
Abstract
Programmed death-ligand 1 (PD-L1) on cancer cells engages with programmed cell death-1 (PD-1) on immune cells, contributing to cancer immune escape. For multiple cancer types, the PD-1/PD-L1 axis is the major speed-limiting step of the anti-cancer immune response. In this context, blocking PD-1/PD-L1 could restore T cells from exhausted status and eradicate cancer cells. However, only a subset of PD-L1 positive patients benefits from α-PD-1/PD-L1 therapies. Actually, PD-L1 expression is regulated by various factors, leading to the diverse significances of PD-L1 positivity. Understanding the mechanisms of PD-L1 regulation is helpful to select patients and enhance the treatment effect. In this review, we focused on PD-L1 regulators at the levels of transcription, post-transcription, post-translation. Besides, we discussed the potential applications of these laboratory findings in the clinic.
Similar content being viewed by others


Background
In physiological conditions, the activities of T cells are intricately regulated. T cell immunity selectively eliminates pathogens and abnormal cells but avoids attacking normal cells, termed immune homeostasis [1]. Programmed cell death-1 (PD-1, which is encoded by PDCD1) and programmed death-ligand 1 (PD-L1, which is encoded by CD274) are vital proteins in maintaining immune homeostasis [2]. The PD-1/PD-L1 pathway restrains the hyperactivation of immune cells and prevents autoimmune diseases [3]. However, in the tumor microenvironment (TME), the PD-1/PD-L1 axis is hijacked by cancer cells to escape immune surveillance [4]. The overexpressed PD-L1 on cancer cells binds to the PD-1 on tumor-infiltrating lymphocytes (TILs), which counteracts the TCR-signaling cascade by phosphorylating SHP-2 [5, 6]. As a result, T cell activation is impaired. Apart from cancer cells, some other types of cells in the TME, such as macrophages, dendritic cells (DCs), activated T cells, as well as cancer-associated fibroblasts, also express PD-L1 [7]. These components orchestrate an immunosuppressive microenvironment, supporting tumor growth.
Inhibiting the PD-1/PD-L1 signaling is a feasible strategy to normalize the dysregulated TME [8]. Up to now, α-PD-1/PD-L1 treatments have exhibited potent antitumor activities in various cancers, such as melanoma, non-small cell lung cancer (NSCLC), gastric cancer, liver cancer, urothelial cancer, lymphoma, and all MSI-high cancers [2, 9,10,11,12,13,14,15,16,17,18,19]. Commonly, the PD-L1 protein level is the primary standard to select patients who are more likely to respond to α-PD-1/PD-L1 treatments [20, 21]. However, the PD-L1 level is determined by several factors, which results in the different significances of PD-L1 positivity or negativity. The PD-L1 positivity might result from immune response-induced PD-L1 expression or oncogenic constructive PD-L1 upregulation [22]. For the latter, in the absence of pre-existing immune response, patients with PD-L1 positive tumors commonly are resistant to α-PD-1/PD-L1 therapies [20].
On the contrary, patients with PD-L1 negative tumors might respond to α-PD-1/PD-L1 treatment when undergoing combination therapies that promote T cell infiltration [22]. Therefore, an in-depth understanding of PD-L1 regulation is valuable for efficacy prediction and patient selection. In this review, we summarized the latest advances of PD-L1 regulation, including genomic alterations, epigenetic modification, transcriptional regulation, post-transcriptional modification, and post-translational modification. Moreover, we discussed the potential applications of these findings in the clinic.
Genomic alterations of CD274
In some cancers such as classical Hodgkin lymphoma and small-cell lung cancer, the copy number of chromosome 9p24.1 (where CD274 resides) was increased [23, 24]. The chromosome rearrangement caused CD274 amplification without influences on the open reading frame (Fig. 1) (Table 1) [24]. Besides, in mediastinal large B-cell lymphoma, the increased transcriptional expression of CD274 was related to an adjacent ectopic promoter or enhancer by translocation [25]. These findings indicated that genomic alterations contributed substantially to cancer immune escape, which might be a potential biomarker for patient selection.

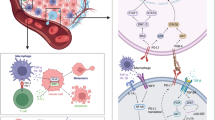
The regulators of PD-L1 expression. PD-L1 abundance is regulated by genomic alterations (amplification or translocation), epigenetic modifications (methylation of histone or CpG island, and histone acetylation), transcriptional regulation (inflammatory stimuli and oncogenic signals), post-transcriptional regulation (miRNA, the status of 3′- UTR, RAS, and Angiotensin II), and post-translational modification (ubiquitination, phosphorylation, glycosylation, palmitoylation). H3K4me3: tri-methylation of histone H3 on lysine 4; H3K27me3: tri-methylation of histone H3 on lysine 27; EGFR: epidermal growth factor receptor; IRF: interferon-responsive factor; IFN: interferon; DSB: double-strand break; GSK3β: glycogen synthase kinase 3β; PI3K: phosphoinositide 3-kinase; NF-κB: Nuclear factor kappa-B; HIF-1α: hypoxia-inducible factor-1α; ALK: Anaplastic lymphoma kinase; ER: endoplasmic reticulum
Epigenetic regulations
Epigenetic regulations such as methylation and histone acetylation determine the PD-L1 expression as well (Fig. 1). Tri-methylation of histone H3 on lysine 4 (H3K4me3) is generally believed as a histone modification promoting gene transcriptions [26]. In pancreatic cancer, MLL1 protein could bind to the CD274 promoter to catalyze H3K4me3, leading to the increased expression of PD-L1 [27]. In agreement, the MLL1 inhibitor had a synergistic effect with α-PD-1/PD-L1 therapy [27]. On the contrary, tri-methylation of histone H3 on lysine 27 (H3K27me3) relates to transcription suppression [28]. In hepatocellular carcinoma, enhancer of zeste homolog 2 negatively regulated PD-L1 expression by promoting H3K27me3 [29].
Besides the methylation of histone, the methylation of DNA at CpG islands regulated PD-L1 expression [30]. Inhibiting methylation of DNA by DNA methyltransferase inhibitors (DNMTis) increased PD-L1 level in cancer cells [31, 32]. The authors assumed that DNMTis elevated the expression of hypermethylated endogenous retroviruses in cancer cells, which might activate the innate immune response and lead to IFN-γ-stimulated PD-L1 expression [30]. Moreover, the methylation of some specific CpG loci in the CD274 promoter determined the level of CD274 mRNA [33,34,35]. In NSCLC, TGF-β1 impaired the activity of DNMTs, demethylated the CD274 promoter, and increased PD-L1 expression [36]. Notably, in patients with recurrent gastric cardia adenocarcinoma, PD-L1 expression was reduced after α-PD-1/PD-L1 treatment [37]. Further investigations indicated that the CD274 promoter was more hypermethylated in the relapsed tumors than in the primary tumors without α-PD-1/PD-L1 treatment [37]. In murine tumor models, the combination therapy of hypomethylating agent azacytidine and α-PD-1 showed a more significant antitumor effect than α-PD-1 monotherapy [37].
Histone acetylation is an epigenetic modification enhancing gene transcription [38]. In some drug-resistant cancer cells, hyperactivated JNK/c-Jun signaling suppressed the histone deacetylase 3 (HADC3) expression, thereby elevating the histone H3 acetylation of the CD274 promoter [39]. The HADC inhibitor had a synergistic effect with α-PD-1 in the B16F10 tumor model [40]. Furthermore, HADC inhibitor-mediated PD-L1 upregulation was observed in other types of cancers [41, 42]. These findings provide a rationale to combine α-PD-1/PD-L1 treatments with HDAC inhibitors.
Transcriptional regulation
Inflammatory Signaling
Interferon (IFN) and IL-6
As a negative feedback for inflammation, PD-L1 could be upregulated by multiple inflammatory signaling pathways to restrain T cells' hyperactivity (Fig. 1). Generally believed, IFN-γ is the prominent stimulator contributing to the inducible PD-L1 expression [43].
During cancer progression, the IFN-γ-derived PD-L1 promotes cancer immune escape [3]. In the TME, activated T cells and NK cells generate most IFN-γ. Then, IFN-γ binds to type II interferon receptor, activating the JAK-STAT signaling (mainly through STAT1) [44, 45]. Subsequently, the expression of several transcriptional factors is upregulated, especially interferon-responsive factors (IRFs). IRF-1 is the vital downstream component of STAT1 upon IFN-γ treatment [46, 47]. In hepatocellular carcinoma, it was identified that two elements (IRE1/2) in the 5′-flanking region of the CD274 promoter were the binding sites of IRF-1, which participated in regulating PD-L1 transcription [48]. Notably, the intactness of JAK-STAT-IRF1 pathway is also related to the response to α-PD-1/PD-L1 therapy. The effect of α-PD-1/PD-L1 treatment is limited in tumors with mutations in JAK1 and JAK2 [49]. It was speculated that these tumors might not rely on the PD-1/PD-L1 pathway to escape immune surveillance [49].
Besides IFN-γ, other inflammatory stimuli such as IFN-α, IFN-β, and IL-6 could induce PD-L1 expression as well. However, IFN-α and IFN-β had a more significant effect on PD-L2 regulation than PD-L1 regulation [44]. In prostate cancer, the IL-6-JAK-STAT3 pathway promoted PD-L1 expression and led to the resistance to immune killing [50]. Moreover, in hepatocellular carcinoma, increased IL-6 activated the STAT3/c-MYC/miR-25-3p pathway, which resulted in the decreased protein tyrosine phosphatase receptor type O (PTPRO) [51]. The downregulated PTPRO-enhanced PD-L1 expression by deregulating the activation of JAK2-STAT1/3 [51]. Furthermore, the glioblastoma-derived IL-6 could induce the local and systemic myeloid PD-L1 expression by STAT3 phosphorylation [52]. Besides, in lung cancer, it was detected that IL-6-derived PD-L1 expression was related to multiple pathways, especially the MEK-ERK signaling [53].
Other inflammatory signals
Tumor necrosis factor-α (TNF-α) increased CD274 mRNA by activating nuclear factor kappa-B (NF-κB) pathway [54]. In renal cell carcinoma, TNF-α cooperated with IL-4 to enhance CD274 transcription by activating NF-κB, IκB, and STAT6 [55]. Moreover, in oral squamous cell carcinoma, the IL-10 level in the TME was positively correlated to the abundance of PD-L1 on tumor-associated macrophages [56]. Blocking IL-10 suppressed PD-L1 expression [56]. Furthermore, in several human cancer cells, IL-27 increased CD274 transcription by promoting the tyrosine phosphorylation of STAT1 and STAT3 [57].
The effect of TGF-β on PD-L1 regulation is still unclear. Although some previous studies indicated that TGF-β downregulated PD-L1 expression in renal tubular epithelial cells and monocytes [58, 59], TGF-β mainly had a positive impact on the PD-L1 expression in the TME. In NSCLC cells, exogenous TGF-β increased the CD274 transcription probably by Smad-binding elements [60]. The expression of phosphorylated-Smad2 was significantly increased in PD-L1 positive NSCLC patients [60]. Apart from cancer cells, TGF-β could increase PD-L1 expression on DCs in the TME [61].
Oncogenic Signaling
Besides inflammatory stimuli, growing evidence suggests that hyperactive oncogenic pathways play a vital role in PD-L1 expression (Fig. 1). Therefore, α-PD-1/PD-L1 therapies might have a synergistic effect with oncogenic signal-targeting treatments.
Epidermal Growth Factor Receptor (EGFR)
In lung epithelial cells, the mutated EGFR pathway (EGFR T790M) increased PD-L1 expression [62]. For lung cancer cells, PD-L1 expression was impaired after EGFR tyrosine kinase inhibitor (TKI) treatment [62]. In murine EGFR-driving lung cancer models, α-PD-1 effectively reversed T cell exhaustion and retarded tumor growth [62]. The results indicated that the mutant EGFR pathway facilitated tumor to escape from immune surveillance [62]. However, a clinical study showed that EGFR-mutant NSCLC patients tended to resist α-PD-1 therapy [63]. The authors found that although some EGFR-mutant NSCLCs were PD-L1 positive, the concurrent PD-L1 upregulation and abundant TILs were rare [63]. The lack of a pre-existing inflammatory TME might limit the effect of α-PD-1/PD-L1 treatment [63]. The low response rate in EGFR-mutant patients was reported by other investigators [64, 65].
Mitogen-activated protein kinase (MAPK)
MAPK is a well-studied oncogenic pathway, which counts for nearly 40% of human cancer cases [66]. According to TCGA database, the CD274 mRNA level was significantly positively related to RAS- or MEK-activation scores in NSCLC patients [67]. In lung adenocarcinoma cells, activating EGF-MAPK signaling increased the mRNA and protein levels of PD-L1 [67]. Inhibiting MAPK signaling by MEK inhibitor (Selumetinib) counteracted the EGF- and IFN-γ-stimulated upregulation of CD274 mRNA and PD-L1 protein [67]. In melanoma cells, the activated NRAS-RAF-MEK1/2-ERK-c-Jun axis enhanced the transcription of CD274 [68]. Moreover, in pancreatic cancer, myeloid cells induced PD-L1 expression on tumor cells by activating EGFR-MAPK pathway [69]. After MEK inhibitor treatment, the levels of p-ERK and PD-L1 were decreased, and this reduced PD-L1 led to a higher sensitivity to α-PD-1 treatment in murine pancreatic tumors [69].
On the contrary, in murine breast cancer cells, suppressing MAPK signaling by Trametinib (a MEK inhibitor) potentiated the IFN-γ-stimulated upregulation of PD-L1 and major histocompatibility complex (MHC) [70]. Furthermore, in cancer cell lines, including KYSE30, TE-1, MKN7, PC-9, SNU-475, OE19, and BT-549, there was no significant alteration when cancer cells were treated with MAPK inhibitor [71]. Besides, the MAPK inhibitor had no significant impact on IFN-γ-stimulated PD-L1 expression [71]. The role of MAPK pathway in PD-L1 regulation might depend on cell types [72].
PTEN/PI3K-AKT pathway
As a well-studied tumor suppressor, PTEN is a vital regulator of the oncogenic signaling pathway PI3K-AKT [73]. PTEN loss and PI3K activation have been identified in multiple types of cancers, including hepatocellular carcinoma, prostate cancer, and breast cancer [73]. Deficient PTEN was detected in nearly half of PD-L1 positive triple-negative breast cancer samples [74]. Knocking down PTEN resulted in a rise of PD-L1 expression [74]. Moreover, activating the PI3K-AKT pathway in gastric cancer cells increased PD-L1 abundance, while PI3K inhibitor (LY294002) reduced PD-L1 level [75]. Besides, in head and neck cancer cells, melanoma cells, colorectal cancer cells, and Her2-amplified cancer cells (SNU216, NCI-N87, and SKBR3), PD-L1 expression was suppressed by PI3K inhibition [68, 76,77,78]. Moreover, inhibiting mTOR (the downstream of PI3K-AKT) by rapamycin reduced PD-L1 level in NSCLC cells [79].
JAK-STAT pathway
Mutations in JAK1, JAK3, and STAT3 were prevalent in mature T-cell lymphomas [80]. Some STAT3 mutations, such as p.E616K, increased the STAT3 phosphorylation and STAT3-mediated transcription [80]. In the meanwhile, silencing STAT3 or STAT3 inhibitor reduced PD-L1 expression [80]. Further chromatin immunoprecipitation qPCR assay indicated the p.E616K mutation might increase the transcription activity of CD274 promoter by a stronger STAT3 binding [80]. Besides, in breast and lung cancer cells, the PD-L1 expression was hampered by JAK and STAT3 inhibitors [81,82,83].
NF-κB pathway
Activated NF-κB signaling was related to the high level of PD-L1 in several cancers [36, 84,85,86,87,88]. Multiple oncogenic signals could impair immune surveillance by activating the NF-κB-PD-L1 axis. In lung cancer cells, overexpressed MUC1-C increased the occupancy of NF-κB p65 in CD274 promoter, which enhanced CD274 transcription [89]. Besides, in breast cancer, reactive oxygen species (ROS) inducers (paclitaxel, glutathione synthesis inhibitor, and buthionine sulphoximine) led to the accumulation of ROS, subsequently activating the downstream NF-κB pathway [90]. In a murine breast cancer model, paclitaxel treatment induced PD-L1 upregulation in tumor-associated macrophages by the NF-κB p65-PD-L1 pathway [90].
Hypoxia-inducible factor-1 (HIF-1)
Hypoxia facilitates the drug resistance and distant metastasis of tumor cells [91]. Besides, a hypoxic TME undermines host immunity activities and contributes to immune escape [92]. Hypoxia upregulated PD-L1 expression by HIF-1α [92]. The hypoxia-induced upregulation of CD274 mRNA was hampered when HIF-1α was silenced [92]. Further investigations suggested the cellular colocalization of PD-L1 and HIF-1α [92]. In the meanwhile, inhibiting HIF-1 signaling could reduce PD-L1 expression in multiple types of cancers [93, 94].
Myc
As a transcription factor regulating cell differentiation, proliferation, and apoptosis, Myc is overexpressed in various cancers [95]. Knocking down or inhibiting Myc in cancer cells reduced CD274 mRNA and PD-L1 protein [96,97,98,99]. The results of the ChIP-seq assay showed that Myc could bind to the CD274 promoter [96]. However, in some particular types of cancer, Myc negatively regulated PD-L1 expression. In hepatocellular carcinoma cells, inhibiting Myc increased the IFN-γ-stimulated PD-L1 expression [100]. Besides, in the murine MycCaP tumor model, Myc inhibitor treatment promoted T cell infiltration, enhanced antitumor immune response, but simultaneously upregulated PD-L1 expression [101]. This PD-L1 upregulation was induced by immune response [101].
Anaplastic lymphoma kinase (ALK)
Chromosomal rearrangements in the ALK gene are an oncogenic driver for NSCLC [102]. In various NSCLC cell lines, the CD274 mRNA and PD-L1 protein levels were higher in cells with echinoderm microtubule-associated protein-like 4 (EML4)-ALK fusion [103]. Ectopic expressing EML4-ALK protein or blocking ALK phosphorylation positively or negatively regulated PD-L1 expression [103]. Besides, inhibiting PI3K-AKT or MEK-ERK pathways reversed the EML4-ALK-induced PD-L1 expression [103]. Apart from NSCLC, the PD-L1 level was higher in ALK-positive systemic anaplastic large cell lymphoma [104].
Met
Alterations in the Met gene were reported in multiple types of cancers [105, 106]. In primary lung cancer tissues, the level of PD-L1 was positively correlated to the Met-amplification [107, 108]. In a microarray assay, inhibiting or knocking down Met substantially reshaped the expression of several immune-related genes, including CD274 [109]. On the contrary, activating Met by hepatocyte growth factor increased PD-L1 expression [109, 110].
BRD4
As a member of the bromodomain and extraterminal (BET) family, BRD4 acts as a super-enhancer of oncogenes [111]. In ovarian cancer cells, BET inhibitor reduced PD-L1 expression in a time- and dose-dependent manner [112]. Further, the ChIP assay showed a significant association of CD274 promoter and BRD4 [112]. After BET inhibitor treatment, the associations of CD274 promoter-BRD4 and CD274 promoter-RNA Pol II were decreased, which contributed to the downregulated CD274 transcription [112]. Besides, it was validated that BET inhibitor suppressed CD274 transcription by reducing the BRD4 occupancy at CD274 promoter, independent of c-Myc [113].
DNA double-strand break (DSB) repair pathway
After inducing DSB by ionizing radiation, PD-L1 expression was increased in multiple cancer cell lines [114]. In contrast, paclitaxel (a non-DNA damaging agent) treatment had no significant impact on PD-L1 expression [114]. DSB-activated ATM-ATR-ChK1-STAT1/3-IRF1 pathway led to the downstream PD-L1 upregulation [114].
Post-transcriptional regulation
microRNA (miRNA)
Cancer-derived miRNA is a vital post-transcriptional regulator for PD-L1 expression in the TME (Fig. 1) [115]. In colorectal cancer cells with mismatch repair deficiency or microsatellite instability-high, miR-148a-3p was decreased while PD-L1 was increased [116]. The results of the co-transfection of miR-148a-3p mimic and wild-type or mutant CD274 3′-untranslated region (UTR) luciferase reporter indicated that CD274 mRNA was the direct target of miR-148a-3p [116]. Furthermore, in breast cancer cells, miR-873 suppressed PD-L1 expression by targeting CD274 mRNA [117]. Up to now, it was identified that CD274 mRNA was the direct target of multiple oncogenic miRNAs such as miR-34a, miR-200 family, miR-142-5p, miR-424, miR-214, miR-497-5p, miR-140 [118,119,120,121,122,123,124,125,126].
Besides, some cancer-derived miRNAs indirectly regulated PD-L1 expression [127,128,129]. In ovarian carcinoma cells, miR-145 downregulated PD-L1 by targeting c-Myc [130]. In cervical cancer cells, increased PD-L1 was related to the upregulation of miR-18a [131]. miR-18a promoted PD-L1 expression by targeting PTEN (inhibitor of PI3K-AKT), WNK2 (inhibitor of MAPK), and SOX6 (inhibitor of Wnt/β-catenin) [131]. Similarly, hepatocellular carcinoma cell-derived miR-23a-3p enhanced PD-L1 expression in macrophages via targeting PTEN [127]. In NSCLC cells, miR-3127-5p promoted PD-L1 expression by activating STAT3 [132]. Moreover, in gastric cancer, miR-BART5-5p increased PD-L1 by targeting PIAS3 (inhibitor of STAT3) [133].
The stability of CD274 mRNA
The variations in the 3′- UTR affected the stability of CD274 mRNA [134]. Disturbing the 3′-UTR of CD274 mRNA by Crisper-Cas9 could stabilize CD274 mRNA [134]. Besides, oncogenic RAS activation inhibited tristetraprolin (AU-rich element-binding protein) by kinase MK2, stabilizing CD274 mRNA [135]. As a result, RAS activation increased PD-L1 expressed on cancer cells [135]. Moreover, in NSCLC, Angiotensin II increased the stability of CD274 mRNA and induced PD-L1 expression by human antigen R (also known as HuR, an AU-rich element-binding protein) [136].
Post-translational modification
Post-translational modifications, including ubiquitination, phosphorylation, glycosylation, palmitoylation, and SUMOylation, play a vital role in regulating protein stability, activation, localization, as well as interaction [137]. Aberrant post-translational modification patterns participated in PD-L1 upregulation in the TME (Fig. 1) [138].
Ubiquitination
Ubiquitination is related to proteasome-mediated protein degradation [139]. In a broad range of cancer cells, CMTM6 maintained PD-L1 expression by reducing PD-L1 ubiquitination and increasing PD-L1 half-life [138, 140]. Moreover, cyclin D/cyclin-dependent kinase 4 (CDK4) promoted PD-L1 ubiquitination by SPOP/Cullin 3-SPOP E3 ligase [141]. CDK4/6 inhibitor treatment increased PD-L1 abundance, which provided a potential for the combining therapy of CDK4/6 inhibitors and α-PD-1/PD-L1 agents [141]. Besides, the TNF-α-NF-κB pathway inhibited PD-L1 ubiquitination via upregulating COP9 signalosome 5 (CSN5) [142]. Inhibiting CSN5 impaired PD-L1 expression and sensitized tumor cells to the following immunotherapy [142].
Phosphorylation
IL-6-activated JAK1 promoted the phosphorylation of PD-L1 protein (Tyr112) [143]. Subsequently, Tyr112-phosphorylated PD-L1 recruited STT3A (N-glycosyltransferase) to catalyze the PD-L1 glycosylation [143]. Activating the IL-6-JAK1 signaling elevated PD-L1 stability by this phosphorylation modification [143]. Blocking the IL-6-JAK1 axis had a synergistic effect with α-Tim-3 treatment in murine tumor models [143]. Besides, metformin-activated AMP-activated protein kinase promoted the phosphorylation of PD-L1 (S195) [144]. The S195 phosphorylation led to the aberrant PD-L1 glycosylation, which undermined the PD-L1 translocation from endoplasmic reticulum to Golgi [144]. Apart from hampering the translocation of PD-L1 to cell membrane, the S195 phosphorylation enhanced endoplasmic reticulum-associated PD-L1 degradation [144]. The combination therapy of metformin and α-cytotoxic T Lymphocyte antigen 4 (CTLA-4) exhibited a robust antitumor activity [144]. Moreover, glycogen synthase kinase 3β (GSK3β) decreased the level of PD-L1 by promoting phosphorylation-dependent proteasome degradation [145, 146].
Glycosylation
Glycosylation modification is related to protein stability [147, 148]. The N192/200/219 glycosylation stabilized PD-L1 and suppressed the formation of GSK3β-β-TrCP-PD-L1 complex [145]. EGF increased PD-L1 expression by promoting glycosylation-induced GSK3β inactivation [145]. Additionally, in epithelial-mesenchymal transition, β-catenin transcriptionally induced the expression of N-glycosyltransferase STT3. The STT3 promoted PD-L1 N-glycosylation, stabilizing and upregulating PD-L1 [149]. Moreover, in glioma, FKBP51s (a co-chaperone) regulated PD-L1 expression by promoting glycosylation modification [150]. Overexpressing or silencing FKBP51s increased or decreased the level of glycosylated-PD-L1 [150].
Palmitoylation
Palmitoylation is a well-studied post-translational lipid modification. Palmitoylation at C272 increased PD-L1 stability by counteracting its ubiquitination [151, 152]. DHHC3 catalyzed C272 palmitoylation of PD-L1 [152]. Silencing DHHC3 enhanced antitumor immune response in vitro and in vivo [152].
Perspectives and conclusion
A growing body of evidence suggests that it is inaccurate to select patients merely by PD-L1 abundance. Understanding the difference between inflammation-induced PD-L1 and oncogenic signal-mediated constitutive PD-L1 is helpful to patient selection. For instance, for EGFR mutant NSCLC patients, α-PD-1 therapy's efficacy was poor despite the high level of PD-L1 [153]. The EGFR mutation-driving NSCLCs commonly harbor lower mutation burdens, and the lower immunogenicity leads to the resistance to α-PD-1 treatments [43]. This oncogenic EGFR-mediated PD-L1 expression could not reflect the real status of the TME. Alternatively, a comprehensive framework containing multiple surrogate markers such as tumor mutation burden would be valuable for selecting patients and predicting outcomes.
Besides, agents regulating PD-L1 expression might have a synergistic effect with the current immune checkpoint inhibitors (Fig. 2). Targeting therapies such as CDK4/6 inhibitor upregulated PD-L1 expression and promoted immune escape [141, 146]. This treatment-induced immune evasion could be overcome by combination therapies containing α-PD-1/PD-L1. Besides, adjuvant treatment regulating PD-L1 expression might elevate the sensitivity to α-PD-1/PD-L1 or other immune checkpoint inhibitors [144, 145]. For example, metformin downregulated PD-L1 by promoting endoplasmic-reticulum-associated degradation, and the combination therapy of metformin and α-CTLA-4 exhibited a synergistic antitumor activity [144].

The current immune checkpoint inhibitor therapy could be further enhanced by regulating PD-L1 expression. Agents regulating PD-L1 expression might have a synergistic effect with the current immune checkpoint inhibitors. For example, targeting therapies such as CDK4/6 inhibitor upregulated PD-L1 expression and promoted immune escape. This treatment-induced immune evasion could be overcome by combination therapies containing α-PD-1/PD-L1 (the left panel). Besides, adjuvant treatment regulating PD-L1 expression might elevate the sensitivity to α-PD-1/PD-L1 or other immune checkpoint inhibitors. For instance, metformin downregulated PD-L1 by promoting endoplasmic-reticulum-associated degradation, and the combination therapy of metformin and α-CTLA-4 had a synergistic antitumor activity (the right panel). CDK: cyclin-dependent kinase; CTLA-4: cytotoxic T Lymphocyte antigen 4; ER: endoplasmic reticulum
Generally, in the TME, the expression of PD-L1 is regulated by numerous factors, including inflammatory stimuli and oncogenic pathways at the levels of transcription, post-transcription, and post-translation. Exploring potential PD-L1 regulators helps select patients and overcome resistance to α-PD-1/PD-L1 treatments. Besides, the agents regulating PD-L1 expression might be possible adjuvant therapies for the current immune checkpoint inhibitors.
Availability of data and materials
Not applicable.
Abbreviations
- PD-1:
-
Programmed cell death-1
- PD-L1:
-
Programmed death-ligand 1
- TME:
-
Tumor microenvironment
- NSCLC:
-
Non-small cell lung cancer
- H3K4me3:
-
Tri-methylation of histone H3 on lysine 4
- H3K27me3:
-
Tri-methylation of histone H3 on lysine 27
- EZH2:
-
Enhancer of zeste homolog 2
- DNMTi:
-
DNA methyltransferase inhibitors
- HADC3:
-
Histone deacetylase 3
- IRF:
-
Interferon-responsive factor
- IFN:
-
Interferon
- PTPRO:
-
Protein tyrosine phosphatase receptor type O
- TNF-α:
-
Tumor necrosis factor-α
- EGFR:
-
Epidermal growth factor receptor
- TKI:
-
Tyrosine kinase inhibitor
- MAPK:
-
Mitogen-activated protein kinase
- MHC:
-
Major histocompatibility complex
- PTEN:
-
Phosphatase and tensin homolog
- PI3K:
-
Phosphoinositide 3-kinase
- NF-κB:
-
Nuclear factor kappa-B
- ROS:
-
Reactive oxygen species
- HIF-1α:
-
Hypoxia-inducible factor-1α
- ALK:
-
Anaplastic lymphoma kinase
- EML4:
-
Echinoderm microtubule-associated protein-like 4
- BET:
-
Bromodomain and extraterminal
- DSB:
-
Double-strand break
- miRNA:
-
MicroRNA
- UTR:
-
Untranslated region
- CDK:
-
Cyclin-dependent kinase
- CTLA-4:
-
Cytotoxic T Lymphocyte antigen 4
- GSK3β:
-
Glycogen synthase kinase 3β
References
Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms controlling PD-L1 expression in cancer. Mol Cell. 2019;76:359–70.
Akinleye A, Rasool Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J Hematol Oncol. 2019;12:92.
Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48:434–52.
Li X, Shao C, Shi Y, Han W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J Hematol Oncol. 2018;11:31.
Bardhan K, Anagnostou T, Boussiotis VA. The PD1:PD-L1/2 pathway from discovery to clinical implementation. Front Immunol. 2016;7:550.
Yi M, Yu S, Qin S, Liu Q, Xu H, Zhao W, et al. Gut microbiome modulates efficacy of immune checkpoint inhibitors. J Hematol Oncol. 2018;11:47.
Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. 2019;18:10.
Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell. 2018;175:313–26.
Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823–33.
Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–13.
Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–32.
Muro K, Chung HC, Shankaran V, Geva R, Catenacci D, Gupta S, et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol. 2016;17:717–26.
El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–502.
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–91.
Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee JL, Fong L, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376:1015–26.
Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372:311–9.
Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856–67.
Chung HC, Ros W, Delord JP, Perets R, Italiano A, Shapira-Frommer R, et al. Efficacy and safety of pembrolizumab in previously treated advanced cervical cancer: results from the Phase II KEYNOTE-158 study. J Clin Oncol. 2019;37:1470–8.
Zhao P, Li L, Jiang X, Li Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J Hematol Oncol. 2019;12:54.
Yi M, Jiao D, Xu H, Liu Q, Zhao W, Han X, et al. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol Cancer. 2018;17:129.
Yu H, Boyle TA, Zhou C, Rimm DL, Hirsch FR. PD-L1 expression in lung cancer. J Thorac Oncol. 2016;11:964–75.
Ribas A, Hu-Lieskovan S. What does PD-L1 positive or negative mean? J Exp Med. 2016;213:2835–40.
Roemer MG, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H, et al. PD-L1 and PD-L2 genetic alterations define classical hodgkin lymphoma and predict outcome. J Clin Oncol. 2016;34:2690–7.
George J, Saito M, Tsuta K, Iwakawa R, Shiraishi K, Scheel AH, et al. Genomic amplification of CD274 (PD-L1) in small-cell lung cancer. Clin Cancer Res. 2017;23:1220–6.
Twa DD, Chan FC, Ben-Neriah S, Woolcock BW, Mottok A, Tan KL, et al. Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood. 2014;123:2062–5.
Howe FS, Fischl H, Murray SC, Mellor J. Is H3K4me3 instructive for transcription activation? BioEssays. 2017;39:1–12.
Lu C, Paschall AV, Shi H, Savage N, Waller JL, Sabbatini ME, et al. The MLL1-H3K4me3 axis-mediated pd-l1 expression and pancreatic cancer immune evasion. J Natl Cancer Inst. 2017;109.
Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications—writers that read. EMBO Rep. 2015;16:1467–81.
Xiao G, Jin LL, Liu CQ, Wang YC, Meng YM, Zhou ZG, et al. EZH2 negatively regulates PD-L1 expression in hepatocellular carcinoma. J Immunother Cancer. 2019;7:300.
Emran AA, Chatterjee A, Rodger EJ, Tiffen JC, Gallagher SJ, Eccles MR, et al. Targeting DNA methylation and EZH2 activity to overcome melanoma resistance to immunotherapy. Trends Immunol. 2019;40:328–44.
Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014;5:587–98.
Chatterjee A, Rodger EJ, Ahn A, Stockwell PA, Parry M, Motwani J, et al. Marked global DNA hypomethylation is associated with constitutive PD-L1 expression in melanoma. iScience. 2018;4:312–25.
Micevic G, Thakral D, McGeary M, Bosenberg MW. PD-L1 methylation regulates PD-L1 expression and is associated with melanoma survival. Pigment Cell Melanoma Res. 2019;32:435–40.
Franzen A, Vogt TJ, Müller T, Dietrich J, Schröck A, Golletz C, et al. PD-L1 (CD274) and PD-L2 (PDCD1LG2) promoter methylation is associated with HPV infection and transcriptional repression in head and neck squamous cell carcinomas. Oncotarget. 2018;9:641–50.
Goltz D, Gevensleben H, Dietrich J, Dietrich D. PD-L1 (CD274) promoter methylation predicts survival in colorectal cancer patients. Oncoimmunology. 2017;6:e1257454.
Asgarova A, Asgarov K, Godet Y, Peixoto P, Nadaradjane A, Boyer-Guittaut M, et al. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. Oncoimmunology. 2018;7:e1423170.
Zhu T, Hu Z, Wang Z, Ding H, Li R, Sun J, et al. Epigenetically silenced PD-L1 confers drug resistance to anti-PD1 therapy in gastric cardia adenocarcinoma. Int Immunopharmacol. 2020;82:106245.
Clayton AL, Hazzalin CA, Mahadevan LC. Enhanced histone acetylation and transcription: a dynamic perspective. Mol Cell. 2006;23:289–96.
Wang H, Fu C, Du J, Wang H, He R, Yin X, et al. Enhanced histone H3 acetylation of the PD-L1 promoter via the COP1/c-Jun/HDAC3 axis is required for PD-L1 expression in drug-resistant cancer cells. J Exp Clin Cancer Res. 2020;39:29.
Woods DM, Sodré AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res. 2015;3:1375–85.
Shi Y, Fu Y, Zhang X, Zhao G, Yao Y, Guo Y, et al. Romidepsin (FK228) regulates the expression of the immune checkpoint ligand PD-L1 and suppresses cellular immune functions in colon cancer. Cancer Immunol Immunother. 2020. https://doi.org/10.1007/s00262-020-02653-1.
Deng S, Hu Q, Zhang H, Yang F, Peng C, Huang C. HDAC3 inhibition upregulates PD-L1 expression in B-cell lymphomas and augments the efficacy of anti-PD-L1 therapy. Mol Cancer Ther. 2019;18:900–8.
Zerdes I, Matikas A, Bergh J, Rassidakis GZ, Foukakis T. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: biology and clinical correlations. Oncogene. 2018;37:4639–61.
Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–201.
Yao S, Jiang L, Moser EK, Jewett LB, Wright J, Du J, et al. Control of pathogenic effector T-cell activities in situ by PD-L1 expression on respiratory inflammatory dendritic cells during respiratory syncytial virus infection. Mucosal Immunol. 2015;8:746–59.
Moon JW, Kong SK, Kim BS, Kim HJ, Lim H, Noh K, et al. IFNγ induces PD-L1 overexpression by JAK2/STAT1/IRF-1 signaling in EBV-positive gastric carcinoma. Sci Rep. 2017;7:17810.
Blazanin N, Cheng T, Carbajal S, DiGiovanni J. Activation of a protumorigenic IFNγ/STAT1/IRF-1 signaling pathway in keratinocytes following exposure to solar ultraviolet light. Mol Carcinog. 2019;58:1656–69.
Yan Y, Zheng L, Du Q, Yan B, Geller DA. Interferon regulatory factor 1 (IRF-1) and IRF-2 regulate PD-L1 expression in hepatocellular carcinoma (HCC) cells. Cancer Immunol Immunother. 2020;69:1891–903.
Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7:188–201.
Xu L, Chen X, Shen M, Yang DR, Fang L, Weng G, et al. Inhibition of IL-6-JAK/Stat3 signaling in castration-resistant prostate cancer cells enhances the NK cell-mediated cytotoxicity via alteration of PD-L1/NKG2D ligand levels. Mol Oncol. 2018;12:269–86.
Zhang W, Liu Y, Yan Z, Yang H, Sun W, Yao Y, et al. IL-6 promotes PD-L1 expression in monocytes and macrophages by decreasing protein tyrosine phosphatase receptor type O expression in human hepatocellular carcinoma. J Immunother Cancer. 2020;8:e000285.
Lamano JB, Lamano JB, Li YD, DiDomenico JD, Choy W, Veliceasa D, et al. Glioblastoma-derived IL6 induces immunosuppressive peripheral myeloid cell PD-L1 and promotes tumor growth. Clin Cancer Res. 2019;25:3643–57.
Shen MJ, Xu LJ, Yang L, Tsai Y, Keng PC, Chen Y, et al. Radiation alters PD-L1/NKG2D ligand levels in lung cancer cells and leads to immune escape from NK cell cytotoxicity via IL-6-MEK/Erk signaling pathway. Oncotarget. 2017;8:80506–20.
Wang X, Yang L, Huang F, Zhang Q, Liu S, Ma L, et al. Inflammatory cytokines IL-17 and TNF-α up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol Lett. 2017;184:7–14.
Quandt D, Jasinski-Bergner S, Müller U, Schulze B, Seliger B. Synergistic effects of IL-4 and TNFα on the induction of B7–H1 in renal cell carcinoma cells inhibiting allogeneic T cell proliferation. J Transl Med. 2014;12:151.
Jiang C, Yuan F, Wang J, Wu L. Oral squamous cell carcinoma suppressed antitumor immunity through induction of PD-L1 expression on tumor-associated macrophages. Immunobiology. 2017;222:651–7.
Carbotti G, Barisione G, Airoldi I, Mezzanzanica D, Bagnoli M, Ferrero S, et al. IL-27 induces the expression of IDO and PD-L1 in human cancer cells. Oncotarget. 2015;6:43267–80.
Starke A, Wüthrich RP, Waeckerle-Men Y. TGF-beta treatment modulates PD-L1 and CD40 expression in proximal renal tubular epithelial cells and enhances CD8 cytotoxic T-cell responses. Nephron Exp Nephrol. 2007;107:e22–9.
Ou JN, Wiedeman AE, Stevens AM. TNF-α and TGF-β counter-regulate PD-L1 expression on monocytes in systemic lupus erythematosus. Sci Rep. 2012;2:295.
David JM, Dominguez C, McCampbell KK, Gulley JL, Schlom J, Palena C. A novel bifunctional anti-PD-L1/TGF-β Trap fusion protein (M7824) efficiently reverts mesenchymalization of human lung cancer cells. Oncoimmunology. 2017;6:e1349589.
Ni XY, Sui HX, Liu Y, Ke SZ, Wang YN, Gao FG. TGF-β of lung cancer microenvironment upregulates B7H1 and GITRL expression in dendritic cells and is associated with regulatory T cell generation. Oncol Rep. 2012;28:615–21.
Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355–63.
Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin Cancer Res. 2016;22:4585–93.
Garassino MC, Gelibter AJ, Grossi F, Chiari R, Soto Parra H, Cascinu S, et al. Italian nivolumab expanded access program in nonsquamous non-small cell lung cancer patients: results in never-smokers and EGFR-mutant patients. J Thorac Oncol. 2018;13:1146–55.
Garassino MC, Cho BC, Kim JH, Mazières J, Vansteenkiste J, Lena H, et al. Durvalumab as third-line or later treatment for advanced non-small-cell lung cancer (ATLANTIC): an open-label, single-arm, phase 2 study. Lancet Oncol. 2018;19:521–36.
Yuan J, Dong X, Yap J, Hu J. The MAPK and AMPK signalings: interplay and implication in targeted cancer therapy. J Hematol Oncol. 2020;13:113.
Stutvoet TS, Kol A, de Vries EG, de Bruyn M, Fehrmann RS, Terwisscha van Scheltinga AG, et al. MAPK pathway activity plays a key role in PD-L1 expression of lung adenocarcinoma cells. J Pathol. 2019;249:52–64.
Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res. 2013;19:598–609.
Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut. 2017;66:124–36.
Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res. 2016;22:1499–509.
Mimura K, Kua LF, Shiraishi K, Kee Siang L, Shabbir A, Komachi M, et al. Inhibition of mitogen-activated protein kinase pathway can induce upregulation of human leukocyte antigen class I without PD-L1-upregulation in contrast to interferon-γ treatment. Cancer Sci. 2014;105:1236–44.
Atefi M, Avramis E, Lassen A, Wong DJ, Robert L, Foulad D, et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res. 2014;20:3446–57.
Haddadi N, Lin Y, Travis G, Simpson AM, Nassif NT, McGowan EM. PTEN/PTENP1: “regulating the regulator of RTK-dependent PI3K/Akt signalling”, new targets for cancer therapy. Mol Cancer. 2018;17:37.
Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res. 2014;2:361–70.
Kim YB, Ahn JM, Bae WJ, Sung CO, Lee D. Functional loss of ARID1A is tightly associated with high PD-L1 expression in gastric cancer. Int J Cancer. 2019;145:916–26.
Suh KJ, Sung JH, Kim JW, Han SH, Lee HS, Min A, et al. EGFR or HER2 inhibition modulates the tumor microenvironment by suppression of PD-L1 and cytokines release. Oncotarget. 2017;8:63901–10.
Huang TY, Chang TC, Chin YT, Pan YS, Chang WJ, Liu FC, et al. NDAT targets PI3K-mediated PD-L1 upregulation to reduce proliferation in gefitinib-resistant colorectal cancer. Cells. 2020;9:1830.
Fiedler M, Schulz D, Piendl G, Brockhoff G, Eichberger J, Menevse AN, et al. Buparlisib modulates PD-L1 expression in head and neck squamous cell carcinoma cell lines. Exp Cell Res. 2020;396:112259.
Lastwika KJ, Wilson W 3rd, Li QK, Norris J, Xu H, Ghazarian SR, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 2016;76:227–38.
Song TL, Nairismägi ML, Laurensia Y, Lim JQ, Tan J, Li ZM, et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood. 2018;132:1146–58.
Ikeda S, Okamoto T, Okano S, Umemoto Y, Tagawa T, Morodomi Y, et al. PD-L1 is upregulated by simultaneous amplification of the PD-L1 and JAK2 genes in non-small cell lung cancer. J Thorac Oncol. 2016;11:62–71.
Shen M, Xu Z, Xu W, Jiang K, Zhang F, Ding Q, et al. Inhibition of ATM reverses EMT and decreases metastatic potential of cisplatin-resistant lung cancer cells through JAK/STAT3/PD-L1 pathway. J Exp Clin Cancer Res. 2019;38:149.
Chen M, Pockaj B, Andreozzi M, Barrett MT, Krishna S, Eaton S, et al. JAK2 and PD-L1 amplification enhance the dynamic expression of PD-L1 in triple-negative breast cancer. Clin Breast Cancer. 2018;18:e1205–15.
Bi XW, Wang H, Zhang WW, Wang JH, Liu WJ, Xia ZJ, et al. PD-L1 is upregulated by EBV-driven LMP1 through NF-κB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J Hematol Oncol. 2016;9:109.
Wang W, Chapman NM, Zhang B, Li M, Fan M, Laribee RN, et al. Upregulation of PD-L1 via HMGB1-activated IRF3 and NF-κB contributes to UV radiation-induced immune suppression. Cancer Res. 2019;79:2909–22.
Jin X, Ding D, Yan Y, Li H, Wang B, Ma L, et al. Phosphorylated RB promotes cancer immunity by inhibiting NF-κB activation and PD-L1 expression. Mol Cell. 2019;73(22–35):e6.
Xu D, Li J, Li RY, Lan T, Xiao C, Gong P. PD-L1 expression is regulated by NF-κB during EMT signaling in gastric carcinoma. Onco Targets Ther. 2019;12:10099–105.
Li H, Xia JQ, Zhu FS, Xi ZH, Pan CY, Gu LM, et al. LPS promotes the expression of PD-L1 in gastric cancer cells through NF-κB activation. J Cell Biochem. 2018;119:9997–10004.
Bouillez A, Rajabi H, Jin C, Samur M, Tagde A, Alam M, et al. MUC1-C integrates PD-L1 induction with repression of immune effectors in non-small-cell lung cancer. Oncogene. 2017;36:4037–46.
Roux C, Jafari SM, Shinde R, Duncan G, Cescon DW, Silvester J, et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc Natl Acad Sci U S A. 2019;116:4326–35.
Teicher BA. Hypoxia and drug resistance. Cancer Metastasis Rev. 1994;13:139–68.
Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014;74:665–74.
Sellam LS, Zappasodi R, Chettibi F, Djennaoui D, Yahi-Ait Mesbah N, Amir-Tidadini ZC, et al. Silibinin down-regulates PD-L1 expression in nasopharyngeal carcinoma by interfering with tumor cell glycolytic metabolism. Arch Biochem Biophys. 2020;690:108479.
Meng L, Cheng Y, Tong X, Gan S, Ding Y, Zhang Y, et al. Tumor oxygenation and hypoxia inducible factor-1 functional inhibition via a reactive oxygen species responsive nanoplatform for enhancing radiation therapy and abscopal effects. ACS Nano. 2018;12:8308–22.
Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35.
Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227–31.
Liang MQ, Yu FQ, Chen C. C-Myc regulates PD-L1 expression in esophageal squamous cell carcinoma. Am J Transl Res. 2020;12:379–88.
Kim EY, Kim A, Kim SK, Chang YS. MYC expression correlates with PD-L1 expression in non-small cell lung cancer. Lung Cancer. 2017;110:63–7.
Pan Y, Fei Q, Xiong P, Yang J, Zhang Z, Lin X, et al. Synergistic inhibition of pancreatic cancer with anti-PD-L1 and c-Myc inhibitor JQ1. Oncoimmunology. 2019;8:e1581529.
Zou J, Zhuang M, Yu X, Li N, Mao R, Wang Z, et al. MYC inhibition increases PD-L1 expression induced by IFN-γ in hepatocellular carcinoma cells. Mol Immunol. 2018;101:203–9.
Han H, Jain AD, Truica MI, Izquierdo-Ferrer J, Anker JF, Lysy B, et al. Small-molecule MYC inhibitors suppress tumor growth and enhance immunotherapy. Cancer Cell. 2019;36(483–97):e15.
Shaw AT, Engelman JA. ALK in lung cancer: past, present, and future. J Clin Oncol. 2013;31:1105–11.
Ota K, Azuma K, Kawahara A, Hattori S, Iwama E, Tanizaki J, et al. Induction of PD-L1 expression by the EML4-ALK oncoprotein and downstream signaling pathways in non-small cell lung cancer. Clin Cancer Res. 2015;21:4014–21.
Shen J, Li S, Medeiros LJ, Lin P, Wang SA, Tang G, et al. PD-L1 expression is associated with ALK positivity and STAT3 activation, but not outcome in patients with systemic anaplastic large cell lymphoma. Mod Pathol. 2020;33:324–33.
Drilon A, Cappuzzo F, Ou SI, Camidge DR. Targeting MET in lung cancer: will expectations finally be MET? J Thorac Oncol. 2017;12:15–26.
Comoglio PM, Trusolino L, Boccaccio C. Known and novel roles of the MET oncogene in cancer: a coherent approach to targeted therapy. Nat Rev Cancer. 2018;18:341–58.
Albitar M, Sudarsanam S, Ma W, Jiang S, Chen W, Funari V, et al. Correlation of MET gene amplification and TP53 mutation with PD-L1 expression in non-small cell lung cancer. Oncotarget. 2018;9:13682–93.
Demuth C, Andersen MN, Jakobsen KR, Madsen AT, Sørensen BS. Increased PD-L1 expression in erlotinib-resistant NSCLC cells with MET gene amplification is reversed upon MET-TKI treatment. Oncotarget. 2017;8:68221–9.
Ahn HK, Kim S, Kwon D, Koh J, Kim YA, Kim K, et al. MET receptor tyrosine kinase regulates the expression of co-stimulatory and co-inhibitory molecules in tumor cells and contributes to PD-L1-mediated suppression of immune cell function. Int J Mol Sci. 2019;20:4287.
Peng S, Wang R, Zhang X, Ma Y, Zhong L, Li K, et al. EGFR-TKI resistance promotes immune escape in lung cancer via increased PD-L1 expression. Mol Cancer. 2019;18:165.
Donati B, Lorenzini E, Ciarrocchi A. BRD4 and Cancer: going beyond transcriptional regulation. Mol Cancer. 2018;17:164.
Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, et al. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. 2016;16:2829–37.
Hogg SJ, Vervoort SJ, Deswal S, Ott CJ, Li J, Cluse LA, et al. BET-bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep. 2017;18:2162–74.
Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun. 2017;8:1751.
Yi M, Xu L, Jiao Y, Luo S, Li A, Wu K. The role of cancer-derived microRNAs in cancer immune escape. J Hematol Oncol. 2020;13:25.
Ashizawa M, Okayama H, Ishigame T, Thar Min AK, Saito K, Ujiie D, et al. miRNA-148a-3p regulates immunosuppression in DNA mismatch repair-deficient colorectal cancer by targeting PD-L1. Mol Cancer Res. 2019;17:1403–13.
Gao L, Guo Q, Li X, Yang X, Ni H, Wang T, et al. MiR-873/PD-L1 axis regulates the stemness of breast cancer cells. EBioMedicine. 2019;41:395–407.
Anastasiadou E, Stroopinsky D, Alimperti S, Jiao AL, Pyzer AR, Cippitelli C, et al. Epstein-Barr virus-encoded EBNA2 alters immune checkpoint PD-L1 expression by downregulating miR-34a in B-cell lymphomas. Leukemia. 2019;33:132–47.
Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5:5241.
Sun C, Lan P, Han Q, Huang M, Zhang Z, Xu G, et al. Oncofetal gene SALL4 reactivation by hepatitis B virus counteracts miR-200c in PD-L1-induced T cell exhaustion. Nat Commun. 2018;9:1241.
Jia L, Xi Q, Wang H, Zhang Z, Liu H, Cheng Y, et al. miR-142-5p regulates tumor cell PD-L1 expression and enhances anti-tumor immunity. Biochem Biophys Res Commun. 2017;488:425–31.
Xu S, Tao Z, Hai B, Liang H, Shi Y, Wang T, et al. miR-424(322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nat Commun. 2016;7:11406.
Sun JR, Zhang X, Zhang Y. MiR-214 prevents the progression of diffuse large B-cell lymphoma by targeting PD-L1. Cell Mol Biol Lett. 2019;24:68.
Qu F, Ye J, Pan X, Wang J, Gan S, Chu C, et al. MicroRNA-497-5p down-regulation increases PD-L1 expression in clear cell renal cell carcinoma. J Drug Target. 2019;27:67–74.
Xie WB, Liang LH, Wu KG, Wang LX, He X, Song C, et al. MiR-140 expression regulates cell proliferation and targets PD-L1 in NSCLC. Cell Physiol Biochem. 2018;46:654–63.
Wang X, Li J, Dong K, Lin F, Long M, Ouyang Y, et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal. 2015;27:443–52.
Liu J, Fan L, Yu H, Zhang J, He Y, Feng D, et al. Endoplasmic reticulum stress causes liver cancer cells to release exosomal miR-23a-3p and up-regulate programmed death ligand 1 expression in macrophages. Hepatology. 2019;70:241–58.
Liu Z, Wen J, Wu C, Hu C, Wang J, Bao Q, et al. MicroRNA-200a induces immunosuppression by promoting PTEN-mediated PD-L1 upregulation in osteosarcoma. Aging (Albany NY). 2020;12:1213–36.
Yao X, Tu Y, Xu Y, Guo Y, Yao F, Zhang X. Endoplasmic reticulum stress-induced exosomal miR-27a-3p promotes immune escape in breast cancer via regulating PD-L1 expression in macrophages. J Cell Mol Med. 2020;24:9560–73.
Sheng Q, Zhang Y, Wang Z, Ding J, Song Y, Zhao W. Cisplatin-mediated down-regulation of miR-145 contributes to up-regulation of PD-L1 via the c-Myc transcription factor in cisplatin-resistant ovarian carcinoma cells. Clin Exp Immunol. 2020;200:45–52.
Dong P, Xiong Y, Yu J, Chen L, Tao T, Yi S, et al. Control of PD-L1 expression by miR-140/142/340/383 and oncogenic activation of the OCT4-miR-18a pathway in cervical cancer. Oncogene. 2018;37:5257–68.
Tang D, Zhao D, Wu Y, Yao R, Zhou L, Lu L, et al. The miR-3127-5p/p-STAT3 axis up-regulates PD-L1 inducing chemoresistance in non-small-cell lung cancer. J Cell Mol Med. 2018;22:3847–56.
Yoon CJ, Chang MS, Kim DH, Kim W, Koo BK, Yun SC, et al. Epstein-Barr virus-encoded miR-BART5-5p upregulates PD-L1 through PIAS3/pSTAT3 modulation, worsening clinical outcomes of PD-L1-positive gastric carcinomas. Gastric Cancer. 2020;23:780–95.
Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3’-UTR disruption in multiple cancers. Nature. 2016;534:402–6.
Coelho MA, de Carné TS, Rana S, Zecchin D, Moore C, Molina-Arcas M, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity. 2017;47(1083–99):e6.
Yang K, Zhou J, Chen Y, Chen Y, Chen L, Zhang P, et al. Angiotensin II contributes to intratumoral immunosuppressionvia induction of PD-L1 expression in non-small cell lung carcinoma. Int Immunopharmacol. 2020;84:106507.
Wu Z, Huang R, Yuan L. Crosstalk of intracellular post-translational modifications in cancer. Arch Biochem Biophys. 2019;676:108138.
Burr ML, Sparbier CE, Chan YC, Williamson JC, Woods K, Beavis PA, et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017;549:101–5.
Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ. 2013;20:21–30.
Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature. 2017;549:106–10.
Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553:91–5.
Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30:925–39.
Chan LC, Li CW, Xia W, Hsu JM, Lee HH, Cha JH, et al. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J Clin Invest. 2019;129:3324–38.
Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell. 2018;71(606–20):e7.
Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632.
Li H, Li CW, Li X, Ding Q, Guo L, Liu S, et al. MET inhibitors promote liver tumor evasion of the immune response by stabilizing PDL1. Gastroenterology. 2019;156(1849–61):e13.
Veillon L, Fakih C, Abou-El-Hassan H, Kobeissy F, Mechref Y. Glycosylation changes in brain cancer. ACS Chem Neurosci. 2018;9:51–72.
Shi X, Zhang D, Li F, Zhang Z, Wang S, Xuan Y, et al. Targeting glycosylation of PD-1 to enhance CAR-T cell cytotoxicity. J Hematol Oncol. 2019;12:127.
Hsu JM, Xia W, Hsu YH, Chan LC, Yu WH, Cha JH, et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun. 2018;9:1908.
D'Arrigo P, Russo M, Rea A, Tufano M, Guadagno E, Del Basso De Caro ML, et al. A regulatory role for the co-chaperone FKBP51s in PD-L1 expression in glioma. Oncotarget. 2017;8:68291–304.
Yang Y, Hsu JM, Sun L, Chan LC, Li CW, Hsu JL, et al. Palmitoylation stabilizes PD-L1 to promote breast tumor growth. Cell Res. 2019;29:83–6.
Yao H, Lan J, Li C, Shi H, Brosseau JP, Wang H, et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat Biomed Eng. 2019;3:306–17.
Lisberg A, Cummings A, Goldman JW, Bornazyan K, Reese N, Wang T, et al. A phase II study of pembrolizumab in EGFR-mutant, PD-L1+, tyrosine kinase inhibitor naïve patients with advanced NSCLC. J Thorac Oncol. 2018;13:1138–45.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81874120, 82073370).
Author information
Authors and Affiliations
Contributions
MY drafted the manuscript and prepared the figures. MN and LX collected the related references and participated in discussion. SL and KW designed this review and revised the manuscript. All authors contributed to this manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yi, M., Niu, M., Xu, L. et al. Regulation of PD-L1 expression in the tumor microenvironment. J Hematol Oncol 14, 10 (2021). https://doi.org/10.1186/s13045-020-01027-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-01027-5