
Abstract
Multiple sclerosis (MS) is the most frequent inflammatory and demyelinating disease of the central nervous system (CNS). The underlying pathophysiology of MS is the destruction of myelin sheath by immune cells. The formation of myelin plaques, inflammation, and injury of neuronal myelin sheath characterizes its neuropathology. MS plaques are multiple focal regions of demyelination disseminated in the brain's white matter, spinal cords, deep grey matter, and cerebral cortex. Fenofibrate is a peroxisome proliferative activated receptor alpha (PPAR-α) that attenuates the inflammatory reactions in MS. Fenofibrate inhibits differentiation of Th17 by inhibiting the expression of pro-inflammatory signaling. According to these findings, this review intended to illuminate the mechanistic immunoinflammatory role of fenofibrate in mitigating MS neuropathology. In conclusion, fenofibrate can attenuate MS neuropathology by modulating different pathways, including oxidative stress, autophagy, mitochondrial dysfunction, inflammatory-signaling pathways, and neuroinflammation.
Similar content being viewed by others
Introduction
Multiple sclerosis (MS) is the most frequent inflammatory and demyelinating disease of the central nervous system (CNS) [1]. MS disrupts motor and sensory neuronal signal transmission, leading to motor and sensory deficits. It is characterized by symptoms, including vision loss in one eye, double vision, muscle weakness, and motor-sensory incoordination [2]. MS patients may have a prodromal phase characterized by cognitive impairments, and neuropsychiatric symptoms continue for years before the manifestation of MS symptoms. Clinical presentations of MS are motor, sensory, and autonomic dysfunctions. A specific feature of MS depends on the site of lesions in the CNS, including visual loss due to optic neuritis, muscle spasm, hyperreflexia due to spinal cord injury, ataxia due to cerebellar involvement and motor-sensory incoordination [3, 4]. Notably, 85% of MS patients presented with acute exacerbations, and 15% of MS patients presented with gradual motor-sensory dysfunction without a period of recovery [5,6,7]. The clinical spectrum of MS includes CIS, RRMS, and SP/PPMS. According to Lublin, there are two main MS phenotypes: relapsing and progressive, additionally modified by the presence or absence of activity-relapses and new changes in MRI (i.e., RRMS active or inactive/stable, SP/PPMS active or inactive) [8,9,10].
MS may be progressive over time or relapsing forms in which the symptoms disappear and return. It has been reported that about one million people in USA will be affected by MS in 2022 [5]. MS affects about 2.8 million people worldwide [6]. MS is more common in women at 20–50 years [7]. Of note, MS was initially identified by Jean-Martin Charcot, a French neurologist, in 1868, who described multiple scars in the brain and spinal cord [8]. To better understand the MS pathophysiology, we focused on the lipids that play an influential role in the disease background. Lipids are not only considerably involved in the formation of myelin sheath but are also involved in cell signaling, communication, and in transport in the CNS. Thus, lipids seem probable candidates for processes underlying the active and progressive phase of MS and potential targets for new, effective, and stage-specific therapeutic interventions [9,10,11,12]. Previous studies illustrated that peroxisome proliferative activated receptor alpha (PPAR-α) agonists such as fenofibrate, gemfibrozil and ciprofibrate could attenuate the inflammatory reactions in MS. It has been reported that gemfibrozil attenuates experimental autoimmune encephalomyelitis (EAE), an animal model of relapsing–remitting multiple sclerosis (RMS) in mice by inhibiting encephalitogenic of myelin basic protein (MBP)-primed T cells and switched the immune response from a Th1 to a Th2 profile independent of PPAR-α [13,14,15,16]. Likewise, ciprofibrate was proposed to be effective in different autoimmune diseases, including MS, by increasing the production of anti-inflammatory cytokines, inhibiting T cells specific for MBP and reducing microglial activity [16].
Oral administration of gemfibrozil, ciprofibrate, and fenofibrate repressed clinical signs of EAE by shifting the cytokine secretion of human T-cell lines by inhibiting interferon-gamma (IFN-γ) and promoting IL-4 secretion [17]. Similarly, fenofibrate inhibits differentiation of Th17 by inhibiting the expression of pro-inflammatory signaling [18,19,20]. These outcomes propose that PPARα agonists may be attractive nominees for use in human inflammatory conditions, such as MS. Compared with gemfibrozil, fenofibrate produced significantly greater reductions in total cholesterol, triglycerides and significantly more significant increases in high-density lipoprotein (HDL). However, fenofibrate is less effective compared to new-generation pemafibrate in amelioration of lipid profile [18]. Moreover, fenofibrate has pleiotropic anti-inflammatory and antioxidant effects that may reduce inflammatory and oxidative stress disorders in different autoimmune disorders, as in MS [17]. According to these findings, this review aimed to clarify the mechanistic immunoinflammatory role of fenofibrate in mitigating MS neuropathology.
Pathophysiology of MS
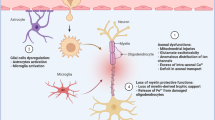
The underlying pathophysiology of MS is the destruction of myelin sheath by immune cells or failure in the production of myelin [4]. The characteristic feature of MS neuropathology is the formation of myelin plaques, inflammation, and injury of neuronal myelin sheath [9]. Myelin plaque represents a clustering of inflammation, myelin breakdown, astrogliosis, oligodendrocyte injury, neurodegeneration, axonal loss, and remyelination [9]. Breakdown of immune response and regulation due to environmental factors and genetic predisposition induce MS neuropathology. Abnormal immune response in genetically susceptible subjects to some environmental factors triggers cell-mediated immunity with the development of demyelination [9].
Of interest, MS is regarded as a hereditary disease, though different genetic variations may increase MS risk [10,11,12]. Different environmental factors trigger the development of MS, including early exposure to infectious agents, which attenuate MS risk. Epstein–Barr virus (EBV), which causes infectious mononucleosis, is implicated in the pathogenesis of MS by 32 folds [14]. Besides, smoking, organic solvents, obesity and certain diets may increase MS risk [15, 18]. However, gout and hyperuricemia are protective against the development of MS [17].
CNS plaques in MS mainly affect the brain stem, basal ganglia, optic nerve, and spinal cord, though peripheral neurons are rarely affected [18, 19]. In the MS plaques, the inflammatory profile is characterized by infiltration of immune cells, including T lymphocytes, monocytes, B and plasma cells [20]. Three different types of MS plaques have been revealed, including type I (macrophages and T lymphocytes dominate lesions), type II (have additional accumulation of activated complement and immunoglobulins) and type III (in which there is additional apoptosis of oligodendrocytes and glial cells) [21, 22]. MS lesions seem identical in the affected patient but vary among patients, reflecting different stages of MS progression rather than different disease subtypes [23]. In MS, oligodendrocytes involved in the myelin sheath synthesis are mainly affected [24]. Myelin sheath is involved in generating action potential and transmitting electrical signals. Progressive loss of myelin sheath with axonal injury leads to neuronal dysfunction [25]. Partial remyelination may occur during the remission state, and demyelination is returned during the relapse state. These changes promote plaque formation in the multiple sites in the CNS. In addition, reactive astrocytosis in response to neuronal injury promote plaque formation [26].
On the other hand, inflammation plays an integral role in the pathogenesis of MS due to the uncontrolled activation of T lymphocytes [27]. Peripheral auto-reactive T lymphocytes trigger inflammatory changes in the MS [28]. However, the underlying mechanism for activating peripheral auto-reactive T lymphocytes is poorly identified. It has been shown that polyclonal activation of peripheral auto-reactive T lymphocytes by viral and bacterial antigens or molecular mimicry could be the possible mechanism [29]. Peripheral auto-reactive T lymphocytes can cross blood–brain barrier (BBB) through binding integrins on the immune cells and VCAM-1 on the endothelial cells [30,31,32,33]. Following entry of peripheral auto-reactive T lymphocytes, these cells bind MHCII expressed by dendritic and antigen-presenting cells, leading to disruption of myelin components and release of other CNS antigens with subsequent recruitment of other immune cells and production of specific myelin autoantibodies, which promote further injury and loss of myelin sheath [34,35,36]. The interaction between auto-reactive T lymphocytes and myelin antigens triggers the release of pro-inflammatory and inflammatory cytokines with the production of antibodies [36]. These immune-inflammatory reactions cause further injury of BBB that promotes entry of auto-reactive T lymphocytes and generation of soluble factors which attack synaptic regions, causing neuronal dysfunction [35, 36]. These neuropathological changes lead to progressive loss of myelin sheath and axonal damage (Fig. 1).

Pathophysiology of MS
Moreover, neurodegeneration is also intricate in MS neuropathology. Mounting evidence highlighted that neurodegeneration occurs early in both gray and white matter of MS patients [37,38,39]. In addition, axonal degeneration analysis in chronic inactive lesions of secondary progressive MS patient spinal cords indicated a 61% reduction in axonal density [40]. Interestingly, gray matter atrophy rates have been correlated with worsening disability in MS. One of the mechanisms hypothesized to explain the diffuse neurodegeneration found in MS patients involves mitochondrial dysfunction [40,41,42,43]. Diffuse mitochondrial dysfunction secondary to MS induces inadequate energy production and intracellular dysregulation. This dysfunction impairs anterograde and retrograde transportation along axons.
Furthermore, progressive inflammation and autoimmunity trigger neurodegeneration in MS. Taken together, autoimmunity, demyelination, inflammation, and neurodegeneration are involved in the pathogenesis of MS. Different studies revealed that fenofibrate is effective in reducing neurodegeneration in patients with diabetic retinopathy [5, 43,44,45] and experimental Parkinson disease (PD) model by inhibiting neuroinflammation and oxidative stress. In addition, fenofibrate has potent anti-inflammatory effects through direct inhibition of pro-inflammatory cytokines or indirectly by reducing dyslipidemia-induced inflammation. Furthermore, fenofibrate can reduce autoimmunity and demyelination in MS through modulation of Th1/Th2 immune response. Thus, fenofibrate can modulate the components of MS neuropathology.
Acute attack of MS is treated by corticosteroids [43], and plasmapheresis is indicated when treatment with corticosteroid is ineffective. Chronic MS is managed by disease-modifying treatments, such as interferons, glatiramer, and mitoxantrone [45, 46]. However, lipid-based therapeutics linked with possible future-based therapeutic interventions such as fenofibrate are proposed in the management of MS [54, 55].
Lipid dysregulation in MS
Furthermore, lipid dysregulation is associated with MS neuropathology as lipid molecules play a dual role in MS, both as target molecules of myelin destruction and as mediators of inflammation [56,57,58]. Altered lipid metabolism with systemic inflammation may contribute to immune activation. Evidence suggests that abnormalities in the lipid-binding proteins of myelin and sphingolipid content that confers increased immunogenicity may cause the autoimmune response against the myelin sheath [59]. CNS is, after all, the second organ richer in lipid content after adipose tissue. In addition, soluble factors secreted by adipose tissue modulate inflammatory responses and contribute to metabolic dysfunction, which may be important in MS pathophysiology. Inflammatory cytokines participate in proatherogenic changes in lipid metabolism by reducing HDL levels and impairing anti-inflammatory and antioxidant functions.
Consequently, the protective actions of HDL can be limited in chronic inflammatory diseases, such as MS [60,61,62,63]. A case–control study illustrated that dysfunctional HDL is correlated with inflammatory mediators in MS patients. Fenofibrate improves circulating HDL and reduces inflammatory disorders. Dyslipidemia with low HDL and high triglyceride (TG) correlated disease activity and disability in MS patients. It remains to be elucidated whether altered lipid metabolism contributes to harmful immune response, possibly through inflammation, or is secondary to neurological disability in MS. A cohort study involving 492 MS patients revealed that serum lipid profile has modest effects on disease progression in MS. Worsening disability is associated with higher levels of LDL, total cholesterol and triglycerides. Higher HDL is associated with lower levels of acute inflammatory activity [63,64,65,66]. Fenofibrate has a potent effect in regulating lipid profiles mainly TG, HDL and LDL. These observations indicated that dysregulation of lipid profile triggers abnormal immune response and increases the risk of autoimmunity as in MS. Fenofibrate through modulation of blood lipid can mitigate the detrimental effects of TG and LDL on MS neuropathology.
Potential beneficial effects of fenofibrate in MS
Pharmacology of fenofibrate
Fenofibrate is a chlorobenzophenone derivative drug (Fig. 2) used to manage hypertriglyceridemia and mixed hyperlipidemia [50, 51].

Chemical structure of fenofibrate
Fenofibrate was initially synthesized from clofibrate in France in 1974; it was known as precetofen, which was renamed fenofibrate according to the WHO no-proprietary guideline [49]. Fenofibrate acts via activation of PPAR-α, leading to activation of lipoprotein lipase and reduction of apolipoprotein CIII, resulting in lipolysis and elimination of TG from plasma. Fenofibrate increases expression of apolipoprotein AI and AII, leading to reducing levels of LDL and VLDL with increment of HDL [50, 51]. Of interest is that fenofibrate use has been shown to be effective in managing diabetic retinopathy [52]. In addition, fenfibrate use in T2DM patients reduces the risk of amputation by 37% regardless of glycemic control [53]. Fenofibrate is subjected to drug interaction with bile acid sequestrants, immunosuppressant agents, warfarin, and statins. The most common adverse effects of fenofibrate are myalgia, headache, arthralgia, and renal stone. Fenofibrate is contraindicated in patients with renal impairment, liver dysfunction, gallbladder diseases, hypothyroidism, and hypersensitivity [50, 51]. Due to its anti-inflammatory and antioxidant effects, fenofibrate was suggested for treating different neurodegenerative disorders. PPAR-α is expressed in the brain and other organs and plays a significant role in oxidative stress, energy homeostasis, mitochondrial fatty acids metabolism and inflammation. PPAR-α contributes to the regulation of genes coding proteins that are intricate in glutamate homeostasis and cholinergic/dopaminergic signaling in the brain.
Furthermore, PPAR-α regulates the expression of genes coding enzymes engaged in amyloid precursor protein (APP) metabolism. It activates gene coding of α secretase, which is responsible for the non-amyloidogenic pathway of APP degradation [67,68,69]. It also down-regulates β secretase (BACE-1), the main enzyme responsible for amyloid beta (Aβ) peptide release in AD. In AD the expression of genes of PPAR-α and PPAR-γ coactivator-1 alpha (PGC-1α) is significantly decreased. PPARs are altered not only in AD but in other neurodegenerative/neurodevelopmental and psychiatric disorders [69,70,71]. PPAR-α downregulation may decrease anti-oxidative and anti-inflammatory processes. It could be responsible for the alteration of fatty acid transport, lipid metabolism and disturbances of mitochondria function in the brain of AD patients. Specific activators of PPAR-α may be important for the improvement of brain cell metabolism and cognitive function in neurodegenerative and neurodevelopmental disorders [71]. However, the potential mechanism of fenofibrate in MS is not fully elucidated.
Role of fenofibrate in MS
PPAR-α agonists have been used over decades to treat human metabolic disorders with little toxicity, making them an attractive candidate for use in the management of MS. PPAR-α agonists, such as fenofibrate, can alter the phenotype of myelin autoreactive T cells and their encephalitogenicity [62, 72].
It has been shown that PPAR-α is expressed in T cells, and its ligands inhibit T-cell proliferation, release of IL-2 and regulation of inflammatory response [63]. Fenofibrate can attenuate autoimmune response in mice with experimental Sjogren syndrome through modulation of T-cell's immune response. PPAR-α-deficient mice had abnormal immune responses to inflammatory mediators, such as prostaglandins and leukotrienes. Expression of adhesion molecules, cyclooxygenase-2 (COX-2) and IL-6 are inhibited by PPAR-α ligands [65]. PPAR-α ligands inhibit expression of NF-κB through increasing expression of NF-κB inhibitor (IκBα) [66]. PPAR-α ligand WY14643 blocks IgG interaction with myelin oligodendrocyte in mice [66]. PPAR-α ligands such as fenofibrate have anti-inflammatory effects by increasing the release of anti-inflammatory cytokines [67]. Interestingly, PPAR-α ligands promote Th2 cytokine production [68].
It has been revealed that fenofibrate can reduce the inflammatory reactions in MS through inhibition expression of IL-2 in lipopolysaccharide (LPS)-induced microglia activation [47]. Moreover, fenofibrate inhibits microglial expression of CD14, which plays a critical role in TLR signaling, signifying a mechanism by which fenofibrate suppresses the release of pro-inflammatory molecules [47]. Prominently, fenofibrate suppression of EAE was associated with decreased expression of IL-12 family cytokine mRNAs and mRNAs encoding TLR4, CD14, and MyD88 known to play critical roles in MyD88-dependent TLR signaling [47]. These findings propose that fenofibrate may modulate the development of EAE by inhibiting the production of IL-12 and MyD88-dependent-signaling pathway [47].
Fenofibrate has the ability to inhibit differentiation of Th17 significantly compared to other PPAR-α agonists, such as bezafibrate. Fenofibrate blocks IL-21 and STAT3 expression is required for Th17 differentiation [48]. It has illustrated that fenofibrate mitigates non-lipid-associated autoimmune diseases, such as autoimmune colitis and MS [48]. It has been hypothesized that fenofibrate reduces the differentiation of Th17 cells and inhibits transforming growth factor-β (TGF-β) and IL-6-induced differentiation of Th17 cells. However, other PPARα agonist's bezafibrate did not affect Th17 differentiation, indicating that this effect of fenofibrate might be PPARα independent [48]. A case–control study revealed that prolonged use of fenofibrate reduces inflammatory biomarkers, including IL-6 and CRP, in patients with dyslipidemia [54], signifying possible fenofibrate effects on systemic inflammation. Janssen et al. [55] observed that IL-6 is involved in the pathogenesis of MS by activating T cells [55]. It has been observed that fenofibrate and other PPAR-α agonists activate the neurons' myelination by increasing expression of sterol regulatory element binding factors (SREBF), which increase sterol biosynthesis [56]. PPARα can stimulate SREBF signaling via multiple mechanisms, including increasing SREBF expression, enhancing SREBF proteolytic cleavage, and increasing SREBF activity through the recruitment of transcriptional co-activators [56]. Gemfibrozil activates PPARα and increases the expression of myelin in human oligodendrocytes. PPARα activation can also stimulate SREBF signaling. Fibrates have been considered as potential therapeutics for diseases associated with impaired oligodendrocytes, such as MS, adrenoleukodystrophy, and traumatic brain injury [56]. Therefore, fenofibrate and gemfibrozil promote myelination by activating SREBFs in MS. Of note, SREBFs are reduced in MS [57]. Fenofibrate and other PPAR-α agonists have been shown to inhibit microglia are involved in MS neuropathology [58].
Collectively, fenofibrate could be effective in managing MS by its anti-inflammatory effect and modulation of SREBFs. Accordingly, fenofibrate regulates two important aspects, including neuroinflammation and neurodegeneration, which are highly intricate in MS neuropathology.
Effect of fenofibrate on neuroinflammation in MS
Neuroinflammation is associated with the progression of different neurodegenerative disorders. T and B cells in the CNS trigger inflammatory disorders and the development of neuroinflammation. Neuroinflammation in the early stage of MS can cause synaptopathy independent of the demyelination process, and this may explain cognitive dysfunction in the early phase of MS patients [73]. In the late phase of MS, exaggeration of immune disturbance and development of neuroinflammation promote MS pathogenesis. It has been shown that cholinergic activity is reduced in MS patients, which regulates the activity and response of immune cells. Reduction of acetylcholine level in the immune cells promotes the release of pro-inflammatory cytokines with the development of neuroinflammation [74, 76]. Therefore, attenuation of neuroinflammation could be a therapeutic strategy in the mitigation of MS neuropathy.
Different preclinical studies revealed a potential role of fenofibrate against the development and progression of neuroinflammation. Fenofibrate inhibits neuroinflammation in traumatic brain injury by suppressing oxidative stress in rats. Fenofibrate has a neuroprotective effect against the development and progression of MS by inhibiting mitochondrial dysfunction, oxidative stress, and neuroinflammation that contribute mutually to neurodegeneration [77]. The underlying mechanism for fenofibrate role against the development of neuroinflammation is related to the inhibition of inflammatory-signaling pathway, antioxidant and anti-inflammatory effects. Remarkably, fenofibrate activates neuronal nicotinic cholinergic receptors with subsequent inhibition of neuroinflammation [78]. These findings indicated that fenofibrate may reduce MS pathogenesis by modulating neuroinflammation.
Toll-like receptors (TLRs) are innate immune sensors that alert the immune system to the presence of external pathogens [79, 80]. Activation of TLR triggers the release of pro-inflammatory cytokines and activation of adaptive immune response to eliminate invading pathogens. TLR can detect danger signals, which are products of inflammation and tissue injury. TLRs are highly expressed by immune cells in the CNS and are involved in the MS neuropathology [81]. Of note, TLR agonists participate in the amplification of harmful inflammatory responses. It has been established that PPAR-α agonists have reciprocal interactions with TLRs, as activation of PPAR-α inhibits expression of TLRs via several mechanisms. Fenofibrate inhibits the expression of CD14, which increases the expression of TLR and the release of pro-inflammatory cytokines. In addition, fenofibrate inhibits the release of IL-12 and the expression of the MyD88–TLR4-signaling pathway [47]. Therefore, PPAR-α agonist fenofibrate can inhibit the primary immune response in MS neuropathology by inhibiting TLRs and their effectors.
Furthermore, different inflammatory-signaling pathways, including NF-κB and nod-like receptor pyrin three receptor (NLRP3) inflammasome, are involved in the pathogenesis of MS [82,83,84,85,86,87]. NF-κB is a DNA-binding protein necessary for transcription of chemokines and pro-inflammatory cytokines. Particularly, immune deregulation encourages the commencement of NF-κB with consequential neuronal injury, neuroinflammation, and development of neurodegeneration [88,89,90,91]. NLRP3 inflammasome is involved in the activation of caspase-1 and maturation of IL-1β and IL-18 [92,93,94]. Diverse stimuli including NF-κB trigger NLRP3 inflammasome. NLRP3 inflammasome is intricate in the pathogenesis of neuroinflammation and development of neurodegeneration [95,96,97,98,99,100]. NF-κB is exaggerated in MS, leading to immune dysregulation and induction release of pro-inflammatory cytokines. Inhibition of the NF-κB-signaling pathway by teriflunomide, fingolimod and dimethyl fumarate may reduce MS severity [100,101,102]. Chen et al. [103] observed that native and memory B cells from MS patients have a higher level of phosphorylated NF-κB, which was inhibited by mycophenolate. In addition, glatiramer attenuates the activation of NF-κB by CD40, which is over-activated in MS [103]. Likewise, NLRP3 inflammasome is also exaggerated and linked with the severity of MS [104]. NLRP3 inflammasome within activated microglia promotes the expression and release of IL-1β and IL-18. Evidence from preclinical and clinical findings illustrated that aberrant activation of NLRP3 inflammasome is associated with the pathogenesis of MS [104]. Over-activation of NLRP3 inflammasome in MS is evident by increasing IL-1β CSF levels in severely affected patients [105]. Targeting NLRP3 inflammasome by specific inhibitors can reduce MS severity [105].
Fenofibrate has a potent anti-inflammatory effect against the development of pulmonary inflammation by inhibiting the expression of NF-κB and NLRP3 inflammasome [106]. Notably, fenofibrate prevents retinal injury and disruption of retinal blood barrier by inhibiting the NF-κB-signaling pathway [107]. Besides, fenofibrate can reduce diabetic retinopathy by inhibiting the expression of NLRP3 inflammasome [108,109,110,111]. Thus, fenofibrate might effectively reduce MS pathogenesis by targeting the most common inflammatory-signaling pathways, including NF-κB and NLRP3 inflammasome [112,113,114,115,116].
Interestingly, TLRs, the release of pro-inflammatory-signaling pathways and inflammatory mediators are involved in the development and progression of neuroinflammation in MS. Inhibition of TLRs and release of pro-inflammatory by fenofibrate result in momentous suppression of neuroinflammation in MS [117,118,119,120,121].
Effect of fenofibrate on neurodegeneration in MS
It has been illustrated that mitochondrial dysfunction and deregulation of neuronal energy balance secondary to MS induce inadequate ATP production and intracellular dysregulation [122,123,124,125,126]. This dysfunction impairs anterograde and retrograde transportation along axons, leading to progressive neurodegeneration in MS [125,126,127]. Moreover, mitochondrial dysfunction contributes to the loss of neurons and axons in MS due to uncontrolled activation of microglia and associated neuronal injury [80]. Impairment of mitochondrial permeability transition pore by Ca2+ dyshomeostasis and ROS is the central mechanism for the development of mitochondrial dysfunction in MS [81]. Pathological opening of mitochondrial permeability transition pore in response to nitrogen species, Ca2+ and ROS, induces an influx of many solutes into the mitochondrial matrix, leading to matrix expansion and mitochondrial rupture with eventual cell deaths [81]. Merlini et al. [82] revealed that mitochondrial dysfunction is regarded as an essential trigger of programmed axon death in MS. Uric acid and serum lactate are considered as potential biomarkers of mitochondrial dysfunction [83]. A case–control study that included 32 MS patients, and 20 healthy controls showed that lactate serum level but not serum uric acid was increased in MS patients compared to the controls [83]. It has been proposed that mitochondrial dysfunction alters lymphocyte homeostasis, leading to a defective apoptotic process of auto-reactive T cells, allowing them to perpetuate within the CNS and continue the inflammation cycle in MS patients [84]. Activation of Th1 cells and their lymphokines, such as interferon-gamma (INF-α) and IL-2, which induce the transformation of B-lymphocytes to plasma cells produce autoantibodies against myelin antigens [84]. Therefore, mitochondrial dysfunction could be a primary cause for MS progression through alteration of lymphocyte activity, or a secondary outcome due to oxidative stress caused by MS. Thus, mitigation of mitochondrial dysfunction could be effective mechanistic way to prevent the progression of MS.
On the other side, PPAR-α agonists have an important role in the modulation of mitochondrial function in diabetic patients [85]. Of interest, fenofibrate improves insulin sensitivity by enhancing mitochondrial β-oxidation [86]. In a similar way, fenofibrate inhibits mitochondrial dysfunction in burn patients [86]. In addition, fenofibrate enhances neurogenesis via modulation of mitochondrial biogenesis in experimental ischemic reperfusion injury [87]. The protective effect of fenofibrate against the development of mitochondrial dysfunction is mediated by increasing the expression of mitochondrial uncoupling protein two, which protects mitochondria from the harmful oxidative stress by reducing the generation of ROS [88]. However, a higher concentration of fenofibrate may induce the development of mitochondrial dysfunction via inhibition of mitochondrial respiratory chain complex I [89]. Therefore, the appropriate dose of fenofibrate could be effective against MS through the modulation of mitochondrial dysfunction.
In addition, oxidative stress plays an integral role in the pathogenesis of MS via the enhancement of the demyelination process and neurodegeneration [73]. ROS promotes peripheral activation of T cells and the development of autoreactive T cells. ROS triggers microglia activation and induces neuronal apoptosis [73]. Inflammatory reactions in MS can provoke oxidative stress bursts in the activated macrophages and microglia, leading to neuronal demyelination. In turn, oxidative stress and released ROS enhance the propagation of inflammation and neurodegeneration in MS [74]. Therefore, there is positive feedback activation between oxidative stress and inflammation in a vicious cycle in MS. A case–control study showed that biomarkers of oxidative stress were increased in patients with RRMS compared to healthy controls [75]. These findings proposed that oxidative stress can aggravate inflammatory reactions and contribute to more neuronal injury and progression of MS. Therefore, the use of antioxidants may hinder the development and progression of MS. Carlson and his colleagues proposed that antioxidants may play a beneficial role in human MS despite conflicting findings in animal MS model [76]. Evidence from preclinical and clinical trials showed that the use of antioxidant alpha lipoic acid reduces brain atrophy and improves the clinical course of MS [77]. It has been demonstrated that PPAR-α agonists have potent antioxidant effects and can ameliorate different neurodegenerative disorders, including AD [78]. PPAR-α agonist GW7647 inhibits lipid peroxidation, oxidative stress, and inflammation in mouse AD models [78]. An experimental study illustrated that fenofibrate improves antioxidant capacity and attenuates hyperglycemia-induced oxidative stress [79]. Oyagbemi et al. [79] revealed that PPAR-α agonist clofibrate attenuates oxidative stress in rats. Therefore, fenofibrate, through inhibition of oxidative stress and potentiating of endogenous antioxidant capacity, can mitigate MS pathogenesis.
Indeed, the propagation of neurodegeneration in MS and other neurodegenerative diseases is related to the reduction of neuroprotective brain-derived neurotrophic factor (BDNF) [61]. It has been illustrated that BDNF level is reduced in MS due to progressive neurodegeneration process [61]. A case control study on 22 MS patients compared to 19 healthy controls revealed that BDNF serum levels were reduced in MS patients compared to the controls [61]. However, a recent study observed that BDNF serum levels were not significantly reduced in MS patients compared to healthy controls [69]. A systemic review and meta-analysis involving 30 studies (689 MS patients and 583 healthy controls) revealed that BDNF serum level was reduced in MS patients compared to healthy controls [70].
Moreover, the fenofibrate neuroprotective effect can attenuate hippocampal insulin resistance and improve cognitive function in rats by increasing the expression of BDNF [48, 59, 127,128,129,130]. The neuroprotective effect of fenofibrate is mediated by increasing expression of BDNF in animal model studies [60]. In addition, fenofibrate upregulates the expression of hippocampal BDNF, which attenuates the neurodegeneration process in MS [60]. BDNF regulates microglia function toward trophic phenotype and prevents microglia-induced neurodegeneration [71, 131,132,133,134]. Notably, naturally derived phytoconstituents, including curcumin, cannabinoids, and genistein, reduce neurodegenerative diseases by increasing expression of BDNF [72]. These observations suggest that PPAR-α agonists such as fenofibrate can reduce MS neuropathology by improving the expression of BDNF.
Notoriously, autophagy plays a critical role in neurodegenerative diseases and could be beneficial in the early stage and detrimental in the late stage, as in AD and PD [90]. Autophagy is an essential intracellular degradative pathway to maintain normal cellular homeostasis by eliminating toxic proteins and injured organelles [115, 128, 135,136,137,138]. Defective autophagy in neurons induces the development of neurodegeneration [90]. Autophagy regulates adaptive and innate immunity, and autophagy abnormality triggers abnormal immune response [139,140,141,142,143,144,145,146]. Neuron function mainly depends on autophagy for its survival and homeostasis [147,148,149]. Defective autophagy contributes to the development and progression of MS. The autophagy process acts as a double-edged sword and could be protective or detrimental [150,151,152,153,154,155]. For example, increasing autophagy of T and B cells promotes the development of neuroinflammation, and inhibition of autophagy in this regard might be effective in treating MS. However, the induction of autophagy in neurons and glial cells improves the remyelination process [90, 91].
Interestingly, restoration of normal autophagy function in the early MS prevents the progression of disease severity [91, 156]. Autophagy-related genes are increased in T cells of MS brains [92]. These findings suggest a controversy regarding the role of autophagy in MS. Interestingly, restoration of normal autophagy function in early MS prevents the progression of disease severity [91]. Thus, targeting neuronal autophagy is important to enhance the remyelination process.
Different studies confirmed that fenofibrate improves the autophagy process [93, 158]. For example, fenofibrate attenuates cardiac injury in diabetic mice by increasing the expression of neuroprotective SIRT1 and autophagy function [93, 159,160,161,162,163]. In addition, fenofibrate attenuates acute kidney injury by regulating the autophagy process through the expression of adenosine monophosphate protein kinase (AMPK) [94, 164]. Both AMPK and SIRT1 activate the autophagy process [95, 165]. Therefore, fenofibrate through modulation of the autophagy process may attenuate the development and progression of MS.
Fenofibrate can attenuate MS neuropathology through modulation of different pathways, including oxidative stress, autophagy, mitochondrial dysfunction, inflammatory-signaling pathways, and neuroinflammation.
Conclusion
MS is the most common inflammatory and demyelinating disease of the CNS. The fundamental pathophysiology of MS is the destruction of myelin sheath by immune cells or failure in the production of myelin. The characteristic feature of MS neuropathology is the formation of CNS plaques, which are multiple focal regions of demyelination distributed in the brain's white matter and spinal cords as well as in the deep grey matter and cerebral cortex. Notably, PPAR-α activator fenofibrate can attenuate the inflammatory reactions in MS by inhibiting the differentiation of Th17 and the expression of pro-inflammatory-signaling. Fenofibrate can reduce MS neuropathology by increasing the expression of BDNF. Fenofibrate, through inhibition of oxidative stress and potentiating of endogenous antioxidant capacity, can mitigate MS pathogenesis. Appropriate doses of fenofibrate could be effective against MS through modulation of mitochondrial dysfunction, autophagy process, inflammatory-signaling pathways, and neuroinflammation. Fenofibrate can reduce MS neuropathology through modulation of different pathways, including oxidative stress, autophagy, mitochondrial dysfunction, inflammatory-signaling pathways, and neuroinflammation. Therefore, preclinical and clinical studies are warranted to elucidate the precise role of fenofibrate in treating MS.
Availability of data and materials
All data are available in the manuscript.
References
Dobson R, Giovannoni G. Multiple sclerosis–a review. Eur J Neurol. 2019;26(1):27–40.
Vecchio F, Miraglia F, Porcaro C, Cottone C, Cancelli A, Rossini PM, Tecchio F. Electroencephalography-derived sensory and motor network topology in multiple sclerosis fatigue. Neurorehabil Neural Repair. 2017;31(1):56–64.
Dutta R, Trapp BD. Relapsing and progressive forms of multiple sclerosis–insights from pathology. Curr Opin Neurol. 2014;27(3):271.
Buscarinu MC, Fornasiero A, Romano S, Ferraldeschi M, Mechelli R, Reniè R, Morena E, Romano C, Pellicciari G, Landi AC, Salvetti M. The contribution of gut barrier changes to multiple sclerosis pathophysiology. Front Immunol. 2019;10:1916.
McGinley MP, Goldschmidt CH, Rae-Grant AD. Diagnosis and treatment of multiple sclerosis: a review. JAMA. 2021;325(8):765–79.
Lane J, Ng HS, Poyser C, Lucas RM, Tremlett H. Multiple sclerosis incidence: A systematic review of change over time by geographical region. Multiple Sclerosis Related Disorders. 2022;63: 103932.
Zeydan B, Kantarci OH. Impact of age on multiple sclerosis disease activity and progression. Curr Neurol Neurosci Rep. 2020;20:1–7.
Clanet M. Jean-Martin Charcot: 1825–1893. The International MS Journal. 2008;15(2):59–62.
Derada Troletti C, Fontijn RD, Gowing E, Charabati M, van Het Hof B, Didouh I, van der Pol SM, Geerts D, Prat A, van Horssen J, Kooij G. Inflammation-induced endothelial to mesenchymal transition promotes brain endothelial cell dysfunction and occurs during multiple sclerosis pathophysiology. Cell Death Dis. 2019;10(2):45.
Hedström AK, Hössjer O, Hillert J, Stridh P, Kockum I, Olsson T, Alfredsson L. The influence of human leukocyte antigen-DRB1* 15: 01 and its interaction with smoking in MS development is dependent on DQA1* 01: 01 status. Mult Scler J. 2020;26(13):1638–46.
Dyment DA, Ebers GC, Sadovnick AD. Genetics of multiple sclerosis. Lancet Neurol. 2004;3(2):104–10.
Gerdes LA, Janoschka C, Eveslage M, Mannig B, Wirth T, Schulte-Mecklenbeck A, Lauks S, Glau L, Gross CC, Tolosa E, Flierl-Hecht A. Immune signatures of prodromal multiple sclerosis in monozygotic twins. Proc Natl Acad Sci. 2020;117(35):21546–56.
International Multiple Sclerosis Genetics Consortium*†, ANZgene, IIBDGC, WTCCC2. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science. 2019;365(6460):eaav7188.
Aloisi F, Cross AH. MINI-review of Epstein-Barr virus involvement in multiple sclerosis etiology and pathogenesis. J Neuroimmunol. 2022:577935.
Coles A. Alastair Compston, Alasdair Coles. Lancet. 2008;372:1502–7.
Marrie RA. Environmental risk factors in multiple sclerosis aetiology. Lancet Neurol. 2004;3(12):709–18.
Bolayir A, Cigdem B, Gokce SF, Yilmaz D. The relationship between neutrophil/lymphocyte ratio and uric acid levels in multiple sclerosis patients. Bratisl Lek Listy. 2021;122(5):357–61.
Nourbakhsh B, Mowry EM. Multiple sclerosis risk factors and pathogenesis. CONTINUUM: Lifelong Learning in Neurology. 2019;25(3):596–610.
Zéphir H. Progress in understanding the pathophysiology of multiple sclerosis. Revue neurologique. 2018;174(6):358–63.
Pinheiro MA, Kooij G, Mizee MR, Kamermans A, Enzmann G, Lyck R, Schwaninger M, Engelhardt B, de Vries HE. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2016;1862(3):461–71.
Grzegorski T, Losy J. Multiple sclerosis–the remarkable story of a baffling disease. Rev Neurosci. 2019;30(5):511–26.
Khatir AA, Hojjati SM, Ahangar AA, Naghshineh H, Saadat P. Multiple sclerosis and its pathophysiology: a narrative review. Tabari Biomedical Student Research Journal. 2020;20:89.
Lassmann H, Brück W, Lucchinetti C. Heterogeneity of multiple sclerosis pathogenesis: implications for diagnosis and therapy. Trends Mol Med. 2001;7(3):115–21.
Lan M, Tang X, Zhang J, Yao Z. Insights in pathogenesis of multiple sclerosis: nitric oxide may induce mitochondrial dysfunction of oligodendrocytes. Reviews in th Lassmann H, Van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nature Reviews Neurology. 2012 Nov;8(11):647–56. e Neurosciences. 2018;29(1):39–53.
Sedel F, Bernard D, Mock DM, Tourbah A. Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology. 2016;110:644–53.
Ponath G, Park C, Pitt D. The role of astrocytes in multiple sclerosis. Front Immunol. 2018;9:217.
Martino G, Adorini L, Rieckmann P, Hillert J, Kallmann B, Comi G, Filippi M. Inflammation in multiple sclerosis: the good, the bad, and the complex. The Lancet Neurology. 2002;1(8):499–509.
Liu GZ, Fang LB, Hjelmström P, Gao XG. Increased CD8+ central memory T cells in patients with multiple sclerosis. Mult Scler J. 2007;13(2):149–55.
Elsayed NS, Aston P, Bayanagari VR, Shukla SK. The gut microbiome molecular mimicry piece in the multiple sclerosis puzzle. Front Immunol. 2022;13: 972160.
Sinha S, Itani FR, Karandikar NJ. Immune regulation of multiple sclerosis by CD8+ T cells. Immunol Res. 2014;59:254–65.
Rice GP, Hartung HP, Calabresi PA. Anti-α4 integrin therapy for multiple sclerosis: mechanisms and rationale. Neurology. 2005;64(8):1336–42.
Haarmann A, Nowak E, Deiß A, van der Pol S, Monoranu CM, Kooij G, Müller N, van der Valk P, Stoll G, de Vries HE, Berberich-Siebelt F. Soluble VCAM-1 impairs human brain endothelial barrier integrity via integrin α-4-transduced outside-in signalling. Acta Neuropathol. 2015;129:639–52.
Mohammadhosayni M, Khosrojerdi A, Lorian K, Aslani S, Imani D, Razi B, Babaie F, Torkamandi S. Matrix metalloproteinases (MMPs) family gene polymorphisms and the risk of multiple sclerosis: systematic review and meta-analysis. BMC Neurol. 2020;20:1.
Martin R, Sospedra M, Eiermann T, Olsson T. Multiple sclerosis: doubling down on MHC. Trends Genet. 2021;37(9):784–97.
Balasa R, Barcutean L, Balasa A, Motataianu A, Roman-Filip C, Manu D. The action of TH17 cells on blood brain barrier in multiple sclerosis and experimental autoimmune encephalomyelitis. Hum Immunol. 2020;81(5):237–43.
James RE, Schalks R, Browne E, Eleftheriadou I, Munoz CP, Mazarakis ND, Reynolds R. Persistent elevation of intrathecal pro-inflammatory cytokines leads to multiple sclerosis-like cortical demyelination and neurodegeneration. Acta Neuropathol Commun. 2020;8(1):1–8.
Batiha GE, Al-Kuraishy HM, Al-Gareeb AI, Elekhnawy E. SIRT1 pathway in Parkinson’s disease: a faraway snapshot but so close. Inflammopharmacology. 2023;31(1):37–56.
Nadwa EH, Al-Kuraishy HM, Al-Gareeb AI, Elekhnawy E, Albogami SM, Alorabi M, Batiha GE, De Waard M. Cholinergic dysfunction in COVID-19: frantic search and hoping for the best. Naunyn Schmiedebergs Arch Pharmacol. 2023;396(3):453–68.
Al-Kuraishy HM, Al-Gareeb AI, Albogami SM, Jean-Marc S, Nadwa EH, Hafiz AA, A. Negm W, Kamal M, Al-Jouboury M, Elekhnawy E, Batiha GE. Potential therapeutic benefits of metformin alone and in combination with sitagliptin in the management of type 2 diabetes patients with COVID-19. Pharmaceuticals. 2022;15(11):1361.
Mayo CD, Miksche K, Attwell-Pope K, Gawryluk JR. The relationship between physical activity and symptoms of fatigue, mood, and perceived cognitive impairment in adults with multiple sclerosis. J Clin Exp Neuropsychol. 2019;41(7):715–22.
Alhossan A, Alaifan NF, Althwaini BT, Ahmad A. Evaluation of antipyretics use and heat sensitivity in patients with multiple sclerosis. Farmacia. 2022;70(4):704–11.
Teoli D, Cabrero FR, Ghassemzadeh S. Lhermitte sign. InStatPearls. 2021. StatPearls Publishing.
Hauser SL, Cree BA. Treatment of multiple sclerosis: a review. Am J Med. 2020;133(12):1380–90.
Pitt D, Lo CH, Gauthier SA, Hickman RA, Longbrake E, Airas LM, Mao-Draayer Y, Riley C, De Jager PL, Wesley S, Boster A. Toward Precision Phenotyping of Multiple Sclerosis. Neurology-Neuroimmunology Neuroinflammation. 2022;9(6):89.
Tintore M, Vidal-Jordana A, Sastre-Garriga J. Treatment of multiple sclerosis—success from bench to bedside. Nat Rev Neurol. 2019;15(1):53–8.
VESPIGNANI M. Integrative Approaches to Multiple Sclerosis. Integrative Neurology. 2020;219.
Xu J, Racke MK, Drew PD. Peroxisome proliferator-activated receptor-α agonist fenofibrate regulates IL-12 family cytokine expression in the CNS: relevance to multiple sclerosis. J Neurochem. 2007;103(5):1801–10.
Zhou Z, Sun W, Liang Y, Gao Y, Kong W, Guan Y, Feng J, Wang X. Fenofibrate inhibited the differentiation of T helper 17 cells in vitro. PPAR research. 2012;2012.
Lalloyer F, Staels B. Fibrates, glitazones, and peroxisome proliferator–activated receptors. Arterioscler Thromb Vasc Biol. 2010;30(5):894–9.
Alkhayyat SS, Al-Kuraishy HM, Al-Gareeb AI, El-Bouseary MM, AboKamer AM, Batiha GE, Simal-Gandara J. Fenofibrate for COVID-19 and related complications as an approach to improve treatment outcomes: the missed key for Holy Grail. Inflamm Res. 2022;89:1–9.
Rasheed HA, Hussien NR, Al-Naimi MS, Al-Kuraishy HM, Al-Gareeb AI. Fenofibrate and Crataegus oxyacantha is an effectual combo for mixed dyslipidemia. Biomedical and Biotechnology Research Journal (BBRJ). 2020;4(3):259.2
Qiu F, Meng T, Chen Q, Zhou K, Shao Y, Matlock G, Ma X, Wu W, Du Y, Wang X, Deng G. Fenofibrate-loaded biodegradable nanoparticles for the treatment of experimental diabetic retinopathy and neovascular age-related macular degeneration. Mol Pharm. 2019;16(5):1958–70.
Ferreira JP, Vasques-Nóvoa F, Ferrão D, Saraiva F, Falcão-Pires I, Neves JS, Sharma A, Rossignol P, Zannad F, Leite-Moreira A. Fenofibrate and heart failure outcomes in patients with type 2 diabetes: analysis from ACCORD. Diabetes Care. 2022;45(7):1584–91.
Aslibekyan S, Kabagambe EK, Irvin MR, Straka RJ, Borecki IB, Tiwari HK, Tsai MY, Hopkins PN, Shen J, Lai CQ, Ordovas JM. A genome-wide association study of inflammatory biomarker changes in response to fenofibrate treatment in the Genetics of Lipid Lowering Drug and Diet Network. Pharmacogenet Genomics. 2012;22(3):191–7.
Janssens K, Slaets H, Hellings N. Immunomodulatory properties of the IL-6 cytokine family in multiple sclerosis. Ann N Y Acad Sci. 2015;1351(1):52–60.
Ashikawa Y, Nishimura Y, Okabe S, Sasagawa S, Murakami S, Yuge M, Kawaguchi K, Kawase R, Tanaka T. Activation of sterol regulatory element binding factors by fenofibrate and gemfibrozil stimulates myelination in zebrafish. Front Pharmacol. 2016;7:206.
Riccio P. The molecular basis of nutritional intervention in multiple sclerosis: a narrative review. Complement Ther Med. 2011;19(4):228–37.
Xu J, Storer PD, Chavis JA, Racke MK, Drew PD. Agonists for the peroxisome proliferator-activated receptor-α and the retinoid X receptor inhibit inflammatory responses of microglia. J Neurosci Res. 2005;81(3):403–11.
Idowu OK, Ajayi SO, Oluyomi OO, Atobatele NM, Okesina AA. Effect of Fenofibrate on Hippocampal Insulin Resistance and Cognitive Function in a Rat Model of Type 2 Diabetes Mellitus. Journal of Krishna Institute of Medical Sciences (JKIMSU). 2021;10(1):23.
Ali EM, Abouelella AM. Brain derived neurotrophic factor has a role in the antidepressant effect of atorvastatin and fenofibrate in Wistar rats. Records of Pharmaceutical and Biomedical Sciences. 2022;6(3):179–94.
Wens I, Keytsman C, Deckx N, Cools N, Dalgas U, Eijnde BO. Brain derived neurotrophic factor in multiple sclerosis: effect of 24 weeks endurance and resistance training. Eur J Neurol. 2016;23(6):1028–35.
Racke MK, Gocke AR, Muir M, Diab A, Drew PD, Lovett-Racke AE. Nuclear receptors and autoimmune disease: the potential of PPAR agonists to treat multiple sclerosis. J Nutr. 2006;136(3):700–3.
Guo X, Dang W, Li N, Wang Y, Sun D, Nian H, Wei R. PPAR-α Agonist Fenofibrate Ameliorates Sjögren Syndrome–Like Dacryoadenitis by Modulating Th1/Th17 and Treg Cell Responses in NOD Mice. Investigative ophthalmology & visual science. 2022;63(6):12-.
Yang Y, Gocke AR, Lovett-Racke A, Drew PD, Racke MK. PPAR alpha regulation of the immune response and autoimmune encephalomyelitis. PPAR research. 2008;2008.
Yu J, Ip E, dela Pena A, Hou JY, Sesha J, Pera N, Hall P, Kirsch R, Leclercq I, Farrell GC. COX-2 induction in mice with experimental nutritional steatohepatitis: role as pro-inflammatory mediator. Hepatology. 2006;43(4):826–36.
Huang D, Zhao Q, Liu H, Guo Y, Xu H. PPAR-α agonist WY-14643 inhibits LPS-induced inflammation in synovial fibroblasts via NF-kB pathway. J Mol Neurosci. 2016;59:544–53.
Gallucci GM, Alsuwayt B, Auclair AM, Boyer JL, Assis DN, Ghonem NS. Fenofibrate Downregulates NF-κB Signaling to Inhibit Pro-inflammatory Cytokine Secretion in Human THP-1 Macrophages and During Primary Biliary Cholangitis. Inflammation. 2022;45(6):2570–81.
Bahrambeigi S, Molaparast M, Sohrabi F, Seifi L, Faraji A, Fani S, Shafiei-Irannejad V. Targeting PPAR ligands as possible approaches for metabolic reprogramming of T cells in cancer immunotherapy. Immunol Lett. 2020;220:32–7.
Naegelin Y, Saeuberli K, Schaedelin S, Dingsdale H, Magon S, Baranzini S, Amann M, Parmar K, Tsagkas C, Calabrese P, Penner IK. Levels of brain-derived neurotrophic factor in patients with multiple sclerosis. Annals of clinical and translational neurology. 2020;7(11):2251–61.
Karimi N, Ashourizadeh H, Pasha BA, Haghshomar M, Jouzdani T, Shobeiri P, Teixeira AL, Rezaei N. Blood levels of brain-derived neurotrophic factor (BDNF) in people with multiple sclerosis (MS): A systematic review and meta-analysis. Multiple Sclerosis and Related Disorders. 2022;65: 103984.
Prowse N, Hayley S. Microglia and BDNF at the crossroads of stressor related disorders: Towards a unique trophic phenotype. Neurosci Biobehav Rev. 2021;131:135–63.
Sharma A, Lee HJ. Role of phytoconstituents as PPAR agonists: implications for neurodegenerative disorders. Biomedicines. 2021;9(12):1914.
Ohl K, Tenbrock K, Kipp M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp Neurol. 2016;277:58–67.
Ortiz GG, Pacheco-Moisés FP, Bitzer-Quintero OK, Ramírez-Anguiano AC, Flores-Alvarado LJ, Ramírez-Ramírez V, Macias-Islas MA, Torres-Sánchez ED. Immunology and oxidative stress in multiple sclerosis: clinical and basic approach. Clinical and developmental immunology. 2013;2013.
Padureanu R, Albu CV, Mititelu RR, Bacanoiu MV, Docea AO, Calina D, Padureanu V, Olaru G, Sandu RE, Malin RD, Buga AM. Oxidative stress and inflammation interdependence in multiple sclerosis. J Clin Med. 2019;8(11):1815.
Carlson NG, Rose JW. Antioxidants in multiple sclerosis: do they have a role in therapy? CNS Drugs. 2006;20:433–41.
Waslo C, Bourdette D, Gray N, Wright K, Spain R. Lipoic acid and other antioxidants as therapies for multiple sclerosis. Curr Treat Options Neurol. 2019;21:1–21.
Qu XX, He JH, Cui ZQ, Yang T, Sun XH. PPAR-α agonist GW7647 protects against oxidative stress and iron deposit via GPx4 in a transgenic mouse model of Alzheimer’s diseases. ACS Chem Neurosci. 2021;13(2):207–16.
Oyagbemi AA, Adebiyi OE, Adigun KO, Ogunpolu BS, Falayi OO, Hassan FO, Folarin OR, Adebayo AK, Adejumobi OA, Asenuga ER, Ola-Davies OE. Clofibrate, a PPAR-α agonist, abrogates sodium fluoride-induced neuroinflammation, oxidative stress, and motor incoordination via modulation of GFAP/Iba-1/anti-calbindin signaling pathways. Environ Toxicol. 2020;35(2):242–53.
Witte ME, Mahad DJ, Lassmann H, van Horssen J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol Med. 2014;20(3):179–87.
Su K, Bourdette D, Forte M. Mitochondrial dysfunction and neurodegeneration in multiple sclerosis. Front Physiol. 2013;4:169.
Merlini E, Coleman MP, Loreto A. Mitochondrial dysfunction as a trigger of programmed axon death. Trends Neurosci. 2022;45(1):53–63.
Hewedi K, Abd El Aziz AF, Essmat A, Faheem M. Laboratory Evaluation for Mitochondrial Dysfunction in Multiple Sclerosis Patients. Al-Azhar International Medical Journal. 2020;1(11):190–2.
Signorile A, Ferretta A, Ruggieri M, Paolicelli D, Lattanzio P, Trojano M, De Rasmo D. Mitochondria, oxidative stress, cAMP signalling and apoptosis: a crossroads in lymphocytes of multiple sclerosis, a possible role of nutraceutics. Antioxidants. 2020;10(1):21.
Lee TW, Bai KJ, Lee TI, Chao TF, Kao YH, Chen YJ. PPARs modulate cardiac metabolism and mitochondrial function in diabetes. J Biomed Sci. 2017;24(1):1–9.
Cree MG, Zwetsloot JJ, Herndon DN, Qian T, Morio B, Fram R. Insulin sensitivity and mitochondrial function are improved in children with burn injury during a randomized controlled trial of fenofibrate. Ann Surg. 2007;245:214–21.
Mohagheghi F, Ahmadiani A, Rahmani B, Moradi F, Romond N, Khalaj L. Gemfibrozil pretreatment resulted in a sexually dimorphic outcome in the Rat models of global cerebral ischemia–reperfusion via modulation of mitochondrial Pro-survival and apoptotic cell death factors as well as MAPKs. J Mol Neurosci. 2013;50:379–93.
Patterson AD, Shah YM, Matsubara T, Krausz KW, Gonzalez FJ. PPARα-dependent induction of uncoupling protein 2 protects against acetaminophen-induced liver toxicity. Hepatology. 2012;56:281–90.
Brunmair B, Lest A, Staniek K, Gras F, Scharf N, Roden M. Fenofibrate impairs Rat mitochondrial function by inhibition of respiratory complex I. J Pharmacol Exp Ther. 2004;311:109–14.
Misrielal C, Mauthe M, Reggiori F, Eggen BJ. Autophagy in multiple sclerosis: two sides of the same coin. Front Cell Neurosci. 2020;14: 603710.
Shen D, Liu K, Wang H, Wang H. Autophagy modulation in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2022;209(2):140–50.
Liang P, Le W. Role of autophagy in the pathogenesis of multiple sclerosis. Neurosci Bull. 2015;31:435–44.
Zhang J, Cheng Y, Gu J, Wang S, Zhou S, Wang Y, Tan Y, Feng W, Fu Y, Mellen N, Cheng R. Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of Type 1 diabetic mice. Clin Sci. 2016;130(8):625–41.
Sohn M, Kim K, Uddin MJ, Lee G, Hwang I, Kang H, Kim H, Lee JH, Ha H. Delayed treatment with fenofibrate protects against high-fat diet-induced kidney injury in mice: the possible role of AMPK autophagy. American Journal of Physiology-Renal Physiology. 2017;312(2):F323–34.
Liu Y, Xu Y, Zhu J, Li H, Zhang J, Yang G, Sun Z. Metformin prevents progression of experimental pulmonary hypertension via inhibition of autophagy and activation of adenosine monophosphate-activated protein kinase. J Vasc Res. 2019;56(3):117–28.
Wu Y, Li X, Zhu JX, Xie W, Le W, Fan Z, Jankovic J, Pan T. Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals. 2011;19(3):163–74.
Al-Kuraishy HM, Al-Fakhrany OM, Elekhnawy E, Al-Gareeb AI, Alorabi M, De Waard M, Albogami SM, Batiha GE. Traditional herbs against COVID-19: back to old weapons to combat the new pandemic. Eur J Med Res. 2022;27(1):186.
Al-Kuraishy HM, Al-Gareeb AI, Fageyinbo MS, Batiha GE. Vinpocetine is the forthcoming adjuvant agent in the management of COVID-19. Future Science OA. 2022;8(5):FSO797.
Batiha GE, Al-Gareeb AI, Rotimi D, Adeyemi OS, Al-Kuraishy HM. Common NLRP3 inflammasome inhibitors and Covid-19: divide and conquer. Scientific African. 2022;18: e01407.
Batiha GE, Al-Gareeb AI, Rotimi D, Adeyemi OS, Al-Kuraishy HM. Common NLRP3 inflammasome inhibitors and Covid-19: Divide and Conquer. Scientific African. 2022;22: e01407.
Holbrook JA, Jarosz-Griffiths HH, Caseley E, Lara-Reyna S, Poulter JA, Williams-Gray CH, Peckham D, McDermott MF. Neurodegenerative disease and the NLRP3 inflammasome. Front Pharmacol. 2021;12: 643254.
Leibowitz SM, Yan J. NF-κB Pathways in the Pathogenesis of Multiple Sclerosis and the Therapeutic Implications. Front Mol Neurosci. 2016;9:8.
Chen D, Ireland SJ, Remington G, Alvarez E, Racke MK, Greenberg B, Frohman EM, Monson NL. CD40-mediated NF-κB activation in B cells is increased in multiple sclerosis and modulated by therapeutics. J Immunol. 2016;197(11):4257–65.
Olcum M, Tastan B, Kiser C, Genc S, Genc K. Microglial NLRP3 inflammasome activation in multiple sclerosis. Adv Protein Chem Struct Biol. 2020;119:247–308.
Shao S, Chen C, Shi G, Zhou Y, Wei Y, Fan N, Yang Y, Wu L, Zhang T. Therapeutic potential of the target on NLRP3 inflammasome in multiple sclerosis. Pharmacol Ther. 2021;227: 107880.
Tien CP, Chang YC, Hung SC, Hsiao M. Abstract B20: Repurposing of fenofibrate to prevent and treat PM-induced pulmonary fibroblast-mediated inflammation: Mechanism involved in SIRT1-SREBP1-PIR/NLRP3 inflammasome axis. Cancer Immunology Research. 2020;8(3_Supplement):B20-.
Garcia-Ramírez M, Hernández C, Palomer X, Vázquez-Carrera M, Simó R. Fenofibrate prevents the disruption of the outer blood retinal barrier through downregulation of NF-κB activity. Acta Diabetol. 2016;53:109–18.
Liu Q, Zhang F, Zhang X, Cheng R, Ma JX, Yi J, Li J. Fenofibrate ameliorates diabetic retinopathy by modulating Nrf2 signaling and NLRP3 inflammasome activation. Mol Cell Biochem. 2018;445:105–15.
Batiha GE, Al-Kuraishy HM, Al-Gareeb AI, Elekhnawy E. SIRT1 pathway in Parkinson’s disease: a faraway snapshot but so close. Inflammopharmacology. 2022;29:1–20.
Alherz FA, Negm WA, Elekhnawy E, El-Masry TA, Haggag EM, Alqahtani MJ, Hussein IA. Silver nanoparticles prepared using encephalartos laurentianus de wild leaf extract have inhibitory activity against candida albicans clinical isolates. Journal of Fungi. 2022;8(10):1005.
Naegele M, Martin R. The good and the bad of neuroinflammation in multiple sclerosis. Handb Clin Neurol. 2014;122:59–87.
Al-kuraishy HM, Al-Gareeb AI, Elekhnawy E, Batiha GE. Dipyridamole and adenosinergic pathway in Covid-19: a juice or holy grail. Egyptian Journal of Medical Human Genetics. 2022;23(1):140.
Gatta V, Mengod G, Reale M, Tata AM. Possible correlation between cholinergic system alterations and neuro/inflammation in multiple sclerosis. Biomedicines. 2020;8(6):153.
Chen XR, Besson VC, Palmier B, Garcia Y, Plotkine M, Marchand-Leroux C. Neurological recovery-promoting, anti-inflammatory, and anti-oxidative effects afforded by fenofibrate, a PPAR alpha agonist, in traumatic brain injury. J Neurotrauma. 2007;24(7):1119–31.
Esmaeili MA, Yadav S, Gupta RK, Waggoner GR, Deloach A, Calingasan NY, Beal MF, Kiaei M. Preferential PPAR-α activation reduces neuroinflammation, and blocks neurodegeneration in vivo. Hum Mol Genet. 2016;25(2):317–27.
Melis M, Scheggi S, Carta G, Madeddu C, Lecca S, Luchicchi A, Cadeddu F, Frau R, Fattore L, Fadda P, Ennas MG. PPARα regulates cholinergic-driven activity of midbrain dopamine neurons via a novel mechanism involving α7 nicotinic acetylcholine receptors. J Neurosci. 2013;33(14):6203–11.
Gran B, Nyirenda MH, Crooks J. The role of Toll-like receptors in multiple sclerosis and experimental autoimmune encephalomyelitis. Multiple Sclerosis Immunology: A Foundation for Current and Future Treatments. 2013:149–76.
Marta M, Meier UC, Lobell A. Regulation of autoimmune encephalomyelitis by toll-like receptors. Autoimmun Rev. 2009;8(6):506–9.
Dana N, Vaseghi G, Javanmard SH. Crosstalk between peroxisome proliferator-activated receptors and toll-like receptors: A systematic review. Advanced pharmaceutical bulletin. 2019;9(1):12.
Farmer BC, Walsh AE, Kluemper JC, Johnson LA. Lipid droplets in neurodegenerative disorders. Front Neurosci. 2020;14:742.
Shen Q, Otoki Y, Sobel RA, Nagra RM, Taha AY. Evidence of increased sequestration of pro-resolving lipid mediators within brain esterified lipid pools of multiple sclerosis patients. Multiple Sclerosis and Related Disorders. 2022;68: 104236.
Dasgupta S, Roy A, Jana M, Hartley DM, Pahan K. Gemfibrozil ameliorates relapsing-remitting experimental autoimmune encephalomyelitis independent of peroxisome proliferator-activated receptor-α. Mol Pharmacol. 2007;72(4):934–46.
Lovett-Racke AE, Hussain RZ, Northrop S, Choy J, Rocchini A, Matthes L, Chavis JA, Diab A, Drew PD, Racke MK. Peroxisome proliferator-activated receptor α agonists as therapy for autoimmune disease. J Immunol. 2004;172(9):5790–8.
Khan MS, Ghumman GM, Baqi A, Shah J, Aziz M, Mir T, Tahir A, Katragadda S, Singh H, Taleb M, Ali SS. Efficacy of pemafibrate versus fenofibrate administration on serum lipid levels in patients with dyslipidemia: Network meta-analysis and systematic review. Am J Cardiovasc Drugs. 2023;23(5):547–58.
Milo R, Korczyn AD, Manouchehri N, Stüve O. The temporal and causal relationship between inflammation and neurodegeneration in multiple sclerosis. Mult Scler J. 2020;26(8):876–86.
Bross M, Hackett M, Bernitsas E. Approved and emerging disease modifying therapies on neurodegeneration in multiple sclerosis. Int J Mol Sci. 2020;21(12):4312.
Celarain N, Tomas-Roig J. Aberrant DNA methylation profile exacerbates inflammation and neurodegeneration in multiple sclerosis patients. J Neuroinflammation. 2020;17(1):1–7.
Bogdanov P, Hernández C, Corraliza L, Carvalho AR, Simó R. Effect of fenofibrate on retinal neurodegeneration in an experimental model of type 2 diabetes. Acta Diabetol. 2015;52:113–22.
Barbiero JK, Ramos DC, Boschen S, Bassani T, Da Cunha C, Vital MA. Fenofibrate promotes neuroprotection in a model of rotenone-induced Parkinson’s disease. Behav Pharmacol. 2022;33(8):513–26.
Jin L, Hua H, Ji Y, Jia Z, Peng M, Huang S. Anti-inflammatory role of fenofibrate in treating diseases. Biomolecules and Biomedicine. 2023;23(3):376.
Reale M, Sanchez-Ramon S. Lipids at the cross-road of autoimmunity in multiple sclerosis. Curr Med Chem. 2017;24(2):176–92.
Rádiková Ž, Penesová A, Vlček M, Havranová A, Siváková M, Šiarnik P, Žitňanová I, Imrich R, Turčáni P, Kollár B. Lipoprotein profiling in early multiple sclerosis patients: effect of chronic inflammation? Lipids Health Dis. 2020;19(1):1.
Flores-Castillo C, Luna-Luna M, Carreón-Torres E, López-Olmos V, Frías S, Juárez-Oropeza MA, Franco M, Fragoso JM, Vargas-Alarcón G, Pérez-Méndez Ó. Atorvastatin and fenofibrate increase the content of unsaturated acyl chains in HDL and modify in vivo kinetics of HDL-cholesteryl esters in New Zealand White Rabbits. Int J Mol Sci. 2019;20(10):2521.
Cho EB, Cho HJ, Choi M, Seok JM, Shin HY, Kim BJ, Min JH. Low high-density lipoprotein cholesterol and high triglycerides lipid profile in neuromyelitis optica spectrum disorder: Associations with disease activity and disability. Multiple Sclerosis and Related Disorders. 2020;40: 101981.
Weinstock-Guttman B, Zivadinov R, Mahfooz N, Carl E, Drake A, Schneider J, Teter B, Hussein S, Mehta B, Weiskopf M, Durfee J. Serum lipid profiles are associated with disability and MRI outcomes in multiple sclerosis. J Neuroinflammation. 2011;8:1–7.
Alkhayyat SS, Al-Kuraishy HM, Al-Gareeb AI, El-Bouseary MM, AboKamer AM, Batiha GE, Simal-Gandara J. Fenofibrate for COVID-19 and related complications as an approach to improve treatment outcomes: the missed key for Holy Grail. Inflamm Res. 2022;71(10–11):1159–67.
Rasheed HA, Hussien NR, Al-Naimi MS, Al-Kuraishy HM, Al-Gareeb AI. Fenofibrate and Crataegus oxyacantha is an effectual combo for mixed dyslipidemia. Biomed Biotechnol Res J. 2020;4(3):259.
Wójtowicz S, Strosznajder AK, Jeżyna M, Strosznajder JB. The novel role of PPAR alpha in the brain: promising target in therapy of Alzheimer’s disease and other neurodegenerative disorders. Neurochem Res. 2020;45:972–88.
Batiha GE, Al-Gareeb AI, Elekhnawy E, Al-Kuraishy HM. Potential role of lipoxin in the management of COVID-19: a narrative review. Inflammopharmacology. 2022;30(6):1993–2001.
Al-Kuraishy HM, Jabir MS, Al-Gareeb AI, Albuhadily AK, Albukhaty S, Sulaiman GM, Batiha GE. Evaluation and targeting of amyloid precursor protein (APP)/amyloid beta (Aβ) axis in amyloidogenic and non-amyloidogenic pathways: A time outside the tunnel. Ageing Res Rev. 2023;4: 102119.
Al-Kuraishy HM, Al-Gareeb AI, Elekhnawy E, Batiha GE. Nitazoxanide and COVID-19: a review. Mol Biol Rep. 2022;49(11):11169–76.
Al-Kuraishy HM, Hamada MT, Al-Samerraie AY. Effects of metformin on omentin levels in a newly diagnosed type II diabetes mellitus: Randomized, placebo controlled study. Mustansiriya Med J. 2016;15:49–55.
Al-Buhadily AK, Al-Uqabi RU, Al-Gareeb AI. Evaluation of Protective Effect of Metformin in Rats with Experimental Osteoarthritis. Mustansiriya Medical Journal. 2023;22(1):50–3.
Al-Kuraishy HM, Jabir MS, Al-Gareeb AI, Albuhadily AK. The conceivable role of prolactin hormone in Parkinson disease: The same goal but with different ways. Ageing Res Rev. 2023;89: 102075.
Almukainzi M, El-Masry TA, Negm WA, Elekhnawy E, Saleh A, Sayed AE, Ahmed HM, Abdelkader DH. Co-delivery of gentiopicroside and thymoquinone using electrospun m-PEG/PVP nanofibers: In-vitro and In vivo studies for antibacterial wound dressing in diabetic rats. Int J Pharm. 2022;625: 122106.
Al-Kuraishy HM, Jabir MS, Albuhadily AK, Al-Gareeb AI, Rafeeq MF. The link between Alzheimer disease and metabolic syndrome: A mutual relationship and long rigorous investigation. Ageing Res Rev. 2023;5: 102084.
Attallah NG, Al-Fakhrany OM, Elekhnawy E, Hussein IA, Shaldam MA, Altwaijry N, Alqahtani MJ, Negm WA. Anti-biofilm and antibacterial activities of Cycas media R. Br secondary metabolites: In silico, in vitro, and in vivo approaches. Antibiotics. 2022;11(8):993.
Elekhnawy E, Negm WA. The potential application of probiotics for the prevention and treatment of COVID-19. Egyptian Journal of Medical Human Genetics. 2022;23(1):1–9.
Alrouji M, Al-Kuraishy HM, Al-Gareeb AI, Zaafar D, Batiha GE. Orexin pathway in Parkinson’s disease: a review. Mol Biol Rep. 2023;8:1–4.
Al-Kuraishy HM, Al-Gareeb AI, Alsayegh AA, Abusudah WF, Almohmadi NH, Eldahshan OA, Ahmed EA, Batiha GE. Insights on benzodiazepines’ potential in Alzheimer’s disease. Life Sci. 2023;8: 121532.
Elekhnawy EA, Sonbol FI, Elbanna TE, Abdelaziz AA. Evaluation of the impact of adaptation of Klebsiella pneumoniae clinical isolates to benzalkonium chloride on biofilm formation. Egyptian Journal of Medical Human Genetics. 2021;22(1):1–6.
Ali NH, Alhamdan NA, Al-Kuraishy HM, Al-Gareeb AI, Elekhnawy E, Batiha GE. Irisin/PGC-1α/FNDC5 pathway in Parkinson’s disease: truth under the throes. Naunyn Schmiedebergs Arch Pharmacol. 2023;11:1–1.
Elekhnawy E, Sonbol F, Abdelaziz A, Elbanna T. An investigation of the impact of triclosan adaptation on Proteus mirabilis clinical isolates from an Egyptian university hospital. Braz J Microbiol. 2021;52:927–37.
Alrouji M, Al-Kuraishy HM, Al-Buhadily AK, Al-Gareeb AI, Elekhnawy E, Batiha GE. DPP-4 inhibitors and type 2 diabetes mellitus in Parkinson’s disease: a mutual relationship. Pharmacol Rep. 2023;3:1–4.
El-Banna T, Abd El-Aziz A, Sonbol F, El-Ekhnawy E. Adaptation of Pseudomonas aeruginosa clinical isolates to benzalkonium chloride retards its growth and enhances biofilm production. Mol Biol Rep. 2019;46:3437–43.
Abdelaziz A, Sonbol F, Elbanna T, El-Ekhnawy E. Exposure to sublethal concentrations of benzalkonium chloride induces antimicrobial resistance and cellular changes in Klebsiellae pneumoniae clinical isolates. Microb Drug Resist. 2019;25(5):631–8.
Sonbol FI, El-Banna TE, Abd El-Aziz AA, El-Ekhnawy E. Impact of triclosan adaptation on membrane properties, efflux and antimicrobial resistance of Escherichia coli clinical isolates. J Appl Microbiol. 2019;126(3):730–9.
Hussien NR, Al-Naimi MS, Rasheed HA, Al-Kuraishy HM, Al-Gareeb AI. Sulfonylurea and neuroprotection: the bright side of the moon. Journal of Advanced Pharmaceutical Technology & Research. 2018;9(4):120–3.
Alkuraishy HM, Al-Gareeb AI, Waheed HJ. Lipoprotein-associated phospholipase A2 is linked with poor cardio-metabolic profile in patients with ischemic stroke: A study of effects of statins. Journal of neurosciences in rural practice. 2018;9(04):496–503.
Al-Kuraishy HM, Al-Gareeb AI. Effect of orlistat alone or in combination with Garcinia cambogia on visceral adiposity index in obese patients. Journal of intercultural ethnopharmacology. 2016;5(4):408.
Batiha GE, Al-Kuraishy HM, Al-Gareeb AI, Welson NN. Pathophysiology of Post-COVID syndromes: a new perspective. Virology Journal. 2022;19(1):158.
Negm WA, El-Aasr M, Kamer AA, Elekhnawy E. Investigation of the antibacterial activity and efflux pump inhibitory effect of Cycas thouarsii R. Br. extract against Klebsiella pneumoniae clinical isolates. Pharmaceuticals. 2021;14(8):756.
Alotaibi B, El-Masry TA, Elekhnawy E, El-Kadem AH, Saleh A, Negm WA, Abdelkader DH. Aqueous core epigallocatechin gallate PLGA nanocapsules: Characterization, antibacterial activity against uropathogens, and in vivo reno-protective effect in cisplatin induced nephrotoxicity. Drug Delivery. 2022;29(1):1848–62.
Al-Kuraishy HM, Al-Gareeb AI, Qusti S, Alshammari EM, Atanu FO, Batiha GE. Arginine vasopressin and pathophysiology of COVID-19: An innovative perspective. Biomed Pharmacother. 2021;143: 112193.
Rasheed HA, Al-Kuraishy HM, Al-Gareeb AI, Hussien NR, Al-Nami MS. Effects of diabetic pharmacotherapy on prolactin hormone in patients with type 2 diabetes mellitus: Bane or Boon. Journal of advanced pharmaceutical technology & research. 2019;10(4):163.
Acknowledgements
Not applicable.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the University of Witten-Herdecke Germany.
Author information
Authors and Affiliations
Contributions
Primary investigation, writing—original draft, conceptualization, writing and editing: HMA, AIA-G, EE, and GE-SB. writing the final manuscript review and editing was performed by AAA, AA, AA, and MP.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Abulaban, A.A., Al-kuraishy, H.M., Al-Gareeb, A.I. et al. Role of fenofibrate in multiple sclerosis. Eur J Med Res 29, 113 (2024). https://doi.org/10.1186/s40001-024-01700-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40001-024-01700-2