
Abstract
Tauopathies are neurodegenerative diseases that typically require postmortem examination for a definitive diagnosis. Detecting neurotoxic tau fragments in cerebrospinal fluid (CSF) and serum provides an opportunity for in vivo diagnosis and disease monitoring. Current assays primarily focus on total tau or phospho-tau, overlooking other post-translational modifications (PTMs). Caspase-cleaved tau is a significant component of AD neuropathological lesions, and experimental studies confirm the high neurotoxicity of these tau species. Recent evidence indicates that certain caspase-cleaved tau species, such as D13 and D402, are abundant in AD brain neurons and only show a modest degree of co-occurrence with phospho-tau, meaning caspase-truncated tau pathology is partially distinct and complementary to phospho-tau pathology. Furthermore, these caspase-cleaved tau species are nearly absent in 4-repeat tauopathies. In this review, we will discuss the significance of caspase-cleaved tau in the development of tauopathies, specifically emphasizing its role in AD. In addition, we will explore the potential of caspase-cleaved tau as a biomarker and the advantages for drug development targeting caspase-6. Developing specific and sensitive assays for caspase-cleaved tau in biofluids holds promise for improving the diagnosis and monitoring of tauopathies, providing valuable insights into disease progression and treatment efficacy.
Similar content being viewed by others


Introduction
Tauopathies are associated with high morbidity and mortality, and lack of effective treatment options [1]. They are classically diagnosed based on the detection of phospho-tau inclusions in the brain, which requires a biopsy or autopsy [2]. In recent years, efforts have been made to create fluid biomarkers based on phospho-tau to non-invasively detect pathology in living patients. These biomarkers have proven useful in diagnostic screening, particularly in the context of Alzheimer’s disease (AD) [3]. Nevertheless, tauopathies exhibit pathological tau inclusions with post-translational modifications (PTMs) beyond phosphorylation. Caspase-cleaved tau is highly neurotoxic [4], and it is present in AD and other tauopathies. Tau protein can undergo cleavage by various caspases, including caspases 1, 2, 3, 6, 7, and 8. Among the caspase-cleaved tau species studied in the context of neurodegenerative diseases, tau D421 is the most investigated. Tau D421 can result from cleavage by multiple caspases. Limited studies on cerebrospinal fluid (CSF) involving caspase-cleaved tau indicate that these fragments are released and can be identified in the CSF, potentially exhibiting a meaningful correlation with clinical deterioration [5, 6]. Recent findings indicate that caspase-6 cleaved tau D13 and D402, commonly present in AD neurons, but rarely in 4-repeat tauopathies, only partially overlap with phospho-tau in the same neuron [7]. This implies that caspase-cleaved tau can signal tau pathology distinct from phospho-tau, highlighting a pathway that cannot be identified solely through phospho-tau-based biomarkers. Despite substantial evidence pointing to the relevance of caspase-cleaved tau in the pathogenesis of certain tauopathies, this disease pathway continues to be inadequately explored. This review will examine the evidence of caspase-cleaved tau contribution to tauopathies, with a specific emphasis on the more recent advancements related to caspase-6 cleaved tau. Furthermore, we will delve into the potential for the development of biomarkers and drugs based on these research findings.
Tauopathies
Tauopathy is an umbrella term encompassing more than 20 well-defined, progressive neurodegenerative entities characterized by abnormal accumulation of protein tau in neurons and glial cells [8]. Sporadic tauopathies are classified based on the pattern of morphological distribution of the inclusions, what types of cells accumulate tau, and the biochemical composition of tau inclusions, namely predominance of three microtubule-binding repeats (3R) tau, four microtubule-binding repeats (4R) tau or a combination of both (3R/4R) [8]. Examples of 3R/4R tauopathies include AD and chronic traumatic encephalopathy (CTE). Pick’s disease (PiD) falls into the 3R category. Progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), globular glial tauopathy (GGT), argyrophilic grain disease (AGD), and aging-related tau astrogliopathy (ARTAG) belong to the 4R category. Familial tauopathies exhibit distinct clinicopathological phenotypes depending on the specific microtubule-associated protein tau (MAPT) mutation [9].
The frequencies of tauopathies vary among the populations and diagnostic criteria applied. AD is the most common neurodegenerative condition with the accumulation of abnormal tau in the brain [10]. AGD and ARTAG are prevalent, affecting up to 50% of individuals who come to autopsy at age 80 and older. However, AGD and ARTAG are considered tau accumulation with minimal clinical correlates [11, 12]. Progressive supranuclear palsy and corticobasal degeneration, the most prevalent pathogenic sporadic 4R-tauopathies, have a pooled prevalence rate of 7.1 and 2.3 per 100,000 individuals, respectively [13], but a recent clinicopathological study suggests that PSP prevalence is much higher [14].
Tau protein isoforms can undergo various post-translational modifications (PTM), such as acetylation, ubiquitination, phosphorylation, glycation, glycosylation, SUMOylation, methylation, oxidation, truncation, and nitration [15], resulting in species with different consequences for tau assembly, function, and accumulation [16]. Some tau PTMs produce neurotoxic fragments of various lengths and divergent pathological effects [17]. Phospho-tau species represent a ubiquitous post-translational modification (PTM) present in all tauopathies. Consequently, the detection of phospho-tau inclusions is the preferred method for the neuropathological diagnosis of tauopathies [8]. While other tau PTMs may be identified in various tauopathies, these inclusions are generally considered to be present only in a subgroup of cells that already exhibit phospho-tau inclusions.
Tau truncation by proteases - such as caspases, the theme of this review - leads to significant alterations in its structure and function, resulting in the loss- or gain of function depending on the truncation site [18]. Some protease-mediated tau truncations have a critical role in molecular events leading to pathological changes in tauopathies [19].
Caspases
Caspases, a large group of cysteine proteases commonly associated with inflammation and apoptosis [20], participate in tau proteolysis. Caspases cleave substrates at specific aspartic acid (Asp) residues [21, 22]. In their inactive proenzyme form, they reside in the cytosol and are activated by dimerization or proteolytic cleavage [23]. Once activated, they proteolytically degrade proteins by altering their cellular structure and functions [23]. The caspase family includes at least 14 enzymes [22]. Classically, caspases are divided into upstream initiators and downstream effectors. Upstream caspases 1, 4, 5, 11, 12, and 13 trigger inflammatory processes by cytokine activation, while 2, 8, 9, and 10 are associated with apoptosis initiation. Upon activation, upstream caspases initiate an amplification cascade that activates downstream effector caspases. Downstream caspases 3, 6, and 7 are effectors of apoptosis, while 14 is involved in cytokine maturation [24].
Caspase-cleaved tau in Alzheimer’s disease
Studies on caspase-cleaved tau have mainly focused on AD and show that tau can be cleaved at multiple sites by caspases resulting in carboxy or amino truncations [24,25,26] (Table 1). Cleavage of tau’s C-terminus or N-terminus by caspases leads to impairments in mitochondrial bioenergetics, weakening of axonal transport, neuronal injury, and cognitive decline [25]. Besides, these truncations contribute to the formation of amyloid-β plaques and intracellular neurofibrillary tangles [27] (Fig. 1).

Pathological Mechanisms induced by Caspase-Cleaved Tau
Caspase-3
Caspase-3 appears to cleave tau protein after Asp25 or Asp421 [31]. Caspase-cleaved tau in its C-terminal tail at Asp421, which removes 20 amino acids from tau C-terminal, also known as TauC3, or tau D421, were the focus of the first studies on tauopathies [28, 33]. In vitro, caspase-3-cleaved tau at Asp421 assembles into filaments more rapidly than wild-type tau [28]. TauC3 may contribute to the propagation of tau pathology by inducing mitochondrial fragmentation and bioenergetics dysfunction in neuronal cells [25, 34, 35], neurite loss in neuronal cultures, and increasing tau polymerization and aggregation in vitro [28, 36]. In vivo, multiphoton imaging in a living tau transgenic mice model (Tg4510 strain), de Calignon et al. detected tau D421, generated by caspase-3-cleavage preceding neurofibrillary tangle pathology and determined that tau D421 promoted the formation of neurofibrillary tangles [37, 38]. Others have also shown that tau cleavage precedes tau tangle pathology [26] and tau oligomer formation in transgenic mice expressing human TauC3 [39]. Moreover, in tau knockout mice, the proportion of caspase-3-cleaved tau at Asp421 doubled in the hippocampus during aging. In this case, cleaved tau induced a toxic gain of function that delayed axonal transport and led to region-specific dendritic atrophy in CA1 neurons [33]. This level of neurotoxicity was confirmed in studies with non-transgenic (male C57BL/6J) mice, caspase-3-cleaved tau increases in the forebrain in an aged-related manner and correlates to cognitive deficits [40]. Likewise, caspase-3-cleaved tau is associated with neurofibrillary tangles and cognitive decline in human brains [26]. Notably, it became clear that caspases 1, 6, 7, and 8 also cleave tau at Asp421 [28](Fig. 2).

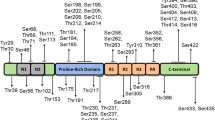
Putative sites caspase-cleaved tau. Caspases 1, 3, 6, 7, and 8 cleave tau at D421. Caspase-2 cleaves tau also at D65 and D314, caspase-3 cleaves tau also at D25, caspase-6 cleaves tau also at D402 and D13. Tau consists of four domains: the projection domain (M1–Y197), a proline-rich region (P1 and P2), the microtubule-binding repeats (R1, R2, R3, R4), and a C-terminus domain (K369–L441). Amino acids 1-441
In humans, tau D421 is found in AD and other tauopathies. Interestingly, E3 ubiquitin ligase CHIP binds the latent C-terminal at tau Asp421. Loss of CHIP expression in AD coincides with the accumulation of tau Asp421, suggesting an interaction between caspases and protein homeostasis in AD and other tauopathies and a therapeutic opportunity [41].
Caspase-6
Evidence that caspase-6 activation is associated with a protracted type of cell death in serum-deprived human primary neurons [42] and renders neurons susceptible to oxidative stress, resulting in either immediate or delayed apoptosis [43], as opposed to the other effector caspases that instead lead to rapid induction of apoptosis. These unique characteristics of caspase-6, less studied than caspase-3, have drawn attention to its potential importance. Caspase-6 in inactivated form is ubiquitous in the human fetal brain and peripheral tissues showing its importance for fetal development [44]. In normal conditions, the human adult brain expresses low levels of caspase 6. However, neuronal activation of caspase-6 is an early event in AD and correlates with adverse clinical outcomes. Increased caspase-6 activity in the anterior olfactory nucleus reflected the degeneration in the entorhinal cortex (affected in Braak stage 1) and correlated with tau pathology in human AD olfactory bulb brain Sect. [45]. Also, in aged non-cognitively impaired individuals, the level of caspase-6 in the entorhinal cortex and CA1 negatively correlates with cognitive domains initially affected in AD [46]. In a study probing the locus coeruleus and dorsal raphe nucleus, brain regions among the first to develop AD-tau pathology, levels of caspase-6 activation in neurons associated with increased Braak staging and burden of neurofibrillary tangles positive for phospho-tau [47]. Levels of caspase-6-cleaved tau are inversely correlated with global cognitive scores in non-demented individuals, supporting tau that cleavage by active caspase-6 may be an early event in AD pathophysiology [21, 48, 49]. In addition to Asp421, caspase-6 cleaves tau at other sites, including Asp402 and Asp13 [23, 32] (Fig. 2). Caspase-6 is particularly efficient in cleaving tau at Asp13 [50]. Caspase-6-cleavage of tau at Asp13 generates two fragments [32]: 1–13 with unknown function and 14–441 with a role in tangle maturation [51, 52]. Caspase-6- cleavage of tau at Asp402 generates a 1-402 fragment, associated with neurodegeneration, and the 403–441 with unknown functions [52]. Altogether, both active caspase-6 and tau truncated at Asp402 and Asp13 are present in neurofibrillary tangles, neuritic plaques, and neuropil threads in sporadic and familial AD but absent in brains without AD pathology [7, 23, 48, 53].
Despite confluent evidence of the role of caspase-6 activation and caspase-6-cleaved in AD pathogenesis [48, 54], only recently monoclonal antibodies against these tau specimens became available to directly investigate the frequency of tau D13 and D402 in tauopathies. Theofilas et al. [7] probed postmortem human brain tissue with a recently developed 5-plex immunohistochemistry with monoclonal novel neoepitope monoclonal antibody against caspase-6 cleaved tau (D402 and D13). The use of multiplex immunostaining allowed these researchers to detect that the number of neurons positive for caspase-6 cleaved tau and phospho-tau in AD is equivalent. However, the overlapping is only 45%. It suggests that currently used antibodies do not flag a significant portion of neurons with tau pathology to label pathological tau in postmortem studies and fluid-based biomarkers based on phospho-tau.
Caspase-1
Caspase-1 has been reported to cleave tau at Asp421 [28], but caspase-1 impact in AD is more likely related to abnormal activation of the NLR family pyrin domain containing 1 (Nlrp1) inflammasome in AD neurons [55]. The Nlrp1 inflammasome is sequentially activated, leading to the activation of caspase-1. Subsequently, caspase-1 triggers caspase-6-mediated neurodegeneration and IL-1β-mediated glial response [56].
Intense active expression of caspase-1 has been detected in the brains of individuals with mild cognitive impairment and dementia due to AD [57]. A selective inhibitor of caspase-1, VX-765, significantly rescued spatial learning, and memory impairments and reduced tau hyperphosphorylation in the brains of senescence-accelerated mouse prone 8 (SAMP8) mice [58]. While the precise role of caspase-1 in AD neuropathology remains unclear, compelling evidence suggests that targeting caspase-1 could be a viable therapeutic approach for addressing AD and possibly other tauopathies [58], alone or combination with drugs targeting Nlrp1 and caspase-6 [59].
Caspase-2
Caspase-2 has been reported to cleave tau at D314 and D65 [29, 30] (Fig. 2). Caspase-2-cleaved tau reversibly impairs memory function in animal and cellular models of tauopathies due to accumulation in dendritic spines and attenuation of synaptic transmission [60]. However, others have shown that caspase-2-cleaved tau shows increased aggregation and accumulation only in vitro, despite strong RNA expression in vivo models, suggesting efficient clearance. In vivo and in vitro, caspase-2-cleaved tau is also recognized by the ubiquitin E3 ligase CHIP, contributing to faster degradation of caspase-2-generated tau fragments [29]. Levels of truncated tau at D314 are elevated in the inferior temporal gyrus of AD and MCI individuals [61]. Incidentally, caspase-2-cleaved tau fragments have been found in other diseases such as Lewy body disease [62], and Huntington’s disease [63], showing that they are not specific to AD or tauopathies [64].
Caspase-cleaved tau in other tauopathies
Only a handful of studies assessed the role of caspase-cleaved tau in non-AD tauopathies, and most studies had focused on tau Asp421 (D421). In PSP, appotosin, a mitochondrial carrier protein, activates caspase-3 and mediates tau cleavage at Asp421 [65]. Ferrer et al. showed TauC3 truncation in PSP in neurons but not in glia [66]. Guillozet-Bongaarts et al. also detected tau Asp421 in neurons but not in glia in AD and PiD [67]. On the other hand, Newman et al. showed tau Asp421 in PiD, PSP, and CBD within regions with neurofibrillary tangles, tufted astrocytes, and Pick bodies [68]. TauC3 was found in FTLD-tau/K317M tufted-like astrocytes and oligodendroglial inclusions [66]. In the same study using multiplex immunohistochemistry and novel tau-cleaved D402 and D13 mentioned above, Theofilas et al. showed that caspase-6 truncated tau is abundant in AD, to a lesser extent in PiD, and almost absent in 4R tauopathies (PSP, CBD, and AGD) in both neurons and glia [7]. Caspase-6 activation levels were also seen at much lower levels in 4R-tauopathies than in AD [7]. Taken together, these limited number of available studies show that although TauC3 is expected in the most common sporadic tauopathies, caspase-6 activation, and caspase-6 cleaved tau fragments are several levels of magnitude more predominant in 3R/4R and 3R tauopathies than in 4R tauopathies, making biomarkers based on caspase-6 cleaved tau potential tools to discriminate between AD and 4R-tauopathies.
Caspase-cleaved tau fragments in CSF
Precise antemortem diagnosis of tauopathies poses a challenge, given the limited predictive accuracy of a neuropathological diagnosis based on the clinical syndrome for most neurodegenerative conditions. As an example, approximately one-third of cases that meet the clinical criteria for corticobasal syndrome exhibit AD pathology as their primary neuropathological feature finding. Furthermore, aside from AD, various conditions can underlie an amnestic syndrome, with limbic predominant age-related TDP-43 encephalopathy (LATE) being one of the most common in aging individuals [69]. The last decade saw an exponential increase in biomarker development aiming to enable differential diagnosis of tauopathies in vivo and monitoring tools to evaluate therapeutics. Most biomarkers for tauopathy are based on total tau and phospho-tau levels. The last decade saw an exponential increase in biomarker development aiming to enable differential diagnosis of tauopathies in vivo and monitoring tools to evaluate therapeutics. Most biomarkers for tauopathy are based on total tau and phospho-tau levels. Phospho-tau-based fluid biomarkers show good specificity and sensibility for detecting AD neuropathology, at least from moderate neuropathological stages [70, 71]. However, phospho-tau-based biomarkers proved to have limited utility in discriminating among tauopathies [72].
Studies on caspase-cleaved tau forms in cerebrospinal fluid (CSF) are limited, yet the emerging findings show promise, underscoring the need for deeper exploration. Using an enzyme-linked immunosorbent assay to detect caspase-6-cleaved tau at Asp402 in postmortem CSF (using a polyclonal antibody), Ramcharitar et al. showed postmortem CSF levels mirror caspase-6-cleaved tau levels and active caspase-6 immunohistochemistry in the hippocampal sections of the same AD individuals and caspase-6-cleaved tau CSF levels correlate with AD severity and lower scores in neuropsychological tests [6]. However, CSF assays based on the tau C-terminal are not ideal because CSF lacks C-terminal tau peptides, making it challenging to detect tau fragments above residue 254 [73,74,75,76,77]. The advancement of immunoassays designed for the detection of N-terminal cleaved-tau fragments holds the potential to serve as a diagnostic tool for Alzheimer’s disease (AD) and distinguish it from other tauopathies [75, 78]. Findings that NT1 fragments (consisting of the N-terminal sequence 6-198) measured in CSF can discriminate between AD and non-AD populations better than full-length tau or tau measured via the middle region alone [5]. Given caspase-6 cleaved tau abundance in AD but scarcity in 4R tauopathies, it is worth testing if a CSF assay for caspase-6 cleaved tau performs better in discriminating AD from 4R tauopathies than phospho-tau based assays. However, the probable most relevant use of a CSF assay for caspase-6 cleaved tau is to detect non-phospho-tau pathology in AD as it seems that neurons with D13 tau are abundant in AD and only partially overlap with phospho-tau in the same neurons [7], making detection of caspase-6 cleaved tau relevance for diagnostic and therapeutic uses. The most likely pertinent application of CSF assay for caspase-6 cleaved tau is identifying non-phospho-tau pathology in AD. This is supported by evidence suggesting an abundance of neurons with D13 tau in AD, which only partially overlaps with phospho-tau in the same neurons [7]. Thus, detecting caspase-6 cleaved tau holds significance for both diagnostic and therapeutic purposes in this context. While a specific biomarker using monoclonal antibodies against caspase-cleaved tau at the N-terminal is currently unavailable, the presented evidence lends strong support to the prospects of its future development. Particularly appealing would be a biomarker centered on caspase-6-cleaved tau at Asp13, given its N-terminal location and the existing availability of a monoclonal antibody [7].
Caspase activation and implications for AD therapeutics
What triggers caspase activation in AD is undefined. Oxidative damage and even accumulation of alpha-synuclein in synucleinopathies result in mitochondrial dysfunction, leading to the release of cytochrome-c and caspase-9 activation, which activate the downstream effector caspase-3 [68]. Also, hydrogen peroxide species induce activation of caspase-3 and − 6, cleaving tau at Asp421 [79]. These two pathways would increase caspase-3 levels and facilitate tau cleavage [80]. When the activity of both caspases was blocked, the amount of cleaved tau was reduced significantly. Amyloid-β can activate caspases and cleave tau contributing to tangle pathology [26]. A study proposed a potential mechanism for activating caspase-8 by amyloid-β peptides in the brain of individuals with AD. The activation occurs via cross-linking with death receptors like Fas.
However, a study using primary human neurons that overexpressed wild-type or mutant APP challenged this model by linking the neurodegeneration process to caspase-6 instead of amyloid-β [42]. This finding suggests that caspase-6 can be activated independently of amyloid-β and at an earlier stage in AD. Moreover, the inhibition of caspase-6 slows caspase-3 activity, indicating a potential interaction between these enzymes. Thus, caspase-6 could potentially participate in activating caspase-3 and promoting the production of truncated tau at Asp421 [79].
Even with the mounting evidence implicating tau toxicity in AD pathogenesis, data from the AlzForum Foundation (www.alzforum.org) suggest that tau-targeting strategies constitute only 10% of the ongoing clinical trials for AD. Among these strategies, efforts to modulate the impact of caspase-truncated tau are relatively limited. For instance, one approach aims to alleviate the toxicity of truncated tau by inhibiting protease activity or selectively weakening protease-substrate interactions [64]. An alternative and attractive method centers around inhibiting caspase activation to reduce tau truncation. Drugs that inhibit caspases, such as minocycline and VX-765, are currently undergoing clinical trials for AD [58, 81,82,83]. Minocycline decreases levels of caspase-cleaved tau by inhibiting caspase-3 activation [83]. VX765, a blood-brain barrier permeable and likely non-toxic Casp1 inhibitor, blocks the Nlrp1-caspase1-caspase6 pathway, attenuating cognitive deficits and microglial activation caused by caspase-6 [59, 81, 82]. Efforts to develop highly selective caspase-6 inhibitors as a therapeutic approach for treating AD are also underway. The challenge in targeting caspase functions arises from the remarkable conservation of their active sites and catalytic machinery. To overcome this limitation, Van Horn and colleagues targeted a non-catalytic cysteine residue (C264) unique to caspase-6 to produce the first generation of a potent and irreversible caspase-6 inhibitor which exhibits selectivity over other caspase family members and high proteome selectivity [84]. Subsequent second and third-generation inhibitors, built upon this initial molecule, are being developed to enhance their potency and bioavailability. This is the starting point for the development of potent and isoform-selective inhibitors for caspase-6 as potential therapeutics. Caspase inhibitors may have the potential to treat tauopathies with significant pathological forms of 3R tau, but their effectiveness in treating 4R tauopathies is uncertain. Inhibiting the enzymes responsible for tau cleavage, such as caspase-6, may promote a significant therapeutic index since caspase-6 knockout mice are more resistant to pro-inflammatory and excitotoxic stimuli, have neuronal damage-induced microglial activation reduced, besides favorable outcomes in memory and neurological hallmarks [85, 86]. Further studies on caspase inhibitors are strongly encouraged.
Conclusion and future directions
The advances in caspase-cleaved tau biomarkers and therapy promise an auspicious future for tauopathies research and move the field toward better diagnoses and disease-modifying events. The potential use of biomarkers in precision medicine is exciting, and caspase-cleaved tau in CSF may add the missing piece to track AD pathology in vivo. CSF D13 caspase-6-cleaved tau is the appealing biofluid biomarker to differentiate AD from 4R-tauopathies.
Our review reveals gaps in knowledge and overlooks significant aspects of the pathology of AD. Further research is needed to investigate the role of fragments produced by caspase-6 cleaved tau at D13 in AD. Additionally, it is crucial to understand in which stage caspase-6 cleaved tau is involved in the progression of AD, as well as the timing of the co-occurrence and dissociation between caspase-6 cleaved tau and phospho-tau pathology. Finally, in addition to developing specific inhibitor drugs for caspase-6, existing drugs could be repurposed to inhibit caspase-6 cleaved tau in AD and other tauopathies.
Data availability
Not applicable.
Change history
12 April 2024
A Correction to this paper has been published: https://doi.org/10.1186/s40478-024-01772-5
Abbreviations
- AD:
-
Alzheimer’s Disease
- AGD:
-
Argyrophilic Grain Disease
- APP:
-
Amyloid Precursor Protein
- ARTAG:
-
Aging-related Tau Astrogliopathy
- Asp:
-
Aspartic Acid
- CBD:
-
Corticobasal Degeneration
- CTE:
-
Chronic Traumatic Encephalopathy
- CSF:
-
Cerebrospinal Fluid
- GGT:
-
Globular Glial Tauopathy
- IL-1β:
-
Interleukin-1 Beta
- MCI:
-
Mild Cognitive Impairment
- MAPT:
-
Microtubule-Associated Protein Tau
- Nlrp1:
-
NLR Family Pyrin Domain Containing 1
- NLR:
-
Nucleotide-Binding Oligomerization Domain, Leucine-Rich Repeat
- NT1:
-
N-Terminal 1
- PiD:
-
Pick’s Disease
- PSP:
-
Progressive Supranuclear Palsy
- PTM:
-
Post-Translational Modification
- SAMP8:
-
Senescence-Accelerated Mouse Prone 8
- 3R:
-
Three Repeat
- 4R:
-
Four Repeat
- 3R/4R:
-
Three and Four Repeats
References
Silva MC, Haggarty SJ (2020) Tauopathies: deciphering Disease mechanisms to develop effective therapies. Int J Mol Sci 21. https://doi.org/10.3390/ijms21238948
Holper S, Watson R, Yassi N (2022) Tau as a biomarker of Neurodegeneration. Int J Mol Sci 23. https://doi.org/10.3390/ijms23137307
Ossenkoppele R, van der Kant R, Hansson O (2022) Tau biomarkers in Alzheimer’s disease: towards implementation in clinical practice and trials. Lancet Neurol 21:726–734. https://doi.org/10.1016/S1474-4422(22)00168-5
Opland CK, Bryan MR, Harris B, McGillion-Moore J, Tian X, Chen Y, Itano MS, Diering GH, Meeker RB, Cohen TJ (2023) Activity-dependent tau cleavage by caspase-3 promotes neuronal dysfunction and synaptotoxicity. iScience 26:106905. https://doi.org/10.1016/j.isci.2023.106905
Chen Z, Mengel D, Keshavan A, Rissman RA, Billinton A, Perkinton M, Percival-Alwyn J, Schultz A, Properzi M, Johnson K al (2019) Learnings about the complexity of extracellular tau aid development of a blood-based screen for Alzheimer’s disease. Alzheimers Dement 15:487–496. https://doi.org/10.1016/j.jalz.2018.09.010
Ramcharitar J, Albrecht S, Afonso VM, Kaushal V, Bennett DA, Leblanc AC (2013) Cerebrospinal fluid tau cleaved by caspase-6 reflects brain levels and cognition in aging and Alzheimer disease. J Neuropathol Exp Neurol 72:824–832. https://doi.org/10.1097/NEN.0b013e3182a0a39f
Theofilas P, Piergies AMH, Oh I, Lee YB, Li SH, Pereira FL, Petersen C, Ehrenberg AJ, Eser RA, Ambrose AJ al (2022) Caspase-6-cleaved tau is relevant in Alzheimer’s disease and marginal in four-repeat tauopathies: diagnostic and therapeutic implications. Neuropathol Appl Neurobiol 48:e12819. https://doi.org/10.1111/nan.12819
Kovacs GG (2017) Tauopathies. Handb Clin Neurol 145:355–368. https://doi.org/10.1016/B978-0-12-802395-200025– 0
Karch CM, Kao AW, Karydas A, Onanuga K, Martinez R, Argouarch A, Wang C, Huang C, Sohn PD Bowles KR (2019) a Comprehensive Resource for Induced Pluripotent Stem cells from patients with primary tauopathies. Stem cell Rep 13: 939–955 https://doi.org/10.1016/j.stemcr.2019.09.006
(2023) Alzheimer’s disease facts and figures. Alzheimers Dement: Doi https://doi.org/10.1002/alz.13016
Nolan A, De Paula Franca Resende E, Petersen C, Neylan K, Spina S, Huang E, Seeley W, Miller Z, Grinberg LT (2019) Astrocytic tau deposition is frequent in typical and atypical Alzheimer Disease presentations. J Neuropathol Exp Neurol 78:1112–1123. https://doi.org/10.1093/jnen/nlz094
Rodriguez RD, Suemoto CK, Molina M, Nascimento CF, Leite RE, de Lucena Ferretti-Rebustini RE, Farfel JM, Heinsen H, Nitrini R, Ueda K et al (2016) Argyrophilic Grain Disease: Demographics, Clinical, and Neuropathological Features From a Large Autopsy Study. J Neuropathol Exp Neurol 75: 628–635 https://doi.org/10.1093/jnen/nlw034
Swallow DMA, Zheng CS, Counsell CE (2022) Systematic review of Prevalence studies of Progressive Supranuclear Palsy and Corticobasal Syndrome. Mov Disord Clin Pract 9:604–613. https://doi.org/10.1002/mdc3.13489
Driver-Dunckley ED, Zhang N, Serrano GE, Dunckley NA, Sue LI, Shill HA, Mehta SH, Belden C, Tremblay C, Atri Aet al et al (2023) Low clinical sensitivity and unexpectedly high incidence for neuropathologically diagnosed progressive supranuclear palsy. J Neuropathol Exp Neurol 82:438–451. https://doi.org/10.1093/jnen/nlad025
Alquezar C, Arya S, Kao AW (2020) Tau post-translational modifications: dynamic transformers of tau function, degradation, and aggregation. Front Neurol 11:595532. https://doi.org/10.3389/fneur.2020.595532
Avila J, Pallas N, Bolós M, Sayas CL, Hernandez F (2016) Intracellular and extracellular microtubule associated protein tau as a therapeutic target in Alzheimer disease and other tauopathies. Expert Opin Ther Targets 20:653–661. https://doi.org/10.1517/14728222.2016.1131269
Lothrop AP, Torres MP, Fuchs SM (2013) Deciphering post-translational modification codes. FEBS Lett 587:1247–1257. https://doi.org/10.1016/j.febslet.2013.01.047
Zilka N, Kovacech B, Barath P, Kontsekova E, Novák M (2012) The self-perpetuating tau truncation circle. Biochem Soc Trans 40:681–686. https://doi.org/10.1042/BST20120015
Boyarko B, Hook V (2021) Human tau isoforms and proteolysis for production of toxic tau fragments in Neurodegeneration. Front Neurosci 15:702788. https://doi.org/10.3389/fnins.2021.702788
Wang XJ, Cao Q, Zhang Y, Su XD (2015) Activation and regulation of caspase-6 and its role in neurodegenerative diseases. Annu Rev Pharmacol Toxicol 55:553–572. https://doi.org/10.1146/annurev-pharmtox-010814-124414
Graham RK, Ehrnhoefer DE, Hayden MR (2011) Caspase-6 and neurodegeneration. Trends Neurosci 34:646–656. https://doi.org/10.1016/j.tins.2011.09.001
Van Opdenbosch N, Lamkanfi M (2019) Caspases in cell death, inflammation, and Disease. Immunity 50:1352–1364. https://doi.org/10.1016/j.immuni.2019.05.020
Guo H, Albrecht S, Bourdeau M, Petzke T, Bergeron C, LeBlanc AC (2004) Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease. Am J Pathol 165:523–531. https://doi.org/10.1016/S0002-9440(10)63317-2
Friedlander RM (2003) Apoptosis and caspases in neurodegenerative diseases. N Engl J Med 348:1365–1375. https://doi.org/10.1056/NEJMra022366
Olesen MA, Quintanilla RA (2023) Pathological impact of tau proteolytical process on neuronal and mitochondrial function: a crucial role in Alzheimer’s Disease. Mol Neurobiol 60:5691–5707. https://doi.org/10.1007/s12035-023-03434-4
Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, LaFerla FM, Rohn TT, Cotman CW (2004) Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest 114:121–130. https://doi.org/10.1172/JCI20640
Cotman CW, Poon WW, Rissman RA, Blurton-Jones M (2005) The role of caspase cleavage of tau in Alzheimer disease neuropathology. J Neuropathol Exp Neurol 64:104–112. https://doi.org/10.1093/jnen/64.2.104
Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N al (2003) Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci U S A 100:10032–10037. https://doi.org/10.1073/pnas.1630428100
Reinhardt L, Musacchio F, Bichmann M, Behrendt A, Ercan-Herbst E, Stein J, Becher I, Haberkant P, Mader J, Schöndorf DC al (2023) Dual truncation of tau by caspase-2 accelerates its CHIP-mediated degradation. Neurobiol Dis 182:106126. https://doi.org/10.1016/j.nbd.2023.106126
Zhao X, Kotilinek LA, Smith B, Hlynialuk C, Zahs K, Ramsden M, Cleary J, Ashe KH (2016) Caspase-2 cleavage of tau reversibly impairs memory. Nat Med 22:1268–1276. https://doi.org/10.1038/nm.4199
Corsetti V, Amadoro G, Gentile A, Capsoni S, Ciotti MT, Cencioni MT, Atlante A, Canu N, Rohn TT, Cattaneo A al (2008) Identification of a caspase-derived N-terminal tau fragment in cellular and animal Alzheimer’s disease models. Mol Cell Neurosci 38:381–392. https://doi.org/10.1016/j.mcn.2008.03.011
Horowitz PM, Patterson KR, Guillozet-Bongaarts AL, Reynolds MR, Carroll CA, Weintraub ST, Bennett DA, Cryns VL, Berry RW, Binder LI (2004) Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer’s disease. J Neurosci 24:7895–7902. https://doi.org/10.1523/JNEUROSCI.1988-04.2004
Conze C, Rierola M, Trushina NI, Peters M, Janning D, Holzer M, Heinisch JJ, Arendt T, Bakota L, Brandt R (2022) Caspase-cleaved tau is senescence-associated and induces a toxic gain of function by putting a brake on axonal transport. Mol Psychiatry 27:3010–3023. https://doi.org/10.1038/s41380-022-01538-2
Pérez MJ, Ibarra-García-Padilla R, Tang M, Porter GA, Johnson GVW, Quintanilla RA (2023) Caspase-3 cleaved tau impairs mitochondrial function through the opening of the mitochondrial permeability transition pore. Biochim Biophys Acta Mol Basis Dis 1870:166898. https://doi.org/10.1016/j.bbadis.2023.166898
Pérez MJ, Vergara-Pulgar K, Jara C, Cabezas-Opazo F, Quintanilla RA (2018) Caspase-cleaved tau Impairs Mitochondrial Dynamics in Alzheimer’s Disease. Mol Neurobiol 55:1004–1018. https://doi.org/10.1007/s12035-017-0385-x
Noël A, Foveau B, LeBlanc AC (2021) Caspase-6-cleaved tau fails to induce tau hyperphosphorylation and aggregation, neurodegeneration, glial inflammation, and cognitive deficits. Cell Death Dis 12:227. https://doi.org/10.1038/s41419-021-03506-0
Avila J (2010) Alzheimer disease: caspases first. Nat Rev Neurol 6:587–588. https://doi.org/10.1038/nrneurol.2010.157
de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT (2010) Caspase activation precedes and leads to tangles. Nature 464:1201–1204. https://doi.org/10.1038/nature08890
Kim Y, Choi H, Lee W, Park H, Kam TI, Hong SH, Nah J, Jung S, Shin B, Lee H al (2016) Caspase-cleaved tau exhibits rapid memory impairment associated with tau oligomers in a transgenic mouse model. Neurobiol Dis 87:19–28. https://doi.org/10.1016/j.nbd.2015.12.006
Means JC, Gerdes BC, Kaja S, Sumien N, Payne AJ, Stark DA, Borden PK, Price JL, Koulen P (2016) Caspase-3-Dependent proteolytic cleavage of tau causes neurofibrillary tangles and results in cognitive impairment during normal aging. Neurochem Res 41:2278–2288. https://doi.org/10.1007/s11064-016-1942-9
Ravalin M, Theofilas P, Basu K, Opoku-Nsiah KA, Assimon VA, Medina-Cleghorn D, Chen YF, Bohn MF, Arkin M, Grinberg LT al (2019) Specificity for latent C termini links the E3 ubiquitin ligase CHIP to caspases. Nat Chem Biol 15:786–794. https://doi.org/10.1038/s41589-019-0322-6
Sivananthan SN, Lee AW, Goodyer CG, LeBlanc AC (2010) Familial amyloid precursor protein mutants cause caspase-6-dependent but amyloid β-peptide-independent neuronal degeneration in primary human neuron cultures. Cell Death Dis 1:e100. https://doi.org/10.1038/cddis.2010.74
Zhang Y, Goodyer C, LeBlanc A (2000) Selective and protracted apoptosis in human primary neurons microinjected with active caspase-3, -6, -7, and– 8. J Neurosci 20:8384–8389. https://doi.org/10.1523/JNEUROSCI.20-22-08384.2000
Godefroy N, Foveau B, Albrecht S, Goodyer CG, LeBlanc AC (2013) Expression and activation of caspase-6 in human fetal and adult tissues. PLoS ONE 8:e79313. https://doi.org/10.1371/journal.pone.0079313
Foveau B, Albrecht S, Bennett DA, Correa JA, LeBlanc AC (2016) Increased Caspase-6 activity in the human anterior olfactory nuclei of the olfactory bulb is associated with cognitive impairment. Acta Neuropathol Commun 4:127. https://doi.org/10.1186/s40478-016-0400-x
Ramcharitar J, Afonso VM, Albrecht S, Bennett DA, LeBlanc AC (2013) Caspase-6 activity predicts lower episodic memory ability in aged individuals. Neurobiol Aging 34:1815–1824. https://doi.org/10.1016/j.neurobiolaging.2013.01.007
Theofilas P, Ehrenberg AJ, Nguy A, Thackrey JM, Dunlop S, Mejia MB, Alho AT, Paraizo Leite RE, Rodriguez RD, Suemoto CK et al (2018) Probing the correlation of neuronal loss, neurofibrillary tangles, and cell death markers across the Alzheimer’s disease Braak stages: a quantitative study in humans. Neurobiol Aging 61: 1–12 https://doi.org/10.1016/j.neurobiolaging.2017.09.007
Albrecht S, Bourdeau M, Bennett D, Mufson EJ, Bhattacharjee M, LeBlanc AC (2007) Activation of caspase-6 in aging and mild cognitive impairment. Am J Pathol 170:1200–1209. https://doi.org/10.2353/ajpath.2007.060974
LeBlanc AC (2013) Caspase-6 as a novel early target in the treatment of Alzheimer’s disease. Eur J Neurosci 37:2005–2018. https://doi.org/10.1111/ejn.12250
Wang Y, Garg S, Mandelkow EM, Mandelkow E (2010) Proteolytic processing of tau. Biochem Soc Trans 38:955–961. https://doi.org/10.1042/BST0380955
Horowitz PM, LaPointe N, Guillozet-Bongaarts AL, Berry RW, Binder LI (2006) N-terminal fragments of tau inhibit full-length tau polymerization in vitro. Biochemistry 45:12859–12866. https://doi.org/10.1021/bi061325g
Quinn JP, Corbett NJ, Kellett KAB, Hooper NM (2018) Tau proteolysis in the pathogenesis of tauopathies: neurotoxic fragments and novel biomarkers. J Alzheimers Dis 63:13–33. https://doi.org/10.3233/JAD-170959
Albrecht S, Bogdanovic N, Ghetti B, Winblad B, LeBlanc AC (2009) Caspase-6 activation in familial alzheimer disease brains carrying amyloid precursor protein or presenilin i or presenilin II mutations. J Neuropathol Exp Neurol 68:1282–1293. https://doi.org/10.1097/NEN.0b013e3181c1da10
Ghoshal N, García-Sierra F, Wuu J, Leurgans S, Bennett DA, Berry RW, Binder LI (2002) Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer’s disease. Exp Neurol 177:475–493. https://doi.org/10.1006/exnr.2002.8014
Kaushal V, Dye R, Pakavathkumar P, Foveau B, Flores J, Hyman B, Ghetti B, Koller BH, LeBlanc AC (2015) Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ 22:1676–1686. https://doi.org/10.1038/cdd.2015.16
Guo H, Pétrin D, Zhang Y, Bergeron C, Goodyer CG, LeBlanc AC (2006) Caspase-1 activation of caspase-6 in human apoptotic neurons. Cell Death Differ 13:285–292. https://doi.org/10.1038/sj.cdd.4401753
Heneka MT, O’Banion MK (2007) Inflammatory processes in Alzheimer’s disease. J Neuroimmunol 184:69–91. https://doi.org/10.1016/j.jneuroim.2006.11.017
Tan MS, Liu Y, Hu H, Tan CC, Tan L (2022) Inhibition of caspase-1 ameliorates tauopathy and rescues cognitive impairment in SAMP8 mice. Metab Brain Dis 37:1197–1205. https://doi.org/10.1007/s11011-022-00914-9
Flores J, Noël A, Fillion ML, LeBlanc AC (2022) Therapeutic potential of Nlrp1 inflammasome, Caspase-1, or Caspase-6 against Alzheimer disease cognitive impairment. Cell Death Differ 29:657–669. https://doi.org/10.1038/s41418-021-00881-1
Pockes S, Walters MA, Ashe KH (2023) Targeting caspase-2 interactions with tau in Alzheimer’s disease and related dementias. Translational Research: J Lab Clin Med 254:34–40. https://doi.org/10.1016/j.trsl.2022.10.009
Liu P, Smith BR, Montonye ML, Kemper LJ, Leinonen-Wright K, Nelson KM, Higgins L, Guerrero CR, Markowski TW, Zhao Xet al et al (2020) A soluble truncated tau species related to cognitive dysfunction is elevated in the brain of cognitively impaired human individuals. Sci Rep 10:3869. https://doi.org/10.1038/s41598-020-60777-x
Smith BR, Nelson KM, Kemper LJ, Leinonen-Wright K, Petersen A, Keene CD, Ashe KH (2019) A soluble tau fragment generated by caspase-2 is associated with dementia in Lewy body disease. Acta Neuropathol Commun 7:124. https://doi.org/10.1186/s40478-019-0765-8
Liu P, Smith BR, Huang ES, Mahesh A, Vonsattel JPG, Petersen AJ, Gomez-Pastor R, Ashe KH (2019) A soluble truncated tau species related to cognitive dysfunction and caspase-2 is elevated in the brain of Huntington’s disease patients. Acta Neuropathol Commun 7:111. https://doi.org/10.1186/s40478-019-0764-9
Yang J, Shen N, Shen J, Yang Y, Li HL (2023) Complicated role of post-translational modification and protease-cleaved fragments of tau in Alzheimer’s Disease and other tauopathies. Mol Neurobiol: Doi. https://doi.org/10.1007/s12035-023-03867-x
Zhao Y, Tseng IC, Heyser CJ, Rockenstein E, Mante M, Adame A, Zheng Q, Huang T, Wang X, Arslan PE al (2015) Appoptosin-mediated caspase cleavage of tau contributes to Progressive Supranuclear Palsy Pathogenesis. Neuron 87:963–975. https://doi.org/10.1016/j.neuron.2015.08.020
Ferrer I, Lopez-Gonzalez I, Carmona M, Arregui L, Dalfo E, Torrejon-Escribano B, Diehl R, Kovacs GG (2014) Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol 73:81–97. https://doi.org/10.1097/NEN.0000000000000030
Guillozet-Bongaarts AL, Glajch KE, Libson EG, Cahill ME, Bigio E, Berry RW, Binder LI (2007) Phosphorylation and cleavage of tau in non-AD tauopathies. Acta Neuropathol 113:513–520. https://doi.org/10.1007/s00401-007-0209-6
Newman J, Rissman RA, Sarsoza F, Kim RC, Dick M, Bennett DA, Cotman CW, Rohn TT, Head E (2005) Caspase-cleaved tau accumulation in neurodegenerative diseases associated with tau and alpha-synuclein pathology. Acta Neuropathol 110:135–144. https://doi.org/10.1007/s00401-005-1027-3
Nelson PT, Brayne C, Flanagan ME, Abner EL, Agrawal S, Attems J, Castellani RJ, Corrada MM, Di Cykowski MD J et al (2022) Frequency of LATE neuropathologic change across the spectrum of Alzheimer’s disease neuropathology: combined data from 13 community-based or population-based autopsy cohorts. Acta Neuropathol 144: 27–44 https://doi.org/10.1007/s00401-022-02444-1
Olsson B, Lautner R, Andreasson U, Ohrfelt A, Portelius E, Bjerke M, Holtta M, Rosen C, Olsson C, Strobel G et al (2016) CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol 15: 673–684 https://doi.org/10.1016/S1474-4422(16)000703
Xia Y, Prokop S, Giasson BI (2021) Don’t Phos over tau: recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Mol Neurodegener 16:37. https://doi.org/10.1186/s13024-021-00460-5
Meeter LHH, Vijverberg EG, Del Campo M, Rozemuller AJM, Donker Kaat L, de Jong FJ, van der Flier WM, Teunissen CE, van Swieten JC, Pijnenburg YAL (2018) Clinical value of neurofilament and phospho-tau/tau ratio in the frontotemporal dementia spectrum. Neurology 90:e1231–e1239. https://doi.org/10.1212/WNL.0000000000005261
Arai T, Ikeda K, Akiyama H, Nonaka T, Hasegawa M, Ishiguro K, Iritani S, Tsuchiya K, Iseki E, Yagishita Set al et al (2004) Identification of amino-terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann Neurol 55:72–79. https://doi.org/10.1002/ana.10793
Cicognola C, Brinkmalm G, Wahlgren J, Portelius E, Gobom J, Cullen NC, Hansson O, Parnetti L, Constantinescu R, Wildsmith K al (2019) Novel tau fragments in cerebrospinal fluid: relation to tangle pathology and cognitive decline in Alzheimer’s disease. Acta Neuropathol 137:279–296. https://doi.org/10.1007/s00401-018-1948-2
Lantero-Rodriguez J, Tissot C, Snellman A, Servaes S, Benedet AL, Rahmouni N, Montoliu-Gaya L, Therriault J, Brum WS, Stevenson Jet al et al (2023) Plasma and CSF concentrations of N-terminal tau fragments associate with in vivo neurofibrillary tangle burden. Alzheimers Dement: https://doi.org/10.1002/alz.13119
Meredith JE, Sankaranarayanan S, Guss V, Lanzetti AJ, Berisha F, Neely RJ, Slemmon JR, Portelius E, Zetterberg H, Blennow Ket al et al (2013) Characterization of novel CSF tau and ptau biomarkers for Alzheimer’s disease. PLoS ONE 8:e76523. https://doi.org/10.1371/journal.pone.0076523
Sato C, Barthélemy NR, Mawuenyega KG, Patterson BW, Gordon BA, Jockel-Balsarotti J, Sullivan M, Crisp MJ, Kasten T, Kirmess KM et al (2018) Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 97: 1284–1298.e1287 https://doi.org/10.1016/j.neuron.2018.02.015
Chu D, Yang X, Wang J, Zhou Y, Gu JH, Miao J, Wu F, Liu F (2024) Tau truncation in the pathogenesis of Alzheimer’s disease: a narrative review. Neural Regen Res 19:1221–1232. https://doi.org/10.4103/1673-5374.385853
Zhao H, Zhao W, Lok K, Wang Z, Yin M (2014) A synergic role of caspase-6 and caspase-3 in tau truncation at D421 induced by H2O 2. Cell Mol Neurobiol 34:369–378. https://doi.org/10.1007/s10571-013-0021-x
Rohn TT, Rissman RA, Davis MC, Kim YE, Cotman CW, Head E (2002) Caspase-9 activation and caspase cleavage of tau in the Alzheimer’s disease brain. Neurobiol Dis 11:341–354. https://doi.org/10.1006/nbdi.2002.0549
Flores J, Noël A, Foveau B, Beauchet O, LeBlanc AC (2020) Pre-symptomatic Caspase-1 inhibitor delays cognitive decline in a mouse model of Alzheimer disease and aging. Nat Commun 11:4571. https://doi.org/10.1038/s41467-020-18405-9
Flores J, Noël A, Foveau B, Lynham J, Lecrux C, LeBlanc AC (2018) Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat Commun 9:3916. https://doi.org/10.1038/s41467-018-06449-x
Noble W, Garwood C, Stephenson J, Kinsey AM, Hanger DP, Anderton BH (2009) Minocycline reduces the development of abnormal tau species in models of Alzheimer’s disease. FASEB J 23:739–750. https://doi.org/10.1096/fj.08-113795
Van Horn KS, Wang D, Medina-Cleghorn D, Lee PS, Bryant C, Altobelli C, Jaishankar P, Leung KK, Ng RA, Ambrose AJet al et al (2023) Engaging a non-catalytic cysteine Residue drives potent and selective inhibition of Caspase-6. J Am Chem Soc 145:10015–10021. https://doi.org/10.1021/jacs.2c12240
Angel A, Volkman R, Royal TG, Offen D (2020) Caspase-6 Knockout in the 5xFAD Model of Alzheimer’s Disease Reveals Favorable Outcome on Memory and Neurological Hallmarks. Int J Mol Sci 21. https://doi.org/10.3390/ijms21031144
Ladha S, Qiu X, Casal L, Caron NS, Ehrnhoefer DE, Hayden MR (2018) Constitutive ablation of caspase-6 reduces the inflammatory response and behavioural changes caused by peripheral pro-inflammatory stimuli. Cell Death Discov 4:40. https://doi.org/10.1038/s41420-018-0043-8
Acknowledgements
Figures were created with BioRender.com.
Funding
This work was supported by National Institutes of Health K24AG053435, R01AG075802, and U54NS123746-01 and by the São Paulo Research Foundation (FAPESP) grant numbers 2022/10299-2 and 2019/23028-4 (Rizzi), Alzheimer Association AARG-16-441514. The funding source had no role in the study design, in the writing of the report, and in the decision to submit the paper for publication.
Author information
Authors and Affiliations
Contributions
LR: writing-original draft; LTG: writing - review & editing, supervision, project administration.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Following the publication of the original article, it was noted that due to a typesetting error the figure legends were paired incorrectly. The figure legends for Figs. 1, 2 were wrongly given as captions for Figs. 2, 1 respectively. The publisher apologizes for the inconvenience caused.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rizzi, L., Grinberg, L.T. Exploring the significance of caspase-cleaved tau in tauopathies and as a complementary pathology to phospho-tau in Alzheimer’s disease: implications for biomarker development and therapeutic targeting. acta neuropathol commun 12, 36 (2024). https://doi.org/10.1186/s40478-024-01744-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-024-01744-9