
Abstract
Long-term cell cultures were used in coffee to study the cytological, genetic and epigenetic changes occurring during cell culture ageing. The objective was to identify the mechanisms associated with somaclonal variation (SV). Three embryogenic cell lines were established in Coffea arabica (2n = 4x = 44) and somatic seedlings were regenerated after 4, 11 and 27 months. Phenotyping and AFLP, MSAP, SSAP molecular markers were performed on 199 and 124 plants, respectively. SV were only observed from the 11 and 27-month-old cultures, affecting 30 and 94 % of regenerated plants, respectively. Chromosome counts performed on 15 plants showed that normal plants systematically displayed normal chromosome numbers and that, conversely, aneuploidy (monosomy) was systematically found in variants. The allopolyploid structure of C. arabica allowed aneuploid cells to survive and regenerate viable plants. No polymorphic fragments were observed between the AFLP and SSAP electrophoretic profiles of mother plants and those of the in vitro progeny. Methylation polymorphism was low and ranged between 0.087 and 0.149 % irrespective of the culture age. The number of methylation changes per plant—normal or variant—was limited and ranged from 0 (55–80 % of the plants) to 4. The three cell lines showed similar SV rate increases during cell culture ageing and produced plants with similar molecular patterns indicating a non random process. The results showed that cell culture ageing is highly mutagenic in coffee and chromosomal rearrangements are directly linked to SV. Conversely, the analysis of methylation and transposable elements changes did not reveal any relation between the epigenetic patterns and SV.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In vitro culture has long been associated with the occurrence of somaclonal variation (SV), ranging from moderate to strikingly high rates (Karp 1991; Skirvin et al. 1994). The term SV defines the phenotypic variation observed in clonally propagated plants derived from tissue cultures. Although some SV have been positively evaluated at agronomic level (Skirvin et al. 1994; Biswas et al. 2009), the loss of genetic fidelity is detrimental for commercial purposes when the main objective is strictly the clonal propagation of elite plant material to ensure the maintenance of selected traits.
Little information is yet available about SV-related mechanisms. Growth regulators such as auxin and/or cytokinin are generally indispensable to sustain cell and embryo multiplication. Unfortunately their involvement in SV occurrence has been reported. Studies have been undertaken either directly on cell cultures (Tanurdzic et al. 2008) or on plants derived from cell cultures (Hao and Deng 2002; Smýkal et al. 2007; Rodriguez-Lopez et al. 2010). Early attempts to explain SV revealed the implication of chromosomal abnormalities (Karp 1991). Mutations affect the primary sequence of DNA and include numerical and structural chromosome changes (chromosome deletions, translocations and changes in ploidy), somatic recombination, point mutations, along with deletions and transpositions also occurring in mitochondrial and chloroplast genomes (Orton 1983; Karp 1991; Kaeppler et al. 2000; Neelakandan and Wang 2012). All of these classes of mutations would be expected to give rise to stable sexually heritable variation (Orton 1983). Chromosomal instabilities and ploidy alterations have been observed in cell cultures (Hao and Deng 2002; Giorgetti et al. 2011).
The involvement of epigenetic events in SV has also been proposed because some variants reverted back to the normal phenotype after some cycles of vegetative propagation or sometime after field planting (Biswas et al. 2009). A wide range of mechanisms that are genetic or supposedly epigenetic in origin have been recently described as possible causes of SV including TE (Transposable Element) transposition and small RNA directed DNA methylation (Smulders and de Klerk 2011; Neelakandan and Wang 2012). DNA methylation in the form of 5-Methylcytosine (5mC) is a major epigenetic mark involved in gene expression and plays an important role in plant regulation and development (Vanyushin and Ashapkin 2011). DNA methylation in plants commonly occurs at cytosine bases in all sequence contexts, the symmetric CG and CHG contexts (where H indicates A, T or C) and the asymmetric CHH contexts. DNA hypomethylation can produce severe phenotypic assignment as noted in the progeny of ddm1 Arabidopsis mutants (Kakutani et al. 1996). Tissue culture induces sequence-specific methylation variation that can be detected by DNA molecular markers such as Met-AFLP (Bednarek et al. 2007; Fiuk et al. 2010) or MSAP (Peraza-Echeverria et al. 2001; Diaz-Martínez et al. 2012).
The loss of DNA methylation in cell cultures can be maintained during plant regeneration, and be stably inherited over subsequent generations (Stroud et al. 2013). However, the effect of DNA methylation deregulation associated with SV remains unclear. An elevated transposition of TEs (class I retrotransposons and class II transposons) has been positively correlated to demethylating events induced by tissue culture in Arabidopsis and maize (Barret et al. 2006; Smith et al. 2012). TEs could potentially alter gene expression when insertions occur inside or near encoding protein genes, finally affecting phenotype (Sato et al. 2011).
Previous studies on Coffea arabica (2n = 4x = 44) have shown that somatic embryogenesis can generate variable and relatively high SV levels (5–90 %) depending on in vitro culture conditions (Söndahl and Bragin 1991; Etienne and Bertrand 2003). Nevertheless, when applied over a short 6-month period in the presence of a low growth regulator supply, embryogenic cell cultures gave rise to very low SV rates, as observed through massive phenotypic observations in field plots (0.74 % from 200,000 plants) (Bobadilla Landey et al. 2013). Molecular analyses performed on the regenerated plants revealed that genetic and epigenetic alterations were particularly limited. The involvement of culture ageing in the induction of SV has been proposed in coffee (Etienne and Bertrand 2003). Whatever the genotype, the degree of SV was low (1.3 %) in plants directly produced from embryogenic callus and increased progressively in line with the cell culture age, reaching 25 % on average in plants produced from 12-month-old suspensions in the presence of 4.52 μM 2,4-D (2,4-dichlorophenoxyacetic acid). However, the mechanisms occurring during cell culture ageing and underlying SV have yet to be elucidated in coffee.
The objectives of this work were to identify the phenotypic, chromosomal, genetic and epigenetic changes associated with SV occurrence during cell culture ageing, as well as the stochastic character of the phenomenon. To that end, by establishing three independent embryogenic cell lines, we investigated the effects of culture ageing on the derived plants after more or less extended cell proliferation periods (up to 27 months). Molecular analyses were performed with AFLP (amplified fragment length polymorphism) and two related techniques, SSAP (sequence-specific amplification polymorphism retrotransposon-based molecular marker) and MSAP (methylation-sensitive amplified polymorphism), in order to investigate changes in the activity of different TEs and DNA methylation patterns.
Materials and methods
Plant material and somatic embryogenesis
The plant material corresponded to somatic embryo-derived plants (somatic seedlings) of C. arabica var. Caturra regenerated from three independent embryogenic cell lines (A, B and C) established with distinct embryogenic calli. Somatic seedlings were regenerated after three different culture periods, i.e. 4, 11 and 27 months from the establishment of the embryogenic cell line (Fig. 1).

Schematic representation of the experimental design based on the establishment and long-term maintenance of independent embryogenic cell lines and further regeneration of somatic embryos-derived plantlets at different culture ages (4, 11 and 27 months)
Establishment of embryogenic cell cultures and plant regeneration
Embryogenic calli were induced from leaf explants as previously described by Etienne (2005). Three long-term cell cultures (Fig. 2a) were successfully established in baby food jars by initially isolating three distinct embryogenic calli that proliferated independently on half-strength MS (Murashige and Skoog 1962) embryogenic callus production medium (Etienne 2005) enriched with 4.5 μM 2,4-D and 12 μM 6-benzylaminopurine (6-BA) and solidified with 2.8 g/L phytagel. Long-term maintenance of the cell cultures was achieved over 3 years by 1-month proliferation cycles in the dark at 27 °C. Plants could be regenerated during the first 27 proliferation months but, after 36 months, all cell cultures lost the ability to regenerate somatic embryos.

Long-term cell cultures of Coffea arabica. a An embryogenic callus belonging to the cellular line A after 11 month proliferation. b Regeneration of globular and heart shaped-somatic embryos from an 11 months old cell line
Cell cultures were subjected to embryo regeneration conditions (Fig. 2b) by subculturing approximately 200 mg callus every 4 weeks twice on ‘R’ regeneration medium (Etienne 2005) containing 17.76 μM 6-BA and twice on ‘M’ maturation medium containing 1.35 μM 6-BA until plantlets developed. Plantlets exhibiting one leaf pair were transferred to the same maturation medium devoid of plant growth regulators and enriched with 1 g/L active charcoal until acclimatization. During 1 month, the plantlets were maintained on a horticultural substrate under a 14 h photoperiod (20 µmol m−2 s−1 light intensity) at 26 °C and 80–90 % relative humidity. Afterwards, they were moved to nursery (2000–4000 lux).
Phenotype characterization
The detection of variants was performed on 199 somatic seedlings from the three cell lines (50–65 plants per cell line) after 12 and 24 months in the nursery. For each culture age, 40–75 somatic seedling plants were observed. Phenotypic characterization consisted in assessing plant height, internode number and length, leaf length, leaf width and collar diameter. Other characteristics such as presence or absence of domatia, leaf shape, canopy shape and orthotropic or plagiotropic development were also observed and compared to those of normal plants.
Slide preparation and chromosome counting
Root tips were collected and placed in an aqueous solution of 8-hydroxyquinoline (2.5 mM) used as the pre-treatment, for 4 h in darkness (2 h at 4 °C plus 2 h at 21 °C). A solution of Carnoy (absolute ethanol and glacial acetic acid, 31 v/v) was used to fix the tissues for at least 24 h at −20 °C. Fixed material was then stored in 70 % ethanol at 4 °C pending slide preparation. The stored root tips were used for slide preparations by employing the technique for cell dissociation of enzymatically macerated roots, as described previously by Herrera et al. (2007). Preparations were frozen in liquid nitrogen to remove the coverslips, stained with 1 μg/mL 4’, 6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI), and mounted in Vectashield (Vector Laboratories, Peterborough, UK). During slide examination, mitotic cells at metaphase or prometaphase stages were used for chromosome counting. Between 4 and 8 mitotic cells from each individual were analysed to determine the chromosome number. The best examples were photomicrographed at metaphase to document the chromosome number and morphology. A Nikon Eclipse 90i epi-fluorescence microscope equipped with a digital, cooled B/W CCD camera (VDS 1300B Vosskühler®) was used with the appropriate filter (UV-2E/C excitation wavelength 340–380).
Molecular analyses
One hundred and twenty four plants derived from the three cell cultures and from all culture ages were subjected to molecular markers to estimate their genetic and epigenetic conformity in comparison with mother plants (Table 1). To that end, AFLP, MSAP and SSAP analyses were performed on both the mother plants and on the somatic embryogenesis progeny to reveal polymorphisms in the molecular marker patterns. Genomic DNA was isolated from 100 mg of lyophilized fully developed young leaves using Dellaporta buffer (Dellaporta et al. 1983) containing sodium dodecyl sulphate (SDS) detergent and sodium bisulphite 1 % w/v to avoid leaf oxidation. DNA was purified on spin-column plates as described in the DNeasy plant kit protocol from QIAGEN.
AFLP markers
AFLP analysis was carried out as described by Vos et al. (1995). Selective amplification included a total of five EcoR1/MspI primer combinations using 5-FAM or 5-HEX fluorescently labelled EcoR1 (+3) and unlabelled MseI (+3) primers (S1). The PCR program for selective amplification was performed under the following conditions: 3 min at 94 °C, followed by 12 cycles of 45 s at 94 °C, 45 s at 65 °C and 1 min at 72 °C; the annealing temperature was decreased by 0.7 °C per cycle from a starting point of 65 °C during this stage (Touchdown), with a final round of 25 cycles of 94 °C for 45 s, 56 °C for 45 s, 72 °C for 1 min and a final extension of 72 °C for 1 min. MSAP and S-SAP molecular markers were used under the same conditions. Two replications using different DNA extractions were performed for each primer combination and for all molecular markers to reduce the possibility of technical artefacts.
MSAP molecular markers
The MSAP protocol (Reyna-López et al. 1997) is an adaptation of the AFLP method for the evaluation of different states of methylation in the symmetric sequence CCGG. In this protocol, the frequent cutting endonuclease (MseI) is replaced by the two restriction enzymes isoschizomers HpaII and MspI with different sensitivities to the methylation state of the CCGG sequence (Table 2). Specifically, HpaII is able to recognize and cut when the CCGG sequence is unmethylated or hemi-methylated on the external cytosine. MspI is able to cut when the CCGG sequence is unmethylated or if the internal cytosine is fully or hemi-methylated. Both HpaII and MspI are unable to cut if the external cytosines are fully methylated. Selective amplification included a total of eight primer combinations per isoschizomer (Supplementary Table S1). HpaII (+2) primers were fluorescently labelled with 5-FAM or 5-HEX while EcoR1 (+3) remained unlabelled. The set of fragments from both digestions was grouped by primer combination, fragment size and sample to determine all individual profiles. After fragment classification three possible EcoRI/HpaII and EcoRI/Msp MSAP patterns were found by presence (1) or absence (0).
SSAP markers
The SSAP protocol (Waugh et al. 1997) is an adaptation of AFLP markers to discover polymorphisms associated with transposable element mobility. The TE sequences used in this study were identified in the C. canephora genome sequence (Denoeud et al. 2014). TE were identified based on sequence similarities (BLAST) against Repbase (Jurka et al. 2005) (http://www.girinst.org/), fine structure analysis using dotter alignments (Sonnhammer and Durbin 1995) or identified through assembly using the AAARF algorithm (DeBarry et al. 2008). Lastly, a sequence database of transposable elements comprising Class I retrotransposons and Class II transposons was constructed and used for SSAP-based primer design. Different elements were used for primer design including 13 different Copia and Gypsy LTR retrotransposon families, one SINE (Short INterspersed Elements and 2 different transposon families (MuLEs and Mariner families; Supplemenatry Table S2). Primers were designed within a maximum distance of 200 bp at both extremities of each type of elements to find nearby polymorphic TE insertions associated with EcoRI restriction sites. Specific TE primers were fluorescently labelled either with 5-FAM, 5-HEX or 5-Tamra while EcoR1 (+1) remained unlabelled (Supplementary Table S2). In all, 35 primers were designed and used in screening prior to final analysis; only clear and repetitive profiles were used in the final analysis.
Capillary electrophoresis and data analysis
PCR products were separated by capillary electrophoresis with Pop 7™ polymer in a 16 capillary 3130 XL Genetic Analyzer from Applied Biosystems. Fragment size was estimated using an internally manufactured 524 ROX fluorophore as the sizing standard. The fragments used for fingerprinting were visualized as electropherograms using applied Biosystems software GeneMapper® version 3.7. Informative fragments were mostly found in the 100–450 bp range. All amplified fragments were classified based on the primer combination and their size. The sample fingerprint data were converted to binary code, with (1) denoting the presence of the fragment and (0) its absence. Different binary matrices were constructed for comparative analysis depending on the kind of molecular marker. Global genetic and epigenetic levels were estimated as total polymorphisms. Total polymorphism = [no. of polymorphic fragments/(no. of fragments x no. emblings)] × 100. A 3σ confidence limit for binomial distribution was calculated to analyse the statistical difference between the global genetic and epigenetic levels derived from different molecular markers using the formula \(p \pm 3\sqrt {p\left( {1 - p} \right)/n}\) with a confidence level of 99 %. The variable analysed was the proportion (p) of polymorphism (p = x/n), where “x” was the number of polymorphic fragments and “n” the total number of fragments observed.
Results
Effect of cell culture ageing on somaclonal variation
Batches of somatic seedlings from three different culture ages (4, 11 and 27-month-old cell cultures) and regenerated from three different cell lines (A, B and C) were phenotyped (Fig. 1). All the three cell lines were able to regenerate plants after 4, 11 and 27 months of proliferation. Based on plant architecture and leaf morphology we identified a clear pattern of phenotypic abnormalities among somatic seedlings after 12 months of development in the nursery (Fig. 3a). We found a predominant aberrant phenotype characterized by smaller size, oval-elliptic leaves and closed canopy (Fig. 3a, b, d, e), corresponding to “Bullata” coffee mutants described by Krug and Carvalho (1951).

Morphological differences between normal and C. arabica variant phenotypes observed in somatic seedlings regenerated after different culture times (4 and 27 months). a Differences in plant size between somatic seedlings derived from 4 (on the right) and 27 months (on the left) old cell cultures. b Size differences after 8 months of parallel development in nursery; Bullata variant from 27 months old cultures (left) and phenotypically normal plant from 4 months old culture (right). c Phenotypically normal somatic seedling from 4 months old cell culture. d Bullata variant phenotype from 27 months old culture. e Leaves from normal and Bullata phenotype. Bullata leaves (left) are rounder and smaller than those from true-to-type coffee somatic seedlings (right)
For the three independent cell lines, somaclonal variants were observed in somatic seedlings from long-term cell cultures i.e. 11 and 27-month-old calli affecting in the latter almost all the regenerated plants (Fig. 3a), although all plants derived from younger cell cultures (4 months) exhibited normal phenotypes (Fig. 3c; Table 3). Thirty percent of somatic seedlings from 11-month-old cultures corresponded to abnormal phenotypes (20 and 40 % for cell lines B and C, respectively). From a total of nine variant plants out of 30 plants observed, eight were Dwarf and one Bullata. Out of 75 somatic seedlings from 27-month-old cultures, ninety-four percent displayed an abnormal phenotype (100, 92 and 92 % for cell lines A, B and C, respectively). Only four plants displayed normal phenotypes. The mutants corresponded to four Dwarf phenotypes, one Angustifolia and the rest of the plants were Bullata phenotypes.
Abnormal chromosome numbers limited to phenotypic variants
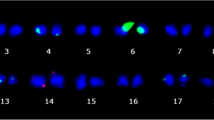
Chromosome counts were performed on 15 somatic seedlings obtained from all cell culture ages (Fig. 4; Table 4). All plants showing a normal phenotype systematically presented normal chromosome numbers. This outcome was even found in the rare case of one phenotypically normal plant regenerated from a 27 month-old cell culture. Conversely, the chromosome counts performed on eight abnormal phenotype plants derived from the two longer cell culture periods revealed aneuploidy. All phenotypic variants presented monosomies i.e. the lack of one or more chromosomes. The Dwarf variants presented 2n-1, 2n-2 and 2n-3 monosomies while the three Bullata phenotypic variants showed 2n-1 monosomy (43 chromosomes).

Mitotic cells at metaphase or prometaphase stages and observed ploidy levels of some normal and variant somatic seedlings from the allotetraploid C. arabica species (2n = 4x = 44). Plants were regenerated from 4 and 27 month old cell cultures. Karyotype analyses were performed by counting chromosomes on four to eight clear metaphase spreads obtained from root tips of 12 month-old nursery plants
No genetic changes revealed by AFLP and SSAP in the somatic seedlings
AFLP analyses were conducted to reveal genetic polymorphisms associated with somatic embryogenesis and more particularly with the age of the embryogenic cell culture. Five primer combinations produced 182 total fragments with an average of 36 fragments per primer combination (Supplementary Table S3). No differences were found between the electrophoretic profiles of mother plants and the 124 somatic seedlings studied. SSAP molecular markers were performed to uncover polymorphisms associated with TE activity (i.e. produced by insertions), possibly activated by tissue culture. In all, 1003 SSAP fragments were generated after 45 selective SSAP amplifications resulting in 22 fragments per primer combination (Supplementary Table S4). No polymorphic fragments were observed between the electrophoretic profiles of mother plants and those of the in vitro progeny (Fig. 5). The number of fragments produced after selective amplification was in a range of 25–133 depending on the element tested. Both SSAP and AFLP analyses of somatic seedlings indicated that the level of genetic variations was not significantly enhanced by either short or long-term cell multiplication periods.

Examples of SSAP electropherograms observed for coffee mother plants and somatic seedlings derived from 4 and 27 months cell cultures. Illustration of monomorphic electrophoretic profiles
Low induction of methylation changes in short and long-term cell cultures
MSAP markers were used on coffee somatic seedlings to investigate the methylation changes induced by tissue culture and more particularly by the cell culture age. Eight primer combinations from the separate EcoRI/HpaII and EcoRI/MspI digestions produced a total of 402 MSAP fragments (Table 5). The pattern distribution resulted in 348 non-methylated sites with pattern 1 (1/1), 52 fragments with internally methylated cytosines with pattern 3 (0/1) and only 2 externally hemi-methylated cytosines with pattern 2 (1/0). The interpretation of the possible methylation state of these patterns indicated that 86.6 % of the fragments corresponded to non-methylated sites (pattern 1) whereas 13.4 % were methylated (patterns 2 and 3).
The mother plants’ profiles and those of somatic seedlings were then compared to find polymorphic methylation fragments (Fig. 6). A total of 58 methylation changes associated with 14 polymorphic fragments were detected in the whole plant population analysed (Table 6). Somatic seedlings derived from 4-month-old cell cultures presented 27 methylation changes accounting for 46.5 % of the overall changes (Table 5). Plant batches from 11 and 27-month-old cell cultures displayed a similar number of changes, i.e. 15 and 16 changes representing 26 and 28 % respectively. The rate of epigenetic variation in somatic seedlings from 4, 11 and 27-month-old cell cultures was calculated as total methylation polymorphism for global estimation. Total polymorphism ranged between 0.087 and 0.149 % whatever the culture age (Table 5). These rates were not statistically different for somatic seedlings derived from short and long-term cell cultures indicating no significant effect of cell culture age. We also noted that methylation changes arose from both methylated and unmethylated sites. The methylation patterns of the 14 polymorphic fragments are indicated in Table 6. Eight fragments showing pattern 3 in mother plants changed into pattern 1 in somatic seedlings indicating demethylation events (the internal methylation of CCMetGG changing to the unmethylated state of the CCGG sequence). The remaining six fragments passed from pattern 1 in mother plants to pattern 3 in somatic seedlings revealing methylation events (the unmethylated state of CCGG changing to the methylated state of the CCMetGG sequence).

Examples of MSAP electropherograms observed for coffee mother plants and somatic seedlings progeny derived from 4 month cell cultures using the isoschizomers HpaII and MspI. Illustration of the variation patterns obtained for the phenotypically normal plants
The number of methylation changes accumulated by somatic seedlings was between 1 and 4 (Table 7). We did not find any somatic seedlings accumulating a large quantity of methylation changes or any effect of cell culture age on their accumulation. The numbers of accumulated methylation changes were similar for somatic seedlings regenerated from all proliferation durations (4, 11, 27 months) (Table 7). Most of the methylation changes appeared and disappeared with no clear relation to a particular cell line or cell culture age (Table 6). Two polymorphic molecular markers (C3 202 bp and C5 454 bp) were observed in some somatic seedlings derived from cell line C and for the three culture ages analysed. However, changes from these two fragments C3 were also shared with other plants derived from different cell lines and without any relation to cell culture age.
Discussion
Elevated levels of SV affecting phenotypes have been frequently reported with long-term cell cultures (Tremblay et al. 1999; Jambhale et al. 2001; Biswas et al. 2009). We clearly showed that the occurrence of SV is closely related to cell culture age. The phenotypic evaluation of regenerated plants derived from three independent cell lines showed that no SV or intermediate levels (20–40 %) were found with short and medium-term cultures, respectively. On the contrary, most of the plants (94 %) were variants when regeneration was induced from 2-year-old cultures. Observation of the same behaviour for the three independent cell lines strongly supported the existence of a non-random mechanism for SV. Low levels of SV (around 1 %) were previously found in C. arabica plants regenerated from recently obtained callus or from 6-month-old cell cultures (Etienne and Bertrand 2001, 2003; Bobadilla Landey et al. 2013).
The first studies on plants derived from tissue culture identified many chromosomal abnormalities as one of the main factors involved in SV (D’Amato 1985; Kaeppler et al. 2000). Many altered mechanisms observed during meiosis and mitosis can produce chromosomal alterations, such as non-disjunction (i.e. the failure of sister chromatids to separate during meiosis or mitosis), multipolar mitotic spindles, lagging anaphase chromosomes and chromosome missegregation into micronuclei (D’Amato 1985; Kaeppler et al. 2000; Holland and Cleveland 2009). Such mechanisms can produce a different extent and severity of chromosomal abnormalities in in vitro plants, including segmental or complete aneuploidy (Kaeppler and Phillips 1993; Tremblay et al. 1999). Aneuploidy has frequently been reported in tissue cultures during the callus induction and cell suspension phases (Hao and Deng 2002; Kumar and Mathur 2004; Giorgetti et al. 2011).
The karyotype analysis of coffee somatic seedlings regenerated from old cell cultures revealed different types of aneuploidy exclusively in the monosomic form. Such abnormalities were systematically found in the analysed somaclonal variants derived from 11 and 27-month-old cultures. Normal ploidy levels were always observed in plants showing a normal phenotype. In coffee, many cytological abnormalities were described in leaf explants and cell cultures by Menéndez-Yuffá et al. (2000), such as non-disjunction, double prophase cells, lagging chromosomes, micronuclei and binucleated cells, also including polyploid and aneuploid cells. Our results showed that cytological abnormalities are enhanced by long-term cultures and that aneuploidy plays a major role in C. arabica SV.
The mechanisms of non-disjunction of chromosomes during anaphase could be the best explanation for the production of aneuploid cells and plants during tissue culture (Holland and Cleveland 2009). The gene imbalance associated with specific altered chromosomes results in well defined and sometimes predictable variant phenotypes (Henry et al. 2010; Makarevitch and Harris 2010). In this study different chromosome numbers can be found for the same Dwarf variant phenotype. We recently reported the same tendency for other variant phenotypes such as Angustifolia, Giant and Variegata observed in commercial batches of coffee somatic seedlings (Bobadilla Landey et al. 2013). Consequently, it is not possible to predict a coffee variant phenotype on the sole basis of karyotype analyses limited to chromosome counting. Moreover, such analyses did not lead to any observation of other chromosomal alterations associated with undetected deletions, duplications, inversions or translocations of specific chromosomal segments.
Different types of molecular markers were used to explore a genetic or epigenetic origin of SV and reveal any accumulation of polymorphisms over time. AFLP markers did not show any polymorphism in the whole plant population analysed, indicating low levels of genetic variation during ageing of coffee cell cultures. Similar approaches with long-term cultures using different kinds of molecular markers found contrasting results. Polymorphisms were not found using RAPD markers in 2-year-old cultures of hop (Peredo et al. 2006). Using SSR markers Smýkal et al. (2007) reported no genetic variation in 24-year-old multiple shoot cultures of pea. However, in the same work, AFLP molecular markers successfully revealed 22 polymorphic and singleton fragments out of a total of 436 fragments.
Extended coffee cell proliferation periods did not reveal TE activity. These findings are in agreement with AFLP results indicating no or low genetic variation. On the contrary, clear evidence of Tos 17 and Tos 19 retrotransposon activity was provided in rice cell cultures (Hirochika et al. 1996). The first hypothesis is that these elements were not reactivated under stressful somatic embryogenesis conditions or are inactive. Although TEs constitute a large percentage of the plant genome, only a few elements were reported active in the literature. A second hypothesis is that the inactivity of the chosen elements resulted from their transcriptional inactivation through methylation or epigenetic post-transcriptional mechanisms preventing their insertion (Mirouze et al. 2009).
Many studies on vitroplants have reported high and unexpected levels of epigenetic variation related to cytosine methylation (Miguel and Marum 2011). A first approach was aimed at exploring specific sequence contexts with methylation-sensitive molecular markers (e.g. CCGG for MSAP) and the other at measuring global methylation levels in all three cytosine contexts CG, CHG and CHH with HPLC methods. In our conditions, the number of methylation changes per plant was small and the resulting total polymorphism was <1 % independently of the age of the cell culture, indicating little disruption to the epigenetic status. Similarly, low levels of methylation variation were found in freesia somatic seedlings derived from short-term cultures (Gao et al. 2010) and pea long-term multiple shoot cultures (Smýkal et al. 2007).
Many studies with methylation-sensitive molecular markers that reported a high level of epigenetic variation reported no effect on phenotype or morpho-agronomic traits (Bednarek et al. 2007; Li et al. 2007; Schellenbaum et al. 2008; Fiuk et al. 2010). Methylation changes contribute poorly to global estimations when they do not occur at high frequencies (Smulders and Klerk 2011). The individual accumulation of methylation changes could be a more reliable indicator of tissue culture stress or plant epigenetic instability. In the epigenetic evaluation made by molecular markers (e.g. MSAP) most in vitro plants do not generally tend to accumulate large numbers of methylation changes (Li et al. 2007; Bednarek et al. 2007; Schellenbaum et al. 2008; Fiuk et al. 2010). However, few plants could individually accumulate many methylation changes indicating higher epigenetic instability. Coffee plants from short and long-term cell cultures accumulated comparable small numbers of methylation changes (1-4 per individual) independently of their normal vs variant phenotypes. Similar observation was done in both normal and variant coffee somatic seedlings propagated industrially (Bobadilla Landey et al. 2013).
The elevated SV rates frequently reported with long-term cell cultures offer a unique opportunity to understand the underlying molecular and physiological mechanisms. Long-term cultures were used to establish the full range of cytological, genetic and epigenetic mechanisms leading to SV in coffee. Cell culture ageing was proved to be highly mutagenic. Although the molecular marker approach used cannot generate exhaustive information, it can be concluded that genetic polymorphisms and epigenetic changes are particularly limited during cell culture ageing, while aneuploidy plays a major role in SV. C. arabica is a young allopolyploid that still has most of its genes in duplicated copies (Cenci et al. 2012). It could be hypothesized that the impact of genetic or epigenetic polymorphisms has been restricted because of the buffer effect due to polyploidy. However the allopolyploid structure also allowed aneuploid cells to survive and regenerate viable plants, which would certainly not be the case with non-polyploid species. The absence of one or more chromosomes was probably buffered by the other homologous chromosomes. Hence, C. arabica is an interesting species for studying the occurrence and consequences of chromosomal abnormalities in relation to SV.
Abbreviations
- 6-BA:
-
6-Benzylaminopurine
- 2,4-D:
-
2,4-Dichlorophenoxyacetic acid
- SV:
-
Somaclonal variation
- TEs:
-
Transposable elements
References
Barret P, Brinkman M, Beckert M (2006) A sequence related to rice Pong transposable element displays transcriptional activation by in vitro culture and reveals somaclonal variations in maize. Genome 49:1399–1407
Bednarek PT, Orłowska R, Koebner RMD, Zimny J (2007) Quantification of tissue-culture induced variation in barley (Hordeum vulgare L.). BMC Plant Biol 7:10
Biswas MK, Dutt M, Roy UK, Islam R, Hossain M (2009) Development and evaluation of in vitro somaclonal variation in strawberry for improved horticultural traits. Sci Hort 22:409–416
Bobadilla Landey R, Cenci A, Georget F, Bertrand B, Camayo G, Dechamp E, Simpson J, Herrera JC, Santoni S, Lashermes P, Etienne H (2013) High genetic and epigenetic stability in Coffea arabica plants derived from embryogenic suspensions and secondary embryogenesis as revealed by AFLP, MSAP and the phenotypic variation rate. PLoS One 8(2):e56372. doi:10.1371/journal.pone.0056372
Cenci A, Combes M-C, Lashermes P (2012) Genome evolution in diploid and tetraploid Coffea species as revealed by comparative analysis of orthologous genome segments. Plant Mol Biol 78:135–145
D’Amato F (1985) Cytogenetics of plant cells and tissue cultures and their regenerates. Plant Sci 3:73–112
DeBarry JD, Liu R, Bennetzen JL (2008) Discovery and assembly of repeat family pseudomolecules from sparse genomics sequence data using the assisted automated assembler of repeat families (AAARF) algorithm. BMC Bioinform 13(9):235. doi:10.1186/1471-2105-9-235
Dellaporta SL, Wood J, Hicks JB (1983) A plant DNA miniprepration: version II. Plant Mol Biol Rep 1:19–21
Denoeud F, Carretero-Paulet L, Dereeper A, Droc G, Guyot R, Pietrella M, Zheng C, Alberti A, Anthony F et al (2014) The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 345:1181–1184. doi:10.1126/science.1255274
Díaz-Martínez M, Nava-Cedillo A, Guzmán-López JA, Escobar-Guzmán R, Simpson J (2012) Polymorphism and methylation patterns in Agave tequilana Weber var. ‘Azul’ plants propagated asexually by three different methods. Plant Sci 185–186:321–330
Etienne H (2005) Protocol of somatic embryogenesis: coffee (Coffea arabica L. and C. canephora P.). In: Jain SM, Gupta PK (eds) Protocols for somatic embryogenesis in woody plants. Series: Forestry Sciences, vol 77. 590 p, Springer, Dordrecht, pp 167–179, ISBN: 1-4020-2984-5
Etienne H, Bertrand B (2001) The effect of the embryogenic cell suspension micropropagation technique on the trueness to type, field performance, bean biochemical content and cup quality of Coffea arabica trees. Tree Physiol 21:1031–1038
Etienne H, Bertrand B (2003) Somaclonal variation in Coffea arabica: effects of genotype and embryogenic cell suspension age on frequency and phenotype of variants. Tree Physiol 23:419–426
Fiuk A, Bednarek PT, Rybczýnski JJ (2010) Flow cytometry, HPLC-RP and met AFLP analyses to assess genetic variability in somatic embryo-derived plantlets of Gentiana pannonica scop. Plant Mol Biol Rep 28:413–420
Gao X, Yang D, Cao D, Ao M, Sui X, Wang Q, Kimatu JN, Wang L (2010) In vitro micropropagation of Freesia hybrida and the assessment of genetic and epigenetic stability in regenerated plantlets. J Plant Growth Reg 29:257–267
Giorgetti L, Castiglione M, Turrini A, Ronchi V, Geri C (2011) Cytogenetic and histological approach for early detection of “mantled” somaclonal variants of oil palm regenerated by somatic embryogenesis: first results on the characterization of regeneration system. Caryologia 64:223–234
Hao YI, Deng XX (2002) Occurrence of chromosomal variations and plant regeneration from long-term-cultured citrus callus. In vitro Cell Dev Biol Plant 38:472–476
Henry IM, Dilkes BP, Miller ES, Burkart-Waco D, Comai L (2010) Phenotypic consequences of aneuploidy in Arabidopsis thaliana. Genetics 186:1231–1245
Herrera JC, D’hont A, Lashermes P (2007) Use of fluorescence in situ hybridization as a tool for introgression analysis and chromosome identification in coffee (Coffea arabica L.). Genome 50:619–626
Hirochika H, Sugimoto K, Otsuki Y, Tsugawa H, Kanda M (1996) Retrotransposons of rice involved in mutations induced by tissue culture. Proc Natl Acad Sci 93:7783–7788
Holland AJ, Cleveland DW (2009) Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol 10:478–487
Jambhale ND, Patil SC, Jadhav AS, Pawar SV, Waghmode BD (2001) Effect of numer of subcultures on in vitro multiplication of four banana clones. Infomusa 10:38–39
Jurka J, Kapitonov VV, Pavlicek A, Klonowsky P, Kohany O, Walichiewicz J (2005) Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110:462–467
Kaeppler SM, Phillips RL (1993) DNA methylation and tissue culture-induced DNA methylation variation in plants. In Vitro Cell Dev Biol Plant 29:125–130
Kaeppler SM, Kaeppler HF, Rhee Y (2000) Epigenetic aspects of somaclonal variation in plants. Plant Mol Biol 43:179–188
Kakutani T, Jeddeloh JA, Flowers SK, Munakata K, Richards EJ (1996) Developmental abnormalities and epimutations associated with DNA hypomethylation mutations. Proc Natl Acad Sci USA 93:12406–12411
Karp A (1991) On the current understanding of somaclonal variation. In: Miflin BJ (ed) Oxford surveys of plant molecular and cell biology, vol 17. Oxford University Press, Oxford, pp 1–58
Krug CA, Carvalho A (1951) The genetics of Coffea. Adv Genet 4:127–158
Kumar PS, Mathur VL (2004) Chromosomal instability in callus culture of Pisum sativum. Plant Cell Tissue Organ Cult 78:267–271
Li X, Yu X, Wang N, Feng Q, Dong Z, Liu L, Shen J, Liu B (2007) Genetic and epigenetic instabilities induced by tissue culture in wild barley (Hordeum brevisubulatum (Trin.) Link). Plant Cell Tissue Organ Cult 90:153–168
Makarevitch I, Harris C (2010) Aneuploidy causes tissue specific qualitative changes in global gene expression patterns in maize. Plant Physiol 152:927–938
Menéndez-Yuffá A, Fernandez R, Rios L, Xena N (2000) Mitotic aberrations in coffee (Coffeea arabica cv. ‘Catimor’) leaf explants and their derived embryogenic calli. Electron J Biotech 3:1–6
Miguel C, Marum L (2011) An epigenetic view of plant cells cultured in vitro: somaclonal variation and beyond. J Exp Bot 62:3713–3725
Mirouze M, Reinders J, Bucher E, Nishimura T, Schneeberger K, Ossowski S, Cao J, Weigel D, Paskowski J, Mathieu O (2009) Selective epigenetic control of retro transposition in Arabidopsis. Nature 461:427–430
Murashige T, Skoog FA (1962) Revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Neelakandan AK, Wang K (2012) Recent progress in the understanding of tissue culture-induced genome level changes in plants and potential applications. Plant Cell Rep 31:597–620
Orton TJ (1983) Experimental approaches to the study of somaclonal variation. Plant Mol Biol Rep 1:67–76
Peraza-Echeverria S, Herrera-Valencia VA, Kay A-J (2001) Detection of DNA methylation changes in micropropagated banana plants using methylation-sensitive amplification polymorphism (MSAP). Plant Sci 161:359–367
Peredo EL, Angeles Revilla M, Arroyo-Garcia R (2006) Assessment of genetic and epigenetic variation in hop plants regenerated from sequential subcultures of organogenic calli. J Plant Physiol 163:1071–1079
Reyna-Lopez GE, Simpson J, Ruiz-Herrera J (1997) Differences in DNA methylation pattern are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mol Gen Genet 253:703–710
Rodríguez López CM, Wetten AC, Wilkinson MJ (2010) Progressive erosion of genetic and epigenetic variation in callus-derived. New Phytol 186:856–868
Sato M, Hosokawa M, Doi M (2011) Somaclonal variation is induced de novo via the tissue culture process: a study quantifying mutated cells in Saintpaulia. PLoS One 6(8):e23541. doi:10.1371/journal.Pone.0023541
Schellenbaum P, Mohler V, Wenzel G, Walter B (2008) Variation in DNA methylation patterns of grapevine somaclones (Vitis vinifera L.). BMC Plant Biol 8:78
Skirvin RM, McPheeters KD, Norton M (1994) Sources and frequency of somaclonal variation. HortScience 29:1232–1237
Smith AM, Hansey CN, Kaeppler SM (2012) TCUP a novel hAT transposon active in maize tissue culture. Front Plant Sci 3:6
Smulders MJM, de Klerk GJ (2011) Epigenetics in plant tissue culture. Plant Growth Reg 63:137–146
Smýkal P, Valledor L, Rodriguez R, Griga M (2007) Assessment of genetic and epigenetic stability in long-term in vitro shoot culture of pea (Pisum sativum L.). Plant Cell Rep 26:1985–1998
Söndahl MR, Bragin A (1991) Somaclonal variation as a breeding tool for coffee improvement. In: Proceedings of 14th Colloquium International Coffee Science Association, San Francisco, USA. ASIC Publishers, Vevey, pp 701–710
Sonnhammer EL, Durbin R (1995) A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 167:1–10
Stroud H, Ding B, Simon SA, Feng S, Bellizzi M, Pellegrini M, Wang GL, Meyers BC, Jacobsen SE (2013) Plants regenerated from tissue culture contain stable epigenome changes in rice. Elife 2:e00354. doi:10.7554/eLife.00354
Tanurdzic M, Vaughn MW, Jiang H, Lee T-J, Slotkin RK, Sosinski B, Thompson WF, Doerge RW, Martienssen RA (2008) Epigenomic consequences of immortalized plant cell suspension culture. PLoS Biol 6:2880–2895
Tremblay L, Levasseur C, Tremblay FM (1999) Frequency of somaclonal variation in black spruce (Picea mariana, Pinaceae) and white spruce (P. glauca, Pinaceae) derived from somatic embryogenesis and identification of some factors involved in genetic instability. Am J Bot 86:1373–1381
Vanyushin BF, Ashapkin VV (2011) DNA methylation in higher plants: past, present and future. Biochim Biophys Acta 1809:360–368
Vos P, Hogers R, Bleeker M, Reijans M, Van de Lee T, Homes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Waugh R, McLean K, Flavell AJ, Pearce SR, Kumar A, Thomas BB, Powell W (1997) Genetic distribution of Bare-1-like retrotransposable elements in the barley genome revealed by sequence-specific amplification polymorphisms (S-SAP). Mol Gen Genet 253:687–694
Acknowledgments
Financial support for this study was provided by the Mexican Government through a grant to Roberto Bobadilla Landey by the CONACyT (CVU:1623391) program and by the CIRAD funds for doctoral support. The authors thank Peter Biggins (CIRAD) for critically reviewing the manuscript.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bobadilla Landey, R., Cenci, A., Guyot, R. et al. Assessment of genetic and epigenetic changes during cell culture ageing and relations with somaclonal variation in Coffea arabica . Plant Cell Tiss Organ Cult 122, 517–531 (2015). https://doi.org/10.1007/s11240-015-0772-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-015-0772-9