
Abstract
The extracellular matrix (ECM) is an important component of the tumor microenvironment (TME), having several important roles related to the hallmarks of cancer. In cancer, multiple components of the ECM have been shown to be altered. Although most of these alterations are represented by the increased or decreased quantity of the ECM components, changes regarding the functional alteration of a particular ECM component or of the ECM as a whole have been described. These alterations can be induced by the cancer cells directly or by the TME cells, with cancer-associated fibroblasts being of particular interest in this regard. Because the ECM has this wide array of functions in the tumor, preclinical and clinical studies have assessed the possibility of targeting the ECM, with some of them showing encouraging results. In the present review, we will highlight the most relevant ECM components presenting a comprehensive description of their physical, cellular and molecular properties which can alter the therapy response of the tumor cells. Lastly, some evidences regarding important biological processes were discussed, offering a more detailed understanding of how to modulate altered signalling pathways and to counteract drug resistance mechanisms in tumor cells.
Similar content being viewed by others

Introduction
The tumor microenvironment (TME) is composed of the extracellular matrix (ECM) and various stromal cell types, including endothelial cells, immune cells, fibroblasts, and adipocytes. Added to this, the cells of the TME interact via soluble factors, intercellular receptor-ligand interactions, and exosomes [1,2,3].
The ECM is composed of approximatively 300 unique matrix macromolecules which are classified into collagens, glycoproteins (laminins, elastin, tenascins and fibronectin) and glycoproteins (heparan sulphate proteoglycans, hyaluronan and versican) [4]. Growth factors as well as ECM-associated proteins, interact with the ECM components, making the ECM to have a key role in regulating intercellular communication [5]. Added to this, the ECM from different tissues exhibit different chemical, mechanical and topographical properties, features which critically influence cell fate and function through different mechanosignalling routes [6, 7]. In this review, we discuss about the importance of ECM and its bidirectional interaction with tumor cells and TME constituents in regulating tumor progression and therapy response.
According to its function, composition and location, the ECM can be divided into two main categories: the interstitial matrix and the basement membrane (Table 1). The interstitial matrix forms a three-dimensional network surrounding the malignant clone interconnecting it with various stromal cells. This matrix assures the structural integrity of tissues and organs and modulates biological processes like cell differentiation and migration. In its composition, the interstitial matrix is specifically composed of collagen, fibronectin and elastin, which varies between tissues [8]. In addition, it is known that, in cancer biology, the remodelling of the interstitial ECM induces several critical changes that affect cell signalling, cell migration and tumor progression [9]. The basement membrane presents as a sheet-like dense structure. This membrane consists of collagens and laminins, being interconnected through several proteins such as heparan sulphate, proteoglycans (HSPGs) and nidogen [10]. In the context of cancer the basement membrane is critical in preventing the initial in situ carcinoma of invading the adjacent tissues [11].
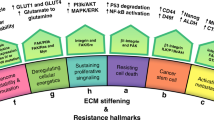
It is known that over 700 proteins are implicated in ECM remodelling which have the ability to modify the abundance, concentration, organization and structure of individual ECM components, causing modification in the spatial topology of the matrix which surrounds the cells, and has an effect on cell fate [24]. Although, the ECM represents the interface between tumor niche and normal tissue being able to present both a pro- and anti-tumorigenic role [25]. ECM can also be altered in the context of premetastatic niche formation, generating an environment that would benefit the malignant clone (Fig. 1) [26,27,28].

ECM components in normal tissue and in the TME. During stiffening, the ECM is remodelled by the ECM components secreted both by cancer and stromal cells. This, in turn, affects the tissue mechanosignalling and its permeability for different therapeutic agents
Extracellular matrix proteins
The ECM is composed of various proteins that create specific structures and properties. The ECM is composed of collagen, laminin, proteoglycans and fibronectin, all of which have been shown to be increased in solid tumors (Table 2) [29].
Collagen represents the main structural element of the ECM having several characteristics such as mechanical strength, activating cell migration and modifying cell adhesion [46]. Collagen can be classified into several types, including types I, III, IV, VII, XI, XVII having been shown to be involved in epithelial-to-mesenchymal transition (EMT), drug resistance and tumor-initiation [47]. Collagen is a helix glycoprotein formed from three homologous or nonhomologous α chains. These α chain amino acids are formed by glycine-X–Y units, where X and Y might be a proline or hydroxyproline. In this regard, hydroxyproline units found in collagens structure are involved in its stability [48]. From a synthesis standpoint, collagen is released into the ECM where it forms a fibril supramolecular structure. This complex starts in Golgi-to-membrane carriers after procollagen excision. In addition, the stability of collagen is controlled by inter- and intramolecular linkages such as covalent bounds (glycosylation, lysyl oxidation and transglutaminase crosslinks) [49].
Proteoglycans represent an essential component of the ECM require enzymes for a proper production and assembly. The structure of proteoglycans consists of a core protein which is glycosylated with multiple chains of glycosaminoglycans (GAGs). In the Golgi apparatus, the attachment of GAGs occurs where the core protein is translocated. In a wide range of tumors, the proteoglycan differs compared to normal tissues. In prostate cancer, CSPG4 and aggrecan are upregulated, while, the expression level of decorin is downregulated [50]. At the same time, in gastric cancer, the expression level for versican and decorin is elevated alongside an increased glycosaminoglycan content [51]. In squamous cell laryngeal carcinoma, versican and decorin are strongly upregulated, while aggrecan is decreased [52]. Further, another study, has shown that versican is increased in the peritumoral stroma of melanoma [53].
Hyaluronic acid, another key component of the ECM is a glycosaminoglycan which is not conjugated to a protein core and not synthesized in the Golgi apparatus. Synthesis of this protein is assured by a family of three transmembrane glycosyltransferase: hyaluronan synthetase 1–3 (HAS1-3) [54]. In a paper published by Schulz et al., it has been shown that hyaluronan synthetases are responsible for extracellular export of the hyaluronic acid macromolecule through a direct extrusion. Conversely, there are others that assume that hyaluronic acid macromolecules are exported through ABC transporters, including MRP5 or MDR1 [55].
Laminin is a component which belongs to the basement membrane, a structure that appears as continuous and well-delineated. In tumors, the basement membrane and laminin is often distorted [56]. The loss of adherence or the disruption of a basement membrane is a main feature of invasiveness. Interestingly, an increased expression level for laminin and its aberrant distribution is associated with invasiveness and poor prognosis [57].
Hemidesmosomes (HDs) are multiprotein structures involved in the attachment of epithelial cells to the basement membrane, mediating cell-to-matrix adhesion [58]. HDs are mainly constituted from plectin isoform 1a, bullous pemphigoid antigen1 isoform e (BP230, also known as BPAG1), integrin α6β4, bullous pemphigoid antigen 2 (BP180, also known as BPAG2 or type XVII collagen) and CD151 (tetraspanin-24) [59, 60], being responsible for keratinocyte adherence, differentiation, spatial organization of tissue architecture, polarization and survival [61]. In pancreas and colon cancer, it has been shown that hemidesmosomes are disrupted, fact leading to an ECM that is easier to invade by the cancer cells [62, 63].
As it is already known, the composition of ECM in tumors is significantly different compared to that of normal tissues, regarding its composition, architecture, and physical properties. Any alterations that modify the ECM can negatively affect the response to therapy. In this regard, the presence of a dense and rigid ECM engulfs the tumor cells by forming a natural capsid and protecting tumor cells from therapeutics. At the same time, it was observed that, this barrier alters the normal circuit of oxygen, nutrients, and metabolites. The antiapoptotic and drug resistance pathways can be activated via an increased level of hypoxia and metabolic stress, while, an increased tissue stiffness might be linked with chemoresistance mediated via integrin and FAK-signalling [57].
The ECMs implication in tumorigenesis and cancer progression
The particular architecture and orientation of ECM constituents in the microenvironment of a specific tissue play a critical role in tumor progression, invasion and metastasis [34, 64]. Therefore, ECM provides a large repertoire of growth factors and cytokines, modulating processes such as cell proliferation and adhesion [65]. An important component of the ECM, collagen, is deeply involved and dictates the primary functional properties of the ECM. Any alterations occurring in the deposition or degradation of collagen alter the ECM [66, 67]. These architectural changes cause an increased secretion of collagen I, III and IV and fibronectin, showing that tumor progression involves a continuous interaction between cancer cells and the ECM [68].
As most of the ECM protein is produced by the tumor stroma, and because both the tumor stroma and the ECM have been shown to be involved in various hallmarks of cancer in a myriad of tumors, there have been studies assessing the prognostic impact of ECM in a multitude of cancers. Specifically, these studies have shown that stroma abundance is different between cancers and the quantification of stroma abundance can be of use in predicting the prognosis of these patients [69].
Deregulation of cell shape and alterations occurring in the TME are considered important hallmarks of cancer and can significantly affect cancer cell proliferation. In different types of cancer, collagen, glycoproteins and proteoglycans may present dual role, pro- or anti-tumorigenic properties [70]. Collagen interacts with tumor suppressor genes. Also, collagen changes have also been shown to occur as a result of mutations occurring in proto-oncogenes, thus having an impact in cancer progression, invasion and metastasis [71]. Thus, collagen seems to act similar to a migrator. In an experiment performed by Bonnans et al., it has been shown that a dense amount of collagen I increased the risk of tumor metastasis, this, in turn, being associated with a worse prognosis [72]. It its known that, in normal tissues, collagen fibres are curly with an irregular arrangement, while, in tumor tissues, these are linear and dense with a directional arrangement [73]. It was observed that collagenases are responsible for collagen remodelling in this case. Thus, matrix metalloproteinase (MMP) and lysine oxidase (LOX) family proteins are of main importance, causing the rearrangement of collagen fibers [74, 75]. An increased collagen cross-linking and deposition cause tumor progression through an increased integrin signalling [64, 76]. Added to this, depletion of fibrillar collagen I and III induces malignant progression as well [77, 78]. Collagen cross-linking is regulated by LOX [46]. Cancer cell-secreted LOX increases matrix stiffness and volume. It is known that, by increasing the stiffness of the ECM, integrins are activated and stimulate Rho-generated cytoskeletal tension [79]. The ECM integrins are modulated by crosstalk with different signalling molecules like receptor molecules on the cell surface or present in the cytoplasm as actin-binding proteins and adaptor proteins [80]. COL11A1 was found to be involved in cancer progression through different biological processes such as apoptosis or EMT [81]. In several studies, it was observed that collagen IV-derived canstatin, tumstatin and tetrastatin, and collagen XIX-derived matrikine act through binding to several integrins, including α3β1, α5β1, or αVβ3. As a result, cellular proliferation and invasion are decreased [82, 83].
In some studies, it was found that an increased secretion of collagenases stimulates an abnormal collagen remodelling in breast cancer and hepatocellular carcinoma [84, 85]. An important aspect is that, in cancer cells, the architecture and content of collagen is modified by mutated tumor suppressor genes. In cancer cells, mutated TP53 affects collagen production [86]. The alteration of collagen in cancer can occur through a multitude of mechanisms, including the alteration of Hsp47, which is linked with collagen deposition [87], but also the overexpression of collagen related genes in cancer cells [88], sustaining resistance to therapy. Added to this, a therapy-resistant ovarian cancer cell line (A2780/A2780cis) was shown to present upregulated COL1A2, COL12A1, COL21A1, LOX, TGFBI, LAMB1, EFEMP1, GPC3, SDC2, MGP, MMP3, and TIMP3 genes. This adds to the evidence that ECM components secreted by cancer cells have an impact in therapy resistance [88].
The collagen production is affected by the presence of mutated TP53 alongside the activation of JAK2-STAT3 signalling [86]. Also, in another study, it has been shown that Arresten, an antiangiogenic factor being localized in the C-terminal non-collagenous domain of COL4A1, is associated with p53 activation [89]. In this regard, p53 increases the level of collagen prolyl-hydroxylase which, in turn, enhances the production of COL4A1 and the content of Arresten [90]. Another study has shown that loss of TP53 leads to the activation of JAK-STAT, which, in turn, is able to affect the number of stellate cells in pancreas tumor stroma, the level of periostin, and, subsequently, the structure of collagen [86].
Fibronectin is involved in tumor progression as well as in drug resistance and metastasis with an increased expression level in many solid tumors [91]. An increased deposition of matrix proteins stimulates tumor progression by interfering with cell polarity, cell-to-cell adhesion as well as growth factor-mediated signalling [92]. Several studies have shown the implication of ECM components in tumor progression.
ECM stiffness plays a critical role in tumor progression by modulating intrinsic properties of tumor cells. The presence of a rigid ECM can determine intracellular contractions further sustaining cell migration [93]. ECM stiffness induces the activation of TGFβ signalling that mediates EMT in cancer cells. Thus, the interaction between stromal and epithelial cells plays a pivotal role in tumor progression [94] (Fig. 2). In pancreatic cancer cells, the genetic alterations which occur in TGFβ signalling change the activity of STAT3 kinase by increasing the deposition and remodelling the tumor ECM. In this regard, the tumor stiffness is increased and enhances tumor growth [95]. It is already known that TGFβ has a dual role in cancer [96]. In the early stages of cancer, the role of TGFβ is to inhibit tumor cell proliferation, while, in the later stages of cancer, it is to stimulate the malignant progression and metastasis of cancer cells [97]. LTBP3, a protein that regulates TGFβ secretion, supports the invasion and metastasis of the primary tumor and is involved in the formation of the fibrillar ECM network. The SNED1 glycoprotein is a component of the basement membrane, and it has been shown to present a functional role in tumor progression and metastasis [98]. Furthermore, a link between the ECM landscape and the genotypic features of the malignant clone has been observed and was shown to be important in tumor growth [80].

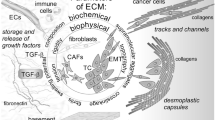
The ECM components implicated in cancer progression and metastasis. Cancer cells are directly exposed to the tumor microenvironment that contains adipocytes when cancer cells break through the basement membrane. Further, adipocytes that surrounded the tumor cells change into CAAs, being characterized by a reduced volume, a gain of an irregular shape with dispersed and small lipid droplets. An important role of these components is involved in sustaining cellular progression and metastasis of cancer cells. Abbreviations: ECM, extracellular matrix, CAAs, cancer-associated adipocytes
Hyaluronic acid has been shown to be heavily altered in the tumor ECM through the alteration of HA synthases, HA receptors, and HYAL-1 hyaluronidase, which lead not only to an altered expression of hyaluronic acid, but, also, to a different signalling pathway involving hyaluronic acid [99]. Moreover, hyaluronic acid production was proven to have an increased expression level in several types of cancer such as pancreatic carcinoma [100], breast cancer, colorectal cancer, prostate cancer and brain tumors [101], with the increased level of hyaluronic acid being associated with poor prognosis [102]. It has been identified that stromal cells, especially fibroblasts, are the main source of hyaluronic acid in the tumor. Added to this, it was found that hyaluronic acid is implicated in EMT induction and can also act as a migration substrate [103]. In the case of hyaluronic acid, it has been shown that HAPLN1, a protein having the role of linking proteoglycans to hyaluronic acid is overexpressed in metastatic melanoma [104].
The ECMs’ stromal components are involved in the regulation of epithelial-to-mesenchymal transition (EMT), invasion and metastasis
To provide a comprehensive understanding about critical biological processes, including EMT, metastasis, the key players involved in these processes have to be better understood. The presence of cancer-associated fibroblasts (CAF) in the TME influences ECM composition, further altering EMT and metastasis. Added to this, it has been shown that EMT is characterized by loss of cell adhesion, increased cell motility, inhibition of E-cadherin expression [105], increased metastasis [106] and chemoresistance [107, 108]. In cancer, EMT is linked with the acquisition of a more stem-cell-like character. In a study performed by Fisher et al., it has been shown that tumor cells resistant to cyclophosphamide acquire a mesenchymal character which promotes the expression of drug efflux pumps (ABCC1 and ABCB1) [109].During cancer progression, tumor cells are able to activate immune cells and fibroblasts which secrete different cytokines affecting cancer development and metastasis [110]. More important, it has been found that several cytokines regulate EMT. One of the most important EMT inducers is TGFβ which is secreted by tumor cells, platelets and CAFs. In prostate cancer, it was observed that TGFβ induces TWIST1 and SNAIL2 expression via IKKα and SMAD signalling [111,112,113]. The activation of the NFkB signalling pathway through TNFα induces the expression of the EMT transcription factors, including SNAIL2, TWIST1 and ZEB1/2 [114, 115]. Also, the activation of NFkB stabilizes SNAIL1 further stimulating cell migration and invasion. In several studies, it has been shown that IL6 increased the expression of TWIST1 and SNAIL1 and promoted EMT in breast and in head and neck cancer [116, 117]. Thus, the inflammatory cytokines from the TME can modulate the expression level and the protein stability of EMT transcription factors leading to the stimulation of EMT and invasion promotion [118].
In addition, tissue stiffness is an aspect that is directly involved in EMT. Increasing evidence shows that an increased stiffness of the surrounding ECM determines EMT by increasing nuclear localization of the transcription factors TWISTs [119], YAP and TAZ [120]. In a study performed by Fattet et al., it was shown that EPHA2/LYN/TWIST1 mechano-transduction pathway, which is associated with collagen fibre alignment, is involved in increasing the ECM stiffness during tumor progression and in stimulating EMT, cell invasion and metastasis [121]. Thus, an increased expression level of vimentin and a decreased level of E-cadherin was observed to be associated with an increased resistance of tumor cells to paclitaxel. In gastric cancer, it has been observed that the interaction occurring between hyaluronic acid and hyaluronan-mediated motility receptor (HMMR) is important in inducing 5-fluorouracil resistance [122]. Hyaluronic acid acts as a ligand for CD44 and plays an important role in EMT, causing an increased in metastasis and invasiveness [123, 124]. By co-culturing tumor cells with fibroblasts, the synthesis of hyaluronic acid is increased. More important, the inhibition of hyaluronic acid reduced the expression of cancer stem cell markers (CD90, CD133, EpCAM) [125].
Another critical aspect is represented by the loss of epithelial polarization in EMT. In a study conducted by Walter et al., it was observed that alterations of the basement membrane and of collagen IV deposition can trigger EMT [126]. The effects of collagen fibres also depend on the collagen type, with collagen IV being important in maintaining an epithelial phenotype, while collagen I being important in stimulating EMT [127]. Also, it was observed that TWIST1 is a direct regulator of collagen alpha1(VI) transcription [128], while, ZEB1 regulates collagen 1 component’s transcription and increases the expression of LOXL2 that is involved in collagen stabilization [129].
Thus, in the pancreatic cancer stellate cells, collagen production is improved through the association between mutated KRAS and EMT regulator SNAIL [49]. Transcription factors can determine an aberrant expression profile of certain transcripts, such as NFkB and STATs which participate in collagen expression and organization. In sarcomatous, COL2A1 is under the transcriptional control of the NFkB subunit p65 [130]. A typical component which belongs to serine/threonine kinase signal transduction, is represented by TGFβ/SMAD signalling involved in collagen modification. Cancer progression is promoted in melanoma cells through the crosstalk between TGFβ and the MAPK/ERK signalling pathway [131]. Added to this, FGFR (fibroblast growth factor receptor) regulates the degradation of COL1, COLII and COLIV by increasing the protein expression of MMP14 in prostate cancer cells [131, 132].
Cartilage oligomeric matrix protein (COMP) also represents a component of the ECM which binds to integrins on the cell surface via the RGD domain (Arg-Gly-Asp) [133]. COMP is a multifunctional protein being expressed in different cell types and tissues [134]. Specifically, it has been shown to promote cancer progression [135, 136], EMT [137] and resistance to therapy [138]. It has been shown in breast cancer epithelial cells, that COMP is significantly elevated being associated with poor survival, invasiveness, and metastasis [136]. In addition, the expression of COMP in breast cancer cells, mediates the interaction between Jagged1 and Notch3, leading to the alteration of the cancer stem cells in vitro and in vivo [139]. In prostate cancer cells, COMP is associated with cancer recurrence. Also, prostate cancer cells which present an increased expression level of COMP are protected against apoptosis and have an enhanced Warburg effect [140]. In colon cancer cells, the knockout of COMP inhibits cell proliferation, reducing 5-Fluorouracil resistance and ultimately induces apoptosis [141].
To create a suitable soil for the colonization of distant sites, metastatic cells are able to create conditions for attachment, survival and growth [26,27,28]. This microenvironment, is also called pre-metastatic niche [142] by involving ECM remodelling [143]. In the primary ECM, the collagen deposition is increased, but fibronectin, glycoproteins and proteoglycans are the key players in the formation of the pre-metastatic niche [144]. Exosomes secreted by primary tumors, activate stromal cells in the pre-metastatic niche in order to secrete new ECM molecules, or to remodel the ECM, by creating a fibrotic and pro-inflammatory environment. Also, there are several soluble factors which sustain the formation of the pre-metastasis niche. At the pre-metastatic niche, bone marrow-derived cells (BMDCs) contribute to ECM remodelling through integrins and pave the way for the arrival of disseminated tumor cells [145]. Neutrophil elastase and MMP9 cleave laminin, create a specific environment that can activate dormant tumor cells, while, circulating tumor cells reach the distant tissue through the disrupted vasculature [146]. Also, periostin and versican induce EMT and promote metastatic niche formation [147], collagen I, III and IV promote metastasis [148]; TGFβ is released during ECM degradation and fibronectin leads to the production of a pre-metastatic niche in the liver [147].
Several studies made on breast cancer, with Hsp47 having been shown to be an important regulator of ECM genes [87]. Other studies on breast cancer ECM have focused on the direct expression of ECM components by breast cancer cells. In invasive ductal breast carcinoma, the cancer cell expression of the protease inhibitor, CSTA and genes involved in adhesion, FAT1, DST, and TMEM45A, were shown to be involved in cancer invasiveness [149]. Further, collagen prolyl hydroxylases are implicated in breast cancer metastasis [150]. These effects have been shown to also extend in the direction of therapy resistance, with therapy-resistant MCF7 having been shown to present upregulated MMP1, MMP9, ADAM9 and TIMP3 genes [151]. More than in breast cancer, other malignancies have been shown to secrete ECM components. Pancreatic circulating tumor cells secrete ECM components, including SPARC, with the knock-out of this gene being associated with reduced invasiveness [152].
The ECMs’ stromal components are involved in the modulation of the response to therapy
ECM was shown to influence drug response (Table 3). It is known that drug distribution of most compounds in tumor tissues occurs mainly via diffusion. In this regard, solid tumors are characterized by the presence of a condensed ECM that significantly reduces drug transport [57]. Lysyl oxidase isoenzymes, including LOX and LOXL1-4, are responsible for the stabilization of collagen networks. Several studies highlighted the importance of this lysyl oxidase in cancer. In a study performed by Schutze et al., it has been shown that diffusion of doxorubicin in tumor spheroids is hampered by the overexpression of LOX and LOXL2. By using 2-aminopropionitril, the lysyl oxidase activity was inhibited and the effect was reversed [153]. Also, lysyl oxidases have a direct effect on VEGF-A expression through oxidation of PDGFR extracellular domain [154]. In tumor tissues, an increased LOXL2 facilitates endothelial invasion, which is a critical step in neo-angiogenesis, most probably through the increased motility of endothelial cells [155]. An in vivo study performed on murine pancreatic ductal adenocarcinoma (PDAC) models, has demonstrated that hyaluronidases reduce the hyaluronic acid content and increase the uptake of gemcitabine and doxorubicin [156]. The same result was observed in osteosarcoma, where, the uptake of liposomal doxorubicin could be improved with hyaluronidase treatment [157].
Cells present a direct interaction with the TME through various cell surface receptors (Fig. 3). These receptors inform the cells of any alterations occurring in the TME regarding ECM composition and stiffness, as well as responses that alter the cells sensitivity to drugs. Many studies showed the implication of these cell surface receptors. In small-cell lung cancer cells, the interaction between ECM and integrin β1 prevents G2/M arrest in response to chemotherapy or radiation by increasing the expression of p21 and p27 and downregulating the expression of cyclin A, B and E [158]. Another study showed that an upregulated expression of survival signals through ILK/Akt/NFkB determines the interaction between fibronectin and integrin β1 and protection against etoposide [158]. FAK signals are also involved in activating pro-survival pathways such as MAPK and AKT, which confer resistance to mTOR inhibition [161]. By silencing FAK in ovarian carcinoma it was observed that cells become sensitive to docetaxel [159], while, in colorectal carcinoma, cells acquired a sensitive phenotype to 5-fluorouracil [160].

The role of ECM in the TME. There is a bidirectional communication between the malignant clone and the TME associated cells, which ultimately leads them to increase their secretion of proteases, crosslinkers and soluble cytokines/chemokines. The altered ECM resulting from these changes in cell secretion subsequently has an impact in affecting the malignant clone and the TME. Specifically, it can be observed how the malignant clone can alter the properties of surrounding fibroblasts, transforming them in cancer associated fibroblasts, which, subsequently secrete VEGFA, with important proangiogenic properties. Another important cell that communicates with the malignant clone and has an influence on the ECM is represented by the tumor associated macrophage, which secretes key inflammatory cytokines which indirectly alter the ECM. Added to this, the malignant clone itself is able to secrete matrix metalloproteinases, which affect the structure of the surrounding ECM, making it more appropriate for tumor development
In tumors, the stiffened matrix is mainly involved in drug resistance by reducing it and presenting a poor therapeutic efficacy of chemo- and immune therapies [162]. In the first step, a physical barrier for drug infiltration is formed by the stiffened matrix. To confirm this aspect, it was shown that decreasing HA deposited in tumor tissue was associated with systemic accumulation of chemotherapy in patients with colorectal cancer [163]. Secondly, a stiff matrix has micro blood vessels causing difficulties for the drugs to reach the central core of the tumor. Third, hypoxia is induced by ECM stiffness [164]. Fourth, in this context, tumor cells are forced to transform to cancer stem cells (CSCs) with the ability to proliferate in a hypoxic environment. In this regard, several studies, revealed that CSCs are more resistant to chemotherapy [165, 166]. It was shown that β1 integrins have an elevated expression profile in several types of cancer, such as colorectal [167] and lung cancer [168]. More important, an increased profile of β1 integrins contribute to tumor cell survival in prostate, pancreatic, melanoma, glioblastoma and colorectal carcinoma after radiation [168,169,170].
Also, the response of matrix stiffness to drug resistance might be linked to the drug and cell type. There are some studies performed on cell lines which tried to highlight the importance of matrix stiffness. In one study Medina et al. showed that, using a matrix to mimic the stiffness of their host tumor, mammary cancer cells acquired an increased drug resistance potential [171]. As a major point of view, by changing the matrix stiffness, drug resistance could be modulated in an advantageous way. A study performed on breast cancer cells, revealed that a stiff 2D matrix induces resistance to sorafenib and lapatinib, while, drug resistance mechanism is reduced in a stiff 3D matrix [172]. To provide a better understanding regarding in vitro drug resistance, studies might focus on 3D culture models using different matrix stiffness for developing new therapeutic strategies [173]
A wide range of studies revealed that collagen might be used as a prognostic factor correlated with cancer invasion and differentiation, lymph node metastasis and clinical stages in cancer patients. It was shown that the content of COLI and COLIV is associated with proliferation, potentially representing future biomarkers [174, 175]. In addition, the expression level of COLI and COLIV can offer information about tumor angiogenesis and progression of glioblastoma [176]. In several types of cancer, including breast cancer, lung adenocarcinoma, squamous cell carcinoma, cervical cancer and neck squamous cell carcinoma, it was observed that hypomethylation of the COL17A1 promoter is associated with an advanced stage and sustained invasion of tumor cells [177].
Neo-epitope biomarkers showed promising results in characterizing localized pathological protein turnover in different diseases. Thus, an important biomarker for bone turnover is represented by carboxy-terminal collagen crosslinked (CTX-1) which is cleaved by cathepsin K[178]. The altered activity of MMP, ADAMTS, calpain and caspase proteases are involved in protein turnover changes in several pathologies. The changes of MMP-generated collagen VI are linked to muscle regrowth following immobilization [179]. The collagen I fragments which are generated by MMP2, 9, and 13 (C1M) [180] or the MMP9 proteolytically showed neo-epitope biomarkers of type III collagen (C3M) used as muscle protein turnover [181]. In a recent study, Groen et al. developed a neo-epitope biomarker for osteoarthritis, based on MMP1 and 13 generated fragment of type II collagen degradation (T2CM) [182]. A well-known marker, type X collagen (Col10) was developed as a new neo-epitope biomarker for radiographic knee osteoarthritis [183]. GPDPLQ1237 was developed as a promising biomarker for cartilage degradation in vitro [184] and PRO-C3 collagen for fibrogenesis [185].
Additionally, collagen plays an essential role in therapy resistance. In esophageal carcinoma, it was shown that an elevated level of collagen is correlated with chemotherapy resistance [186]. The increased content of collagen is associated with an elevated accumulation of hyaluronan, causing resistance to doxorubicin in pancreatic cancer [187]. In several studies, it was observed that COLI induces resistance to cisplatin and mitoxantrone in ER-positive cancer cells, by activating β1 integrin followed by the FAK/PI3K/AKT pathway, in triple negative cancer cells by MAPK pathway and in ovarian cancer through the co-expression of LOX with COL1A2 [188, 189]. The upregulation of COLI caused paclitaxel resistance in ovarian carcinoma [190], as well as COL11A1 by mediating the transcriptional activation of NFkB to increase the expression of Twist family [191]. Anoikis resistance is caused by an upregulated COLVI profile, affecting the response of colorectal cancer to adjuvant chemotherapy and of salivary gland cancer to radiotherapy [192, 193]. The response of cancer cells to antiangiogenic therapy is strongly associate with COLIV expression. Thus, the resistance to VEGF therapy through TGFβ signalling, is causing by the reduction of collagen to PDAC cell surface receptors [194]. In addition, collagen can be used in clinical applications as a therapy resistance biomarker, as a predictor of prognostic and recurrence, as a drug carrier and as a therapy target [49].
Cancer-associated fibroblasts
CAFs are one of the most dominant cell types in the TME and are found in all solid tumors [195]. CAFs mediate the onset of a pro-invasive TME through multiple epigenetic, transcriptomic or genetic alterations [196]. CAFs are different between each type of tumor, influence angiogenesis, tumour mechanics, drug access and therapy responses [197]. In this regard, normal fibroblasts, epithelial cells, endothelial cells [198], adipose tissue-mesenchymal stem cells, stellate cells, bone marrow-derived mesenchymal stem cells, as well as resident quiescent fibroblasts were found to be the cells of origin of CAFs [199, 200]. According to their origin, CAFs exhibit several differences in morphology, cell-to-cell interaction and expression profile [201]. CAFs are able to secrete proteases which increase their ability to migrate and remodel the ECM, as well as inflammatory molecules and growth factors [202].
In breast cancer, it has been observed that the connection between tumor cells and fibroblasts promotes the CAFs phonotype via Notch signalling [197, 203]. Several inflammatory modulators are able to activate CAFs through IL1 acting via NFkB and IL6 acting on STAT factors [197, 204]. Also, the contractile cytoskeleton and alterations in histone acetylation stimulate CAF activation through JAK-STAT signalling [196]. Physical changes occurring in the ECM also play an important role in CAFs activation [205, 206]. In this regard, the association between CTGF and CYR61, and contractile cytoskeleton have the role to increase tissue stiffness causing SRF-dependent and YAP1-dependent transcriptional programmes, which further lock CAFs into a self-sustaining positive-feedback loop [207]. The presence of HSP1 is also required for CAFs activation [208]. Alongside physical changes, physiological and genomic stress are also important for changes occurring in fibroblasts. In some cases, the production of IL6 and TGFβ caused a non-proliferative state of fibroblasts called senescence [209]. The senescent fibroblasts are a minor component of the TME [210] and their absence has substantial consequences for disease relapse [209]. In CAFs it was observed that YAP establishes and maintains tumor-promoting functions, including invasion, angiogenesis, and stromal sclerosis. An increased level of YAP is associated with positive feedback between mechanical conduction and CAF-driven matrix hardening [207]. Also, YAP is activated in CAFs in response to mechanical stress, ECM stiffness and alterations of the actin cytoskeleton [211].
Following therapy, cancer cell activities are affected through multiple signalling pathways, which alter TME components. In this regard, CAFs secrete various cytokines that activate signalling cascades to hinder the elimination of the cancer cells. Thus, CAFs contribute to drug resistance through several mechanisms, via PAI-1 which suppresses caspase-3 activation and activates Erk/Akt signalling [212]. Through the production of IL6, CAFs induce the expression of CXCR7 through the STAT3/NFkB pathway in tumor cells, which is responsible for drug resistance [213]. The co-culture of CAFs in conditioned media from gastric cancer cells showed an increased level of phosphorylated AKT, PI3K, p65, lkb and ABCB1, together with the activation of NFkB that increased the resistance to cisplatin in cancer cells [214]. Performing a cytokine antibody array on a multidrug-resistant breast cancer cell line, MCF-7/R, it was observed that IL6 and IL8 exhibited a significantly increased level of these cytokines, suggesting their implication in drug resistance [215]. Under hypoxic conditions, it was observed that CAFs secrete an increased level of TGFβ, causing stem cell-like properties and increased chemoresistance in colon cancer cell lines [216]. In order to counteract drug resistance mechanisms, CAFs are reprogramed to a quiescent phenotype by inhibiting NFkB signalling. In a study performed on ovarian cancer, it was demonstrated that inhibition of NFkB reversed the typical CAFs phenotype leading to an improved response to cisplatin in tumor cells [217]. Added to this, NFkB inhibition reduced the deposition of collagen in the TME after cisplatin therapy. It has been shown that WNT16B is regulated by NFkB, after the activation of DNA damage mechanisms, which might be induced by TNFα and radiotherapy. This mechanism targets the canonical Wnt pathway leading to drug resistance [218]. In pancreatic cancer, it was observed that CAFs protect cancer cells from cell death induced by gemcitabine in an NFkB-dependent manner. The apoptosis process was regulated by IL1β and IRAK4 where, by knocking down IRAK4 or inhibiting IL1β, drug resistance is increased, but fibrosis is decreased [219]. Due to the fact that CAFs express vitamin D receptor, using calcipotriol, CAFs were converted to stellate cells causing an increased concentration of gemcitabine in the tumor site improving the response to therapy [220]. In breast cancer, it was observed that collagen density decreased the number of CD8 + tumor-infiltrating T cells (TILs) and modulated T-cell cytotoxic activity [221]. In the same malignancy, using an in vivo model, it has been found that CAFs reprogramming is mediated through hedgehog ligand, leading to stemness and drug resistance through the expression of FGF5 and production of fibrillar collagen [222]. Smoothened inhibitors (SMOi) can reduce the expression of cancer stem cell markers and sensitize cells to taxanes by reducing metastasis, thus improving the survival rate [222]. In addition, it has been observed that CAFs regulate EMT and promote chemoresistance by secreting IL6 and HGF [223]. In non-small cell lung cancer (NSCLC), CAFs increase the expression of TGFβ1 by secreting IL6, further increasing the resistance to cisplatin [224]. A study performed by Ying et al., showed that an increased expression of HGF secreted by CAFs enhanced the activation of PI3K/AKT pathway and GRP78, causing resistance in NSCLC to paclitaxel. Also, CAFs induce EMT in NSCLC causing resistance to chemotherapy [225].
Another important factor that sustains drug resistance is the maintenance of CSCs. In colorectal cancer, drug resistance is promoted in cancer cells through IL17A secreted from CAFs [226]. It was found that Wnt signalling is critical for establishing drug resistance during cancer progression, but exosomal Wnt from CAFs are able to induce a drug resistant phenotype to cancer cells [227]. The microvesicles secreted from CAFs transfer miR-221 to breast cancer cells that activate an ERlow/Notchhigh feed-forward loop responsible for CD133high CSCs generation. Drug resistance was observed to occur when using patient-derived xenograft (PDX) models [228]. The presence of non-coding RNA H19 from CAFs promotes cancer stemness and drug resistance in colorectal cancer through the activation of the β-catenin pathway [229]. In colorectal cancer, it was shown that hypoxic TME stimulates the stemness and resistance of CSCs to chemotherapy. Thus, a hypoxic TME can induce the aggregation of HIF-1α and TGFβ2 secreted from CAFs, as well as promoting the expression of GLI2 in CSCs, which are responsible of enhancing stemness and therapy resistance in CSCs. An increased expression of HIF-1α/TGFβ2/GLI2 was shown to be related to the risk of recurrence in patients undergoing chemotherapy [216]. Specifically, it was found that the TME and CAFs are the major concern for drug resistance development in tumors. In colorectal cancer, cetuximab can induce CAFs to secrete a large amount of EGF which mediates MAPK signal transduction, ultimately leading to cetuximab resistance [230]. Patients diagnosed with pancreatic cancer which exhibited an increased expression on miR-21 are more likely to be resistant to gemcitabine therapy. An elevated expression of miR-21 promotes the activation of CAFs leading to an increased resistance to gemcitabine therapy, while a downregulation of miR-21 expression is associated with an improved efficacy of gemcitabine therapy [231].
Cancer-associated adipocytes
Cancer-associated adipocytes (CAA) are important players of the TME being involved in tumor progression, angiogenesis, invasion, metastasis, and drug resistance through the secretion of adipokines such as leptin, adiponectin, CCL2, CCL5 and IL6 [232, 233]. An increased expression of leptin which was found in CAAs can stimulate cell proliferation and angiogenesis by increasing the expression of cyclin D1 and VEGF/VEGFR, upregulation of lysyl hydroxylase enzyme, as well as the activation of several signalling pathways such as ER, JAK/STAT and PI3K/AKT [234, 235]. In addition, it was observed that adiponectin is downregulated in CAAs. It is known that adiponectin has an anti-tumorigenic role by suppressing several biological processes, including growth, and the invasion process of tumor cells [232]. Another study showed that leptin has the ability to activate the focal adhesion kinase, increase metalloproteinase (MMP2 and MMP9) secretion needed for the ECM remodelling and cell migration, and diminish cell-to-cell adhesion [236]. Also, IL6 is secreted by CAAs and was observed to promote cancer progression when adipocytes were co-cultured with breast cancer cells [237]. Interestingly, some have shown that the increased expression of MMP2 and MMP9 might inhibit breast cancer invasion and metastasis through emodin by down-regulating CCL5 in adipocytes [225]. Collagen IV is essential in defining the ECM environment for normal and tumor cells and contributes significantly to tumor growth [238]. In a study performed by Park et al., the effect of collagen IV-derived endotrophin was investigated, which showed a strong effect on growth, angiogenesis, and tissue fibrosis in breast cancer. Also, endotrophin promotes EMT by enhancing TGFβ signalling, causing aggressive and high metastatic breast cancer growth [239]. Therefore, the production of IL6 regulates cell survival, immune suppression and drug resistance through the activation of JAK/STAT [240].
Another study has shown that the protein level of MMP3 in lung tumors is higher in obese patients compared to non-obese patients. Thus, adipocyte-derived exosomes increase the MMP3 expression in lung cancer cells. At the same time, in lung cancer cells, MMP3 increases the activity of MMP9, causing the invasion of cancer cells in vitro and in vivo [241]. An increased expression profile of miR-21 in CAAs was also observed. MiR-21 is transferred via exosomes from CAAs to cancer cells. In this aspect, exosomal miR-21 inhibits apoptosis in ovarian cancer cells and promotes drug resistance by combining with APAF1 [242].
Endothelial cells
Endothelial cells interact with the ECM and offer the support for angiogenesis. The role of angiogenesis is to assure nutrients and to remove waste, both required for tumor growth. The key activator of endothelial cells is VEGF. This molecule stimulates endothelial cell migration into a normal tissue, and initiates several processes such as cell division and self-organization into vessels [243]. Meanwhile, in tumor tissues, these cells are leaky and are able to form a denser network [244]. At the tumor site, it is known that the ECM is altered, suggesting that endothelial cells present an altered sprouting angiogenesis and cell-to-cell adhesion formation [245]. By controlling the physiological properties of the ECM such as density and mechanics, it is possible to modulate the vascular network [246]. When subjected to an ECM with increased stiffness, tumor-associated endothelial cells generate vessels in an irregular manner, fact caused by their inability to orient themselves [247]. Therefore, the elasticity property of the ECM modulates cell proliferation and angiogenesis by controlling the level of transcription factor that regulates the expression of VEGFR2 [248].
Glycocalyx (GCX) is represented by a mixture of carbohydrates attached to proteins, which have the ability to regulate the access of cells and bioactive molecules to the endothelium in the blood stream. Therefore, CGX modulates essential processes such as microvascular homeostasis and pathogenesis [249]. Recently, the attention was focused on the endothelial GCX (eGCX) which might serve as a key factor in disease progression [249]. The eGCX marks the luminal surface of the vascular endothelium and interacts with plasma proteins and lipids. Being an important component of the vascular wall, the eGCX modulates vascular homeostasis and hydraulic conductivity [250], maintains the endothelial barrier function by acting as a molecular filter for the endothelium. The eGCX modulates the molecular binding to the endothelial cells surface, according to the high density of anionic charges on its GAGs side chains. Sulfate residues carry the negative charge of eGCX, favouring the docking of positively charged molecules [251]. Specifically, because endothelial GCX can influence endothelial cells stability, it can also be involved in angiogenesis and influence therapy response [249, 252].
The effects of ECM on immunotherapy
Immunotherapy started to gain attention in the oncology field by using several therapeutic approaches based on patient’s immune system against cancer. In the present, there are several clinical trials based on the ECM in order to improve the response to therapy, and not only related to the functioning of immune cells (Table S1). Checkpoint inhibitors are widely used in the field of oncology, with good results. Nonetheless, tumor cells can express checkpoint inhibitors, like PD-L1, and inhibit T-cell activity. The most well-characterized interaction in cancer immunotherapy is between CD80/CD86-CTLA4 and PDL1-PD1 [253, 254]. An increased number of evidences showed that several types of cancer, including melanoma and NSCLC, present a high rate of response to immunotherapy and checkpoint inhibitors [255, 256]. In some cancer patients, the application of immune therapy is impeded due to the low T-cell infiltrate. Added to this, the infiltration rate is also blocked by the ECM which acts as a protective shield. Thus, an increased density of the tumor ECM is responsible for the distribution of immunomodulatory drugs, as well as the infiltration of immune cells into the tumor [257]. A poor diffusion in tumor ECM causes increased hypoxia and metabolic stress, which determines an upregulation of immunosuppressive factors, including CCL18, CCL22, IL10, TGFβ, and prostaglandin E2 and VEGFA [257]. It is known that TGFβ in the TME acts as a suppressor of infiltrating CD8+ cytotoxic lymphocytes (CTL) and natural killer (NK) cells [258]. The aim of VEGFA in the TME is to recruit regulatory T cells (Tregs) that express a VEGFA co-receptor, called NRP1, further suppressing the activation of CTLs [259]. In a study performed by Salmon et al. it was observed that, in lung cancer, the migration and distribution of T cells is controlled by aligned collagen fibers that surround the tumor site, as well as perivascular regions in the tumor stroma. Thus, T cells are trapped in the stroma area without being able to reach the cells targeted for destruction. Nonetheless, when using collagenase treatment, an increased T cell infiltration was observed [260]. Another study performed on pancreatic ductal adenocarcinomas, showed that the dense architecture formed by hyaluronan acts in a similar manner as collagen, by preventing infiltration of effector immune cells [156]. A hypoxic TME, caused by a dense architecture of ECM, induces an increased expression of VEGF. The CTL infiltration is reduced and cannot attach on the cell surface proteins of endothelial cells, due to the fact that endothelial cells reduce the expression of cell surface glycoproteins in these conditions, including selectins and adhesion molecules (VCAM1, ICAM1, ICAM2), which are needed for T cell homing [261, 262]. Another study showed that VEGFA promotes immune escape by inducing Treg and PD1 expression on CD8+T cells expressing VEGFR2 [263]. Added to this, CAFs release various chemokines which modulate the immune landscape within the TME [264].
The ECM components are able to control the differentiation process of the Treg cells. For example, osteopontin inhibit Treg differentiation through CD44, hyaluronan stimulates the production of IL10, which, is in turn responsible of Treg stimulation [265]. Moreover, it has been shown that the ECM rigidity strongly affects the activation, proliferation, and differentiation of T cells [266]. In a study performed on pancreatic cancer, it has been shown that, by reducing the tumor tissue stiffness and total collagen, myofibroblast depletion caused an extensive remodelling of the tumor ECM. Thus, myofibroblast depletion is linked with Teff/ Treg reduction, causing suppression of immune surveillance. In myofibroblasts depleted mouse tumors, it was observed that suppressed immune surveillance is associated with the stimulation of Tregs [77].
Immune and inflammatory responses can be modulated by metalloproteinases through degradation of the ECM. It is known that metalloproteinases are responsible for the release of immunoactive factors from the cells surface. For example, MMP9, ADAM10 and ADAM17 are involved in MICA shedding from tumor cells [267]. In several malignancies, including prostate and breast cancer, as well as osteosarcoma, an elevated expression profile of metalloproteinase allows the tumor cells to escape immune surveillance [268, 269]. Degradation of collagen I by MMP8 and MMP9 produces the acetylated proline-glycine-proline tripeptide that mimics CXCL8 thus recruiting neutrophils [270]. In chronically inflamed lung, it was found that peptides resulting from elastin digestion by MMP12 act as chemoattractants for monocytes [271]. Fibronectin, tenascin and hyaluronan bind TLR2/4 promoting an inflammatory response [57].
In the TME, the most frequent immune cells are tumor-associated macrophages (TAMs), which mediate adaptative immune responses in cancer. The ECM components are involved in TAM polarization. Thus, it was observed that collagen and hyaluronic acid cause M2 polarization, while fibronectin skews macrophages to an M1 polarization, enhancing the cytotoxic activity of macrophages towards tumor cells [57]. Also, it was found that NK cells are supressed by transmembrane collagens such as Collagen XVII [272].
Although there have been various approaches to targeting ECM in cancer, there still has not been the breakthrough that scientists and clinicians have been looking for. There is no one correct answer for this fact, but there are some hypotheses that have been launched in the community. Firstly, targeting ECM alone might lead to the development of compensatory mechanisms that would offer the tumor another way to thrive [57]. Secondly, because the ECM is composed of a myriad of molecules and because it differs between different tissues, it is rather difficult to choose the optimal target [57].
Conclusions
The ECM is strongly involved in determining the TME architecture and the different hallmarks of cancer acquired by the malignant cells. Due to the fact that the ECM is a complex mixture of different macromolecules further studies are still needed to better investigate the effect of all ECM components and the interactions between them and the malignant clone. Further, other components are also present in the TME besides the malignant clone and the ECM, represented by the associated cells. These are not only able to influence the malignant clone, but to also have a bidirectional communication with the ECM. Dysregulation of ECM components sustain the design of a favourable environmental niche for tumor cells. Beside critical biological processes such as tumor growth and proliferation, invasion and migration, apoptosis, and metastasis, TME is a key player in the drug resistance of cancer cells. To provide a comprehensive understanding regarding TME modulation of drug resistance activation in different cancer types, advanced researches are needed to identify critical signalling pathways. The ECM components are critical for tumor sustainability and progression. Thus, the better understanding of these can not only lead to more insights into the biology of tumors but can also lead to more therapeutic options for various cancers.
Availability of data and materials
Not applicable.
References
Sontheimer-Phelps A, Hassell BA, Ingber DE. Modelling cancer in microfluidic human organs-on-chips. Nat Rev Cancer. 2019;19(2):65–81.
Jurj A, Zanoaga O, Braicu C, Lazar V, Tomuleasa C, Irimie A, Berindan-Neagoe I. A Comprehensive Picture of Extracellular Vesicles and Their Contents. Molecular Transfer to Cancer Cells. Cancers (Basel). 2020;12(2):298.
Braicu C, Tomuleasa C, Monroig P, Cucuianu A, Berindan-Neagoe I, Calin GA. Exosomes as divine messengers: are they the Hermes of modern molecular oncology? Cell Death Differ. 2015;22(1):34–45.
Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA, Hynes RO. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016;49:10–24.
Hastings JF, Skhinas JN, Fey D, Croucher DR, Cox TR. The extracellular matrix as a key regulator of intracellular signalling networks. Br J Pharmacol. 2019;176(1):82–92.
Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol. 2014;15(12):802–12.
Huang J, Zhang L, Wan D, Zhou L, Zheng S, Lin S, Qiao Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct Target Ther. 2021;6(1):153.
Mouw JK, Ou G, Weaver VM. Extracellular matrix assembly: a multiscale deconstruction. Nat Rev Mol Cell Biol. 2014;15(12):771–85.
Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol. 2010;22(5):697–706.
Pozzi A, Yurchenco PD, Iozzo RV. The nature and biology of basement membranes. Matrix Biol. 2017;57–58:1–11.
Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67.
Walma DAC, Yamada KM. The extracellular matrix in development. Development. 2020;147(10):dev175596.
Karamanos NK, Theocharis AD, Piperigkou Z, Manou D, Passi A, Skandalis SS, Vynios DH, Orian-Rousseau V, Ricard-Blum S, Schmelzer CEH, et al. A guide to the composition and functions of the extracellular matrix. Febs j. 2021;288(24):6850–912.
Domogatskaya A, Rodin S, Tryggvason K. Functional diversity of laminins. Annu Rev Cell Dev Biol. 2012;28:523–53.
Hayes AJ, Farrugia BL, Biose IJ, Bix GJ, Melrose J. Perlecan, A Multi-Functional, Cell-Instructive, Matrix-Stabilizing Proteoglycan With Roles in Tissue Development Has Relevance to Connective Tissue Repair and Regeneration. Front Cell Dev Biol. 2022;10:856261.
Sekiguchi R, Yamada KM. Basement Membranes in Development and Disease. Curr Top Dev Biol. 2018;130:143–91.
Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular matrix structure. Adv Drug Deliv Rev. 2016;97:4–27.
Hu M, Ling Z, Ren X. Extracellular matrix dynamics: tracking in biological systems and their implications. J Biol Eng. 2022;16(1):13.
Ozsvar J, Yang C, Cain SA, Baldock C, Tarakanova A, Weiss AS. Tropoelastin and Elastin Assembly. Front Bioeng Biotechnol. 2021;9:643110.
Hopf M, Göhring W, Mann K, Timpl R. Mapping of binding sites for nidogens, fibulin-2, fibronectin and heparin to different IG modules of perlecan11Edited by M. F Moody. J Mol Biol. 2001;311(3):529–41.
Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115(Pt 20):3861–3.
Mythreye K, Blobe GC. Proteoglycan signaling co-receptors: roles in cell adhesion, migration and invasion. Cell Signal. 2009;21(11):1548–58.
Zhu J, Clark RAF. Fibronectin at select sites binds multiple growth factors and enhances their activity: expansion of the collaborative ECM-GF paradigm. J Invest Dermatol. 2014;134(4):895–901.
Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. 2020;11(1):5120.
Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol. 2004;16(5):558–64.
Chiarugi P, Giannoni E. Anoikis: a necessary death program for anchorage-dependent cells. Biochem Pharmacol. 2008;76(11):1352–64.
Shibue T, Weinberg RA. Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci U S A. 2009;106(25):10290–5.
Cox TR, Bird D, Baker AM, Barker HE, Ho MW, Lang G, Erler JT. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013;73(6):1721–32.
Henke E, Nandigama R, Ergün S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front Mol Biosci. 2020;6:160.
Kirkland SC. Type I collagen inhibits differentiation and promotes a stem cell-like phenotype in human colorectal carcinoma cells. Br J Cancer. 2009;101(2):320–6.
Medici D, Nawshad A. Type I collagen promotes epithelial-mesenchymal transition through ILK-dependent activation of NF-kappaB and LEF-1. Matrix Biol. 2010;29(3):161–5.
Januchowski R, Świerczewska M, Sterzyńska K, Wojtowicz K, Nowicki M, Zabel M. Increased Expression of Several Collagen Genes is Associated with Drug Resistance in Ovarian Cancer Cell Lines. J Cancer. 2016;7(10):1295–310.
Lim YC, Oh SY, Kim H. Cellular characteristics of head and neck cancer stem cells in type IV collagen-coated adherent cultures. Exp Cell Res. 2012;318(10):1104–11.
Sun B, Huang Z, Wang B, Yu Y, Lin S, Luo L, Wang Y, Huang Z. Significance of Glypican-3 (GPC3) Expression in Hepatocellular Cancer Diagnosis. Med Sci Monit. 2017;23:850–5.
Wang X, Zuo D, Chen Y, Li W, Liu R, He Y, Ren L, Zhou L, Deng T, Wang X, et al. Shed Syndecan-1 is involved in chemotherapy resistance via the EGFR pathway in colorectal cancer. Br J Cancer. 2014;111(10):1965–76.
Du WW, Fang L, Yang X, Sheng W, Yang BL, Seth A, Zhang Y, Yang BB, Yee AJ. The role of versican in modulating breast cancer cell self-renewal. Mol Cancer Res. 2013;11(5):443–55.
Chanmee T, Ontong P, Mochizuki N, Kongtawelert P, Konno K, Itano N. Excessive hyaluronan production promotes acquisition of cancer stem cell signatures through the coordinated regulation of Twist and the transforming growth factor β (TGF-β)-Snail signaling axis. J Biol Chem. 2014;289(38):26038–56.
Domogatskaya A, Rodin S, Boutaud A, Tryggvason K. Laminin-511 but not -332, -111, or -411 enables mouse embryonic stem cell self-renewal in vitro. Stem Cells. 2008;26(11):2800–9.
Li CL, Yang D, Cao X, Wang F, Hong DY, Wang J, Shen XC, Chen Y. Fibronectin induces epithelial-mesenchymal transition in human breast cancer MCF-7 cells via activation of calpain. Oncol Lett. 2017;13(5):3889–95.
Soteriou D, Iskender B, Byron A, Humphries JD, Borg-Bartolo S, Haddock MC, Baxter MA, Knight D, Humphries MJ, Kimber SJ. Comparative proteomic analysis of supportive and unsupportive extracellular matrix substrates for human embryonic stem cell maintenance. J Biol Chem. 2013;288(26):18716–31.
Pupa SM, Giuffré S, Castiglioni F, Bertola L, Cantú M, Bongarzone I, Baldassari P, Mortarini R, Argraves WS, Anichini A, et al. Regulation of breast cancer response to chemotherapy by fibulin-1. Cancer Res. 2007;67(9):4271–7.
Liu J, Tan Y, Zhang H, Zhang Y, Xu P, Chen J, Poh YC, Tang K, Wang N, Huang B. Soft fibrin gels promote selection and growth of tumorigenic cells. Nat Mater. 2012;11(8):734–41.
Braam SR, Zeinstra L, Litjens S, Ward-van Oostwaard D, van den Brink S, van Laake L, Lebrin F, Kats P, Hochstenbach R, Passier R, et al. Recombinant vitronectin is a functionally defined substrate that supports human embryonic stem cell self-renewal via alphavbeta5 integrin. Stem Cells. 2008;26(9):2257–65.
Zhou Y, Zhu Y, Fan X, Zhang C, Wang Y, Zhang L, Zhang H, Wen T, Zhang K, Huo X, et al. NID1, a new regulator of EMT required for metastasis and chemoresistance of ovarian cancer cells. Oncotarget. 2017;8(20):33110–21.
Das S, Rachagani S, Torres-Gonzalez MP, Lakshmanan I, Majhi PD, Smith LM, Wagner KU, Batra SK. Carboxyl-terminal domain of MUC16 imparts tumorigenic and metastatic functions through nuclear translocation of JAK2 to pancreatic cancer cells. Oncotarget. 2015;6(8):5772–87.
Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123(Pt 24):4195–200.
Nallanthighal S, Heiserman JP, Cheon DJ. The Role of the Extracellular Matrix in Cancer Stemness. Front Cell Dev Biol. 2019;7:86.
Gu L, Shan T, Ma YX, Tay FR, Niu L. Novel Biomedical Applications of Crosslinked Collagen. Trends Biotechnol. 2019;37(5):464–91.
Xu S, Xu H, Wang W, Li S, Li H, Li T, Zhang W, Yu X, Liu L. The role of collagen in cancer: from bench to bedside. J Transl Med. 2019;17(1):309.
Suhovskih AV, Mostovich LA, Kunin IS, Boboev MM, Nepomnyashchikh GI, Aidagulova SV, Grigorieva EV. Proteoglycan expression in normal human prostate tissue and prostate cancer. ISRN Oncol. 2013;2013:680136.
Theocharis AD, Vynios DH, Papageorgakopoulou N, Skandalis SS, Theocharis DA. Altered content composition and structure of glycosaminoglycans and proteoglycans in gastric carcinoma. Int J Biochem Cell Biol. 2003;35(3):376–90.
Vynios DH, Theocharis DA, Papageorgakopoulou N, Papadas TA, Mastronikolis NS, Goumas PD, Stylianou M, Skandalis SS. Biochemical changes of extracellular proteoglycans in squamous cell laryngeal carcinoma. Connect Tissue Res. 2008;49(3):239–43.
Gambichler T, Kreuter A, Grothe S, Altmeyer P, Brockmeyer NH, Rotterdam S. Versican overexpression in cutaneous malignant melanoma. Eur J Med Res. 2008;13(11):500–4.
Passi A, Vigetti D, Buraschi S, Iozzo RV. Dissecting the role of hyaluronan synthases in the tumor microenvironment. Febs j. 2019;286(15):2937–49.
Schulz T, Schumacher U, Prehm P. Hyaluronan export by the ABC transporter MRP5 and its modulation by intracellular cGMP. J Biol Chem. 2007;282(29):20999–1004.
Qiu X, Tan H, Fu D, Zhu Y, Zhang J. Laminin is over expressed in breast cancer and facilitate cancer cell metastasis. J Cancer Res Ther. 2018;14(Supplement):S1170-s1172.
Henke E, Nandigama R, Ergün S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front Mol Biosci. 2019;6:160.
Lu X, Watsky MA. Influence of Vitamin D on Corneal Epithelial Cell Desmosomes and Hemidesmosomes. Invest Ophthalmol Vis Sci. 2019;60(13):4074–83.
Turcan I, Jonkman MF. Structure of Hemidesmosomes and the Epidermal Basement Membrane Zone. In: Jonkman MF, editor. Autoimmune Bullous Diseases: Text and Review. Cham: Springer International Publishing; 2016. p. 113–7.
Schreurs O, Balta MG, Karatsaidis A, Schenck K. Composition of hemidesmosomes in basal keratinocytes of normal buccal mucosa and oral lichen planus. Eur J Oral Sci. 2020;128(5):369–78.
Roig-Rosello E, Rousselle P. The Human Epidermal Basement Membrane: A Shaped and Cell Instructive Platform That Aging Slowly Alters. Biomolecules. 2020;10(12):1607.
Laval S, Laklai H, Fanjul M, Pucelle M, Laurell H, Billon-Galés A, Le Guellec S, Delisle MB, Sonnenberg A, Susini C, et al. Dual roles of hemidesmosomal proteins in the pancreatic epithelium: the phosphoinositide 3-kinase decides. Oncogene. 2014;33(15):1934–44.
De Arcangelis A, Hamade H, Alpy F, Normand S, Bruyère E, Lefebvre O, Méchine-Neuville A, Siebert S, Pfister V, Lepage P, et al. Hemidesmosome integrity protects the colon against colitis and colorectal cancer. Gut. 2017;66(10):1748–60.
Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906.
Geiger B, Yamada KM. Molecular architecture and function of matrix adhesions. Cold Spring Harb Perspect Biol. 2011;3(5):a005033.
Fang M, Yuan J, Peng C, Li Y. Collagen as a double-edged sword in tumor progression. Tumour Biol. 2014;35(4):2871–82.
Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4(1):38.
Malik R, Lelkes PI, Cukierman E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol. 2015;33(4):230–6.
Micke P, Strell C, Mattsson J, Martín-Bernabé A, Brunnström H, Huvila J, Sund M, Wärnberg F, Ponten F, Glimelius B, et al. The prognostic impact of the tumour stroma fraction: A machine learning-based analysis in 16 human solid tumour types. EBioMedicine. 2021;65:103269.
Brassart-Pasco S, Brézillon S, Brassart B, Ramont L, Oudart JB, Monboisse JC. Tumor Microenvironment: Extracellular Matrix Alterations Influence Tumor Progression. Front Oncol. 2020;10:397.
Liang Y, Lv Z, Huang G, Qin J, Li H, Nong F, Wen B. Prognostic significance of abnormal matrix collagen remodeling in colorectal cancer based on histologic and bioinformatics analysis. Oncol Rep. 2020;44(4):1671–85.
Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15(12):786–801.
Paul CD, Hruska A, Staunton JR, Burr HA, Daly KM, Kim J, Jiang N, Tanner K. Probing cellular response to topography in three dimensions. Biomaterials. 2019;197:101–18.
Hua H, Li M, Luo T, Yin Y, Jiang Y. Matrix metalloproteinases in tumorigenesis: an evolving paradigm. Cell Mol Life Sci. 2011;68(23):3853–68.
Tjin G, White ES, Faiz A, Sicard D, Tschumperlin DJ, Mahar A, Kable EPW, Burgess JK. Lysyl oxidases regulate fibrillar collagen remodelling in idiopathic pulmonary fibrosis. Dis Model Mech. 2017;10(11):1301–12.
Karagiannis GS, Poutahidis T, Erdman SE, Kirsch R, Riddell RH, Diamandis EP. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol Cancer Res. 2012;10(11):1403–18.
Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–34.
Arnold SA, Rivera LB, Miller AF, Carbon JG, Dineen SP, Xie Y, Castrillon DH, Sage EH, Puolakkainen P, Bradshaw AD, et al. Lack of host SPARC enhances vascular function and tumor spread in an orthotopic murine model of pancreatic carcinoma. Dis Model Mech. 2010;3(1–2):57–72.
Xiao Q, Ge G. Lysyl oxidase, extracellular matrix remodeling and cancer metastasis. Cancer Microenviron. 2012;5(3):261–73.
Jinka R, Kapoor R, Sistla PG, Raj TA, Pande G. Alterations in Cell-Extracellular Matrix Interactions during Progression of Cancers. Int J Cell Biol. 2012;2012:219196.
Liu Z, Lai J, Jiang H, Ma C, Huang H. Collagen XI alpha 1 chain, a potential therapeutic target for cancer. FASEB J. 2021;35(6):e21603.
Magnon C, Galaup A, Mullan B, Rouffiac V, Bouquet C, Bidart JM, Griscelli F, Opolon P, Perricaudet M. Canstatin acts on endothelial and tumor cells via mitochondrial damage initiated through interaction with alphavbeta3 and alphavbeta5 integrins. Cancer Res. 2005;65(10):4353–61.
Lambert E, Fuselier E, Ramont L, Brassart B, Dukic S, Oudart JB, Dupont-Deshorgue A, Sellier C, Machado C, Dauchez M, et al. Conformation-dependent binding of a Tetrastatin peptide to α(v)β(3) integrin decreases melanoma progression through FAK/PI(3)K/Akt pathway inhibition. Sci Rep. 2018;8(1):9837.
Wong CC, Tse AP, Huang YP, Zhu YT, Chiu DK, Lai RK, Au SL, Kai AK, Lee JM, Wei LL, et al. Lysyl oxidase-like 2 is critical to tumor microenvironment and metastatic niche formation in hepatocellular carcinoma. Hepatology. 2014;60(5):1645–58.
Moreno-Bueno G, Salvador F, Martín A, Floristán A, Cuevas EP, Santos V, Montes A, Morales S, Castilla MA, Rojo-Sebastián A, et al. Lysyl oxidase-like 2 (LOXL2), a new regulator of cell polarity required for metastatic dissemination of basal-like breast carcinomas. EMBO Mol Med. 2011;3(9):528–44.
Wörmann SM, Song L, Ai J, Diakopoulos KN, Kurkowski MU, Görgülü K, Ruess D, Campbell A, Doglioni C, Jodrell D, et al. Loss of P53 Function Activates JAK2-STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated With Patient Survival. Gastroenterology. 2016;151(1):180-193.e112.
Zhu J, Xiong G, Fu H, Evers BM, Zhou BP, Xu R. Chaperone Hsp47 Drives Malignant Growth and Invasion by Modulating an ECM Gene Network. Cancer Res. 2015;75(8):1580–91.
Januchowski R, Zawierucha P, Ruciński M, Nowicki M, Zabel M. Extracellular Matrix Proteins Expression Profiling in Chemoresistant Variants of the A2780 Ovarian Cancer Cell Line. Biomed Res Int. 2014;2014:365867.
Teodoro JG, Parker AE, Zhu X, Green MR. p53-mediated inhibition of angiogenesis through up-regulation of a collagen prolyl hydroxylase. Science. 2006;313(5789):968–71.
Assadian S, El-Assaad W, Wang XQ, Gannon PO, Barrès V, Latour M, Mes-Masson AM, Saad F, Sado Y, Dostie J, et al. p53 inhibits angiogenesis by inducing the production of Arresten. Cancer Res. 2012;72(5):1270–9.
da Silva PHR, Borges BC, Uehara IA, Soldi LR, de Araújo RA, Silva MJB. Chemokines and the extracellular matrix: Set of targets for tumor development and treatment. Cytokine. 2021;144:155548.
Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8(3):241–54.
Paavolainen O, Peuhu E. Integrin-mediated adhesion and mechanosensing in the mammary gland. Semin Cell Dev Biol. 2021;114:113–25.
Martinez J, Smith PC. The Dynamic Interaction between Extracellular Matrix Remodeling and Breast Tumor Progression. Cells. 2021;10(5):1046.
Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, Hill RC, Lakins JN, Schlaepfer DD, Mouw JK, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med. 2016;22(5):497–505.
Ciocan-Cȃrtiţă CA, Jurj A, Raduly L, Cojocneanu R, Moldovan A, Pileczki V, Pop LA, Budişan L, Braicu C, Korban SS, et al. New perspectives in triple-negative breast cancer therapy based on treatments with TGFβ1 siRNA and doxorubicin. Mol Cell Biochem. 2020;475(1–2):285–99.
Colak S, Ten Dijke P. Targeting TGF-β Signaling in Cancer. Trends Cancer. 2017;3(1):56–71.
Naba A, Clauser KR, Lamar JM, Carr SA, Hynes RO. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. Elife. 2014;3:e01308.
Lokeshwar VB, Mirza S, Jordan A. Targeting hyaluronic acid family for cancer chemoprevention and therapy. Adv Cancer Res. 2014;123:35–65.
Cheng XB, Sato N, Kohi S, Yamaguchi K. Prognostic impact of hyaluronan and its regulators in pancreatic ductal adenocarcinoma. PLoS ONE. 2013;8(11):e80765.
Jadin L, Pastorino S, Symons R, Nomura N, Jiang P, Juarez T, Makale M, Kesari S. Hyaluronan expression in primary and secondary brain tumors. Ann Transl Med. 2015;3(6):80.
Josefsson A, Adamo H, Hammarsten P, Granfors T, Stattin P, Egevad L, Laurent AE, Wikström P, Bergh A. Prostate cancer increases hyaluronan in surrounding nonmalignant stroma, and this response is associated with tumor growth and an unfavorable outcome. Am J Pathol. 2011;179(4):1961–8.
McAtee CO, Barycki JJ, Simpson MA. Emerging roles for hyaluronidase in cancer metastasis and therapy. Adv Cancer Res. 2014;123:1–34.
Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics. 2012;11(4):M111.014647.
Cichon MA, Degnim AC, Visscher DW, Radisky DC. Microenvironmental influences that drive progression from benign breast disease to invasive breast cancer. J Mammary Gland Biol Neoplasia. 2010;15(4):389–97.
Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, Massagué J, Benezra R. TGF-β-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 2013;5(5):1228–42.
Li H, Li J, Chen L, Qi S, Yu S, Weng Z, Hu Z, Zhou Q, Xin Z, Shi L, et al. HERC3-Mediated SMAD7 Ubiquitination Degradation Promotes Autophagy-Induced EMT and Chemoresistance in Glioblastoma. Clin Cancer Res. 2019;25(12):3602–16.
Li N, Babaei-Jadidi R, Lorenzi F, Spencer-Dene B, Clarke P, Domingo E, Tulchinsky E, Vries RGJ, Kerr D, Pan Y, et al. An FBXW7-ZEB2 axis links EMT and tumour microenvironment to promote colorectal cancer stem cells and chemoresistance. Oncogenesis. 2019;8(3):13.
Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527(7579):472–6.
Rakoff-Nahoum S. Why cancer and inflammation? Yale J Biol Med. 2006;79(3–4):123–30.
Shiota M, Zardan A, Takeuchi A, Kumano M, Beraldi E, Naito S, Zoubeidi A, Gleave ME. Clusterin mediates TGF-β-induced epithelial-mesenchymal transition and metastasis via Twist1 in prostate cancer cells. Cancer Res. 2012;72(20):5261–72.
Slabáková E, Pernicová Z, Slavíčková E, Staršíchová A, Kozubík A, Souček K. TGF-β1-induced EMT of non-transformed prostate hyperplasia cells is characterized by early induction of SNAI2/Slug. Prostate. 2011;71(12):1332–43.
Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL, et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol. 2009;11(8):943–50.
Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, Chao CH, Yamaguchi H, Yang NK, Ding Q, et al. Epithelial-mesenchymal transition induced by TNF-α requires NF-κB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012;72(5):1290–300.
Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007;26(5):711–24.
Su YW, Xie TX, Sano D, Myers JN. IL-6 stabilizes Twist and enhances tumor cell motility in head and neck cancer cells through activation of casein kinase 2. PLoS ONE. 2011;6(4):e19412.
Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, Oberyszyn TM, Hall BM. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene. 2009;28(33):2940–7.
Jung H-Y, Fattet L, Yang J. Molecular pathways: linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin Cancer Res. 2015;21(5):962–8.
Wei SC, Fattet L, Tsai JH, Guo Y, Pai VH, Majeski HE, Chen AC, Sah RL, Taylor SS, Engler AJ, et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat Cell Biol. 2015;17(5):678–88.
Rice AJ, Cortes E, Lachowski D, Cheung BCH, Karim SA, Morton JP, Del Río HA. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis. 2017;6(7):e352.
Fattet L, Jung HY, Matsumoto MW, Aubol BE, Kumar A, Adams JA, Chen AC, Sah RL, Engler AJ, Pasquale EB, et al. Matrix Rigidity Controls Epithelial-Mesenchymal Plasticity and Tumor Metastasis via a Mechanoresponsive EPHA2/LYN Complex. Dev Cell. 2020;54(3):302-316.e307.
Zhang H, Ren L, Ding Y, Li F, Chen X, Ouyang Y, Zhang Y, Zhang D. Hyaluronan-mediated motility receptor confers resistance to chemotherapy via TGFβ/Smad2-induced epithelial-mesenchymal transition in gastric cancer. Faseb j. 2019;33(5):6365–77.
Ghatak S, Hascall VC, Markwald RR, Misra S. Stromal hyaluronan interaction with epithelial CD44 variants promotes prostate cancer invasiveness by augmenting expression and function of hepatocyte growth factor and androgen receptor. J Biol Chem. 2010;285(26):19821–32.
Heldin P, Basu K, Kozlova I, Porsch H. HAS2 and CD44 in breast tumorigenesis. Adv Cancer Res. 2014;123:211–29.
Sukowati CHC, Anfuso B, Fiore E, Ie SI, Raseni A, Vascotto F, Avellini C, Mazzolini G, Tiribelli C. Hyaluronic acid inhibition by 4-methylumbelliferone reduces the expression of cancer stem cells markers during hepatocarcinogenesis. Sci Rep. 2019;9(1):4026.
Walter C, Davis JT, Mathur J, Pathak A. Physical defects in basement membrane-mimicking collagen-IV matrices trigger cellular EMT and invasion. Integr Biol (Camb). 2018;10(6):342–55.
Zeisberg M, Bonner G, Maeshima Y, Colorado P, Müller GA, Strutz F, Kalluri R. Renal fibrosis: collagen composition and assembly regulates epithelial-mesenchymal transdifferentiation. Am J Pathol. 2001;159(4):1313–21.
García-Palmero I, Torres S, Bartolomé RA, Peláez-García A, Larriba MJ, Lopez-Lucendo M, Peña C, Escudero-Paniagua B, Muñoz A, Casal JI. Twist1-induced activation of human fibroblasts promotes matrix stiffness by upregulating palladin and collagen α1(VI). Oncogene. 2016;35(40):5224–36.
Peng DH, Ungewiss C, Tong P, Byers LA, Wang J, Canales JR, Villalobos PA, Uraoka N, Mino B, Behrens C, et al. ZEB1 induces LOXL2-mediated collagen stabilization and deposition in the extracellular matrix to drive lung cancer invasion and metastasis. Oncogene. 2017;36(14):1925–38.
Yoshida T, Hashimura M, Kuwata T, Matsumoto T, Suzuki E, Tazo Y, Nakajima H, Inukai M, Saegusa M. Transcriptional regulation of the alpha-1 type II collagen gene by nuclear factor B/p65 and Sox9 in the chondrocytic phenotype of uterine carcinosarcomas. Hum Pathol. 2013;44(9):1780–8.
Jenkins MH, Croteau W, Mullins DW, Brinckerhoff CE. The BRAF(V600E) inhibitor, PLX4032, increases type I collagen synthesis in melanoma cells. Matrix Biol. 2015;48:66–77.
Sugiyama N, Varjosalo M, Meller P, Lohi J, Hyytiäinen M, Kilpinen S, Kallioniemi O, Ingvarsen S, Engelholm LH, Taipale J, et al. Fibroblast growth factor receptor 4 regulates tumor invasion by coupling fibroblast growth factor signaling to extracellular matrix degradation. Cancer Res. 2010;70(20):7851–61.
Rock MJ, Holden P, Horton WA, Cohn DH. Cartilage oligomeric matrix protein promotes cell attachment via two independent mechanisms involving CD47 and alphaVbeta3 integrin. Mol Cell Biochem. 2010;338(1–2):215–24.
Iozzo RV, Gubbiotti MA. Extracellular matrix: The driving force of mammalian diseases. Matrix Biol. 2018;71–72:1–9.
Nfonsam VN, Jecius HC, Janda J, Omesiete PN, Elquza E, Scott AJ, Nfonsam LE, Jandova J. Cartilage oligomeric matrix protein (COMP) promotes cell proliferation in early-onset colon cancer tumorigenesis. Surg Endosc. 2020;34(9):3992–8.
Englund E, Bartoschek M, Reitsma B, Jacobsson L, Escudero-Esparza A, Orimo A, Leandersson K, Hagerling C, Aspberg A, Storm P, et al. Cartilage oligomeric matrix protein contributes to the development and metastasis of breast cancer. Oncogene. 2016;35(43):5585–96.
Zhong W, Hou H, Liu T, Su S, Xi X, Liao Y, Xie R, Jin G, Liu X, Zhu L, et al. Cartilage Oligomeric Matrix Protein promotes epithelial-mesenchymal transition by interacting with Transgelin in Colorectal Cancer. Theranostics. 2020;10(19):8790–806.
Rafaeva M, Erler JT. Framing cancer progression: influence of the organ- and tumour-specific matrisome. Febs j. 2020;287(8):1454–77.
Papadakos KS, Bartoschek M, Rodriguez C, Gialeli C, Jin SB, Lendahl U, Pietras K, Blom AM. Cartilage Oligomeric Matrix Protein initiates cancer stem cells through activation of Jagged1-Notch3 signaling. Matrix Biol. 2019;81:107–21.
Englund E, Canesin G, Papadakos KS, Vishnu N, Persson E, Reitsma B, Anand A, Jacobsson L, Helczynski L, Mulder H, et al. Cartilage oligomeric matrix protein promotes prostate cancer progression by enhancing invasion and disrupting intracellular calcium homeostasis. Oncotarget. 2017;8(58):98298–311.
Liu TT, Liu XS, Zhang M, Liu XN, Zhu FX, Zhu FM, Ouyang SW, Li SB, Song CL, Sun HM, et al. Cartilage oligomeric matrix protein is a prognostic factor and biomarker of colon cancer and promotes cell proliferation by activating the Akt pathway. J Cancer Res Clin Oncol. 2018;144(6):1049–63.
Chin AR, Wang SE. Cancer Tills the Premetastatic Field: Mechanistic Basis and Clinical Implications. Clin Cancer Res. 2016;22(15):3725–33.
Høye AM, Erler JT. Structural ECM components in the premetastatic and metastatic niche. Am J Physiol Cell Physiol. 2016;310(11):C955-967.
He X, Lee B, Jiang Y. Extracellular matrix in cancer progression and therapy. Med Rev. 2022;2(2):125–39. https://doi.org/10.1515/mr-2021-0028.
Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur Basant K, Becker A, Hoshino A, Mark MT, Molina H, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17(6):816–26.
Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, Upadhyay P, Uyeminami DL, Pommier A, Küttner V, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361(6409):eaao4227.
Paolillo M, Schinelli S. Extracellular Matrix Alterations in Metastatic Processes. Int J Mol Sci. 2019;20(19):4947.
Williamson T, Sultanpuram N, Sendi H. The role of liver microenvironment in hepatic metastasis. Clin Transl Med. 2019;8(1):21.
Lee S, Stewart S, Nagtegaal I, Luo J, Wu Y, Colditz G, Medina D, Allred DC. Differentially expressed genes regulating the progression of ductal carcinoma in situ to invasive breast cancer. Cancer Res. 2012;72(17):4574–86.
Gilkes DM, Chaturvedi P, Bajpai S, Wong CC, Wei H, Pitcairn S, Hubbi ME, Wirtz D, Semenza GL. Collagen prolyl hydroxylases are essential for breast cancer metastasis. Cancer Res. 2013;73(11):3285–96.
Işeri ÖD, Kars MD, Arpaci F, Gündüz U. Gene expression analysis of drug-resistant MCF-7 cells: implications for relation to extracellular matrix proteins. Cancer Chemother Pharmacol. 2010;65(3):447–55.
Ting DT, Wittner BS, Ligorio M, Vincent Jordan N, Shah AM, Miyamoto DT, Aceto N, Bersani F, Brannigan BW, Xega K, et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep. 2014;8(6):1905–18.
Schütze F, Röhrig F, Vorlová S, Gätzner S, Kuhn A, Ergün S, Henke E. Inhibition of Lysyl Oxidases Improves Drug Diffusion and Increases Efficacy of Cytotoxic Treatment in 3D Tumor Models. Sci Rep. 2015;5(1):17576.
Baker A-M, Bird D, Welti JC, Gourlaouen M, Lang G, Murray GI, Reynolds AR, Cox TR, Erler JT. Lysyl Oxidase Plays a Critical Role in Endothelial Cell Stimulation to Drive Tumor Angiogenesis. Can Res. 2013;73(2):583–94.
Zaffryar-Eilot S, Marshall D, Voloshin T, Bar-Zion A, Spangler R, Kessler O, Ghermazien H, Brekhman V, Suss-Toby E, Adam D, et al. Lysyl oxidase-like-2 promotes tumour angiogenesis and is a potential therapeutic target in angiogenic tumours. Carcinogenesis. 2013;34(10):2370–9.
Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62(1):112–20.
Eikenes L, Tari M, Tufto I, Bruland OS, de Lange DC. Hyaluronidase induces a transcapillary pressure gradient and improves the distribution and uptake of liposomal doxorubicin (Caelyx) in human osteosarcoma xenografts. Br J Cancer. 2005;93(1):81–8.
Hodkinson PS, Elliott T, Wong WS, Rintoul RC, Mackinnon AC, Haslett C, Sethi T. ECM overrides DNA damage-induced cell cycle arrest and apoptosis in small-cell lung cancer cells through beta1 integrin-dependent activation of PI3-kinase. Cell Death Differ. 2006;13(10):1776–88.
Halder J, Landen CN Jr, Lutgendorf SK, Li Y, Jennings NB, Fan D, Nelkin GM, Schmandt R, Schaller MD, Sood AK. Focal adhesion kinase silencing augments docetaxel-mediated apoptosis in ovarian cancer cells. Clin Cancer Res. 2005;11(24 Pt 1):8829–36.
Chen Y, Wang Z, Chang P, Xiang L, Pan F, Li J, Jiang J, Zou L, Yang L, Bian Z, et al. The effect of focal adhesion kinase gene silencing on 5-fluorouracil chemosensitivity involves an Akt/NF-kappaB signaling pathway in colorectal carcinomas. Int J Cancer. 2010;127(1):195–206.
Yoon SO, Shin S, Karreth FA, Buel GR, Jedrychowski MP, Plas DR, Dedhar S, Gygi SP, Roux PP, Dephoure N, et al. Focal Adhesion- and IGF1R-Dependent Survival and Migratory Pathways Mediate Tumor Resistance to mTORC1/2 Inhibition. Mol Cell. 2017;67(3):512-527.e514.
Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000;60(9):2497–503.
Rahbari NN, Kedrin D, Incio J, Liu H, Ho WW, Nia HT, Edrich CM, Jung K, Daubriac J, Chen I, et al. Anti-VEGF therapy induces ECM remodeling and mechanical barriers to therapy in colorectal cancer liver metastases. Sci Transl Med. 2016;8(360):360ra135.
Ramjiawan RR, Griffioen AW, Duda DG. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis. 2017;20(2):185–204.
Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611–29.
Al-Hajj M, Becker MW, Wicha M, Weissman I, Clarke MF. Therapeutic implications of cancer stem cells. Curr Opin Genet Dev. 2004;14(1):43–7.
Meineke V, Gilbertz K-P, Schilperoort K, Cordes N, Sendler A, Moede T, van Beuningen D. Ionizing Radiation Modulates Cell Surface Integrin Expression and Adhesion of COLO-320 Cells to Collagen and Fibronectin in Vitro. Strahlenther Onkol. 2002;178(12):709–14.
Cordes N, Blaese MA, Meineke V, Beuningen DV. Ionizing radiation induces up-regulation of functional β 1-integrin in human lung tumour cell lines in vitro. Int J Radiat Biol. 2002;78(5):347–57.
Cordes N, Frick S, Brunner TB, Pilarsky C, Grützmann R, Sipos B, Klöppel G, McKenna WG, Bernhard EJ. Human pancreatic tumor cells are sensitized to ionizing radiation by knockdown of caveolin-1. Oncogene. 2007;26(48):6851–62.
Cordes N, Seidler J, Durzok R, Geinitz H, Brakebusch C. β1-integrin-mediated signaling essentially contributes to cell survival after radiation-induced genotoxic injury. Oncogene. 2006;25(9):1378–90.
Medina SH, Bush B, Cam M, Sevcik E, DelRio FW, Nandy K, Schneider JP. Identification of a mechanogenetic link between substrate stiffness and chemotherapeutic response in breast cancer. Biomaterials. 2019;202:1–11.
Schwartz AD, Barney LE, Jansen LE, Nguyen TV, Hall CL, Meyer AS, Peyton SR. A biomaterial screening approach reveals microenvironmental mechanisms of drug resistance. Integr Biol. 2017;9(12):912–24.
Ishihara S, Haga H. Matrix Stiffness Contributes to Cancer Progression by Regulating Transcription Factors. Cancers. 2022;14(4):1049.
Jang M, Koh I, Lee JE, Lim JY, Cheong J-H, Kim P. Increased extracellular matrix density disrupts E-cadherin/β-catenin complex in gastric cancer cells. Biomaterials Science. 2018;6(10):2704–13.
Wegner CS, Gaustad J-V, Andersen LMK, Simonsen TG, Rofstad EK. Diffusion-weighted and dynamic contrast-enhanced MRI of pancreatic adenocarcinoma xenografts: associations with tumor differentiation and collagen content. J Transl Med. 2016;14(1):161.
Mammoto T, Jiang A, Jiang E, Panigrahy D, Kieran MW, Mammoto A. Role of Collagen Matrix in Tumor Angiogenesis and Glioblastoma Multiforme Progression. Am J Pathol. 2013;183(4):1293–305.
Thangavelu PU, Krenács T, Dray E, Duijf PHG. In epithelial cancers, aberrant COL17A1 promoter methylation predicts its misexpression and increased invasion. Clin Epigenetics. 2016;8(1):120.
Arvanitidis A, Henriksen K, Karsdal MA, Nedergaard A. Neo-epitope Peptides as Biomarkers of Disease Progression for Muscular Dystrophies and Other Myopathies. J Neuromuscul Dis. 2016;3:333–46.
Nedergaard A, Sun S, Karsdal MA, Henriksen K, Kjær M, Lou Y, He Y, Zheng Q, Suetta C. Type VI collagen turnover-related peptides—novel serological biomarkers of muscle mass and anabolic response to loading in young men. J Cachexia Sarcopenia Muscle. 2013;4(4):267–75.
Sivakumar P, Kitson C, Jarai G. Modeling and measuring extracellular matrix alterations in fibrosis: challenges and perspectives for antifibrotic drug discovery. Connect Tissue Res. 2019;60(1):62–70.
Barascuk N, Veidal SS, Larsen L, Larsen DV, Larsen MR, Wang J, Zheng Q, Xing R, Cao Y, Rasmussen LM, et al. A novel assay for extracellular matrix remodeling associated with liver fibrosis: An enzyme-linked immunosorbent assay (ELISA) for a MMP-9 proteolytically revealed neo-epitope of type III collagen. Clin Biochem. 2010;43(10–11):899–904.
Groen SS, Sinkeviciute D, Bay-Jensen A-C, Thudium CS, Karsdal MA, Thomsen SF, Lindemann S, Werkmann D, Blair J, Staunstrup LM, et al. A serological type II collagen neoepitope biomarker reflects cartilage breakdown in patients with osteoarthritis. Osteoarthr Cartil Open. 2021;3(4):100207.
He Y, Manon-Jensen T, Arendt-Nielsen L, Petersen KK, Christiansen T, Samuels J, Abramson S, Karsdal MA, Attur M, Bay-Jensen AC. Potential diagnostic value of a type X collagen neo-epitope biomarker for knee osteoarthritis. Osteoarthr Cartil. 2019;27(4):611–20.
Löfvall H, Katri A, Dąbrowska A, Karsdal MA, Luo Y, He Y, Manon-Jensen T, Dziegiel MH, Bay-Jensen AC, Thudium CS, et al. GPDPLQ(1237)-A Type II Collagen Neo-Epitope Biomarker of Osteoclast- and Inflammation-Derived Cartilage Degradation in vitro. Sci Rep. 2019;9(1):3050.
Boyle M, Tiniakos D, Schattenberg JM, Ratziu V, Bugianessi E, Petta S, Oliveira CP, Govaere O, Younes R, McPherson S, et al. Performance of the PRO-C3 collagen neo-epitope biomarker in non-alcoholic fatty liver disease. JHEP Rep. 2019;1(3):188–98.
Senthebane DA, Jonker T, Rowe A, Thomford NE, Munro D, Dandara C, Wonkam A, Govender D, Calder B, Soares NC, et al. The Role of Tumor Microenvironment in Chemoresistance: 3D Extracellular Matrices as Accomplices. Int J Mol Sci. 2018;19(10):2861.
Li X, Shepard HM, Cowell JA, Zhao C, Osgood RJ, Rosengren S, Blouw B, Garrovillo SA, Pagel MD, Whatcott CJ, et al. Parallel Accumulation of Tumor Hyaluronan, Collagen, and Other Drivers of Tumor Progression. Clin Cancer Res. 2018;24(19):4798–807.
Jakubzig B, Baltes F, Henze S, Schlesinger M, Bendas G. Mechanisms of Matrix-Induced Chemoresistance of Breast Cancer Cells-Deciphering Novel Potential Targets for a Cell Sensitization. Cancers (Basel). 2018;10(12):495.
Sterzyńska K, Klejewski A, Wojtowicz K, Świerczewska M, Nowacka M, Kaźmierczak D, Andrzejewska M, Rusek D, Brązert M, Brązert J, et al. Mutual Expression of ALDH1A1, LOX, and Collagens in Ovarian Cancer Cell Lines as Combined CSCs- and ECM-Related Models of Drug Resistance Development. Int J Mol Sci. 2019;20(1):54.
Gurler H, Yu Y, Choi J, Kajdacsy-Balla AA, Barbolina MV. Three-Dimensional Collagen Type I Matrix Up-Regulates Nuclear Isoforms of the Microtubule Associated Protein Tau Implicated in Resistance to Paclitaxel Therapy in Ovarian Carcinoma. Int J Mol Sci. 2015;16(2):3419–33.
Wu Y-H, Huang Y-F, Chang T-H, Chou C-Y. Activation of TWIST1 by COL11A1 promotes chemoresistance and inhibits apoptosis in ovarian cancer cells by modulating NF-κB-mediated IKKβ expression. Int J Cancer. 2017;141(11):2305–17.
Angenendt L, Mikesch J-H, Görlich D, Busch A, Arnhold I, Rudack C, Hartmann W, Wardelmann E, Berdel WE, Stenner M, et al. Stromal collagen type VI associates with features of malignancy and predicts poor prognosis in salivary gland cancer. Cell Oncol. 2018;41(5):517–25.
Rolff HC, Christensen IJ, Vainer B, Svendsen LB, Eefsen RL, Wilhelmsen M, Lund IK, Høyer-Hansen G, Nielsen HJ, Illemann M. The Prognostic and Predictive Value of Soluble Type IV Collagen in Colorectal Cancer: A Retrospective Multicenter Study. Clin Cancer Res. 2016;22(10):2427–34.
Aguilera KY, Rivera LB, Hur H, Carbon JG, Toombs JE, Goldstein CD, Dellinger MT, Castrillon DH, Brekken RA. Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma. Cancer Res. 2014;74(4):1032–44.
Zhao Q, Eichten A, Parveen A, Adler C, Huang Y, Wang W, Ding Y, Adler A, Nevins T, Ni M, et al. Single-Cell Transcriptome Analyses Reveal Endothelial Cell Heterogeneity in Tumors and Changes following Antiangiogenic Treatment. Cancer Res. 2018;78(9):2370–82.
Albrengues J, Bertero T, Grasset E, Bonan S, Maiel M, Bourget I, Philippe C, Herraiz Serrano C, Benamar S, Croce O, et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat Commun. 2015;6:10204.
Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20(3):174–86.
Bu L, Baba H, Yasuda T, Uchihara T, Ishimoto T. Functional diversity of cancer-associated fibroblasts in modulating drug resistance. Cancer Sci. 2020;111(10):3468–77.
Borriello L, Nakata R, Sheard MA, Fernandez GE, Sposto R, Malvar J, Blavier L, Shimada H, Asgharzadeh S, Seeger RC, et al. Cancer-Associated Fibroblasts Share Characteristics and Protumorigenic Activity with Mesenchymal Stromal Cells. Cancer Res. 2017;77(18):5142–57.
Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA, Lee EJ, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214(3):579–96.
Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–98.
Annaratone L, Cascardi E, Vissio E, Sarotto I, Chmielik E, Sapino A, Berrino E, Marchiò C. The Multifaceted Nature of Tumor Microenvironment in Breast Carcinomas. Pathobiology. 2020;87(2):125–42.
Strell C, Paulsson J, Jin SB, Tobin NP, Mezheyeuski A, Roswall P, Mutgan C, Mitsios N, Johansson H, Wickberg SM, et al. Impact of Epithelial-Stromal Interactions on Peritumoral Fibroblasts in Ductal Carcinoma in Situ. J Natl Cancer Inst. 2019;111(9):983–95.
Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell. 2010;17(2):135–47.
Avery D, Govindaraju P, Jacob M, Todd L, Monslow J, Puré E. Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts. Matrix Biol. 2018;67:90–106.
Malik R, Luong T, Cao X, Han B, Shah N, Franco-Barraza J, Han L, Shenoy VB, Lelkes PI, Cukierman E. Rigidity controls human desmoplastic matrix anisotropy to enable pancreatic cancer cell spread via extracellular signal-regulated kinase 2. Matrix Biol. 2019;81:50–69.
Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15(6):637–46.
Ferrari N, Ranftl R, Chicherova I, Slaven ND, Moeendarbary E, Farrugia AJ, Lam M, Semiannikova M, Westergaard MCW, Tchou J, et al. Dickkopf-3 links HSF1 and YAP/TAZ signalling to control aggressive behaviours in cancer-associated fibroblasts. Nat Commun. 2019;10(1):130.
Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017;7(2):165–76.
Mellone M, Hanley CJ, Thirdborough S, Mellows T, Garcia E, Woo J, Tod J, Frampton S, Jenei V, Moutasim KA, et al. Induction of fibroblast senescence generates a non-fibrogenic myofibroblast phenotype that differentially impacts on cancer prognosis. Aging (Albany NY). 2016;9(1):114–32.
Yoshida GJ. Regulation of heterogeneous cancer-associated fibroblasts: the molecular pathology of activated signaling pathways. J Exp Clin Cancer Res. 2020;39(1):112.
Che Y, Wang J, Li Y, Lu Z, Huang J, Sun S, Mao S, Lei Y, Zang R, Sun N, et al. Cisplatin-activated PAI-1 secretion in the cancer-associated fibroblasts with paracrine effects promoting esophageal squamous cell carcinoma progression and causing chemoresistance. Cell Death Dis. 2018;9(7):759.
Qiao Y, Zhang C, Li A, Wang D, Luo Z, Ping Y, Zhou B, Liu S, Li H, Yue D, et al. IL6 derived from cancer-associated fibroblasts promotes chemoresistance via CXCR7 in esophageal squamous cell carcinoma. Oncogene. 2018;37(7):873–83.
Zhai J, Shen J, Xie G, Wu J, He M, Gao L, Zhang Y, Yao X, Shen L. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett. 2019;454:37–43.
Shi Z, Yang WM, Chen LP, Yang DH, Zhou Q, Zhu J, Chen JJ, Huang RC, Chen ZS, Huang RP. Enhanced chemosensitization in multidrug-resistant human breast cancer cells by inhibition of IL-6 and IL-8 production. Breast Cancer Res Treat. 2012;135(3):737–47.
Tang YA, Chen YF, Bao Y, Mahara S, Yatim S, Oguz G, Lee PL, Feng M, Cai Y, Tan EY, et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc Natl Acad Sci U S A. 2018;115(26):E5990-e5999.
Xu S, Yang Z, Jin P, Yang X, Li X, Wei X, Wang Y, Long S, Zhang T, Chen G, et al. Metformin Suppresses Tumor Progression by Inactivating Stromal Fibroblasts in Ovarian Cancer. Mol Cancer Ther. 2018;17(6):1291–302.
Sun Y, Campisi J, Higano C, Beer TM, Porter P, Coleman I, True L, Nelson PS. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med. 2012;18(9):1359–68.
Zhang D, Li L, Jiang H, Li Q, Wang-Gillam A, Yu J, Head R, Liu J, Ruzinova MB, Lim KH. Tumor-Stroma IL1β-IRAK4 Feedforward Circuitry Drives Tumor Fibrosis, Chemoresistance, and Poor Prognosis in Pancreatic Cancer. Cancer Res. 2018;78(7):1700–12.
Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159(1):80–93.
Kuczek DE, Larsen AMH, Thorseth ML, Carretta M, Kalvisa A, Siersbæk MS, Simões AMC, Roslind A, Engelholm LH, Noessner E, et al. Collagen density regulates the activity of tumor-infiltrating T cells. J Immunother Cancer. 2019;7(1):68.
Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan CL, Skhinas JN, Collot R, Yang J, Harvey K, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. 2018;9(1):2897.
Yoshida GJ, Azuma A, Miura Y, Orimo A. Activated Fibroblast Program Orchestrates Tumor Initiation and Progression; Molecular Mechanisms and the Associated Therapeutic Strategies. Int J Mol Sci. 2019;20(9):2256.
Bochet L, Lehuédé C, Dauvillier S, Wang YY, Dirat B, Laurent V, Dray C, Guiet R, Maridonneau-Parini I, Le Gonidec S, et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013;73(18):5657–68.
Zhao C, Wu M, Zeng N, Xiong M, Hu W, Lv W, Yi Y, Zhang Q, Wu Y. Cancer-associated adipocytes: emerging supporters in breast cancer. J Exp Clin Cancer Res. 2020;39(1):156.
Lotti F, Jarrar AM, Pai RK, Hitomi M, Lathia J, Mace A, Gantt GA Jr, Sukhdeo K, DeVecchio J, Vasanji A, et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J Exp Med. 2013;210(13):2851–72.
Hu YB, Yan C, Mu L, Mi YL, Zhao H, Hu H, Li XL, Tao DD, Wu YQ, Gong JP, et al. Exosomal Wnt-induced dedifferentiation of colorectal cancer cells contributes to chemotherapy resistance. Oncogene. 2019;38(11):1951–65.
Sansone P, Berishaj M, Rajasekhar VK, Ceccarelli C, Chang Q, Strillacci A, Savini C, Shapiro L, Bowman RL, Mastroleo C, et al. Evolution of Cancer Stem-like Cells in Endocrine-Resistant Metastatic Breast Cancers Is Mediated by Stromal Microvesicles. Cancer Res. 2017;77(8):1927–41.
Ren J, Ding L, Zhang D, Shi G, Xu Q, Shen S, Wang Y, Wang T, Hou Y. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics. 2018;8(14):3932–48.
Garvey CM, Lau R, Sanchez A, Sun RX, Fong EJ, Doche ME, Chen O, Jusuf A, Lenz HJ, Larson B, et al. Anti-EGFR Therapy Induces EGF Secretion by Cancer-Associated Fibroblasts to Confer Colorectal Cancer Chemoresistance. Cancers (Basel). 2020;12(6):1393.
Zhang L, Yao J, Li W, Zhang C. Micro-RNA-21 Regulates Cancer-Associated Fibroblast-Mediated Drug Resistance in Pancreatic Cancer. Oncol Res. 2018;26(6):827–35.
Choi J, Cha YJ, Koo JS. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog Lipid Res. 2018;69:11–20.
Elliott BE, Tam SP, Dexter D, Chen ZQ. Capacity of adipose tissue to promote growth and metastasis of a murine mammary carcinoma: effect of estrogen and progesterone. Int J Cancer. 1992;51(3):416–24.
Wu Q, Li B, Li Z, Li J, Sun S, Sun S. Cancer-associated adipocytes: key players in breast cancer progression. J Hematol Oncol. 2019;12(1):95.
He JY, Wei XH, Li SJ, Liu Y, Hu HL, Li ZZ, Kuang XH, Wang L, Shi X, Yuan ST, et al. Adipocyte-derived IL-6 and leptin promote breast Cancer metastasis via upregulation of Lysyl Hydroxylase-2 expression. Cell Commun Signal. 2018;16(1):100.
Juárez-Cruz JC, Zuñiga-Eulogio MD, Olea-Flores M, Castañeda-Saucedo E, Mendoza-Catalán M, Ortuño-Pineda C, Moreno-Godínez ME, Villegas-Comonfort S, Padilla-Benavides T, Navarro-Tito N. Leptin induces cell migration and invasion in a FAK-Src-dependent manner in breast cancer cells. Endocr Connect. 2019;8(11):1539–52.
Kim HS, Jung M, Choi SK, Woo J, Piao YJ, Hwang EH, Kim H, Kim SJ, Moon WK. IL-6-mediated cross-talk between human preadipocytes and ductal carcinoma in situ in breast cancer progression. J Exp Clin Cancer Res. 2018;37(1):200.
Iyengar P, Espina V, Williams TW, Lin Y, Berry D, Jelicks LA, Lee H, Temple K, Graves R, Pollard J, et al. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J Clin Invest. 2005;115(5):1163–76.
Park J, Scherer PE. Adipocyte-derived endotrophin promotes malignant tumor progression. J Clin Invest. 2012;122(11):4243–56.
Gyamfi J, Eom M, Koo JS, Choi J. Multifaceted Roles of Interleukin-6 in Adipocyte-Breast Cancer Cell Interaction. Transl Oncol. 2018;11(2):275–85.
Wang J, Wu Y, Guo J, Fei X, Yu L, Ma S. Adipocyte-derived exosomes promote lung cancer metastasis by increasing MMP9 activity via transferring MMP3 to lung cancer cells. Oncotarget. 2017;8(47):81880–91.
Au Yeung CL, Co NN, Tsuruga T, Yeung TL, Kwan SY, Leung CS, Li Y, Lu ES, Kwan K, Wong KK, et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat Commun. 2016;7:11150.
Leight JL, Drain AP, Weaver VM. Extracellular Matrix Remodeling and Stiffening Modulate Tumor Phenotype and Treatment Response. Ann Rev Cancer Biol. 2017;1(1):313–34.
Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3(6):401–10.
Birukova AA, Tian X, Cokic I, Beckham Y, Gardel ML, Birukov KG. Endothelial barrier disruption and recovery is controlled by substrate stiffness. Microvasc Res. 2013;87:50–7.
Kniazeva E, Putnam AJ. Endothelial cell traction and ECM density influence both capillary morphogenesis and maintenance in 3-D. Am J Physiol Cell Physiol. 2009;297(1):C179-187.
Ghosh K, Pan Z, Guan E, Ge S, Liu Y, Nakamura T, Ren XD, Rafailovich M, Clark RA. Cell adaptation to a physiologically relevant ECM mimic with different viscoelastic properties. Biomaterials. 2007;28(4):671–9.
Liu J, Agarwal S. Mechanical signals activate vascular endothelial growth factor receptor-2 to upregulate endothelial cell proliferation during inflammation. J Immunol. 2010;185(2):1215–21.
Villalba N, Baby S, Yuan SY. The Endothelial Glycocalyx as a Double-Edged Sword in Microvascular Homeostasis and Pathogenesis. Front Cell Dev Biol. 2021;9:711003.
Curry FE, Adamson RH. Endothelial glycocalyx: permeability barrier and mechanosensor. Ann Biomed Eng. 2012;40(4):828–39.
Lieleg O, Baumgärtel RM, Bausch AR. Selective filtering of particles by the extracellular matrix: an electrostatic bandpass. Biophys J. 2009;97(6):1569–77.
Hu Z, Cano I, D’Amore PA. Update on the Role of the Endothelial Glycocalyx in Angiogenesis and Vascular Inflammation. Front Cell Dev Biol. 2021;9:734276.
Eggermont AMM, Blank CU, Mandala M, Long GV, Atkinson V, Dalle S, Haydon A, Lichinitser M, Khattak A, Carlino MS, et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N Engl J Med. 2018;378(19):1789–801.
Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF, et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med. 2018;378(22):2078–92.
Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521–32.
Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gümüş M, Mazières J, Hermes B, Çay Şenler F, Csőszi T, Fülöp A, et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N Engl J Med. 2018;379(21):2040–51.
Hallmann R, Zhang X, Di Russo J, Li L, Song J, Hannocks MJ, Sorokin L. The regulation of immune cell trafficking by the extracellular matrix. Curr Opin Cell Biol. 2015;36:54–61.
Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, Wang H, Fang R, Bu X, Cai S, et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget. 2016;7(32):52294–306.
Gavalas NG, Tsiatas M, Tsitsilonis O, Politi E, Ioannou K, Ziogas AC, Rodolakis A, Vlahos G, Thomakos N, Haidopoulos D, et al. VEGF directly suppresses activation of T cells from ascites secondary to ovarian cancer via VEGF receptor type 2. Br J Cancer. 2012;107(11):1869–75.
Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, Mami-Chouaib F, Donnadieu E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. 2012;122(3):899–910.
Castermans K, Griffioen AW. Tumor blood vessels, a difficult hurdle for infiltrating leukocytes. Biochim Biophys Acta. 2007;1776(2):160–74.
Lund AW, Swartz MA. Role of lymphatic vessels in tumor immunity: passive conduits or active participants? J Mammary Gland Biol Neoplasia. 2010;15(3):341–52.
Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, Latreche S, Bergaya S, Benhamouda N, Tanchot C, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212(2):139–48.
Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol. 2015;15(11):669–82.
Bollyky PL, Wu RP, Falk BA, Lord JD, Long SA, Preisinger A, Teng B, Holt GE, Standifer NE, Braun KR, et al. ECM components guide IL-10 producing regulatory T-cell (TR1) induction from effector memory T-cell precursors. Proc Natl Acad Sci U S A. 2011;108(19):7938–43.
O’Connor RS, Hao X, Shen K, Bashour K, Akimova T, Hancock WW, Kam LC, Milone MC. Substrate rigidity regulates human T cell activation and proliferation. J Immunol. 2012;189(3):1330–9.
Chitadze G, Lettau M, Bhat J, Wesch D, Steinle A, Fürst D, Mytilineos J, Kalthoff H, Janssen O, Oberg HH, et al. Shedding of endogenous MHC class I-related chain molecules A and B from different human tumor entities: heterogeneous involvement of the “a disintegrin and metalloproteases” 10 and 17. Int J Cancer. 2013;133(7):1557–66.
Barsoum IB, Hamilton TK, Li X, Cotechini T, Miles EA, Siemens DR, Graham CH. Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: role of nitric oxide. Cancer Res. 2011;71(24):7433–41.
Sun D, Wang X, Zhang H, Deng L, Zhang Y. MMP9 mediates MICA shedding in human osteosarcomas. Cell Biol Int. 2011;35(6):569–74.
Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, Folkerts G, Nijkamp FP, Blalock JE. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med. 2006;12(3):317–23.
Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, Mecham RP, Senior RM, Shapiro SD. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest. 2006;116(3):753–9.
Rygiel TP, Stolte EH, de Ruiter T, van de Weijer ML, Meyaard L. Tumor-expressed collagens can modulate immune cell function through the inhibitory collagen receptor LAIR-1. Mol Immunol. 2011;49(1–2):402–6.
Acknowledgements
Not applicable.
Funding
This research was partially funded by the project PDI-PFE-CDI 2021, entitled Increasing the Performance of Scientific Research, Supporting Excellence in Medical Research and Innovation, PROGRES, no. 40PFE/20.12.2021 and PN-III-P2-2.1-PED-2019–3640: Advanced preclinical validation of microRNA-205-5p-based therapeutic model for inhibition of epithelial to mesenchymal transition în melanoma metastasis MELAMET, 539PED. Dr. Ancuta Jurj won a national fellowship program L’Oréal – UNESCO “For Women in Science”.
Author information
Authors and Affiliations
Contributions
AJ and CB wrote the paper; IC and IBN assisted in the preparation of the manuscript and editing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Clinical trials of cancer therapies that target ECM and molecules linked with ECM.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jurj, A., Ionescu, C., Berindan-Neagoe, I. et al. The extracellular matrix alteration, implication in modulation of drug resistance mechanism: friends or foes?. J Exp Clin Cancer Res 41, 276 (2022). https://doi.org/10.1186/s13046-022-02484-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-022-02484-1