
Abstract
Background
The unambiguous identification of individual chromosomes is a key part of the genomic characterization of any species. In this respect, the development and application of chromosome banding techniques has revolutionised mammalian and especially, human genomics. However, partly because of the traditional use of chromosome squash preparations, consistent fluorescence banding has rarely been achieved in plants. Here, successful fluorescence chromosome banding has been achieved for the first time in perennial ryegrass (Lolium perenne), a forage and turf grass with a large genome and a symmetrical karyotype with chromosomes that are difficult to distinguish.
Results
Based on flame-dried chromosome preparations instead of squashes, a simple fluorescence Q-banding technique using quinacrine mustard, unambiguously identified each chromosome and enabled the development of a banded karyotype and ideogram of the species. This Q-banding technique was also shown to be compatible with sequential FISH mapping enabling labelled genes and molecular markers to be precisely assigned to specific cytogenetic bands. A technique for DAPI-banding, which gave a similar pattern to Q-banding, was also introduced. This was compatible with FISH mapping and was used to anchor a single copy gene from an earlier mapped linkage group of L. perenne, thus providing a step towards integration of the genetic and cytogenetic maps.
Conclusions
By enabling the allocation of genes mapped by other methods to physically identified chromosome positions, this work will contribute to a better understanding of genomic structures and functions in grasses.
Similar content being viewed by others

Background
The development and application of genomic methods to plant genetics and breeding involves the integration of information across the spectrum from DNA sequences to chromosomes, the units of transmission of genetic information [1]. A complete genomic analysis thus requires an ability to unambiguously identify the individual chromosomes of a species. Before 1970, the only available parameters for chromosome identification were the relative lengths and arm ratios [2]. Such parameters were not sufficient to identify chromosomes in species where chromosomes exhibited gross morphological similarities. The development of chromosome banding techniques in the 1970’s resolved this limitation to a large extent and promoted rapid advances in mammalian and human cytogenetics [3]. The patterns of longitudinal differentiation along chromosomes, produced through G-, Q- or R-banding protocols, are specific for each pair of chromosomes in a given species. This unambiguous chromosome identification has led to the development of standardized banded karyotypes incorporating a chromosome numbering system and banding nomenclature for several mammalian species including human [4], cattle [5], sheep [6] and dog [7]. As modern cytogenetics now incorporates several molecular-based technologies, fluorescence in situ hybridization (FISH), which allows direct visualisation of specific DNA sequences on chromosomes, plays an important role in bridging the gap between classical cytogenetics and molecular genetics. The application of a ‘toolbox’ of standardized banded karyotypes in combination with FISH has revolutionised structural, functional and comparative genomics of several mammalian species. On the rare occasions when the chromosome banding pattern cannot resolve chromosome pairs with similar morphologies, FISH mapping of molecular marker sequences can be applied [8].
Similar chromosome banding protocols have not yet been developed for plant species. Several variations of chromosome banding techniques have been tried with plant chromosomes [9–12] but consistent patterns necessary for sequential mapping have not been achieved [13]. It has been argued that the relatively enhanced compactness of the chromatin of plant metaphase chromosomes and/or the adherence of cytoplasmic debris over chromosome preparations obtained from traditional squash preparations obliterates the resolution of G-bands [9, 14]. A fluorescence DAPI (4’, 6-diamidino-2-phenylindole)-banding technique has recently been reported for plant chromosomes which is compatible with FISH mapping experiments using repetitive DNA sequences [15]. The banding patterns were resolved by digital deconvolution and reconstruction of 3D images of chromosomes.
Perennial ryegrass, Lolium perenne (2n = 2x = 14) (subfamily Pooideae; tribe Poeae) is native to the temperate parts of Asia, Europe and North Africa. It belongs to the same grass family as several important food crop plants including wheat (Triticum spp.), barley (Hordeum vulgare), rice (Oryza sativa), maize (Zea mays), sorghum (Sorghum bicolor), as well as the model grass, Brachypodium distachyon. It is widely cultivated for forage and turf because of its rapid establishment from seed, high yield, and good digestibility. However, perennial ryegrass has only limited variation for tolerance to biotic and abiotic stresses [16, 17]. Genomics-supported breeding strategies provide a promising approach to the improvement of the agronomic performances and stress tolerances of many crops, including L. perenne. The complexity of the perennial ryegrass genome, posed by its large size (2.6 Gbp/1Cx; [18]) and consisting of a significant proportion of repetitive DNA sequences, has hampered progress in sequencing of this genome. Nevertheless, ryegrass genomics is advancing rapidly, with the availability of a draft genome sequence [19] and several linkage maps [20–23]. Despite these advances, it has not so far been possible to unambiguously relate LGs with individual L. perenne chromosomes. A key reason is that all seven pairs of conventionally stained chromosomes of ryegrass, biarmed in morphology, display a symmetrical karyotype [24, 25] which makes their individual identification extremely difficult [1].
In this work, we have successfully obtained for the first time fluorescence Q-banding patterns using quinacrine mustard (QM) on ryegrass chromosomes prepared by an improved flame drying technique previously developed in our laboratory [26]. Importantly, the banding method is compatible with the FISH technique for regional chromosomal mapping of DNA sequences so that the banding and the FISH signals can be visualised in the same cell. These results have subsequently led us to produce a banded karyotype for the first time and develop a corresponding banded ideogram for perennial ryegrass. We have used this resource as a tool to map 18S rDNA and 5S rDNA sequences with FISH to precise chromosome bands. In addition, we have mapped a single copy gene to a L. perenne cytogenetic band, and so have demonstrated the anchorage of a linkage map marker to a chromosome region. Potential applications are discussed, including the combining of genetic and cytological information into an integrated cytogenetic map.
Results
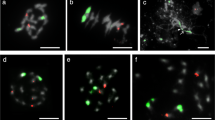
Quinacrine mustard staining displayed distinct and reproducible Q-banding patterns on flame-dried preparations of the somatic chromosomes of both diploid and haploid L. perenne. The same preparations were then subjected to FISH procedures to achieve successful regional mapping of 5S and 18S rDNA sequences (Fig. 1). The availability of large numbers of suitable cells allowed us to produce a Q-banded karyotype for L. perenne (Fig. 2). Individual chromosome pairs in each cell were identified based on morphology, FISH mapping of rDNA markers and banding patterns. Thereafter, morphometric analysis was carried out based on the 10 selected cells (Table 1). The seven pairs of diploid L. perenne chromosomes displayed a gradation in size and have been numbered according to their decreasing size order (Fig. 3). The chromosome sizes ranged from chromosome 1, with 17.93% of the total haploid set to chromosome 7, with 10.83% of the total haploid set. Based on the estimated nuclear genome size of L. perenne (2.623 Gbp/1Cx; [18]), the molecular sizes of the chromosomes ranged from 470 Mbp for chromosome 1 to 284 Mbp for chromosome 7. FISH mapping provided marker status to three chromosome pairs in the diploid L. perenne. Chromosomes 2, 3 and 5 displayed proximal hybridization of 18S rDNA representing nucleolus organizer regions (NORs) or secondary constrictions. While 18S rDNA hybridized on the long arm in chromosomes 3 and 5, it was localised on the short arm in chromosome 2 (Fig. 2). Among these three NOR carrying chromosomes, chromosome 3, with a secondary constriction on the long arm, co-localized with 5S rDNA on the short arm. In our chromosome preparations, the GC-rich NOR chromatin was often decondensed and occasionally stretched, generally with condensed flanking ends (Fig. 1). Consequently, in DAPI staining the two condensed components of the same chromosome often appeared to be separated, without any visible connection. FISH showed that these parts were joined by cloudy decondensed 18S rDNA (Figs. 1d and 4b) and that there was no chromosome breakage. In some cells the FISH signals of the highly decondensed 18S rDNA were not picked up by the camera, but they were always visible under the microscope.

Q-banding and sequential FISH mapping in L. perenne. Early metaphase cells of diploid (a, b) and haploid (c, d) L. perenne after Q-banding (a, c) and sequential FISH mapping (b, d) with 5S and 18S rDNA sequences. Dotted lines in (a) and (c) denote decondensed NORs. Bar represents 5 μm

Q-band karyotype standardization of L. perenne with sequential FISH mapping of cytogenetic markers, Representative Q-banded karyotype (grayscale) and sequential FISH mapping of 5S (red) and 18S (green) rDNA sequences. Diagrammatic representation of Q-banding on left of each chromosome. Lines across chromosomes represent centromeric positions

Q-banded ideogram of L. perenne

DAPI-banding and FISH mapping of LpGI gene in L. perenne. Metaphase cell after (a) DAPI staining and (b) FISH mapping with LpGI (red) and 18S rDNA (green) sequences. In (c), the left inset shows an example of Q-banded chromosome 2 (grayscale) with sequential hybridization of 18S rDNA. The right inset shows DAPI-banded chromosome 2 in grayscale along with LpGI (red) and 18S rDNA (green) hybridizations from two cells at different condensation levels. At the bottom right, LpGI is positioned on the banded ideogram of chromosome 2. Red arrows in (b) and (c) indicate LpGI FISH signals. Lines across chromosomes in (c) represent centromeric positions. Bar represents 5 μm
The seven chromosomes of the haploid L. perenne plant displayed identical Q-banding patterns to those of the seven pairs of the diploid plant. However, instead of three marker chromosomes, four chromosomes displayed 18S rDNA hybridization (Fig. 1) with an additional NOR being detected proximally on the long arm of chromosome 1 (Fig. 2). Regional hybridization of the 18S and/or 5S rDNA sequences on chromosomes 2, 3 and 5 was identical to that observed in the diploid plant.
All seven chromosomes from each cell of the haploid plant were identified and karyotyped as for the diploid cells. An ideogram was drawn based on the data provided in Table 1. The Q-banding patterns for all seven chromosomes were analysed from cells selected from both the diploid and haploid plants. Landmark bands were identified, and the banding patterns and their nomenclature were diagrammatically represented in the banded ideogram with 156 bands (Fig. 3). This enabled us to assign the 5S and 18S rDNA loci in L. perenne to a specific chromosome, chromosome arm and band position. The 18S rDNA sequences have been assigned to chromosome positions 1q21, 2p21, 3q21 and 5q21 while 5S rDNA was on 3p21 (Fig. 2).
To test if DAPI-banding on the flame-dried chromosome preparations could also be produced while maintaining compatibility with the FISH procedure, a modified protocol was developed to map a circadian clock-associated gene of L. perenne, GIGANTEA (LpGI), using genomic as well as cDNA sequences as probes. Although a defined DAPI differential staining was not observed on mid-metaphase chromosomes, low levels of Q-like DAPI-banding were generated in pro-metaphase and early metaphase chromosomes. Specific and identical hybridization of both the probes was observed on a chromosome arm with 18S rDNA. Using the combined banding and marker patterns, LpGI was unambiguously assigned to 2p31 (Fig. 4).
Discussion
A simple fluorescence based Q-banding technique using quinacrine mustard for plant chromosomes, which is compatible with sequential FISH mapping of cloned DNA sequences, is reported here for the first time. This technique enabled the visualization of chromosome banding patterns and FISH signals in the same cell. The longitudinal differentiation of Q-bands produced along the length of each chromosome facilitated unambiguous identification of perennial ryegrass chromosomes. Based on these results, a banded karyotype and ideogram of perennial ryegrass was developed and used as a cytogenomic tool. Based on only two individuals from different cultivars, this represents a first step, and analyses of more populations is needed before a standardized karyotype can be established.
Success was also achieved in producing DAPI-banding patterns while maintaining compatibility with FISH mapping of a single copy sequence on the same cytological preparation. While the DAPI-bands could be visualised only in pro-metaphase and early metaphase cells (Fig. 4), the banding patterns were similar to Q-banding as would be expected because both QM and DAPI are AT-specific fluorochromes [27]. Although less defined than Q-bands, the DAPI bands were sufficiently clear to identify commonality. DAPI-bands were not observed by Rocha et al. [25] who restricted their observations to metaphase cells of L. perenne. Cytochemical pre-treatments for the production of bands on fixed chromosomes may cause minor destruction or loss of chromosomal DNA [15, 28]. Such preparations may then be incompatible with FISH mapping, especially with low-copy targets. Because the Q- or DAPI-banding techniques presented here did not involve any specific pre-treatment before staining with QM or DAPI, they were compatible with FISH mapping.
Technical limitations in chromosome preparation have previously hampered progress in developing plant chromosome banding techniques. The traditional squash method causes adherence of cytoplasmic debris from clumped meristematic tissue and may have been the main barrier to generating resolvable bands on plant chromosomes [9, 29]. The flame-drying method reported previously [26], and adopted in this work, did not involve the traditional squashing of tissue. Instead, the enzymatically macerated meristematic tissue was teased into a droplet of cell suspension, given a temperature shock with chilled fixative and then flame-dried. This left the chromosomes well-spread and completely clean on the glass slide without any cytoplasmic debris. An additional advantage of the well-spread and debris-free chromosome preparation has been the enhanced sensitivity of FISH signals [30, 31]. Here, we have successfully mapped a small target sequence, LpGI, onto a specific chromosome band of L. perenne using both genomic DNA and cDNA probes.
Variability in the number of NOR loci in L. perenne has been described in earlier reports [25, 32–34]. Six NOR loci have been common and an additional NOR FISH signal has been recorded in a few reports [25, 33, 34], but eight NOR loci have not been encountered. The chromosomes carrying these sequences were either not numbered or were inconsistently numbered. In the present work, the three pairs of chromosomes in the diploid L. perenne plant consistently carrying NOR loci were designated according to the proposed karyotype. The NOR (18S rDNA) loci and the single 5S rDNA locus were then regionally mapped and assigned to cytogenetic bands. Identical assignments were made in the haploid L. perenne but with the addition of a fourth 18S rDNA signal on the largest chromosome. This was consistent with the previous observation [25] that a hemizygous 18S rDNA locus occurred on the largest chromosome.
Seven pairs of biarmed chromosomes of L. perenne, were numbered according to decreasing size order in the karyotype presented here. A combination of gross chromosome morphology, Q-banding patterns and molecular cytogenetic markers (two rDNA sequences) has helped in individualizing all seven pairs for the first time (Fig. 2). An estimation of the molecular size of each chromosome has also been presented, based on the nuclear genome size determined earlier by flow cytometry [18]. The chromosome designations given here (Table 1) are similar, but not identical, to those assigned by Rocha et al. [25]. Chromosomes 1–5 are probably the same in both classifications, but the potential confusion of chromosomes 6 and 7, which are of similar morphology and size, can now be resolved by Q-bands.
L. perenne exhibits extensive conserved synteny with the Triticeae and other members of subfamily Pooideae [21, 35]. Various gene maps of L. perenne have been developed through comparative mapping, mainly with Triticeae [20–22]. The numbering system of linkage groups (LGs) in these maps have directly been adapted from Triticeae [21, 22, 36, 37]. Due to the lack of a standardized karyotype of L. perenne, no correlation between LGs and individual chromosomes has been established so far through FISH mapping. Neither the analysis of alien substitution lines [38] nor alien introgression lines [39] has provided unambiguous identification of all L. perenne chromosomes. Indeed, as noted by Kopecky and Studer [1], only one of the seven chromosomes had so far been discriminated. GIGANTEA (LpGI) has been mapped to LG3 of L. perenne [40]. Here, this single copy gene has been precisely assigned through FISH mapping to a cytogenetic band on the short arm of chromosome 2 (Fig. 4). LG3 has been intensively analyzed by introgression mapping and the presence of an NOR as noted previously [41], was also found in the present work. Thus Q-banding combined with FISH mapping has enabled LG3 to be identified as chromosome 2 and described by its size, banding pattern and morphology. The accumulation of many such regional assignments should lead to the development of an integrative cytogenetic map. Such an integrative cytogenetic map will provide an alternative approach for analyses of recombination (cM) along the chromosome [39] and reveal the extent of unmapped genomic regions. It could also help to resolve contradictory chromosome assignments of markers [21] and LG map discrepancies [22]. Chromosomal mapping will also allow the placement of markers which cannot be genetically mapped [42].
To facilitate DNA sequencing of large grass genomes, individual chromosomes can be isolated by flow cytometry [31, 43, 44]. DNA preparations from flow-sorted chromosomes can then be used for the generation of chromosome-specific sequences and assemblies [45, 46]. Application of the chromosome banding technique presented here should provide unambiguous identification of flow sorted chromosomes and help to reveal contamination. Flow-sorted chromosomes can also be identified by FISH if appropriate markers are available [18]. However, the chromosome banding method is faster, cost effective and will also reveal the intact status of the sorted chromosomes.
Introgression of alien chromosome or chromosome segments have produced cytogenetic stocks of addition or substitution lines in a number of Pooideae, including Festulolium hybrids [38, 47, 48]. Application of the Q-banding technique in combination with sequential FISH and/or genomic in situ hybridization (GISH) should provide identification of specific chromosomes and, by extension, the nature and extent of introgression.
Transgenesis is often used by agricultural biotechnologists to introduce traits of agronomic interest into crops [49–54] but instability in the expression of the introduced transgene is common [55–57]. The transgenes may unpredictably integrate at different locations in the host genome and be subject to “position effect” variation in expression patterns [51, 58]. The chromosome banding technique developed here with the concomitant visualization of the integrated transgene through FISH should reveal the number of insertion events and the hemi- or homozygosity of each insertion. Further, the precise cytogenetic determination of transgene integration site(s) may throw light on the relationship between transgene expression pattern and the location of integration. Additionally, it should detect tissue culture-mediated chromosomal abnormalities which frequently occur in transformed plants [59, 60].
Conclusions
The Q-banding method described here has enabled the unambiguous identification of all seven chromosomes (and all 14 chromosome arms) of L. perenne. Further, the technique has been demonstrated to be compatible with FISH mapping, enabling the anchoring of a locus mapped by linkage analysis to a specific chromosome position on an identifiable chromosome arm. Similar work should quickly enable the anchoring of many more loci currently located by linkage analysis to specific chromosome positions, thus integrating cytogenetics and genetics and enabling LGs to be associated with identifiable chromosomes. Extension of the method to the close relatives of L. perenne is likely to greatly facilitate the development of addition/substitution lines and, by extension, the integration of introgression bins with known chromosome positions. The work thus provides an integrative cytogenomic resource that will contribute to a better understanding of genomic structures and functions in grasses.
Methods
Plant materials and chromosome preparation
Diploid perennial ryegrass (accession no. A10642, a late-flowering selection of New Zealand x Spanish origin) and haploid perennial ryegrass derived from anther culture [61] of cultivar Option WH-1, were obtained from the Margot Forde Forage Germplasm Centre at AgResearch Grasslands, Palmerston North, New Zealand and grown under glasshouse conditions. Actively growing root tips from single individuals of each were processed for somatic chromosome preparations according to the flame drying technique described previously [26] with minor modifications. Briefly, freshly harvested root tips were incubated with 3 mM 8-hydroxyquinoline for 2 h at 22 °C and then for 6 h at 4 °C. These were fixed in 3:1 methanol-acetic acid solution and stored at 4 °C. Root tips were washed in citrate buffer (4 mM citric acid, 6 mM sodium citrate, pH 4.8) and then macerated in 2% (w/v) cellulase (1.6% cellulase Calbiochem 515883 + 0.4% cellulase Onozuka R-10) in citrate buffer at pH 4.8 and 20% (v/v) pectinase (from Aspergillus niger in 40% glycerol, Sigma P-0690) for from 50 min to 1 h at 37 °C. After washing in citrate buffer, root tips were placed on a clean glass slide in a droplet of the buffer and apical meristematic tissue was gently extruded from the surrounding tissues under a stereomicroscope. Excess buffer was aspirated out, the meristematic tissue was dissociated into a droplet of cell suspension using fine needles and, thereafter, 1 drop of 48% acetic acid was placed on the cell suspension. After 2 min of incubation at room temperature, 2–3 drops of chilled methanol-acetic acid (3:1) fixative (stored at -20 °C) was gently placed on the slide and quickly flame-dried. Slides were screened using phase contrast optics to assess the quality of cytological preparations.
Chromosome banding, FISH mapping and karyotype analysis
For Q-banding, cytological preparations were stained with 0.002% quinacrine mustard for 30 s, rinsed with distilled water and mounted in M/15 Sorensen phosphate buffer at pH 6.8. Photomicrographs were taken immediately using appropriate Nikon filters. Coverslips were removed and the slides were rinsed with distilled water and incubated in methanol:acetic acid fixative. After air drying, the slides were ready for sequential FISH procedures.
Three DNA sequences were used as probes for FISH mapping. pTr18S (GenBank accession no. AF071069), a 1.8 kb fragment from Trifolium repens containing almost the entire 18S rDNA sequence representing the nucleolus organizer region (NOR), and pTr5S (GenBank accession no. AF072692), a 596 bp fragment encoding T. repens 5S rDNA, were labelled with either Fluor-X-dCTP (Amersham) or Cy3-dCTP (Amersham), using nick translation according to the manufacturer’s specifications. A 5.5 kb genomic fragment encompassing a circadian clock-associated gene of L. perenne, GIGANTEA (LpGI) (GenBank accession DQ534010) and its cDNA complement of 3.44 kb were labelled with Cy3-dCTP.
Sequential double target FISH using the 5S and 18S rDNA probes was performed according to a previously reported method [26] on slides previously used for Q-banding. Here, counter staining of chromosomes subjected to sequential FISH was carried out using DAPI (1 μg/ml in McIlvaine’s buffer, pH 7) for 6 min before mounting in Vectashield (Vector Laboratories). The whole process facilitated visualisation of banding as well as probe localisation in the same cell, allowing regional assignments of DNA probes in relation to chromosome bands. FISH mapping of each LpGI probe was performed on separate preparations in combination with 18S rDNA according to the earlier protocol except that these preparations were not subjected to QM staining. In this case, after the FISH procedures, DAPI-banding was achieved by staining the chromosomes with DAPI (0.5 μg/ml in McIlvaine’s buffer, pH 7) for 30 s before mounting in Vectashield (Vector Laboratories). Photomicrographs were taken using a Zeiss monochrome CCD camera on a Nikon epifluorescence microscope Microphot-SA and images were processed with ISIS FISH Imaging System (MetaSystems, Germany).
Ten well spread and good quality Q-banded prometaphase to mid-metaphase cells from each of the diploid and haploid plants were selected for analysis. After chromosome identification based on gross morphology, banding patterns and molecular markers, individual karyotypes were prepared. Morphometric analysis (Table 1) based on 10 cells selected from the diploid plant was carried out according to an established method [2] The banded ideogram was drawn based on morphometric data (Table 1). Chromosomes were arranged in decreasing size order. Relative staining intensity of the bands was represented in the ideogram using one of the three shades from white (no fluorescence), light stipple (medium fluorescence) to black (bright fluorescence). The designation of landmarks and nomenclature of chromosomal regions and bands followed the recommendations of ISCN (1985) [4] for human chromosomes. Briefly, short and long chromosome arms were designated p and q, respectively. Chromosome arms were divided into regions and bands which were numbered outward from the centromere. Consistent and conspicuous banding landmarks were used for delineating the regions. A landmark band was designated as the first band of the region next to the landmark. Each region was divided into bands depending on the staining patterns of the region. A specific chromosomal band was designated by the chromosome number from the standardised karyotype, the arm symbol, the region number and the band number within that region written in continuation without spaces. Occasionally, bands were subdivided at prometaphase or late prophase stages. In these cases, decimal points were used to designate the sub-bands.
References
Kopecký D, Studer B. Emerging technologies advancing forage and turf grass genomics. Biotechnol Adv. 2014;32:190–9.
Levan A, Fredga K, Sandberg AA. Nomenclature for centromeric position on chromosomes. Hereditas. 1964;52:201–20.
Hsu T-C. Human and Mammalian Cytogenetics: An Historical Perspective. New York: Springer; 1979.
ISCN. An international system for human cytogenetic nomenclature: report of the Standing Committee on Human Cytogenetic Nomenclature. Harnden DG, Klinger HP, editors. Basel: Karger; 1985.
DiBerardino D, Hayes H, Fries R, Long S. International system for cytogenetic nomenclature of domestic animals. The Second International Conference on Standardization of Domestic Animal Karyotypes, INRA, Jouy-en Josas, France, 22nd-26th May, 1989. Cytogenet Cell Genet. 1990;53:65–79.
Ansari HA, Bosma AA, Broad TE, Bunch TD, Long SE, Maher DW, Pearce PD, Popescu CP. Standard G-, Q-, and R-banded ideograms of the domestic sheep (Ovis aries): homology with cattle (Bos taurus). Report of the Committee for the Standardization of the Sheep Karyotype. Cytogenet Cell Genet. 1999;87:134–42.
Świtoński M, Reimann N, Bosma AA, Long S, Bartnitzke S, Pieńkowska A, Moreno-Milan MM, Fischer P. Report on the progress of standardization of the G-banded canine (Canis familiaris) karyotype. Chromosome Res. 1996;4:306–9.
Breen M, Bullerdiek J, Langford CF. The DAPI banded karyotype of the domestic dog (Canis familiaris) generated using chromosome-specific paint probes. Chromosome Res. 1999;7:401–6.
Griesbach RJ. The use of confocal microscopy to study chromosome banding in Ornithogalum. J Hered. 1998;89:184–8.
Murata M, Orton TJ. G-band-like differentiation in mitotic prometaphase chromosomes of celery. J Hered. 1984;75:225–8.
Muravenko OV, Amosova AV, Samatadze TE, Popov KV, Poletaev AI, Zelenin AV. 9-Aminoacridine: an efficient reagent to improve human and plant chromosome banding patterns and to standardize chromosome image analysis. Cytometry Part A. 2003;51:52–7.
Song YC, Liu LH, Ding Y, Tian XB, Yao Q, Meng L, He CR, Xu MS. Comparisons of G-banding patterns in six species of the Poaceae. Hereditas. 1994;121:31–8.
Chen R, Song W, Li X, An Z. Chromosome G-banding in plants by inducing with trypsin and urea. Cell Res. 1994;4:79–87.
Greilhuber J. Why plant chromosomes do not show G-bands. Theor Appl Genet. 1977;50:121–4.
Liu J, She C, Hu Z, Xiong Z, Liu L, Song Y. A new chromosome fluorescence banding technique combining DAPI staining with image analysis in plants. Chromosoma. 2004;113:16–21.
Humphreys M, Feuerstein U, Vanderwalle M, Baert J. Ryegrasses. In: Boller B, Posselt UK, Veronesi F, editors. Fodder crops and amenity grasses: handbook of plant breeding. New York: Springer; 2010. p. 221–60.
Williams WM, Easton HS, Jones CS. Future options and targets for pasture plant breeding in New Zealand. N Z J Agric Res. 2007;50:223–48.
Kopecký D, Havránková M, Loureiro J, Castro S, Lukaszewski AJ, Bartoš J, Kopecká J, Doležel J. Physical distribution of homoeologous recombination in individual chromosomes of Festuca pratensis in Lolium multiflorum. Cytogenet Genome Res. 2010;129:162–72.
Byrne SL, Nagy I, Pfeifer M, Armstead I, Swain S, Studer B, Mayer K, Campbell JD, Czaban A, Hentrup S, et al. A synteny-based draft genome sequence of the forage grass Lolium perenne. Plant J. 2015;84:816–26.
Faville MJ, Vecchies AC, Schreiber M, Drayton MC, Hughes LJ, Jones ES, Guthridge KM, Smith KF, Sawbridge T, Spangenberg GC, et al. Functionally associated molecular genetic marker map construction in perennial ryegrass (Lolium perenne L.). Theor Appl Genet. 2004;110:12–32.
Pfeifer M, Martis M, Asp T, Mayer KFX, Lübberstedt T, Byrne S, Frei U, Studer B. The perennial ryegrass GenomeZipper: targeted use of genome resources for comparative grass genomics. Plant Physiol. 2013;161:571–82.
King J, Thomas A, James C, King I, Armstead I. A DArT marker genetic map of perennial ryegrass (Lolium perenne L.) integrated with detailed comparative mapping information; comparison with existing DArT marker genetic maps of Lolium perenne, L. multiflorum and Festuca pratensis. BMC Genomics. 2013;14:437.
Studer B, Byrne S, Nielsen RO, Panitz F, Bendixen C, Islam MS, Pfeifer M, Lübberstedt T, Asp T. A transcriptome map of perennial ryegrass (Lolium perenne L.). BMC Genomics. 2012;13:140.
Malik CP, Thomas PT. Karyotypic studies in some Lolium and Festuca species. Caryologia. 1966;19:167–96.
Rocha LC, de Oliveira Bustamante F, Silveira RAD, Torres GA, Mittelmann A, Techio VH. Functional repetitive sequences and fragile sites in chromosomes of Lolium perenne L. Protoplasma. 2015;252:451–60.
Ansari HA, Ellison NW, Reader SM, Badaeva ED, Friebe B, Miller TE, Williams WM. Molecular cytogenetic organization of 5S and 18S-26S rDNA loci in white clover (Trifolium repens L.) and related species. Ann Bot. 1999;83:199–206.
Saitoh Y, Laemmli UK. Metaphase chromosome structure: bands arise from a differential folding path of the highly AT-rich scaffold. Cell. 1994;76:609–22.
Chaves R, Adega F, Santos S, Guedes-Pinto H, Heslop-Harrison JS. In situ hybridization and chromosome banding in mammalian species. Cytogenet Genome Res. 2002;96:113–6.
Anderson LK, Stack SM, Mitchell JB. An investigation of the basis of a current hypothesis for the lack of G-banding in plant chromosomes. Exp Cell Res. 1982;138:433–6.
Feng J, Liu Z, Cai X, Jan CC. Toward a molecular cytogenetic map for cultivated sunflower (Helianthus annuus L.) by landed BAC/BIBAC clones. G3 Genes Genome Genet. 2013;3:31–40.
Kopecký D, Martis M, Číhalíková J, Hřibová E, Vrána J, Bartoš J, Kopecká J, Cattonaro F, Stočes Š, Novák P, et al. Flow sorting and sequencing meadow fescue chromosome 4 F. Plant Physiol. 2013;163:1323–37.
Inda LA, Wolny E. Fluorescent in situ hybridization of the ribosomal RNA genes (5S and 35S) in the genus Lolium: Lolium canariense, the missing link with Festuca? An Jard Bot Madrid. 2013;70:97–102.
Ksia̧zczyk T, Taciak M, Zwierzykowski Z. Variability of ribosomal DNA sites in Festuca pratensis, Lolium perenne, and their intergeneric hybrids, revealed by FISH and GISH. J Appl Genet. 2010;51:449–60.
Thomas HM, Harper JA, Meredith MR, Morgan WG, Thomas ID, Timms E, King IP. Comparison of ribosomal DNA sites in Lolium species by fluorescence in situ hybridization. Chromosome Res. 1996;4:486–90.
Hasterok R, Marasek A, Donnison IS, Armstead I, Thomas A, King IP, Wolny E, Idziak D, Draper J, Jenkins G. Alignment of the genomes of Brachypodium distachyon and temperate cereals and grasses using bacterial artificial chromosome landing with fluorescence in situ hybridization. Genetics. 2006;173:349–62.
Jones ES, Mahoney NL, Hayward MD, Armstead IP, Jones JG, Humphreys MO, King IP, Kishida T, Yamada T, Balfourier F, et al. An enhanced molecular marker based genetic map of perennial ryegrass (Lolium perenne) reveals comparative relationships with other Poaceae genomes. Genome. 2002;45:282–95.
Sim S, Chang T, Curley J, Warnke SE, Barker RE, Jung G. Chromosomal rearrangements differentiating the ryegrass genome from the Triticeae, oat, and rice genomes using common heterologous RFLP probes. Theor Appl Genet. 2005;110:1011–9.
Harper J, Armstead I, Thomas A, James C, Gasior D, Bisaga M, Roberts L, King I, King J. Alien introgression in the grasses Lolium perenne (perennial ryegrass) and Festuca pratensis (meadow fescue): the development of seven monosomic substitution lines and their molecular and cytological characterization. Ann Bot. 2011;107:1313–21.
King J, Armstead I, Harper J, Ramsey L, Snape J, Waugh R, James C, Thomas A, Gasior D, Kelly R, et al. Exploitation of interspecific diversity for monocot crop improvement. Heredity. 2013;110:475–83.
Gagic M, Faville M, Kardailsky I, Putterill J. Comparative genomics and functional characterisation of the GIGANTEA gene from the temperate forage perennial ryegrass Lolium perenne. Plant Mol Biol Report. 2015;33:1098–106.
King J, Armstead IP, Donnison IS, Harper JA, Roberts LA, Thomas H, Ougham H, Thomas A, Huang L, King IP. Introgression mapping in the grasses. Chromosome Res. 2007;15:105–13.
Harper LC, Cande WZ. Mapping a new frontier; development of integrated cytogenetic maps in plants. Funct Integr Genomics. 2000;1:89–98.
Doležel J, Kubaláková M, Paux E, Bartoš J, Feuillet C. Chromosome-based genomics in the cereals. Chromosome Res. 2007;15:51–66.
Šafář J, Bartoš J, Janda J, Bellec A, Kubaláková M, Valárik M, Pateyron S, Weiserová J, Tušková R, Číhalíková J, et al. Dissecting large and complex genomes: flow sorting and BAC cloning of individual chromosomes from bread wheat. Plant J. 2004;39:960–8.
Molnár I, Vrána J, Farkas A, Kubaláková M, Cseh A, Molnár-Láng M, Doležel J. Flow sorting of C-genome chromosomes from wild relatives of wheat Aegilops markgrafii, Ae. triuncialis and Ae. cylindrica, and their molecular organization. Ann Bot. 2015;116:189–200.
Tanaka T, Kobayashi F, Joshi GP, Onuki R, Sakai H, Kanamori H, Wu J, Šimková H, Nasuda S, Endo TR, et al. Next-generation survey sequencing and the molecular organization of wheat chromosome 6B. DNA Res. 2014;21:103–14.
Kopecký D, Lukaszewski AJ, Doležel J. Meiotic behaviour of individual chromosomes of Festuca pratensis in tetraploid Lolium multiflorum. Chromosome Res. 2008;16:987–98.
Kopecký D, Lukaszewski AJ, Doležel J. Cytogenetics of Festulolium (Festuca x Lolium hybrids). Cytogenet Genome Res. 2008;120:370–83.
Choi HW, Lemaux PG, Cho MJ. Use of fluorescence in situ hybridization for gross mapping of transgenes and screening for homozygous plants in transgenic barley (Hordeum vulgare L.). Theor Appl Genet. 2002;106:92–100.
Leggett JM, Perret SJ, Harper J, Morris P. Chromosomal localization of cotransformed transgenes in the hexaploid cultivated oat Avena sativa L. using fluorescence in situ hybridization. Heredity. 2000;84:46–53.
Moeller L, Wang K. Engineering with precision: tools for the new generation of transgenic crops. Bioscience. 2008;58:391–401.
Pedersen C, Zimny J, Becker D, Jähne-Gärtner A, Lörz H. Localization of introduced genes on the chromosomes of transgenic barley, wheat and triticale by fluorescence in situ hybridization. Theor Appl Genet. 1997;94:749–57.
Salvo-Garrido H, Travella S, Billiam LJ, Harwood WA, Snape JW. The distribution of transgene insertion sites in barley determined by physical and genetic mapping. Genetics. 2004;167:1371–9.
Svitashev S, Ananiev E, Pawlowski WP, Somers DA. Association of transgene integration sites with chromosome rearrangements in hexaploid oat. Theor Appl Genet. 2000;100:872–80.
Iyer LM, Kumpatla SP, Chandrasekharan MB, Hall TC. Transgene silencing in monocots. Plant Mol Biol. 2000;43:323–46.
Shou H, Frame BR, Whitham SA, Wang K. Assessment of transgenic maize events produced by particle bombardment or Agrobacterium-mediated transformation. Mol Breed. 2004;13:201–8.
Travella S, Ross SM, Harden J, Everett C, Snape JW, Harwood WA. A comparison of transgenic barley lines produced by particle bombardment and Agrobacterium-mediated techniques. Plant Cell Rep. 2005;23:780–9.
Tang W, Newton RJ, Weidner DA. Genetic transformation and gene silencing mediated by multiple copies of a transgene in eastern white pine. J Exp Bot. 2007;58:545–54.
Barro F, Martin A, Cabrera A. Transgene integration and chromosome alterations in two transgenic lines of tritordeum. Chromosome Res. 2003;11:565–72.
Choi HW, Lemaux PG, Cho MJ. Increased chromosomal variation in transgenic versus nontransgenic barley (Hordeum vulgare L.) plants. Crop Sci. 2000;40:524–33.
Hussain SW, Richardson K, Faville M, Woodfield D. Production of haploids and double haploids in annual (Lolium multiflorum) and prennial (L. perenne) ryegrasses. In: Mercer CF, editor. Advances in Pasture Plant Breeding: Papers from the 13th Australasian Plant Breeding Conference, 18-21 April 2006, Christchurch, New Zealand. Dunedin: New Zealand Grassland Association; 2006. p. 45–8.
Acknowledgements
The contributions of Joy Dick and Jaspreet Singh Sidhu to improving the manuscript are gratefully acknowledged. This work was supported by the New Zealand Ministry of Business, Innovation and Employment (contract no. C10X1202).
Availability of data and materials
The sequence data of the DNA probes have been submitted to GenBank and the accession numbers are provided in the manuscript. Seeds of the plant material and the raw data used for the morphometric analysis are available from AgResearch.
Authors’ contributions
HAA designed the study with the help of WMW and GTB. HAA performed experiments, analysed the data and co-wrote the manuscript. NWE isolated the rDNA and labelled all the probes for FISH. WMW co-wrote the manuscript and NWE provided major help in improving the manuscript. SAB prepared genomic and cDNA constructs for the LpGI gene to be used as probes for FISH. SWH developed the haploid ryegrass plant and provided it for experimental use. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ansari, H.A., Ellison, N.W., Bassett, S.A. et al. Fluorescence chromosome banding and FISH mapping in perennial ryegrass, Lolium perenne L.. BMC Genomics 17, 977 (2016). https://doi.org/10.1186/s12864-016-3231-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-016-3231-z