
Abstract
Imbalance between amyloid-beta (Aβ) peptide synthesis and clearance results in Aβ deregulation. Failure to clear these peptides appears to cause the development of Alzheimer’s disease (AD). In recent years, microRNAs have become established key regulators of biological processes that relate among others to the development and progression of neurodegenerative diseases, such as AD. This review article gives an overview on microRNAs that are involved in the Aβ cascade and discusses their inhibitory impact on their target mRNAs whose products participate in Aβ clearance. Understanding of the mechanism of microRNA in the associated signal pathways could identify novel therapeutic targets for the treatment of AD.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD)—the most common form of dementia—is a devastating diagnosis that accounts for 93,541 deaths in the United States in 2014 [1]. Clinical manifestation of AD is often a loss of memory and cognitive skills. AD comprises two types: early-onset AD (EOAD), the familial type of AD which is inherited in an autosomal dominant pattern, and sporadic late-onset AD (LOAD), the most prevalent form of AD which develops at a later age [2]. The main pathological characteristics in the brains of AD patients are extracellular senile plaques composed of Aβ peptides [3] and intracellular neurofibrillary tangles (NFTs) formed by the accumulation of hyperphosphorylated tau [4].
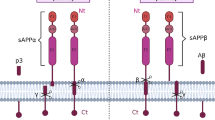
Aβ is cleaved from the amyloid precursor protein (APP) by β-secretase (BACE1) and γ-secretase in the amyloidogenic pathway [5], while in the non-pathological stage, APP is cleaved to non-toxic proteins by α-secretase [6]. Aβ has two major forms: Aβ40 and Aβ42, which are 40 and 42 amino acid-long fragments, respectively. Since Aβ42 is more hydrophobic than Aβ40, it is more prone to aggregate and scaffold for oligomeric and fibrillar forms [7]. The microtubule-associated protein tau regulates the assembly of microtubules and maintains its structural stability. Thus, it plays an important role in microtubule dynamics. In AD, however, tau becomes abnormally hyperphosphorylated leading to its dissociation from microtubules. Then, the unbound tau molecules aggregate as insoluble filaments, which accumulate and form neurofibrillary tangles (NFT) [8]. The accumulation of Aβ and NFTs in brain can trigger a cascade of events that may lead to AD.
According to the Aβ hypothesis, Aβ accumulation arises from a failure of clearance rather than over-production [9]. Indeed, Bateman et al. [10] demonstrated that the clearance rate of Aβ is impaired by approximately 30% in the cerebrospinal fluid of patients with LOAD. Mawuenyega et al. [11] found that the clearance rate of Aβ40 and Aβ42 is reduced by 25% and 30%, respectively in AD patients. The study by Cirrito et al. [12] showed the effect of age on the clearance rate of Aβ and found that the half-life of Aβ doubled within the interstitial fluid of older animal models of AD. These studies definitely established that defects in Aβ clearance have a fundamental role in AD pathology. Mechanisms that are involved in Aβ clearance include the ubiquitin–proteasome system (UPS), autophagic processes, proteolytic enzymes, transportation across the blood brain barrier (BBB), cellular uptake and heat shock protein (HSP)-mediated clearance, as illustrated in Fig. 1. The relative contributions of each of these procedures resulting in the overall clearance of Aβ are unknown.

Balanced Aβ clearance pathways. UPS ubiquitin–proteasome system, AβDPs Aβ degrading proteases, BBB blood brain barrier, HSP heat shock proteins
MicroRNAs (miRNAs) have emerged as essential post-transcriptional regulators of gene expression. These small, non-coding RNAs regulate mRNA stability and transcription by binding to the 3′-UTR region of their targets [13]. The dysregulation of miRNAs leads to an altered protein expression which in turn results in a pathogenic signaling network connected with the imbalance between Aβ peptide synthesis and clearance causing AD. The involvement of miRNAs in these pathways may provide information about the molecular mechanism of AD. To survey and overcome the imbalance between synthesis and clearing, the research field on miRNAs may be promising, and is eligible for establishing a continuous monitoring of disease progression and therapeutic interventions, not only for AD but also for other diseases.
To date, miRNAs described above document their usefulness as diagnostic and predictive markers for AD. For the assessment of miRNAs, real-time PCR, microarrays or even sequencing could be applied in tissues and body fluids, such as plasma or serum. The development of miRNA-based therapies anticipates restoring normal miRNA expression levels. In clinical settings, the levels of down-regulated tumor suppressor miRNAs could be normalized by their re-expression using synthetic or viral vectors encoded for miRNA or synthetic double strand RNA molecules (mimics), whereas the up-regulated oncogenic miRNAs could be silenced by antisense-mediated inhibition, miRNA sponges and anti-miRNA peptides. As delivery vehicles of miRNAs could serve polymer-based, lipid or viral vesicles or MSCs [14]. However, to reach their destination, miRNAs (mimics or antisense) have to cross the blood–brain barrier. To overcome this limitation, strategies, such as the use of conjugated nanoparticle or intracerebroventricular infusion have been shown to improve the transport through the blood–brain barrier [15]. Further challenges for an efficient miRNA-based gene therapy are the potential degradation of miRNAs by cellular nucleases and poor cellular uptake. In particular, miRNAs elicit unspecific effects, toxicity and/or unfavorable immune response, since they only partially bind to their target mRNA. In addition, they participate in several signaling pathways and consequently, have different regulatory functions which require further research. For example, with respect to the treatment of cancer, in September 2016, the sponsoring company (Mirna Therapeutic, Inc.) stopped the enrollment and dosing of miR-34 (MRX34) in a clinical study after numerous immune-related severe adverse effects in patients dosed with MRX34 [16]. Therefore, to realize their therapeutic application, it is essential to intensely investigate the biology and functions of miRNAs. As described above, numerous efforts have already made to identify miRNAs for introducing them into the clinical practice of AD. Most notably in animal models, these miRNAs appeared to be well tolerated with promising outcomes. For example, the intracerebroventricular infusion of anti-miR-33 inhibited the brain-specifically expressed miR-33 and in turn decreased Aβ levels in the cortex of mice [17].
On the other hand, a disruption of miRNA biogenesis is to avoid since it is assumed to cause neurodegeneration. For example, the onset of a neurodegenerative disease may happen by the loss of Dicer, an enzyme which cleaves pre-miRNA into a double-stranded miRNA duplex [18]. Such investigations show that miRNAs play an important role in long-term brain integrity and highlight their clinical relevance in AD. As up to 80% of all human genes are regulated by miRNAs [19] and their potential utility as AD biomarkers have been reported, we introduce potential miRNA-regulated targets in Aβ clearance pathways that will provide insights into the role of miRNAs in AD pathology.
Ubiquitin–proteasome system
The ubiquitin–proteasome system (UPS) is the main intracellular proteolytic pathway in eukaryotic cells. The pathway degrades more than 70–80% of intracellular proteins, including damaged and misfolded proteins [20]. At first, in the tagging reaction of the UPS-mediated protein degradation, a polyubiquitin chain is added to target proteins through three steps: (1) in an ATP-dependent process, an ubiquitin-activating enzyme (E1) activates an ubiquitin (Ub) monomer, a 76-amino acid peptide; (2) the activated Ub binds to an ubiquitin-conjugating enzyme (E2); and (3) ubiquitin ligase (E3) then transfers Ub to the target protein. In some cases, an additional ubiquitination enzyme, the chain elongation factor E4, is required to extend a polyubiquitin chain. Finally, the polyubiquitinated proteins are recognized and degraded in the 26S proteasome, a system that is composed of a 20S catalytic core and two 19S regulatory subunits [21].
After the detection of Ub in senile plaques in 1987 [22] and the observation that Aβ can bind to proteasomes [23], it was suggested that UPS is involved in the clearance of Aβ. Later studies substantiated this hypothesis. Lopez et al. [24] demonstrated that inhibition of the proteolytic activity of the 26S proteasome in neurons and astrocytes led to a reduction in Aβ degradation. Chadwick et al. [25] showed that a mutant form of Ub capped by polyubiquitin chains inhibited 26S proteasome and interfered with Aβ clearance. Furthermore, proteolytic activities of the 26S proteasome can also be inhibited by Aβ [26].
MiRNAs and their targets in UPS
Usually, in neocortex and hippocampal regions of AD brain tissues, the E2 family member UBE2A is down-regulated. In this regard, Zhao et al. [27] showed that the over-expression of miR-7 led to UBE2A down-regulation in the brain tissues of AD patients. In addition, the E2 isoforms UBE2B, UBE2D3 and UBCH10 that were down-regulated by miR-455-5p [28], miR-21-5p [29] and miR-631 [30] respectively, were identified as AD-related genes in a study conducted by Libro et al. [31]. Finally, the expression of UBC9 (UBE2I) was inversely correlated with miR-30a and miR-214 expression [32, 33] (Table 1).
There are several hundred E3 ligases in mammals, and this class shows the greatest diversity among the enzymes. E3 ligases are divided into two classes: E3 ligases with homology to the E6-AP carboxyl terminus (HECT), and the new RING ligases [34]. Singh et al. showed that the decreased levels of E3 ligase UBE3A caused by miR-375 over-expression [35], could influence the progression of AD [36]. Christie et al. showed that the levels of E3 ligase XIAP which were down-regulated by miR-497 and miR-7 [37, 38], were higher in AD patients than control cases [39]. Similarly, miR-24 over-expression decreased XIAP expression [40] (Table 1).
There are ~ 95 deubiquitinating enzymes (DUBs) in the human genome. DUBs are classified into five classes including: ubiquitin C-terminal hydrolase (UCH), ubiquitin-specific protease (USP), Machado-Joseph disease protease (MJD), otubain protease (OTU) and JAB1/MPN/Mov34 metalloenzyme (JAMM) [41]. Ubiquitin C-terminal hydrolase L1 (UCHL1) appears to be the only DUB playing a role in AD. It constitutes 1–5% of total neuronal protein, and stabilizes monoubiquitin by binding to it [42]. MiR-922 and miR-181b decreased UCHL1 expression in kidney and neuroblastoma cells, respectively [43, 44] (Table 1; Fig. 2).

The inhibitory effect of miRNAs on their target molecules in the UPS pathway. Ubiquitin is transferred to the E2 enzyme after activation by the E1 enzyme, and is then transferred to the substrate by E3 enzyme. E4 enzyme is required for the formation of the polyubiquitin chain. After the recognition process, substrates are degraded by the 26S proteasome or their polyubiquitin monomers are removed by DUB. Ub ubiquitin, E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme, E3 ubiquitin ligase, DUB deubiquitinating enzyme, UBE2A ubiquitin conjugating enzyme E2 A, UBE2B ubiquitin conjugating enzyme E2 B, UBE2C ubiquitin conjugating enzyme E2 C, UBE2I ubiquitin conjugating enzyme E2 I, UBE3A E3 ubiquitin-protein ligase A, XIAP E3 ubiquitin-protein ligase XIAP, CBX4 E3 SUMO-protein ligase CBX4, ITCH E3 ubiquitin-protein ligase Itchy, SMURF1 E3 ubiquitin-protein ligase SMURF1, UCHL1 ubiquitin carboxyl-terminal hydrolase isozyme L1, BAP1 ubiquitin carboxyl-terminal hydrolase BAP1, USP2 ubiquitin specific peptidase 2, USP13 ubiquitin specific peptidase 13; CSN5 COP9 signalosome complex subunit 5
Autophagy
Autophagy is a highly conserved catabolic process which has a key role in maintaining cell hemostasis through recycling nutrients and degrading aggregated proteins or damaged organelles [91]. Autophagy has distinct stages: formation of an isolation membrane (phagophore) and initiation of autophagy, vesicle nucleation, elongation and expansion of the autophagosome membrane, sequestration of aggregated proteins and cytoplasmic organelles into an autophagosome, and finally fusion of autophagosomes with endosomes or lysosomes for content degradation.
The first step in the autophagy process is the fusion of vesicles that originate from different membrane sources, such as the plasma membrane, endoplasmic reticulum (ER), Golgi apparatus and mitochondria [92]. Integration of these vesicles leads to the formation of an isolation membrane, called the phagophore. Autophagy initiation begins with the activation of a complex comprised of ULK1, ULK2, ATG13, ATG101 and the family interacting protein of 200 kD (FIP200) [93]. The mechanistic target of the rapamycin complex 1 (mTORC1) which is comprised of mTOR, RAPTOR, mLST8, and DEPTOR inhibits autophagy by phosphorylating ULK1 and ATG13 [94], while the adenosine monophosphate activated protein kinase (AMPK) activates autophagy by phosphorylating ULK1 at other sites [94].
The ULK1 complex controls vesicle nucleation through the class III phosphatidylinositol 3-kinase (PI3 K) complex. This complex is comprised of vacuolar protein sorting 34 (VPS34), VPS15, ATG14, and ultraviolet irradiation resistance-associated gene (UVRAG), all of which are scaffolded by Beclin 1 [95]. There are two ubiquitin-like conjugation steps that are involved in autophagosome elongation: (1) formation of a complex between ATG5, ATG12 and ATG16L1 that requires the catalytic activities of ATG7 (E1-like enzyme) and ATG10 (E2-like enzyme), (2) processing of microtubule-associated protein 1 light chain 3 (LC3). Initially, LC3 is cleaved by ATG4B, to form LC3-I which is then conjugated to phosphatidylethanolamine (PE) by ATG7 (E1-like enzyme) and ATG3 (E2-like enzyme), to form LC3-II [96]. After the formation of autophagosomes, the ATG5-ATG12-ATG16L1 complex separates from the outer membrane, while LC3-II remains attached with the completed autophagosomes, to facilitate their identification. Finally, double-membraned autophagosomes fuse with lysosomes for content degradation.
Growing evidence indicates that autophagy plays a role in AD pathology. For example it has been reported that autophagic vacuoles are abundant in AD brains [97] and that their clearance is impaired in AD [98]. Furthermore, restoring autophagy reduced Aβ accumulation in a TgCRND8 mouse model of AD and ameliorated memory deficits [99]. In their study, Wu et al. [100] validated miRNA-binding sequences for miR-20a and miR-106b in the 3′-UTR region of ULK1 and found that these two miRNAs negatively regulated autophagy through suppressing ULK1 expression in mouse myoblast cell lines. Korkmaz et al. [101] found that miR-376b attenuated the luciferase activity of the BECN1 3′-UTR, and thus, decreased mRNA levels of BECN1 in human breast and hepatocellular carcinoma cell lines leading to autophagy inhibition. A number of miRNAs that regulate the autophagy cascade are summarized in Table 2, Fig. 3.

MiRNAs inhibit autophagy by down-regulating their target molecules. AMPK and mTORC1 are key modulators of autophagy and exert their effects by regulating ULK1 and ATG13. Activation of the ULK1 complex initiates autophagy and regulates vesicle nucleation through the class III phosphatidylinositol 3-kinase (PI3K) complex. The final step in the autophagosome formation requires the two ubiquitin-like conjugation systems. AMPK adenosine monophosphate activated protein kinase, mTOR mechanistic target of rapamycin, RAPTOR regulatory-associated protein of Mtor, mLST8 MTOR associated protein, LST8 homolog, DEPTOR DEP domain containing MTOR interacting protein, ULK1 unc-51 like autophagy activating kinase 1, ULK2 unc-51 like autophagy activating kinase 2, ATG autophagy-related gene, FIP200 family interacting protein of 200 kD, UVRAG UV radiation resistance-associated gene protein, LC3 microtubule-associated protein 1 light chain 3, PE phosphatidylethanolamine
Degrading enzymes
Aβ is degraded by various types of proteases collectively known as Aβ-degrading proteases (AβDPs), e.g., by neprilysin, myelin basic protein, matrix metallopeptidase, angiotensin converting enzyme and cathepsins.
Neprilysin
Neprilysin (NEP) is a zinc-dependent membrane metalloendopeptidase (MME) belonging to the M13 family of metallopeptidases. After the introduction of Neprilysin as one of the major AβDPs [132], Iwata et al. [133] showed that in Neprilysin knockout-mice the vulnerability of the hippocampus was caused by Aβ accumulation. In this regard, neprilysin was shown to degrade both monomeric and oligomeric forms of Aβ [134]. Moreover, a meta-analysis documented that mRNA and protein levels of Neprilysin, as well as the enzymatic activity of neprilysin are decreased in AD patients [135].
Myelin basic protein
Myelin basic protein (MBP), an 18.5 kD protein is the main protein component of myelin, and participates in the formation and maintenance of the myelin sheath. MBP has serine protease activity and degrades Aβ40 and Aβ42 peptides [136]. Hoos et al. [137] found that MBP inhibited fibrillar assembly of Aβ, and Liao et al. [138] demonstrated that this was mediated by the N-terminal domain of MBP. Furthermore, Wang et al. [139] showed that miR-212 reduced the expression of MBP, and thus, promoted the assembly.
Matrix metallopeptidase
Matrix metalloproteinases (MMPs) that belong to the metzincin family have at least two domains: the pro-domain which is ~ 80 amino acids long and the catalytic-domain which contains a zinc ion in the active site. They degrade both soluble and fibrillar Aβ peptides [140]. Zhang et al. [141] reported that miR-9 directly targeted the MMP-14 3′-UTR and decreased transcriptional and consequently, protein levels of MMP-14 in neuroblastoma cells reducing adhesion, migration, invasion and angiogenesis of these cells. Multiple MMPs are implicated in Aβ degradation and their repression by miRNAs is shown in Table 3.
Angiotensin converting enzyme
Angiotensin-converting enzyme (ACE) is a zinc-dependent dipeptidase that catalyzes the conversion of angiotensin I to angiotensin II. Hu et al. [155] found that ACE degraded Aβ40 by cleaving the peptide bond between Asp7 and Ser8 residues, and found that ACE prevented the accumulation of amyloid plaques by degrading Aβ in vivo. Following studies indicated that the N-terminal domain of ACE was responsible for Aβ degradation [156] and pharmacological inhibition of ACE enhanced the accumulation of Aβ in APP expressing cells [157]. Several miRNAs are implicated in inhibiting ACE expression, as listed in Table 3.
Cathepsins
Cathepsin B, a major representative of cysteine proteases, acts as either an exopeptidase or an endopeptidase. It is present in lysosomes from all cell types, and participates in lysosomal turnover of proteins. Sun et al. [158] indicated that Cathepsin B was able to induce Aβ degradation in vivo. Moreover, lysosomal Cathepsin B is essential in microglial clearance of Aβ [159] and its up-regulation promotes Aβ42 degradation in AD monocytes [160]. By using homology modeling, Dhanavade et al. [161] found that Cathepsin B cleaved Aβ peptide from the carboxylic end of Glu11. Cathepsin D, an aspartyl protease is present in lysosomes from most mammalian cells, and engages in the degradation of intracellular and endocytosed proteins. It cleaves Aβ peptide at Phe19-Phe20 and Leu34-Met35 [162], and is down-regulated in monocytes of AD patients [163]. Overexpression of miR-128 down-regulated the expression of Cathepsin B and Cathepsin D. Consequently, miR-128 inhibition enhanced Aβ42 degradation in monocytes from AD patients [164].
Blood–brain barrier clearance of Aβ
The blood–brain barrier (BBB) is a physical barrier that separates peripheral circulation from the central nervous system (CNS). The BBB, which is formed by endothelial cells connected by tight junctions, plays a significant role in controlling brain homeostasis by eliminating toxic metabolites from the brain into the blood, such as Aβ aggregates. It has two sides, the luminal side facing the blood circulation, and the abluminal side facing the brain parenchyma. Transporters and receptors which are expressed on the two sides are involved in the transportation and clearance of Aβ. Aβ efflux and influx through the BBB are regulated by several miRNAs, some of which are illustrated in Fig. 4 and listed in Table 4.

MiRNA-dependent regulation of Aβ clearance through the BBB. Aβ efflux and influx through the BBB. Aβ entrance to the brain is mediated by RAGE, but several transporters are expressed on both the luminal and abluminal sides of the BBB can eliminate Aβ from the brain. LRP1 is one of the major receptors for Aβ efflux through the BBB. Due to the sink hypothesis, it is also implicated in Aβ removal. Presence of Clusterin is essential for Aβ clearance by LRP2 receptor. ABCA1 indirectly facilitates Aβ clearance by lipidating ApoE. The drug pumps ABCB1 and ABCG2 are also involved in Aβ transport across the BBB. RAGE receptor for advanced glycation end products, LRP1 LDL receptor related protein 1, LRP2 LDL receptor related protein 2, ABCA1 ATP binding cassette subfamily A member 1, ABCB1 multidrug resistance protein, P-glycoprotein, ABCG2 breast cancer resistance protein, ABCG4 ATP binding cassette subfamily G member 4
Receptor-mediated Aβ influx
Receptor for advanced glycation end products
The receptor for advanced glycation end products (RAGE) belongs to the immunoglobulin family, and is expressed on the luminal surface of brain vessels. RAGE is a multi-ligand receptor that binds a range of ligands, including Aβ [165]. By using an in vitro BBB model, Mackic et al. [166] showed that RAGE is involved in the internalization of soluble monomeric forms of Aβ40. Candela et al. [167] reported that RAGE inhibitors mediated a significant decrease in Aβ40 and Aβ42 transport through the brain endothelium. Similarly, Takuma et al. [168] found that a genetic deletion of RAGE suppressed Aβ uptake in neurons. Mice studies confirmed these findings and showed the influx of circulating Aβ into the brain as a receptor-mediated transport depending on RAGE [169]. Furthermore, the inhibition of the RAGE/Aβ interaction repressed Aβ accumulation in the brain of PD-hAPP mice [170]. Also in line with these data, Ma et al. showed that RAGE up-regulation contributed to the accumulation of Aβ and cognitive impairment in rats [171]. Finally, Choi et al. [172] reported the elevated RAGE expression in a mouse model of AD.
Receptor-mediated Aβ efflux
Low-density lipoprotein receptor (LDLR) family
The LDLR family are cell surface receptors and includes LDLR, VLDLR, LRP1, LRP1B, LRP2 (megalin), LRP3, LRP4, LRP5, LRP6 and LRP8. The main function of this receptor family is receptor-mediated endocytosis. In APP/PS1/LDLR transgenic mice, LDLR over-expression was reported to promote Aβ clearance [173].
Initial studies identified LRP1 as an abluminal receptor that mediated Aβ transport across the BBB [174], and subsequent studies proved a role for LRP1 in brain-to-blood Aβ clearance [175]. In a mouse model of AD, LRP1 deletion resulted in decreased Aβ levels in plasma and enhanced soluble Aβ in brain endothelial cells [176]. Moreover, LRP1 oligodeoxynucleotide antisense impaired recognition memory in mice by reducing BBB clearance of Aβ [177]. Several studies proved that ApoE had suppressive effects on LRP1-mediated BBB clearance of Aβ as preincubation with ApoE reduced Aβ40 clearance [178]. Moreover, ApoE suppressed soluble Aβ (sAβ) clearance by competing with sAβ for interaction with LRP1 [179]. Further studies showed an isoform-specific effect for ApoE since ApoE4-Aβ complexes were not cleared by the rapid LRP1 receptor, and their clearance was mediated by VLDLR which has a significant slower rate of endocytosis compared to LRP1. However, both LRP1 and VLDLR are involved in the clearance of ApoE2- and ApoE3-Aβ complexes [180]. Wang et al. [181] found that miR-1908 reduced mRNA levels of ApoE by targeting its 3′-UTR, and thereby inhibited ApoE-mediated Aβ clearance in astrocytoma and human macrophage cell lines.
Based on the sink hypothesis, it is assumed that expression of LRP1 in peripheral tissues affects Aβ clearance through the BBB. According to this hypothesis, an equilibrium exists between the levels of Aβ in the brain and peripheral tissues. Thus, Aβ elimination by peripheral tissues causes brain Aβ to move into the blood through the BBB in order to maintain this balance [175, 182]. By expressing LRP1, the liver is able to clear plasma Aβ [183], therefore, LRP1 suppression in the liver reduced the Aβ uptake as reported by Tamaki et al. [184]. Clearance of plasma Aβ by the liver is saturable and age-related [184]. Investigations showed that soluble LRP1 which is produced from the cleavage of LRP1 by β-secretase [185], is the main peripheral Aβ-binding protein and reduced the load of Aβ in mice brain by acting as a peripheral sink [186].
LRP2 (megalin) is expressed on the abluminal side of the BBB, and also involved in the BBB clearance of Aβ [187]. Aβ does not directly bind to LRP2, and needs ApoJ for the interaction with LRP2 [188]. Only, ApoJ-bounded Aβ can be cleared from the brain by this receptor [189]. Interestingly, a recent study indicated that Clusterin administration reduced Aβ accumulation in a mouse model of AD by increasing LRP2 levels [190]. Zhang et al. [191] identified LRP2 mRNA 3′-UTR as a direct target of miR-146a and indicated that LRP2 protein levels were significantly inhibited by miR-146a in human neuroblastoma cell line. MiR-146a also elevated the rate of apoptosis in human neuroblastoma cells exposed to Aβ, and thus, may contribute to AD progression.
ATP-binding cassette transporters (ABC transporters)
The ABC transporter, one of the most common transmembrane proteins exists in all living organisms and is divided into subfamilies A to G based on its sequence homology and functional similarity. ABC transporters use the energy generated by ATP hydrolysis to transport substrates across cell-membranes, playing an important role in many physiological processes. Recent evidence showed that ABC transporters are involved in Aβ clearance, especially ABCA1, ABCB1 (multidrug resistance protein, MDR1 or P-glycoprotein), ABCG2 (breast cancer resistance protein, BCRP), and ABCG4.
ABCA1 is a transmembrane protein that is expressed on the abluminal side of the BBB. It transports cholesterol and phospholipids to ApoE in order to form high-density lipoproteins (HDL). Analyses showed that ABCA1 indirectly facilitated Aβ clearance through ApoE lipidation in the brain as no significant differentiation was seen in Aβ elimination between ABCA1-deficient and wild-type mice [192]. Mouse studies indicated that ABCA1 deficiency reduced ApoE levels and its lipidation state in the brain which were accompanied by Aβ accumulation [193, 194] and co-deposition of poorly lipidated ApoE with Aβ [195]. Thus, ABCA1-mediated ApoE lipidation reduced Aβ accumulation [196]. Similarly, Corona et al. [197] revealed that ABCA1-mediated ApoE lipidation is essential in Aβ clearance. The role of ABCA1 and ApoE in Aβ clearance is not fully elucidated as Aβ clearance was reduced in APP/ABCA1+/− mice expressing ApoE4 but not ApoE3 [198]. While ABCA1 expression was reduced in the brain of APP/PS1 mice [199], it was up-regulated in 3xTg-AD mice [200]. Further studies showed that ABCA1-mediated cholesterol efflux was reduced in the CSF of AD patients [201]. Nordestgaard et al. [202] found that a loss-of-function mutation in ABCA1 was associated with a higher risk of AD. In neuroblastoma and liver cells, miR-106b prevented Aβ clearance by suppressing ABCA1 expression [203], while inhibition of miR-33a increased lipidated ApoE levels, and reduced Aβ levels mediated by the re-expression of ABCA1 [17]. Liang et al. [204] found that miR-20a/b reduced mRNA and protein expression of ABCA1 in human and mouse macrophage-derived foam cells. MiR-20a/b over-expression decreased cholesterol efflux to ApoA-I, and thus, may interfere with Aβ clearance.
The ABCB1 transporter that is expressed on the luminal side of the BBB acts as an efflux pump of exogenous molecules, and is involved in Aβ clearance, as shown in ABCB1-knockout mice [205]. Other in vitro and in vivo studies also proved that P-glycoprotein had efflux activity since ABCB1 up-regulation enhanced the efflux of Aβ40 from cells [206] and led to a reduction in parenchymal Aβ40 and Aβ42 levels [207]. Moreover, previous studies showed that peripherally-injected Aβ accumulated in the brain of ABCB1-knockout mice [208], and ABCB1 deficiency increased Aβ burden in a mouse model of AD [209]. Consistent with these results, Aβ accumulation was inversely correlated with ABCB1 expression in AD patients [210]. Notably, Aβ42 down-regulated the expression of P-glycoprotein [211].
ABCG2 is also expressed at the luminal side of the BBB, and is also involved in Aβ efflux from brain to blood circulation [212] since Aβ levels were reported to be higher in the brain of ABCG2 knock-out mice than in the brain of wild type mice [208]. Shen et al. [213] also proved that ABCG2 had efflux activity, since ABCG2 deficiency led to Aβ accumulation in mice brain. Moreover, ABCG2 levels were age-dependently increased in a mice model of AD [200], and Xiong et al. [214] reported its up-regulation in AD brains.
The ABCG4 transporter participates in the cholesterol and desmosterol efflux. Do et al. [200] identified ABCG4 as a receptor that controls Aβ efflux through the BBB. Other in vivo studies proved its role in Aβ clearance by disclosing that ABCG4 contributes to Aβ40 elimination across the mouse BBB [212], and that Aβ efflux was decreased in ABCG4-knockout mice [215]. Finally, a mouse model showed that ABCG4 is expressed in the cerebral cortex and medulla regions of the brain [216], while a human study demonstrated that ABCG4 was up-regulated in the microglia-surrounded senile plaques in AD brains [217].
Glymphatic clearance
Aquaporin-4 (AQP4), a water-channel protein is expressed in astrocytes, and plays a key role in Aβ clearance by regulating the glymphatic pathway. AQP4 is involved in the clearance of soluble Aβ from the brain [218]. Yang et al. [219] revealed that AQP4 was up-regulated in areas of senile plaques, predominantly at later stages of plaque formation. In AQP4 knockout mice, glymphatic clearance of Aβ was reduced compared with wild-type mice [218].
Receptor-mediated Aβ phagocytosis
Phagocytosis is an evolutionarily conserved process, critical for innate immunity. It has been shown that impaired immune response in AD negatively affects Aβ elimination [239]. Similarly, macrophage-dependent phagocytosis of Aβ is impaired in AD [240]. In this section we introduce receptors that are expressed on the surface of phagocytic cells, and involved in Aβ phagocytosis. These surface receptors are regulated by several miRNAs, some of which are shown in Fig. 5 and Table 5.

Receptor-mediated Aβ phagocytosis. The immune microglial cells and astrocytes reduce the load of Aβ in the brain by phagocytosis mediated by surface receptors. Aβ is directly phagocytosed by toll-like receptors and scavenger receptors, while the presence of LDL is crucial for TREM2-dependent phagocytosis. TLR2 toll like receptor 2, TLR4 toll like receptor 4, TLR9 toll like receptor 9, TREM2 triggering receptor expressed on myeloid cells 2, LDL low density lipoprotein, SR-A scavenger receptor class A, SR-B1 scavenger receptor class B type 1
Toll-like receptors
Toll-like receptors (TLRs) are a family of pattern recognition receptors (PRRs), and involved in innate immune recognition. There are at least ten TLRs in mammals, and though they have a high degree of structural similarity, their functions are distinct. TLRs are involved in the clearance of diffuse and fibrillar forms of Aβ through microglial activation [241]. Song et al. [242] showed that TLR2 deletion increased Aβ levels in the brain of APP transgenic mice which was accompanied with memory deficits. Consistent with these results, TLR4 mutation caused Aβ deposition and cognitive deficits in a mouse model of AD [243]. Frank et al. [244] detected increased mRNA levels of TLR2, TLR4, and TLR9 in a transgenic mouse model. Zhang et al. [245] found that miR-181c suppressed the activity of the luciferase reporter plasmid containing TLR4 3′-UTR by reducing TLR4 mRNA and protein expression in microglial cells. Consequently, miR-181c inhibited the downstream production of proinflammatory mediators. Table 5 listed the miRNAs that inhibit the expression of TLR2 and TLR4.
Triggering receptor expressed on myeloid cells 2
Triggering receptor expressed on myeloid cells 2 (TREM2) is expressed on microglial cells and belongs to the immunoglobulin superfamily. This surface receptor has several ligands, including low density lipoproteins (LDL), ApoJ and ApoE. Yeh et al. [246] showed that microglial cells are capable of uptaking LDL-Aβ complexes in a TREM2-dependent manner. In a mouse model of AD, TREM2 enhanced Aβ42 phagocytosis in the primary microglia [247]. Thus Aβ levels were higher in TREM2-deficient mice [248]. Kober et al. [249] found that the ligand affinity of LDL-Aβ complex was reduced in the R47H and R62H variants of TREM2, leading to phagocytosis impairment and Aβ accumulation [246]. Jay et al. [250] detected that TREM2 was up-regulated on microglial cells that were clustered around Aβ deposits in a mouse model of AD and human AD tissues. Alexandrov et al. [251] showed that miR-34a down-regulated TREM2 expression leading to Aβ accumulation by impairing phagocytosis.
Scavenger receptors
Scavenger receptors (SRs) are cell surface receptors that participate in the uptake of various polyanionic ligands. Based on their protein sequence, SRs are classified into 10 families (A-J). It has been shown that scavenger receptor class A (SR-A) and class B type 1 (SR-B1), as well as CD36 participate in Aβ clearance [252,253,254]. SR-A which is expressed on microglial cells and macrophages is implicated in Aβ phagocytosis [255]. Therefore SR-A deficiency reduced phagocytic activity of microglia and macrophages [256, 257], accelerated Aβ accumulation and consequently led to increased mortality in a mouse model of AD [258]. SR-B1 is expressed on microglial cells and astrocytes, mediates the binding of Aβ to microglia [259] and is implicated in the astrocyte-mediated clearance of Aβ [260]. In vivo studies indicated that SR-B1 deficiency promoted Aβ deposition [261]. CD36 which is found in a variety of cell types mediates macrophage and microglial response to Aβ [262]. In vitro studies demonstrated that CD36 deficiency decreased Aβ phagocytosis [263], while PPARγ-induced CD36 up-regulation enhanced Aβ phagocytosis in microglia [264]. Kouadir et al. [265] reported the increases in SR-B1 and CD36 expression by Aβ42, while Giunta et al. [266] reported the downregulation of CD36 in AD patients. Li et al. [267] showed that miR-758-5p significantly reduced mRNA and protein levels of CD36, and therefore attenuated cellular uptake of cholesterol.
Heat shock proteins
Heat shock proteins (HSPs), a group of molecular chaperones repress molecular denaturation under stressful conditions. HSPs also prevent protein aggregation by binding to newly synthesized or misfolded proteins, thereby helping maintain protein homeostasis. According to their size and function, HSPs can be divided into two different families: classic HSPs with a molecular weight of 60 kD or more that possess an ATP-binding site, e.g., HSP90 and HSP70, and small HSPs with a molecular weight of 40 kD or less that are ATP-independent, e.g., HSP27. Initial studies showed that HSPs regulated microglial interactions with Aβ, substantiating the role of HSP90 and HSP70 in phagocytosis-dependent Aβ clearance [276]. Subsequent in vivo studies showed similar data, and demonstrated that microglial clearance of Aβ was facilitated by HSP90 in a rat brain [277], and that HSP70 over-expression decreased Aβ levels in a mouse model of AD [278]. Furthermore, Evans et al. [279] demonstrated that HSP90 and HSP70 could induce structural changes in Aβ oligomers that suppressed self-assembly. Similarly, Rivera et al. [280] found that HSP70 prevented Aβ oligomerization and consequently reduced Aβ-induced toxicity in cultured neurons. HSP27 was also able to bind Aβ40, reducing its formation into mature fibrils [281]. Therefore, HSP27 protects neurons against Aβ [282]. On the other hand, Aβ could enhance the expression of HSP27 and HSP70 in neuronal cultures [283, 284]. Table 6 specified the miRNAs that inhibit HSPs expression.
Conclusion
Emerging evidences indicate that impaired Aβ clearance plays a crucial role in both EOAD and LOAD. Thus, understanding how Aβ is cleared from the brain might be of clinical relevance. Aβ removal from the brain occurs via various pathways: UPS, autophagy, proteolytic enzymes, transportation across the BBB and cellular uptake. Any disturbance of these pathways may lead to Aβ accumulation, resulting in the pathological process driving AD. Our present review shows that numerous miRNAs inhibit the translation of key molecules in these pathways, promoting the Aβ accumulation. This ability of miRNAs to target multiple mRNAs in the network of Aβ clearance make them to valuable therapeutic target molecules in AD. In particular, those miRNAs should be selected as target molecules that are involved in several pathways. As shown above, miR-34a and miR-29b may be attractive candidates for AD treatment because they inhibit at least three pathways leading to Aβ clearance. In the adult mammalian brain, miR-34a is highly expressed, and has been implicated in a range of neurodevelopmental and neuropathological processes. MiR-34a was reported to regulate neural stem/progenitor cell differentiation. High levels of this miRNA have been detected during epileptic seizures and ischemic stroke contributing to neuronal injury and death [290]. MiR-29b has been identified as a putative regulator of immunity. Moreover, ectopic expression of miR-29b promoted neuronal cell death, whereas its repression decreased cell death [291]. In summary, the research field on miRNAs is promising for therapeutic applications, not only for the treatment of AD but also for regenerative medicine. However, several obstacles prevent their utility in the clinic, of which the accurate determination of their expression levels might be a critical point [292]. Indeed, due to the lack of consensus on the reference controls, the appropriate normalization approach should be validated in each experimental study [293, 294].
Availability of data and materials
Not applicable.
References
Association As. 2017 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2017;13(4):325–73.
Reitz C, Mayeux R. Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem Pharmacol. 2014;88(4):640–51.
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and down syndrome. Proc Natl Acad Sci. 1985;82(12):4245–9.
Grundke-Iqbal I, Iqbal K, Tung Y-C, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci. 1986;83(13):4913–7.
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, et al. β-Secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–41.
Kojro E, Fahrenholz F. The non-amyloidogenic pathway: structure and function of α-secretases. Alzheimer’s disease. Boston: Springer; 2005. p. 105–27.
Jarrett JT, Berger EP, Lansbury PT Jr. The carboxy terminus of the.beta. amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 1993;32(18):4693–7.
Medeiros R, Baglietto-Vargas D, LaFerla FM. The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci Ther. 2011;17(5):514–24.
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6.
Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. Quantifying CNS protein production and clearance rates in humans using in vivo stable isotope labeling, immunoprecipitation, and tandem mass spectrometry. Nat Med. 2006;12(7):856.
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science. 2010;330(6012):1774.
Cirrito JR, May PC, O’Dell MA, Taylor JW, Parsadanian M, Cramer JW, et al. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-β metabolism and half-life. J Neurosci. 2003;23(26):8844–53.
Hammond SM. An overview of microRNAs. Adv Drug Deliv Rev. 2015;87:3–14.
Smith B, Agarwal P, Bhowmick NA. MicroRNA applications for prostate, ovarian and breast cancer in the era of precision medicine. Endocr Relat Cancer. 2017;24(5):R157.
Petrescu GE, Sabo AA, Torsin LI, Calin GA, Dragomir MP. MicroRNA based theranostics for brain cancer: basic principles. J Exp Clin Cancer Res. 2019;38(1):231.
Beg MS, Brenner AJ, Sachdev J, Borad M, Kang Y-K, Stoudemire J, et al. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest New Drugs. 2017;35(2):180–8.
Kim J, Yoon H, Horie T, Burchett JM, Restivo JL, Rotllan N, et al. microRNA-33 regulates ApoE lipidation and amyloid-β metabolism in the brain. J Neurosci. 2015;35(44):14717–26.
Abe M, Bonini NM. MicroRNAs and neurodegeneration: role and impact. Trends Cell Biol. 2013;23(1):30–6.
Lu J, Clark AG. Impact of microRNA regulation on variation in human gene expression. Genome Res. 2012;22(7):1243–54.
Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. J Am Soc Nephrol. 2006;17(7):1807–19.
Bedford L, Paine S, Sheppard PW, Mayer RJ, Roelofs J. Assembly, structure, and function of the 26S proteasome. Trends Cell Biol. 2010;20(7):391–401.
Perry G, Friedman R, Shaw G, Chau V. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc Natl Acad Sci. 1987;84(9):3033–6.
Gregori L, Hainfeld JF, Simon MN, Goldgaber D. Binding of amyloid β protein to the 20 S proteasome. J Biol Chem. 1997;272(1):58–62.
Salon ML, Pasquini L, Moreno MB, Pasquini J, Soto E. Relationship between β-amyloid degradation and the 26S proteasome in neural cells. Exp Neurol. 2003;180(2):131–43.
Chadwick L, Gentle L, Strachan J, Layfield R. Unchained maladie–a reassessment of the role of Ubb+1-capped polyubiquitin chains in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2012;38(2):118–31.
Almeida CG, Takahashi RH, Gouras GK. β-Amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci. 2006;26(16):4277–88.
Zhao Y, Alexandrov PN, Jaber V, Lukiw WJ. Deficiency in the ubiquitin conjugating enzyme UBE2A in Alzheimer’s disease (AD) is linked to deficits in a natural circular miRNA-7 sponge (circRNA; ciRS-7). Genes. 2016;7(12):116.
Cheng CM, Shiah SG, Huang CC, Hsiao JR, Chang JY. Up-regulation of miR-455-5p by the TGF-β-SMAD signalling axis promotes the proliferation of oral squamous cancer cells by targeting UBE2B. J Pathol. 2016;240(1):38–49.
Chang JT, Wang F, Chapin W, Huang RS. Identification of MicroRNAs as breast cancer prognosis markers through the cancer genome atlas. PLoS ONE. 2016;11(12):e0168284.
Xi H, Li L, Du J, An R, Fan R, Lu J, et al. hsa-miR-631 resensitizes bortezomib-resistant multiple myeloma cell lines by inhibiting UbcH10. Oncol Rep. 2017;37(2):961–8.
Libro R, Diomede F, Scionti D, Piattelli A, Grassi G, Pollastro F, et al. Cannabidiol modulates the expression of Alzheimer’s disease-related genes in mesenchymal stem cells. Int J Mol Sci. 2016;18(1):26.
Koh EH, Chen Y, Bader DA, Hamilton MP, He B, York B, et al. Mitochondrial activity in human white adipocytes is regulated by the ubiquitin carrier protein 9/microRNA-30a Axis. J Biol Chem. 2016;291(47):24747–55.
Wang S, Jiao B, Geng S, Ma S, Liang Z, Lu S. Combined aberrant expression of microRNA-214 and UBC9 is an independent unfavorable prognostic factor for patients with gliomas. Med Oncol. 2014;31(1):767.
Stewart MD, Ritterhoff T, Klevit RE, Brzovic PS. E2 enzymes: more than just middle men. Cell Res. 2016;26(4):423.
Song L, Liu S, Zeng S, Zhang L, Li X. miR-375 modulates radiosensitivity of HR-HPV-positive cervical cancer cells by targeting UBE3A through the p53 pathway. Med Sci Monit. 2015;21:2210.
Singh BK, Vatsa N, Kumar V, Shekhar S, Sharma A, Jana NR. Ube3a deficiency inhibits amyloid plaque formation in APPswe/PS1δE9 mouse model of Alzheimer’s disease. Hum Mol Genet. 2017;26(20):4042–54.
Zhang X, Liu H. MicroRNA-497 induces apoptosis through downregulating XIAP in hepatic cancer. Int J Clin Exp Med. 2017;10(9):13188–93.
Liu S, Zhang P, Chen Z, Liu M, Li X, Tang H. MicroRNA-7 downregulates XIAP expression to suppress cell growth and promote apoptosis in cervical cancer cells. FEBS Lett. 2013;587(14):2247–53.
Christie L-A, Su JH, Tu CH, Dick MC, Zhou J, Cotman CW. Differential regulation of inhibitors of apoptosis proteins in Alzheimer’s disease brains. Neurobiol Dis. 2007;26(1):165–73.
Xie Y, Tobin LA, Camps J, Wangsa D, Yang J, Rao M, et al. MicroRNA-24 regulates XIAP to reduce the apoptosis threshold in cancer cells. Oncogene. 2013;32(19):2442.
Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123(5):773–86.
Osaka H, Wang Y-L, Takada K, Takizawa S, Setsuie R, Li H, et al. Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum Mol Genet. 2003;12(16):1945–58.
Zhao Z-B, Wu L, Xiong R, Wang L-L, Zhang B, Wang C, et al. MicroRNA-922 promotes tau phosphorylation by downregulating ubiquitin carboxy-terminal hydrolase L1 (UCHL1) expression in the pathogenesis of Alzheimer’s disease. Neuroscience. 2014;275:232–7.
Peng Z, Li J, Li Y, Yang X, Feng S, Han S, et al. Downregulation of miR-181b in mouse brain following ischemic stroke induces neuroprotection against ischemic injury through targeting heat shock protein A5 and ubiquitin carboxyl-terminal hydrolase isozyme L1. J Neurosci Res. 2013;91(10):1349–62.
Haghikia A, Missol-Kolka E, Tsikas D, Venturini L, Brundiers S, Castoldi M, et al. Signal transducer and activator of transcription 3-mediated regulation of miR-199a-5p links cardiomyocyte and endothelial cell function in the heart: a key role for ubiquitin-conjugating enzymes. Eur Heart J. 2010;32(10):1287–97.
Manvati S, Mangalhara KC, Kalaiarasan P, Srivastava N, Kumar B, Bamezai R. MiR-101 induces senescence and prevents apoptosis in the background of DNA damage in MCF7 cells. PLoS ONE. 2014;9(10):e111177.
Devor EJ, Schickling BM, Reyes HD, Warrier A, Lindsay B, Goodheart MJ, et al. Cullin-5, a ubiquitin ligase scaffold protein, is significantly underexpressed in endometrial adenocarcinomas and is a target of miR-182. Oncol Rep. 2016;35(4):2461–5.
Gao F, Sun X, Wang L, Tang S, Yan C. Downregulation of microRNA-145 caused by hepatitis B virus X protein promotes expression of CUL5 and contributes to pathogenesis of hepatitis B virus-associated hepatocellular carcinoma. Cell Physiol Biochem. 2015;37(4):1547–59.
Xu X-M, Wang X-B, Chen M-M, Liu T, Li Y-X, Jia W-H, et al. MicroRNA-19a and-19b regulate cervical carcinoma cell proliferation and invasion by targeting CUL5. Cancer Lett. 2012;322(2):148–58.
Zheng C, Li J, Wang Q, Liu W, Zhou J, Liu R, et al. MicroRNA-195 functions as a tumor suppressor by inhibiting CBX4 in hepatocellular carcinoma. Oncol Rep. 2015;33(3):1115–22.
Sun T, Wang X, He H, Sweeney C, Liu S, Brown M, et al. MiR-221 promotes the development of androgen independence in prostate cancer cells via downregulation of HECTD2 and RAB1A. Oncogene. 2014;33(21):2790.
Wu X, Li L, Li Y, Liu Z. MiR-153 promotes breast cancer cell apoptosis by targeting HECTD3. Am J Cancer Res. 2016;6(7):1563.
Cheng DD, Yu T, Hu T, Yao M, Fan CY, Yang QC. MiR-542-5p is a negative prognostic factor and promotes osteosarcoma tumorigenesis by targeting HUWE1. Oncotarget. 2015;6(40):42761.
Luo ZL, Luo HJ, Fang C, Cheng L, Huang Z, Dai R, et al. Negative correlation of ITCH E3 ubiquitin ligase and miRNA-106b dictates metastatic progression in pancreatic cancer. Oncotarget. 2016;7(2):1477.
Xia K, Zhang Y, Cao S, Wu Y, Guo W, Yuan W, et al. miR-411 regulated ITCH expression and promoted cell proliferation in human hepatocellular carcinoma cells. Biomed Pharmacother. 2015;70:158–63.
Qu M-H, Han C, Srivastava AK, Cui T, Zou N, Gao Z-Q, et al. MiR-93 promotes TGF-β-induced epithelial-to-mesenchymal transition through downregulation of NEDD4L in lung cancer cells. Tumor Biol. 2016;37(4):5645–51.
Pan Y, Dai J, Liao Y, Yu Q. MicroRNA-137 promotes hepatitis B virus gene expression and replication via targeting the protein inhibitor of activated STAT 2. Die Pharmazie. 2017;72(9):550–4.
Wang C, Ba X, Guo Y, Sun D, Jiang H, Li W, et al. MicroRNA-199a-5p promotes tumour growth by dual-targeting PIAS3 and p27 in human osteosarcoma. Sci Rep. 2017;7:41456.
Tang X, Yin K, Zhu H, Tian J, Shen D, Yi L, et al. Correlation between the expression of microRNA-301a-3p and the proportion of Th17 cells in patients with rheumatoid arthritis. Inflammation. 2016;39(2):759–67.
Liu T, Qin A-P, Liao B, Shao H-G, Guo L-J, Xie G-Q, et al. A novel microRNA regulates osteoclast differentiation via targeting protein inhibitor of activated STAT3 (PIAS3). Bone. 2014;67:156–65.
Wang Z, Han J, Cui Y, Zhou X, Fan K. miRNA-21 inhibition enhances RANTES and IP-10 release in MCF-7 via PIAS3 and STAT3 signalling and causes increased lymphocyte migration. Biochem Biophys Res Commun. 2013;439(3):384–9.
Wu W, Takanashi M, Borjigin N, Ohno S, Fujita K, Hoshino S, et al. MicroRNA-18a modulates STAT3 activity through negative regulation of PIAS3 during gastric adenocarcinogenesis. Br J Cancer. 2013;108(3):653.
Chen X, Wang Y, Zang W, Du Y, Li M, Zhao G. miR-194 targets RBX1 gene to modulate proliferation and migration of gastric cancer cells. Tumor Biol. 2015;36(4):2393–401.
Sun Y, Xu J, Xu L, Zhang J, Chan K, Pan X, et al. MiR-503 promotes bone formation in distraction osteogenesis through suppressing Smurf1 expression. Sci Rep. 2017;7(1):409.
Garros RF, Paul R, Connolly M, Lewis A, Garfield BE, Natanek SA, et al. MicroRNA-542 promotes mitochondrial dysfunction and SMAD activity and is elevated in intensive care unit—acquired weakness. Am J Respir Crit Care Med. 2017;196(11):1422–33.
Wang W, Ren F, Wu Q, Jiang D, Li H, Peng Z, et al. MicroRNA-497 inhibition of ovarian cancer cell migration and invasion through targeting of SMAD specific E3 ubiquitin protein ligase 1. Biochem Biophys Res Commun. 2014;449(4):432–7.
Vimalraj S, Partridge NC, Selvamurugan N. A positive role of microRNA-15b on regulation of osteoblast differentiation. J Cell Physiol. 2014;229(9):1236–44.
Song R, Fullerton DA, Ao L, Zhao KS, Meng X. An epigenetic regulatory loop controls pro-osteogenic activation by TGF-β1 or bone morphogenetic protein 2 in human aortic valve interstitial cells. J Biol Chem. 2017;292(21):8657–66.
Xiao X, Huang C, Zhao C, Gou X, Senavirathna LK, Hinsdale M, et al. Regulation of myofibroblast differentiation by miR-424 during epithelial-to-mesenchymal transition. Arch Biochem Biophys. 2015;566:49–57.
Cao S, Xiao L, Rao JN, Zou T, Liu L, Zhang D, et al. Inhibition of Smurf2 translation by miR-322/503 modulates TGF-β/Smad2 signaling and intestinal epithelial homeostasis. Mol Biol Cell. 2014;25(8):1234–43.
Liu X, Gu X, Sun L, Flowers AB, Rademaker AW, Zhou Y, et al. Downregulation of Smurf2, a tumor-suppressive ubiquitin ligase, in triple-negative breast cancers: involvement of the RB-microRNA axis. BMC Cancer. 2014;14(1):57.
Tao J, Liu Z, Wang Y, Wang L, Yao B, Li Q, et al. MiR-542-3p inhibits metastasis and epithelial-mesenchymal transition of hepatocellular carcinoma by targeting UBE3C. Biomed Pharmacother. 2017;93:420–8.
Li Q, Li Z, Wei S, Wang W, Chen Z, Zhang L, et al. Overexpression of miR-584-5p inhibits proliferation and induces apoptosis by targeting WW domain-containing E3 ubiquitin protein ligase 1 in gastric cancer. J Exp Clin Cancer Res. 2017;36(1):59.
Yang S, Banerjee S, Freitas AD, Cui H, Xie N, Abraham E, et al. miR-21 regulates chronic hypoxia-induced pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol. 2012;302(6):L521–9.
Wang Z, Yin H, Zhang Y, Feng Y, Yan Z, Jiang X, et al. miR-214-mediated downregulation of RNF8 induces chromosomal instability in ovarian cancer cells. Cell Cycle. 2014;13(22):3519–28.
Zhao L, Zhao Y, He Y, Mao Y. miR-19b promotes breast cancer metastasis through targeting MYLIP and its related cell adhesion molecules. Oncotarget. 2017;8(38):64330.
Zhang J, Su B, Gong C, Xi Q, Chao T. miR-214 promotes apoptosis and sensitizes breast cancer cells to doxorubicin by targeting the RFWD2-p53 cascade. Biochem Biophys Res Commun. 2016;478(1):337–42.
Yu M, Liang H, Fu Z, Wang X, Liao Z, Zhou Y, et al. BAP1 suppresses lung cancer progression and is inhibited by miR-31. Oncotarget. 2016;7(12):13742.
Akhtar N, Singh AK, Ahmed S. MicroRNA-17 suppresses TNF-α signaling by interfering with TRAF2 and cIAP2 association in rheumatoid arthritis synovial fibroblasts. J Immunol. 2016;197(6):2219–28.
Zhang L, Xing M, Wang X, Cao W, Wang H. MiR-148a suppresses invasion and induces apoptosis of breast cancer cells by regulating USP4 and BIM expression. Int J Clin Exp Pathol. 2017;10(8):8361–8.
Zhu L, Liu R, Zhang W, Qian S, Wang JH. MicroRNA-205 regulates ubiquitin specific peptidase 7 protein expression in hepatocellular carcinoma cells. Mol Med Rep. 2015;12(3):4652–6.
Xiang S, Fang J, Wang S, Deng B, Zhu L. MicroRNA-135b regulates the stability of PTEN and promotes glycolysis by targeting USP13 in human colorectal cancers. Oncol Rep. 2015;33(3):1342–8.
Zhu Y, Zhang Y, Sui Z, Zhang Y, Liu M, Tang H. USP14 de-ubiquitinates vimentin and miR-320a modulates USP14 and vimentin to contribute to malignancy in gastric cancer cells. Oncotarget. 2017;8(30):48725.
Xiao J, Li Y, Zhang W, Jiang Y, Du B, Tan Y. miR-34b inhibits nasopharyngeal carcinoma cell proliferation by targeting ubiquitin-specific peptidase 22. OncoTargets Ther. 2016;9:1525.
Li J, Tan Q, Yan M, Liu L, Lin H, Zhao F, et al. miRNA-200c inhibits invasion and metastasis of human non-small cell lung cancer by directly targeting ubiquitin specific peptidase 25. Mol Cancer. 2014;13(1):166.
Han H, Sun D, Li W, Shen H, Zhu Y, Li C, et al. A c-Myc-MicroRNA functional feedback loop affects hepatocarcinogenesis. Hepatology. 2013;57(6):2378–89.
Zhang B, Yin Y, Hu Y, Zhang J, Bian Z, Song M, et al. MicroRNA-204-5p inhibits gastric cancer cell proliferation by downregulating USP47 and RAB22A. Med Oncol. 2015;32(1):331.
Huang F, Zhang L, Long Z, Chen Z, Hou X, Wang C, et al. miR-25 alleviates polyQ-mediated cytotoxicity by silencing ATXN3. FEBS Lett. 2014;588(24):4791–8.
Liu N, Wang L, Sun C, Yang L, Sun W, Peng Q. MicroRNA-125b-5p suppresses Brucella abortus intracellular survival via control of A20 expression. BMC Microbiol. 2016;16(1):171.
Wang S, Pan Y, Zhang R, Xu T, Wu W, Wang C, et al. Hsa-miR-24-3p increases nasopharyngeal carcinoma radiosensitivity by targeting both the 3′ UTR and 5′ UTR of Jab1/CSN5. Oncogene. 2016;35(47):6096.
Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–31.
Nascimbeni AC, Giordano F, Dupont N, Grasso D, Vaccaro MI, Codogno P, et al. ER–plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. EMBO J. 2017;36(14):2018–33.
Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14(12):759.
Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132.
Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol. 2009;186(6):773–82.
Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, et al. A protein conjugation system essential for autophagy. Nature. 1998;395(6700):395.
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64(2):113–22.
Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28(27):6926–37.
Yang D-S, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain. 2010;134(1):258–77.
Wu H, Wang F, Hu S, Yin C, Li X, Zhao S, et al. MiR-20a and miR-106b negatively regulate autophagy induced by leucine deprivation via suppression of ULK1 expression in C2C12 myoblasts. Cell Signal. 2012;24(11):2179–86.
Korkmaz G, Le Sage C, Tekirdag KA, Agami R, Gozuacik D. miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1. Autophagy. 2012;8(2):165–76.
Ren YF, Zhang TH, Zhong S, Zhao YT, Lv YN. miR-144 suppresses proliferation and induces apoptosis of osteosarcoma cells via direct regulation of mTOR expression. Oncol Lett. 2018;15(1):1163–9.
Li W, Chang J, Wang S, Liu X, Peng J, Huang D, et al. miRNA-99b-5p suppresses liver metastasis of colorectal cancer by down-regulating mTOR. Oncotarget. 2015;6(27):24448.
Wu D, Huang HJ, He CN, Wang KY. MicroRNA-199a-3p regulates endometrial cancer cell proliferation by targeting mammalian target of rapamycin (mTOR). Int J Gynecol Cancer. 2013;23(7):1191–7.
Pankratz F, Hohnloser C, Bemtgen X, Jaenich C, Kreuzaler S, Hoefer IE, et al. MicroRNA-100 suppresses chronic vascular inflammation by stimulation of endothelial autophagy. Circ Res. 2017. https://doi.org/10.1161/CIRCRESAHA.117.31142.
Lin Z, Li X, Zhan X, Sun L, Gao J, Cao Y, et al. Construction of competitive endogenous RNA network reveals regulatory role of long non-coding RNAs in type 2 diabetes mellitus. J Cell Mol Med. 2017;21(12):3204–13.
Chen S, Zheng Y, Zhang S, Jia L, Zhou Y. Promotion effects of mir-375 on the osteogenic differentiation of human adipose-derived mesenchymal stem cells. Stem Cell Rep. 2017;8(3):773–86.
Wang Z, Wang N, Liu P, Chen Q, Situ H, Xie T, et al. MicroRNA-25 regulates chemoresistance-associated autophagy in breast cancer cells, a process modulated by the natural autophagy inducer isoliquiritigenin. Oncotarget. 2014;5(16):7013.
Clotaire DZJ, Zhang B, Wei N, Gao R, Zhao F, Wang Y, et al. miR-26b inhibits autophagy by targeting ULK2 in prostate cancer cells. Biochem Biophys Res Commun. 2016;472(1):194–200.
Lu W, Han L, Su L, Zhao J, Zhang Y, Zhang S, et al. A 3′ UTR-associated RNA, FLJ11812 maintains stemness of human embryonic stem cells by targeting miR-4459. Stem Cells Dev. 2014;24(9):1133–40.
Guo X, Xue H, Guo X, Gao X, Xu S, Yan S, et al. MiR224-3p inhibits hypoxia-induced autophagy by targeting autophagy-related genes in human glioblastoma cells. Oncotarget. 2015;6(39):41620.
Chatterjee A, Chattopadhyay D, Chakrabarti G. miR-17-5p downregulation contributes to paclitaxel resistance of lung cancer cells through altering beclin1 expression. PLoS ONE. 2014;9(4):e95716.
Xu R, Liu S, Chen H, Lao L. MicroRNA-30a downregulation contributes to chemoresistance of osteosarcoma cells through activating Beclin-1-mediated autophagy. Oncol Rep. 2016;35(3):1757–63.
Shi G, Shi J, Liu K, Liu N, Wang Y, Fu Z, et al. Increased miR-195 aggravates neuropathic pain by inhibiting autophagy following peripheral nerve injury. Glia. 2013;61(4):504–12.
He J, Yu J-J, Xu Q, Wang L, Zheng JZ, Liu L-Z, et al. Downregulation of ATG14 by EGR1-MIR152 sensitizes ovarian cancer cells to cisplatin-induced apoptosis by inhibiting cyto-protective autophagy. Autophagy. 2015;11(2):373–84.
Wang H, Sun T, Hu J, Zhang R, Rao Y, Wang S, et al. miR-33a promotes glioma-initiating cell self-renewal via PKA and NOTCH pathways. J Clin Invest. 2014;124(10):4489–502.
Huangfu L, Liang H, Wang G, Su X, Li L, Du Z, et al. miR-183 regulates autophagy and apoptosis in colorectal cancer through targeting of UVRAG. Oncotarget. 2016;7(4):4735.
Seca H, Lima RT, Lopes-Rodrigues V, Guimaraes JE, Gabriela GM, Vasconcelos MH. Targeting miR-21 induces autophagy and chemosensitivity of leukemia cells. Curr Drug Targets. 2013;14(10):1135–43.
Zhang Y, Liu C, Wang J, Li Q, Ping H, Gao S, et al. MiR-299-5p regulates apoptosis through autophagy in neurons and ameliorates cognitive capacity in APPswe/PS1dE9 mice. Sci Rep. 2016;6:24566.
Li X, Li Y, Fang S, Su J, Jiang J, Liang B, et al. Downregulation of autophagy-related gene ATG5 and GABARAP expression by IFN-λ1 contributes to its anti-HCV activity in human hepatoma cells. Antiviral Res. 2017;140:83–94.
Wang P, Zhang J, Zhang L, Zhu Z, Fan J, Chen L, et al. MicroRNA 23b regulates autophagy associated with radioresistance of pancreatic cancer cells. Gastroenterology. 2013;145(5):1133.e12–1143.e12.
Pan B, Feng B, Chen Y, Huang G, Wang R, Chen L, et al. MiR-200b regulates autophagy associated with chemoresistance in human lung adenocarcinoma. Oncotarget. 2015;6(32):32805.
Zhai Z, Wu F, Dong F, Chuang AY, Messer JS, Boone DL, et al. Human autophagy gene ATG16L1 is post-transcriptionally regulated by MIR142-3p. Autophagy. 2014;10(3):468–79.
Chen R, Li X, He B, Hu W. MicroRNA-410 regulates autophagy-related gene ATG16L1 expression and enhances chemosensitivity via autophagy inhibition in osteosarcoma. Mol Med Rep. 2017;15(3):1326–34.
Wang K, Liu C-Y, Zhou L-Y, Wang J-X, Wang M, Zhao B, et al. APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nat Commun. 2015;6:6779.
Comincini S, Allavena G, Palumbo S, Morini M, Durando F, Angeletti F, et al. microRNA-17 regulates the expression of ATG7 and modulates the autophagy process, improving the sensitivity to temozolomide and low-dose ionizing radiation treatments in human glioblastoma cells. Cancer Biol Ther. 2013;14(7):574–86.
Li Q, Fang Y, Zhu P, Ren CY, Chen H, Gu J, et al. Burkholderia pseudomallei survival in lung epithelial cells benefits from miRNA-mediated suppression of ATG10. Autophagy. 2015;11(8):1293–307.
Rothe K, Lin H, Lin KB, Leung A, Wang HM, Malekesmaeili M, et al. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood. 2014;123(23):3622–34.
D’Adamo S, Alvarez-Garcia O, Muramatsu Y, Flamigni F, Lotz MK. MicroRNA-155 suppresses autophagy in chondrocytes by modulating expression of autophagy proteins. Osteoarthr Cartil. 2016;24(6):1082–91.
Xiao J, Zhu X, He B, Zhang Y, Kang B, Wang Z, et al. MiR-204 regulates cardiomyocyte autophagy induced by ischemia-reperfusion through LC3-II. J Biomed Sci. 2011;18(1):35.
Li X, Zeng Z, Li Q, Xu Q, Xie J, Hao H, et al. Inhibition of microRNA-497 ameliorates anoxia/reoxygenation injury in cardiomyocytes by suppressing cell apoptosis and enhancing autophagy. Oncotarget. 2015;6(22):18829.
Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, et al. Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6(2):143.
Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, et al. Metabolic regulation of brain Aβ by neprilysin. Science. 2001;292(5521):1550–2.
Kanemitsu H, Tomiyama T, Mori H. Human neprilysin is capable of degrading amyloid β peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci Lett. 2003;350(2):113–6.
Zhang H, Liu D, Wang Y, Huang H, Zhao Y, Zhou H. Meta-analysis of expression and function of neprilysin in Alzheimer’s disease. Neurosci Lett. 2017;657:69–76.
Liao M-C, Ahmed M, Smith SO, Van Nostrand WE. Degradation of amyloid β protein by purified myelin basic protein. J Biol Chem. 2009;284(42):28917–25.
Hoos MD, Ahmed M, Smith SO, Van Nostrand WE. Inhibition of familial cerebral amyloid angiopathy mutant amyloid β-protein fibril assembly by myelin basic protein. J Biol Chem. 2007;282(13):9952–61.
Liao M-C, Hoos MD, Aucoin D, Ahmed M, Davis J, Smith SO, et al. N-terminal domain of myelin basic protein inhibits amyloid β-protein fibril assembly. J Biol Chem. 2010;285(46):35590–8.
Wang CY, Deneen B, Tzeng SF. MicroRNA-212 inhibits oligodendrocytes during maturation by downregulation of differentiation-associated gene expression. J Neurochem. 2017;143(1):112–25.
Liao M-C, Van Nostrand WE. Degradation of soluble and fibrillar amyloid β-protein by matrix metalloproteinase (MT1-MMP) in vitro. Biochemistry. 2010;49(6):1127–36.
Zhang H, Qi M, Li S, Qi T, Mei H, Huang K, et al. microRNA-9 targets matrix metalloproteinase 14 to inhibit invasion, metastasis, and angiogenesis of neuroblastoma cells. Mol Cancer Ther. 2012;11(7):1454–66.
Di Gregoli K, Jenkins N, Salter R, White S, Newby AC, Johnson JL. MicroRNA-24 regulates macrophage behavior and retards atherosclerosis. Arterioscler Thromb Vasc Biol. 2014;34(9):1990–2000.
Li Y, Kuscu C, Banach A, Zhang Q, Pulkoski-Gross A, Kim D, et al. miR-181a-5p inhibits cancer cell migration and angiogenesis via downregulation of matrix metalloproteinase-14. Can Res. 2015;75(13):2674–85.
Jasińska M, Miłek J, Cymerman IA, Łęski S, Kaczmarek L, Dziembowska M. miR-132 regulates dendritic spine structure by direct targeting of matrix metalloproteinase 9 mRNA. Mol Neurobiol. 2016;53(7):4701–12.
Yang L, Song X, Zhu J, Li M, Ji Y, Wu F, et al. Tumor suppressor microRNA-34a inhibits cell migration and invasion by targeting mmp-2/mmp-9/fndc3b in esophageal squamous cell carcinoma. Int J Oncol. 2017;51(1):378–88.
Zhu J, Zhang Y, Yang X, Jin L. Clinical significance and tumor-suppressive function of miR-516b in nonsmall cell lung cancer. Cancer Biother Radiopharm. 2017;32(4):115–23.
Hibino Y, Sakamoto N, Naito Y, Goto K, Oo HZ, Sentani K, et al. Significance of miR-148a in colorectal neoplasia: downregulation of miR-148a contributes to the carcinogenesis and cell invasion of colorectal cancer. Pathobiology. 2015;82(5):233–41.
Jiang Q, He M, Guan S, Ma M, Wu H, Yu Z, et al. MicroRNA-100 suppresses the migration and invasion of breast cancer cells by targeting FZD-8 and inhibiting Wnt/β-catenin signaling pathway. Tumor Biol. 2016;37(4):5001–11.
Fang JH, Zhou HC, Zeng C, Yang J, Liu Y, Huang X, et al. MicroRNA-29b suppresses tumor angiogenesis, invasion, and metastasis by regulating matrix metalloproteinase 2 expression. Hepatology. 2011;54(5):1729–40.
Pan Q, Niu H, Cheng L, Li X, Zhang Q, Ning Y. Invasion of trophoblast cell lines is inhibited by miR-93 via MMP-2. Placenta. 2017;53:48–53.
Luo L-J, Zhang L-P, Duan C-Y, Wang B, He N-N, Abulimiti P, et al. The inhibition role of miR-22 in hepatocellular carcinoma cell migration and invasion via targeting CD147. Cancer Cell Int. 2017;17(1):17.
Sun X, Liu Y, Li M, Wang M, Wang Y. Involvement of miR-485-5p in hepatocellular carcinoma progression targeting EMMPRIN. Biomed Pharmacother. 2015;72:58–65.
Peng L, Zhu H, Wang J, Sui H, Zhang H, Jin C, et al. MiR-492 is functionally involved in Oxaliplatin resistance in colon cancer cells LS174T via its regulating the expression of CD147. Mol Cell Biochem. 2015;405(1–2):73–9.
Kohlstedt K, Trouvain C, Boettger T, Shi L, Fisslthaler B, Fleming I. The AMP-activated protein kinase regulates endothelial cell angiotensin-converting enzyme expression via p53 and the post-transcriptional regulation of microRNA-143/145. Circ Res. 2013. https://doi.org/10.1161/CIRCRESAHA.113.301282.
Hu J, Igarashi A, Kamata M, Nakagawa H. Angiotensin-converting enzyme degrades Alzheimer amyloid β-peptide (Aβ); retards Aβ aggregation, deposition, fibril formation; and inhibits cytotoxicity. J Biol Chem. 2001;276(51):47863–8.
Oba R, Igarashi A, Kamata M, Nagata K, Takano S, Nakagawa H. The N-terminal active centre of human angiotensin-converting enzyme degrades Alzheimer amyloid β-peptide. Eur J Neurosci. 2005;21(3):733–40.
Hemming ML, Selkoe DJ. Amyloid β-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J Biol Chem. 2005;280(45):37644–50.
Sun B, Zhou Y, Halabisky B, Lo I, Cho S-H, Mueller-Steiner S, et al. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer’s disease. Neuron. 2008;60(2):247–57.
Yang C-N, Shiao Y-J, Shie F-S, Guo B-S, Chen P-H, Cho C-Y, et al. Mechanism mediating oligomeric Aβ clearance by naïve primary microglia. Neurobiol Dis. 2011;42(3):221–30.
Tiribuzi R, Crispoltoni L, Chiurchiù V, Casella A, Montecchiani C, Del Pino AM, et al. Trans-crocetin improves amyloid-β degradation in monocytes from Alzheimer’s disease patients. J Neurol Sci. 2017;372:408–12.
Dhanavade MJ, Parulekar RS, Kamble SA, Sonawane KD. Molecular modeling approach to explore the role of cathepsin B from Hordeum vulgare in the degradation of Aβ peptides. Mol BioSyst. 2016;12(1):162–8.
Sakamoto T, Saito H, Ishii K, Takahashi H, Tanabe S, Ogasawara Y. Aluminum inhibits proteolytic degradation of amyloid β peptide by cathepsin D: a potential link between aluminum accumulation and neuritic plaque deposition. FEBS Lett. 2006;580(28–29):6543–9.
Tian L, Zhang K, Tian Z-Y, Wang T, Shang D-S, Li B, et al. Decreased expression of cathepsin D in monocytes is related to the defective degradation of amyloid-β in Alzheimer’s disease. J Alzheimer’s Dis. 2014;42(2):511–20.
Tiribuzi R, Crispoltoni L, Porcellati S, Di Lullo M, Florenzano F, Pirro M, et al. miR128 up-regulation correlates with impaired amyloid β (1-42) degradation in monocytes from patients with sporadic Alzheimer’s disease. Neurobiol Aging. 2014;35(2):345–56.
Malherbe P, Richards JG, Gaillard H, Thompson A, Diener C, Schuler A, et al. cDNA cloning of a novel secreted isoform of the human receptor for advanced glycation end products and characterization of cells co-expressing cell-surface scavenger receptors and Swedish mutant amyloid precursor protein. Mol Brain Res. 1999;71(2):159–70.
Mackic JB, Stins M, McComb JG, Calero M, Ghiso J, Kim KS, et al. Human blood-brain barrier receptors for Alzheimer’s amyloid-beta 1-40. Asymmetrical binding, endocytosis, and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J Clin Invest. 1998;102(4):734–43.
Candela P, Gosselet F, Saint-Pol J, Sevin E, Boucau M-C, Boulanger E, et al. Apical-to-basolateral transport of amyloid-β peptides through blood-brain barrier cells is mediated by the receptor for advanced glycation end-products and is restricted by P-glycoprotein. J Alzheimer’s Dis. 2010;22(3):849–59.
Takuma K, Fang F, Zhang W, Yan S, Fukuzaki E, Du H, et al. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-β and neuronal dysfunction. Proc Natl Acad Sci. 2009;106(47):20021–6.
Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, et al. A multimodal RAGE-specific inhibitor reduces amyloid β–mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest. 2012;122(4):1377–92.
Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9(7):907.
Ma L-Y, Fei Y-L, Wang X-Y, Wu S-D, Du J-H, Zhu M, et al. The research on the relationship of RAGE, LRP-1, and Aβ accumulation in the hippocampus, prefrontal lobe, and amygdala of STZ-induced diabetic rats. J Mol Neurosci. 2017;62(1):1–10.
Choi B-R, Cho W-H, Kim J, Lee HJ, Chung C, Jeon WK, et al. Increased expression of the receptor for advanced glycation end products in neurons and astrocytes in a triple transgenic mouse model of Alzheimer’s disease. Exp Mol Med. 2015;46(2):e75.
Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, et al. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular Aβ clearance. Neuron. 2009;64(5):632–44.
Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer’s amyloid-β 1-40 peptide from brain by LDL receptor–related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106(12):1489–99.
Zlokovic BV, Deane R, Sagare AP, Bell RD, Winkler EA. Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling Alzheimer’s amyloid β-peptide elimination from the brain. J Neurochem. 2010;115(5):1077–89.
Storck SE, Meister S, Nahrath J, Meißner JN, Schubert N, Di Spiezio A, et al. Endothelial LRP1 transports amyloid-β 1–42 across the blood–brain barrier. J Clin Invest. 2016;126(1):123–36.
Jaeger LB, Dohgu S, Hwang MC, Farr SA, Murphy MP, Fleegal-DeMotta MA, et al. Testing the neurovascular hypothesis of Alzheimer’s disease: LRP-1 antisense reduces blood-brain barrier clearance, increases brain levels of amyloid-β protein, and impairs cognition. J Alzheimer’s Dis. 2009;17(3):553–70.
Ito S, Ohtsuki S, Kamiie J, Nezu Y, Terasaki T. Cerebral clearance of human amyloid-β peptide (1–40) across the blood–brain barrier is reduced by self-aggregation and formation of low-density lipoprotein receptor-related protein-1 ligand complexes. J Neurochem. 2007;103(6):2482–90.
Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, et al. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc Natl Acad Sci. 2013;110(19):E1807–16.
Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, et al. apoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J Clin Invest. 2008;118(12):4002–13.
Wang Z, Qin W, Wei C, Tang Y, Zhao L, Jin H, et al. The microRNA-1908 up-regulation in the peripheral blood cells impairs amyloid clearance by targeting ApoE. Int J Geriatr Psychiatry. 2018;33(7):980–6.
DeMattos RB, Bales KR, Cummins DJ, Dodart J-C, Paul SM, Holtzman DM. Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci. 2001;98(15):8850–5.
Ghiso J, Shayo M, Calero M, Ng D, Tomidokoro Y, Gandy S, et al. Systemic catabolism of Alzheimer’s Aβ40 and Aβ42. J Biol Chem. 2004;279(44):45897–908.
Tamaki C, Ohtsuki S, Iwatsubo T, Hashimoto T, Yamada K, Yabuki C, et al. Major involvement of low-density lipoprotein receptor-related protein 1 in the clearance of plasma free amyloid β-peptide by the liver. Pharm Res. 2006;23(7):1407–16.
von Einem B, Schwanzar D, Rehn F, Beyer A-S, Weber P, Wagner M, et al. The role of low-density receptor-related protein 1 (LRP1) as a competitive substrate of the amyloid precursor protein (APP) for BACE1. Exp Neurol. 2010;225(1):85–93.
Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, et al. Clearance of amyloid-β by circulating lipoprotein receptors. Nat Med. 2007;13(9):1029.
Matsumoto K, Chiba Y, Fujihara R, Kubo H, Sakamoto H, Ueno M. Immunohistochemical analysis of transporters related to clearance of amyloid-β peptides through blood–cerebrospinal fluid barrier in human brain. Histochem Cell Biol. 2015;144(6):597–611.
Hammad SM, Ranganathan S, Loukinova E, Twal WO, Argraves WS. Interaction of apolipoprotein J-amyloid β-peptide complex with low density lipoprotein receptor-related protein-2/megalin. A mechanism to prevent pathological accumulation of amyloid β-peptide. J Biol Chem. 1997;272(30):18644–9.
Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, et al. Transport pathways for clearance of human Alzheimer’s amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab. 2007;27(5):909–18.
Qi X-M, Wang C, Chu X-K, Li G, Ma J-F. Intraventricular infusion of clusterin ameliorated cognition and pathology in Tg6799 model of Alzheimer’s disease. BMC Neurosci. 2018;19(1):2.
Zhang B, Wang LL, Ren RJ, Dammer EB, Zhang YF, Huang Y, et al. Micro RNA-146a represses LRP 2 translation and leads to cell apoptosis in Alzheimer’s disease. FEBS Lett. 2016;590(14):2190–200.
Akanuma SI, Ohtsuki S, Doi Y, Tachikawa M, Ito S, Hori S, et al. ATP-binding cassette transporter A1 (ABCA1) deficiency does not attenuate the brain-to-blood efflux transport of human amyloid-β peptide (1–40) at the blood–brain barrier. Neurochem Int. 2008;52(6):956–61.
Hirsch-Reinshagen V, Maia LF, Burgess BL, Blain J-F, Naus KE, McIsaac SA, et al. The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J Biol Chem. 2005;280(52):43243–56.
Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem. 2005;280(52):43224–35.
Wahrle SE, Jiang H, Parsadanian M, Hartman RE, Bales KR, Paul SM, et al. Deletion of Abca1 increases Aβ deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem. 2005;280(52):43236–42.
Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008;118(2):671–82.
Corona AW, Kodoma N, Casali BT, Landreth GE. ABCA1 is necessary for bexarotene-mediated clearance of soluble amyloid beta from the hippocampus of APP/PS1 mice. J Neuroimmune Pharmacol. 2016;11(1):61–72.
Fitz NF, Cronican AA, Saleem M, Fauq AH, Chapman R, Lefterov I, et al. Abca1 deficiency affects Alzheimer’s disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J Neurosci. 2012;32(38):13125–36.
Canepa E, Borghi R, Viña J, Traverso N, Gambini J, Domenicotti C, et al. Cholesterol and amyloid-β: evidence for a cross-talk between astrocytes and neuronal cells. J Alzheimer’s Dis. 2011;25(4):645–53.
Do TM, Dodacki A, Alata W, Calon F, Nicolic S, Scherrmann J-M, et al. Age-dependent regulation of the blood–brain barrier influx/efflux equilibrium of amyloid-β peptide in a mouse model of Alzheimer’s disease (3xTg-AD). J Alzheimer’s Dis. 2016;49(2):287–300.
Yassine HN, Feng Q, Chiang J, Petrosspour LM, Fonteh AN, Chui HC, et al. ABCA1-mediated cholesterol efflux capacity to cerebrospinal fluid is reduced in patients with mild cognitive impairment and Alzheimer’s disease. J Am Heart Assoc. 2016;5(2):e002886.
Nordestgaard LT, Tybjærg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Loss-of-function mutation in ABCA1 and risk of Alzheimer’s disease and cerebrovascular disease. Alzheimer’s Dement. 2015;11(12):1430–8.
Kim J, Yoon H, Ramírez CM, Lee S-M, Hoe H-S, Fernández-Hernando C, et al. MiR-106b impairs cholesterol efflux and increases Aβ levels by repressing ABCA1 expression. Exp Neurol. 2012;235(2):476–83.
Liang B, Wang X, Song X, Bai R, Yang H, Yang Z, et al. MicroRNA-20a/b regulates cholesterol efflux through post-transcriptional repression of ATP-binding cassette transporter A1. Biochim Biophys Acta (BBA) Mol Cell Biol Lipids. 2017;1862(9):929–38.
Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-β deposition in an Alzheimer disease mouse model. J Clin Invest. 2005;115(11):3285–90.
Abuznait AH, Cain C, Ingram D, Burk D, Kaddoumi A. Up-regulation of P-glycoprotein reduces intracellular accumulation of beta amyloid: investigation of P-glycoprotein as a novel therapeutic target for Alzheimer’s disease. J Pharm Pharmacol. 2011;63(8):1111–8.
Brenn A, Grube M, Jedlitschky G, Fischer A, Strohmeier B, Eiden M, et al. St. John’s Wort reduces beta-amyloid accumulation in a double transgenic Alzheimer’s disease mouse model—role of P-glycoprotein. Brain Pathol. 2014;24(1):18–24.
Zhang W, Xiong H, Callaghan D, Liu H, Jones A, Pei K, et al. Blood–brain barrier transport of amyloid beta peptides in efflux pump knock-out animals evaluated by in vivo optical imaging. Fluids Barriers CNS. 2013;10(1):13.
Wang W, Bodles-Brakhop AM, Barger SW. A role for P-glycoprotein in clearance of Alzheimer amyloid β-peptide from the brain. Curr Alzheimer Res. 2016;13(6):615–20.
Vogelgesang S, Warzok RW, Cascorbi I, Kunert-Keil C, Schroeder E, Kroemer HK, et al. The role of P-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer’s disease. Curr Alzheimer Res. 2004;1(2):121–5.
Brenn A, Grube M, Peters M, Fischer A, Jedlitschky G, Kroemer HK, et al. Beta-amyloid downregulates MDR1-P-glycoprotein (Abcb1) expression at the blood-brain barrier in mice. Int J Alzheimer’s Dis. 2011. https://doi.org/10.4061/2011/690121.
Do TM, Noel-Hudson M-S, Ribes S, Besengez C, Smirnova M, Cisternino S, et al. ABCG2-and ABCG4-mediated efflux of amyloid-β peptide 1-40 at the mouse blood-brain barrier. J Alzheimer’s Dis. 2012;30(1):155–66.
Shen S, Callaghan D, Juzwik C, Xiong H, Huang P, Zhang W. ABCG2 reduces ROS-mediated toxicity and inflammation: a potential role in Alzheimer’s disease. J Neurochem. 2010;114(6):1590–604.
Xiong H, Callaghan D, Jones A, Bai J, Rasquinha I, Smith C, et al. ABCG2 is upregulated in Alzheimer’s brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood–brain barrier for Aβ1–40 peptides. J Neurosci. 2009;29(17):5463–75.
Dodacki A, Wortman M, Saubaméa B, Chasseigneaux S, Nicolic S, Prince N, et al. Expression and function of Abcg4 in the mouse blood-brain barrier: role in restricting the brain entry of amyloid-β peptide. Sci Rep. 2017;7(1):13393.
Koshiba S, Ito T, Shiota A, Wakabayashi K, Ueda M, Ichinose H, et al. Development of polyclonal antibodies specific to ATP-binding cassette transporters human ABCG4 and mouse Abcg4: site-specific expression of mouse Abcg4 in brain. J Exp Ther Oncol. 2007;6(4):321–33.
Uehara Y, Yamada T, Baba Y, Miura SI, Abe S, Kitajima K, et al. ATP-binding cassette transporter G4 is highly expressed in microglia in Alzheimer’s brain. Brain Res. 2008;1217:239–46.
Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Trans Med. 2012;4(147):147ra11-ra11.
Yang J, Zhang R, Shi C, Mao C, Yang Z, Suo Z, et al. AQP4 association with amyloid deposition and astrocyte pathology in the Tg-ArcSwe mouse model of Alzheimer’s disease. J Alzheimer’s Dis. 2017;57(1):157–69.
Jing R, Chen W, Wang H, Ju S, Cong H, Sun B, et al. Plasma miR-185 is decreased in patients with esophageal squamous cell carcinoma and might suppress tumor migration and invasion by targeting RAGE. Am J Physiol Gastrointest Liver Physiol. 2015;309(9):G719–29.
Luo T, Yan Y, He Q, Ma X, Wang W. miR-328-5p inhibits MDA-MB-231 breast cancer cell proliferation by targeting RAGE. Oncol Rep. 2018;39(6):2906–14.
Song H, Bu G. MicroRNA-205 inhibits tumor cell migration through down-regulating the expression of the LDL receptor-related protein 1. Biochem Biophys Res Commun. 2009;388(2):400–5.
Wen L, Andersen PK, Husum DM, Nørregaard R, Zhao Z, Liu Z, et al. MicroRNA-148b regulates megalin expression and is associated with receptor downregulation in mice with unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2017;313(2):F210–7.
Sun D, Zhang J, Xie J, Wei W, Chen M, Zhao X. MiR-26 controls LXR-dependent cholesterol efflux by targeting ABCA1 and ARL7. FEBS Lett. 2012;586(10):1472–9.
Lv Y-C, Tang Y-Y, Peng J, Zhao G-J, Yang J, Yao F, et al. MicroRNA-19b promotes macrophage cholesterol accumulation and aortic atherosclerosis by targeting ATP-binding cassette transporter A1. Atherosclerosis. 2014;236(1):215–26.
Meiler S, Baumer Y, Toulmin E, Seng K, Boisvert WA. MicroRNA 302a is a novel modulator of cholesterol homeostasis and atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35(2):323–31.
Chen J, Tian W, Cai H, He H, Deng Y. Down-regulation of microRNA-200c is associated with drug resistance in human breast cancer. Med Oncol. 2012;29(4):2527–34.
Li XS, Meng X-N, Yan J, Zong ZH. MicroRNA-873 mediates multidrug resistance in ovarian cancer cells by targeting ABCB1. Tumor Biol. 2016;37(8):10499–506.
Zhao Y, Qi X, Chen J, Wei W, Yu C, Yan H, et al. The miR-491-3p/Sp3/ABCB1 axis attenuates multidrug resistance of hepatocellular carcinoma. Cancer Lett. 2017;408:102–11.
Yang T, Zheng ZM, Li XN, Li ZF, Wang Y, Geng YF, et al. MiR-223 modulates multidrug resistance via downregulation of ABCB1 in hepatocellular carcinoma cells. Exp Biol Med. 2013;238(9):1024–32.
Pan Y-Z, Morris ME, Yu A-M. MicroRNA-328 negatively regulates the expression of breast cancer resistance protein (BCRP/ABCG2) in human cancer cells. Mol Pharmacol. 2009;75(6):1374–9.
Wang Y, Zhao L, Xiao Q, Jiang L, He M, Bai X, et al. miR-302a/b/c/d cooperatively inhibit BCRP expression to increase drug sensitivity in breast cancer cells. Gynecol Oncol. 2016;141(3):592–601.
Jia M, Wei Z, Liu P, Zhao X. Silencing of ABCG2 by microRNA-3163 inhibits multidrug resistance in retinoblastoma cancer stem cells. J Korean Med Sci. 2016;31(6):836–42.
Jiao X, Zhao L, Ma M, Bai X, He M, Yan Y, et al. MiR-181a enhances drug sensitivity in mitoxantone-resistant breast cancer cells by targeting breast cancer resistance protein (BCRP/ABCG2). Breast Cancer Res Treat. 2013;139(3):717–30.
Deng L, Wang X, Jiang L, Yang J, Zhou X, Lu Z, et al. Modulation of miR-185-5p expression by EBV-miR-BART6 contributes to developmental differences in ABCG4 gene expression in human megakaryocytes. Int J Biochem Cell Biol. 2016;81:105–11.
Hou X, Wu W, Yin B, Liu X, Ren F. Micro RNA-463-3p/ABCG 4: a new axis in glucose-stimulated insulin secretion. Obesity. 2016;24(11):2368–76.
Wang Y, Huang J, Ma Y, Tang G, Liu Y, Chen X, et al. MicroRNA-29b is a therapeutic target in cerebral ischemia associated with aquaporin 4. J Cereb Blood Flow Metab. 2015;35(12):1977–84.
Zheng Y, Wang L, Chen M, Pei A, Xie L, Zhu S. Upregulation of miR-130b protects against cerebral ischemic injury by targeting water channel protein aquaporin 4 (AQP4). Am J Transl Res. 2017;9(7):3452.
Trieb K, Ransmayr G, Sgonc R, Lassmann H, Grubeck-Loebenstein B. APP peptides stimulate lymphocyte proliferation in normals, but not in patients with Alzheimer’s disease. Neurobiol Aging. 1996;17(4):541–7.
Fiala M, Lin J, Ringman J, Kermani-Arab V, Tsao G, Patel A, et al. Ineffective phagocytosis of amyloid-β by macrophages of Alzheimer’s disease patients. J Alzheimer’s Dis. 2005;7(3):221–32.
Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi KI. Role of toll-like receptor signalling in Aβ uptake and clearance. Brain. 2006;129(11):3006–19.
Richard KL, Filali M, Préfontaine P, Rivest S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid β1–42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. J Neurosci. 2008;28(22):5784–93.
Song M, Jin J, Lim J-E, Kou J, Pattanayak A, Rehman JA, et al. TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J Neuroinflam. 2011;8(1):92.
Frank S, Copanaki E, Burbach GJ, Müller UC, Deller T. Differential regulation of toll-like receptor mRNAs in amyloid plaque-associated brain tissue of aged APP23 transgenic mice. Neurosci Lett. 2009;453(1):41–4.
Zhang L, Li YJ, Wu XY, Hong Z, Wei WS. Micro RNA-181c negatively regulates the inflammatory response in oxygen-glucose-deprived microglia by targeting Toll-like receptor 4. J Neurochem. 2015;132(6):713–23.
Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. 2016;91(2):328–40.
Jiang T, Tan L, Zhu X-C, Zhang Q-Q, Cao L, Tan M-S, et al. Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology. 2014;39(13):2949.
Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160(6):1061–71.
Kober DL, Alexander-Brett JM, Karch CM, Cruchaga C, Colonna M, Holtzman MJ, et al. Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. elife. 2016;5:e20391.
Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, et al. TREM2 deficiency eliminates TREM2 + inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med. 2015;212(3):287–95.
Alexandrov PN, Zhao Y, Jones BM, Bhattacharjee S, Lukiw WJ. Expression of the phagocytosis-essential protein TREM2 is down-regulated by an aluminum-induced miRNA-34a in a murine microglial cell line. J Inorg Biochem. 2013;128:267–9.
Tsay H-J, Huang Y-C, Chen Y-J, Lee Y-H, Hsu S-M, Tsai K-C, et al. Identifying N-linked glycan moiety and motifs in the cysteine-rich domain critical for N-glycosylation and intracellular trafficking of SR-AI and MARCO. J Biomed Sci. 2016;23(1):27.
Srivastava RAK, Jain JC. Scavenger receptor class B type I expression and elemental analysis in cerebellum and parietal cortex regions of the Alzheimer’s disease brain. J Neurol Sci. 2002;196(1):45–52.
Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, et al. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to β-amyloid fibrils. Am J Pathol. 2002;160(1):101–12.
Zhang H, Su YJ, Zhou WW, Wang SW, Xu PX, Yu XL, et al. Activated scavenger receptor A promotes glial internalization of Aβ. PLoS ONE. 2014;9(4):e94197.
Cornejo F, Vruwink M, Metz C, Muñoz P, Salgado N, Poblete J, et al. Scavenger receptor-A deficiency impairs immune response of microglia and astrocytes potentiating Alzheimer’s disease pathophysiology. Brain Behav Immun. 2017;69:336–50.
Lifshitz V, Weiss R, Levy H, Frenkel D. Scavenger receptor A deficiency accelerates cerebrovascular amyloidosis in an animal model. J Mol Neurosci. 2013;50(1):198–203.
Frenkel D, Wilkinson K, Zhao L, Hickman SE, Means TK, Puckett L, et al. Scara1 deficiency impairs clearance of soluble amyloid-β by mononuclear phagocytes and accelerates Alzheimer’s-like disease progression. Nat Commun. 2013;4:2030.
Husemann J, Loike J, Kodama T, Silverstein S. Scavenger receptor class B type I (SR-BI) mediates adhesion of neonatal murine microglia to fibrillar β-amyloid. J Neuroimmunol. 2001;114(1):142–50.
Iram T, Trudler D, Kain D, Kanner S, Galron R, Vassar R, et al. Astrocytes from old Alzheimer’s disease mice are impaired in Aβ uptake and in neuroprotection. Neurobiol Dis. 2016;96:84–94.
Thanopoulou K, Fragkouli A, Stylianopoulou F, Georgopoulos S. Scavenger receptor class B type I (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proc Natl Acad Sci. 2010;107(48):20816–21.
El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, et al. CD36 mediates the innate host response to β-amyloid. J Exp Med. 2003;197(12):1657–66.
Li X, Melief E, Postupna N, Montine KS, Keene CD, Montine TJ. Prostaglandin E2 receptor subtype 2 regulation of scavenger receptor CD36 modulates microglial Aβ42 phagocytosis. Am J Pathol. 2015;185(1):230–9.
Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer MP, Heneka MT. PPARγ/RXRα-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci. 2012;32(48):17321–31.
Kouadir M, Yang L, Tu J, Yin X, Zhou X, Zhao D. Comparison of mRNA expression patterns of class B scavenger receptors in BV2 microglia upon exposure to amyloidogenic fragments of beta-amyloid and prion proteins. DNA Cell Biol. 2011;30(11):893–7.
Giunta M, Rigamonti A, Scarpini E, Galimberti D, Bonomo S, Venturelli E, et al. The leukocyte expression of CD36 is low in patients with Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging. 2007;28(4):515–8.
Li B-R, Xia L-Q, Liu J, Zhang Y, Deng M, Zhong H-J, et al. miR-758-5p regulates cholesterol uptake via targeting the CD36 3′ UTR. Biochem Biophys Res Commun. 2017;494(1–2):384–9.
Zhou M, Chen J, Zhou L, Chen W, Ding G, Cao L. Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell Immunol. 2014;292(1–2):65–9.
Lv Y-n, Ou-yang A-j, Fu L-s. MicroRNA-27a negatively modulates the inflammatory response in lipopolysaccharide-stimulated microglia by targeting TLR4 and IRAK4. Cell Mol Neurobiol. 2017;37(2):195–210.
Xia X, Li Z, Liu K, Wu Y, Jiang D, Lai Y. Staphylococcal LTA-induced miR-143 inhibits Propionibacterium acnes-mediated inflammatory response in skin. J Investig Dermatol. 2016;136(3):621–30.
Li Z, Cai J, Cao X. MiR-19 suppresses fibroblast-like synoviocytes cytokine release by targeting toll like receptor 2 in rheumatoid arthritis. Am J Transl Res. 2016;8(12):5512.
Zhang B, Wang A, Xia C, Lin Q, Chen C. A single nucleotide polymorphism in primary-microRNA-146a reduces the expression of mature microRNA-146a in patients with Alzheimer’s disease and is associated with the pathogenesis of Alzheimer’s disease. Mol Med Rep. 2015;12(3):4037–42.
Chen T, Yan H, Li Z, Jing T, Zhu W, Ge J, et al. MicroRNA-155 regulates lipid uptake, adhesion/chemokine marker secretion and SCG2 expression in oxLDL-stimulated dendritic cells/macrophages. Int J Cardiol. 2011;147(3):446–7.
Wang L, Jia X-J, Jiang H-J, Du Y, Yang F, Si S-Y, et al. MicroRNAs 185, 96, and 223 repress selective high-density lipoprotein cholesterol uptake through posttranscriptional inhibition. Mol Cell Biol. 2013;33(10):1956–64.
He P-P, OuYang X-P, Li Y, Lv Y-C, Wang Z-B, Yao F, et al. MicroRNA-590 inhibits lipoprotein lipase expression and prevents atherosclerosis in apoE knockout mice. PLoS ONE. 2015;10(9):e0138788.
Kakimura J-I, Kitamura Y, Takata K, Umeki M, Suzuki S, Shibagaki K, et al. Microglial activation and amyloid-β clearance induced by exogenous heat-shock proteins. FASEB J. 2002;16(6):601–3.
Takata K, Kitamura Y, Tsuchiya D, Kawasaki T, Taniguchi T, Shimohama S. Heat shock protein-90-induced microglial clearance of exogenous amyloid-β1–42 in rat hippocampus in vivo. Neurosci Lett. 2003;344(2):87–90.
Hoshino T, Murao N, Namba T, Takehara M, Adachi H, Katsuno M, et al. Suppression of Alzheimer’s disease-related phenotypes by expression of heat shock protein 70 in mice. J Neurosci. 2011;31(14):5225–34.
Evans CG, Wisén S, Gestwicki JE. Heat shock proteins 70 and 90 inhibit early stages of amyloid β-(1–42) aggregation in vitro. J Biol Chem. 2006;281(44):33182–91.
Rivera I, Capone R, Cauvi DM, Arispe N, De Maio A. Modulation of Alzheimer’s amyloid β peptide oligomerization and toxicity by extracellular Hsp70. Cell Stress Chaperones. 2017;23(2):269–79.
Wilhelmus MM, Boelens WC, Otte-Höller I, Kamps B, de Waal RM, Verbeek MM. Small heat shock proteins inhibit amyloid-β protein aggregation and cerebrovascular amyloid-β protein toxicity. Brain Res. 2006;1089(1):67–78.
King M, Nafar F, Clarke J, Mearow K. The small heat shock protein Hsp27 protects cortical neurons against the toxic effects of β-amyloid peptide. J Neurosci Res. 2009;87(14):3161–75.
Yang T-T, Hsu C-T, Kuo Y-M. Cell-derived soluble oligomers of human amyloid-β peptides disturb cellular homeostasis and induce apoptosis in primary hippocampal neurons. J Neural Transm. 2009;116(12):1561.
Magrané J, Smith RC, Walsh K, Querfurth HW. Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed β-amyloid in neurons. J Neurosci. 2004;24(7):1700–6.
Zhu WS, Guo W, Zhu JN, Tang CM, Fu YH, Lin QX, et al. Hsp90aa1: a novel target gene of miR-1 in cardiac ischemia/reperfusion injury. Sci Rep. 2016;6:24498.
O’Brien K, Lowry MC, Corcoran C, Martinez VG, Daly M, Rani S, et al. miR-134 in extracellular vesicles reduces triple-negative breast cancer aggression and increases drug sensitivity. Oncotarget. 2015;6(32):32774.
MacKenzie TN, Mujumdar N, Banerjee S, Sangwan V, Sarver A, Vickers S, et al. Triptolide induces the expression of miR-142-3p: a negative regulator of heat shock protein 70 and pancreatic cancer cell proliferation. Mol Cancer Ther. 2013;12(7):1266–75.
Shehata RH, Abdelmoneim SS, Osman OA, Hasanain AF, Osama A, Abdelmoneim SS, et al. Deregulation of miR-34a and Its chaperon Hsp70 in hepatitis C virus-induced liver cirrhosis and hepatocellular carcinoma patients. Asian Pac J Cancer Prev. 2017;18(9):2395.
Huan Y, He Y, Liu B, Li Y, Jia L, Qu C, et al. Zhenbao Pill reduces the percentage of Treg cells by inducing HSP27 expression. Biomed Pharmacother. 2017;96:818–24.
Chua CEL, Tang BL. miR-34a in neurophysiology and neuropathology. J Mol Neurosci. 2018;67(2):235–46.
Di Y, Lei Y, Yu F, Changfeng F, Song W, Xuming M. MicroRNAs expression and function in cerebral ischemia reperfusion injury. J Mol Neurosci. 2014;53(2):242–50.
Madadi S, Soleimani M. Plasma microRNA investigation: the impact of selecting a suitable internal control on data normalization in pancreatic cancer. J Hepato-Biliary Pancreat Sci. 2019;26(2):E1.
Madadi S, Schwarzenbach H, Lorenzen J, Soleimani M. MicroRNA expression studies: challenge of selecting reliable reference controls for data normalization. Cell Mol Life Sci. 2019;76(18):3497–514.
Madadi S, Soleimani M. Study of serum microrna expression in an amyotrophic lateral sclerosis patient: challenge of selecting suitable internal control for normalization. Muscle Nerve. 2019;59(1):E2–3.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
SM and MS wrote the manuscript. HS, MS and RM revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Madadi, S., Schwarzenbach, H., Saidijam, M. et al. Potential microRNA-related targets in clearance pathways of amyloid-β: novel therapeutic approach for the treatment of Alzheimer’s disease. Cell Biosci 9, 91 (2019). https://doi.org/10.1186/s13578-019-0354-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-019-0354-3