
Abstract
Phosphorylation is one of the most prevalent post-translational modifications found in aggregated tau isolated from Alzheimer’s disease (AD) patient brains. In tauopathies like AD, increased phosphorylation or hyperphosphorylation can contribute to microtubule dysfunction and is associated with tau aggregation. In this review, we provide an overview of the structure and functions of tau protein as well as the physiologic roles of tau phosphorylation. We also extensively survey tau phosphorylation sites identified in brain tissue and cerebrospinal fluid from AD patients compared to age-matched healthy controls, which may serve as disease-specific biomarkers. Recently, new assays have been developed to measure minute amounts of specific forms of phosphorylated tau in both cerebrospinal fluid and plasma, which could potentially be useful for aiding clinical diagnosis and monitoring disease progression. Additionally, multiple therapies targeting phosphorylated tau are in various stages of clinical trials including kinase inhibitors, phosphatase activators, and tau immunotherapy. With promising early results, therapies that target phosphorylated tau could be useful at slowing tau hyperphosphorylation and aggregation in AD and other tauopathies.
Similar content being viewed by others


Main text
Overview of tau structure and function
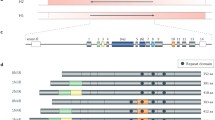
Tau protein is encoded in the microtubule associated protein tau (MAPT) gene on chromosome 17. In the human brain, alternative RNA splicing of exons 2, 3, and 10 lead to the expression of six major tau isoforms: 0N3R, 1N3R, 2N3R, 0N4R, 1N4R, and 2N4R (Fig. 1) [6,7,8]. Exon 2 and 3 encode two different N-terminal domains of 29 amino acids and their presence or absence results in either 0 N, 1 N, or 2 N isoforms. Exon 10 encodes the second microtubule-associated binding repeat (MTBR) and its alternative splicing differentiates between 3R and 4R tau isoforms [9]. 3R tau isoforms contain the first, third, and fourth MTBR, while 4R tau isoforms contain all four MTBR. In the adult human brain, the six major tau isoforms are expressed at different abundance: 0 N isoforms make up ~ 40% of all isoforms, while ~ 50% are 1 N isoforms and ~ 10% are 2 N isoforms [10, 11]. Expression of 3R and 4R isoforms is relatively equal throughout the brain [10, 11].

Schematic showing 2N4R tau (441 amino acids), the longest isoform expressed in human brain. Tau protein contains major structural domains including N-terminal domain with N1 and N2 inserts, proline rich region, four major microtubule-binding repeats (R1-R4), and C-terminal domain. The N1, N2 and R2 regions can be alternatively spliced in the human brain resulting in 6 isoforms: 0N3R, 1N3R, 2N3R, 0N4R, 1N4R, and 2N4R. The position of identified phosphorylation sites found in AD brains are shown [1,2,3,4,5]
Tau is expressed and localized primarily in the axons of neurons and at lower levels in glial cells [12, 13]. Tau is overall hydrophilic and basic, consisting of several structural regions including the N-terminal projection region, proline-rich domain, MTBR, and the C-terminal region [14] (shown in Fig. 1). The MTBR region is positively charged facilitating binding with negatively charged microtubules (MT) [15, 16]. The proline rich domain is also involved in MT binding and regulation; its partial or complete deletion can lead to MT impairment [14].
Since its discovery in 1975, tau was named as a tubulin associated unit due to its functions in promoting the growth and extension of MT [17]. The MTBR within tau have a high homology with other microtubule-associated proteins (MAPs) that maintain MT. Tau can bind to the interface between alpha-tubulin and beta-tubulin heterodimer and stabilize the MT structure [18,19,20]. Recently, it has been proposed that the effects of tau may be more indirect, and tau elongates the labile domain rather than stabilize MT [21]. Nonetheless, tau is important for the homeostatic growing and shrinking of MT along the neuronal axon. Tau can also regulate motor proteins such as dynein and kinesin and affect the rates of axonal transport [22, 23].
Tau is involved in neuronal maturation and maintenance of cytoarchitecture, evidenced by smaller axons and dendrites in tau knockout mice [24, 25]. Some aged tau knockout mice developed spatial memory deficits [26], while another lineage of tau null mice developed motor deficits without cognitive changes [27]. Differences in genetic background may explain the divergence in behavioral phenotypes [28]. Interestingly, tau knockdown by AAV-delivered RNA interference in adult mice impaired spatial learning and memory and lead to decreased synaptic proteins and dendritic spine density [29], while restoration of tau expression rescued the behavioral deficits in these mice. Overall, these studies suggest that loss of tau function is likely detrimental for cognition and memory.
Physiologic roles of tau phosphorylation
Tau protein can be post-translationally modified by enzymatic additions of acetylation, methylation, glycosylation, ubiquitination, and many others [30, 31]. As discussed below, of these post-translation modifications (PTMs), phosphorylation is one of the earliest and most prevalent modifications associated the formation of pathological inclusions. In the longest tau isoform expressed in the human CNS (2N4R isoform), there are over 85 potential phosphorylation sites represented by either serine, threonine, or tyrosine residues [8]. Phosphorylation is performed by kinases that add a phosphate group from adenosine triphosphate. Three major classes of kinases can phosphorylate tau [32]: proline-directed kinases such as glycogen synthase kinase 3 beta (GSK-3β) or cyclin dependent kinase 5 (CDK5) [33, 34], non-proline-directed kinases such as tau-tubulin kinases (TTBK) or microtubule affinity regulated kinases (MARK) [35, 36], and tyrosine kinases such as Fyn or Abl kinases [37, 38]. Dephosphorylation or the removal of a phosphate group is performed by phosphatases. Protein phosphatase 2A (PP2A) is the major enzyme that accounts for ~ 71% of the total tau dephosphorylation activity [39, 40], while other phosphatases involved include PP1, PP5, and PP2B [40].
Tau phosphorylation is elevated during some physiologic processes such as development, hibernation, and hypothermia. During development, fetal tau is highly phosphorylated at multiple sites including Thr181, Ser199, Ser202, Thr205, Thr231, Ser396 and Ser404 [41,42,43]. After the developmental period, tau phosphorylation returns to steady state levels in part because mature brains have higher phosphatase activity [43]. Tau phosphorylation at multiple sites has been found in mammals that experience hibernation such as arctic ground squirrels, Syrian hamsters and American black bears [44,45,46]. Increased tau phosphorylation during hibernation is physiologic and likely due to decreased metabolic rate, which can affect both kinase and phosphatase activity [46]. This process is reversible since phosphorylation levels return to normal levels after arousal. Similarly, drug-induced anesthesia can also lead to hypothermia and induce hyperphosphorylation by either activating kinases or by inhibiting phosphatase activity, which returns to normal levels after anesthesia is removed [47, 48].
Site-specific phosphorylation can regulate tau-MT interactions and MT assembly. The MTBR region is generally positively charged to improve interactions with negatively charged MT and the addition of a negative phosphate group via phosphorylation generally results in decreased MT binding. Some studies have shown that tau phosphorylation at Ser231 or Ser262 can directly inhibit MT interactions [49, 50]. Site specific tau phosphorylation of other sites such as Ser208 lead to increased MT binding, suggesting that the regulation of MT binding greatly depends on the phosphorylation site [51]. In addition to MT binding, phosphorylation at Thr231, Ser262, Ser396, or Ser404 can decrease the ability of tau to promote MT assembly and regulate MT dynamic stability [52]. Interestingly, multi-site phosphorylation can restore normal MT assembly and may be inherently complex in its modulation of MT function [52]. The consequences of MT regulation also can affect axonal transport: tau phosphorylation at Tyr18 can regulate tau’s inhibition of motor proteins such as kinesin-1 and is generally a protective mechanism that promotes physiological axonal trafficking [53, 54]. Conversely, phosphorylation of Ser422 can overall inhibit anterograde and retrograde axonal transport [55]. These studies demonstrate that the physiologic MT regulation by tau is bidirectionally modulated by the sum of multiple phosphorylation sites.
Evidence of elevated tau phosphorylation in AD
Alzheimer’s disease (AD) is the most common form of dementia and a devastating neurodegenerative disorder that affects over 5.8 million Americans [56]. By 2050, this number is projected to increase to 13.8 million [56]. Along with Aβ amyloid plaques, neurofibrillary tangles (NFT) made of phosphorylated tau are one of the major pathological hallmarks of AD and are cornerstones of the current NIA criteria for the post-mortem diagnosis of AD neuropathologic change [57, 58]. NFT directly correlate with progressive decline in dementia scores and development of cognitive deficits, making phosphorylated tau (p-tau) a potential driver of neurotoxicity in AD [59, 60].
In early studies, aggregated tau isolated from AD brains was found to have 3–4 fold higher overall phosphorylation levels (8 mol per protein) compared to healthy controls (2–3 mol per protein) [61]. This increase in tau phosphorylation within pathological inclusions allows for their detection with tau phosphorylation-specific antibodies (Fig. 2). Numerous mass spectrometry studies have mapped phosphorylation sites in detergent-insoluble tau isolated from AD and healthy control brains [1,2,3,4]. A recent quantitative mass spectrometry study examined a cohort of 49 AD patients and 42 AD control subjects and greatly expanded the number of potential phosphorylation sites found in AD and control brains [5]. A compilation of these results reveals at least 59 phosphorylation sites found in AD compared to at least 19 sites in control brains (Listed in Table 1 and shown in Fig. 1). There is considerable overlap between many sites found in AD and control brains.

Tau pathological inclusions associated with neurodegenerative diseases stained with antibodies specific for p-tau. Neurofibrillary tangles (A) and neuritic plaques (B) in AD stained with antibody AT8 that reacts with tau phosphorylated at Ser202 and Thr205. Tufted astrocytes (C) in PSP stained with antibody AT8. Antibody 3G12 specific for tau phosphorylated at Ser208 depicts neurofibrillary tangles (D) and neuritic plaques (E, asterisks) in AD. Astrocytic plaques (F) in CBD stained with antibody AT8. Scale bar = 60 μm
Since hyperphosphorylation is a major component of tau pathological inclusions, phosphorylation-specific antibodies have been very useful for tracking the progression of tau pathology. Braak staging of NFT pathology in AD was traditionally performed with Gallyas silver stain [64]; however, the AT8 antibody against tau phosphorylated at Ser202/Thr205 was found to be just as effective at tracking the progression of tau pathology [65, 66]. According to the Braak staging scheme, tau spreads in a stereotypical manner in most patients: NFT composed of hyperphosphorylated tau are found in the transentorhinal region in early stages (stage I-II), in limbic regions such as hippocampus in middle stages (stage III-IV), and throughout the neocortex in late stages (stage V-VI) [64, 65]. Phosphorylation at other sites has been found to be distinctively increased in more advanced Braak stages (stages V/VI) including Tyr18, Ser199, Thr231, and Ser422 [67]. Interestingly Tyr18 and Thr231 are found to be phosphorylated in earlier Braak stages (stages III/IV) [67].
In a follow up study, Braak and colleagues also found that some cases had unique types of AT8 positive and Gallyas negative pre-tangles in the brainstem and some cortical areas, even before any bona fide NFT were detectable in the transentorhinal region. These cases were designated as prodromal stages with pre-tangles that contain phosphorylated tau [68]. Antibodies against other phosphorylation sites such as Thr153, Thr231, and Ser262 have stained pre-tangles early in disease [69]. These findings suggest that different phosphorylation sites are distinctively involved in early versus late stages of NFT progression in AD.
Effects of tau hyperphosphorylation on microtubule interactions, aggregation and prion-like spread
In pathologic states like AD, hyperphosphorylated tau exhibits impaired MT binding and is less capable of promoting MT assembly contributing to a breakdown of MT in late stages of AD [70]. Another consequence of decreased MT binding is the mislocalization of tau from the axon to the cytosol. In a tau knock-in mouse model with 27 different phosphomimetics that model hyperphosphorylation, axonal tau was mislocalized into the somatodendritic compartment [71], similar to what is observed in early tau lesions in human brains.
Multi-site phosphorylation can lead to conformational changes that make tau protein prone to misfolding and aggregation [72]. Experimental tau pseudophosphorylation at Ser199, Ser202, Thr205, Ser396, and Ser404 led to disruption of the paperclip conformation and induction of a pathological conformation that may be more prone to aggregation [73]. Multiple phosphorylation sites such as Thr175, Ser202, Thr205, Thr212, and Ser422 have been shown to promote aggregation and lead to the formation of tau filaments in vitro [55, 74,75,76]. Triple phosphorylation at Ser202/Thr205/Ser208 has been proposed to be a combination that leads to rapid tau aggregation [51, 77]. On the other hand, phosphorylation sites such as Ser214, Ser262, and Ser305 have been demonstrated to inhibit tau aggregation and may be neuroprotective [78, 79]. In late disease stages, the hyperphosphorylation of multiple pro-aggregation sites may overcome protective ones. It is also unclear whether certain specific tau phospho-epitopes are added before or after the formation of tau inclusions, which may mitigate potential neuroprotective effects.
Like other proteins involved in neurodegeneration, tau has been proposed to spread by prion-like mechanisms where tau seeds can induce aggregation of wild type tau and spread from neuron to neuron [80,81,82]. Both unmodified tau fibrils [83] and phosphorylated tau seeds [84] can induce the spread of tau pathology when injected into the brains of tau transgenic mice. When these seeds are dephosphorylated by phosphatases, they are less effective at inducing tau pathology. Similarly, tau seeds that were immunodepleted with phosphorylation-specific antibodies were less capable of inducing tau aggregation, suggesting that phosphorylation may be a characteristic of potent tau seeds [85]. Specific phosphorylated tau residues including Ser262, and combined Thr231/Ser235 found in AD brain lysate enhance tau seeding efficiency, while other sites such as Ser198/Ser199/Ser202 and Ser400/Thr403/Ser404 can be associated with lower seeding efficiency [86]. This suggests that tau phosphorylation can heterogeneously modulate tau seeding capacity by either enhancing or diminishing tau spread.
Heterogeneity of tau species in different Tauopathies
In addition to AD, tauopathies encompass a group of heterogeneous neurological diseases that share the common feature of brain-related tau pathological inclusions. These include subtypes of Frontotemporal lobar degeneration (FTLD)-tau, such as corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), Pick’s Disease (PiD), globular glial tauopathy (GGT) and argyrophilic brain disease (AGD), as well as the distinct entity of chronic traumatic encephalopathy (CTE). Various missense, silent and intronic MAPT mutations cause familial forms of frontotemporal dementia with parkinsonism [87, 88] and these mutations are associated with tau hyperphosphorylation, MT dysfunction, and aggregation [87, 89,90,91,92]. Intronic, silent and some of the missense mutations cause disease by altering the splicing efficiency of tau exon 10 and changing the ratio of 3R to 4R isoforms [93]. Some MAPT missense mutations reduce MT binding and increase phosphorylation, while a small subset of them promote tau aggregation [87, 89,90,91,92]. Cases with MAPT mutations have traditionally been referred to as frontotemporal degeneration and parkinsonism linked to chromosome 17 (FTDP)-17, but recent nomenclature schemes re-classify them under familial forms of FTLD-tau since patients with familial tau mutations have similar tau pathology as sporadic FTLD cases [94] and to avoid confusion with cases of FTLD-TDP associated with mutations in the GRN gene which is also found on chromosome 17.
Within the group of tauopathies, there is diversity in the morphology, regional distribution and cell type specificity of tau inclusions [95]. Several of the major tauopathies including AD, CBD, and PSP are shown in Fig. 2 with staining by AT8 (pSer202, pThr205) or 3G12 (pSer208) antibodies. In AD, tau pathology consists mostly of neuronal pathology including NFT (Fig. 2A, D), neuropil threads, and dystrophic neurites within senile plaques (Fig. 2B, E). In addition to neuronal tau pathology in the form of globose tangles, subtypes of FTLD-tau are associated with glial tau inclusions including tufted astrocytes in PSP (Fig. 2C) and astrocytic plaques in CBD (Fig. 2F) [96]. PiD has distinctive cytoplasmic Pick bodies in neurons [97], while GGT has characteristic globular glial inclusions [98]. CTE, a secondary tauopathy associated with repetitive brain injuries, develops pathognomonic p-tau positive inclusions in neurons, astrocytes and cell processes mostly around blood vessels and distinct cortical regions [99].
The differences in tau pathology are also reflected structurally within different tau filament folds. Recent cryo-electron microscopy studies have uncovered differential tau structure for AD, PiD, CTE, and CBD [100,101,102,103]. PTMs may partially contribute to conformational differences of tau filaments. Ubiquitination and acetylation of lysine sites may differ between AD and CBD tau filaments [104]. Different phosphorylation sites may also help differentiate AD from other tauopathies. For example, pSer208 antibody which strongly stained neuronal NFT, but barely detected astrocytic pathology in PSP and CBD [51]. This may be due to the different kinase environment in various forms of tau inclusions.
Due to the structural and PTM differences, tau filaments from different tauopathies may represent separate tau prion-like strains. Human brain lysates from AD, PSP, CBD, AGD and GGT induce different forms of tau pathology when injected into the brains of tau transgenic mice and even non-transgenic mice [105,106,107,108]. AD brain lysate results in primarily neuronal pathology, while PSP, CBD, AGD and GGT lead to disease-specific glial pathology. Since tauopathies may represent different tau strains, there is potential for biomarkers like phosphorylation sites that could differentiate AD from other tauopathies.
Development of p-tau CSF and plasma biomarkers
The National Institute on Aging and Alzheimer’s Association recommended defining AD by neuropathology and biomarkers for tracking of disease progression [109]. Biomarkers have been grouped in the ATN framework as either detecting Aβ aggregation (A), tau aggregation (T), or neurodegeneration (N). Currently, the gold standards for tau-based biomarkers include pThr181 tau within the cerebrospinal fluid (CSF) and tau detected by positron emission tomography (PET) [109]. Multiple studies are ongoing to evaluate different tau-specific PET tracers [110]. However, there is a need for the expansion of multiple biomarkers. Here, we present an overview of recent developments in novel biomarker assays for the detection of different p-tau sites in CSF and blood.
CSF p-tau biomarkers
CSF is produced by ependymal cells of the choroid plexus and can be sampled by lumbar puncture. The development of an effective CSF tau biomarker could help track the progression of AD and symptoms. Overall tau protein levels and p-tau are significantly elevated in AD compared to controls. Much of the tau proteins in CSF are truncated tau fragments that are often phosphorylated [62, 63]. Most of the phosphorylation sites within the CSF are clustered in the proline rich domain, which suggests a unique set of kinases that are responsible for phosphorylation of tau secreted into CSF. Table 1 summarizes at least 19 major phosphorylation sites that are significantly elevated in the CSF of AD compared to healthy adults.
Soluble phosphorylated tau in CSF has been identified to be a potential early marker of AD disease progression. Increased pThr217 and pThr181 appear simultaneously during the formation of Aβ amyloid plaques, about 20 years before the onset of AD [111]. pThr205 was found to be elevated later when decreased brain metabolism and neuronal dysfunction occur about 13 years before AD onset. In the final stages, NFT formation is detected by PET-tau imaging during AD onset. These findings show that soluble hyperphosphorylated tau species are distinctively elevated within the CSF decades before AD diagnosis.
CSF pThr181 tau
From a meta-analysis of p-tau CSF studies in AD, pThr181 is one of the most evaluated CSF tau biomarkers and is consistently elevated in AD compared to age-matched healthy controls [112, 113]. It is considered one of the gold standards and is included within the biomarker definition of AD [109]. pThr181 is also elevated in patients with mild cognitive impairment (MCI) [114]. Interestingly, pThr181 is increased in AD, but in non-AD dementia such as FTD, pThr181 is significantly decreased compared to healthy controls [115, 116]. In a cohort of 361 participants, a low ratio of pThr181 to total tau levels can differentiate FTD from healthy controls with 73% sensitivity and 93% specificity [115]. However, pThr181 is not significantly different between PSP, CBD, or other FTD variants [115].
CSF pThr217 tau
Since pThr217 was found to be elevated early along with pThr181, several new immunoassays have been developed to directly compare pThr217 with pThr181. Eli Lilly developed a new ELISA to detect pThr217. In AD, pThr217 had a higher range of 7.3–9.6 fold change versus 3.6–3.7 fold change in pThr181 [117]. Compared to Thr181, pThr217 correlated better with progressive NFT pathology as determined by PET imaging. pThr217 was slightly better at differentiating AD from non-AD with a 91% sensitivity and 91% specificity, compared to pThr181 which had 79% sensitivity and 96% specificity [117].
Another ELISA assay detects pThr217 using validated antibodies, DC2E2 and DC2E7, that both detect tau pathology in AD and other tauopathies [118]. In a direct comparison, they showed that pThr217 can distinguish AD from healthy controls with 98% specificity and 93% sensitivity [118]. This is slightly better than pThr181 that identified AD with specificity and 86% sensitivity. pThr217 was also better at differentiating AD from tauopathies like PiD, PSP, and CBD with a 90% accuracy, while pThr181 had lower accuracy of 78% [118].
A different approach is to use quantitative mass spectrometry to determine p-tau changes in CSF [119]. With this method, pThr217 had 6-fold change in AD patients compared controls, while pThr181 was elevated with 1.3-fold change. Overall levels of pThr217 may be higher than pThr181, which could make it easier to detect by immunoassays.
CSF pThr231 tau
Another early epitope is pThr231, which has been detected to be elevated in CSF of patients with AD and patients with MCI when compared to healthy controls [120, 121]. pThr231 can differentiate patients with AD from other conditions such as FTD, vascular dementia, and LBD with 90.2% sensitivity and 80% specificity [122]. For identifying patients with AD, pThr231 may have better sensitivity than pThr181 and detect less false positives [123]. Interestingly, AD patients who are ApoE ε4 carriers have higher levels of pThr231, but no difference in pThr181 [123, 124]. Another study found that pThr231, pThr181, and pThr217 were similarly elevated in preclinical AD, before the appearance significant Aβ pathology [125]. All three of these phosphorylation sites are likely elevated during early stages of AD and can be detected in the CSF.
Many of the recent studies have been designed to determine the best biomarker for AD and other tauopathies. However, it can be difficult to compare the results of different studies due to the variety of antibodies used in different immunoassays. The choice of detection and capture antibody in sandwich ELISAs greatly affects the efficacy of these assays. For instance, many immunoassays use similar antibodies against pThr181 and pThr217 as the capture antibody but may differ in the choice of the detection antibody. Instead of using the standard mid-domain tau antibody for detection (amino acid residues 159–163), another group used an N-terminal tau antibody (amino acid residues 6–18) as the detection antibody to bind a longer tau peptide. The N-terminal directed pThr181 and pThr217 immunoassays were better than the mid-domain directed pThr181 assay at differentiating patients with AD and MCI [126]. There are many additional p-tau sites found within the CSF (Table 1) that could be new potential sites that predict different stages of AD progression. There may also be phosphorylation signatures specific to other tauopathies that are not found in AD. In the future, expanding the repertoire of p-tau biomarkers and using a combination of multiple assays will improve the diagnostic accuracy and tracking of AD and other tauopathies.
Plasma p-tau biomarkers
Blood-based p-tau biomarkers are currently in development and evaluated for their prognostic value primarily in AD. Blood tests are advantageous over CSF tests, because they are minimally invasive and less expensive to evaluate in a large population. Since there are lower levels of tau protein in the blood compared to CSF, blood tests may require a highly sensitive assay to detect minute amounts of p-tau. Other cell types and proteins found in blood may further complicate detection. Overall plasma tau levels are elevated in AD compared to patients with MCI and healthy controls [127]. Due to the success of p-tau in CSF, p-tau epitopes such as pThr181 and pThr217 were also evaluated in plasma ELISA assays.
Plasma pThr181 tau
Plasma pThr181 levels are significantly elevated in AD at 3.5 times more compared to healthy controls [128]. It generally correlates with CSF pTau181 levels and Aβ levels measured by PET imaging [128, 129]. Additionally, Plasma pThr181 has diagnostic value and can differentiate AD from other neurodegenerative diseases such as FTLD with 70–80% sensitivity and specificity [128]. A longitudinal prospective study tracked a cohort of 1113 participants with follow up 8 years later and found that plasma pThr181 tau levels is associated with progressive decline in cognitive symptoms and brain atrophy [130]. This association was also found in patients with MCI, suggesting that plasma pThr181 could be an early predictor of neurodegeneration and cognitive decline.
Plasma pThr217 tau
Similar to plasma pThr181, Plasma pThr217 is increased in AD and can distinguish AD from other neurodegenerative diseases such as PD, PSP, and FTD in a cohort of 1402 participants [131]. Comparable to CSF pThr217 assays, plasma levels were also associated with worsening cognitive scores and overall brain atrophy [131, 132]. Plasma pThr217 may be a good predictor of early disease progression prior to AD. In a cohort of 490 participants of healthy controls and patients with MCI, both plasma and CSF pThr217 tau increased before major changes on tau PET imaging [132]. As an early marker, plasma pThr217 can predict the progression of AD in a longitudinal study tracked a cohort of 250 participants up to 6 years. Patients with MCI who eventually developed AD had higher baseline pThr217.
Recent advancements in plasma p-tau assays have demonstrated that these can be as effective as their CSF counterparts. Both plasma pThr181 and pThr217 tau are good early biomarkers of AD and predictor of disease progression in multiple longitudinal studies. Plasma p-tau is highly elevated in AD compared to other related neurodegenerative diseases such as FTD. Further studies with larger sample sizes will better validate the efficacy of plasma p-tau levels compared to other markers in AD. A combination of multiple p-tau biomarkers in addition to pThr181 and pThr217 could improve the sensitivity and specificity of tracking early AD progression.
Therapies that target tau phosphorylation
Currently, there is a lack of effective disease-modifying treatments for AD. Tau and particularly tau phosphorylation, has become an attractive target, because it is involved in early disease progression and can be tracked by various biomarkers. Recently, several clinical trials primarily in Phase I/II have been completed or ongoing involving several therapeutic strategies to reduce tau phosphorylation: kinase inhibitors, phosphatase activators, and p-tau immunotherapy (Fig. 3). Kinase inhibitors and phosphatase activators aim to decrease tau phosphorylation levels early on (Table 2) and p-tau immunotherapy will lead to the clearance of phosphorylated tau (Table 3), thus achieving a similar goal.

Summary of therapies targeting p-tau. A Kinase inhibitors such as Tideglusib, lithium, valproate and nilotinib act to prevent hyperphosphorylation. Phosphatase activators such as sodium selenate increases dephosphorylation activity. B Passive p-tau immunotherapy are specific antibodies that target p-tau epitopes for degradation. Active p-tau immunotherapy involves immunization with a p-tau peptide to generate antibodies. Figure was made with Biorender
Kinase inhibitors and phosphatase activators
Tideglusib
Tideglusib is a thiadiazolidinone-based drug that can competitively inhibit the enzymatic activity of glycogen synthase kinase 3-β (GSK3-β) [133, 134]. In transgenic mouse models that express both tau and Aβ pathology, Tideglusib reduced tau phosphorylation and Aβ levels, ameliorated neuronal death, and rescued cognitive memory deficits [135].
Due to these positive preclinical results, a pilot Phase I trial of Tideglusib tested safety and different doses (400, 600, 800, and 1000 mg) given daily for 4–6 weeks in patients with mild or moderate AD [136]. There were no major side effects except for elevated serum transaminases found on liver function tests. Patients on the 1000 mg daily dose schedule showed an uptrend in dementia scores, but due to the small sample size of 30 participants, it may be difficult to extrapolate the results.
A follow-up Phase II study expanded the number of participants to 306 patients with mild or moderate AD, testing 500 mg versus 1000 mg daily doses [137]. Similar to the previous study, there were no major side effects found although 14–18% of patients experienced diarrhea, and a few had transaminase elevations. Overall, the results showed that there was no significant clinical benefit as determined by the primary outcome of dementia inventory scores. There was a slight uptrend for the 500 mg dose group with improvements in different dementia scores and word fluency.
Another phase II trial tested the effects of Tideglusib in 37 patients with PSP at 600 or 800 mg daily for a year [138]. Tideglusib treatment reduced brain atrophy throughout the brain, especially for the parietal and occipital lobes as measured by MRI imaging. There were no major changes in clinical dementia scores, but the reduction of brain atrophy is very promising and the extension of treatment time to 1 year may have a stronger effect.
Lithium
Lithium salts are oral drugs approved for treatment of mood disorders including bipolar disorder and major depressive disorder [139]. Lithium was initially mainly considered for the treatment of agitation and psychosis in AD [140]. Preclinical studies suggest that lithium is also a direct reversible inhibitor of GSK-3β activity and may be capable of decreasing tau hyperphosphorylation and slowing AD disease progression [141]. In transgenic mouse models, chronic treatment with lithium prevented tau phosphorylation and slowed disease progression [142, 143], but existing mature NFT were not significantly removed [142].
In an initial trial of 71 mild AD patients, 10 weeks of lithium (titrated to a maximum of 336 mg daily dose) did not significantly alter CSF p-tau or lymphocyte GSK-3β activity [144]. The other primary outcomes of dementia scores were also not changed. Subsequent trials had more success with longer treatment periods and in patients with early MCI. Forty-five patients with MCI received 150 to 600 mg daily doses for 1 year, which significantly decreased CSF pThr181 tau and improved cognitive symptoms as measured by the clinical dementia rating (CDR) [145]. Similar results were positive in another trial of 61 patients with MCI that received lithium for 2 years [146]. Overall, they received more stable scores on Alzheimer’s disease assessment scale (ADAS) and CDR dementia scales. Even lower doses at 300 μg daily for 15 months in AD patients was able to prevent declining MMSE scores [147]. Longer treatment schedules and use in patients with early symptoms like MCI showed promising results for biomarker and dementia score improvements. A current phase IV clinical trial of lithium in MCI patients is ongoing until 2023 [148].
Valproate
Valproate is approved for treatment of bipolar disorder and epilepsy [149, 150]. In the context of tau, valproate has been demonstrated to be an inhibitor of both GSK3-β and cyclin dependent kinase 5 (cdk5), thus decreasing tau phosphorylation [151]. This was confirmed in transgenic mouse models where valproate beneficially restored synaptic loss and decreased tau phosphorylation [152]. Another study found that valproate inhibits Aβ plaque formation and improved cognitive deficits [153].
Initially, valproate was considered for clinical use of treating agitation and aggression in AD [154, 155], but it could also potentially slow AD progression by targeting tau and Aβ pathology. A phase II trial found that AD disease progression and cognitive decline was not significantly altered by valproate treatment [156]. Another phase II trial showed that valproate led to worsening dementia scores as assessed by the mini-mental state examination (MMSE) [157]. Both studies detected that valproate treatment resulted in increased brain atrophy on MRI imaging [156, 157]. This is a rare side effect of valproate that leads to reversible brain atrophy and has been previously reported [158, 159]. Due to the serious side effects and lack of efficacy, valproate may not be the best candidate compared to other kinase inhibitors.
Nilotinib
Nilotinib is a pyrimidinyl benzamide inhibitor of the tyrosine kinase Abl and is primarily used to treat chronic myeloid leukemia [160]. Recently, Nilotinib has been considered for use in neurological disease such as AD. Abl tyrosine kinase is involved in tau phosphorylation and its inhibition could slow hyperphosphorylation. Nilotinib showed other benefits such as autophagic clearance of phosphorylated tau and improvements in neurotransmitter imbalance in tau transgenic mice [161]. Nilotinib may also help clear Aβ amyloid and improve cognitive deficits by affecting soluble parkin [162].
A phase II trial tested safety and tolerance of nilotinib in 37 patients with mild or moderate AD and measured different biomarkers [163]. Nilotinib was given daily for 26 weeks in either 150 or 300 mg doses. These treatments were generally well tolerated with no significant side effects except for mood swings with agitation and irritability in the higher dosed 300 mg group. Measurement of biomarkers showed reduction of pThr181 tau and Aβ in CSF and slowing of hippocampal atrophy as measured by imaging. These results support the potential benefit of nilotinib on reducing both tau and Aβ pathology.
Sodium selenate
Sodium selenate has been identified as a potent activator of PP2A phosphatase enzymatic activity, leading to tau dephosphorylation [164, 165]. In tau transgenic mouse models, oral treatment with sodium selenate reduces tau hyperphosphorylation, tau pathology progression, and behavioral and cognitive deficits [164,165,166]. By targeting tau phosphorylation in the early stages, it could be possible to slow or prevent the formation of tau inclusions.
In Australia, a phase II clinical trial examined supranutritional levels of VEL015, a formulation of sodium selenate (10 mg taken three times daily), for 24 weeks in patients with mild or moderate AD [167]. Sodium selenate was generally safe with some minor side effects of headache, fatigue and nausea. There were no major changes in the dementia scores, but there was a significant improvement in MRI diffusivity, indicating increased membrane integrity. The exploratory measurement of total tau and pThr181 tau in CSF showed no major differences, but there was a downward trend.
A follow-up study collected data from the previous trial and measured serum and CSF selenium levels, showing that selenate uptake into CSF varied greatly from patient to patient [168]. In a group with high CSF selenium levels, these patients showed improvement in MMSE cognitive scores. Future studies that better control CSF selenate uptake could show better improvement in dementia scores and changes in CSF tau biomarkers. A new phase II trial on sodium selenate in behavioral variant of FTD will use higher doses (15 mg taken three times daily) in a target cohort of 120 patients [169]. This trial is currently in recruitment and is expected to be completed by 2024.
Overall, kinase inhibitors and phosphatase activators have shown some effect at altering tau biomarkers, but effects on cognitive symptoms were mixed. Some kinase inhibitors like valproate have serious side effects. Lower doses and slower titration schedules could help improve tolerance and decrease complications. Targeting kinase and phosphatase activity likely will likely have a larger clinical effect during early disease, such as in patients with MCI or mild AD.
Phospho-tau immunotherapy
Tau immunotherapy is one of the most promising tau-targeting therapeutics in recent years due to much greater target specificity. There are two major strategies: the first is active immunotherapy which involves the injection of a peptide to generate an intrinsic immune response against that epitope. An advantage is that multiple antibodies may arise from a given vaccine and offer sustained protection, but this may not be as effective in older patients with weaker immune systems. The second strategy is the use of passive immunotherapy or the peripheral infusion of an antibody that can bind a specific epitope, but this would require repeat infusions. Passive immunotherapy has proved to be more popular, since preclinical studies in different animal models have demonstrated that tau-targeting antibodies can lead to clearance of toxic tau aggregates and inclusions [85, 170,171,172,173,174]. This strategy also allows an active pipeline for clinical development since an antibody made in other mammals such as mice or rabbits can be screened and humanized in a cost-efficient way. Humanized antibodies retain its variable binding domain and epitope specificity, but its constant domain is human to reduce unwanted immune responses to the antibody. In addition to tau antibodies, tau intrabodies to target intracellular tau have also been effective in preclinical studies, but have not been tested in clinical trials [175, 176].
Peripheral administration such as intravenous routes may only deliver small amounts of antibodies to the central nervous system across the blood brain barrier (BBB). In MCI and dementia, the BBB can be slightly leaky [177, 178] allowing for higher levels of antibodies to enter the CNS [170, 179]. Tau-targeting antibodies may be taken up by neurons via clathrin-dependent endocytosis and lead to autophagy-dependent protein degradation of tau aggregates [180]. Extracellularly, antibodies can bind to secreted tau and activate microglia to engulf and help degrade tau aggregates based on cell culture experiments [181]. This could inhibit transcellular spread of tau from neuron to neuron, thus slowing the disease progression of different tauopathies [85, 182].
The efficacy of passive immunotherapy will largely depend on choice of epitope and properties of the antibody such as binding affinity. Many antibodies target total tau and aim to lower overall tau levels, but this may also decrease physiologic functional tau. The alternative is to target phosphorylation-specific epitopes, which can allow for more specific clearance of pathogenic tau with less removal of functional tau that may be needed to maintain MT and axonal transport. However, specific phospho-tau antibodies are unlikely to capture all tau species. In recent years, many antibodies tested in preclinical models have been humanized and taken to phase I and phase II clinical trials. Many of these immunotherapies target phosphorylation epitopes in the proline rich region or N-terminus of tau. Here, we will summarize the current progress of p-tau specific immunotherapy and ongoing clinical trials (listed in Table 3).
JNJ-63733657 antibody against pThr217 tau
JNJ-63733657 is a humanized IgG1 antibody that is specific for tau phosphorylated at Thr217 and is under development by Janssen Biotech, a subsidiary of Johnson and Johnson. It was found that targeting phosphorylated tau epitopes near the proline-rich domain was effective at blocking the spread of tau seeds in cell-based assays and in animal models [183]. Based on these positive results, the antibody was humanized and tested in two Phase I trials [184, 185]. In the initial Phase I trial, healthy subjects from 55 to 75 years of age were treated with intravenous injections of JNJ-63733657 [186, 187]. The antibody was generally well tolerated without significant side effects, though some patients experienced back pain and headache. Antibody concentrations increased linearly with the dosage amount and can enter the CSF. Based on these positive results, a Phase II trial is started in 2021 to recruit up to 420 participants with early AD to test a low and high dose injection every 4 weeks for several years [188]. The trial is expected to conclude in 2025.
PNT001 against cis p-Thr231 tau
Tau phosphorylation at Thr231 can exist in either a cis or trans isomer and its interconversion is catalyzed by Pin1, a propyl isomerase [189]. Cis-pThr231 tau but not trans-pThr231 tau impairs MT assembly and is prone to tau aggregation in AD and CTE [172, 190]. A monoclonal cis-pThr231 tau antibody (clone 113) was developed to be cis isomer specific at pThr231 [171]. In mouse models of traumatic brain injury (TBI), peripheral delivery of cis- pThr231 antibody prevented short term and long-term outcomes of TBI, leading to reduced spread of tau axonal pathology, decreased neuroinflammation, and improved motor coordination deficits [171, 172]. On the other hand, the trans-pThr231 tau antibody was not effective, suggesting that cis-pThr231 tau is the primary driver of tau pathology and neurodegeneration in TBI. The cis-pThr231 tau antibody was humanized as PNT001 by Pinteon Therapeutics and will evaluated in ongoing Phase I trials of healthy adults and patient with acute TBI, which are scheduled to be completed in 2021 and 2022 [191, 192].
Lu AF87908 antibody against pSer396 tau
To help inhibit cell-to-cell spread of hyperphosphorylated tau, C10.2 antibody was screened as a mouse monoclonal antibody that can specifically target tau phosphorylated at Ser396. Tau seeds immunodepleted by C10.2 are less competent at inducing tau aggregation and spread in cellular assays and in rTg4510 tau transgenic mice [85]. Specifically, the C10.2 antibody has been demonstrated to help mediate lysosomal degradation of pathogenic tau by microglia in cell culture experiments [181], and in general, antibody uptake is dependent on its isotype and other characteristics [193]. This antibody has been humanized as Lu AF87908 by H. Lundbeck company and will be tested in a Phase I clinical trial, which is expected to be completed in May of 2021 [194].
RG7345 antibody against pSer422 tau
RG7345 is the humanized form of the Mab86 antibody and is thought to be specific for tau phosphorylated Ser422. Mab86 was made by immunizing rabbits with a tau peptide from 416 to 430 residues that contains phosphorylated Ser422. Peripheral administration of the antibody in triple transgenic mice with both tau and Aβ pathology resulted in significant reduction of tau inclusions [170]. The antibody was confirmed to enter neurons and lysosomes to help clear p-tau. Roche started a Phase I trial on RG7345 to compare the safety profile and serum levels; however, the use of RG7345 has been discontinued and no further trials have been announced [195].
ACI-35 vaccine
AC Immune SA developed ACI-35 as a liposome-based vaccine that consists of a C-terminal tau fragment (393–408 residues) containing phosphorylated Ser396 and Ser404. In C57/BL6 and tau-P301L transgenic mice [196], injection of the ACI-35 peptide generated a robust immune response, yielding multiple antibodies that are specific for p-tau and react with tau inclusions [197]. A preliminary Phase I trial of ACI-35 in AD patients showed no major safety concerns, but the vaccine elicited a weak immune response [198, 199]. A new improved version of ACI-35.030 was made with additional epitopes to improve T cell responses, which was tested and confirmed in rhesus monkeys. A Phase I and II trial in patients with early AD is ongoing to determine the safety and immune response of ACI-35.030 based on three different doses [200]. The trial is expected to complete by 2022.
Overall, initial clinical results from p-tau specific immunotherapies have been promising and have a good safety profile without significant side effects. Many of these Phase I and II trials are ongoing in the next few years and these results will be important for future development of other immunotherapies. It will be prudent to develop tau immunotherapies against multiple tau epitopes. There is considerable heterogeneity in the degree of post-translational modifications such as phosphorylation in different patient populations. Different tauopathies may also harbor various tau strains that have variations in phosphorylation signature. The development of a cocktail of p-tau immunotherapies may be the most effective method for efficient removal of pathogenic tau species.
Conclusion
P-tau is a hallmark of tau pathological inclusions in AD and other tauopathies. During late disease stages, tau aggregates isolated from AD brains are hyperphosphorylated at multiple residues (Fig. 1 and Table 1). Tau hyperphosphorylation can lead to MT dysfunction, mislocalization, and increased propensity for aggregation. However, it is still not completely understood how and if tau hyperphosphorylation directly leads to aggregation and neuronal death. Nonetheless, specific sites are phosphorylated in early disease stages and can provide insight as potential biomarkers. Recent advancements in the development of blood and plasma biomarkers including pThr181 and pThr217 have been good predictors of disease progression from MCI to symptomatic AD and may help differentiate AD from FTD. These non-invasive tests combined with imaging and clinical dementia scores will improve early detection and tracking of potential AD, FTD, and other tauopathies.
Tau phosphorylation is also a potential target for clinical therapies. Several drug classes are currently considered for clinical trials: kinase inhibitors, phosphatase activators, and p-tau immunotherapy (Fig. 3). Kinase inhibitors and phosphatase activators are promising and may work better in patients with MCI or early AD to prevent or slow progression, but are more likely to have off-target effects. Likely, a combination of multiple drugs will be necessary to prevent tau phosphorylation since many kinases are involved and all phosphorylate multiple sites. Many antibodies used for p-tau immunotherapy are still in Phase I and II but have fewer side effects and are relatively safer compared to kinase inhibitors. In the next few decades, more clinical trial data will offer a better picture on whether drugs against p-tau will have disease-modifying effects in AD and other tauopathies.
Availability of data and materials
Not applicable.
Abbreviations
- Aβ:
-
Beta-amyloid
- AD:
-
Alzheimer’s disease
- ADAS :
-
Alzheimer’s disease assessment score
- AGD:
-
Argyrophilic grain disease
- CBD:
-
Corticobasal degeneration
- CDK5:
-
Cyclin dependent kinase 5
- CDR:
-
Clinical dementia rating
- CSF:
-
Cerebrospinal fluid
- CTE:
-
Chronic traumatic encephalopathy
- FTDP-17:
-
Frontotemporal degeneration and parkinsonism linked to chromosome 17
- FTLD:
-
Frontotemporal lobar degeneration
- GGT:
-
Globular glial tauopathy
- GSK-3β:
-
Glycogen synthase kinase 3β
- MAP:
-
Microtubule-associated protein
- MAPT:
-
Microtubule-associated protein tau
- MARK:
-
Microtubule affinity regulated kinases
- MCI:
-
Mild cognitive impairment
- MT:
-
Microtubule
- MTBD:
-
Microtubule binding domain
- NFTs:
-
Neurofibrillary tangles
- PET:
-
Positron emission tomography
- PHFs:
-
Paired helical filaments
- PiD:
-
Pick’s disease
- PP2A:
-
Protein phosphatase 2A
- PSP:
-
Progressive supranuclear palsy
- p-tau:
-
Phosphorylated tau
- TBI:
-
Traumatic brain injury
- TTBK:
-
Tau-tubulin kinase
References
Hanger DP, Byers HL, Wray S, Leung K-Y, Saxton MJ, Seereeram A, et al. Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J Biol Chem. 2007;282(32):23645–54. https://doi.org/10.1074/jbc.M703269200.
Hasegawa M, Morishima-Kawashima M, Takio K, Suzuki M, Titani K, Ihara Y. Protein sequence and mass spectrometric analyses of tau in the Alzheimer’s disease brain. J Biol Chem. 1992;267(24):17047–54. https://doi.org/10.1016/S0021-9258(18)41890-X.
Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Titani K, et al. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem. 1995;270(2):823–9. https://doi.org/10.1074/jbc.270.2.823.
Hanger DP, Betts JC, Loviny TLF, Blackstock WP, Anderton BH. New phosphorylation sites identified in Hyperphosphorylated tau (paired helical filament-tau) from Alzheimer’s disease brain using Nanoelectrospray mass spectrometry. J Neurochem. 2002;71(6):2465–76. https://doi.org/10.1046/j.1471-4159.1998.71062465.x.
Wesseling H, Mair W, Kumar M, Schlaffner CN, Tang S, Beerepoot P, et al. Tau PTM profiles identify patient heterogeneity and stages of Alzheimer’s disease. Cell. 2020;183:1699–1713.e13. https://doi.org/10.1016/j.cell.2020.10.029.
Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci. 1988;85(11):4051–5. https://doi.org/10.1073/pnas.85.11.4051.
Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 1989;8(2):393–9. https://doi.org/10.1002/j.1460-2075.1989.tb03390.x.
Goedert M, Spillantini MGG, Jakes R, Rutherford D, Crowther RAA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3(4):519–26. https://doi.org/10.1016/0896-6273(89)90210-9.
Lee G, Neve RL, Kosik KS. The microtubule binding domain of tau protein. Neuron. 1989;2(6):1615–24. https://doi.org/10.1016/0896-6273(89)90050-0.
Trabzuni D, Wray S, Vandrovcova J, Ramasamy A, Walker R, Smith C, et al. MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies. Hum Mol Genet. 2012;21(18):4094–103. https://doi.org/10.1093/hmg/dds238.
Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–7. https://doi.org/10.1126/science.282.5395.1914.
Black MM, Slaughter T, Moshiach S, Obrocka M, Fischer I. Tau is enriched on dynamic microtubules in the distal region of growing axons. J Neurosci. 1996;16(11):3601–19. https://doi.org/10.1523/JNEUROSCI.16-11-03601.1996.
LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP, Binder LI. Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. Proc Natl Acad Sci. 1995;92(22):10369–73. https://doi.org/10.1073/pnas.92.22.10369.
Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. Domains of τ protein and interactions with microtubules. Biochemistry. 1994;33(32):9511–22. https://doi.org/10.1021/bi00198a017.
Quraishe S, Sealey M, Cranfield L, Mudher A. Microtubule stabilising peptides rescue tau phenotypes in-vivo. Sci Rep. 2016;6(1):1–10. https://doi.org/10.1038/srep38224.
Mukrasch MD, Von Bergen M, Biernat J, Fischer D, Griesinger C, Mandelkow E, et al. The “jaws” of the tau-microtubule interaction. J Biol Chem. 2007;282(16):12230–9. https://doi.org/10.1074/jbc.M607159200.
Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72(5):1858–62. https://doi.org/10.1073/pnas.72.5.1858.
Kellogg EH, Hejab NMAA, Poepsel S, Downing KH, DiMaio F, Nogales E. Near-atomic model of microtubule-tau interactions. Science. 2018;360(6394):1242–6. https://doi.org/10.1126/science.aat1780.
Kadavath H, Hofele RV, Biernat J, Kumar S, Tepper K, Urlaub H, et al. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc Natl Acad Sci U S A. 2015;112(24):7501–6. https://doi.org/10.1073/pnas.1504081112.
Santarella RA, Skiniotis G, Goldie KN, Tittmann P, Gross H, Mandelkow EM, et al. Surface-decoration of microtubules by human tau. J Mol Biol. 2004;339(3):539–53. https://doi.org/10.1016/j.jmb.2004.04.008.
Qiang L, Sun X, Austin TO, Muralidharan H, Jean DC, Liu M, et al. Tau does not stabilize axonal microtubules but rather enables them to have long labile domains. Curr Biol. 2018;28:2181–2189.e4. https://doi.org/10.1016/j.cub.2018.05.045.
Chaudhary AR, Berger F, Berger CL, Hendricks AG. Tau directs intracellular trafficking by regulating the forces exerted by kinesin and dynein teams. Traffic. 2018;19(2):111–21. https://doi.org/10.1111/tra.12537.
Dixit R, Ross JL, Goldman YE, Holzbaur ELF. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–9. https://doi.org/10.1126/science.1152993.
Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, et al. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369(6480):488–91. https://doi.org/10.1038/369488a0.
Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci. 2001;114(6):1179–87. https://doi.org/10.1242/jcs.114.6.1179.
Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. 2012;18(2):291–5. https://doi.org/10.1038/nm.2613.
Morris M, Hamto P, Adame A, Devidze N, Masliah E, Mucke L. Age-appropriate cognition and subtle dopamine-independent motor deficits in aged tau knockout mice. Neurobiol Aging. 2013;34(6):1523–9. https://doi.org/10.1016/j.neurobiolaging.2012.12.003.
Lei P, Ayton S, Moon S, Zhang Q, Volitakis I, Finkelstein DI, et al. Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol Neurodegener. 2014;9(1):29. https://doi.org/10.1186/1750-1326-9-29.
Velazquez R, Ferreira E, Tran A, Turner EC, Belfiore R, Branca C, et al. Acute tau knockdown in the hippocampus of adult mice causes learning and memory deficits. Aging Cell. 2018;17(4):e12775. https://doi.org/10.1111/acel.12775.
Kontaxi C, Piccardo P, Gill AC. Lysine-directed post-translational modifications of tau protein in Alzheimer’s disease and related Tauopathies. Front Mol Biosci. 2017;4:56. https://doi.org/10.3389/fmolb.2017.00056.
Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. 2017;133(5):665–704. https://doi.org/10.1007/s00401-017-1707-9.
Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin M-L, Yardin C, et al. Tau protein kinases: involvement in Alzheimer’s disease. Ageing Res Rev. 2013;12(1):289–309. https://doi.org/10.1016/j.arr.2012.06.003.
Llorens-Martín M, Jurado J, Hernández F, Avila J. GSK-3β, a pivotal kinase in Alzheimer disease. Front Mol Neurosci. 2014;7:46. https://doi.org/10.3389/fnmol.2014.00046.
Kimura T, Ishiguro K, Hisanaga S. Physiological and pathological phosphorylation of tau by Cdk5. Front Mol Neurosci. 2014;7:65. https://doi.org/10.3389/fnmol.2014.00065.
Tomizawa K, Omori A, Ohtake A, Sato K, Takahashi M. Tau-tubulin kinase phosphorylates tau at Ser-208 and Ser-210, sites found in paired helical filament-tau. FEBS Lett. 2001;492(3):221–7. https://doi.org/10.1016/S0014-5793(01)02256-6.
Matenia D, Mandelkow E-M. The tau of MARK: a polarized view of the cytoskeleton. Trends Biochem Sci. 2009;34(7):332–42. https://doi.org/10.1016/j.tibs.2009.03.008.
Lee G. Phosphorylation of tau by Fyn: implications for Alzheimer’s disease. J Neurosci. 2004;24(9):2304–12. https://doi.org/10.1523/JNEUROSCI.4162-03.2004.
Derkinderen P. Tyrosine 394 is phosphorylated in Alzheimer’s paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase. J Neurosci. 2005;25(28):6584–93. https://doi.org/10.1523/JNEUROSCI.1487-05.2005.
Gong C-X, Grundke-Iqbal I, Iqbal K. Dephosphorylation of Alzheimer’s disease abnormally phosphorylated tau by protein phosphatase-2A. Neuroscience. 1994;61(4):765–72. https://doi.org/10.1016/0306-4522(94)90400-6.
Liu F, Grundke-Iqbal I, Iqbal K, Gong C-X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22(8):1942–50. https://doi.org/10.1111/j.1460-9568.2005.04391.x.
Brion J-P, Smith C, Couck A-M, Gallo J-M, Anderton BH. Developmental changes in τ phosphorylation: fetal τ is transiently phosphorylated in a manner similar to paired helical filament-τ characteristic of Alzheimer’s disease. J Neurochem. 1993;61(6):2071–80. https://doi.org/10.1111/j.1471-4159.1993.tb07444.x.
Kenessey A, Yen S-HC. The extent of phosphorylation of fetal tau is comparable to that of PHF-tau from Alzheimer paired helical filaments. Brain Res. 1993;629(1):40–6. https://doi.org/10.1016/0006-8993(93)90478-6.
Yu Y, Run X, Liang Z, Li Y, Liu F, Liu Y, et al. Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J Neurochem. 2009;108(6):1480–94. https://doi.org/10.1111/j.1471-4159.2009.05882.x.
Arendt T, Stieler J, Strijkstra AM, Hut RA, Rüdiger J, Van der Zee EA, et al. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci. 2003;23(18):6972–81. https://doi.org/10.1523/JNEUROSCI.23-18-06972.2003.
Su B, Wang X, Drew KL, Perry G, Smith MA, Zhu X. Physiological regulation of tau phosphorylation during hibernation. J Neurochem. 2008;105(6):2098–108. https://doi.org/10.1111/j.1471-4159.2008.05294.x.
Stieler JT, Bullmann T, Kohl F, Tøien Ø, Brückner MK, Härtig W, et al. The physiological link between metabolic rate depression and tau phosphorylation in mammalian hibernation. PLoS One. 2011;6(1):e14530. https://doi.org/10.1371/journal.pone.0014530.
Planel E, Richter KEG, Nolan CE, Finley JE, Liu L, Wen Y, et al. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci. 2007;27(12):3090–7. https://doi.org/10.1523/JNEUROSCI.4854-06.2007.
Run X, Liang Z, Zhang L, Iqbal K, Grundke-Iqbal I, Gong C-X. Anesthesia induces phosphorylation of tau. J Alzheimers Dis. 2009;16(3):619–26. https://doi.org/10.3233/JAD-2009-1003.
Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys. 1998;357(2):299–309. https://doi.org/10.1006/abbi.1998.0813.
Fischer D, Mukrasch MD, Biernat J, Bibow S, Blackledge M, Griesinger C, et al. Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules. Biochemistry. 2009;48(42):10047–55. https://doi.org/10.1021/bi901090m.
Xia Y, Prokop S, Gorion K-MM, Kim JD, Sorrentino ZA, Bell BM, et al. Tau Ser208 phosphorylation promotes aggregation and reveals neuropathologic diversity in Alzheimer’s disease and other tauopathies. Acta Neuropathol Commun. 2020;8(1):88. https://doi.org/10.1186/s40478-020-00967-w.
Kiris E, Ventimiglia D, Sargin ME, Gaylord MR, Altinok A, Rose K, et al. Combinatorial tau pseudophosphorylation: markedly different regulatory effects on microtubule assembly and dynamic instability than the sum of the individual parts. J Biol Chem. 2011;286(16):14257–70. https://doi.org/10.1074/jbc.M111.219311.
Stern JL, Lessard DV, Hoeprich GJ, Morfini GA, Berger CL. Phosphoregulation of Tau modulates inhibition of kinesin-1 motility. Mol Biol Cell. 2017;28:1079–87. https://doi.org/10.1091/mbc.E16-10-0728.
Kanaan NM, Morfini G, Pigino G, LaPointe NE, Andreadis A, Song Y, et al. Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiol Aging. 2012;33:826.e15–30. https://doi.org/10.1016/j.neurobiolaging.2011.06.006.
Linte CA, Camp JJ, Rettmann ME, Iii DRH, Richard A. Pseudophosphorylation of tau at S422 enhances SDS-stable dimer formation and impairs both anterograde and retrograde fast axonal transport. Exp Neurol. 2015;283:1–13. https://doi.org/10.1117/12.2008529.Image-based.
2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020;16(3):391–460. https://doi.org/10.1002/alz.12068.
Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123(1):1–11. https://doi.org/10.1007/s00401-011-0910-3.
Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8(1):1–13. https://doi.org/10.1016/j.jalz.2011.10.007.
Nelson PT, Jicha GA, Schmitt FA, Liu H, Davis DG, Mendiondo MS, et al. Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol. 2007;66(12):1136–46. https://doi.org/10.1097/nen.0b013e31815c5efb.
Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362–81. https://doi.org/10.1097/NEN.0b013e31825018f7.
Köpke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem. 1993;268(32):24374–84. https://doi.org/10.1016/S0021-9258(20)80536-5.
Barthélemy NR, Mallipeddi N, Moiseyev P, Sato C, Bateman RJ. Tau phosphorylation rates measured by mass spectrometry differ in the intracellular brain vs. extracellular cerebrospinal fluid compartments and are differentially affected by Alzheimer’s disease. Front Aging Neurosci. 2019;11:121. https://doi.org/10.3389/fnagi.2019.00121.
Russell CL, Mitra V, Hansson K, Blennow K, Gobom J, Zetterberg H, et al. Comprehensive quantitative profiling of tau and phosphorylated tau peptides in cerebrospinal fluid by mass spectrometry provides new biomarker candidates. J Alzheimers Dis. 2016;55(1):303–13. https://doi.org/10.3233/JAD-160633.
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–59. https://doi.org/10.1007/BF00308809.
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112(4):389–404. https://doi.org/10.1007/s00401-006-0127-z.
Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189(3):167–9. https://doi.org/10.1016/0304-3940(95)11484-e.
Neddens J, Temmel M, Flunkert S, Kerschbaumer B, Hoeller C, Loeffler T, et al. Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol Commun. 2018;6(1):52. https://doi.org/10.1186/s40478-018-0557-6.
Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70(11):960–9. https://doi.org/10.1097/NEN.0b013e318232a379.
Augustinack JC, Schneider A, Mandelkow E-M, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002;103(1):26–35. https://doi.org/10.1007/s004010100423.
Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci. 2006;91(12):5562–6. https://doi.org/10.1073/pnas.91.12.5562.
Gilley J, Ando K, Seereeram A, Rodríguez-Martín T, Pooler AM, Sturdee L, et al. Mislocalization of neuronal tau in the absence of tangle pathology in phosphomutant tau knockin mice. Neurobiol Aging. 2016;39:1–18. https://doi.org/10.1016/j.neurobiolaging.2015.11.028.
Alonso AC, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci. 2001;98(12):6923–8. https://doi.org/10.1073/pnas.121119298.
Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of tau and generates a pathological (MC-1) conformation. J Biol Chem. 2008;283(46):32066–76. https://doi.org/10.1074/jbc.M805300200.
Gohar M, Yang W, Strong W, Volkening K, Leystra-Lantz C, Strong MJ. Tau phosphorylation at threonine-175 leads to fibril formation and enhanced cell death: implications for amyotrophic lateral sclerosis with cognitive impairment. J Neurochem. 2009;108(3):634–43. https://doi.org/10.1111/j.1471-4159.2008.05791.x.
Rankin CA, Sun Q, Gamblin TC. Pseudo-phosphorylation of tau at Ser202 and Thr205 affects tau filament formation. Mol Brain Res. 2005;138(1):84–93. https://doi.org/10.1016/j.molbrainres.2005.04.012.
Chang E, Kim S, Schafer KN, Kuret J. Pseudophosphorylation of tau protein directly modulates its aggregation kinetics. Biochim Biophys Acta - Proteins Proteomics. 1814;2011(2):388–95. https://doi.org/10.1016/j.bbapap.2010.10.005.
Despres C, Byrne C, Qi H, Cantrelle F-X, Huvent I, Chambraud B, et al. Identification of the tau phosphorylation pattern that drives its aggregation. Proc Natl Acad Sci. 2017;114(34):9080–5. https://doi.org/10.1073/pnas.1708448114.
Strang KH, Sorrentino ZA, Riffe CJ, Gorion K-MM, Vijayaraghavan N, Golde TE, et al. Phosphorylation of serine 305 in tau inhibits aggregation. Neurosci Lett. 2019;692:187–92. https://doi.org/10.1016/j.neulet.2018.11.011.
Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow E-M. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments †. Biochemistry. 1999;38(12):3549–58. https://doi.org/10.1021/bi981874p.
Lewis J, Dickson DW. Propagation of tau pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016;131(1):27–48. https://doi.org/10.1007/s00401-015-1507-z.
Goedert M, Eisenberg DS, Crowther RA. Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci. 2017;40(1):189–210. https://doi.org/10.1146/annurev-neuro-072116-031153.
Ayers JI, Giasson BI, Borchelt DR. Prion-like spreading in Tauopathies. Biol Psychiatry. 2018;83(4):337–46. https://doi.org/10.1016/j.biopsych.2017.04.003.
Peeraer E, Bottelbergs A, Van Kolen K, Stancu IC, Vasconcelos B, Mahieu M, et al. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol Dis. 2015;73:83–95. https://doi.org/10.1016/j.nbd.2014.08.032.
Hu W, Zhang X, Tung YC, Xie S, Liu F, Iqbal K. Hyperphosphorylation determines both the spread and the morphology of tau pathology. Alzheimers Dement. 2016;12(10):1066–77. https://doi.org/10.1016/j.jalz.2016.01.014.
Rosenqvist N, Asuni AA, Andersson CR, Christensen S, Daechsel JA, Egebjerg J, et al. Highly specific and selective anti-pS396-tau antibody C10.2 targets seeding-competent tau. Alzheimer’s Dement Transl Res Clin Interv. 2018;4(1):521–34. https://doi.org/10.1016/j.trci.2018.09.005.
Dujardin S, Commins C, Lathuiliere A, Beerepoot P, Fernandes AR, Kamath TV, et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat Med. 2020;26(8):1256–63. https://doi.org/10.1038/s41591-020-0938-9.
Strang KH, Golde TE, Giasson BI. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab Investig. 2019;99(7):912–28. https://doi.org/10.1038/s41374-019-0197-x.
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–5. https://doi.org/10.1038/31508.
Alonso ADC, Mederlyova A, Novak M, Grundke-Iqbal I, Iqbal K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem. 2004;279(33):34873–81. https://doi.org/10.1074/jbc.M405131200.
Combs B, Gamblin TC. FTDP-17 tau mutations induce distinct effects on aggregation and microtubule interactions. Biochemistry. 2012;51(43):8597–607. https://doi.org/10.1021/bi3010818.
Xia Y, Sorrentino ZA, Kim JD, Strang KH, Riffe CJ, Giasson BI. Impaired tau-microtubule interactions are prevalent among pathogenic tau variants arising from missense mutations. J Biol Chem. 2019;294(48):18488–503. https://doi.org/10.1074/jbc.RA119.010178.
Strang KH, Croft CL, Sorrentino ZA, Chakrabarty P, Golde TE, Giasson BI. Distinct differences in prion-like seeding and aggregation between tau protein variants provide mechanistic insights into tauopathies. J Biol Chem. 2018;293(7):2408–21. https://doi.org/10.1074/jbc.M117.815357.
Liu F, Gong C-XX. Tau exon 10 alternative splicing and tauopathies. Mol Neurodegener. 2008;3(1):1–10. https://doi.org/10.1186/1750-1326-3-8.
Forrest SL, Kril JJ, Stevens CH, Kwok JB, Hallupp M, Kim WS, et al. Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain. 2018;141(2):521–34. https://doi.org/10.1093/brain/awx328.
Götz J, Halliday G, Nisbet RM. Molecular pathogenesis of the Tauopathies. Annu Rev Pathol Mech Dis. 2019;14(1):239–61. https://doi.org/10.1146/annurev-pathmechdis-012418-012936.
Dickson DW. Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol. 1999;246(S2):s006–15. https://doi.org/10.1007/PL00007746.
Probst A, Tolnay M, Langui D, Goedert M, Spillantini MG. Pick’s disease: Hyperphosphorylated tau protein segregates to the somatoaxonal compartment. Acta Neuropathol. 1996;92(6):588–96. https://doi.org/10.1007/s004010050565.
Ahmed Z, Doherty KM, Silveira-Moriyama L, Bandopadhyay R, Lashley T, Mamais A, et al. Globular glial tauopathies (GGT) presenting with motor neuron disease or frontotemporal dementia: an emerging group of 4-repeat tauopathies. Acta Neuropathol. 2011;122(4):415–28. https://doi.org/10.1007/s00401-011-0857-4.
McKee AC, Stein TD, Kiernan PT, Alvarez VE. The neuropathology of chronic traumatic encephalopathy. Brain Pathol. 2015;25:350–64. Blackwell Publishing Ltd. https://doi.org/10.1111/bpa.12248.
Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, et al. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature. 2018;561(7721):137–40. https://doi.org/10.1038/s41586-018-0454-y.
Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature. 2017;547(7662):185–90. https://doi.org/10.1038/nature23002.
Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature. 2019;568(7752):420–3. https://doi.org/10.1038/s41586-019-1026-5.
Zhang W, Tarutani A, Newell KL, Murzin AG, Matsubara T, Falcon B, et al. Novel tau filament fold in corticobasal degeneration. Nature. 2020;580(7802):283–7. https://doi.org/10.1038/s41586-020-2043-0.
Arakhamia T, Lee CE, Carlomagno Y, Duong DM, Kundinger SR, Wang K, et al. Posttranslational modifications mediate the structural diversity of tauopathy strains. Cell. 2020;180:633–644.e12. https://doi.org/10.1016/j.cell.2020.01.027.
Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci. 2013;110(23):9535–40. https://doi.org/10.1073/pnas.1301175110.
Narasimhan S, Guo JL, Changolkar L, Stieber A, McBride JD, Silva LV, et al. Pathological tau strains from human brains recapitulate the diversity of Tauopathies in nontransgenic mouse brain. J Neurosci. 2017;37(47):11406–23. https://doi.org/10.1523/JNEUROSCI.1230-17.2017.
Boluda S, Iba M, Zhang B, Raible KM, Lee VMY, Trojanowski JQ. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol. 2015;129(2):221–37. https://doi.org/10.1007/s00401-014-1373-0.
Chung DEC, Carlomagno Y, Cook CN, Jansen-West K, Daughrity L, Lewis-Tuffin LJ, et al. Tau exhibits unique seeding properties in globular glial tauopathy. Acta Neuropathol Commun. 2019;7(1):36. https://doi.org/10.1186/s40478-019-0691-9.
Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535–62. https://doi.org/10.1016/j.jalz.2018.02.018.
Leuzy A, Chiotis K, Lemoine L, Gillberg PG, Almkvist O, Rodriguez-Vieitez E, et al. Tau PET imaging in neurodegenerative tauopathies—still a challenge. Mol Psychiatry. 2019;24(8):1112–34. https://doi.org/10.1038/s41380-018-0342-8.
Barthélemy NR, Li Y, Joseph-Mathurin N, Gordon BA, Hassenstab J, Benzinger TLS, et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat Med. 2020;26(3):398–407. https://doi.org/10.1038/s41591-020-0781-z.
Vanmechelen E, Vanderstichele H, Davidsson P, Van Kerschaver E, Van Der Perre B, Sjögren M, et al. Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: a sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci Lett. 2000;285(1):49–52. https://doi.org/10.1016/S0304-3940(00)01036-3.
Olsson B, Lautner R, Andreasson U, Öhrfelt A, Portelius E, Bjerke M, et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol. 2016;15(7):673–84. https://doi.org/10.1016/S1474-4422(16)00070-3.
Tondelli M, Bedin R, Chiari A, Molinari MA, Bonifacio G, Lelli N, et al. Role of cerebrospinal fluid biomarkers to predict conversion to dementia in patients with mild cognitive impairment: a clinical cohort study. Clin Chem Lab Med. 2015;53(3):453–60. https://doi.org/10.1515/cclm-2014-0414.
Meeter LHH, Vijverberg EG, Del Campo M, Rozemuller AJM, Donker Kaat L, de Jong FJ, et al. Clinical value of neurofilament and phospho-tau/tau ratio in the frontotemporal dementia spectrum. Neurology. 2018;90(14):e1231–9. https://doi.org/10.1212/WNL.0000000000005261.
Rojas JC, Bang J, Lobach IV, Tsai RM, Rabinovici GD, Miller BL, et al. CSF neurofilament light chain and phosphorylated tau 181 predict disease progression in PSP. Neurology. 2018;90(4):E273–81. https://doi.org/10.1212/WNL.0000000000004859.
Janelidze S, Stomrud E, Smith R, Palmqvist S, Mattsson N, Airey DC, et al. Cerebrospinal fluid p-tau217 performs better than p-tau181 as a biomarker of Alzheimer’s disease. Nat Commun. 2020;11(1):1–12. https://doi.org/10.1038/s41467-020-15436-0.
Hanes J, Kovac A, Kvartsberg H, Kontsekova E, Fialova L, Katina S, et al. Evaluation of a novel immunoassay to detect p-tau Thr217 in the CSF to distinguish Alzheimer disease from other dementias. Neurology. 2020;95(22):e3026–35. https://doi.org/10.1212/WNL.0000000000010814.
Barthélemy NR, Bateman RJ, Hirtz C, Marin P, Becher F, Sato C, et al. Cerebrospinal fluid phospho-tau T217 outperforms T181 as a biomarker for the differential diagnosis of Alzheimer’s disease and PET amyloid-positive patient identification. Alzheimers Res Ther. 2020;12(1):26. https://doi.org/10.1186/s13195-020-00596-4.
Kohnken R, Buerger K, Zinkowski R, Miller C, Kerkman D, DeBernardis J, et al. Detection of tau phosphorylated at threonine 231 in cerebrospinal fluid of Alzheimer’s disease patients. Neurosci Lett. 2000;287(3):187–90. https://doi.org/10.1016/S0304-3940(00)01178-2.
Buerger K, Teipel SJ, Zinkowski R, Blennow K, Arai H, Engel R, et al. CSF tau protein phosphorylated at threonine 231 correlates with cognitive decline in MCI subjects. Neurology. 2002;59(4):627–9. https://doi.org/10.1212/WNL.59.4.627.
Buerger K, Zinkowski R, Teipel SJ, Tapiola T, Arai H, Blennow K, et al. Differential diagnosis of Alzheimer disease with cerebrospinal fluid levels of tau protein phosphorylated at threonine 231. Arch Neurol. 2002;59(8):1267–72. https://doi.org/10.1001/archneur.59.8.1267.
Spiegel J, Pirraglia E, Osorio RS, Glodzik L, Li Y, Tsui W, et al. Greater specificity for cerebrospinal fluid P-tau231 over P-tau181 in the differentiation of healthy controls from Alzheimer’s disease. J Alzheimers Dis. 2015;49(1):93–100. https://doi.org/10.3233/JAD-150167.
Santos JRF, Bauer C, Schuchhardt J, Wedekind D, Waniek K, Lachmann I, et al. Validation of a prototype tau Thr231 phosphorylation CSF ELISA as a potential biomarker for Alzheimer’s disease. J Neural Transm. 2019;126(3):339–48. https://doi.org/10.1007/s00702-019-01982-5.
Suárez-Calvet M, Karikari TK, Ashton NJ, Lantero Rodríguez J, Milà-Alomà M, Gispert JD, et al. Novel tau biomarkers phosphorylated at T181, T217 or T231 rise in the initial stages of the preclinical Alzheimer’s continuum when only subtle changes in Aβ pathology are detected. EMBO Mol Med. 2020;12(12):e12921. https://doi.org/10.15252/emmm.202012921.
Karikari TK, Emeršič A, Vrillon A, Lantero-Rodriguez J, Ashton NJ, Kramberger MG, et al. Head-to-head comparison of clinical performance of CSF phospho-tau T181 and T217 biomarkers for Alzheimer’s disease diagnosis. Alzheimers Dement. 2020;17(5):755–67. https://doi.org/10.1002/alz.12236.
Mattsson N, Zetterberg H, Janelidze S, Insel PS, Andreasson U, Stomrud E, et al. Plasma tau in Alzheimer disease. Neurology. 2016;87(17):1827–35. https://doi.org/10.1212/WNL.0000000000003246.
Thijssen EH, La Joie R, Wolf A, Strom A, Wang P, Iaccarino L, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat Med. 2020;26(3):387–97. https://doi.org/10.1038/s41591-020-0762-2.
Janelidze S, Mattsson N, Palmqvist S, Smith R, Beach TG, Serrano GE, et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med. 2020;26(3):379–86. https://doi.org/10.1038/s41591-020-0755-1.
Moscoso A, Grothe MJ, Ashton NJ, Karikari TK, Lantero Rodríguez J, Snellman A, et al. Longitudinal associations of blood phosphorylated Tau181 and Neurofilament light chain with neurodegeneration in Alzheimer disease. JAMA Neurol. 2021;78(4):396–406. https://doi.org/10.1001/jamaneurol.2020.4986.
Palmqvist S, Janelidze S, Quiroz YT, Zetterberg H, Lopera F, Stomrud E, et al. Discriminative accuracy of plasma Phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA. 2020;324(8):772–81. https://doi.org/10.1001/jama.2020.12134.
Janelidze S, Berron D, Smith R, Strandberg O, Proctor NK, Dage JL, et al. Associations of plasma Phospho-Tau217 levels with tau positron emission tomography in early Alzheimer disease. JAMA Neurol. 2020;78(2):149–56. https://doi.org/10.1001/jamaneurol.2020.4201.
Martinez A, Alonso M, Castro A, Pérez C, Moreno FJ. First non-ATP competitive glycogen synthase kinase 3 β (GSK-3β) inhibitors: Thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease. J Med Chem. 2002;45(6):1292–9. https://doi.org/10.1021/jm011020u.
Domínguez JM, Fuertes A, Orozco L, del Monte-Millán M, Delgado E, Medina M. Evidence for irreversible inhibition of glycogen synthase kinase-3β by Tideglusib*. J Biol Chem. 2012;287(2):893–904. https://doi.org/10.1074/jbc.M111.306472.
Serenó L, Coma M, Rodríguez M, Sánchez-Ferrer P, Sánchez MB, Gich I, et al. A novel GSK-3β inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol Dis. 2009;35(3):359–67. https://doi.org/10.1016/j.nbd.2009.05.025.
del Ser T, Steinwachs KC, Gertz HJ, Andrés MV, Gómez-Carrillo B, Medina M, et al. Treatment of Alzheimer’s disease with the GSK-3 inhibitor Tideglusib: a pilot study. J Alzheimers Dis. 2012;33(1):205–15. https://doi.org/10.3233/JAD-2012-120805.
Lovestone S, Boada M, Dubois B, Hüll M, Rinne JO, Huppertz H-J, et al. A phase II trial of Tideglusib in Alzheimer’s disease. J Alzheimers Dis. 2015;45(1):75–88. https://doi.org/10.3233/JAD-141959.
Höglinger GU, Huppertz H-J, Wagenpfeil S, Andrés MV, Belloch V, León T, et al. Tideglusib reduces progression of brain atrophy in progressive supranuclear palsy in a randomized trial. Mov Disord. 2014;29(4):479–87. https://doi.org/10.1002/mds.25815.
Smith KA, Cipriani A. Lithium and suicide in mood disorders: updated meta-review of the scientific literature. Bipolar Disord. 2017;19(7):575–86. https://doi.org/10.1111/bdi.12543.
Devanand DP, Strickler JG, Huey ED, Crocco E, Forester BP, Husain MM, et al. Lithium treatment for agitation in Alzheimer’s disease (lit-AD): clinical rationale and study design. Contemp Clin Trials. 2018;71:33–9. https://doi.org/10.1016/j.cct.2018.05.019.
Hong M, Chen DCR, Klein PS, Lee VMY. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem. 1997;272(40):25326–32. https://doi.org/10.1074/jbc.272.40.25326.
Engel T, Goñi-Oliver P, Lucas JJ, Avila J, Hernández F. Chronic lithium administration to FTDP-17 tau and GSK-3? Overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J Neurochem. 2006;99(6):1445–55. https://doi.org/10.1111/j.1471-4159.2006.04139.x.
Leroy K, Ando K, Héraud C, Yilmaz Z, Authelet M, Boeynaems JM, et al. Lithium treatment arrests the development of neurofibrillary tangles in mutant tau transgenic mice with advanced neurofibrillary pathology. J Alzheimers Dis. 2010;19(2):705–19. https://doi.org/10.3233/JAD-2010-1276.
Hampel H, Ewers M, Bürger K, Annas P, Mörtberg A, Bogstedt A, et al. Lithium trial in Alzheimer’s disease. J Clin Psychiatry. 2009;70(6):922–31. https://doi.org/10.4088/JCP.08m04606.
Forlenza OV, Diniz BS, Radanovic M, Santos FS, Talib LL, Gattaz WF. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351–6. https://doi.org/10.1192/bjp.bp.110.080044.
Forlenza OV, Radanovic M, Talib LL, Gattaz WF. Clinical and biological effects of long-term lithium treatment in older adults with amnestic mild cognitive impairment: randomised clinical trial. Br J Psychiatry. 2019;215(5):668–74. https://doi.org/10.1192/bjp.2019.76.
Andrade Nunes M, Araujo Viel T, Sousa BH. Microdose lithium treatment stabilized cognitive impairment in patients with Alzheimer’s disease. Curr Alzheimer Res. 2013;10(1):104–7. https://doi.org/10.2174/1567205011310010014.
Medicine UNL of. Lithium As a Treatment to Prevent Impairment of Cognition in Elders. ClinicaltrialsGov n.d.
Löscher W. Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy. CNS Drugs. 2002;16(10):669–94. https://doi.org/10.2165/00023210-200216100-00003.
Cipriani A, Reid K, Young AH, Macritchie K, Geddes J. Valproic acid, valproate and divalproex in the maintenance treatment of bipolar disorder. Cochrane Database Syst Rev. 2013;2013. https://doi.org/10.1002/14651858.CD003196.pub2.
Hu J-P, Xie J-W, Wang C-Y, Wang T, Wang X, Wang S-L, et al. Valproate reduces tau phosphorylation via cyclin-dependent kinase 5 and glycogen synthase kinase 3 signaling pathways. Brain Res Bull. 2011;85(3-4):194–200. https://doi.org/10.1016/j.brainresbull.2011.03.006.
Long Z-M, Zhao L, Jiang R, Wang K-J, Luo S-F, Zheng M, et al. Valproic acid modifies synaptic structure and accelerates neurite outgrowth via the glycogen synthase kinase-3β signaling pathway in an Alzheimer’s disease model. CNS Neurosci Ther. 2015;21(11):887–97. https://doi.org/10.1111/cns.12445.
Qing H, He G, Ly PTT, Fox CJ, Staufenbiel M, Cai F, et al. Valproic acid inhibits Aβ production, neuritic plaque formation, and behavioral deficits in Alzheimer’s disease mouse models. J Exp Med. 2008;205(12):2781–9. https://doi.org/10.1084/jem.20081588.
Herrmann N, Lanctôt KL, Rothenburg LS, Eryavec G. A placebo-controlled trial of valproate for agitation and aggression in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2007;23(2):116–9. https://doi.org/10.1159/000097757.
Dolder C, Mckinsey J. Low-dose divalproex in agitated patients with Alzheimerʼs disease. J Psychiatr Pract. 2010;16(1):63–7. https://doi.org/10.1097/01.pra.0000367781.26507.86.
Tariot PN. Chronic divalproex sodium to attenuate agitation and clinical progression of Alzheimer disease. Arch Gen Psychiatry. 2011;68(8):853–61. https://doi.org/10.1001/archgenpsychiatry.2011.72.
Fleisher AS, Truran D, Mai JT, Langbaum JBS, Aisen PS, Cummings JL, et al. Chronic divalproex sodium use and brain atrophy in Alzheimer disease. Neurology. 2011;77(13):1263–71. https://doi.org/10.1212/WNL.0b013e318230a16c.
Pardoe HR, Berg AT, Jackson GD. Sodium valproate use is associated with reduced parietal lobe thickness and brain volume. Neurology. 2013;80(20):1895–900. https://doi.org/10.1212/WNL.0b013e318292a2e5.
Papazian O, Canizales E, Alfonso I, Archila R, Duchowny M, Aicardi J. Reversible dementia and apparent brain atrophy during valproate therapy. Ann Neurol. 1995;38(4):687–91. https://doi.org/10.1002/ana.410380423.
Sacha T, Saglio G. Nilotinib in the treatment of chronic myeloid leukemia. Future Oncol. 2019;15(9):953–65. https://doi.org/10.2217/fon-2018-0468.
Hebron ML, Javidnia M, Moussa CEH. Tau clearance improves astrocytic function and brain glutamate-glutamine cycle. J Neurol Sci. 2018;391:90–9. https://doi.org/10.1016/j.jns.2018.06.005.
Lonskaya I, Hebron ML, Desforges NM, Franjie A, Moussa CE-H. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol Med. 2013;5(8):1247–62. https://doi.org/10.1002/emmm.201302771.
Turner RS, Hebron ML, Lawler A, Mundel EE, Yusuf N, Starr JN, et al. Nilotinib effects on safety, tolerability, and biomarkers in Alzheimer’s disease. Ann Neurol. 2020;88(1):183–94. https://doi.org/10.1002/ana.25775.
van Eersel J, Ke YD, Liu X, Delerue F, Kril JJ, Gotz J, et al. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc Natl Acad Sci. 2010;107(31):13888–93. https://doi.org/10.1073/pnas.1009038107.
Corcoran NM, Martin D, Hutter-Paier B, Windisch M, Nguyen T, Nheu L, et al. Sodium selenate specifically activates PP2A phosphatase, dephosphorylates tau and reverses memory deficits in an Alzheimer’s disease model. J Clin Neurosci. 2010;17(8):1025–33. https://doi.org/10.1016/j.jocn.2010.04.020.
Ahmed T, Van der Jeugd A, Caillierez R, Buée L, Blum D, D’Hooge R, et al. Chronic sodium Selenate treatment restores deficits in cognition and synaptic plasticity in a murine model of Tauopathy. Front Mol Neurosci. 2020;13:181. https://doi.org/10.3389/fnmol.2020.570223.
Malpas CB, Vivash L, Genc S, Saling MM, Desmond P, Steward C, et al. A phase IIa randomized control trial of VEL015 (sodium Selenate) in mild-moderate Alzheimer’s disease. J Alzheimers Dis. 2016;54(1):223–32. https://doi.org/10.3233/JAD-160544.
Cardoso BR, Roberts BR, Malpas CB, Vivash L, Genc S, Saling MM, et al. Supranutritional sodium Selenate supplementation delivers selenium to the central nervous system: results from a randomized controlled pilot trial in Alzheimer’s disease. Neurotherapeutics. 2019;16(1):192–202. https://doi.org/10.1007/s13311-018-0662-z.
Vivash L, Malpas CB, Churilov L, Walterfang M, Brodtmann A, Piguet O, et al. A study protocol for a phase II randomised, double-blind, placebo-controlled trial of sodium selenate as a disease-modifying treatment for behavioural variant frontotemporal dementia. BMJ Open. 2020;10(11):e040100. https://doi.org/10.1136/bmjopen-2020-040100.
Collin L, Bohrmann B, Göpfert U, Oroszlan-Szovik K, Ozmen L, Grüninger F. Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer‘s disease. Brain. 2014;137(10):2834–46. https://doi.org/10.1093/brain/awu213.
Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen C-H, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature. 2015;523(7561):431–6. https://doi.org/10.1038/nature14658.
Albayram O, Kondo A, Mannix R, Smith C, Tsai C-Y, Li C, et al. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun. 2017;8(1):1000. https://doi.org/10.1038/s41467-017-01068-4.
d’Abramo C, Acker CM, Jimenez H, Davies P. Passive immunization in JNPL3 transgenic mice using an Array of Phospho-tau specific antibodies. PLoS One. 2015;10(8):e0135774. https://doi.org/10.1371/journal.pone.0135774.
Sankaranarayanan S, Barten DM, Vana L, Devidze N, Yang L, Cadelina G, et al. Passive immunization with Phospho-tau antibodies reduces tau pathology and functional deficits in two distinct mouse Tauopathy models. PLoS One. 2015;10(5):e0125614. https://doi.org/10.1371/journal.pone.0125614.
Goodwin MS, Sinyavskaya O, Burg F, O’Neal V, Ceballos-Diaz C, Cruz PE, et al. Anti-tau scFvs targeted to the cytoplasm or secretory pathway variably modify pathology and neurodegenerative phenotypes. Mol Ther. 2021;29(2):859–72. https://doi.org/10.1016/j.ymthe.2020.10.007.
Nisbet RM, Van Der Jeugd A, Leinenga G, Evans HT, Janowicz PW, Götz J. Combined effects of scanning ultrasound and a tau-specific single chain antibody in a tau transgenic mouse model. Brain. 2017;140(5):1220–30. https://doi.org/10.1093/brain/awx052.
van de Haar HJ, Burgmans S, Jansen JFA, van Osch MJP, van Buchem MA, Muller M, et al. Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology. 2016;281(2):527–35. https://doi.org/10.1148/radiol.2016152244.
Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14(3):133–50. https://doi.org/10.1038/nrneurol.2017.188.
Wu Q, Lin Y, Gu J, Sigurdsson EM. Dynamic assessment of tau immunotherapies in the brains of live animals by two-photon imaging. EBioMedicine. 2018;35:270–8. https://doi.org/10.1016/j.ebiom.2018.08.041.
Congdon EE, Gu J, Sait HBR, Sigurdsson EM. Antibody uptake into neurons occurs primarily via clathrin-dependent Fcγ receptor endocytosis and is a prerequisite for acute tau protein clearance. J Biol Chem. 2013;288(49):35452–65. https://doi.org/10.1074/jbc.M113.491001.
Andersson CR, Falsig J, Stavenhagen JB, Christensen S, Kartberg F, Rosenqvist N, et al. Antibody-mediated clearance of tau in primary mouse microglial cultures requires Fcγ-receptor binding and functional lysosomes. Sci Rep. 2019;9(1):4658. https://doi.org/10.1038/s41598-019-41105-4.
Nobuhara CK, DeVos SL, Commins C, Wegmann S, Moore BD, Roe AD, et al. Tau antibody targeting pathological species blocks neuronal uptake and interneuron propagation of tau in vitro. Am J Pathol. 2017;187(6):1399–412. https://doi.org/10.1016/j.ajpath.2017.01.022.
To Block Tau’s Proteopathic Spread, Antibody Must Attack its Mid-Region. Alzforum n.d. https://www.alzforum.org/news/conference-coverage/block-taus-proteopathic-spread-antibody-must-attack-its-mid-region.
Medicine UNL of. A Study of JNJ-63733657 in Healthy Japanese Participants. ClinicalTrialsGov n.d.
Medicine UNL of. A Study to Investigate Safety and Tolerability, Pharmacokinetics and Pharmacodynamics of JNJ-63733657 in Healthy Subjects and Subjects With Alzheimer’s Disease. ClinicalTrialsGov n.d.
Galpern WR, Mercken M, Van Kolen K, Timmers M, Haeverans K, Janssens L, et al. P1-052: a single ascending dose study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of the anti-Phospho-tau antibody JNJ-63733657 in healthy subjects. Alzheimers Dement. 2019;15:P252–3. https://doi.org/10.1016/j.jalz.2019.06.077.
JNJ-63733657. Alzforum n.d. https://www.alzforum.org/therapeutics/jnj-63733657.
Medicine UNL of. A Study of JNJ-63733657 in Participants With Early Alzheimer’s Disease. ClinicalTrialsGov n.d.
Zhou XZ, Kops O, Werner A, Lu P-J, Shen M, Stoller G, et al. Pin1-dependent prolyl isomerization regulates Dephosphorylation of Cdc25C and tau proteins. Mol Cell. 2000;6(4):873–83. https://doi.org/10.1016/S1097-2765(05)00083-3.
Nakamura K, Greenwood A, Binder L, Bigio EH, Denial S, Nicholson L, et al. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cell. 2012;149(1):232–44. https://doi.org/10.1016/j.cell.2012.02.016.
Medicine UNL of. Safety and Tolerability of PNT001 in Healthy Adults. ClinicalTrialsGov n.d.
Medicine UNL of. Safety and Tolerability of PNT001 in Patients With Acute Traumatic Brain Injury (TBI). ClinicalTrialsGov n.d.
Bajracharya R, Brici D, Bodea L-G, Janowicz PW, Götz J, Nisbet RM. Tau antibody isotype induces differential effects following passive immunisation of tau transgenic mice. Acta Neuropathol Commun. 2021;9(1):42. https://doi.org/10.1186/s40478-021-01147-0.
Medicine UNL of. Study with Lu AF87908 in healthy subjects and patients with Alzheimer’s disease. ClinicalTrialsGov n.d.
Medicine UNL of. A Study of RO6926496 in Healthy Volunteers. ClinicalTrialsGov n.d. https://clinicaltrials.gov/ct2/show/NCT02281786 (Accessed 19 Jan 2021).
Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van Haesendonck C, Borghgraef P, et al. Changed conformation of mutant tau-P301L underlies the moribund Tauopathy, absent in progressive, nonlethal Axonopathy of tau-4R/2N transgenic mice. J Biol Chem. 2005;280(5):3963–73. https://doi.org/10.1074/jbc.M409876200.
Theunis C, Crespo-Biel N, Gafner V, Pihlgren M, López-Deber MP, Reis P, et al. Efficacy and Safety of A Liposome-Based Vaccine against Protein Tau, Assessed in Tau.P301L Mice That Model Tauopathy. PLoS One. 2013;8(8):e72301. https://doi.org/10.1371/journal.pone.0072301.
A study comparing the safety and effects of a new compound, ACI-35 with placebo in patients with mild to moderate Alzheimer’s disease. ISRCTN n.d.
Active Tau Vaccine: Hints of Slowing Neurodegeneration. Alzforum n.d. https://www.alzforum.org/news/conference-coverage/active-tau-vaccine-hints-slowing-neurodegeneration.
Medicine UNL of. A Study to Evaluate the Safety, Tolerability and Immunogenicity of Tau Targeted Vaccines in Participants With Early Alzheimer’s Disease. ClinicalTrialsGov n.d.
Funding
Human brain tissue was obtained from the University of Florida Neuromedicine Human Brain and Tissue Bank that is funded in part by a grant from the National Institute on Aging (P30AG066506). YX is supported by a fellowship from National Institute on Aging (F30AG067673).
Author information
Authors and Affiliations
Contributions
YX, SP and BIG co-wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Post mortem human brain tissue was obtained through the University of Florida Neuromedicine Human Brain and Tissue Bank according to protocols approved by the Institutional Review Board.
Consent for publication
Not applicable.
Competing interests
We declare no conflict of interest in this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xia, Y., Prokop, S. & Giasson, B.I. “Don’t Phos Over Tau”: recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Mol Neurodegeneration 16, 37 (2021). https://doi.org/10.1186/s13024-021-00460-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-021-00460-5