
Abstract
Background
Spinosad is a macrolide antibiotic produced by Saccharopolyspora spinosa with aerobic fermentation. However, the wild strain has a low productivity. In this article, a computational guided engineering approach was adopted in order to improve the yield of spinosad in S. spinosa.
Results
Firstly, a genome-scale metabolic network reconstruction (GSMR) for S.spinosa based on its genome information, literature data and experimental data was extablished. The model was consists of 1,577 reactions, 1,726 metabolites, and 733 enzymes after manually refined. Then, amino acids supplying experiments were performed in order to test the capabilities of the model, and the results showed a high consistency. Subsequently, transhydrogenase (PntAB, EC 1.6.1.2) was chosen as the potential target for spinosad yield improvement based on the in silico metabolic network models. Furthermore, the target gene was manipulated in the parent strain in order to validate the model predictions. At last, shake flask fermentation was carried out which led to spinosad production of 75.32 mg/L, 86.5% higher than the parent strain (40.39 mg/L).
Conclusions
Results confirmed the model had a high potential in engineering S. spinosa for spinosad production. It is the first GSMM for S.spinosa, it has significance for a better understanding of the comprehensive metabolism and guiding strain designing of Saccharopolyspora spinosa in the future.
Similar content being viewed by others

Background
Spinosad is a macrolide antibiotic produced by Saccharopolyspora spinosa with aerobic fermentation. Due to its unique chemical structure and mechanism, it can control diamondback moth; beet armyworm and other lepidopteron pests effectively, while have non-toxic to mammals and birds [1]. Therefore spinosad has been considered to be the most effective biological pesticides after avermectin, becoming a new hot issue in the research and development of biological pesticides. The compounds got the "Presidential Green Chemistry Challenge Award" of the United States in 1999, because it has the chemical pesticide’s fast-acting and the biopesticide’s safety, low residual properties. So its derivative got the award again in 2008 [2].
Spinosyns A and D, the two major components in the S. spinosa fermentation,are defined as spinosad [3]. It mainly contains a 21-carbon tetracyclic with two deoxysugars: tri-O-methylated rhamnose and forosamine [4]. Waldron firstly analyzed the proposed spinosyn biosynthetic pathway [5], In recent years, the spinosad biosynthetic pathway has been clarified more accuracy: SpnA, spnB, spnC, spnD, and spnE responsible for type I polyketide synthase; spnF,spnJ, spnL, and spnM for modifying the polyketide synthase product [6]; spnG, spnH, spnI, and spnK for rhamnose attachment and methylation [7]; spnP, spnO, spnN, spnQ, spnR, and spnS for forosamine biosynthesis; gtt, gdh, epi, and kre for rhamnose biosynthesis [8] and beside the spinosad gene cluster four genes ORF-L16, ORF-R1, and ORF-R2, have no effect on spinosad biosynthesis.
The wild stain of Saccharopolyspora spinosa has low spinosad productivity, so lots of work has been done in strain modification. Three strategies have been mainly used in the spinosad improvement: (a) Traditional physical and chemical mutagenesis [9]; (b) Optimization of fermentation process [10, 11]; (c) Improving the spinosad production by the genetic engineering method. Pan [12] got a over threefold spinosad production by introducing the additional gtt and gdh genes under the control of PermE* promoter. Tang [13] increased the spinosyn production by 3.8-fold through overexpression of the spinosyn biosynthetic genes participating in the conversion of the cyclized polyketide to spinosyn, obtained by direct cloning via Red/ET recombination. Xue [14] duplicated spnP, spnQ, spnN, spnQ, spnR, spnS, spnK, gtt, gdh and kre genes under the control of PermE* promoter, spinosad production was a 5.0-fold enhancement compared with the wild-type S. spinosa. However, the spinosad is a secondary metabolite which has complex metabolic pathways. It is difficult to satisfy the industrial demands if we only pay attention to a few gene modifications.
In recent years, more and more whole genomes have been sequenced with the development of high-throughput sequencing technology and reduction of sequencing prices. It is a hot topic to study how to use such a large genome database. The genome-scale metabolic network (GSM) constructed based on genome annotation information provide an integral level to review organism metabolism. Now Genome-Scale-Metabolic-Model (GSMM) reconstruction technology has a great progress no matter in speed but quality [15, 16]. At present, many articles has elaborated GSMM establishment process [17, 18], these provided foundation to build a precision standardized GSMM. GSMM is widely used to aid strain transformation in metabolic engineering as a systems biology tool [19, 20]. Brochado [21]used GSMM successfully predict several genes targets in Bread yeast, which increased the vanillin yield about 5-fold. Huang used the GSMM of Streptomyces tsukubaensis to improve the FK506 production which led to a 1.47 fold increase [22]. The similar way was also used to increase the expression of the green fluorescent protein in lactococcus lactis[23]. GSMM reconstruction is a time consuming work, the metabolic reconstruction for secondary metabolite producing still requires extensive manual refinement, because the lack of genome annotation information.
There were no reports on the refined GSMM for Saccharopolyspora spinosa. In this work, the first manually refined GSMM for Saccharopolyspora spinosa was established. The amino acid supplementation experiments were conducted to verify the module. Then a potential target gene transhydrogenase was assessed. Overexpression of this gene led to 86.5% higher spinosad yield than that of the wild stain.
Results and discussion
Character of the GSMM
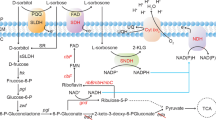
The first GSMM for Saccharopolyspora spinosa using a highthroughput protocol was constructed. The GSMM contained 1577 metabolites and 1736 reactions and 733 enzymes (Additional file 1 shows the network in more detail). Given that the spinosad was produced alongside biomass, 42 reactions were required to fill the model gaps. Major metabolic pathways for S.spinosa, including the glycolytic pathway, pentose phosphate pathway (PPP), tricarboxylic acid (TCA) cycle, and spinosad biosynthesis, are illustrated in Figure 1. Additionally five subsystems were divided, containing: gluconeogenesis, amino acid synthesis pathway, fatty acid synthesis pathway, phospholipid synthesis pathways and nucleotide synthesis pathways. The Character was summarized in Table 1.

Schematic diagram of metabolic pathways for S.spinosa. The NADH/NADPH generation and consumption are showed in red. The enhanced pathway is showed in blue (solid line represents gene overexpression, dotted line means possible enhanced pathways). The weakened pathway is showed in purple.
Amino acid addition in silico simulation and experiments
To validate the spinosad biosynthetic pathway, we used the GSMR model to predict the potential benefit of supplementing the media with individual amino acid (AA) in terms of spinosad production. Eight AAs (Leu, Gly, Met, Ala, Asp, Cys, Lys, Pro) were individually added to study the effect on spinosad production. The experiments and in silico simulation results were showed respectively in Figures 2B and A. The experiment dates showed that Gly was the best amino acid to spinosad production and following Leu, Ala. Pro had a negative effect on spinosad production. The remaining four amino acids (Asp, Met, Cys, Lys) displayed no significant effects on spinosad production.

Amino acids supplement experiment. A: In silico simulations of the effect on spinosad biosynthesis after supplementing individual amino acid. B: Spinosad production after supplementing each individual amino acid.
Amino acids play an important role in antibiotic synthesis and cell growth in the fermentation process as important biological small molecules. AA can directly participate in the biosynthesis of antibiotics as the precursor [24, 25]; AA or its peptide structure influence the biosynthesis of antibiotics by regulating the activity of the enzymes in the pathway as activators or inhibitors and its hydrolysates can change microbial growth environment by altering the PH in the medium. The main product in glycine metabolism is acetyl-COA which is the precursor of spinosad [26], this may explain the results in our work. Hinnebusch [27] proved that the GCN4 gene which encodes a positive regulator of unlinked amino acid biosynthetic genes in yeast is itself regulated by amino acid availability and that the regulation occurs at the translational level. Proline inhibit the spinosad synthesis may due to it or its metabolites participate in the regulation process. The GSMM in our work has no regulation systems, so the result was not consistent with the experiments.
Impact of PntAB gene on spinosad synthesis
Spinosad is a secondary metabolite. Energy and reducing power NADPH were required in its synthesis process. Cellular reducing power level, such as NADPH/NADP+ and NADH/NAD+, has an important role in the metabolic regulation in the cell. Therefore the reaction R00112 (NADPH + NAD+ = NADP+ + NADH) was used as control variables, the spinosad synthesis reaction as a target reaction to study the effect of transhydrogenase activity on spinosad synthesis. The results were exhibited in Figure 3A.

The effect of reducing power level on spinosad synthesis. A: Impact of transhydrogenase activity on spinosad synthesis (in silico) B: The experimental results of shake flask fermentation.
In the Figure 2A, when the reaction R00112 flux is 0, the spinosad synthesis rate didn’t reach the peak. When the reaction flux is positive (NADPH → NADH), the spinosad synthesis rate decreased. In contrast, the spinosad synthesis rate climb up and then decline when the the reaction flux is negative. Further conclude can be obtained, spinosad was produced in the stationary phase, at this phase, transhydrogenase in the cell kept the balance of the reducing power(NADPH/NADH), but the balance point was not the best for spinosad synthesis. In order to prove the simulation results, pntAB was over expressed, the yield of spinosad in S.spinosa 261P increased to 37.9 mg/L which was little higher than the wild strain(34.4 mg/L). Then 0.1% rhamnose was added, the yield increased to 75.32 mg/L which was 0.86-fold higher than that in the wild-type S. spinosa (40.39 mg/L).
Efficient regeneration of NADPH which mainly comes from PPP and TCA pathways plays an important role in biotransformation processes. The biotransformation is often limited by cofactor regeneration because cells would rather grow better than intend to sustain the biotransformation [28, 29]. The NADPH regeneration system needs to be enhanced in order to satisfy biotransformation. Three ways were modified to increase the NADPH level in the cell including oxidative part of the pentose phosphate pathway (PPP) [30, 31], the tricarboxylic acid cycle (TCA) [32], and the transhydrogenases system. Overexpression of pntAB in E. coli got a 3.5-fold conversion of acetophenone to (R)-phenylethanol [33]. The similar way was conducted in Corynebacterium glutamicum to get a high yield of L-lysine [34]. Bastian [35] obtained a 6.5-fold enhancement of isobutanol production under anaerobic conditions in E. coli by overexpression of pntAB. In our work, two reasons may explain the results that the engineered strain has a low production in general fermentation medium: Firstly, spinosad is a secondary metabolite and it has a low synthesis rate; Secondly, NADPH was mainly used in biomass synthesis.
Rhamnose is both the precursor of spinosad and the cell wall ingredient, but the Saccharopolyspora spinosa only have one set of rhamonose gene [8, 36]. The yield of spinosad had a 3.5-fold enhancement by duplication of the rhamnose biosynthetic genes (gtt and gdh) in the previous study [14]. So the rhamnose plays an important role in spinosad synthesis. The rhamnose in the fermentation medium can accelerate the use of pseudoaglycones (PSA). Then PSA synthesis made more NADPH flow into spinosad synthesis. Reducing power balance changed by over expressing pntAB, more NADPH was produced from NADH. When the rhamnose was added, more NADPH flowed into spinosad synthesis pathway, so the engineered strain had a high productivity in rhamnose medium.
Conclusions
In this work, the first Genome-scale metabolic network of Saccharopolyspora spinosa was constructed following a systematic workflow. Then it was validated by amino acids supplementation experiments. Finally based on the GSMM, PntAB was identified as the target gene. The strain was engineered according to the predicted target. Fermentation characterization of the engineered strain showed the improved capacities of spinosad production. The final yield of spinosad is 75.32 mg/L which is increased by 86.5% than the parental strain. Our results show the genome-scale metabolic network model is a powerful tool to guide strain engineering towards improved bio-production in S.spinosa, as well as other microorganisms.
Materials and methods
Genome-scale metabolic network reconstruction
The genome of Saccharopolyspora spinosa was sequenced and annotated in 2011 by Yuanlong Pan [37], the results can be got in NCBI [38]. The complete sequence of the 8.809 Mb Saccharopolyspora spinosa NRRL 18395 genome has a 67.94% GC content. The model reconstruction followed the standard protocols described previously [17]. The process of the genome-scale metabolic network reconstruction for the Saccharopolyspora spinosa can be described as follows:
Firstly, a draft reconstruction was created. The necessary information, containing genome annotation information and the biochemistry information of the enzymes, can be searched in KEGG and NCBI. The following factors were included in the draft: (1) the basic reaction information (ID, name, equation and reversibility), (2) the enzymes and genes relevant to particular reaction. (3) the subsystems. All the information above was saved in an Excel which contains 1830 reactions and 1849 metabolites.
Secondly, refine the draft model got above. Many reactions were redundant in the draft while some necessary ones were absent, so a lot of work was done in the refine process. The reactions and metabolites IDs may different in different database, only one ID was remained; The reactions which synthesize or modify the macromolecules were replaced by the biomass formulation reactions; Only one form of the multi-step reaction was remained in the model. For example, the overall reaction R004385 was kept while its sub-step reactions R00174 were removed; At last, reactions’ reversibility and cofactors were determined and verified.
At the last stage, spontaneous reactions, exchange reactions, transport reactions, biomass reactions and spinosad synthesis reactions were added to get a complete GSMM. Biomass formation was mainly based on the chemicals of the cell. The reactions containing the metabolites which were verified to be taken up from the medium and the metabolite which can diffuse through the membranes were added as the transport reaction. Exchange reactions were added for all extracellular metabolites, which represented the system boundaries.
Biomass equation
The biomass equation information was mainly got from available literature date. It is divided into proteins, RNA, DNA, lipid cell wall peptidoglycanpolysaccharide muramic acid. DNA and RNA composition were determined based on genomic data from the Saccharopolyspora spinosa genome. Protein composition was determined based on open reading frames (ORFs). Other components were adapted from S. coelicolor[39]. The biomass composition is described in detail in the Additional file 2.
Constraint-Based Flux Analysis
Constraint-Based Flux Analysis (FBA) has a wildly use in flux distribution calculations [40, 41]. The resulting model was analyzed using Constraint-Based Reconstruction and Analysis (COBRA). Robustness prediction was performed using the COBRAToolbox-2.0 in MATLAB, with GLPK and CPLEX as the optimization programming solvers [42]. The stoichiometric matrix (S) and reaction flux constrains were extracted from the SBML file. At the model curation process, FBA was used to fill the gaps until the model can use the nature medium to synthesize biomass and spinosad. Robustness [43] analysis is an algorithm based on FBA, the flux through a selected reaction is varied and the optimal objective value is calculated as a function of this flux. This method can be used to simulate the influence of the biological metabolic state caused by internal or external disturbance.
Bacterial strains, plasmids, media and culture conditions
Escherichia coli DH5α was used for all plasmid constructions and amplification. E. coli ET-12567 was used as the door strain in biparental intergeneric conjugations. S. spinosa ATCC49460 was used as the parent strain. The pOJ261, which is pOJ260 with ermE* promoter, was used as expression plasmid. The ermE* promoter was digested from pIB139. Plasmids and stains used in this study are summarized in Table 2.
E. coli strains were cultured in Luria-Bertani (LB) medium at 37°C. The slant and seed medium contained (g/L): trypticase soy broth, 30; yeast extract, 3; MgSO4 · 7H2O, 2; glucose, 10; and maltose, 4, pH 7.2. In the slant medium 20 g/L agar was added. The fermentation medium contained (g/L): glucose, 68; cottonseed flour, 22; peptone C, 25; corn seed liquor, 14.5; methyloleate, 40; and CaCO3, 5, pH 7.2. ABB13 medium contained (g/L): soy peptone, 5; soluble starch; CaCO3, 3; MOPS, 2.1; agar 20.
Strains were cultured on a 220 rpm rotary shaker at 29°C in a 250 mL flask containing 30 mL of seed medium for 3 days. Then 3 mL of seed medium was injected into 30 mL fermentation media in a 250 mL flask. Strains were cultured for 9 days at 29°C on a 220 rpm rotary shaker. The fermentations were run in triplicate [14].
Amino acids addition
After 72 h fermentation, 0.05% of amino acids (Leu, Gly, Met, Ala, Asp, Cys, Lys, Pro) Standards was added by 0.22 μ filter membrane filtration.
Gene cloning, plasmid construction and transformation
General DNA manipulation was performed according to the standard protocols [46]. PntAB was amplified from genomic DNA of S. spinosa using primer pairs of F-CCTATCTAGA CGCCGGACAAGGACGl-XbaI,R-GGATCCATATG ACCTCTCCGGAGAGC-NdeI(The italics parts represent the restriction enzyme cutting site). The PCR product of PntAB was digested by Ndel and Xbal. Then, it was cloned into pOJ261 which was also digested by Ndel and Xbal to get pOJ261P.
The constructed plasmid was introduced into S. spinosa strains by conjugation from E. coli ET12567 and homologous recombination into the chromosome as described as [44].The recombination stain S.spinosa 261P was confirmed by PCR amplification with vector primers V-F:CCGTGATTTTGTAGCCCTGG and V-R: GGCCTACTTCA CCTATCCTGC, and the result was positive.
Analysis of spinosad biosynthesis in fermentation cultures
Spinosad in the fermentation broth was determined as described [10]. Every level in the experiment was repeated three times, so the figures in the article were the mean value of the experiment, Standard deviations were calculated from triplicate flasks.
References
Kirst HA, Michel KH, Mynderase JS, Chio EH, Yao RC, Nakasukasa WM, Boeck LD, Occlowitz JL, Paschal JW, Deeter JB, Thompson GD: Discovery, isolation, and structure elucidation of a family of structurally unique, fermentation-derived tetracyclic macrolides. Am Chem Soc. 1992, 504: 214-214.
Kirst HA: The spinosyn family of insecticides: realizing the potential of natural products research. J Antibiot. 2010, 63: 101-111. 10.1038/ja.2010.5.
Thompson GD, Dutton R, Sparks TC: Spinosad–a case study: an example from a natural products discovery programme[J]. Pest Manag Sci. 2000, 56: 696-702. 10.1002/1526-4998(200008)56:8<696::AID-PS182>3.0.CO;2-5.
Donnelly TH, Shergold JH, Southgate PN: Anomalous geochemical signals from phosphatic Middle Cambrian rocks in the southern Georgina Basin, Australia[J]. Sedimentology. 1988, 35: 549-570. 10.1111/j.1365-3091.1988.tb01235.x.
Waldron C, Matsushima P, Rosteck PR, Broughton MC, Turner J, Madduri K, Crawford KP, Merlo DJ, Baltz RH: Cloning and analysis of the spinosad biosynthetic gene cluster of Saccharopolyspora spinosa [J]. Chem Biol. 2001, 8: 487-499. 10.1016/S1074-5521(01)00029-1.
Kim HJ, Ruszczycky MW, Choi S, Liu Y, Liu H: Enzyme-catalysed 4+ 2 cycloaddition is a key step in the biosynthesis of spinosyn A[J]. Nature. 2011, 473: 109-112. 10.1038/nature09981.
Kim HJ, White-Phillip JA, Ogasawara Y, Shin N, Isiorho EA, Liu HW: Biosynthesis of spinosyn in Saccharopolyspora spinosa: synthesis of permethylated rhamnose and characterization of the functions of SpnH, SpnI and SpnK[J]. J Am Chem Soc. 2010, 132: 2901-2903. 10.1021/ja910223x.
Madduri K, Waldron C, Merlo DJ: Rhamnose biosynthesis pathway supplies precursors for primary and secondary metabolism in Saccharopolyspora spinosa[J]. J Bacteriol. 2001, 183: 5632-5638. 10.1128/JB.183.19.5632-5638.2001.
Liang Y, Lu WY, Wen JP: Improvement of Saccharopolyspora spinosa and the kinetic analysis for spinosad production. Appl Biochem Biotechnol. 2009, 152: 440-448. 10.1007/s12010-008-8281-5.
Strobel RJ, Nakatsukasa WM: Response surface methods for optimizing Saccharopolyspora spinosa, a novel macrolide producer. J Ind Microbiol. 1993, 11: 121-127. 10.1007/BF01583684.
JIN Z, CHENG X, CEN P: Effects of Glucose and Phosphate on Spinosad Fermentation by Saccharopolyspora spinosa. Chin J Chem Eng. 2006, 14: 542-546. 10.1016/S1004-9541(06)60111-0.
Pan HX, Li JA, He NJ, Chen JY, Zhou YM, Shao L, Chen DJ: Improvement of spinosad production by overexpression of gtt and gdh controlled by promoter PermE* in Saccharopolyspora spinosa SIPI-A2090[J]. Biotechnol Lett. 2011, 33: 733-739. 10.1007/s10529-010-0481-8.
Tang Y, Xia L, Ding X, Luo Y, Huang F, Jiang Y: Duplication of partial spinosyn biosynthetic gene cluster in Saccharopolyspora spinosa enhances spinosyn production [J]. FEMS Microbiol Lett. 2011, 325: 22-29. 10.1111/j.1574-6968.2011.02405.x.
Xue C, Duan Y, Zhao F, Lu W: Stepwise increase of spinosad production in Saccharopolyspora spinosa by metabolic engineering. Biochem Eng J. 2013, 72: 90-95.
Kim TY, Sohn SB, Kim YB, Kim WJ, Lee SY: Recent advances in reconstruction and applications of genome-scale metabolic models. Curr Opin Biotechnol. 2011, 23: 1-7. 10.1016/j.ceb.2010.12.003.
Notebaart RA, van Enckevort FH, Francke C, Siezen RJ, Teusink B: Accelerating the reconstruction of genome-scale metabolic networks. BMC Bioinforma. 2006, 7: 296-10.1186/1471-2105-7-296. 10.1186/1471-2105-7-296.
Thiele I, Palsson BO: A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 2010, 5: 93-121. 10.1038/nprot.2009.203.
Xu C, Liu L, Zhang Z, Jin DF, Qiu J, Chen M: Genome-scale metabolic model in guiding metabolic engineering of microbial improvement. Appl Microbiol Biotechnol. 2013, 97: 519-539. 10.1007/s00253-012-4543-9.
Lee JW, Kim TY, Jang YS, Choi S, Lee SY: Systems metabolic engineering for chemicals and materials. Trends Biotechnol. 2010, 29: 370-378.
Park JH, Lee SY: Towards systems metabolic engineering of microorganisms for amino acid production. Curr Opin Biotechnol. 2008, 19: 454-460. 10.1016/j.copbio.2008.08.007.
Brochado AR, Matos C, Møller BL, Hansen J, Mortensen UH, Patil KP: Improved vanillin production in baker's yeast through in silico design. Microb Cell Fact. 2010, 9: 84- 10.1186/1475-2859-9-84.
Huang D, Li S, Xia M, Wen JP, Jia XQ: Genome-scale metabolic network guided engineering of Streptomyces tsukubaensis for FK506 production improvement. Microb Cell Fact. 2013, 12: 1-18. 10.1186/1475-2859-12-1.
Oddone GM, Mills DA, Block DE: A dynamic, genome-scale flux model of Lactococcus lactis to increase specific recombinant protein expression. Metab Eng. 2009, 11: 367-381. 10.1016/j.ymben.2009.07.007.
Katz E, Weissbach H: Incorporation of C14-labeled amino acids into actinomycin and protein by Streptomyces antibioticus. J Biol Chem. 1963, 238: 666-675.
Cheng YR, Fang A, Demain AL: Effect of amino acids on rapamycin biosynthesis by Streptomyces hygroscopicus. Appl Microbiol Biotechnol. 1995, 43: 1096-1098. 10.1007/BF00166931.
O'Hagan D: Biosynthesis of polyketide metabolites. Nat Prod Rep. 1992, 9: 447-479. 10.1039/np9920900447.
Hinnebusch AG: Evidence for translational regulation of the activator of general amino acid control in yeast. Proc Natl Acad Sci. 1984, 81: 6442-6446. 10.1073/pnas.81.20.6442.
Duetz WA, Van Beilen JB, Witholt B: Using proteins in their natural environment: potential and limitations of microbial whole-cell hydroxylations in applied biocatalysis. Curr Opin Biotechnol. 2001, 12: 419-425. 10.1016/S0958-1669(00)00237-8.
Park JB: Oxygenase-based whole-cell biocatalysis in organic synthesis. J Microbiol Biotechnol. 2007, 17: 379-392.
Siedler S, Bringer S, Bott M: Increased NADPH availability in Escherichia coli: improvement of the product per glucose ratio in reductive whole-cell biotransformation. Appl Microbiol Biotechnol. 2011, 92: 929-937. 10.1007/s00253-011-3374-4.
Lee HC, Kim JS, Jang W, Kim SY: High NADPH/NADP ratio improves thymidine production by a metabolically engineered Escherichia coli strain. J Biotechnol. 2010, 149: 24-32. 10.1016/j.jbiotec.2010.06.011.
Chin JW, Khankal R, Monroe CA, Maranas CD, Cirino PC: Analysis of NADPH supply during xylitol production by engineered Escherichia coli. Biotechnol Bioeng. 2009, 102: 209-220. 10.1002/bit.22060.
Weckbecker A, Hummel W: Improved synthesis of chiral alcohols with Escherichia coli cells co-expressing pyridine nucleotide transhydrogenase, NADP+-dependent alcohol dehydrogenase and NAD+-dependent formate dehydrogenase. Biotechnol Lett. 2004, 26: 1739-1744. 10.1007/s10529-004-3746-2.
Kabus A, Georgi T, Wendisch VF, Bott M: Expression of the Escherichia coli pntAB genes encoding a membrane-bound transhydrogenase in Corynebacterium glutamicum improves L-lysine formation. Appl Microbiol Biotechnol. 2007, 75: 47-53. 10.1007/s00253-006-0804-9.
Bastian S, Liu X, Meyerowitz JT, Snow SD, Chen MMY, Arnold FH: Engineered ketol-acid reductoisomerase and alcohol dehydrogenase enable anaerobic 2-methylpropan-1-ol production at theoretical yield in Escherichia coli. Metab Eng. 2011, 13: 345-352. 10.1016/j.ymben.2011.02.004.
Madduri K, Waldron C, Matsushima P, Broughton MC, Crawford K, Merlo DJ, Baltz RH: Genes for the biosynthesis of spinosyns: applications for yield improvement in Saccharopolyspora spinosa. J Ind Microbiol Biotechnol. 2001, 27: 399-402. 10.1038/sj.jim.7000180.
Pan Y, Yang X, Li J, Zhang R, Hu Y, Zhou Y, Wang J, Zhu B: Genome Sequence of the Spinosyns-Producing Bacterium Saccharopolyspora spinosa NRRL 18395. J Bacteriol. 2011, 193: 3150-3151. 10.1128/JB.00344-11.
Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Landsman D, Lipman D, Lu Z, Madden TL, Madej T, Maglott DR, Marchler-Bauer A, Miller V, Mizrachi I, Ostell J, Panchenko A, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M: Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2010, 39: D38-D51. Database issue
Borodina I, Krabben P, Nielsen J: Genome-scale analysis of Streptomyces coelicolor A3 (2) metabolism. Genome Res. 2005, 15: 820-829. 10.1101/gr.3364705.
Orth JD, Thiele I, Palsson B: What is flux balance analysis?. Nat Biotechnol. 2010, 28: 245-248. 10.1038/nbt.1614.
Price ND, Reed JL, Palsson B: Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat Rev Microbiol. 2004, 2: 886-897. 10.1038/nrmicro1023.
Becker SA, Feist AM, Mo ML, Hannum G, Palsson BØ, Herrgard MJ: Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox. Nat Protoc. 2007, 2: 727-738. 10.1038/nprot.2007.99.
Edwards JS, Palsson BO: Robustness analysis of the Escherichia coli metabolic network. Biotechnol Prog. 2000, 16: 927-939. 10.1021/bp0000712.
Flett F, Mersinias V, Smith CP: High efficiency intergeneric conjugal transfer of plasmid DNA from Escherichia coli to methyl DNA‒restricting streptomycetes. FEMS Microbiol Lett. 1997, 155: 223-229. 10.1111/j.1574-6968.1997.tb13882.x.
Matsushima P, Broughton MC, Turner JR, Baltz RH: Conjugal transfer of cosmid DNA from Escherichia coli to Saccharopolyspora spinosa: effects of chromosomal insertions on macrolide A83543 production. Gene. 1994, 146 (1): 39-45. 10.1016/0378-1119(94)90831-1.
Sambrook J, Russell DW: Molecular cloning: a laboratory manual. 2001, New York: Cold Spring Harbor Laboratory Press, 3
Acknowledgements
We thank the financial supported by Natural Science Foundation of China (No. 31270087 and 21076148), Program for New Century Excellent Talents in University (NCET-10-0616), Plan of Tianjin Science and Technology support (No. 11ZCKFSY0100) and Project supported by the Specialized Research Fund for the Doctoral Program of Higher Education of China (No. 20100032120012).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
WYL and XYW conceived and designed the research. XYW and CBZ performed the model construction and calculation. Amino acids supplement experiments and strain engineering was conducted by MLW and XYW respectively. CBZ analyzed the date and drafted the manuscript. WYL supervised the research and revised the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Wang, X., Zhang, C., Wang, M. et al. Genome-scale metabolic network reconstruction of Saccharopolyspora spinosa for Spinosad Production improvement. Microb Cell Fact 13, 41 (2014). https://doi.org/10.1186/1475-2859-13-41
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2859-13-41