these two populations. In addition, the intracellular localization of FOXP3 requires cell permeabilization for its detection, which makes the isolation of viable Tregs challenging. Tregs also express high levels of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and glucocorticoid-induced tumor necrosis factor receptor family-related protein (GITR) [98, 99].
Treg cells display great heterogeneity within the population, thus can be divided into phenotypically and functionally distinct subpopulations based on their localization, origin and expression profile of markers [100]. CD25+CD4+ Tregs arising in the thymus, termed natural regulatory T cells, express the FOXP3 transcription factor constitutively and are crucial for the maintenance of self-tolerance. In contrast, peripheral CD25+CD4+ Tregs can differentiate from conventional mature CD4+ T cells outside of the thymus, thus are called induced or adaptive Tregs. These T cells require activation in the presence of cytokines, such as IL-2 and TGF-β, to upregulate FOXP3 [101] and their main function is to prevent local inflammation.
Since their discovery, the molecular mechanisms by which Tregs exert their suppressor function have been intensely studied. It has been observed Tregs can influence the immune system via either contact-dependent or contact-independent mechanisms. Vignali et al. arranged these mechanisms into four modes of action: (1) suppression by inhibitory cytokines (such as IL-10, IL-35 and TGF-β), (2) suppression by cytolysis via granzyme-A/B-dependent and perforin-dependent killing of target cells, (3) suppression of effector T cells by metabolic disruption via depletion of IL-2, and (4) suppression by modulation of dendritic-cell (DC) maturation or function [102].
Platelets
Platelets, also known as thrombocytes, are anucleated cells arising as fragments of megakaryocytes in the bone marrow, that serve as another major cellular group of first responders at the site of injury. It has been thought the primary function of platelets is thrombosis, wound healing and maintaining of homeostasis, but in recent years numerous studies started to focus on the role of blood platelets in regard of cancerogenesis, tumor biology and inflammation.
Platelets mediate the tumor microenvironment via three types of secretory granules - dense granules, lysosomes and α-granules. During platelet activation, the cargo from these granules is released into the extracellular environment, leading to platelet aggregation, vasoconstriction and regulation of cell proliferation through secretion of numerous growth factors [103]. The dense granules contain mainly small molecules, including ADP, ATP, calcium, 5-HT (5-hydroxytryptamine, also known as serotonin) and pyrophosphate [104,105,106]. Dense granules also contain membrane proteins CD63 and lysosomal-associated membrane protein 1/2 (LAMP1/2), glycoprotein-(GP)-Ib, P-selectin, and integrin αII-β3 [107]. Lysosomes represent another type of platelet granules. The function of these granules has not yet been fully eluciated, however they contain an acidic pH with acid hydrolases, which are able to degrade and remodel the ECM and vasculature. Also similarly to dense granules, lysosomes express membrane proteins CD63 and LAMP1/2 [108]. The most abundant group, α-granules, contains a vast number of proteins and factors important in hemostasis, thrombosis and adhesion, including vitronectin, thrombospondin, fibrinogen, fibronectin and von Willebrand factor (VWF). In addition, α-granules contain proteins involved in inflammation and angiogenesis, many mitogenic growth factors, a variety of chemokines and various MMPs [109,110,111,112,113]. The release of these factors from α-granules attracts other cells to form tumor cell–platelet emboli, stimulating tumor cell growth and angiogenesis. α-granules also express number of transmembrane proteins, such as integrins, GP αIIbβ3, CD36, glucose transporter 3 (GLUT3), GPVI and P-selectin [114,115,116,117]. P-selectin, a surface protein translocated during platelet activation, is responsible for mediating platelet-leukocyte interactions via binding to leukocyte P-selectin glycoprotein ligand-1 (PSGL-1) [118].
Mast cells
Mast cells (MCs) represent another important myeloid component of the immune system that contributes to both innate and acquired immune responses. Like other immune cells, mast cells originate from pluripotent progenitor cells in the bone marrow, which they exit undifferentiated and migrate to target peripheral tissues to complete maturation. This terminal differentiation is strongly regulated by various factors provided by the microenvironment, including stem-cell factor (SCF) and IL-3 [119]. The activation of a mast cell is mediated by the cross-linkage of the IgE receptor (FcεRI) expressed on their surface, which leads to the release of the granule inflammatory cargo into the extracellular space, including histamine, TNF-α, heparin, chondroitin sulfate E, prostaglandin D2 (PGD2), tryptase, chymase, cathepsin G, carboxypeptidase A (CPA1), leukotriene C4 (LTC4), various interleukins and GM-CSF [120]. In addition to the rapid secretion of the granule content through exocytosis, mast cells release their contents selectively via piecemeal degranulation [121]. Interestingly, piecemeal degranulation has been particularly detected in areas of chronic inflammation or tumors and has been reported to be a preferred secretory pathway of tumour-associated mast cells (TAMCs) [122].
The aforementioned profile of mediators secreted by TAMCs suggests that TAMCs can play both pro- and anti-tumorigenic roles in cancer development. Tumor-promoting functions of TAMCs include angiogenesis through the production of VEGF and fibroblast growth factor-(FGF)-2 [123], ECM degradation via production of MMPs and various proteases, which results in tumor cell invasion and migration [124], and induction of tumor cell proliferation via production of histamine [125]. In addition, mast cells produce a variety of chemotactic factors in order to recruit other immune cells into the tumor [126, 127]. In contrast, in some types of tumors, the tumor suppressive effects of TAMCs have been reported, mainly by supporting tumor rejection [128] and mediating tumor cell apoptosis via the production of IL-4 and TNF-α [129, 130].
Natural killer cells
Natural killer cells (NK cells) play a crucial role in the innate immune system, since their main function in the organism is the ability to quickly detect and kill virus-infected or malignant cells. NK cells are characterized as large granular CD3- lymphocytes that can be classified into two subsets, depending on their expression levels of surface markers CD16 and CD56. CD56dim/CD16bright subpopulation constitutes the majority, approximately 90% of all peripheral blood NK cells, and is responsible for high natural cytotoxicity [131]. CD56bright/CD16dim subpopulation is characterized by higher expression levels of variety of immunomodulatory cytokines. The most prominent cytokines secreted by NK cells are IFN-γ and TNF-α. However, NK cells have been reported to produce a variety of other important factors, including GM-SCF, IL-5, IL-8, IL-10, IL-13, CCL2, CCL3, CCL4, CCL5 and CXCL10 [132,133,134,135].
NK cell function is tightly regulated by the ratio of signals from two different types of receptors present on the cell surface – activating and inhibitory receptors. The self-MHC class I molecules expressed on healthy cells act as inhibitory stimuli preventing NK cell activation [136]. Malignant or virus-infected cells downregulate MHC-I expression in order to escape cytotoxic T cells; this however, results in recognition by NK cells. In addition, activating receptors on the target cells’ surface are upregulated in response to the virus infection or their malignant transformation [137]. The activation of NK cells is then followed by number of possible inductions of apoptosis of target cell, including exocytosis of perforin and granzymes, Fas ligand (FasL), TNF-related apoptosis-inducing ligand (TRAIL) activation or antibody-dependent cellular cytotoxicity (ADCC) [138,139,140,141].
In contrast to cytotoxic T cells, NK cells do not require prior sensitization or stimulation for their effector function. However, some recent studies provide evidence that a subpopulation of NK-like cells, termed natural killer T cells (NKT cells), may play an important role in the immune response, since this subpopulation lies at the interface between innate and adaptive immune systems [142]. NKT cells are of lymphoid lineage and they share many morphological and functional characteristics of T cells and NK cells since they are defined by the expression of both T cell and NK cell surface markers [143]. NKT cells require prior priming for their function and can develop antigen-specific immunological memory [144,145,146]. One subset of NKT cells, the invariant natural killer T cells (iNKT cells), express a highly restricted invariant aβ T cell receptor (TCR) and low levels of these iNKT cells in peripheral blood predict poor outcome in HNSCC patients [147, 148].
These and other aforementioned subpopulations are summarized in Table 1.
Extracellular matrix
The extracellular matrix (ECM) is a non-cellular network of macromolecules, including fibrous structural proteins, glycoproteins, growth factors and proteoglycans that form a structure providing other surrounding cells with physical and biochemical support. In cancer, ECM becomes frequently deregulated and disorganized, which directly stimulates malignant cell transformation [149, 150]. ECM produces high amounts of MMPs. MMPs are a group of zinc-dependent protein and peptide hydrolases secreted and activated by malignant cells, capable of degradation of ECM proteins of the basement membrane, as well as other important molecules, such as growth factors, cell surface receptors and adhesion molecules [151,152,153,154,155].
The first hypothesis surrounding the function of MPPs has been attributed to their capability of degrading ECM and helping tumor cells migrate to local and distant sites. In recent years, it has been observed the crucial function of MMPs in the ECM is activating growth factors or releasing them from the matrix, thus promoting the initiation and proliferation of primary tumors. MMPs are also involved in tumor angiogenesis by activating basic fibroblasts growth factor (bFGF), VEGF and TGF-β [156,157,158]. Although tumor cells were considered to be the source of MMPs in the stroma to help degrade the surrounding ECM, it is now becoming clear that most of the MMPs are produced by the stromal cells in the tumor microenvironment, such as fibroblasts and inflammatory cells [159, 160].
Proteins of ECM, such as collagen, elastin, fibronectin, laminin and tenascin influence cell adhesion and proliferation as well as provide a structural support along which cells migrate out of and into the TME. Increased production of collagen, laminin and elastin also results in elevated stiffness of tumor compared to surrounding normal tissue [161,162,163]. Increased tumor stiffness has a strong impact on cancer progression by activating oncogenic intracellular signaling, such as Akt, β-catenin, focal adhesion kinase (FAK) and phosphatidylinositol 3-kinase (PI3K) pathways, while simultaneously inhibiting tumor suppressor genes for phosphatase and tensin homolog (PTEN) and glycogen synthase kinase 3α/β (GSK3α/β) [164]. Increased matrix stiffness also promotes the activation of surrounding fibroblasts to a CAF phenotype, which is maintained via mechanosensitive transcription factor yes-associated protein (YAP) [165].
Up to the 30% of the ECM protein mass constitutes of collagen, which provides the cell with tensile strength and support for migration, therefore playing an important role in the regulation of the cell behavior and development [166]. Besides the mechanical and structural contributions, collagens also play a crucial role in a wide range of biological functions, such as tissue scaffolding, cell adhesion, cell differentiation, cell migration and wound repair [167,168,169,170]. Along with collagen, one of the most abundant glycoproteins of the ECM is fibronectin (Fn), which is produced by various different cell types, such as fibroblasts and endothelial cells [171, 172]. Fibronectin structure contains binding and interaction sites for several other molecules present in the ECM, such as integrins, fibrin, heparin, tenascin, collagen, gelatin and syndecan [173,174,175,176,177]. In regards to cancer development, increased levels of fibronectin have been associated with tumor progression, migration, invasion and reduced responsiveness to treatment [178,179,180,181,182]. Moreover, CAF-derived matrices exhibit aligned fibronectin organization, which mediates directional migration of cancer cells [183].
Metabolic reprogramming of TME
A common feature of the rapid progression of solid tumors is intratumoral hypoxia, which arises as a consequence of insufficient oxygen supply to the tissue. Rapidly growing tumors quickly exhaust the available oxygen, which stimulates an upregulation of production of pro-angiogenic factors, such as VEGF, to form new vessels. However, these newly formed blood vessels are often characteristic of high leakage and irregular structure, which impair their function [184]. Hypoxic microenvironment has also been implicated as a crucial contributor to radio- and multidrug-resistance [185, 186]. Hypoxia leads to upregulation of hypoxia-inducible factor 1 (HIF-1) [187]. HIF-1 represents a key player in mediating the adaptive cellular response to low oxygen levels in the microenvironment. As a major transcription factor, HIF-1 has been implicated in the regulation of the expression of various genes associated with tumor cell growth, survival and proliferation [188,189,190], including genes involved in cellular energy metabolism. HIF-1 induces upregulation of many glucose transporters (GLUTs) and enzymes (such as lactate dehydrogenase A) [191], thus triggering the shift from oxidative phosphorylation (OXPHOS) to less energetically efficient glycolytic pathway in tumor cells, a process known as the Warburg effect.
Warburg effect describes an observation, in which glucose taken up by the tumor tends to get metabolized into lactate to generate ATP even in a sufficient presence of oxygen via aerobic glycolysis instead of oxidative phosphorylation [192]. It has been suggested, that the Warburg effect may promote the creation of more advantageous TME for cancer cell proliferation, survival and invasion. Due to these metabolic alterations, tumor cells produce elevated amounts of lactate, H+ and CO2, which results in enhanced acidification of the TME, thus increasing the tumor metastatic potential and resistance to treatment [193,194,195]. Interestingly, tumor-derived lactate has been reported to contribute to the polarization of TAMs into the M2 phenotype [196]. In addition to glucose, tumor cells can utilize L-lactate as an alternative energy source via lactate shuttle, which is regulated by the conversion of lactate into pyruvate by the lactate dehydrogenase (LDH) as well as by the transport of lactate across the tumor cell plasma membrane [197, 198]. The proton-linked transport of L-lactate, pyruvate, acetate and ketone bodies across the plasma membrane is facilitated by monocarboxylic acid transporters MCT1-MCT4 [199]. In tumors, the influx and efflux of excessive levels of L-lactate into and out of tumor cells are directed by MCT1 and MCT4. The overexpression of these two MCTs has been reported in several tumors, including HNSCC, and has been associated with poor prognosis [200,201,202,203]. Many types of cancer, including HNSCC, exhibit a metabolic symbiosis between tumor cells and surrounding stroma, CAFs in particular. A recent study demonstrated that the glycolytic switch in HNSCC cancer cells is induced by CAF-derived HGF and in turn HNSCC-secreted bFGF promotes lactate consumption by CAFs [204].
TME in the pathogenesis of HNSCC
Premalignant lesion
HNSCC is associated with severe immunosuppression, however, the milieu of the premalignant lesion has yet to be well defined. It has been reported that oral leukoplakia shows a significant infiltration of proinflammatory immune cells, such as TAMs, CD8+ T cells and NK cells [205,206,207]. Costa et al. conducted a study to examine the differences in the immunological phenotype of the premalignant and malignant stages of HNSCC using a mouse model of 4-nitroquinoline 1-oxide (4-NQO)-induced oral carcinogenesis [208]. It was observed that the premalignant stage is associated with elevated levels of inflammatory Th1, Tc1 and Th17 cells compared to controls and HNSCC-bearing mice, while the number of Tregs increased in HNSCC-bearing mice. The same mouse model was utilized to investigate the shift in the inflammatory cytokine profile depending on the malignant progression [209]. It has been observed, that premalignant oral lesions are associated with an increased level of IL-17, as well as IL-23, compared to controls or HNSCC, thus promoting the Th17 phenotype. In contrast, HNSCC tissues showed a downregulation of IL-23 and upregulation of TGF-β, most likely to skew the Th17 phenotype toward the Treg phenotype. Another study showed that premalignant lesions secrete many proinflammatory mediators, such as CCL5 (also known as RANTES), monocyte chemoattractant protein 1 (MCP-1), granulocyte-colony stimulating factor (G-CSF) and prostaglandin-E2 (PGE2) compared to HNSCC cells, suggesting the premalignant microenvironment to be more immune stimulatory than the microenvironment of an established HNSCC [210]. Some research has also been conducted on saliva samples of patients with premalignant oral lesions, which showed increased levels of proinflammatory cytokines TNF-α and IL-6 [211,212,213]. Several studies investigated the effect of immune cell infiltration on the progression of the premalignant lesion to malignant phenotype through angiogenesis. Immunohistochemical analyses have shown the total number of immune cells infiltration to be significantly elevated depending on the severity of the lesion, with lowest numbers observed in normal gingival tissue. In addition, the mast cell density (MCD) significantly correlated with microvessel density (MVD) depending on the progression of the malignancy [214,215,216,217].
In addition to the immune cell infiltration, the contribution of CAFs to the progression from the premalignant lesion to oral squamous cell carcinoma (OSCC) has been investigated. These studies have focused on the distribution of the CAFs marker α-SMA, which has been detected in samples of premalignant lesions, while absent in normal epithelium [218,219,220]. Interestingly, increased frequency of CAFs correlated with the progression from normal mucosa and potentially malignant disorders to an invasive phenotype. Potentially malignant oral leukoplakia also shows elevated expression of ECM components tenascin, MMP-2, as well as FGF-2 and its receptors FGFR-2 and FGFR-3, which are predictive of progression to OSCC [221,222,223].
Primary tumor
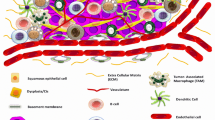
The growth of primary tumor is associated with the presence of immune cells, which cause inflammation frequently observed in HNSCC (shown in Fig. 1). Several studies investigated the significance of the overall population of tumor-infiltrating lymphocytes (TILs) as a prognostic marker of HNSCC. In these studies, various representative subsets of TILs, such as CD8+ cytotoxic T cells, CD4+ helper T cells, CD68+ macrophages and MDSCs, CD163+ macrophages, CD57+ NK cells and FOXP3+ Tregs, were evaluated and correlated with clinicopathologic characteristics of HNSCC patients. Immunohistochemical analysis revealed that tumors heavily infiltrated by TILs were associated with better outcome [224,225,226,227].

Cellular constituents within the tumor microenvironment. In addition to the cancer cells, the tumor stroma is comprised of many other supporting cell populations as well as the extracellular matrix, which crucially contribute to the tumor progression. The characteristics and function of individual cell populations are described in the Table 1. Abbreviations: TAM tumor-associated macrophage, TAN tumor-associated neutrophil, MDSC myeloid-derived suppressor cell, NK natural killer cell, Treg regulatory T cell, CAF cancer-associated fibroblast, ECM extracellular matrix
The infiltration of TAMs is a major contributor to the inflammation in HNSCC and is associated with poor prognosis, lymph node metastasis and low survival [228,229,230,231,232]. Kross et al. found the level of monocyte-derived IL-6 predicted recurrence and survival of HNSCC patients using an in vitro coculture system of monocytes with spheroids derived from HNSCC patients [233]. Costa et al. reported a predominance of M2 macrophages expressing TGF-β and IL-10 in oral squamous cell carcinoma (OSCC) group compared with healthy controls, which was further correlated with worse prognosis [234]. A recent study by Jiang et al. showed that compared to peritumoral macrophages OSCC-derived TAMs expressed higher levels of PD-L1, which correlated with increased T cell apoptosis [235], and this has been confirmed by other studies [236]. Beside tumor cells, macrophages also constitute an important source of VEGF, thus may contribute to tumor development via neovascularization [237,238,239,240]. Several studies also evaluated the prognostic significance of CD68+ macrophage infiltration regarding HPV status of HNSCC, which show that higher macrophage infiltration in HPV+ compared with HPV- HNSCC correlated with better prognosis [56, 241]. Also, high infiltration of neutrophils in OSCC is associated with poor clinical outcomes. A study by Trellakis et al. showed that high neutrophil infiltration correlated with poor patient survival [242]. This was confirmed by Wang et al., who correlated high neutrophil infiltration with high tumor stage, recurrence and lymph node metastases [243]. An in vitro study by Trellakis et al. investigated the interaction of neutrophils and HNSCC cancer cells, which reported that HNSCC-conditioned medium reduced neutrophil apoptosis, increased chemotaxis of neutrophils and induced the production of MMP-9 and CCL4 by neutrophils [244]. Mast cells influence the primary tumor mainly by the production of many pro-angiogenic factors, such as VEGF, bFGF, TGF, TNF-α, tryptase, heparin and various MMPs, which are associated with ECM degradation, angiogenesis, progression and growth of OSCC [245, 246]. Mast cell and microvessel densities are increased in OSCC compared with normal mucosa, however, no significant correlation has been found [247,248,249,250,251]. Various studies focused on the presence of NK cells in HNSCC patients, in which an increased number of NK cells predicted improved survival [252, 253]. Korrer et al. found that NK cells derived from HNSCC primary tumors significantly downregulated activating receptors NKG2D, DNAM-1, NKp30, CD16 and 2B4, and upregulated their inhibitory receptors NKG2A and PD-1 compared with NK cells from the blood of the same patients [254]. A significantly increased number of Tregs in peripheral blood, lymph nodes and tumors in HNSCC patients have been observed [255,256,257,258], which has been correlated with cancer recurrence [259]. Although HNSCC patients show increased levels of Tregs compared to healthy controls, various studies provide conflicting results in terms of the prognostic significance of Tregs [260,261,262]. In addition, Tregs are increased in HNSCC patients after treatment [263]. HNSCC displays a high abundance of circulating MDSCs, which correlates with advanced stages of HNSCC [264]. Although the main function of MDSCs is the inhibition of T cell activation, a study by Zheng et al. demonstrated that MDSCs-derived caspase-1 promotes HNSCC cancer cell proliferation in a T cell-independent manner both in vitro and in vivo [265]. Moreover, several studies demonstrated that targeting MDSCs leads to enhanced antitumor immunity via increasing the number of CD8+ cytotoxic T cells in HNSCC [266,267,268].
Immunohistochemical analyses of primary OSCC report higher density of CAFs in over 60% cases, while healthy tissues and adjacent stroma of premalignant lesions show no staining [269,270,271]. It has been observed that increased numbers of CAFs within the primary tumor correlate with worse prognosis of HNSCC patients [272,273,274,275]. Several studies have shown that CAFs reside in the vicinity of tumor cells, thus the reciprocal interaction between CAFs and cancer cells has been suggested as the main force driving tumor development. Coculture systems of CAFs and HNSCC cancer cells revealed that tumor-CAFs crosstalk enhances the production of various tumor-promoting cytokines, chemokines, components of ECM, growth factors and MMPs. Jung et al. demonstrated that OSCC cancer cells induced upregulation of several molecules in CAFs after coculture, such as CCL7, CXCL1, CXCL2, CXCL3 and IL-8 [17]. A recent study by Álvarez-Teijeiro et al. identified several proteins differentially secreted in CAF-conditioned medium compared to normal fibroblasts, including EGF containing fibulin-like extracellular matrix protein 1 (EFEMP1), platelet derived growth factor D (PDGFD) and insulin-like growth factor binding proteins 5/7 (IBP5/IBP7) that may be responsible for sustaining the cancer stem cell phenotype in HNSCC [276]. Several studies found that HNSCC-derived CAFs express elevated levels of various molecules, such as TGF-β [277], HGF [278] and MMPs [279] compared to normal fibroblasts. Takahashi et al. demonstrated that, compared to normal fibroblasts, CAFs suppressed T cell proliferation and induced T cell apoptosis and the differentiation of PBMCs into Tregs more efficiently, which suggests an important role of HNSCC-derived CAFs in immunosuppression. Their results also showed an increased expression of IL-6, CXCL8, TNF, TGFB1, and VEGFA in CAFs compared to normal fibroblasts [280]. Bagordakis et al. identified number of overexpressed proteins related to ECM organization, ECM disassembly and metabolic processing of collagen in the CAFs secretome compared to normal oral fibroblasts, such as fibronectin type III domain-containing protein 1 (FNDC1), serpin peptidase inhibitor type 1 (SERPINE1) and stanniocalcin 2 (STC2) [281].
It is well known that ECM plays a crucial role in HNSCC development. Reportedly, the major ECM proteins involved in HNSCC development and progression are collagen, laminin and fibronectin [282]. Immunohistological studies of different histological grades of HNSCC show that distribution of ECM proteins, such as collagen and laminin, decrease depending on increased grade [283,284,285]. Harada et al. found that decreased expression of laminin, collagen type IV and vitronectin, and increased expression of fibronectin and tenascin correlated with the invasive phenotype of primary OSCC tumors [286]. In addition, an immunohistochemical analysis by Fabricius et al. investigated the expression of integrins αvβ3, αvβ5, α5β1 and their ligands osteopontin, vitronectin, fibronectin and fibrinogen in primary HNSCC tissues. Their results suggest that interactions αvβ3-osteopontin, αvβ3-fibronectin and α5β1-fibronectin play a role in HNSCC angiogenesis and interactions α5β1-fibronectin and αvβ5-vitronectin in HNSCC cancer cell behavior [287].
Epithelial-mesenchymal transition
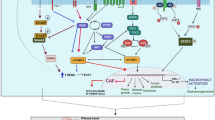
Epithelial-mesenchymal transition (EMT) is a dynamic process in cancer development, during which polarized epithelial tumor cells acquire a mesenchymal phenotype. This shift to a mesenchymal phenotype is characterized by the loss of cell adhesion and upregulation of various components of the extracellular matrix, followed by increased migratory potential and enhanced invasiveness (shown in Fig. 2). EMT is associated with the loss of proteins involved in cell junctions, such as E-cadherin and β-catenin, and with an upregulated expression of mesenchymal markers such as α-SMA, vimentin, FSP-1 and N-cadherin [288,289,290]. The loss of E-cadherin and high vimentin levels have been associated with tumor progression and an increase of metastases in HNSCC patients [291].

Metastatic cascade. a Acquisition of metastatic potential via epithelial-mesenchymal transition, degradation of the ECM (secretion of MMPs) and invasion through the basement membrane. Immune cells are recruited to the primary tumor site via cancer cell-derived and CAF-derived factors and cytokines. b Intravasation of cancer cells via invadopodia formation. Cancer cells acquire the resistance to anoikis. c Survival in the circulation. Cancer cells mediate the so-called tumor cell-induced platelet aggregation (TCIPA) to form a “platelet cloak” in order to be protected from TNF-α and to escape NK cells. Cancer cells evade the immune system by upregulation of indoleamine 2,3-dioxygenase (IDO). d Extravasation and formation of a secondary tumor site. Arrest of tumor cells on the endothelium, sequestration of tumor cells via NET formation, followed by transendothelial migration and invasion into the surrounding tissue. Abbreviations: TAM tumor-associated macrophage, TAN tumor-associated neutrophil, MDSC myeloid-derived suppressor cell, NK natural killer cell, Treg regulatory T cell, CAF cancer-associated fibroblast, ECM extracellular matrix, MMPs matrix metalloproteinases, MIF migration inhibitory factor, TGF-β transforming growth factor-β, EGF epithelial growth factor, HGF hepapocyte growth factor, TNF- α tumor necrosis factor-α, IDO indoleamine 2,3-dioxygenase, NET neutrophil extracellular trap
In order for tumor cells to migrate to local and distant sites, tumor and the surrounding stroma cells acquire the ability to proteolytically degrade the basement membrane and underlying collagen matrix. This degradation of, and invasion through, ECM largely depends on the function of filament-like protrusions formed on invading tumor cells, termed invadopodia, and many recent studies suggest a crucial involvement of invadopodia-mediated ECM remodeling during EMT. These structures contain various proteins such as actin regulators cortactin, dynamin and neural Wiskott–Aldrich syndrome protein (N-WASP) [292]; adhesion proteins including many integrins [293]; adaptor proteins Tyr kinase substrate with four SH3 domains (TKS4) and Tyr kinase substrate with five SH3 domains (TKS5) [294]; and many MMPs such as MT1-MMP and MMP-2 [295]. It has been observed many types of cancer cells, including HNSCC, form invadopodia, which has been correlated to their invasive phenotype in vitro and in vivo [296,297,298,299,300]. Invadopodia facilitate the ECM degradation in a variety of cancers through the regulation of various MMPs, primarily MMP-14 (also known as MT1-MMP), MMP-2 and MMP-9 [301, 302]. MMPs commonly overexpressed in HNSCC include MMP-1, MMP-2, MMP-3, MMP-7, MMP-8, MMP-9, MMP-10, MMP-11, MMP-13, and MT1-MMP. The expression of secreted MMP-1, MMP-2, MMP-9 and transmembrane protease membrane type 1 MMP are commonly associated with HNSCC progression. MMP-2 and MMP-9 levels have been reported in correlation with local invasion, cervical nodal metastasis, tumor progression and prognosis of HNSCC patients. In addition, high levels of MMP-9 have been detected at the invasive tumor front (ITF), thus many studies describe MMP-9 as a potential marker of invasive OSCC [303,304,305]. MT1-MMP, which is involved in the regulation of MMP-2 activity, has been considered a crucial protease in HNSCC, since its expression is dysregulated in 75% to 100% of HNSCC tumors. The activity of MMPs is regulated by tissue inhibitors of metalloproteases (TIMPs) [306], secreted mainly by fibroblasts in the stroma. These molecules serve as inhibitors of the catalytic activity of MMPs, as well as activators of pro-MMPs, the latter represented by TIMP-2 required for activation of pro-MMP-2. Among the most commonly identified TIMPs in HNSCC have been TIMP-1 and TIMP-2. Upregulated levels of TIMP-1 expression have been associated with poor survival, while levels of TIMP-2 have been often reported to be unchanged between HNSCC tumors and adjacent tissue. Regarding the invasion and migration of cancer cells, invadopodia formation and secretion of MMPs, the overexpression of neural precursor cell expressed developmentally downregulated 9 (NEDD9) has been suggested as a biomarker of tumor agressiveness in many types of cancer, including oral cancer. Lucas et al. demonstrated that VEGF-stimulated HNSCC cell migration and invasion was NEDD9-dependent, while immunohistochemical analysis revealed that NEDD9 co-localized to invadopodia with MT1-MMP [307]. Their following studies investigated the role of NEDD9 in secretion of MMPs, MMP-9 and MMP-2 in particular, the formation of invadopodia, as well as the interactions of NEDD9 with vimentin and non-muscle myosin IIA [308, 309]. Consistent with their findings, the high-throughput gene expression profiling of HNSCC tumor samples has shown that overexpression of NEDD9 is associated with invasive HNSCC [310]. Recent studies examined the potential involvement of stromal cells on the invadopodia formation and EMT induction in HNSCC. A study conducted by Gao et al. demonstrated that HNSCC cells were able to recruit and educate monocytes into M2 macrophages in a co-culture system via the CCL2/CCR2 axis, and these M2 macrophages then enhanced the invadopodia formation, thus invasion and migration of HNSCC cells. This study also implicated macrophages to be crucial for the induction of EMT in HNSCC cells, since the majority of macrophages have been detected at the leading front of the scratch during the wound healing assay [311]. In a follow-up study Gao et al. implicated that upregulated levels of EGF and TGF-β secreted by TAMs in direct and indirect co-culture systems with HNSCC cells induce EMT of HNSCC cells via activation of the EGFR/ERK1/2 signaling pathway [312]. Another study investigated the role of M1 and M2 macrophages in EMT induction in a co-culture system with tongue carcinoma cells, in which they showed that the interaction between cancer cells and M2 macrophages induces migration and invasion in 3D model. Macrophages as well as cancer cells exhibited altered secretome, such as upregulated expression of TGF-β, EGF and M-CSF [313]. In contrast, a study by Smirnova et al. showed that although macrophages invade together with tumor cells in vivo, the invasion of HNSCC cells was not macrophage-dependent [314]. TAMs produce macrophage migration inhibitory factor (MIF), which has been associated with EMT in many types of cancer including HNSCC. Zheng et al. demonstrated that knock-down of MIF inhibited proliferation and migration of OSCC cells [315]. Another study showed that neutrophils can be recruited by HNSCC-derived MIF via a CXCR2 mechanism in vitro. In addition, MIF promoted invasive phenotype of HNSCC cells via neutrophil-secreted CCL4 and MMP9 [316]. Trellakis et al. observed that neutrophils from HNSCC patients displayed reduced apoptosis compared with healthy donors, which has been associated with upregulated secretion of HNSCC-derived MIF [317]. Furthermore, neutrophils have been linked to the invadopodia formation in HNSCC cancer cells. Glogauer et al. demonstrated that a co-culture system of neutrophils and OSCC cancer cells increased the invasiveness of OSCC, invadopodia formation and matrix degradation through increased secretion of TNF-α and IL-8 in a contact-independent manner [318]. Also, a study conducted by Dumitru et al. has shown that neutrophils promote migration of HNSCC by increasing cortactin phosphorylation in cancer cells in vitro [319]. The role of MDSCs in EMT induction of HNSCC has not yet been extensively studied. However, being a major source of MMP-9, EGF, bFGF and TGF-β, MDSCs have been heavily implicated with the EMT promotion and neoangiogenesis in several other types of cancer [320,321,322,323]. Furthermore, there is increasing evidence MDSCs may play a crucial role in establishing the pre-metastatic niche. The exact mechanism of the pre-metastatic niche formation has not yet been fully described, however, it has been suggested the microenvironment of the distant organ site can be altered by the primary tumor itself prior to tumor cell dissemination. Primary tumor cells promote the formation of supportive metastatic microenvironment via secretion of various cytokines and growth factors, such as VEGF, placental growth factor (PlGF), TGF-β and TNF-α, granulocyte-colony forming factor (G-CSF), versican and lysyl oxidase (LOX) into the circulation to mobilize and recruit other supporting cells that interact with stromal cells and ECM of the secondary site, thus establishing the microenvironment suitable for the formation of metastases [324]. Sceneay at al. suggested that tumor-derived monocyte chemoattractant protein-1 (MCP-1) regulates the accumulation of MDSC in the pre-metastatic niche. In addition, although also the number the NK cells in the pre-metastatic niche was increased, their cytotoxic effector function was compromised, which resulted in metastasis formation [325]. Another study conducted by Wang et al. demonstrated that VEGFA secreted by cancer cells stimulates TAMs to produce CXCL1, which results in the recruitment of MDSCs to form the pre-metastatic niche [326]. Shi et al. reported that mo-MDSCs accumulate in the lungs of tumor-bearing mice before the arrival of tumor cells and that these cells secrete IL-1β to stimulate expression of E-selectin, which results in metastasis formation [327]. The mechanism of pre-metastatic niche formation in HNSCC, however, has not yet been extensively investigated. It has been demonstrated that MDSCs, as well as neutrophils and macrophages, can be recruited to the tumor site via inflammatory protein calprotectin (S100A8/A9; MRP8/14) [328,329,330]. During inflammation, calprotectin is actively secreted by many types of cells in the microenvironment, such as neutrophils, macrophages, monocytes and MDSCs to modulate the inflammatory response by pro-inflammatory cytokine secretion, reactive oxygen species (ROS) and nitric oxide (NO) [331,332,333]. The role of calprotectin in EMT has not yet been fully elucidated; however, it has been implicated in the promotion of metastatic spread by MDSCs [334]. It has been reported, calprotectin activates the MAPK and NF-κB signaling in cancer cells, thus promoting metastasis [335,336,337] and is strongly upregulated in several types of cancer [338]. However, the levels of expression of calprotectin in primary HNSCC are downregulated compared with other types of cancer [339,340,341,342]. Silva et al. reported, that in HNSCC calprotectin contributes to the regulation of MMP-2 expression and secretion in the 3D cell culture, thus inhibiting invasion and migration of cancer cells [343].
Representing the most abundant cell type within the tumor microenvironment, the role of CAFs in the process of EMT in many types of cancer, including HNSCC, has been intensely researched. Many studies show that the presence of CAFs promotes cancer cell invasion [22, 344,345,346,347,348,349]. It has been reported CAFs enhance the invasion of cancer cells via various mechanisms, such as MMP-mediated ECM degradation and subsequent release of latent growth factors [22]; matrix stiffening through integrin-mediated mechanotransduction and through actomyosin contractility [150, 350]; secretion of soluble factors, including HGF and TGF-β [345, 351, 352]; secretion of exosomes [55]; and direct cell-cell contact [353]. The stimulating effect of CAFs on HNSCC invasion has been described by various in vitro assays [354,355,356]. The possible contribution of CAFs to the EMT induction in HNSCC carcinoma cells has been implicated by immunohistochemical analyses, in which markers associated with EMT in CAFs in paired primary and metastatic OSCC showed that Ki-67+ metastatic carcinoma cells downregulate E-cadherin when in direct contact with CAFs [357]. In addition, various in vitro studies demonstrated that EMT in HNSCC cells can be induced by CAF-derived molecules, such as SDF-1 via activation of the PI3K-Akt/PKB signaling pathway [358], TGF-β1 via the TGF-β/Smad signaling pathway [359], endothelin-1 [360] and CCL-7 [17]. Richter et al. demonstrated that TGFβ1/ EGF long-term co-stimulation enhances the invasive phenotype of OSCC, such as significantly upregulated expression of MMP-2 and MMP-9, compared with single growth factor stimulation [361]. A study conducted by Wu et al. examined the effect of Gal-1 on OSCC cell invasion and migration. It has been observed that blocking Gal-1 expression inhibits cancer cell migration and invasion induced by CAF-conditioned medium via MCP-1/CCR2 signaling pathway. Furthermore, in vivo study revealed that Gal-1 knockdown in CAFs efficiently inhibits metastasis in vivo [362]. Knowles et al. reported that HNSCC-derived CAFs contribute to the HNSCC invasion and metastasis via activation of the HGF/c-Met signaling axis in vitro [363]. Their following study showed the effects of CAFs on HNSCC metastasis in a mouse model. The co-injection of CAFs with HNSCC cells resulted in increased tumor growth, disease spread to the lymph nodes and lung metastases when compared to the injection of HNSCC cells alone [364]. Several studies also report that IL-1 secretion of OSCC cells stimulates TGF-β and HGF production by CAFs, which promotes invasion of cancer cells in vitro [365, 366]. In addition, Lewis et al. show that cancer cell-derived TGF-β1 directly induced the activated phenotype in CAF, which in turn stimulate the OSCC invasion via the HGF production [367].
Beside the stromal components of tumor environment, it is reasonable to assume that also hypoxia, a crucial hallmark of cancer, may play a major role in the formation of invadopodia, in the induction of EMT and in promotion of migration and invasion of cancer cells. It has been reported that expressions of EMT promoters, Snail, Slug, TWIST and SMAD nuclear interacting protein-1(SNIP1), which are regulated by HIF-1α, correlate with induction of EMT phenotype in OSCC cells in vitro [368,369,370]. A study by Huang et al. reported that SLUG regulated the expression of MT4-MMP under hypoxia, which promoted the invasiveness of HNSCC cell lines [371]. Yang et al. demonstrated that hypoxia-induced TWIST activated BMI1 expression and a knock down of TWIST reversed the EMT and invasive phenotype in HNSCC under hypoxia in vitro [372]. It has been suggested that hypoxia induces EMT in OSCC via activation of the Notch signaling pathway and the inhibition of the Notch signaling pathway suppresses EMT [373]. These results are consistent with a study by Diaz et al. showing that hypoxia potentiates the invadopodia formation and ECM degradation in HNSCC in a HIF-1α-dependent manner. Furthermore, their results also implicate that the invasive phenotype of cancer cells is regulated by cell contact-dependent hypoxia-mediated Notch signaling coupled with the paracrine activation of the EGFR, which is mediated by the ADAM12-dependent secretion of HB-EGF [374]. A recent study suggests that hypoxic conditions promote EMT, metastasis and glycolysis in HNSCC via positive feedback loop between metadherin (MTDH) and HIF-1α. The study showed that hypoxia increased the expression levels of genes associated with glycolysis, such as MCT1, MCT4, GLUT1 and LDHA in HNSCC cells and stimulated uptake of glucose, production of lactate and cell invasion in vitro [375]. Several studies suggest that targeting the pathways associated with altered tumor metabolism impairs EMT, migration and invasion of HNSCC. A recent study by Li et al. demonstrated that blockage of glycolysis via targeting PFKFB3 suppressed the migration and invasion of HNSCC cells by inhibiting the invadopodia formation of HNSCC cancer cells in vitro and in vivo [376]. A study by Xu et al. showed that blockage of glycolysis by 2-DG reversed EGF-induced EMT in OSCC in vitro and moreover, the treatment of 2-DG reduced the metastatic spread to regional lymph nodes in vivo [377]. A report by Wang et al. indicates that HNSCC cell invasion and glucose metabolism is regulated via the transcription factor tripartite motif containing 24 (TRIM24)-mediated GLUT3 induction [378]. Similar results were shown in a study by Chang et al. which provided evidence that the HNSCC cell migration and invasion are regulated by the activation of the GLUT4-TRIM24 axis [379].
Survival in the circulation
Normal epithelial cells require direct contact with the basement membrane via integrins in order to survive and proliferate. When normal cells lose contact with the surrounding ECM or other neighboring cells, these cells undergo programmed cell death, termed anoikis, to reduce the development of metastases. However, in the case of a metastatic cascade, to develop a resistance to anoikis is a crucial step for tumor cells to disseminate from the primary tumor, survive in the circulation in an adhesion-independent manner, travel to the secondary site, extravasate and form metastases. A study by Neiva et al. described that a crosstalk between tumor-associated endothelial cells and tumor cells protected the tumor cells from anoikis. Their results demonstrated that endothelial cell-secreted factors IL-6, IL-8 and EGF induced the activation of the STAT3/Akt/ERK signaling pathways in HNSCC cells in a contact-independent manner, which lead to increased tumor cell survival and migration [380]. Several studies examined the role of the pro-survival signaling pathway c-Met/Akt in anoikis in HNSCC. It has been reported that CAF-derived HGF activated the c-Met/Akt pathway in HNSCC cells in vitro [363]. The effect of HGF on anchorage-independent tumor cell survival has been investigated in a study by Zheng et al., which showed that HGF-induced anoikis resistance was dependent on ERK and Akt pathways and the blockage of either pathway resulted in apoptosis of tumor cells. Furthermore, it has been reported the HGF-induced anoikis was independent of NFκB [381]. Their following study revealed that COX-2 provided resistance to HGF-induced anoikis in HNSCC via the activation of activator protein-1 (AP-1) through the ERK signaling pathway [382]. The neurotrophic tyrosine kinase receptor B (TrkB), which is frequently overexpressed in many cancer types including HNSCC, has been suggested as one of the major inducers of anoikis resistance [383,384,385]. A study by Jiffar et al. revealed that CAFs contribute to the invasive OSCC phenotype via brain-derived neurotrophic factor (BDNF)-mediated TrkB signaling axis cascade, which has been then further supported in vivo [386]. Also, the ECM proteins including collagen, fibronectin and laminin, which are major regulators of tumor cell differentiation, invasion, migration and survival, have been implicated in promoting the anoikis resistance [387]. Among matrix proteins collagen type I is the most effective in delaying anoikis in cancer cells [388]. A study by Koontongkaew et al. showed that metastatic cells plated on collagen I gel significantly upregulated their cytokine secretion, which activated MMP-2 and MMP-9 and enhanced HNSCC cell invasion [389]. Fibronectin has been also implicated in playing a role in anoikis resistance in HNSCC. Zhang et al. found that OSCC cells escape p53-induced anoikis by forming multicellular aggregates followed by integrin αv-mediated upregulation of fibronectin [390]. Their following study demonstrated that alternatively spliced V region and function-perturbing point mutations in the high-affinity heparin-binding domain of fibronectin induce anoikis in OSCC via integrin αv-mediated phosphorylation of FAK and ERK [391].
After entering the circulation, tumor cells exploit many mechanisms of immunoevasion. Numerous studies show that cancer cells acquire the ability to aggregate platelets in order to survive in the circulation, a process known as tumor cell-induced platelet aggregation (TCIPA). The formation of this “platelet cloak” provides many advantages to tumor cells, such as a shield which enables tumor cells to evade the immune systems, since platelets protect tumor cells from TNF-α [392] and NK-mediated cytotoxicity [393]; an increased extravasation of the tumor cells by the adhesion to the vascular endothelium [394]; a protection from high shear forces in the bloodstream; and a secretion of various growth factors for tumor cells to utilize [395]. Reportedly, the “platelet cloak” can also transfer platelet-derived normal MHC class I onto the tumor cell surface to help escape the T cell-mediated immunity [396]. The mechanism by which tumor cells activate platelets to form TCIPA includes the stimulation of the release of various molecules, such as ADP, MMP-2 and PGE2, and generation of thromboxane A2 (TXA2). This process is often stimulated by tumor cell-derived proteases, such as thrombin, cathepsin B, cancer procoagulant (EC 3.4.22.26), MMP-2 and MMP-14 [397, 398]. The interaction leads to the activation of major platelet adhesion molecules, such as integrin receptors GPIb-IX-V and GPIIb/IIIa, P-selectin and Toll-like receptor 4 (TLR4) [351, 399, 400]. Huang et al. demonstrated increased platelet aggregation in HNSCC patients, which was correlated with the tumor stage [401]. Although many studies investigated the role of the tumor cell-induced platelet aggregation in various types of cancer, the contribution of platelet aggregation to the process of immunoevasion in HNSCC has not yet been studied. Another suggested mechanism by which cancer cells survive in the circulation and evade the immune system is by upregulation of indoleamine 2,3-dioxygenase (IDO), a tryptophan-catabolising enzyme. Studies report the upregulation of IDO correlates with metastasis and worse prognosis in various types of cancers including OSCC [402]. The increased expression has been correlated with decreased numbers of CD3+ infiltrating T cells and with an upregulation of Tregs [403, 404]. Moreover, various studies report that iNOS production by peripheral blood neutrophils is significantly reduced in OSCC patients and depends on tumor stage [405, 406].
Extravasation
After the successful arrival at the secondary metastatic site, it is crucial for tumor cells to escape the hostile intravascular environment and extravasate into the tissue. The predominant mechanism of extravasation involves the arrest of tumor cells on the endothelium, which is followed by transendothelial migration (TEM) and invasion into the surrounding tissue. This process is characterized by alterations in endothelial cell-cell junctions. In vitro studies show, that the attachment of tumor cells onto the luminal side of the endothelial cell [407] is enabled by various adhesion ligands and receptors, such as selectins, intergrins, cadherins, immunoglobulins and CD44 [408]. However, the exact mechanism in vivo has yet to be elucidated. Using transgenic zebrafish that uniformly express GFP throughout their vasculature, Stoletov et al. confirmed the extravasation cascade and further demonstrated, that this process is mediated by Twist, VEGFA and integrin β (ITGB1) expression [409].
Besides the interaction between tumor cells and endothelial cells (ECs), the tumor-promoting immune cells also assist in successful extravasation. Suggested mechanisms, by which platelets promote extravasation and transendothelial migration, include induction of the EMT and invasiveness in tumor cells via TGF-β-mediated activation of Smad and NF-κB signaling pathways [410], as well as modulation of endothelial junctions and cytoskeleton via ATP secreted by platelets after tumor cell activation, which interacts with endothelial P2Y2 receptor in order to open the EC junctions [411]. Weber et al. demonstrated that platelets promote endothelial permeability and extravasation of tumor cells when activated by integrin αvβ3 expressed on tumor cells in vivo [412]. Furthermore, platelets contribute to the extravasation of tumor cells by recruitment of granulocytes via production of CXCL5 and CXCL7 [413]. Upon activation, neutrophils form NETs, which have been shown to promote extravasation by tumor cells sequestration [414] and MMP-9-mediated degradation of ECM [415]. Monocytes/macrophages can be recruited to the metastatic site by tumor cell-derived molecules. Reportedly, after the arrival into the lung (pulmonary metastases account for 66% of distant metastases in HNSCC [416]) tumor–platelet aggregates attached to ECs express tissue factor (TF) to stimulate the expression of VCAM-1 and VAP-1. These inflammatory mediators trigger the recruitment of macrophages, which then promote tumor cell survival and increase the vascular permeability, possibly by transmitting the pro-survival signals via VCAM-1 expressed on tumor cell surface [417,418,419]. Moreover, VEGF produced by macrophages and tumor cells has been shown to induce vascular permeability and transendothelial migration [420, 421].
Since HNSCC metastasizes primarily via the lymphatic invasion, Fennewald et al. investigated the interaction of HNSCC cancer cell and ECM components of lymph node parenchyma, such as laminin, fibronectin, vitronectin and hyaluronic acid in low fluid shear conditions. Their results show that HNSCC cell lines bound to laminin via α2β1, α3β1, and α6β1 integrins in a presence of lymphodynamic low shear stress, which resulted in activation of calcium signaling [422]. A study by Yen et al. demonstrated that integrin β1 promotes migration and transendothelial migration of OSCC cells via insulin-like growth factor (IGF)-independent insulin-like growth binding protein 3 (IGFBP3) [423]. The mechanisms of extravasation of tumor cells have been well described in many types of cancer; however, the effect of microenvironmental factors on extravasation in HNSCC has yet to be investigated.
Mesenchymal-epithelial transition
Mesenchymal-epithelial transition (MET), also known as mesenchymal-to-epithelial reverting transtition (MErT), describes a process, by which cancer cells revert back from the EMT-induced mesenchymal phenotype. The disseminated cancer cells undergo this process in order to adapt to the microenvironment of the secondary metastatic site to allow the colonization, as metastases recapitulate the primary tumor pathology. Although the precise mechanism of MET has not yet been elucidated, several studies highlight the importance of E-cadherin re-expression in the metastatic tissue. Several studies studied the molecular mechanisms of MET in HNSCC [424,425,426,427], however, the role of the cells within the surrounding microenvironment of the secondary metastatic site in HNSCC has not yet been investigated.
Conclusions
The emerging evidence of crucial contribution of different stromal components to the regulation of the HNSCC development implicates a fundamental role of the tumor microenvironment in providing a supportive niche, thus substantially promoting HNSCC development and metastasis. While the research has previously focused mainly on altered expression of genes and aberrant genetic and epigenetic mutations in tumor cells, it is becoming clear that investigation of differences in stromal composition of the HNSCC tumor microenvironment and their impact on cancer development and progression may help better understand the mechanisms behind different responses to therapy, thus help define possible targets for clinical intervention.
Abbreviations
- 4-NQO:
-
4-nitroquinoline 1-oxide
- 5-HT:
-
5-hydroxytryptamine
- ADAM:
-
A disintegrin and metalloproteinase
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- AP:
-
Activator protein
- BDMC:
-
Bone marrow-derived mesenchymal cell
- BDNF:
-
Brain-derived neurotrophic factor
- CAF:
-
Cancer-associated fibroblasts
- CCL:
-
Chemokine (C-C motif) ligand;
- COX:
-
cyclooxygenase
- CPA:
-
Carboxypeptidase A
- CTLA:
-
Cytotoxic T-lymphocyte-associated protein
- CXCL:
-
Chemokine (C-X-C motif) ligand
- CXCR:
-
Chemokine (C-X-C motif) receptor
- DC:
-
Dendritic cell
- EC:
-
Endothelial cell
- ECM:
-
Extracellular matrix
- EdMT:
-
Endothelial-mesenchymal transition
- EFEMP1:
-
EGF containing fibulin-like extracellular matrix protein
- EGF:
-
Epithelial growth factor
- EMT:
-
Epithelial-mesenchymal transition
- FAK:
-
Focal adhesion kinase
- FAP:
-
Fibroblast-activation protein
- FGF:
-
Fibroblast growth factor
- FNDC:
-
Fibronectin type III domain-containing protein
- FOXP3:
-
Forkhead box P3
- FSP:
-
Fibroblast-specific protein
- Gal:
-
Galectin
- GITR:
-
Glucocorticoid-induced tumor necrosis factor receptor family-related protein
- GLUT:
-
Glucose transporter
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- GP:
-
Glycoprotein
- GSK:
-
Glycogen synthase kinase
- HGF:
-
Hepatocyte growth factor
- HIF:
-
Hypoxia inducible factor
- HLA-DR:
-
Human leukocyte antigen DR isotype
- HNC:
-
Head and neck cancer
- HNSCC:
-
Head and neck squamous cell carcinoma
- HPV:
-
Human papillomavirus
- IBP:
-
Insulin-like binding protein
- ICAM:
-
Intercellular adhesion molecule
- IDO:
-
Indoleamine 2,3-dioxygenase
- IFN:
-
Interferon
- IL:
-
Interleukin
- iNKT:
-
Invariant neutral killer T cell
- iNOS:
-
Inducible nitric oxid synthase
- ITF:
-
Invasive tumor front
- ITGB:
-
Integrin beta
- LAMP:
-
Lysosomal-associated membrane protein
- LDH:
-
Lactate dehydrogease
- LOX:
-
Lysyl oxidase
- LPS:
-
Lipopolysacharide
- LT:
-
Leukotrien
- MAPK:
-
Mitogen-activated protein kinase
- MC:
-
Mast cell
- MCD:
-
Mast cell density
- MCP:
-
Monocyte chemoattractant protein
- MCT:
-
Monocarboxylate transporter
- MDSC:
-
Myeloid-derived suppressor cell
- MErT:
-
Mesenchymal-epithelial reverting transition
- MET:
-
Mesenchymal-epithelial transition
- MHC:
-
Major histocompatibility complex
- MIF:
-
Migration inhibitory factor
- MMP:
-
Matrix metalloproteinase
- MMT:
-
Mesothelial-mesenchymal transition
- MTDH:
-
Metadherin
- MVD:
-
Microvessel density
- NEDD9:
-
Neural precursor cell expressed developmentally downregulated 9
- NET:
-
Neutrophil extracellular trap
- NF-κB:
-
Nuclear factor κB
- NK:
-
Neutral killer cell
- NO:
-
Nitric oxid
- OSCC:
-
Oral squamous cell carcinoma
- OXPHOS:
-
Oxidative phosphorylation
- PBMC:
-
Peripheral blood mononuclear cell
- PDGFR:
-
Platelet-derived growth factor receptor
- PD-L1:
-
Programmed death receptor ligand 1
- PFKFB3:
-
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3
- PG:
-
Prostaglandin
- PI3K:
-
Phosphatidylinositol 3-kinase
- PlGF:
-
Placental growth factor
- PMN:
-
Polymorphonuclear leukocyte
- PPAR:
-
Peroxisome proliferator-activated receptor
- PSGL:
-
P-selectin glycoprotein ligand
- PTEN:
-
Phosphatase and tensin homolog
- RANTES:
-
Regulated on activation, normal T cell expressed and secreted
- ROS:
-
Reactive oxygen species
- SCF:
-
Stem-cell factor
- SDF:
-
Stromal cell-derived factor
- SERPINE:
-
Serpin peptidase inhibitor
- SMA:
-
Smooth muscle actin
- STAT:
-
Signal transducer and activator of transcription
- STC:
-
Stanniocalcin
- TAM:
-
Tumor-associated macrophage
- TAMC:
-
Tumor-associated mast cell
- TAN:
-
Tumor-associated neutrophil
- TCIPA:
-
tumor cell-induced platelet aggregation
- TCR:
-
T cell receptor
- TEM:
-
Transendothelial migration
- TF:
-
Tissue factor
- TGF:
-
Transforming growth factor
- Th:
-
Helper T cell
- TIL:
-
Tumor infiltrating leukocyte
- TIMP:
-
Tissue inhibitor of metalloproteases
- TKS:
-
Tyr kinase substrate
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- TRAIL:
-
TNF-Related apoptosis-inducing ligand
- Treg:
-
Regulatory T cell
- TRIM24:
-
Tripartite motif containing 24
- TrkB:
-
Tropomyosin receptor kinase B
- TXA2:
-
Thromboxane A2
- VAP:
-
Vascular adhesion protein
- VCAM:
-
Vascular cell adhesion protein
- VEGF:
-
Vascular endothelial growth factor
- VTE:
-
Venous tromboembolism
- VWF:
-
Von Willebrand factor
- YAP:
-
Yes-associated protein
References
Vermorken JB, et al. Overview of the efficacy of cetuximab in recurrent and/or metastatic squamous cell carcinoma of the head and neck in patients who previously failed platinum-based therapies. Cancer. 2008;112(12):2710–9.
Filho MRM, et al. Quality of life of patients with head and neck cancer. Brazilian Journal of Otorhinolaryngology. 2013;79(1):82–8.
Skvortsov S, et al. Rac1 as a potential therapeutic target for chemo-radioresistant head and neck squamous cell carcinomas (HNSCC). Br J Cancer. 2014;110(11):2677–87.
Stewart BW, Wild CP. World Cancer Report 2014; 2014.
Parkin DM, et al. Global Cancer Statistics, 2002. A Cancer Journal for Clinicians. 2005;55(2):74–108.
Chuang SC, et al. Risk of second primary cancer among patients with head and neck cancers: A pooled analysis of 13 cancer registries. Int J Cancer. 2008;123(10):2390–6.
Brockstein B, et al. Patterns of failure, prognostic factors and survival in locoregionally advanced head and neck cancer treated with concomitant chemoradiotherapy: a 9-year, 337-patient, multi-institutional experience. Ann Oncol. 2004;15(8):1179–86.
Castellsague X, et al. The role of type of tobacco and type of alcoholic beverage in oral carcinogenesis. Int J Cancer. 2004;108(5):741–9.
Carlander AF, et al. Continuing rise in oropharyngeal cancer in a high HPV prevalence area: A Danish population-based study from 2011 to 2014. Eur J Cancer. 2017;70:75–82.
Mork J, et al. Human papillomavirus infection as a risk factor for squamous-cell carcinoma of the head and neck. The New England Journal of Medicine. 2001;344(15):1125–31.
D’Souza G, et al. Case–Control Study of Human Papillomavirus and Oropharyngeal Cancer. The new engl and journal of medicine. 2007;356(19):1944–56.
Jia CC, et al. Cancer-associated fibroblasts from hepatocellular carcinoma promote malignant cell proliferation by HGF secretion. PLoS One. 2013;8(5):e63243.
Luker KE, et al. Scavenging of CXCL12 by CXCR7 promotes tumor growth and metastasis of CXCR4-positive breast cancer cells. Oncogene. 2012:1476–5594 (Electronic)).
Augsten M, et al. Cancer-associated fibroblasts expressing CXCL14 rely upon NOS1-derived nitric oxide signaling for their tumor-supporting properties. Cancer Res. 2014:1538–7445 (Electronic)).
Bello IO, et al. Cancer-associated fibroblasts, a parameter of the tumor microenvironment, overcomes carcinoma-associated parameters in the prognosis of patients with mobile tongue cancer. Oral Oncol. 2011;47:1879–0593 (Electronic)).
Wang X, et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition through secreted cytokines in endometrial cancer cells. Oncology Letters. 2018;15(4):5694–702.
Jung D-W, et al. Tumor-stromal crosstalk in invasion of oral squamous cell carcinoma: a pivotal role of CCL7. International Journal of Cancer. 2009;127(2):332–44.
Lotti F, et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. The Journal of Experimental Medicine. 2013;210(13):2851–72.
Calon A, Tauriello DV, Batlle E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin Cancer Biol. 2014:1096–3650 (Electronic)).
Glentis A, et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nature Communications. 2017;8(1):924.
Hawinkels LJ, et al. Interaction with colon cancer cells hyperactivates TGF-beta signaling in cancer-associated fibroblasts. Oncogene. 2014:1476–5594 (Electronic)).
Gaggioli C, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007:1465–7392 (Print)).
De Wever O, et al. Stromal myofibroblasts are drivers of invasive cancer growth. Int J Cancer. 2008;123(10):2229–38.
Council, L., Hameed O. Differential expression of immunohistochemical markers in bladder smooth muscle and myofibroblasts, and the potential utility of desmin, smoothelin, and vimentin in staging of bladder carcinoma. Mod Pathol. 2009;22(5):639–50.
Ohlund D, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214(3):579–96.
Kim HM, Jung WH, Koo JS. Expression of cancer-associated fibroblast related proteins in metastatic breast cancer: an immunohistochemical analysis. J Transl Med. 2015;13:222.
Sandoval P, et al. Carcinoma-associated fibroblasts derive from mesothelial cells via mesothelial-to-mesenchymal transition in peritoneal metastasis. J Pathol. 2013;231(4):517–31.
Zeisberg EM, et al. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67(21):10123–8.
Iwano M, et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. Journal of Clinical Investigation. 2002;110(3):341–50.
Jotzu C, et al. Adipose tissue-derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor-derived factors. Anal Cell Pathol (Amst). 2010;33(2):61–79.
Quante M, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19(2):257–72.
Skalli O, et al. A Monoclonal Antibody against a-Smooth Muscle Actin: A New Probe for Smooth Muscle Differentiation. The Journal of Cell Biology. 1986;103(6):2787–96.
Lim KP, et al. Fibroblast gene expression profile reflects the stage of tumour progression in oral squamous cell carcinoma. The Journal of Pathology. 2010;223(4):459–69.
Park JE, et al. Fibroblast Activation Protein, a Dual Specificity Serine Protease Expressed in Reactive Human Tumor Stromal Fibroblasts. THE JOURNAL OF BIOLOGICAL CHEMISTRY. 1999;274(51):36505–12.
Wonganu B, Berger BW. A specific, transmembrane interface regulates fibroblast activation protein (FAP) homodimerization, trafficking and exopeptidase activity. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2016;1858(8):1876–82.
Sukowati CHC, et al. The role of multipotent cancer associated fibroblasts in hepatocarcinogenesis. BMC Cancer. 2015;15:188.
Zhou B, et al. A role for cancer-associated fibroblasts in inducing the epithelial-to-mesenchymal transition in human tongue squamous cell carcinoma. Journal of Oral Pathology & Medicine. 2014;43(8):585–92.
Netea MG, Mantovani A. In: Biswas SK, Mantovani A, editors. Adaptive Characteristics of Innate Immune Responses in Macrophages, in Macrophages: Biology and Role in the Pathology of Diseases. New York: Springer New York; 2014. p. 339–48.
Stout RD, et al. Macrophages Sequentially Change Their Functional Phenotype in Response to Changes in Microenvironmental Influences. The Journal of Immunology. 2005;175(1):342.
Murray PJ, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20.
Melton DW, et al. Temporal Phenotypic Features Distinguish Polarized Macrophages In Vitro. Autoimmunity. 2015;48(3):161–76.
Stein M, et al. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. The Journal of Experimental Medicine. 1992;176(1):287.
Verreck FAW, et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(13):4560–5.
Duluc D, et al. Interferon-γ reverses the immunosuppressive and protumoral properties and prevents the generation of human tumor-associated macrophages. International Journal of Cancer. 2009;125(2):367–73.
Zhang S, et al. Delineation of Diverse Macrophage Activation Programs in Response to Intracellular Parasites and Cytokines. PLOS Neglected Tropical Diseases. 2010;4(3):e648.
Lacavé-Lapalun J-V, Benderitter M, Linard C. Flagellin or Lipopolysaccharide Treatment Modified Macrophage Populations after Colorectal Radiation of Rats. Journal of Pharmacology and Experimental Therapeutics. 2013;346(1):75.
Weber M, et al. Small oral squamous cell carcinomas with nodal lymphogenic metastasis show increased infiltration of M2 polarized macrophages – An immunohistochemical analysis. Journal of Cranio-Maxillofacial Surgery. 2014;42(7):1087–94.
Kurahara H, et al. Significance of M2-Polarized Tumor-Associated Macrophage in Pancreatic Cancer. Journal of Surgical Research. 2011;167(2):e211–9.
Mantovani A, et al. Macrophage plasticity and polarization in tissue repair and remodelling. The Journal of Pathology. 2013;229(2):176–85.
Chunyan L, et al. Expression of M2-Polarized Macrophages is Associated with Poor Prognosis for Advanced Epithelial Ovarian Cancer. Technology in Cancer Research & Treatment. 2013;12(3):259–67.
Makita N, et al. IL-10 enhances the phenotype of M2 macrophages induced by IL-4 and confers the ability to increase eosinophil migration. International Immunology. 2015;27(3):131–41.
Müller U, et al. IL-13 Induces Disease-Promoting Type 2 Cytokines, Alternatively Activated Macrophages and Allergic Inflammation during Pulmonary Infection of Mice with Cryptococcus neoformans. The Journal of Immunology. 2007;179(8):5367.
Awad F, et al. Impact of human monocyte and macrophage polarization on NLR expression and NLRP3 inflammasome activation. PLOS ONE. 2017;12(4):e0175336.
Hu Y, et al. Tumor-associated macrophages correlate with the clinicopathological features and poor outcomes via inducing epithelial to mesenchymal transition in oral squamous cell carcinoma. J Exp Clin Cancer Res. 2016:1756–9966 (Electronic)).
Li W, et al. TGFβ1 in fibroblasts-derived exosomes promotes epithelial-mesenchymal transition of ovarian cancer cells. Oncotarget. 2017;8(56):96035–47.
Seminerio I, et al. High infiltration of CD68+ macrophages is associated with poor prognoses of head and neck squamous cell carcinoma patients and is influenced by human papillomavirus. Oncotarget. 2018;9(13):11046–59.
Liu C-Y, et al. M2-polarized tumor-associated macrophages promoted epithelial–mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Laboratory Investigation. 2013;93:844.
Rodriguez PC, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. The Journal of Experimental Medicine. 2005;202(7):931.
Van Ginderachter JA, et al. Peroxisome proliferator-activated receptor γ (PPARγ) ligands reverse CTL suppression by alternatively activated (M2) macrophages in cancer. Blood. 2006;108(2):525.
Kratochvill F, et al. TNF Counterbalances the Emergence of M2 Tumor Macrophages. Cell Reports. 2015;12(11):1902–14.
Zhou X, Qi Y. Larynx carcinoma regulates tumor-associated macrophages through PLGF signaling. Scientific Reports. 2015;5:10071.
Biswas SK, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-κB and enhanced IRF-3/STAT1 activation). Blood. 2006;107(5):2112.
Welch DR, et al. Tumor-elicited polymorphonuclear cells, in contrast to "normal" circulating polymorphonuclear cells, stimulate invasive and metastatic potentials of rat mammary adenocarcinoma cells. Proceedings of the National Academy of Sciences. 1989;86(15):5859–63.
Uhl B, et al. Aged neutrophils contribute to the first line of defense in the acute inflammatory response. Blood. 2016;128(19):2327–37.
Tangye SG, Brink R. A helping hand from neutrophils in T cell–independent antibody responses? Nature Immunology. 2012;13:111.
Sandilands GP, et al. Cross-linking of neutrophil CD11b results in rapid cell surface expression of molecules required for antigen presentation and T-cell activation. Immunology. 2005;114(3):354–68.
Beauvillain C, et al. Neutrophils efficiently cross-prime naive T cells in vivo. Blood. 2007;110(8):2965.
Brinkmann V, et al. Neutrophil Extracellular Traps Kill Bacteria. Science. 2004;303(5663):1532.
Fuchs TA, et al. Novel cell death program leads to neutrophil extracellular traps. The Journal of Cell Biology. 2007;176(2):231.
Yipp BG, et al. Dynamic NETosis is Carried Out by Live Neutrophils in Human and Mouse Bacterial Abscesses and During Severe Gram-Positive Infection. Nature medicine. 2012;18(9):1386–93.
Fuchs TA, et al. Extracellular DNA traps promote thrombosis. Proceedings of the National Academy of Sciences. 2010;107(36):15880.
Demers M, et al. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proceedings of the National Academy of Sciences. 2012;109(32):13076.
Paneesha S, et al. Frequency, demographics and risk (according to tumour type or site) of cancer-associated thrombosis among patients seen at outpatient DVT clinics. Thrombosis and Haemostasis. 2010;103(2):338–43.
Pillay J, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. The Journal of Clinical Investigation. 2012;122(1):327–36.
Elghetany MT, et al. Flow cytometric study of neutrophilic granulopoiesis in normal bone marrow using an expanded panel of antibodies: Correlation with morphologic assessments. Journal of Clinical Laboratory Analysis. 2004;18(1):36–41.
Lakschevitz FS, et al. Identification of neutrophil surface marker changes in health and inflammation using high-throughput screening flow cytometry. Experimental Cell Research. 2016;342(2):200–9.
Fridlender ZG, et al. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell. 2009;16(3):183–94.
Jablonska J, et al. Neutrophils responsive to endogenous IFN-β regulate tumor angiogenesis and growth in a mouse tumor model. The Journal of Clinical Investigation. 2010;120(4):1151–64.
Andzinski L, et al. Type IIFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. International Journal of Cancer. 2016;138(8):1982–93.
Youn JI, et al. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008:1550–6606 Electronic.
Lechner MG, Liebertz DJ, Epstein AL. CHARACTERIZATION OF CYTOKINE-INDUCED MYELOID-DERIVED SUPPRESSOR CELLS FROM NORMAL HUMAN PERIPHERAL BLOOD MONONUCLEAR CELLS. Journal of immunology. 2010;185(4):2273–84.
Srivastava MK, et al. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010:1538–7445 Electronic.
Marigo I, et al. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008:1600–065X Electronic.
Cimen Bozkus C, et al. Expression of Cationic Amino Acid Transporter 2 Is Required for Myeloid-Derived Suppressor Cell-Mediated Control of T Cell Immunity. J Immunol. 2015:1550–6606 Electronic.
Raber PL, et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer. 2014:1097–0215 Electronic.
Corzo CA, et al. Mechanism regulating reactive oxygen species in tumor induced myeloid-derived suppressor cells: MDSC and ROS in cancer. Journal of immunology. 2009;182(9):5693–701.
Zhang H, et al. Myeloid-derived suppressor cells inhibit T cell proliferation in human extranodal NK/T cell lymphoma: a novel prognostic indicator. Cancer Immunology, Immunotherapy. 2015;64(12):1587–99.
Noman MZ, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. The Journal of Experimental Medicine. 2014;211(5):781.
Huang B, et al. Gr-1+CD115+ Immature Myeloid Suppressor Cells Mediate the Development of Tumor-Induced T Regulatory Cells and T-Cell Anergy in Tumor-Bearing Host. Cancer Research. 2006;66(2):1123.
Hoechst B, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009:1527–3350 Electronic.
Pak AS, et al. Mechanisms of immune suppression in patients with head and neck cancer: presence of CD34(+) cells which suppress immune functions within cancers that secrete granulocyte-macrophage colony-stimulating factor. Clinical Cancer Research. 1995;1(1):95.
Garrity T, et al. Increased presence of CD34+ cells in the peripheral blood of head and neck cancer patients and their differentiation into dendritic cells. International Journal of Cancer. 1998;73(5):663–9.
Young MRI, et al. Increased recurrence and metastasis in patients whose primary head and neck squamous cell carcinomas secreted granulocyte-macrophage colony-stimulating factor and contained CD34+ natural suppressor cells. International Journal of Cancer. 1998;74(1):69–74.
Du R, et al. HIF1α Induces the Recruitment of Bone Marrow-Derived Vascular Modulatory Cells to Regulate Tumor Angiogenesis and Invasion. Cancer cell. 2008;13(3):206–20.
Grunewald M, et al. VEGF-Induced Adult Neovascularization: Recruitment, Retention, and Role of Accessory Cells. Cell. 2006;124(1):175–89.
Lahl K, et al. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med. 2007:0022–1007 Print.
Sakaguchi S, et al. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010:1474–741 Electronic.
McHugh RS, et al. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002:1074–7613 Print.
Tang AL, et al. CTLA4 expression is an indicator and regulator of steady-state CD4+ FoxP3+ T cell homeostasis. J Immunol. 2008:1550–6606 Electronic.
Mason GM, et al. Phenotypic Complexity of the Human Regulatory T Cell Compartment Revealed by Mass Cytometry. J Immunol. 2015:1550–6606 Electronic.
Zheng SG, et al. IL-2 is essential for TGF-beta to convert naive CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol. 2007:0022–1767 Print.
Vignali DAA, Collison LW, Workman CJ. How regulatory T cells work. Nature reviews. Immunology. 2008;8(7):523–32.
Shah BH, et al. Molecular mechanisms involved in human platelet aggregation by synergistic interaction of platelet-activating factor and 5-hydroxytryptamine. Experimental &Amp. Molecular Medicine. 2001;33:226.
Holmsen H, Weiss HJ. Secretable Storage Pools in Platelets. Annual Review of Medicine. 1979;30(1):119–34.
Youssefian T, et al. Platelet and Megakaryocyte Dense Granules Contain Glycoproteins Ib and IIb-IIIa. Blood. 1997;89(11):4047.
Ruiz FA, et al. Human Platelet Dense Granules Contain Polyphosphate and Are Similar to Acidocalcisomes of Bacteria and Unicellular Eukaryotes. Journal of Biological Chemistry. 2004;279(43):44250–7.
Israels SJ, et al. Platelet dense granule membranes contain both granulophysin and P- selectin (GMP-140). Blood. 1992;80(1):143.
Metzelaar MJ, et al. CD63 antigen. A novel lysosomal membrane glycoprotein, cloned by a screening procedure for intracellular antigens in eukaryotic cells. Journal of Biological Chemistry. 1991;266(5):3239–45.
Sander HJ, et al. Immunocytochemical localization of fibrinogen, platelet factor 4, and beta thromboglobulin in thin frozen sections of human blood platelets. The Journal of Clinical Investigation. 1983;72(4):1277–87.
Wencel-Drake JD, et al. Ultrastructural localization of human platelet thrombospondin, fibrinogen, fibronectin, and von Willebrand factor in frozen thin section. Blood. 1985;65(4):929.
Gleissner CA, von Hundelshausen P, Ley K. Platelet chemokines in vascular disease. Arteriosclerosis, thrombosis, and vascular biology. 2008;28(11):1920–7.
King SM, Reed GL. Development of platelet secretory granules. Seminars in Cell & Developmental Biology. 2002;13(4):293–302.
Santos-Martínez MJ, et al. Role of metalloproteinases in platelet function. Thrombosis Research. 2008;121(4):535–42.
Maynard DM, et al. Proteomic analysis of platelet α-granules using mass spectrometry. Journal of Thrombosis and Haemostasis. 2007;5(9):1945–55.
Trikha M, et al. Multiple Roles for Platelet GPIIb/IIIa and αvβ3 Integrins in Tumor Growth, Angiogenesis, and Metastasis. Cancer Research. 2002;62(10):2824.
Suzuki H, et al. Intracellular localization of glycoprotein VI in human platelets and its surface expression upon activation. British Journal of Haematology. 2003;121(6):904–12.
Berger G, et al. Ultrastructural demonstration of CD36 in the alpha-granule membrane of human platelets and megakaryocytes. Blood. 1993;82(10):3034.
Larsen E, et al. PADGEM protein: A receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell. 1989;59(2):305–12.
Farrell AM, et al. Signaling pathways activated in a unique mast cell line where interleukin-3 supports survival and stem cell factor is required for a proliferative response. Blood. 1996;87(9):3655.
Prussin C, Metcalfe DD. 4. IgE, mast cells, basophils, and eosinophils. Journal of Allergy and Clinical Immunology. 2003;111(2, Supplement 2):S486–94.
Hugle T. Beyond allergy: the role of mast cells in fibrosis. Swiss Med Wkly. 2014:1424–3997 Electronic.
Dvorak M. A. Basophil and mast cell degranulation and recovery, vol. 4; 1991.
Norrby, KJ. Jakobsson A Fau-Sorbo, and J. Sorbo, Mast-cell secretion and angiogenesis, a quantitative study in rats and mice. Virchows Arch B Cell Pathol Incl Mol Pathol., 1989(0340-6075 (Print)).
Baram D, et al. Human Mast Cells Release Metalloproteinase-9 on Contact with Activated T Cells: Juxtacrine Regulation by TNF-α. The Journal of Immunology. 2001;167(7):4008.
Stoyanov E, et al. Mast cells and histamine enhance the proliferation of non-small cell lung cancer cells. Lung Cancer. 2012;75(1):38–44.
Saleem SJ, et al. Cutting edge: mast cells critically augment myeloid-derived suppressor cell activity. J Immunol. 2012:1550–6606 Electronic).
Yang Z, et al. Mast Cells Mobilize Myeloid-Derived Suppressor Cells and Treg Cells in Tumor Microenvironment via IL-17 Pathway in Murine Hepatocarcinoma Model. PLOS ONE. 2010;5(1):e8922.
Tanooka H, et al. Evidence for Involvement of Mast Cells in Tumor Suppression in Mice23. JNCI: Journal of the National Cancer Institute. 1982;69(6):1305–9.
Gooch JL, Lee AV, Yee D. Interleukin 4 Inhibits Growth and Induces Apoptosis in Human Breast Cancer Cells. Cancer Research. 1998;58(18):4199.
Lätti S, et al. Mast cell-mediated apoptosis of endothelial cells in vitro: A paracrine mechanism involving TNF-α-mediated down-regulation of bcl-2 expression. Journal of Cellular Physiology. 2003;195(1):130–8.
Lanier LL, et al. The relationship of CD16 (Leu-11) and Leu-19 (NKH-1) antigen expression on human peripheral blood NK cells and cytotoxic T lymphocytes. The Journal of Immunology. 1986;136(12):4480.
Cuturi MC, Sherman AIF, Loudon R, Clark SC, Perussia B, Trinchieri G. Production of hematopoietic colony-stimulating factors by human natural killer cells. The Journal of Experimental Medicine. 1989;169(2):569–83.
Smyth MJ, et al. IL-8 gene expression and production in human peripheral blood lymphocyte subsets. J Immunol. 1991:0022–1767 Print.
Warren HS, et al. Production of IL-5 by human NK cells and regulation of IL-5 secretion by IL-4, IL-10, and IL-12. J Immunol. 1995:0022–1767 Print.
Fauriat C, et al. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood. 2010;115(11):2167–76.
Orr MT, Lanier LL. Natural Killer Cell Education and Tolerance. Cell. 2010;142(6):847–56.
Chan CJ, Smyth MJ, Martinet L. Molecular mechanisms of natural killer cell activation in response to cellular stress. Cell Death And Differentiation. 2013;21:5.
Eischen CM, Leibson PJ. Role for NK-cell-associated Fas ligand in cell-mediated cytotoxicity and apoptosis. Research in Immunology. 1997;148(3):164–9.
Zamai L, et al. Natural killer (NK) cell-mediated cytotoxicity: differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J Exp Med. 1998:0022–1007 Print.
Topham NJ, Hewitt EW. Natural killer cell cytotoxicity: how do they pull the trigger? Immunology. 2009;128(1):7–15.
Wang W, et al. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Frontiers in Immunology. 2015;6:368.
McEwen-Smith RM, Salio M, Cerundolo V. The regulatory role of invariant NKT cells in tumor immunity. Cancer immunology research. 2015;3(5):425–35.
Bendelac A, Fau-Teyton LSP, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007:0732–582 Print.
Shimizu K, et al. KLRG(+) invariant natural killer T cells are long-lived effectors. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(34):12474–9.
O'Leary JG, et al. T cell– and B cell–independent adaptive immunity mediated by natural killer cells. Nature Immunology. 2006;7:507.
Paust S, et al. Critical role for the chemokine receptor CXCR6 in NK cell–mediated antigen-specific memory of haptens and viruses. Nature Immunology. 2010;11:1127.
RCG SF F-d B, et al. Circulating invariant natural killer T-cell numbers predict outcome in head and neck squamous cell carcinoma: updated analysis with 10-year follow-up. J Clin Oncol. 2012:1527–7755 Electronic.
Molling JW, et al. Low levels of circulating invariant natural killer T cells predict poor clinical outcome in patients with head and neck squamous cell carcinoma. J Clin Oncol. 2007:1527–7755 Electronic.
Provenzano PP, et al. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Medicine. 2006;4(1):38.
Levental KR, et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell. 2009;139(5):891–906.
Tallant C, Marrero A, Gomis-Rüth FX. Matrix metalloproteinases: Fold and function of their catalytic domains. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2010;1803(1):20–8.
Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nature Reviews Cancer. 2002;2:161.
Chandler S, et al. Macrophage Metalloelastase Degrades Matrix and Myelin Proteins and Processes a Tumour Necrosis Factor-α Fusion Protein. Biochemical and Biophysical Research Communications. 1996;228(2):421–9.
Black RA, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature. 1997;385:729.
Suzuki M, et al. Matrix Metalloproteinase-3 Releases Active Heparin-binding EGF-like Growth Factor by Cleavage at a Specific Juxtamembrane Site. Journal of Biological Chemistry. 1997;272(50):31730–7.
Whitelock JM, et al. The Degradation of Human Endothelial Cell-derived Perlecan and Release of Bound Basic Fibroblast Growth Factor by Stromelysin, Collagenase, Plasmin, and Heparanases. Journal of Biological Chemistry. 1996;271(17):10079–86.
Bergers G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell Biology. 2000;2:737.
Tatti O, et al. MT1-MMP releases latent TGF-β1 from endothelial cell extracellular matrix via proteolytic processing of LTBP-1. Experimental Cell Research. 2008;314(13):2501–14.
Rosenthal EL, et al. Expression of proteolytic enzymes in head and neck cancer–associated fibroblasts. Archives of Otolaryngology–Head & Neck Surgery. 2004;130(8):943–7.
Iwata H, et al. Production of Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Human Breast Carcinomas. Japanese Journal of Cancer Research. 1996;87(6):602–11.
Acerbi I, et al. Human Breast Cancer Invasion and Aggression Correlates with ECM Stiffening and Immune Cell Infiltration. Integrative biology : quantitative biosciences from nano to macro. 2015;7(10):1120–34.
García-Palmero I, et al. Twist1-induced activation of human fibroblasts promotes matrix stiffness by upregulating palladin and collagen α1(VI). Oncogene. 2016;35:5224.
Chaudhuri O, et al. Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nature Materials. 2014;13:970.
Mouw JK, et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nature Medicine. 2014;20:360.
Calvo F, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15(6):637–46.
Rozario T, DeSimone DW. The Extracellular Matrix In Development and Morphogenesis: A Dynamic View. Developmental biology. 2010;341(1):126–40.
Jokinen J, et al. Integrin-mediated Cell Adhesion to Type I Collagen Fibrils. Journal of Biological Chemistry. 2004;279(30):31956–63.
Hill RC, et al. Quantification of Extracellular Matrix Proteins from a Rat Lung Scaffold to Provide a Molecular Readout for Tissue Engineering. Molecular & Cellular Proteomics. 2015;14(4):961–73.
Schenke-Layland K, et al. Reprogrammed Mouse Fibroblasts Differentiate into Cells of the Cardiovascular and Hematopoietic Lineages. STEM CELLS. 2008;26(6):1537–46.
Huang R, et al. Biomimetic LBL structured nanofibrous matrices assembled by chitosan/collagen for promoting wound healing. Biomaterials. 2015;53(Supplement C):58–75.
Peters JH, et al. Human endothelial cells synthesize, process, and secrete fibronectin molecules bearing an alternatively spliced type III homology (ED1). Blood. 1990;75(9):1801.
Adachi Y, et al. Fibronectin production by cultured human lung fibroblasts in three-dimensional collagen gel culture. In Vitro Cellular & Developmental Biology - Animal. 1998;34(3):203–10.
Garciapardo A, Gold LI. Further Characterization of the Binding of Fibronectin to Gelatin Reveals the Presence of Different Binding Interactions. Archives of Biochemistry and Biophysics. 1993;304(1):181–8.
Garcia AJ, Boettiger D. Integrin–fibronectin interactions at the cell-material interface: initial integrin binding and signaling. Biomaterials. 1999;20(23):2427–33.
Ingham KC, Brew SA, Erickson HP. Localization of a Cryptic Binding Site for Tenascin on Fibronectin. Journal of Biological Chemistry. 2004;279(27):28132–5.
Huang W, et al. Interference of Tenascin-C with Syndecan-4 Binding to Fibronectin Blocks Cell Adhesion and Stimulates Tumor Cell Proliferation. Cancer Research. 2001;61(23):8586.
Brown AC, et al. Integrin α3β1 binding to fibronectin is dependent on the 9th type III repeat. Journal of Biological Chemistry. 2015.
Nam J-M, et al. Breast Cancer Cells in Three-dimensional Culture Display an Enhanced Radioresponse after Coordinate Targeting of Integrin α5β1 and Fibronectin. Cancer research. 2010;70(13):5238–48.
Lou X, et al. SOX2 Targets Fibronectin 1 to Promote Cell Migration and Invasion in Ovarian Cancer: New Molecular Leads for Therapeutic Intervention. OMICS : a Journal of Integrative Biology. 2013;17(10):510–8.
Knowles LM, et al. Integrin αvβ3 and fibronectin upregulate Slug in cancer cells to promote clot invasion and metastasis. Cancer research. 2013;73(20):10.1158/0008–5472 CAN-13-0602.
Eke I, et al. Cetuximab Attenuates Its Cytotoxic and Radiosensitizing Potential by Inducing Fibronectin Biosynthesis. Cancer Research. 2013;73(19):5869.
Pontiggia O, et al. The tumor microenvironment modulates tamoxifen resistance in breast cancer: a role for soluble stromal factors and fibronectin through β1 integrin. Breast cancer research and treatment. 2012;133(2):459–71.
Erdogan B, et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. The Journal of Cell Biology. 2017;216(11):3799.
Blouw B, et al. The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell. 2003;4(2):133–46.
Rockwell S, et al. Hypoxia and radiation therapy: Past history, ongoing research, and future promise. Current molecular medicine. 2009;9(4):442–58.
Teicher, B.A., Hypoxia and drug resistance. Cancer Metastasis Rev. , 1994(0167-7659 (Print)).
Nam SY, et al. A hypoxia-dependent upregulation of hypoxia-inducible factor-1 by nuclear factor-κB promotes gastric tumour growth and angiogenesis. British Journal Of Cancer. 2010;104:166.
Semenza GL. HIF-1 and mechanisms of hypoxia sensing. Current Opinion in Cell Biology. 2001;13(2):167–71.
Royds JA, et al. Response of tumour cells to hypoxia: role of p53 and NFkB. Mol Pathol. 1998; 1366-8714 (Print).
Giatromanolaki A, Harris AL. Tumour hypoxia, hypoxia signaling pathways and hypoxia inducible factor expression in human cancer. Anticancer Research. 2001; 0250-7005 (Print).
Marin-Hernandez A, et al. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem. 2009; 1389-5575 (Print).
Warburg O. On respiratory impairment in cancer cells. Science. 1956; 0036-8075 (Print).
Wojtkowiak JW, et al. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Molecular pharmaceutics. 2011;8(6):2032–8.
Greijer AE, et al. Hypoxia-induced acidification causes mitoxantrone resistance not mediated by drug transporters in human breast cancer cells. Cell Oncol. 2005; 1570-5870 (Print).
Estrella V, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer research. 2013;73(5):1524–35.
Colegio OR, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014; 1476-4687 (Electronic).
Sonveaux P, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008; 0021-9738 (Print).
Whitaker-Menezes D, et al. Evidence for a stromal-epithelial "lactate shuttle" in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011; 1551-4005 (Electronic).
Halestrap AP. The monocarboxylate transporter family--Structure and functional characterization. IUBMB Life. 2012; 1521-6551 (Electronic).
Curry JM, et al. Cancer metabolism, stemness and tumor recurrence: MCT1 and MCT4 are functional biomarkers of metabolic symbiosis in head and neck cancer. Cell Cycle. 2013; 1551-4005 (Electronic).
Pinheiro C, et al. Expression of monocarboxylate transporters 1, 2, and 4 in human tumours and their association with CD147 and CD44. J Biomed Biotechnol. 2010; 1110-7251 (Electronic).
Zhu J, et al. Monocarboxylate transporter 4 facilitates cell proliferation and migration and is associated with poor prognosis in oral squamous cell carcinoma patients. PLoS One. 2014; 1932-6203 (Electronic).
Simoes-Sousa S, et al. Prognostic significance of monocarboxylate transporter expression in oral cavity tumors. Cell Cycle. 2016; 1551-4005 (Electronic).
Kumar DA, et al. Cancer-Associated Fibroblasts Drive Glycolysis in a Targetable Signaling Loop Implicated in Head and Neck Squamous Cell Carcinoma Progression. Cancer Res. 2018; 1538-7445 (Electronic).
Stasikowska-Kanicka O, Wagrowska-Danilewicz M, Danilewicz M. CD8+ and CD163+ infiltrating cells and PD-L1 immunoexpression in oral leukoplakia and oral carcinoma. APMIS. 2018;126:732–8.
Mori K, et al. Tumor-associated macrophages in oral premalignant lesions coexpress CD163 and STAT1 in a Th1-dominated microenvironment. BMC Cancer. 2015;15(1):573.
Bondad-Palmario GG. Histological and immunochemical studies of oral leukoplakia: phenotype and distribution of immunocompetent cells. J Philipp Dent Assoc. 1995; 0031-7497 (Print).
De Costa AA-M, et al. Characterization of the evolution of immune phenotype during the development and progression of squamous cell carcinoma of the head and neck. Cancer immunology, immunotherapy : CII. 2012;61(6):927–39.
Woodford D, et al. An Inflammatory Cytokine Milieu is Prominent in Premalignant Oral Lesions, but Subsides when Lesions Progress to Squamous Cell Carcinoma. Journal of clinical & cellular immunology. 2014;5(3):230.
Johnson SD, De Costa A-MA, Young MRI. Effect of the Premalignant and Tumor Microenvironment on Immune Cell Cytokine Production in Head and Neck Cancer. Cancers. 2014;6(2):756–70.
Juretic M, et al. Salivary levels of TNF-alpha and IL-6 in patients with oral premalignant and malignant lesions. Folia Biol. 2013; 0015-5500 (Print).
Rhodus NL, et al. The feasibility of monitoring NF-kappaB associated cytokines: TNF-alpha, IL-1alpha, IL-6, and IL-8 in whole saliva for the malignant transformation of oral lichen planus. Mol Carcinog. 2005; 0899-1987 (Print).
Rhodus NL, et al. NF-kappaB dependent cytokine levels in saliva of patients with oral preneoplastic lesions and oral squamous cell carcinoma. Cancer Detect Prev. 2005; 0361-090X (Print).
Michailidou EZ, Fau-Antoniades DZMA, Antoniades DZ. Mast cells and angiogenesis in oral malignant and premalignant lesions. Open Dent J. 2008; 1874-2106 (Electronic).
Laishram D, et al. Mast cells and angiogenesis in malignant and premalignant oral lesions: An immunohistochemical study. J Oral Maxillofac Pathol. 2017; 0973-029X (Print).
Jyothsna M, Rammanohar M, Kumar K. Histomorphometric Analysis of Angiogenesis using CD31 Immunomarker and Mast Cell Density in Oral Premalignant and Malignant Lesions: A Pilot Study. J Clin Diagn Res. 2017; 2249-782X (Print).
Ingaleshwar PS, et al. Immunohistochemical analysis of angiogenesis by CD34 and mast cells by toluidine blue in different grades of oral squamous cell carcinoma. J Oral Maxillofac Pathol. 2016; 0973-029X (Print).
Chaudhary M, et al. Comparison of myofibroblasts expression in oral squamous cell carcinoma, verrucous carcinoma, high risk epithelial dysplasia, low risk epithelial dysplasia and normal oral mucosa. Head Neck Pathol. 2012; 1936-0568 (Electronic).
Kapse SC, et al. Quantitative assessment of myofibroblast in severe dysplasia, microinvasion and oral squamous cell carcinoma: an immunohistochemical study. J Contemp Dent Pract. 2013; 1526-3711 (Electronic).
Gupta K, Fau-Gupta JMR, Gupta J. Evaluation of stromal myofibroblasts in oral leukoplakia, oral submucous fibrosis, and oral squamous cell carcinoma--an immunohistochemical study. Journal of Cancer Research and Therapeutics. 2015; 1998-4138 (Electronic).
Nayak S, et al. Fibroblast Growth Factor (FGF-2) and Its Receptors FGFR-2 and FGFR-3 May Be Putative Biomarkers of Malignant Transformation of Potentially Malignant Oral Lesions into Oral Squamous Cell Carcinoma. PLoS One. 2015; 1932-6203 (Electronic).
Mane DR, et al. Immunoexpression of tenascin as a predictor of the malignancy potential of oral leukoplakia associated with a tobacco habit. Biotech Histochem. 2015; 1473-7760 (Electronic).
Bajracharya D, et al. Immunohistochemical correlation of matrix metalloproteinase-2 and tissue inhibitors of metalloproteinase-2 in tobacco associated epithelial dysplasia. Disease Markers. 2014; 1875-8630 (Electronic).
Fang J, et al. Prognostic significance of tumor infiltrating immune cells in oral squamous cell carcinoma. BMC cancer. 2017;17(1):375.
Nguyen N, et al. Tumor infiltrating lymphocytes and survival in patients with head and neck squamous cell carcinoma. Head & neck. 2016;38(7):1074–84.
De Meulenaere A, et al. Prognostic markers in oropharyngeal squamous cell carcinoma: focus on CD70 and tumour infiltrating lymphocytes. Pathology. 2017;49(4):397–404.
Karpathiou G, et al. Prognostic impact of immune microenvironment in laryngeal and pharyngeal squamous cell carcinoma: Immune cell subtypes, immuno-suppressive pathways and clinicopathologic characteristics. Oncotarget. 2016;8(12):19310–22.
Ni YH, et al. Microlocalization of CD68+ tumor-associated macrophages in tumor stroma correlated with poor clinical outcomes in oral squamous cell carcinoma patients. Tumour Biol. 2015; 1423-0380 (Electronic).
He K-F, et al. CD163+ Tumor-Associated Macrophages Correlated with Poor Prognosis and Cancer Stem Cells in Oral Squamous Cell Carcinoma. BioMed Research International. 2014;2014:9.
Weber M, et al. Prognostic significance of macrophage polarization in early stage oral squamous cell carcinomas. Oral Oncol. 2016; 1879-0593 (Electronic).
Marcus B, et al. Prognostic factors in oral cavity and oropharyngeal squamous cell carcinoma. Cancer. 2004; 0008-543X (Print).
Liu SY, et al. Clinicopathologic significance of tumor cell-lined vessel and microenvironment in oral squamous cell carcinoma. Oral Oncol. 2008; 1368-8375 (Print).
Kross KW, et al. Co-culture of Head and Neck Squamous Cell Carcinoma Spheroids with Autologous Monocytes Predicts Prognosis. Scandinavian Journal of Immunology. 2008;67(4):392–9.
Costa NL, et al. Tumor-associated macrophages and the profile of inflammatory cytokines in oral squamous cell carcinoma. Oral Oncology. 2013;49(3):216–23.
Jiang C, et al. Oral squamous cell carcinoma suppressed antitumor immunity through induction of PD-L1 expression on tumor-associated macrophages. Immunobiology. 2017;222(4):651–7.
Kubota K, et al. CD163+CD204+ tumor-associated macrophages contribute to T cell regulation via interleukin-10 and PD-L1 production in oral squamous cell carcinoma. Scientific Reports. 2017;7(1):1755.
Salven P, et al. Vascular endothelial growth factor in squamous cell head and neck carcinoma: expression and prognostic significance. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 1997;10(11):1128–33.
Neuchrist C. Vascular Endothelial Growth Factor (VEGF) and Microvessel Density in Squamous Cell Carcinomas of the Larynx: an Immunohistochemical Study. Acta Oto-Laryngologica. 1999;119(6):732–8.
Sun H, et al. TGF-beta1/TbetaRII/Smad3 signaling pathway promotes VEGF expression in oral squamous cell carcinoma tumor-associated macrophages. Biochem Biophys Res Commun. 2018;497(2):583–90 1090-2104 (Electronic).
Li C, et al. Infiltration of tumor-associated macrophages in human oral squamous cell carcinoma. Oncol Rep. 2002;9(6):1219–23 1021-335X (Print).
Oguejiofor K, et al. Distinct patterns of infiltrating CD8+ T cells in HPV+ and CD68 macrophages in HPV- oropharyngeal squamous cell carcinomas are associated with better clinical outcome but PD-L1 expression is not prognostic. Oncotarget. 2017;8(9):14416–27 1949-2553 (Electronic).
Trellakis S, et al. Polymorphonuclear granulocytes in human head and neck cancer: enhanced inflammatory activity, modulation by cancer cells and expansion in advanced disease. Int J Cancer. 2011;129(9):2183–93 1097-0215 (Electronic).
Wang N, et al. Neutrophils infiltration in the tongue squamous cell carcinoma and its correlation with CEACAM1 expression on tumor cells. PLoS One. 2014;9(2):e89991 1932-6203 (Electronic).
Trellakis S, et al. Polymorphonuclear granulocytes in human head and neck cancer: Enhanced inflammatory activity, modulation by cancer cells and expansion in advanced disease. International Journal of Cancer. 2010;129(9):2183–93.
Iamaroon A, et al. Increase of mast cells and tumor angiogenesis in oral squamous cell carcinoma. J Oral Pathol Med. 2003;32(4):195–9 0904-2512 (Print).
Ciurea R, et al. VEGF and his R1 and R2 receptors expression in mast cells of oral squamous cells carcinomas and their involvement in tumoral angiogenesis. Rom J Morphol Embryol. 2011;52:1227 1220-0522 (Print).
Sharma B, et al. Immunohistochemical evaluation of mast cells and angiogenesis in oral squamous cell carcinoma. Indian J Dent Res. 2010;21:260 1998-3603 (Electronic).
Telagi N, et al. The master switch: Comparative study of mast cell in oral epithelial dysplasia, oral submucous fibrosis and oral squamous cells carcinoma and their association with inflammation and angiogenesis. J Oral Maxillofac Pathol. 2015;19:25 0973-029X (Print).
Zaidi M, Mallick A. A study on assessment of mast cells in oral squamous cell carcinoma. Ann Med Health Sci Res. 2014;4:457 2141-9248 (Print).
Anuradha A, et al. Incidence of mast cells in oral squamous cell carcinoma: a short study. Journal of Oncology. 2014;2014:3 1687-8450 (Print).
Kathuriya PT, et al. Cd34 and Mast Cell Analysis in Normal Oral Mucosa and Different Grades of Oral Squamous Cell Carcinoma: A Comparative Study. J Clin Diagn Res. 2015;9:ZC61 2249-782X (Print).
Wagner S, et al. CD56-positive lymphocyte infiltration in relation to human papillomavirus association and prognostic significance in oropharyngeal squamous cell carcinoma. International Journal of Cancer. 2015;138(9):2263–73.
Agarwal R, et al. Evaluation of natural killer cell (CD57) as a prognostic marker in oral squamous cell carcinoma: An immunohistochemistry study. Journal of oral and maxillofacial pathology : JOMFP. 2016;20(2):173–7.
Korrer MJ, Kim Y. Natural Killer cells from primary human head and neck squamous cell carcinomas upregulate NKG2A. The Journal of Immunology. 2017;198(1 Supplement):130.18.
Gaur P, et al. Inter-relation of Th1, Th2, Th17 and Treg cytokines in oral cancer patients and their clinical significance. Human Immunology. 2014;75(4):330–7.
Hussaini HM, et al. Forkhead box-P3(+) regulatory T cells and toll-like receptor 2 co-expression in oral squamous cell carcinoma. Acta Histochem. 2017;119:205 1618-0372 (Electronic).
Song JJ, et al. Foxp3 overexpression in tumor cells predicts poor survival in oral squamous cell carcinoma. BMC Cancer. 2016;16:530 1471-2407 (Electronic).
Stasikowska-Kanicka O, Wągrowska-Danilewicz M, Danilewicz M. Immunohistochemical Analysis of Foxp3+, CD4+, CD8+ Cell Infiltrates and PD-L1 in Oral Squamous Cell Carcinoma. Pathology & Oncology Research. 2018;24(3):497–505.
Weed DT, et al. FOXP3 subcellular localization predicts recurrence in oral squamous cell carcinoma. PloS one. 2013;8(8):–e71908.
Schott AK, Fau-Wollenberg BPR, Wollenberg B. Permanent up-regulation of regulatory T-lymphocytes in patients with head and neck cancer. Int J Mol Med. 2010;26:67 1791-244X (Electronic).
Drennan S, et al. Increased frequency and suppressive activity of CD127low/− regulatory T cells in the peripheral circulation of patients with head and neck squamous cell carcinoma are associated with advanced stage and nodal involvement. Immunology. 2013;140(3):335–43.
Ihara F, et al. CD45RA(-)Foxp3(high) regulatory T cells have a negative impact on the clinical outcome of head and neck squamous cell carcinoma. Cancer Immunol Immunother. 2017;66:1275 1432-0851 (Electronic).
Strauss L, et al. The frequency and suppressor function of CD4+CD25highFoxp3+ T cells in the circulation of patients with squamous cell carcinoma of the head and neck. Clin Cancer Res. 2007;13:6301 1078-0432 (Print).
Vasquez-Dunddel D, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. The Journal of clinical investigation. 2013;123(4):1580–9.
Zeng Q, et al. Caspase-1 from Human Myeloid-Derived Suppressor Cells Can Promote T Cell–Independent Tumor Proliferation. Cancer Immunology Research. 2018;6(5):566.
Weed DT, et al. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res. 2015;21:39 1078-0432 (Print).
Mao L, et al. Selective blockade of B7-H3 enhances antitumour immune activity by reducing immature myeloid cells in head and neck squamous cell carcinoma. Journal of cellular and molecular medicine. 2017;21(9):2199–210.
Younis RH, Han KL, Webb TJ. Human Head and Neck Squamous Cell Carcinoma-Associated Semaphorin 4D Induces Expansion of Myeloid-Derived Suppressor Cells. Journal of immunology. 2016;196(3):1419–29.
Kellermann MG, et al. Myofibroblasts in the stroma of oral squamous cell carcinoma are associated with poor prognosis. Histopathology. 2007;51(6):849–53.
Rodrigues PC, et al. Stromal myofibroblasts in potentially malignant and malignant lesions of the oral cavity. Oncol Lett. 2015;9(2):667 1792-1074 (Print).
Kellermann MG, et al. Mutual paracrine effects of oral squamous cell carcinoma cells and normal oral fibroblasts: Induction of fibroblast to myofibroblast transdifferentiation and modulation of tumor cell proliferation. Oral Oncology. 2008;44(5):509–17.
Bello IO, et al. Cancer-associated fibroblasts, a parameter of the tumor microenvironment, overcomes carcinoma-associated parameters in the prognosis of patients with mobile tongue cancer. Oral Oncol. 2011;47:33 1879-0593 (Electronic).
Marsh D, et al. Stromal features are predictive of disease mortality in oral cancer patients. The Journal of Pathology. 2010;223(4):470–81.
Kawashiri S, et al. Significance of stromal desmoplasia and myofibroblast appearance at the invasive front in squamous cell carcinoma of the oral cavity. Head & Neck. 2009;31(10):1346–53.
Fujii N, et al. Cancer-associated fibroblasts and CD163-positive macrophages in oral squamous cell carcinoma: their clinicopathological and prognostic significance. Journal of Oral Pathology & Medicine. 2012;41(6):444–51.
Álvarez-Teijeiro S, et al. Factors Secreted by Cancer-Associated Fibroblasts that Sustain Cancer Stem Properties in Head and Neck Squamous Carcinoma Cells as Potential Therapeutic Targets. Cancers. 2018;10(9):334.
Rosenthal E, et al. Elevated expression of TGF-beta1 in head and neck cancer-associated fibroblasts. Mol Carcinog. 2004;40:116 0899-1987 (Print).
Knowles LM, et al. HGF and c-Met participate in paracrine tumorigenic pathways in head and neck squamous cell cancer. Clin Cancer Res. 2009;15(11):3740 1078-0432 (Print).
Johansson AC, et al. Cancer-associated fibroblasts induce matrix metalloproteinase-mediated cetuximab resistance in head and neck squamous cell carcinoma cells. Mol Cancer Res. 2012;10(9):1158 1557-3125 (Electronic).
Takahashi H, et al. Immunosuppressive activity of cancer-associated fibroblasts in head and neck squamous cell carcinoma. Cancer Immunology, Immunotherapy. 2015;64(11):1407–17.
Bagordakis E, et al. Secretome profiling of oral squamous cell carcinoma-associated fibroblasts reveals organization and disassembly of extracellular matrix and collagen metabolic process signatures. Tumor Biology. 2016;37(7):9045–57.
Ziober AF, Falls EM, Ziober BL. The extracellular matrix in oral squamous cell carcinoma: Friend or foe? Head & Neck. 2006;28(8):740–9.
Agarwal P, Ballabh R. Expression of type IV collagen in different histological grades of oral squamous cell carcinoma: an immunohistochemical study. Journal of Cancer Research and Therapeutics. 2013;9(2):272 1998-4138 (Electronic).
Shruthy R, et al. Immunohistochemical expression of basement membrane laminin in histological grades of oral squamous cell carcinoma: A semiquantitative analysis. Journal of oral and maxillofacial pathology : JOMFP. 2013;17(2):185–9.
Firth NA, Reade PC. The prognosis of oral mucosal squamous cell carcinomas: a comparison of clinical and histopathological grading and of laminin and type IV collagen staining. Aust Dent J. 1996;41(2):83 0045-0421 (Print).
Harada T, et al. An immunohistochemical study of the extracellular matrix in oral squamous cell carcinoma and its association with invasive and metastatic potential. Virchows Arch. 1994;424(3):257 0945-6317 (Print).
Fabricius E-M, et al. Immunohistochemical analysis of integrins αvβ3, αvβ5 and α5β1, and their ligands, fibrinogen, fibronectin, osteopontin and vitronectin, in frozen sections of human oral head and neck squamous cell carcinomas. Experimental and therapeutic medicine. 2011;2(1):9–19.
Rygiel KA, et al. Epithelial–mesenchymal transition contributes to portal tract fibrogenesis during human chronic liver disease. Laboratory Investigation. 2007;88:112.
Okada H, et al. Early role of Fsp1 in epithelial-mesenchymal transformation. Am J Physiol. 1997;273(4 Pt 2):F563 0002-9513 (Print).
Myong NH. Loss of E-cadherin and Acquisition of Vimentin in Epithelial-Mesenchymal Transition are Noble Indicators of Uterine Cervix Cancer Progression. Korean J Pathol. 2012;46(4):341 2092-8920 (Electronic).
Nijkamp MM, et al. Expression of E-cadherin and vimentin correlates with metastasis formation in head and neck squamous cell carcinoma patients. Radiotherapy and Oncology. 2011;99(3):344–8.
Oser M, et al. Cortactin regulates cofilin and N-WASp activities to control the stages of invadopodium assembly and maturation. The Journal of Cell Biology. 2009;186(4):571.
Peláez R, et al. β3 integrin expression is required for invadopodia-mediated ECM degradation in lung carcinoma cells. PLOS ONE. 2017;12(8):e0181579.
Iizuka S, et al. The role of Tks adaptor proteins in invadopodia formation, growth and metastasis of melanoma. Oncotarget. 2016;7(48):78473–86.
Artym VV, et al. Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res. 2006;66(6):3034 0008-5472 (Print).
Blouw B, et al. A role for the podosome/invadopodia scaffold protein Tks5 in tumor growth in vivo. Eur J Cell Biol. 2008;87:555 0171-9335 (Print).
Hayes KE, et al. Ableson kinases negatively regulate invadopodia function and invasion in head and neck squamous cell carcinoma by inhibiting an HB-EGF autocrine loop. Oncogene. 2013;32(40):4766–77.
Kelley LC, et al. Oncogenic Src requires a wild-type counterpart to regulate invadopodia maturation. Journal of cell science. 2010;123(Pt 22):3923–32.
Clark ES, et al. Cortactin Is an Essential Regulator of Matrix Metalloproteinase Secretion and Extracellular Matrix Degradation in Invadopodia. Cancer Research. 2007;67(9):4227.
Bowden ET, et al. An invasion-related complex of cortactin, paxillin and PKCμ associates with invadopodia at sites of extracellular matrix degradation. Oncogene. 1999;18:4440.
Nakahara H, et al. Transmembrane/cytoplasmic domain-mediated membrane type 1-matrix metalloprotease docking to invadopodia is required for cell invasion. Proc Natl Acad Sci U S A. 1997;94(15):7959 0027-8424 (Print).
Jacob A, et al. Rab40b regulates trafficking of MMP2 and MMP9 during invadopodia formation and invasion of breast cancer cells. J Cell Sci. 2013;126:4647 1477-9137 (Electronic).
Mohtasham N, et al. Immunohistochemical study of p53, Ki-67, MMP-2 and MMP-9 expression at invasive front of squamous cell and verrucous carcinoma in oral cavity. Pathol Res Pract. 2013;209:110 1618-0631 (Electronic).
Sterz CM, et al. A basal-cell-like compartment in head and neck squamous cell carcinomas represents the invasive front of the tumor and is expressing MMP-9. Oral Oncol. 2010;46:116 1879-0593 (Electronic).
Sterz, C. and R. Mandic, [Cells of the tumor front : a potential therapeutic target in head and neck cancer therapy?]. (1433-0458 (Electronic)).
Herrmann D, et al. Three-dimensional cancer models mimic cell-matrix interactions in the tumour microenvironment. Carcinogenesis. 2014;35(8):1671–9.
Lucas JT Jr, et al. Regulation of invasive behavior by vascular endothelial growth factor is HEF1-dependent. Oncogene. 2010;29(31):4449–59.
Grauzam S, et al. NEDD9 stimulated MMP9 secretion is required for invadopodia formation in oral squamous cell carcinoma. Oncotarget. 2018;9(39):25503–16.
Semelakova M, et al. Vimentin and Non-Muscle Myosin IIA are Members of the Neural Precursor Cell Expressed Developmentally Down-Regulated 9 (NEDD9) Interactome in Head and Neck Squamous Cell Carcinoma Cells. Translational oncology. 2018;12(1):49–61.
Loudig O, et al. Illumina whole-genome complementary DNA–mediated annealing, selection, extension and ligation platform: assessing its performance in formalin-fixed, paraffin-embedded samples and identifying invasion pattern–related genes in oral squamous cell carcinoma. Human Pathology. 2011;42(12):1911–22.
Gao L, et al. CCL2/EGF positive feedback loop between cancer cells and macrophages promotes cell migration and invasion in head and neck squamous cell carcinoma. Oncotarget. 2016;7(52):87037–51.
Gao L, et al. Tumor associated macrophages induce epithelial to mesenchymal transition via the EGFR/ERK1/2 pathway in head and neck squamous cell carcinoma. Oncol Rep. 2018;40:2558 1791-2431 (Electronic).
Pirilä E, et al. Macrophages Modulate Migration and Invasion of Human Tongue Squamous Cell Carcinoma. PLOS ONE. 2015;10(3):e0120895.
Smirnova T, et al. In vivo invasion of head and neck squamous cell carcinoma cells does not require macrophages. The American journal of pathology. 2011;178(6):2857–65.
Zeng J, Quan J, Xia X. Transient transfection of macrophage migration inhibitory factor small interfering RNA disrupts the biological behavior of oral squamous carcinoma cells. Molecular medicine reports. 2016;13(1):174–80.
Dumitru CA, et al. Tumor-derived macrophage migration inhibitory factor modulates the biology of head and neck cancer cells via neutrophil activation. International Journal of Cancer. 2011;129(4):859–69.
Trellakis S, et al. Peripheral blood neutrophil granulocytes from patients with head and neck squamous cell carcinoma functionally differ from their counterparts in healthy donors. Int J Immunopathol Pharmacol. 2011;24:683 0394-6320 (Print).
Glogauer JE, et al. Neutrophils Increase Oral Squamous Cell Carcinoma Invasion through an Invadopodia-Dependent Pathway. Cancer Immunology Research. 2015;3(11):1218.
Dumitru CA, et al. Neutrophils Activate Tumoral CORTACTIN to Enhance Progression of Orohypopharynx Carcinoma. Frontiers in immunology. 2013;4:33.
Finke J, et al. MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. Int Immunopharmacol. Int Immunopharmacol. 2011;11:856 1878-1705 (Electronic).
Toh B, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS biology. 2011;9(9):e1001162.
Zhu H, et al. CXCR2(+) MDSCs promote breast cancer progression by inducing EMT and activated T cell exhaustion. Oncotarget. 2017;8(70):114554–67.
Ouzounova M, et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nature Communications. 2017;8:14979.
Peinado H, et al. Pre-metastatic niches: organ-specific homes for metastases. Nature Reviews Cancer. 2017;17:302.
Sceneay J, et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. 2012; 1538-7445 (Electronic).
Wang D, et al. CXCL1 Is Critical for Premetastatic Niche Formation and Metastasis in Colorectal Cancer. Cancer Res. 2017; 1538-7445 (Electronic).
Shi H, et al. Recruited monocytic myeloid-derived suppressor cells promote the arrest of tumor cells in the premetastatic niche through an IL-1β-mediated increase in E-selectin expression. International Journal of Cancer. 2016;140(6):1370–83.
Sinha P, et al. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. Journal of immunology. 2008;181(7):4666–75.
Gebhardt C, et al. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med. 2008; 1540-9538 (Electronic).
Grebhardt S, et al. Impact of S100A8/A9 expression on prostate cancer progression in vitro and in vivo. J Cell Physiol. 2014; 1097-4652 (Electronic).
Simard J-C, et al. S100A8 and S100A9 Induce Cytokine Expression and Regulate the NLRP3 Inflammasome via ROS-Dependent Activation of NF-κB1. PLOS ONE. 2013;8(8):e72138.
Ichikawa M, et al. S100A8/A9 activate key genes and pathways in colon tumor progression. Mol Cancer Res. 2011; 1557-3125 (Electronic).
Tardif M, et al. Secretion of S100A8, S100A9, and S100A12 by Neutrophils Involves Reactive Oxygen Species and Potassium Efflux. Journal of Immunology Research. 2015;2015:16.
Hiratsuka S, et al. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006; 1465-7392 (Print).
Hermani A, et al. S100A8 and S100A9 activate MAP kinase and NF-kappaB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp Cell Res, 2006. 0014-4827 (Print).
Sinha P, et al. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008; 1550-6606 (Electronic).
Hiratsuka S, et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008; 1476-4679 (Electronic).
Cross SS, et al. Expression of S100 proteins in normal human tissues and common cancers using tissue microarrays: S100A6, S100A8, S100A9 and S100A11 are all overexpressed in common cancers. Histopathology. 2005;46(3):256–69.
Tugizov S, et al. Inhibition of human papillomavirus type 16 E7 phosphorylation by the S100 MRP-8/14 protein complex. J Virol. 2005; 0022-538X (Print).
Kong JP, et al. Loss of myeloid-related proteins 8 and myeloid-related proteins 14 expression in human esophageal squamous cell carcinoma correlates with poor differentiation. World J Gastroenterol. 2004; 1007-9327 (Print).
Wang J, et al. Expression of MRP14 gene is frequently down-regulated in Chinese human esophageal cancer. Cell Res. 2004; 1001-0602 (Print).
Khammanivong A, et al. Involvement of calprotectin (S100A8/A9) in molecular pathways associated with HNSCC. Oncotarget. 2016;7(12):14029–47.
Silva EJ, et al. S100A8/A9 regulates MMP-2 expression and invasion and migration by carcinoma cells. The international journal of biochemistry & cell biology. 2014;55:279–87.
Shan T, et al. Prometastatic mechanisms of CAF-mediated EMT regulation in pancreatic cancer cells. Int J Oncol. 2017; 1791-2423 (Electronic).
Zhuang J, et al. TGFβ1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Scientific Reports. 2015;5:11924.
Giannoni E, et al. Reciprocal Activation of Prostate Cancer Cells and Cancer-Associated Fibroblasts Stimulates Epithelial-Mesenchymal Transition and Cancer Stemness. Cancer Research. 2010;70(17):6945.
Ren Y, et al. Paracrine and epigenetic control of CAF-induced metastasis: the role of HOTAIR stimulated by TGF-ß1 secretion. Molecular Cancer. 2018;17(1):5.
Orimo A, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005; 0092-8674 (Print).
Goetz JG, et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 2011; 1097-4172 (Electronic).
Maller O, DuFort CC, Weaver VM. YAP forces fibroblasts to feel the tension. Nature Cell Biology. 2013;15:570.
Yu L-X, et al. Platelets promote tumour metastasis via interaction between TLR4 and tumour cell-released high-mobility group box1 protein. Nature Communications. 2014;5:5256.
Lau EY, et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016; 2211-1247 (Electronic).
Otomo R, et al. TSPAN12 is a critical factor for cancer-fibroblast cell contact-mediated cancer invasion. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(52):18691–6.
Berndt A, et al. Oral squamous cell carcinoma invasion is associated with a laminin-5 matrix re-organization but independent of basement membrane and hemidesmosome formation. clues from an in vitro invasion model. Invasion & metastasis. 1997;17(5):251–8.
Satoh S, et al. A new in vitro model for analyzing the biological behavior of well-differentiated squamous cell carcinoma. Pathology - Research and Practice. 2005;201(1):27–35.
Nyström ML, et al. Development of a quantitative method to analyse tumour cell invasion in organotypic culture. The Journal of Pathology. 2005;205(4):468–75.
Vered M, et al. Cancer-associated fibroblasts and epithelial-mesenchymal transition in metastatic oral tongue squamous cell carcinoma. International Journal of Cancer. 2010;127(6):1356–62.
Onoue T, et al. Epithelial-mesenchymal transition induced by the stromal cell-derived factor-1/CXCR4 system in oral squamous cell carcinoma cells. Int J Oncol. 2006;29:1133–8.
Yu C, et al. TGF-β1 mediates epithelial to mesenchymal transition via the TGF-β/Smad pathway in squamous cell carcinoma of the head and neck. Oncol Rep. 2011;25:1581–7.
Hinsley EE, et al. Endothelin-1 stimulates oral fibroblasts to promote oral cancer invasion. Life Sciences. 2012;91(13):557–61.
Richter P, et al. EGF/TGFβ1 co-stimulation of oral squamous cell carcinoma cells causes an epithelial–mesenchymal transition cell phenotype expressing laminin 332. Journal of Oral Pathology & Medicine. 2010;40(1):46–54.
Wu M-H, et al. Targeting Galectin-1 in Carcinoma-Associated Fibroblasts Inhibits Oral Squamous Cell Carcinoma Metastasis by Downregulating MCP-1/CCL2 Expression. Clinical Cancer Research. 2011;17(6):1306.
Knowles LM, et al. HGF and c-Met participate in paracrine tumorigenic pathways in head and neck squamous cell cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(11):3740–50.
Wheeler SE, et al. Enhancement of head and neck squamous cell carcinoma proliferation, invasion, and metastasis by tumor-associated fibroblasts in preclinical models. Head & neck. 2014;36(3):385–92.
Hwang YS, et al. Functional invadopodia formation through stabilization of the PDPN transcript by IMP-3 and cancer-stromal crosstalk for PDPN expression. Carcinogenesis. 2012;33(11):2135–46.
Hasina R, et al. Autocrine and paracrine motility factors and their involvement in invasiveness in a human oral carcinoma cell line. British Journal Of Cancer. 1999;80:1708.
Lewis MP, et al. Tumour-derived TGF-β1 modulates myofibroblast differentiation and promotes HGF/SF-dependent invasion of squamous carcinoma cells. British Journal Of Cancer. 2004;90:822.
Takkunen M, et al. Snail-dependent and -independent Epithelial-Mesenchymal Transition in Oral Squamous Carcinoma Cells. Journal of Histochemistry & Cytochemistry. 2006;54(11):1263–75.
Liang X, et al. Hypoxia-inducible factor-1 alpha, in association with TWIST2 and SNIP1, is a critical prognostic factor in patients with tongue squamous cell carcinoma. Oral Oncology. 2011;47(2):92–7.
Yokoyama K, et al. Reverse correlation of E-cadherin and snail expression in oral squamous cell carcinoma cells in vitro. Oral Oncology. 2001;37(1):65–71.
Huang C-H, et al. Regulation of membrane-type 4 matrix metalloproteinase by SLUG contributes to hypoxia-mediated metastasis. Neoplasia. 2009;11(12):1371–82.
Yang M-H, et al. Bmi1 is essential in Twist1-induced epithelial–mesenchymal transition. Nature Cell Biology. 2010;12:982.
Ishida T, et al. Notch signaling induces EMT in OSCC cell lines in a hypoxic environment. Oncology letters. 2013;6(5):1201–6.
Díaz B, et al. Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. The Journal of cell biology. 2013;201(2):279–92.
Zhu G, et al. Hypoxia promotes migration/invasion and glycolysis in head and neck squamous cell carcinoma via an HIF-1α-MTDH loop. Oncol Rep. 2017;38.
Li H-M, et al. Blockage of glycolysis by targeting PFKFB3 suppresses tumor growth and metastasis in head and neck squamous cell carcinoma. Journal of Experimental & Clinical Cancer Research. 2017;36(1):7.
Xu Q, et al. EGF induces epithelial-mesenchymal transition and cancer stem-like cell properties in human oral cancer cells via promoting Warburg effect. Oncotarget. 2016;8(6):9557–71.
Wang H, Xue W, Jiang X. Overexpression of TRIM24 Stimulates Proliferation and Glucose Metabolism of Head and Neck Squamous Cell Carcinoma. BioMed research international. 2018;2018:–6142843.
Chang Y-C, et al. Glucose transporter 4 promotes head and neck squamous cell carcinoma metastasis through the TRIM24-DDX58 axis. Journal of Hematology & Oncology. 2017;10(1):11.
Neiva KG, et al. Cross talk initiated by endothelial cells enhances migration and inhibits anoikis of squamous cell carcinoma cells through STAT3/Akt/ERK signaling. Neoplasia. 2009;11(6):583–93.
Zeng Q, et al. Hepatocyte Growth Factor Inhibits Anoikis in Head and Neck Squamous Cell Carcinoma Cells by Activation of ERK and Akt Signaling Independent of NFκB. Journal of Biological Chemistry. 2002;277(28):25203–8.
Zeng Q, McCauley LK, Wang C-Y. Hepatocyte Growth Factor Inhibits Anoikis by Induction of Activator Protein 1-dependent Cyclooxygenase-2: IMPLICATION IN HEAD AND NECK SQUAMOUS CELL CARCINOMA PROGRESSION. Journal of Biological Chemistry. 2002;277(51):50137–42.
Geiger TR, Peeper DS. Critical Role for TrkB Kinase Function in Anoikis Suppression, Tumorigenesis, and Metastasis. Cancer Research. 2007;67(13):6221.
Moriwaki K, et al. TRKB tyrosine kinase receptor is a potential therapeutic target for poorly differentiated oral squamous cell carcinoma. Oncotarget. 2018;9(38):25225–43.
Kupferman ME, et al. TrkB induces EMT and has a key role in invasion of head and neck squamous cell carcinoma. Oncogene. 2010;29(14):2047–59.
Jiffar T, et al. Brain derived neutrophic factor (BDNF) coordinates lympho-vascular metastasis through a fibroblast-governed paracrine axis in the tumor microenvironment. Cancer Cell Microenviron. 2017;4.
Ziober BL, Silverman SS, Kramer RH. Adhesive Mechanisms Regulating Invasion and Metastasis in Oral Cancer. Critical Reviews in Oral Biology & Medicine. 2001;12(6):499–510.
Bozzo C, et al. Activation of caspase-8 triggers anoikis in human neuroblastoma cells. Neuroscience Research. 2006;56(2):145–53.
Koontongkaew S, et al. Fibroblasts and extracellular matrix differently modulate MMP activation by primary and metastatic head and neck cancer cells. Medical Oncology. 2012;29(2):690–703.
Zhang Y, et al. Squamous Cell Carcinoma Cell Aggregates Escape Suspension-induced, p53-mediated Anoikis: FIBRONECTIN AND INTEGRIN αv MEDIATE SURVIVAL SIGNALS THROUGH FOCAL ADHESION KINASE. Journal of Biological Chemistry. 2004;279(46):48342–9.
Kamarajan P, Kapila YL. An altered fibronectin matrix induces anoikis of human squamous cell carcinoma cells by suppressing integrin alpha v levels and phosphorylation of FAK and ERK. Apoptosis. 2007;12(12):2221–31.
Philippe C, et al. Protection from tumor necrosis factor-mediated cytolysis by platelets. American Journal of Pathology. 1993;143(6):1713–23.
Nieswandt B, et al. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Research. 1999;59(6):1295–300.
Rickles FR, Falanga A. Molecular Basis for the Relationship Between Thrombosis and Cancer. Thrombosis Research. 2001;102(6):V215–24.
Takagi S, et al. Platelets promote osteosarcoma cell growth through activation of the platelet-derived growth factor receptor-Akt signaling axis. Cancer Science. 2014;105(8):983–8.
Placke T, et al. Platelet-Derived MHC Class I Confers a Pseudonormal Phenotype to Cancer Cells That Subverts the Antitumor Reactivity of Natural Killer Immune Cells. Cancer Research. 2012;72(2):440.
Lian L, et al. Inhibition of MCF-7 breast cancer cell-induced platelet aggregation using a combination of antiplatelet drugs. Oncology letters. 2013;5(2):675–80.
Jurasz P, et al. Matrix Metalloproteinase 2 in Tumor Cell-induced Platelet Aggregation: Regulation by Nitric Oxide. Cancer Research. 2001;61(1):376.
Steinert BW, et al. Studies on the role of platelet eicosanoid metabolism and integrin αIIbβ3 in tumor-cell-induced platelet aggregation. International Journal of Cancer. 1993;54(1):92–101.
Borsig L, et al. Heparin and cancer revisited: Mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proceedings of the National Academy of Sciences. 2001;98(6):3352.
Huang G-W, et al. Platelet Aggregation in Head and Neck Tumors in China. The Laryngoscope. 2009;107(8):1142–5.
Laimer K, et al. Expression and prognostic impact of indoleamine 2,3-dioxygenase in oral squamous cell carcinomas. Oral Oncology. 2011;47(5):352–7.
Brandacher G, et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin Cancer Res. 2006; 1078-0432 (Print).
Witkiewicz A, et al. Expression of indoleamine 2,3-dioxygenase in metastatic pancreatic ductal adenocarcinoma recruits regulatory T cells to avoid immune detection. J Am Coll Surg. 2008; 1879-1190 (Electronic).
Jabłońska E, et al. iNOS expression and NO production by neutrophils in cancer patients. Arch Immunol Ther Exp. 2005;53(March–April (2)):175–9.
Ratajczak-Wrona W, et al. Role of p38 MAPK pathway in induction of iNOS expression in neutrophils and peripheral blood mononuclear cells in patients with squamous cell carcinoma of the oral cavity. Journal of Oral and Maxillofacial Surgery. 2009;67(11):2354–63.
Miles FL, et al. Stepping out of the flow: capillary extravasation in cancer metastasis. Clinical & Experimental Metastasis. 2008;25(4):305–24.
Bendas G, Borsig L. Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. International journal of cell biology. 2012;2012:–676731.
Stoletov K, et al. Visualizing extravasation dynamics of metastatic tumor cells. Journal of Cell Science. 2010;123(13):2332.
Labelle, M., Begum S Fau - Hynes, Hynes, Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011(1878-3686 (Electronic)).
Schumacher D, et al. Platelet-Derived Nucleotides Promote Tumor-Cell Transendothelial Migration and Metastasis via P2Y2 Receptor. Cancer Cell. 2013;24(1):130–7.
Weber MR, et al. Activated tumor cell integrin αvβ3 cooperates with platelets to promote extravasation and metastasis from the blood stream. Thrombosis research. 2016;140(Suppl 1):S27–36.
Labelle M, Begum S, Hynes RO. Platelets guide the formation of early metastatic niches. Proceedings of the National Academy of Sciences. 2014;111(30):E3053.
Cools-Lartigue J, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. The Journal of Clinical Investigation. 2013;123(8):3446–58.
Spiegel A, et al. Neutrophils Suppress Intraluminal NK Cell–Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer Discovery. 2016;6(6):630.
Ferlito, A., et al., Incidence and sites of distant metastases from head and neck cancer. ORL J Otorhinolaryngol Relat Spec., 2001(0301-1569 (Print)).
Ferjančič Š, et al. VCAM-1 and VAP-1 recruit myeloid cells that promote pulmonary metastasis in mice. Blood. 2013;121(16):3289.
Gil-Bernabé AM, et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood. 2012;119(13):3164.
Chen Q, Xiang HF. Zhang, and J. Massagué, Macrophage Binding to Receptor VCAM-1 Transmits Survival Signals in Breast Cancer Cells that Invade the Lungs. Cancer Cell. 2011;20(4):538–49.
Qian B, et al. A Distinct Macrophage Population Mediates Metastatic Breast Cancer Cell Extravasation. Establishment and Growth. PLOS ONE. 2009;4(8):e6562.
Prager GW, et al. Targeting of VEGF-dependent transendothelial migration of cancer cells by bevacizumab. Molecular Oncology. 2010;4(2):150–60.
Fennewald SM, et al. Laminin Interactions with Head and Neck Cancer Cells under Low Fluid Shear Conditions Lead to Integrin Activation and Binding. Journal of Biological Chemistry. 2012;287(25):21058–66.
Yen Y-C, et al. Insulin-like growth factor-independent insulin-like growth factor binding protein 3 promotes cell migration and lymph node metastasis of oral squamous cell carcinoma cells by requirement of integrin β1. Oncotarget. 2015;6(39):41837–55.
Hong K-O, et al. Inhibition of Akt activity induces the mesenchymal-to-epithelial reverting transition with restoring E-cadherin expression in KB and KOSCC-25B oral squamous cell carcinoma cells. Journal of Experimental & Clinical Cancer Research. 2009;28(1):28.
Nguyen PT, et al. The FGFR1 inhibitor PD173074 induces mesenchymal-epithelial transition through the transcription factor AP-1. Br J Cancer. 2013:1532–827 (Electronic)).
Chang CC, et al. Connective tissue growth factor activates pluripotency genes and mesenchymal-epithelial transition in head and neck cancer cells. Cancer Res. 2013:1538–7445 (Electronic)).
Cheng CW, et al. Loss of GDF10/BMP3b as a prognostic marker collaborates with TGFBR3 to enhance chemotherapy resistance and epithelial-mesenchymal transition in oral squamous cell carcinoma. Mol Carcinog. 2016:1098–2744 (Electronic)).
Acknowledgements
Not applicable
Funding
This work was supported by funds from Specific University Research Grant, as provided by the Ministry of Education, Youth and Sports of the Czech Republic in the year 2018 (MUNI/A/1297/2017), by funds from the Faculty of Medicine, Masaryk University to junior researcher (Martina Raudenska), by the Grant Agency of the Czech Republic (18-03978S), by the Ministry of Health of the Czech Republic (NV18-08-00229) and by the “Center for Tumor Ecology – Research of the Cancer Microenvironment Supporting Cancer Growth and Spread” (reg. no. CZ.02.1.01/0.0/0.0/16_019/0000785) is supported by the Operational Program Research, Development and Education.
Availability of data and materials
Not applicable
Author information
Authors and Affiliations
Contributions
BP wrote the manuscript, MR conceived the structure and revised the manuscript, MM revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Peltanova, B., Raudenska, M. & Masarik, M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: a systematic review. Mol Cancer 18, 63 (2019). https://doi.org/10.1186/s12943-019-0983-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-019-0983-5