
Abstract
Background
Yarrowia lipolytica is an ascomycete yeast used in biotechnological research for its abilities to secrete high concentrations of proteins and accumulate lipids. Genetic tools have been made in a variety of backgrounds with varying similarity to a comprehensively sequenced strain.
Results
We have developed a set of genetic and molecular tools in order to expand capabilities of Y. lipolytica for both biological research and industrial bioengineering applications. In this work, we generated a set of isogenic auxotrophic strains with decreased non-homologous end joining for targeted DNA incorporation. Genome sequencing, assembly, and annotation of this genetic background uncovers previously unidentified genes in Y. lipolytica. To complement these strains, we constructed plasmids with Y. lipolytica-optimized superfolder GFP for targeted overexpression and fluorescent tagging. We used these tools to build the “Yarrowia lipolytica Cell Atlas,” a collection of strains with endogenous fluorescently tagged organelles in the same genetic background, in order to define organelle morphology in live cells.
Conclusions
These molecular and isogenetic tools are useful for live assessment of organelle-specific protein expression, and for localization of lipid biosynthetic enzymes or other proteins in Y. lipolytica. This work provides the Yarrowia community with tools for cell biology and metabolism research in Y. lipolytica for further development of biofuels and natural products.
Similar content being viewed by others

Background
Within a single fungal species, chromosome number and content can vary widely [1]. Genomic instability has been documented in chromosomal changes in yeasts [2], induced in Candida albicans by centromere removal [3], and observed in the fragmentary gene order conservation of filamentous Ascomycetes [4]. Yarrowia lipolytica also has evidence of inter-strain differences, demonstrated by CHEF gel [5]. Studies in Y. lipolytica have primarily been done in three popular genetic backgrounds: W29 (Wild-type French strain ATCC20460™), H222 (wild-type German strain), and CBS6142-2 (the wild-type American strain) [6]. The Po1 series, derived of a set of backcrosses between W29 and CBS6142-2 [7], have been used for a number of studies. CLIB122, or E150, the reference genome sequence, is derived of W29 in a cross with YB423-12, isolated from milled corn fiber tailings [8]. Genome sequencing efforts have covered some original isolates and additional progeny from genetic studies, including strain W29 [9], and one of a backcrossed series, Po1f [10]. Molecular genetic tools in Yarrowia include ablation of the ku70 ortholog, done in the citric acid producer H222, and in Po1d, which increases the rate of homologous recombination during transformation [11, 12]. Further genome sequencing is needed, as included for Po1g below, to clarify gene content and regulatory region differences between Y. lipolytica strains.
Yarrowia lipolytica has a precedence of organelle studies, particularly for peroxisome biogenesis and dynamics, including six stages of microbody development with differing size and contents [13]. Study of catabolism in the peroxisome, and by lipases [14], modeling efforts [15, 16], and perturbation of both beta-oxidation and elements of the lipid biosynthetic pathways [17] have contributed to engineering desired products such as carotenoids [18], and omega-3 fatty acids [19]. The availability of tools to identify organelle compartments would facilitate studies of this type.
A variety of stains are available for visualizing different intracellular compartments in yeast. FUN-1, Nile Red, MitoTracker, ER-Tracker, and DAPI among others can be used to visualize the vacuole, lipid droplet, mitochondria, endoplasmic reticulum, and nucleus, respectively. Compendia of cell staining techniques are available for specific organisms [20] or specific organelles [21, 22]. However, in Y. lipolytica, we observed poor penetration, toxicity, the need for fixation, and resistance to stain uptake, consistent with many fungi [23–26]. Fixation frequently alters intracellular morphology and staining effectiveness when using these stains. Similarly, changes in growth conditions, or incubation buffer may perturb stain application, as stain efflux differences can be pH dependent [27]. The cell wall changes thickness in an age- and nutrient-dependent manner [28], and may provide an additional barrier to permeability. Probe or stain molecule size requiring a chemical vehicle which increases permeability of cell membranes (e.g., DMSO) may also affect cell health and cogency of data, just as complications of dye delivery under nutrient specific conditions and removal of excess dye may perturb cell integrity. Organelle-specific proteins tagged with green fluorescent protein (GFP) overcome many of these limitations, particularly for life cell imaging.
The technique of using GFP labeled proteins for localization and expression is more than two decades old [29]. The Yarrowia community has developed plasmids for over-expression, or carbon-induced expression using a non-integrating cassette for secretion or exogenous protein expression [7, 30, 31]. Their use is dependent upon having the correct auxotrophs and efficient transformation methods [17, 32]. Fluorescent protein studies in Y. lipolytica have encompassed tagging for localization of Fat1p, Fat4p, and Faa1p using plasmids [33, 34]. Similarly, hybrid promoter studies have utilized fluorescent proteins [35], and transcription factors have been localized using a GFP-fusion expressed from a plasmid [36]. However, sets of Y. lipolytica strains with GFP tagged organelles are not available.
Tools presented here will allow definition of pathways, localization of biosynthetic enzymes, and organelle dynamics in living cells. We developed an isogenic ku70::hph strain set for improved homologous recombination efficiency when transforming PCR products, and assessing localization of proteins within a cell by fluorescent tagging under a high expression promoter using auxotrophic selection of transformants or integrants. This genetic background was sequenced and annotated to facilitate genetic studies. A superfolder GFP gene which shows bright fluorescence [37], was codon optimized for multi-modal use in Y. lipolytica, demonstrated by an enzyme over-expression collection using lipid biosynthesis proteins. To query organelle dynamics in response to changing environmental conditions and build a tool set for co-localization studies, we built the Y. lipolytica Cell Atlas composed of seven strains with different cell compartment labels in both auxotrophic and prototrophic backgrounds. This work provides a consistent set of strains and tools for genetics and cell biology in Y. lipolytica and demonstrates the dynamic nature of organelles important for energy metabolism under conditions relevant to industrial biofuel production.
Results and discussion
Construction of isogenic NHEJ-deficient auxotrophic strains
Previous work has shown that a deletion of the ku70 ortholog increases transformation efficiency and rate of recovery of transformants targeted to specific loci [11, 12]. This removes a non-homologous DNA repair process which allows random integration of DNA and so decreases mis-localization of constructs intended for a particular locus. The Y. lipolytica ku70 ortholog was identified as YALI0C08701g by BLAST. We set out to construct a set of isogenic strains in which ku70 was replaced with a gene conferring hygromycin resistance (hygromycin phosphotransferase; hph) in the Po1g genetic background, commonly used for studies in fungal biotechnology [7, 30] (Table 1). The majority of selective markers available in Y. lipolytica are amino acid and nucleobase auxotrophies. Hygromycin B, derived from Streptomyces hygroscopicus, is a translation inhibitor effective in other fungal systems. It has been demonstrated to be useful in Y. lipolytica in a loxP Cre-removable system [11, 38]. Hygromycin B is an archival and successful selective agent for ascomycetes and hemiascomycetes [39–42]. The wild-type strains ATCC20460™ and ATCC18944 ™ as well as Po1g have different natural resistance levels to the antibiotic Hygromycin B (see Additional file 1: Figure S1). Efficient expression of hph is key for use as a selective marker. We therefore codon optimized hph to enable stricter selection. In order to produce auxotrophic markers leu2 and ura3 separately and together, we replaced ku70 (YALI0C08701g) with hph driven by the tef promoter in the leu2 auxotroph Po1g as a starting point to make FKP355 (matA, ku70::hph+, leu2-270) (Fig. 1b). Successful replacement was confirmed by Southern blot. Figure S2E (see Additional file 1) shows the proper length of all genomic DNA fragments after digestion with BglII, EcoRV, or PvuII restriction endonucleases. We then complemented leu2-270 by replacement with leu2+ to make FKP391 (matA, ku70::hph+, leu2-270::leu2+), providing a prototroph. To generate a double auxotroph from leucine-requiring FKP355, we selected for disruption of ura3 by selection on 5-FOA containing medium to make FKP393 (matA, ku70::hph+, leu2-270, ura3 −). The complicated ura3 locus in FKP355 contains ura3::suc2+ adjacent to an intact copy of ura3. The disrupted and intact copies of ura3 recombined leaving only ura3::suc2+ in FKP393. Analysis of RNA suggests that the plasmid sequence between the disrupted and intact copies of ura3 was lost, indicative of a loop out event while genes adjacent to ura3 (YALI0E26653g and YALI0E26785g) are intact (RNA data not shown). We then complemented leu2-270 in FKP393 by replacement with leu2+ to make FEB130 (matA, ku70::hph+, ura3 −) using the gene from wild-type strain W29 (ATCC 20460™) [9]. These strains represent a complimented prototroph and a set of auxotrophs in an isogenic NHEJ-deficient background for biological investigation (Fig. 1b).

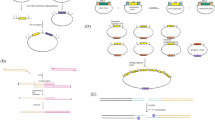
FKP355 genomic background and identified transcripts. a SNP frequency map of FKP355 versus CLIB122 genome shows genomic regions that differ from the available reference. Chromosome A is almost wholly from another background, while other chromosomes show distinct blocks of variation. b Heritage of the strains generated in this work derived from the widely used Po1 series of progeny in an inbred W29 background. c New transcripts undiscovered in the reference CLIB122 strain have been annotated in FKP355 by RNA sequencing. See Additional file 2 for a table of annotations in FKP355
Genome sequencing, assembly, and annotation of NHEJ-deficient base strain
Yarrowia lipolytica strains from the Po1 series of auxotrophs [7, 31, 43] are a commonly used genetic background for experimental studies. However, the available CLIB122 reference genome was produced from an outcrossed strain such that approximately half the reference genome is representative of the sequence of the Po1 series. Recently, the Po1f strain was sequenced [10], and we have sequenced strains derived from W29 [9] to shed additional light on this genetic background. In this study, we sequenced the genome of the ku70::hph strain (FKP355, ku70 mutant of Po1g) from which all the strains described herein are derived. We assembled 680 contains from 23,305,816 paired-end 150 nucleotide reads. The resulting assembly has a size of 20.3 Mb (N50 162,550 bp, NMax 562,182 bp, median coverage 45×) and GC content of 48.2%. By analyzing SNPs, we identified which regions in this strain are derived from a genetic background distinct from the CLIB122 reference genome based on SNP frequency. We identified few conserved SNPs (0.0034 ± 0.0075 SNP/kb) in regions of CLIB122 derived from W29 while divergent regions have 0.916 ± 0.483 SNP/kb (Fig. 1a). Comparison of CLIB122 gene models to the FKP355 assembly identified 26 genes with low (less than 10%) homology to FKP355. Sixteen of these are transposable elements and six are ORFs without known functional domains or homology to any other species, suggesting they may be pseudogenes. Two belong to the matb mating-type locus of CLIB122 and would not be expected in FKP355 which is mata. We did not identify ku70 since it was replaced with hph in FKP355, leaving the predicted acid phosphatase Yali0E35222g as the only gene likely to be physiologically relevant that is present in CLIB122 but not FKP355. We identified 8423 transcripts based on alignment of RNA-sequencing data [15] from which 8571 proteins were predicted and annotated (Additional file 2: Table S1–Protein annotations). Among the 8571 annotations, there are examples of proteins with RNA-seq coverage [15] which do not have a reciprocal best BLAST hit in the CLIB122 genome (Fig. 1c). Thirty-seven transcripts with low homology to the CLIB122 reference genome [44], that contain long open reading frames, were identified as potentially novel protein coding genes in FKP355 (Table 2, New proteins). Most of the novel proteins are predicted from transcripts that are clustered in particular genomic regions suggesting that small stretches of DNA containing multiple genes are not present or were not assembled in strain CLIB122. Within these regions are a variety of important enzymes that are not found in the CLIB122 genome including homogentisate dioxygenase, fumarylacetoacetase, and argininosuccinate synthase. Also present within the new proteins are those belonging to multi-gene families with low homology to species other than Y. lipolytica. Within these multi-gene families are an uncharacterized group of five general substrate transporters, and a group of six cytochrome p450 enzymes that may endow Y. lipolytica with, as yet undocumented, metabolic capabilities. The ura3 gene is known to have a large indel in the CLIB122 strain, rendering it a uracil auxotroph, but has been reintroduced into the FKP355 genetic background. We identified the transcript with the coding gene for Ura3p in this list as a demonstration of its utility.
Construction of a multipurpose vector for rapid expression of fluorescently tagged proteins in Yarrowia lipolytica
We constructed a multipurpose expression vector pYL15 (GenBank accession: KU378202) containing a codon optimized version of the superfolder GFP gene (sfGFP) [37] preceded by a 10× glycine linker and driven by the high expression exp1 promoter [35] to enable rapid analysis of protein localization by expression of GFP fusion proteins in Y. lipolytica. Coding sequences of interest are amplified by PCR from genomic or complementary DNA with primers containing short overhangs homologous to either side of a SmaI site present in the pYL15 vector, which contains the full length leu2 gene from Y. lipolytica. The vector and coding sequence are then assembled by Gibson assembly [45] to reduce the chances of producing vector without an insert (Fig. 2). The vector contains centromere and origin of replication sequences from ARS68 [46] enabling autonomous replication when transformed as a circular plasmid but when linear integrates at the leu2-270 locus present in strains commonly used for Y. lipolytica genetic studies [7, 43] (Fig. 2). Transformed cells are typically grown in yeast nitrogen base medium with ammonium sulfate (YNB) requiring biosynthesis of leucine to select for maintenance of the plasmid. This system enables rapid assembly and high-level expression of sfGFP-tagged proteins in Y. lipolytica. The ability to clone any coding sequence without the use of restriction enzymes and using a single linear plasmid makes this construct amenable to moderate- and high-throughput protein localization studies. To demonstrate the usefulness of pYL15, we cloned enzymes involved in triglyceride biosynthesis and expressed them extra-chromosomally with sfGFP tags to determine their localization.

Utilization of vector pYL15 for expression of sfGFP fusion proteins. Vector pYL15 is linearized by digestion with SmaI. Coding sequences of interest are amplified with primers containing short 5′ and 3′ overhangs homologous to the pYL15 cut site. The vector and coding sequence are assembled and transformed into E. coli. The pYL15 vector contains Y. lipolytica centromere and origin of replication sequences derived from ARS68 [46] as well as leu2 for selection. Map is not drawn to scale to accommodate text
Expression of sfGFP-tagged lipid biosynthetic enzymes reveals their localization
Yarrowia lipolytica is a model organism for the study of metabolism in the context of lipid production for biofuels and other value added lipid products. A number of compartmentalized metabolic models have been constructed for Y. lipolytica that incorporate data derived primarily from homologous proteins in other organisms [15, 16, 47]. We therefore assessed the localization of enzymes involved in triglyceride biosynthesis from glucose in Y. lipolytica to determine where key metabolic reactions occur and to improve on location-dependent metabolic models in Y. lipolytica (Fig. 3; Table 3). The initial building block for fatty acids is Acetyl-CoA, which is produced from citrate by ATP-ctirate lyase (ACL). The presence of ACL in Y. lipolytica is a major distinction between its metabolic network and that of non-oleaginous organisms [48] and deletion results in reduced ability to accumulate lipids [49]. Both subunits of this enzyme localize to the cytosol where they presumably interact to drive lipid accumulation from citrate. Conversion of Acetyl-CoA to Malonyl-CoA by Acetyl-CoA Carboxylase (ACC) is the committed step in fatty acid production and has been successfully used to produce higher quantities of lipids in Y. lipolytica [50, 51]. In Saccharomyces cerevisiae, ACC is expressed by two genes, acc1 and hfa1, which encode cytosol- [52], and mitochondria-localized [53] variants. Y. lipolytica has a single copy of acc1 with a putative mitochondrial targeting signal that exhibits punctate localization that partially overlaps with mitochondria (Figs. 3, 4). Acetyl-CoA and Malonyl-CoA are then utilized by the Type I Fatty Acid Synthase complex, which in S. cerevisiae consists of six copies each of cytoplasmically located Fas1p and Fas2p [54, 55] and Beta-ketoacyl-ACP synthase to make acyl-CoA molecules which primarily consist of 16:0, 18:1, 18:2 chains when grown on glucose [28, 56]. The genes encoding homologs of fas1 and fas2 in Y. lipolytica are both predicted to have short introns. Intriguingly, the splice junction at the 5′ end of the intron is within the translation start site of fas2 and directly adjacent to the translation start site of fas1, respectively, potentially enabling splicing as a strong regulator of expression for these genes. We were unable to detect a green fluorescent protein signal in fas1-sfGFP or fas2-sfGFP strains constructed with or without the short leader introns (data not shown) suggesting that translation of fas1 and fas2 may be tightly controlled by an as yet unknown mechanism in Y. lipolytica. Acyl-CoA molecules produced by the fatty acid synthase complex are utilized by enzymes during lipid biosynthesis. The initial acyl group is attached to the lipid backbone by the glycerol-3-phosphate sn-1 acyltransferase sct1, which we were unable to detect green fluorescent protein signal from. A second acyl chain can be added to lyso-phosphatidic acid by a number of different enzymes in S. cerevisiae including Slc1p [57], Ale1p [58, 59], and Loa1p [60], to produce phosphatidic acid. Interestingly, we found that Slc1-sfGFPp localizes to both the endoplasmic reticulum and the periphery of lipid droplets while Ale1-sfGFPp and Loa1-sfGFPp localize exclusively to the endoplasmic reticulum, consistent with results in S. cerevisiae [61, 62] (Figs. 3, 5). Utilization of phosphatidic acid represents a major regulatory point in lipid homeostasis (for review see: [62–64]). CDP-Diacylglycerol Synthase (CDS) activates phosphatidic acid by addition of CDP prior to release of CMP during the construction of the polar head groups for phospholipid synthesis [65], localized in the ER in Y. lipolytica (Fig. 3). Alternatively, phosphatidic acid is dephosphorylated to form diacylglycerol by Phosphatidate Phosphatase. In S. cerevisiae, the Phosphatidate Phosphatase Pah1p is regulated by phosphorylation to maintain lipid homeostasis between storage and phospholipids [66–69], and the homolog of Pah1p in Y. lipolytica resides in the cytosol (Fig. 3). The endoplasmic reticulum localized enzyme Diacylglycerol Kinase (Dgk1-sfGFPp) phosphorylates diacylglycerol to produce phosphatidic acid (Fig. 3). Alternately, acylation of diacylglycerol to produce storage lipids in the form of triglycerides is performed by Dga1p, Dga2p, and Lro1p. Dga1p and Dga2p utilize acyl-CoA as the acyl chain donor while the lecithin cholesterol acyltransferase Lro1p utilizes phospholipids [70–72]. Lro1-sfGFPp localizes to the endoplasmic reticulum and like Loa1-sfGFPp and Dgk1-sfGFPp is found primarily at the nuclear periphery (Fig. 3). In contrast, Dga1-sfGFPp localizes to the endoplasmic reticulum and the periphery of lipid droplets (Figs. 3, 5), suggesting it is the major producer of lipid droplet stored triglycerides in Y. lipolytica, while Dga2-sfGFPp localizes exclusively to the endoplasmic reticulum (Fig. 3). Acyl-CoA is also used for the esterification of sterols by Are1p [73] which localizes to the endoplasmic reticulum in Y. lipolytica (Fig. 3). We are also interested in the localization of enzymes that utilize triglycerides. Y. lipolytica has four triglyceride lipases that catalyze the cleavage of triglycerides to produce free fatty acids. We found that three of them localize to lipid droplets (Tgl1-sfGFPp, Tgl3-sfGFPp, and Tgl4-sfGFPp; Fig. 5) in a manner consistent with previous findings for Tgl3p and Tgl4p [74], while Tgl2-sfGFPp localizes to the mitochondria (Figs. 3, 4) as in S. cerevisiae [75].

Localization patterns of lipid biosynthetic enzymes in Y. lipolytica. Coding sequences for lipid biosynthetic enzymes were cloned into plasmid pYL15 to create C-terminal fusion proteins with sfGFP. Plasmids were transformed into strain FKP355 and green fluorescence visualized by laser scanning confocal microscopy

Mitochondria localized enzymes. Y. lipolytica cultures expressing sfGFP fusion proteins with a punctate pattern were grown to late log phase in YNB, fixed briefly with formaldehyde, and stained with MitoTracker deep red to visualize mitochondria. Co-localization with green fluorescent proteins was assessed by laser scanning confocal microscopy

Lipid droplet localized enzymes. Y. lipolytica cultures expressing sfGFP fusion proteins with a punctate pattern were grown to late log phase in YNB, fixed briefly with formaldehyde, and stained with LipidTOX red to visualize lipid droplets. Co-localization with green fluorescent proteins was assessed by laser scanning confocal microscopy
The locations we determined are frequently consistent with homologs of enzymes in S. cerevisiae [61, 62] with all five of the exceptions being enzymes where we identified only a single location in Y. lipolytica but for which multiple locations have been determined in S. cerevisiae (Fig. 3). Additional experimentation in Y. lipolytica is likely to augment these additional locations. For example, we determined that Pah1-sfGFPp is localized to the cytosol; however, its substrate, phosphatidic acid, resides within the endoplasmic reticulum membrane suggesting that Pah1p must interact at least transiently with the endoplasmic reticulum. We found that many of the lipid biosynthesis enzymes localize to the endoplasmic reticulum; however, GFP data alone cannot determine ER lumen or cytosol direction of enzyme activity. In summary, we identified the location of 17 enzymes involved in lipid metabolism in Y. lipolytica by plasmid-based expression, many of which localize to multiple organelles (Fig. 6). These data provide evidence for additional locations of Dga1p and Slc1p and previously undetermined locations for Dga2p and Loa1p that can be used to improve compartmentalized metabolic models for Y. lipolytica [15].

Lipid biosynthesis in Y. lipolytica. The localization of enzymes involved in production of lipids from citrate is shown along with results from our analysis of localization. Enzyme names with multiple colors indicate localization to more than one compartment. ACL ATPcitrate lyase, ACC Acetyl-CoA carboxylase, ALE1 1-acyl-sn-glycerol-3-P acyltransferase, ARE Acyl-CoA:sterol acyltransferase, CDS CDP-diglyceride synthase, CEM1 β-ketoacyl synthase, DGA Diacylglycerol acyltransferase, DGK Diacylglycerol kinase, FAS Fatty acid synthase, LOA1 1-acyl-sn-glycerol-3-P acyltransferase, LRO1 Phospholipid:diacylglycerol acyltransferase, PAH1 Phosphatidate phosphatase, SLC1 1-acyl-sn-glycerol-3-P acyltransferase, SCT1 Glycerol-3-P o-acyltransferase, TGL Triacylglycerol lipase. G3P glycerol-3-phosphate, LPA lyso-phosphatidic acid, PA phosphatidic acid, DAG diacylglycerol, TAG triacylglycerol, FFA free fatty acid
Construction of an atlas of endogenous sfGFP-tagged organelles
To overcome limitations of organelle identification by staining and fixation, we generated an atlas of strains with green fluorescent organelles by tagging genes with sfGFP at their endogenous locus (Figs. 7, 8). These intracellular markers respond to native promoter control, rather than being constitutively expressed from plasmids, and avoid overexpression toxicity, which has been suggested in other eukaryotic systems [76]. We chose non-essential proteins which show high and consistent expression, membrane association to define the organelle, DNA or rRNA association for the nucleus, and successful tagging in the S. cerevisiae GFP protein libraries [77, 78]. We identified genes encoding Y. lipolytica homologs of proteins known to be specific to particular organelles in S. cerevisiae including proteins that localize to the nucleus (hH1, YALI0B16280g; arx1, YALI0C05599g), mitochondrion (rdl2, YALI0F29667g; aim17, YALI0F16357g), peroxisome (pex13, YALI0C05775g), lipid droplet (erg6, YALI0F08701g), endoplasmic reticulum (emc2, YALI023188g), vacuole (cpy1, YALI0A18810g), and golgi apparatus (vrg4, YALI0F21791g) (Table 4). We constructed the green fluorescent organelle atlas in a prototrophic and auxotrophic (ura3−) ku70::hph genetic background to enable observation of experimental perturbations in mutant and wild-type–sfGFP strains, and to provide a convenient tool for co-localization studies.

Cell atlas prototrophs. The strains labeling 7 organelle compartments within cells in a prototrophic background. FEB130 was transformed with an endogenously targeted PCR products containing fragments to C-terminally tag organelle-specific proteins followed by a complimenting URA3 gene. Increased lipid droplet accumulation is accompanied by loss of peroxisomes, increase in hH1 signal, and other cellular changes tracked in a live cell without further processing steps. Strains were imaged at 16 h in YPD (t0) and after 20 h in Y-D media (t1). Top GFP channel; bottom GFP with transmission PMT overlay

Cell atlas auxotrophs. An atlas showing cell compartment-specific proteins at 16 h in YPD (t0) and after 20 h of nitrogen limitation in Y-D media, lacking a nitrogen source (t1). Localization is the same as in the prototrophs, leaving open a ura3 marker for further strain construction. Top GFP channel; bottom GFP with transmission PMT overlay
For all markers tested, localization in Y. lipolytica is consistent with S. cerevisiae orthologs and previous work [77, 78]. The function of the endoplasmic reticulum marker Emc2p is unrelated to lipid biogenesis, to avoid bias to a process of primary interest in Y. lipolytica. Emc2p is a member of a transmembrane complex; when members are deleted or perturbed, S. cerevisiae cells show signs of the unfolded protein response [79]. Vrg4p is a golgi GDP-mannose transporter needed for protein glycosylation [80]. We tagged two proteins predicted to localize to the nucleus. The linker histone H1 directly interacts with DNA throughout the interior of the nucleus while the ribosomal export protein Arx1p shuttles the pre-60S subunit for export, and interacts with a number of nucleoporin subunits [81, 82]. The primary localization of Arx1p-sfGFP is also nuclear, at times sub-localized, presumably at the nucleolus (Figs. 7, 8). We tagged two mitochondrial proteins. Rdl2p is a rhodanese-like protein which has thiosulfate sulfurtransferase activity [83]. Rdl2p function in S. cerevisiae is linked to H2O2 detoxification, which contributes to life span in longer-lived wine fermentation yeast strains, where this protein is highly expressed in the late stages of fermentation [84]. Aim17p is implicated in mitochondrial biogenesis in S. cerevisiae [85, 86]. Additionally, we report specific and active localization through the course of experiments to characterize these proteins with existing tools and techniques. Specific localization of the mitochondrial markers Rdl2p and Aim17p was more discrete than the commercially available MitoTracker (Fig. 9), which contains a reactive thiol and is delivered in dimethylsulfoxide to perforate cell membranes [87]. Nitrogen limitation in a carbon-replete environment is a condition used to stimulate lipid accumulation in microalgal, algal, cyanobacterial, and fungal organisms, including Y. lipolytica, for biofuel development [88–92]. We evaluated the atlas proteins for expression and organelle-specific localization in YPD and lower nitrogen (Y-D, YPD lacking peptone) media in both our prototroph and auxotroph strains (Figs. 7, 8). Localization not only was consistent with the expected organelle compartments but also reflects responses to different nutrient types. We found several interesting patterns after transfer to nitrogen-deficient (Y-D) medium that may be associated with stationary phase such as histone H1, important for DNA packaging [93]. Erg6 is a protein in the ergosterol biosynthetic pathway, used as a lipid droplet marker, and a target of antifungals [94, 95]. It has also been identified by proteomics in Y. lipolytica lipid droplets [56], and here shows changing lipid droplet size and number. Cyp1p, a nonspecific carboxypeptidase involved in protein degradation [96], also exhibits altered vacuole size and number consistent with a role in degradation. In total, we have tagged proteins covering seven cellular compartments.

Mitochondrial marker co-localization with Mito-Tracker deep red. The cells of the two prototrophic mitochondrial strains both co-occur with the mitochondrial stain. The GFP signal is limited to mitochondrial membranes, while Mito-Tracker shows nonspecific signal around nuclei and other cytosolic structures, indicated by gray arrows. tPMT transmission PMT, GFP emission at 495–540 nm, MT Mito-Tracker deep red (Invitrogen), overlay GFP overlaid on Mito-Tracker
Mitochondrial response to reactive oxygen species is growth phase dependent
We wanted to assess the response of Rdl2p to hydrogen peroxide stress. It was previously observed that S. cerevisiae sensitivity to hydrogen peroxide is enhanced in minimal medium compared with the presence of amino acids and nucleobases [97], a susceptibility also confirmed in Y. lipolytica [98]. Further, it has been noted that prior exposure of yeast to an oxidative stress increases tolerance to higher concentrations of ROS (reactive oxygen species) [97] suggesting a physiological response which may be based in protein expression. ROS tolerance is also increased in stationary vs. exponential phase cells [99]. Susceptibility to and tolerance of oxidative stress is relevant to lipid biogenesis in several respects: lipids are a target for peroxidation by ROS, and the oxidative state of the cell may change the level of available NADPH, which is used directly in lipid biogenesis.
To determine the response of organelle tags involved in detoxification of ROS, we grew cells expressing Rdl2-sfGFPp, Aim17-sfGFPp, and Pex13-sfGFPp to exponential and stationary phases. They were then treated with hydrogen peroxide and imaged immediately and again after 24 h. We observed a difference in growth phase susceptibility, and that Rdl2-sfGFPp signal intensity changes after H2O2 treatment (Fig. 10). Stationary phase cells have emblematic mitochondrial signal, which is distributed as seen in other growth conditions and stains (Figs. 7, 8, 9) after 24 h of stress. Exponential phase cells are somewhat elongated in all samples, indicative of stress [100], and have punctate mitochondrial and peroxisomal protein signals. These results suggest that Rdl2-sfGFPp distribution, such as between exponential and stationary phase cells (Fig. 10), has the potential to be used as a relative marker of oxidative stress. Consistent with Rdl2 expression levels correlating with stress tolerance in S. cerevisiae [84], monitored expression of Rdl2 under controlled and experimental growth conditions will aid optimization of bioproduct production in this obligate aerobe.

Mitochondrial and peroxisomal proteins under hydrogen peroxide stress. Organelle prototrophic atlas strains were grown for 16 h, after which an aliquot was removed to a fresh tube for the exponential samples. Live cells were imaged at time points t0 = prior to stressor, t1 = 1–2 h after transfer to 40 mM hydrogen peroxide YPD media, and t2 = 20 h after transfer. Images are internally normalized to the tagged protein in Fiji [136]
Pex13-sfGFPp signal is altered by carbon source and nitrogen limitation
The dynamics of Y. lipolytica peroxisomes have been followed by immunostaining [101, 102], by biochemical sedimentation gradients [13, 103], and mass spectrometry [104]. Expression of a limited number of peroxisomal protein components have been developed in other backgrounds [105, 106]. However, previously studied proteins are involved in beta-oxidation (Mfe1p), or are not a constant peroxisome resident (Pex3p) [106]. Malate synthase has been studied for alternative splicing, with one form localized by an N-terminal GFP to the peroxisome matrix by plasmid-based expression [107]. Peroxisome maturation has been studied in detail in Y. lipolytica, following a series of microbody structures which contain peroxisome associated proteins (Pex6p and Pex2p) and enzyme functions such as beta-oxidation and hydrogen peroxide detoxification [13]. Loss of peroxisome-localized beta-oxidation enzyme function resulting in increased lipid accumulation shows the importance of this organelle to biofuel development [108].
Pex13p in S. cerevisiae is a protein with a C-terminal SH3 domain used for docking peroxisomal targeting signal (PTS)-containing proteins, which are chaperones for peroxisome bound cargo [109, 110]. To demonstrate peroxisome labeling, Pex13-sfGFPp was grown with oleate as the sole carbon source (Fig. 11), which induces peroxisome formation. Pex13-sfGFPp signal is generally punctate but disperses somewhat after nitrogen limitation in the auxotrophic and prototrophic atlas strains (Figs. 7, 8). This suggests that Pex13p, an essential part of the protein import complex of Pex13/14/17 for movement into the peroxisomal matrix [111], may be redistributed or broken down in different media [105, 112]. To test this, we grew the Pex13-sfGFPp strain in rich medium (YPD) and then transferred it to rich and minimal (YNB) medium with and without (YNBnoN or Y-D) a nitrogen source for 16 h before microscopy. In both media types with reduced nitrogen, Pex13-sfGFPp signal is increased. Peroxisome signal is lost in minimal medium (YNB), and is greatest in nitrogen-deficient rich medium (Y-D) (Fig. 12). The signal quantification is in contrast to S. cerevisiae, which experiences induction of pexophagy by transfer from oleate media into glucose with nitrogen [113]. However, it is difficult to interpret total signal, as this could indicate redistribution or breakdown of the organelles. These data are evidence of different peroxisome behaviors in oleaginous organisms as compared to conventional yeast models. This strain set will allow minimization of peroxisome production and activity specific to Y. lipolytica without major biochemical pathway loss in the mutation of mfe1p.

Peroxisome induction in carbon sources as visualized by pex13-gfp. Peroxisome development in untagged and pex13-GFP strains is followed showing specific localization. Top images GFP; Middle images GFP and calcofluor white staining; Bottom images transmission PMT overlaid with GFP

Pex13-GFP signal in media with nitrogen limitation. a Cells were inoculated from a single flask after growth into 10 mL each of 4 different media [YNB with 2% glucose and 0.5% ammonium sulfate (w/v), YNB with no ammonium sulfate, YPD, and yeast extract-dextrose with no peptone added (Y-D)]. The cultures were grown shaking at 200 rpm for 16 h followed by imaging on a Leica LSM710 scanning confocal microscope. Upper and lower panels are replicate images for each condition above. b Images were quantified for integrated intensity using CellProfiler [135] with calcofluor signal to find cell area, and statistical analysis done by Statistica. All groups are statistically different except YNBnoN and YPD
Conclusions
The cell biology of lipid accumulation is of industrial interest. Here, we have developed a suite of isogenic strains and codon optimized plasmids to enable cell biology studies in Y. lipolytica. We have sequenced, assembled, and annotated the initial non-homologous end-joining mutant strain used for construction of all the strains described herein allowing for accurate design of genetic constructs. Although some of the resources we have developed have been previously described in other genetic backgrounds in Y. lipolytica [31], including auxotrophs [7], utilization of hph [114] and gfp [115], and replicating and integrating plasmids, they have guided the design of these strains and plasmids. Having all these tools, along with the fluorescent organelle atlas in an isogenic non-homologous end-joining mutant will remove data differences caused by genetic background, and expedite functional genetics in Y. lipolytica. Organelle-specific GFP tagging can identify specific membrane structures under conditions relevant to industrial use, and parallel analysis in an isogenic background with live GFP-labeled proteins expressed from endogenous loci to monitor the size, shape, and quantity of a particular organelle. The atlas strains presented allow rapid and efficient targeted disruption, tagging, overexpression, multi-protein labeling with additional fluorescent tags, and an indicator of cellular stress as suggested by Rdl2-sfGFPp expression in response to reactive oxygen species (Fig. 10). Biofuel organism engineering efforts require consideration not only of total lipid accumulation, but also of cell health and growth rate. Continual drain on lipid resources may make senescence an issue, as the balance of triglyceride synthesis has been shown to confer an energy dependence increase in longevity to S. cerevisiae [116].
We present these tools as a compliment to the holistic approach on the global level of –omic analyses. The rapid increase in –omic studies in Y. lipolytica, which inquire multiple levels of regulation, has generated many hypotheses [15, 28, 117–119] that require more efficient tools for functional genomics. For example, during lipid accumulation experiments, membrane bound cellular substructures can be distinguished by confocal microscopy. While lipid droplets are frequently notable due to a difference in bright field diffraction, quantifying and evaluating their growth in live cells are a GFP functionality of interest for exploring lipid and metabolic phenotypes. We found previously that protein degradation, peroxisome activity, and alcohol sugar secretion were characteristics of Y. lipolytica during nitrogen starvation [28]. In order to study compartmentalized activities, we have included atlas strains demarking membrane-bound compartments relevant to these processes, including the vacuole (nitrogen storage and material degradation), mitochondria (producer of ATP and NADH and NADPH for lipid biosynthesis), golgi (protein modification, sorting, and secretion), peroxisome (oxidative detoxification, beta-oxidation), nucleus (transcriptional activity), and the endoplasmic reticulum (translation and lipid droplet biogenesis) which will enable new routes for the investigation of energy relevant organelles by the Yarrowia research community.
Methods
Chemicals
All chemicals and reagents were purchased from Thermo Fisher Scientific (Waltham, MA) unless otherwise noted. The water used was of Milli-Q grade purified by a Millipore (Bedford, MA) Milli-Q UV Purification System. PCR amplification was performed using Q5 high fidelity DNA polymerase (New England Biolabs; Ipswich, MA). Digestions used FastDigest enzymes (Thermo Fisher Scientific; Waltham, MA) and ligations used T4 DNA ligase (Life Tech.; Carlsbad, CA).
Yeast strains and cultivation
Strains were maintained on YPD medium (1% yeast extract, 1% peptone, 2% glucose) or YNB medium (1.71 g/L Yeast Nitrogen base lacking amino acids, 5 g/L ammonium sulfate, 5 g/L glucose) at 28 °C unless otherwise noted. The widely used Y. lipolytica strain Po1g [7] is from Yeastern Biotech (Taipei, Taiwan). Wild-type matA (W29; ATCC20460™) and matB (CBS6124-2; ATCC18944™) Y. lipolytica strains were from American Type Tissue Culture (Manassas, VA).
Generation of ku70::hph construct
To create the ku70 gene deletion construct, 1.9 kb upstream and downstream of the ku70 gene and 0.55 kb of the Y. lipolytica tef1 promoter and the bacterial hygromycin phosphotransferase gene (hph) were amplified by PCR with oligo pairs (Additional file 3, Oligos) 1313/1314 (ku70 upstream), 1315/1321 (hph), 1322/1318 (tef1), and 1319/1320 (ku70 downstream), respectively. During PCR amplification, restriction sites for the endonucleases XhoI and HindIII were introduced at the 5′-end of ku70 upstream and 3′-end of ku70 downstream for further cloning. All four fragments were fused together by yeast gap repair with pRS426 vector as previously described [120] and then cloned into the T-DNA binary vector pZD663 (see Additional file 1: Figure S2). The fragment was confirmed by DNA sequencing. The pZD663 binary vector, a derivative of the pBI121 binary Ti plasmid [121], was constructed by replacing the whole DNA fragment between left and right borders of the pBI121 T-DNA region (GenBank: AF485783.1) with a synthetic DNA fragment containing ten unique multiple cloning sites.
Agrobacterium-mediated transformation
The transgene expression T-DNA binary vector pZD663-ku70 downstream-tef1-hph-ku70 upstream was mobilized into the A. tumefaciens EHA105 strain by a freeze–thaw technique [122]. A. tumefaciens was grown in YEP (10 g/L yeast extract, 10 g/L peptone and 5 g/L NaCl) overnight. Agrobacterium cells were aliquoted into 5-mL induction medium [123] with and without 0.2 mM AS (acetosyringone) to a final density of 0.2 at A600 for an additional 5–6 h growth to final OD600 of 0.4–0.5 (~2 × 109 cells/mL determined by plate growth count). Three different amounts (5 × 106, 1 × 107, and 5 × 107 cells) of overnight Y. lipolytica cells grown in YPD medium were aliquoted into microcentrifuge tubes and washed twice with IM buffer. Five different ratios of Y. lipolytica and Agrobacterium cells (i.e., 5 × 106, 1 × 107, and 5 × 107 Y. lipolytica:100 µL (~2 × 108) Agrobacterium cells with 0.2 mM AS; 1 × 107 Y. lipolytica:300 µL (~6 × 108) of Agrobacterium cells with 0.2 mM AS; and 1 × 107 Y. lipolytica:100 µL (~2 × 108) of Agrobacterium cells without AS) were mixed well in a final volume of 200 µL and spread onto the 25 × 30 mm sterile 0.45 µm Hybond-N + nylon membrane (GE Healthcare Bio-Sciences, Pittsburgh, Pennsylvania) laid on the IM agar plate with or without AS. After 2 days of incubation at room temperature (~23 °C), the transformed Y. lipolytica cells were washed from the nylon membrane with 5 mL sterile distilled water and 1/10 of the volume was spread onto the YPD agar plate with the 300 mg/L hygromycin B and 250 mg/L cefotaxime. The transformed Y. lipolytica cells were visible on the plate after incubation at 28 °C for 2 days. Individual colonies were picked and streaked onto a new YPD agar plate containing 300 mg/L hygromycin B and 250 mg/L cefotaxime (see Additional file 1: Figure S1). Single colonies were grown in 2 mL of YPD liquid medium containing the same set of antibiotics at 28 °C and 200 rpm for 18–24 h, which was used for strain storage and genomic DNA isolation.
Identification of Y. lipolytica ku70::hph clones
The genomic DNA of selected Y. lipolytica transgenic clones was isolated by CTAB method [124] with modifications. Overnight cultures were transferred into a 2-mL screw-cap micro-tubes (Mikro-Schraubrohre) and centrifuged for 2 min at 17,000g and 20 °C. The supernatant was removed and 250 μL of CTAB and 250 µL of 0.5 mm zirconia/silica beads were added to each tube. The cells were homogenized in a mini-beadbeater (Biospec) for 2 min. The homogenized cell mixtures were incubated at 58 °C for 1 h. Genomic DNA was extracted with 170 µL of phenol:chloroform:isoamyl alcohol (25:24:1, pH 8.0) once and centrifuged for 8 min at 17,000g. After transfer, 500 μL of 95% ethanol was mixed with the supernatants, and incubated at room temperature for 10 min. The DNA was pelleted at 17,000g and 15 °C for 10 min, then re-suspended in 200 µL of 50× TE (50 mM TrisHCl/10 mM EDTA, pH8.0) containing 20 µg of RNase A per sample and incubated at 55 °C for 30~60 min. The DNA mixture was extracted twice with 200 µL of phenol:chloroform:isoamyl alcohol and centrifuged at 17,000g for 10 min. For precipitation, 20 µL of 3 M sodium acetate (pH 5.2) and 500 μL of 95% ethanol were added to each DNA sample, mixed gently and kept at room temperature for 10~30 min. The genomic DNA was pelleted by centrifugation at 10,000g and 15 °C for 10 min, then washed once with 1 mL of 70% ethanol. The dried DNA pellets were re-suspended with 50 µL of 10 mM Tris–HCl (pH 8.0). The genomic DNA was used for PCR screening with oligo pairs located outside the transgene deletion fragment of ku70 (see Additional file 1: Figure S2B). The PCR product was digested with PvuII and separated by agarose gel electrophoresis to test for ku70 gene replacement by hph. After screening about 120 individual transformants, we identified a clone in which ku70 was replaced by hph (see Additional file 1: Figure S2C).
Southern blotting analysis
For Southern blotting, one microgram of total genomic DNA from Po1g or ku70::hph strains was digested with the restriction endonuclease BglII, EcoRV, and PvuII, respectively. The genomic DNA fragments were separated by electrophoresis in a 1% agarose gel, then transferred onto the Amersham Hybond-N+ membrane (GE Healthcare Life Sci, Pittsburgh, PA) by alkaline capillary transfer. The 2.0 kb genomic DNA fragment of ku70 gene upstream region used for the ku70 deletion construct was used to prepare a biotin-labeled probe (see Additional file 1: Figure S2D). The genomic DNA on the Hybond-N+ membrane was hybridized with the biotin-labeled probe overnight at 60 °C in a Problot Hybridization Oven (Labnet International, Edison, NJ, USA) and visualized with a North2South chemiluminescent detection kit (Pierce Protein Research Products, Rockford, IL) in a Koda Imaging Station 2000R (Eastman Kodak Company, Rochester, NY, USA).
Generation of isogenetic ku70::hph auxotrophs
Ura3 mutants derived from FKP355 were selected on YNB with 5 g/L uracil and 5 g/L 5-FOA, and supplemented with 0.1 g/L leucine to make FKP393. Leu2 was complemented in FKP355 resulting in FKP391 and also in FKP393, yielding FEB130 by transformation with a leu2 PCR product amplified with primers OKP443 and OKP444.
Genome sequencing, assembly, and annotation
Genomic DNA was isolated from FKP355 using the yeast genomic DNA purification kit (AMRESCO, Solon, OH) followed by 150 bp paired-end sequencing on an Illumina MiSeq instrument. The reads were assembled using Velvet v1.2.10 [125] with a k-mer length of 81 chosen to optimize for the highest N 50. Raw RNA-seq data from previously described strains of similar genetic background [15, 51] were mapped to the assembled genome using TopHat v2.0.13 [126] to predict transcripts from which coding regions were predicted. Predicted proteins were functionally annotated using Blast2GO [127]. Full length transcripts were aligned to contigs from strain CLIB122 [44] using Blat v32×1 [128] to determine their position and percent identity. Reads were aligned to the reference genome using Bowtie v0.12.9 [129] and SNPs identified with custom Perl scripts.
Plasmid construction
A codon optimized superfolder GFP (sfGFP) gene [37] followed by a codon optimized hph gene from Escherichia coli [130] driven by the high expression Yarrowia lipolytica tef promoter along with the centromere and origin of replication sequences from ARS68 [46] were synthesized and cloned into pMK-RQ to make pYL1. The high expression exp1 promoter was amplified from Y. lipolytica strain Po1g with primer pairs OKP189/190 and ligated into pYL1 following digestion with KpnI and EcoRI to make pYL2 (Genbank: KU378203). The Y. lipolytica leu2 gene was amplified with primers OKP193/194 and ligated into pYL2 following digestion with BamHI to make plasmid pYL4. Additional leu2 5′ flanking sequence was amplified with primer pair OKP451/452 and ligated into pYL4 following digestion with HindIII to make pYL15 (Genbank: KU378202). Homologs of enzymes predicted to be directly involved in the biosynthesis of triglycerides from glucose were predicted using BlastP. The coding region for each enzyme was PCR amplified from Y. lipolytica strain FKP355 genomic DNA using Q5 DNA polymerase (New England Biolabs, Ipswich, MA) and oligos (Life Technologies, Carlsbad, CA) designed with 5′ (5′-ATATCTACAGCGGTACCCCC-3′) and 3′ (5′-CCGCCTCCGCCGATATCCCCC-3′) overhangs homologous to plasmid pYL15 (listed in Additional file 3). Plasmid pYL15 digested with SmaI (Fermentas, Waltham, MA) and the PCR products were purified using a GeneJET purification kit (Thermo Fisher Scientific, Waltham, MA) and assembled using the NEBuilder HiFi assembly kit (New England Biolabs, Ipswich, MA) to produce the replicating plasmids listed in Additional file 4. FKP355 was transformed with pYL15 derived plasmids using the lithium acetate method [131] followed by selection on YNB agar. Transformants were verified by PCR and microscopy and maintained at −80 °C in 15% glycerol.
Generation of organelle GFP library
Selection of candidate organelle-targeted proteins was done using quantitative and image data in S. cerevisiae GFP libraries [77, 78, 132]. For transformation, we utilized a PEG-lithium acetate buffer method developed for S. cerevisiae with modifications [133]. Y. lipolytica cells were grown for 1-3 days before transformation. DNA fragments of approximately 1 kb upstream (5′ flank) and downstream (3′ flank) of the stop codon of the gene for C-terminal GFP tagging were amplified (oligos used in Additional file 3). The leu2 gene was amplified from pYL4 using OEB 170/171, and the codon optimized sfGFP from pYL2 using OKP31/OEB169 which append a synthetic linker for attachment to the selective gene. Targeted proteins selected by orthologous Blast (Table 4). Overlap PCRs were used to assemble the 5′ flank and sfGFP plus a partial region in the selection gene, and separately, another portion of the selection gene and the 3′ flank (OEB3/4 or OKP 211/212). These extension PCRs were run using a 15-cycle program to allow the fragments to prime against one another (98 for 2:00, with cycles of 98 for 0:30, 60 for 0:20, 72 for 2:00, followed by 72 for 5:00). Primers were added (5′ flank forward or 3′ flank reverse primer with the appropriate split marker primer), and the PCRs continued for 30 cycles. Gel purified fragments were extracted using a GeneJET Micro kit (K0832). Agar plates containing YNB with appropriate Leucine or Uracil supplementation (5 g/L) were used for selection [131, 133]. For organelle response to nitrogen limitation, strains were grown to turbidity shaking in YPD for 16 h (t0) followed by washing in YNB salts, and transfer to Y-D media lacking a nitrogen source for 20 h (t1).
Microscopy
For microscopy, cells were visualized using a Zeiss LSM710 confocal laser-scanning microscope (Carl Zeiss MicroImaging GmbH, Munchen, Germany) with a Plan-Apochromate 100×/1.4 Oil objective. For co-localization studies, the cell cultures were stained with MitoTracker deep red (Molecular Probes-Thermo-Fisher, M22426, Eugene, OR) or lipidTOX red (Molecular Probes) according to the manufacturer’s instructions. Cells were grown overnight in 4 mL of YPD in a shake tube, then 700 μL was pelleted gently and transferred to 2 mL fresh YPD for 1 h containing MitoTracker Deep Red added to a 1:5000 dilution. Calcofluor white was added to the cells at the time of imaging with the addition of 1 μL of 1:150 dilution of a 1 mg/mL stock solution to every 4 μL of cells. Images were processed using imageJ [134] and CellProfiler [135]. Images were normalized within the tagged strain relative to wild-type or within time point as appropriate.
Hydrogen peroxide stress
Atlas strains FEB64, FEB92, FEB93 were grown 16 h in YPD with shaking. For exponential phase cells, 1 mL of culture was transferred to fresh media, while the remaining stationary cells were used in parallel at the 24 h mark for stress treatment. Briefly, cells were collected by centrifugation at 200 rpm for 1 min. Reserving some of the initial culture for imaging (t0), strains were transferred to YPD containing 40 mM hydrogen peroxide, then imaged at 1–2 h (t1) and 20 h (t2).
Peroxisome media assay
Strain FEB64 was grown overnight, collected by centrifugation, washed 1X in PBS, and then equally inoculated into 10 mL of rich (YPD), minimal (YNB; 1.7 g/L yeast nitrogen base without amino acids and ammonium sulfate, 20 g/L glucose, 5 g/L ammonium sulfate), nitrogen-deficient rich (Y-D; 10 g/L yeast extract, 20 g/L glucose) and nitrogen-deficient minimal medium (YNBnoN; 1.7 g/L yeast nitrogen base without amino acids and ammonium sulfate, 20 g/L glucose). Cultures were grown shaking at 200 rpm for 16 h, and then imaged. Calcofluor staining was used for definition of cellular space in Cell Profiler.
Abbreviations
- ACC:
-
acetyl-CoA carboxylase
- ACL:
-
ATP-citrate lyase
- AS:
-
acetosyringone
- bp:
-
base pair(s)
- Mb:
-
megabases
- CDS:
-
CDP-Diacylglycerol Synthase
- GFP:
-
green fluorescent protein
- sfGFP:
-
superfolder green fluorescent protein
- FKP:
-
FEB-fungal strain numbers by author initials
- hph:
-
hygromycin phosphotrasferase, gene confers resistance to hygromycin B
- PTS:
-
peroxisome targeting signal
- ROS:
-
reactive oxygen species
- SNP:
-
single nucleotide polymorphism
- YPD:
-
yeast extract, peptone, dextrose media, see “Methods” section
- YNB:
-
yeast nitrogen base media, see “Methods” section
References
Vlaardingerbroek I, Beerens B, Rose L, Fokkens L, Cornelissen BJC, Rep M. Exchange of core chromosomes and horizontal transfer of lineage-specific chromosomes in Fusarium oxysporum: chromosome transfer and exchange in F. oxysporum. Environ Microbiol. 2016. doi:10.1111/1462-2920.13281.
Gordon JL, Byrne KP, Wolfe KH. Mechanisms of chromosome number evolution in yeast. PLoS Genet. 2011;7:e1002190 (Fay JC, editor).
Ketel C, Wang HSW, McClellan M, Bouchonville K, Selmecki A, Lahav T, et al. Neocentromeres form efficiently at multiple possible loci in Candida albicans. PLoS Genet. 2009;5:e1000400 (Copenhaver GP, editor).
Hane JK, Rouxel T, Howlett BJ, Kema GH, Goodwin SB, Oliver RP. A novel mode of chromosomal evolution peculiar to filamentous Ascomycete fungi. Genome Biol. 2011;12:R45.
Naumova E, Naumov G, Fournier P, Nguyen HV, Gaillardin C. Chromosomal polymorphism of the yeast Yarrowia lipolytica and related species: electrophoretic karyotyping and hybridization with cloned genes. Curr Genet. 1993;23:450–4.
Beopoulos A, Desfougeres T, Sabirova J, Zinjarde S, Neuvéglise C, Nicaud J-M. The hydrocarbon-degrading oleaginous yeast Yarrowia lipolytica. In: Timmis KN, editor. Handb. Hydrocarb. Lipid Microbiol. [Internet]. Berlin, Heidelberg: Springer Berlin Heidelberg; 2010. p. 2111–2121. http://link.springer.com/10.1007/978-3-540-77587-4_152. Accessed 10 Jun 2016.
Madzak C, Tréton B, Blanchin-Roland S. Strong hybrid promoters and integrative expression/secretion vectors for quasi-constitutive expression of heterologous proteins in the yeast Yarrowia lipolytica. J Mol Microbiol Biotechnol. 2000;2:207–16.
Wickerham LJ, Kurtzman CP, Herman AI. Sexual reproduction in Candida lipolytica. Science. 1970;167:1141.
Pomraning KR, Baker SE. Draft genome sequence of the dimorphic yeast Yarrowia lipolytica strain W29. Genome Announc. 2015;3:e01211–5.
Liu L, Alper HS. Draft genome sequence of the oleaginous yeast Yarrowia lipolytica PO1f, a commonly used metabolic engineering host. Genome Announc. 2014;2:e00652-14.
Kretzschmar A, Otto C, Holz M, Werner S, Hübner L, Barth G. Increased homologous integration frequency in Yarrowia lipolytica strains defective in non-homologous end-joining. Curr Genet. 2013;59:63–72.
Verbeke J, Beopoulos A, Nicaud J-M. Efficient homologous recombination with short length flanking fragments in Ku70 deficient Yarrowia lipolytica strains. Biotechnol Lett. 2013;35:571–6.
Titorenko VI, Chan H, Rachubinski RA. Fusion of small peroxisomal vesicles in vitro reconstructs an early step in the in vivo multistep peroxisome assembly pathway of Yarrowia lipolytica. J Cell Biol. 2000;148:29–44.
Fickers P, Marty A, Nicaud JM. The lipases from Yarrowia lipolytica: genetics, production, regulation, biochemical characterization and biotechnological applications. Biotechnol Adv. 2011;29:632–44.
Kerkhoven EJ, Pomraning KR, Baker SE, Nielsen J. Regulation of amino-acid metabolism controls flux to lipid accumulation in Yarrowia lipolytica. Npj Syst Biol Appl. 2016;2:16005.
Kavšček M, Bhutada G, Madl T, Natter K. Optimization of lipid production with a genome-scale model of Yarrowia lipolytica. BMC Syst Biol [Internet]. 2015; 9. http://www.biomedcentral.com/1752-0509/9/72. Accessed 15 Jan 2016.
Blazeck J, Hill A, Liu L, Knight R, Miller J, Pan A, et al. Harnessing Yarrowia lipolytica lipogenesis to create a platform for lipid and biofuel production. Nat Commun [Internet]. 2014; 5. http://www.nature.com/doifinder/10.1038/ncomms4131. Accessed 15 Jan 2016.
Matthaus F, Ketelhot M, Gatter M, Barth G. Production of lycopene in the non-carotenoid-producing yeast Yarrowia lipolytica. Appl Environ Microbiol. 2014;80:1660–9.
Xue Z, Sharpe PL, Hong S-P, Yadav NS, Xie D, Short DR, et al. Production of omega-3 eicosapentaenoic acid by metabolic engineering of Yarrowia lipolytica. Nat Biotechnol. 2013;31:734–40.
Wolinski H, Kohlwein SD. Single yeast cell imaging. Methods Mol Biol Clifton NJ. 2014;1205:91–109.
Dimmer KS. Fluorescence Staining of Mitochondria for morphology analysis in Saccharomyces cerevisiae. In: Xiao W, editor. Yeast Protoc [Internet]. New York: Springer New York; 2014. p. 131–52. http://link.springer.com/10.1007/978-1-4939-0799-1_9. Accessed 28 Mar 2016.
Wolinski H, Kohlwein SD. Microscopic and spectroscopic techniques to investigate lipid droplet formation and turnover in yeast. Methods Mol Biol Clifton NJ. 2015;1270:289–305.
Chaffin WL, López-Ribot JL, Casanova M, Gozalbo D, Martínez JP. Cell wall and secreted proteins of Candida albicans: identification, function, and expression. Microbiol Mol Biol Rev. 1998;62:130–80.
Ram AFJ, Klis FM. Identification of fungal cell wall mutants using susceptibility assays based on Calcofluor white and Congo red. Nat Protoc. 2006;1:2253–6.
Gulshan K, Moye-Rowley WS. Multidrug resistance in fungi. Eukaryot Cell. 2007;6:1933–42.
Fotopoulos V. Never say dye: new roles for an old fluorochrome. Plant Signal Behav. 2012;7:342–4.
Aguedo M, Waché Y, Belin J-M. Intracellular pH-dependent efflux of the fluorescent probe pyranine in the yeast Yarrowia lipolytica. FEMS Microbiol Lett. 2001;200:185–9.
Pomraning KR, Wei S, Karagiosis SA, Kim Y-M, Dohnalkova AC, Arey BW, et al. Comprehensive metabolomic, lipidomic and microscopic profiling of Yarrowia lipolytica during lipid accumulation identifies targets for increased lipogenesis. PLOS ONE. 2015;10:e0123188 (Nowrousian M, editor).
Chalfie M, Tu Y, Euskirchen G, Ward W, Prasher D. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–5.
Nicaud J-M, Madzak C, van den Broek P, Gysler C, Duboc P, Niederberger P, et al. Protein expression and secretion in the yeast Yarrowia lipolytica. FEMS Yeast Res. 2002;2:371–9.
Barth G, Gaillardin C. Yarrowia lipolytica. Nonconv Yeasts Biotechnol [Internet]. Berlin, Heidelberg: Springer Berlin Heidelberg; 1996. p. 313–88. http://www.springerlink.com/index/10.1007/978-3-642-79856-6_10. Accessed 4 May 2016.
Wang Z-P, Xu H-M, Wang G-Y, Chi Z, Chi Z-M. Disruption of the MIG1 gene enhances lipid biosynthesis in the oleaginous yeast Yarrowia lipolytica ACA-DC 50109. Biochim Biophys Acta BBA-Mol Cell Biol Lipids. 2013;1831:675–82.
Dulermo R, Gamboa-Meléndez H, Dulermo T, Thevenieau F, Nicaud J-M. The fatty acid transport protein Fat1p is involved in the export of fatty acids from lipid bodies in Yarrowia lipolytica. FEMS Yeast Res. 2014;14:883–96.
Tenagy, Park JS, Iwama R, Kobayashi S, Ohta A, Horiuchi H, et al. Involvement of acyl-CoA synthetase genes in n-alkane assimilation and fatty acid utilization in yeast Yarrowia lipolytica. FEMS Yeast Res. 2015;15:fov031 (Nielsen J, editor).
Blazeck J, Liu L, Redden H, Alper H. Tuning gene expression in Yarrowia lipolytica by a hybrid promoter approach. Appl Environ Microbiol. 2011;77:7905–14.
Martinez-Vazquez A, Gonzalez-Hernandez A, Domínguez Á, Rachubinski R, Riquelme M, Cuellar-Mata P, et al. Identification of the transcription factor Znc1p, which regulates the yeast-to-hypha transition in the dimorphic yeast Yarrowia lipolytica. PLoS ONE. 2013;8:e66790 (Bassilana M, editor).
Pédelacq J-D, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24:79–88.
Fickers P, Le Dall M, Gaillardin C, Thonart P, Nicaud J. New disruption cassettes for rapid gene disruption and marker rescue in the yeast Yarrowia lipolytica. J Microbiol Methods. 2003;55:727–37.
Yang J, Nie L, Chen B, Liu Y, Kong Y, Wang H, et al. Hygromycin-resistance vectors for gene expression in Pichia pastoris. Yeast Chichester Engl. 2014;31:115–25.
Fennessy D, Grallert A, Krapp A, Cokoja A, Bridge AJ, Petersen J, et al. Extending the Schizosaccharomyces pombe molecular genetic toolbox. PLoS ONE. 2014;9:e97683.
Carroll Anne M, Sweigard James A. Barbara valent. Improved vectors for selecting resistance to hygromycin. Fungal Genet Newsl. 1994;41:22.
Punt PJ, Oliver RP, Dingemanse MA, Pouwels PH, van den Hondel CA. Transformation of Aspergillus based on the hygromycin B resistance marker from Escherichia coli. Gene. 1987;56:117–24.
Le Dall MT, Nicaud JM, Gaillardin C. Multiple-copy integration in the yeast Yarrowia lipolytica. Curr Genet. 1994;26:38–44.
Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, Lafontaine I, et al. Genome evolution in yeasts. Nature. 2004;430:35–44.
Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–5.
Fournier P, Abbas A, Chasles M, Kudla B, Ogrydziak DM, Yaver D, et al. Colocalization of centromeric and replicative functions on autonomously replicating sequences isolated from the yeast Yarrowia lipolytica. Proc Natl Acad Sci USA. 1993;90:4912–6.
Pan P, Hua Q. Reconstruction and in silico analysis of metabolic network for an oleaginous yeast, Yarrowia lipolytica. PLoS ONE. 2012;7:e51535.
Ratledge C, Wynn JP. The biochemistry and molecular biology of lipid accumulation in oleaginous microorganisms. Adv Appl Microbiol. 2002;51:1–51.
Dulermo T, Lazar Z, Dulermo R, Rakicka M, Haddouche R, Nicaud J-M. Analysis of ATP-citrate lyase and malic enzyme mutants of Yarrowia lipolytica points out the importance of mannitol metabolism in fatty acid synthesis. Biochim Biophys Acta. 2015;1851:1107–17.
Qiao K, Imam Abidi SH, Liu H, Zhang H, Chakraborty S, Watson N, et al. Engineering lipid overproduction in the oleaginous yeast Yarrowia lipolytica. Metab Eng. 2015;29:56–65.
Tai M, Stephanopoulos G. Engineering the push and pull of lipid biosynthesis in oleaginous yeast Yarrowia lipolytica for biofuel production. Metab Eng. 2013;15:1–9.
Ivessa AS, Schneiter R, Kohlwein SD. Yeast acetyl-CoA carboxylase is associated with the cytoplasmic surface of the endoplasmic reticulum. Eur J Cell Biol. 1997;74:399–406.
Hoja U, Marthol S, Hofmann J, Stegner S, Schulz R, Meier S, et al. HFA1 encoding an organelle-specific acetyl-CoA carboxylase controls mitochondrial fatty acid synthesis in Saccharomyces cerevisiae. J Biol Chem. 2004;279:21779–86.
Kolodziej SJ, Penczek PA, Schroeter JP, Stoops JK. Structure-function relationships of the Saccharomyces cerevisiae fatty acid synthase. Three-dimensional structure. J Biol Chem. 1996;271:28422–9.
Tehlivets O, Scheuringer K, Kohlwein SD. Fatty acid synthesis and elongation in yeast. Biochim Biophys Acta. 2007;1771:255–70.
Athenstaedt K, Jolivet P, Boulard C, Zivy M, Negroni L, Nicaud J-M, et al. Lipid particle composition of the yeast Yarrowia lipolytica depends on the carbon source. Proteomics. 2006;6:1450–9.
Athenstaedt K, Daum G. Biosynthesis of phosphatidic acid in lipid particles and endoplasmic reticulum of Saccharomyces cerevisiae. J Bacteriol. 1997;179:7611–6.
Benghezal M, Roubaty C, Veepuri V, Knudsen J, Conzelmann A. SLC1 and SLC4 encode partially redundant acyl-coenzyme A 1-acylglycerol-3-phosphate O-acyltransferases of budding yeast. J Biol Chem. 2007;282:30845–55.
Jain S, Stanford N, Bhagwat N, Seiler B, Costanzo M, Boone C, et al. Identification of a novel lysophospholipid acyltransferase in Saccharomyces cerevisiae. J Biol Chem. 2007;282:30562–9.
Ayciriex S, Le Guédard M, Camougrand N, Velours G, Schoene M, Leone S, et al. YPR139c/LOA1 encodes a novel lysophosphatidic acid acyltransferase associated with lipid droplets and involved in TAG homeostasis. Mol Biol Cell. 2012;23:233–46.
Henry SA, Kohlwein SD, Carman GM. Metabolism and regulation of glycerolipids in the yeast Saccharomyces cerevisiae. Genetics. 2012;190:317–49.
Kohlwein SD, Veenhuis M, van der Klei IJ. Lipid droplets and peroxisomes: key players in cellular lipid homeostasis or a matter of fat–store ‘em up or burn’em down. Genetics. 2013;193:1–50.
Carman GM, Han G-S. Regulation of phospholipid synthesis in the yeast Saccharomyces cerevisiae. Annu Rev Biochem. 2011;80:859–83.
Pascual F, Carman GM. Phosphatidate phosphatase, a key regulator of lipid homeostasis. Biochim Biophys Acta. 2013;1831:514–22.
Raetz CR. Enzymology, genetics, and regulation of membrane phospholipid synthesis in Escherichia coli. Microbiol Rev. 1978;42:614–59.
Carman GM, Han G-S. Phosphatidic acid phosphatase, a key enzyme in the regulation of lipid synthesis. J Biol Chem. 2009;284:2593–7.
Su W-M, Han G-S, Casciano J, Carman GM. Protein kinase A-mediated phosphorylation of Pah1p phosphatidate phosphatase functions in conjunction with the Pho85p-Pho80p and Cdc28p-cyclin B kinases to regulate lipid synthesis in yeast. J Biol Chem. 2012;287:33364–76.
Su W-M, Han G-S, Carman GM. Cross-talk phosphorylations by protein kinase C and Pho85p-Pho80p protein kinase regulate Pah1p phosphatidate phosphatase abundance in Saccharomyces cerevisiae. J Biol Chem. 2014;289:18818–30.
Choi H-S, Su W-M, Han G-S, Plote D, Xu Z, Carman GM. Pho85p-Pho80p phosphorylation of yeast Pah1p phosphatidate phosphatase regulates its activity, location, abundance, and function in lipid metabolism. J Biol Chem. 2012;287:11290–301.
Athenstaedt K. YALI0E32769 g (DGA1) and YALI0E16797 g (LRO1) encode major triacylglycerol synthases of the oleaginous yeast Yarrowia lipolytica. Biochim Biophys Acta. 2011;1811:587–96.
Beopoulos A, Haddouche R, Kabran P, Dulermo T, Chardot T, Nicaud J-M. Identification and characterization of DGA2, an acyltransferase of the DGAT1 acyl-CoA:diacylglycerol acyltransferase family in the oleaginous yeast Yarrowia lipolytica. New insights into the storage lipid metabolism of oleaginous yeasts. Appl Microbiol Biotechnol. 2012;93:1523–37.
Oelkers P, Tinkelenberg A, Erdeniz N, Cromley D, Billheimer JT, Sturley SL. A lecithin cholesterol acyltransferase-like gene mediates diacylglycerol esterification in yeast. J Biol Chem. 2000;275:15609–12.
Zweytick D, Leitner E, Kohlwein SD, Yu C, Rothblatt J, Daum G. Contribution of Are1p and Are2p to steryl ester synthesis in the yeast Saccharomyces cerevisiae. Eur J Biochem FEBS. 2000;267:1075–82.
Dulermo T, Tréton B, Beopoulos A, Kabran Gnankon AP, Haddouche R, Nicaud J-M. Characterization of the two intracellular lipases of Y. lipolytica encoded by TGL3 and TGL4 genes: new insights into the role of intracellular lipases and lipid body organisation. Biochim Biophys Acta. 2013;1831:1486–95.
Ham HJ, Rho HJ, Shin SK, Yoon H-J. The TGL2 gene of Saccharomyces cerevisiae encodes an active acylglycerol lipase located in the mitochondria. J Biol Chem. 2010;285:3005–13.
Ansari AM, Ahmed AK, Matsangos AE, Lay F, Born LJ, Marti G, et al. Cellular GFP toxicity and immunogenicity: potential confounders in in vivo cell tracking experiments. Stem Cell Rev Rep. 2016;12:553–9.
Breker M, Gymrek M, Moldavski O, Schuldiner M. LoQAtE–localization and quantitation ATlas of the yeast proteomE. A new tool for multiparametric dissection of single-protein behavior in response to biological perturbations in yeast. Nucleic Acids Res. 2014;42:D726–30.
Huh W-K, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, et al. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–91.
Jonikas MC, Collins SR, Denic V, Oh E, Quan EM, Schmid V, et al. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science. 2009;323:1693–7.
Dean N, Zhang YB, Poster JB. The VRG4 gene is required for GDP-mannose transport into the lumen of the Golgi in the yeast, Saccharomyces cerevisiae. J Biol Chem. 1997;272:31908–14.
Hung N-J, Johnson AW. Nuclear recycling of the Pre-60S ribosomal subunit-associated factor Arx1 depends on Rei1 in Saccharomyces cerevisiae. Mol Cell Biol. 2006;26:3718–27.
Hung N-J, Lo K-Y, Patel SS, Helmke K, Johnson AW. Arx1 is a nuclear export receptor for the 60S ribosomal subunit in yeast. Mol Biol Cell. 2008;19:735–44.
Foster MW, Forrester MT, Stamler JS. A protein microarray-based analysis of S-nitrosylation. Proc Natl Acad Sci USA. 2009;106:18948–53.
Orozco H, Matallana E, Aranda A. Oxidative stress tolerance, adenylate cyclase, and autophagy are key players in the chronological life span of Saccharomyces cerevisiae during Winemaking. Appl Environ Microbiol. 2012;78:2748–57.
Reinders J, Zahedi RP, Pfanner N, Meisinger C, Sickmann A. Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics. J Proteome Res. 2006;5:1543–54.
Hess DC, Myers CL, Huttenhower C, Hibbs MA, Hayes AP, Paw J, et al. Computationally driven, quantitative experiments discover genes required for mitochondrial biogenesis. PLoS Genet. 2009;5:e1000407 (Kim SK, editor).
Delmer DP. Dimethylsulfoxide as a potential tool for analysis of compartmentation in living plant cells. Plant Physiol. 1979;64:623–9.
Evans Christopher T, Ratledge Colin. Effect of nitrogen source on lipid accumulation in Oleaginous yeasts. J Gen Microbiol. 1984;130:1693–704.
Park WS, Murphy PA, Glatz BA. Lipid metabolism and cell composition of the oleaginous yeast Apiotrichum curvatum grown at different carbon to nitrogen ratios. Can J Microbiol. 1990;36:318–26.
Cescut J, Fillaudeau L, Molina-Jouve C, Uribelarrea J-L. Carbon accumulation in Rhodotorula glutinis induced by nitrogen limitation. Biotechnol Biofuels. 2014;7:164.
Calvey CH, Su Y-K, Willis LB, McGee M, Jeffries TW. Nitrogen limitation, oxygen limitation, and lipid accumulation in Lipomyces starkeyi. Bioresour Technol. 2016;200:780–8.
Liu T, Li Y, Liu F, Wang C. The enhanced lipid accumulation in oleaginous microalga by the potential continuous nitrogen-limitation (CNL) strategy. Bioresour Technol. 2016;203:150–9.
Ushinsky SC, Bussey H, Ahmed AA, Wang Y, Friesen J, Williams BA, et al. Histone H1 in Saccharomyces cerevisiae. Yeast Chichester Engl. 1997;13:151–61.
Sorger D, Athenstaedt K, Hrastnik C, Daum G. A yeast strain lacking lipid particles bears a defect in ergosterol formation. J Biol Chem. 2004;279:31190–6.
Chung S-K, Lee K-W, Kang HI, Yamashita C, Kudo M, Yoshida Y. Design and synthesis of potential inhibitors of the ergosterol biosynthesis as antifungal agents. Bioorg Med Chem. 2000;8:2475–86.
Van Den Hazel HB, Kielland-Brandt MC, Winther JR. Review: biosynthesis and function of yeast vacuolar proteases. Yeast Chichester Engl. 1996;12:1–16.
Spencer J, Phister TG, Smart KA, Greetham D. Tolerance of pentose utilising yeast to hydrogen peroxide-induced oxidative stress. BMC Res. Notes. 2014;7:151.
Lopes M, Mota M, Belo I. Comparison of Yarrowia lipolytica and Pichia pastoris cellular response to different agents of oxidative stress. Appl Biochem Biotechnol. 2013;170:448–58.
Biryukova EN, Medentsev AG, Arinbasarova AY, Akimenko VK. Tolerance of the yeast Yarrowia lipolytica to oxidative stress. Microbiology. 2006;75:243–7.
Kawasse FM, Amaral PF, Rocha-Leão MHM, Amaral AL, Ferreira EC, Coelho MAZ. Morphological analysis of Yarrowia lipolytica under stress conditions through image processing. Bioprocess Biosyst Eng. 2003;25:371–5.
Eitzen GA, Szilard RK, Rachubinski RA. Enlarged peroxisomes are present in oleic acid-grown Yarrowia lipolytica overexpressing the PEX16 gene encoding an intraperoxisomal peripheral membrane peroxin. J Cell Biol. 1997;137:1265–78.
Szilard RK, Titorenko VI, Veenhuis M, Rachubinski RA. Pay32p of the yeast Yarrowia lipolytica is an intraperoxisomal component of the matrix protein translocation machinery. J Cell Biol. 1995;131:1453–69.
Titorenko VI, Smith JJ, Szilard RK, Rachubinski RA. Pex20p of the Yeast Yarrowia lipolytica is required for the oligomerization of thiolase in the cytosol and for its targeting to the peroxisome. J Cell Biol. 1998;142:403–20.
Wriessnegger T, Gübitz G, Leitner E, Ingolic E, Cregg J, de la Cruz BJ, et al. Lipid composition of peroxisomes from the yeast Pichia pastoris grown on different carbon sources. Biochim Biophys Acta. 2007;1771:455–61.
Nazarko T, Nicaud J, Sibirny A. Observation of the peroxisome-vacuole dynamics by fluorescence microscopy with a single filter set. Cell Biol Int. 2005;29:65–70.
Chang J, Mast FD, Fagarasanu A, Rachubinski DA, Eitzen GA, Dacks JB, et al. Pex3 peroxisome biogenesis proteins function in peroxisome inheritance as class V myosin receptors. J Cell Biol. 2009;187:233–46.
Kabran P, Rossignol T, Gaillardin C, Nicaud J-M, Neuveglise C. Alternative splicing regulates targeting of malate dehydrogenase in Yarrowia lipolytica. DNA Res. 2012;19:231–44.
Dulermo T, Nicaud J-M. Involvement of the G3P shuttle and β-oxidation pathway in the control of TAG synthesis and lipid accumulation in Yarrowia lipolytica. Metab Eng. 2011;13:482–91.
Gould SJ, Kalish JE, Morrell JC, Bjorkman J, Urquhart AJ, Crane DI. Pex13p is an SH3 protein of the peroxisome membrane and a docking factor for the predominantly cytoplasmic PTs1 receptor. J Cell Biol. 1996;135:85–95.
Elgersma Y, Kwast L, Klein A, Voorn-Brouwer T, van den Berg M, Metzig B, et al. The SH3 domain of the Saccharomyces cerevisiae peroxisomal membrane protein Pex13p functions as a docking site for Pex5p, a mobile receptor for the import PTS1-containing proteins. J Cell Biol. 1996;135:97–109.
Smith JJ, Aitchison JD. Peroxisomes take shape. Nat Rev Mol Cell Biol. 2013;14:803–17.
Manjithaya R, Nazarko TY, Farré J-C, Subramani S. Molecular mechanism and physiological role of pexophagy. FEBS Lett. 2010;584:1367–73.
Hutchins MU, Veenhuis M, Klionsky DJ. Peroxisome degradation in Saccharomyces cerevisiae is dependent on machinery of macroautophagy and the Cvt pathway. J Cell Sci. 1999;112(Pt 22):4079–87.
Cordero Otero R, Gaillardin C. Efficient selection of hygromycin-B-resistant Yarrowia lipolytica transformants. Appl Microbiol Biotechnol. 1996;46:143–8.
Ruiz-Pavón L, Domínguez A. Characterization of the Yarrowia lipolytica YlSRP72 gene, a component of the yeast signal recognition particle. Int Microbiol Off J Span Soc Microbiol. 2007;10:283–9.
Handee W, Li X, Hall KW, Deng X, Li P, Benning C, et al. An energy-independent pro-longevity function of triacylglycerol in yeast. PLOS Genet. 2016;12:e1005878 (Longo VD, editor).
Pomraning KR, Kim Y-M, Nicora CD, Chu RK, Bredeweg EL, Purvine SO, et al. Multi-omics analysis reveals regulators of the response to nitrogen limitation in Yarrowia lipolytica. BMC Genomics [Internet]. 2016; 17. http://www.biomedcentral.com/1471-2164/17/138. Accessed 30 Mar 2016.
Mekouar M, Blanc-Lenfle I, Ozanne C, Da Silva C, Cruaud C, Wincker P, et al. Detection and analysis of alternative splicing in Yarrowia lipolytica reveal structural constraints facilitating nonsense-mediated decay of intron-retaining transcripts. Genome Biol. 2010;11:R65.
Morin N, Cescut J, Beopoulos A, Lelandais G, Le Berre V, Uribelarrea J-L, et al. Transcriptomic analyses during the transition from biomass production to lipid accumulation in the oleaginous yeast Yarrowia lipolytica. PLoS ONE. 2011;6:e27966.
Colot HV, Park G, Turner GE, Ringelberg C, Crew CM, Litvinkova L, et al. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc Natl Acad Sci. 2006;103:10352–7.
Jefferson RA, Kavanagh TA, Bevan MW. GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 1987;6:3901–7.
Holsters M, de Waele D, Depicker A, Messens E, van Montagu M, Schell J. Transfection and transformation of Agrobacterium tumefaciens. Mol Gen Genet MGG. 1978;163:181–7.
de Groot MJA, Bundock P, Hooykaas PJ, Beijersbergen AGM. Agrobacterium tumefaciens-mediated transformation of filamentous fungi. Nat Biotechnol. 1998;16:839–42.
Murray MG, Thompson WF. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980;8:4321–6.
Zerbino DR. Using the Velvet de novo assembler for short-read sequencing technologies. Curr Protoc Bioinforma. Ed. Board Andreas Baxevanis Al. 2010; Chapter 11: Unit 11.5.
Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinforma Oxf Engl. 2009;25:1105–11.
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–35.
Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12:656–64.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25.
Gritz L, Davies J. Plasmid-encoded hygromycin B resistance: the sequence of hygromycin B phosphotransferase gene and its expression in Escherichia coli and Saccharomyces cerevisiae. Gene. 1983;25:179–88.
Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96.
Ghaemmaghami S, Huh W-K, Bower K, Howson RW, Belle A, Dephoure N, et al. Global analysis of protein expression in yeast. Nature. 2003;425:737–41.
Gietz RD. Yeast transformation by the LiAc/SS carrier DNA/PEG method. Methods Mol Biol Clifton NJ. 2014;1163:33–44.
Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5.
Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, et al. Cell profiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82.
Gaillardin CM, Charoy V, Heslot H. A study of copulation, sporulation and meiotic segregation in Candida lipolytica. Arch Mikrobiol. 1973;92(1):69–83.
Authors’ contributions
ELB, KRP, ZD, EJK, JN, and SEB conceived and designed the experiments, and wrote the paper. ELB, SEB, KRP, ZD performed the experiments and analyzed the data. All authors read and approved the final manuscript.
Acknowledgements
Part of this research was performed at the Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility sponsored by the U.S. DOE OBER, and located at PNNL. PNNL is a multiprogram national laboratory operated by Battelle for the DOE under Contract DE-AC05-76RLO 1830.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Yarrowia lipolytica FKP355, FEB130, FKP391, FKP393, and atlas strains are available at the Fungal Genetics Stock Center (Manhattan, KS, http://www.fgsc.net).
Plasmid sequences are available for pYL2 (GenBank Accession: KU378203), and pYL15 (GenBank Accession: KU378202).
Funding
This material is based upon work supported by the U.S. Department of Energy (DOE), Office of Science, Office of Biological and Environmental Research (OBER), Genomic Science program, under Award Number DE-SC0008744. Support was also provided by a William Wiley postdoctoral fellowship.
Author information
Authors and Affiliations
Corresponding authors
Additional information
An erratum to this article is available at http://dx.doi.org/10.1186/s13068-017-0731-2.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bredeweg, E.L., Pomraning, K.R., Dai, Z. et al. A molecular genetic toolbox for Yarrowia lipolytica . Biotechnol Biofuels 10, 2 (2017). https://doi.org/10.1186/s13068-016-0687-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-016-0687-7