
Abstract
Tumor immune escape is an important strategy of tumor survival. There are many mechanisms of tumor immune escape, including immunosuppression, which has become a research hotspot in recent years. The programmed death ligand-1/programmed death-1 (PD-L1/PD-1) signaling pathway is an important component of tumor immunosuppression, which can inhibit the activation of T lymphocytes and enhance the immune tolerance of tumor cells, thereby achieving tumor immune escape. Therefore, targeting the PD-L1/PD-1 pathway is an attractive strategy for cancer treatment; however, the therapeutic effectiveness of PD-L1/PD-1 remains poor. This situation requires gaining a deeper understanding of the complex and varied molecular mechanisms and factors driving the expression and activation of the PD-L1/PD-1 signaling pathway. In this review, we summarize the regulation mechanisms of the PD-L1/PD-1 signaling pathway in the tumor microenvironment and their roles in mediating tumor escape. Overall, the evidence accumulated to date suggests that induction of PD-L1 by inflammatory factors in the tumor microenvironment may be one of the most important factors affecting the therapeutic efficiency of PD-L1/PD-1 blocking.
Similar content being viewed by others

Background
Tumor immune escape refers to the phenomenon by which tumor cells can grow and metastasize by avoiding recognition and attack by the immune system through various mechanisms, which is an important strategy for tumor survival and development [1].The are many inducible factors of tumor immune escape, including the low immunogenicity of tumor cells, recognition of tumor-specific antibodies as autoantigens, tumor surface antigen modulation, tumor-induced exemption regions, and tumor-induced immunosuppression, the latter of which has been the most extensively studied mechanism to date. Tumor-induced immunosuppression operates in two main ways. The first occurs by inducing immunosuppressive cells to accumulate around the tumor and secrete immunosuppressive factors, which inactivate cytolytic T lymphocytes (CTL) to decrease the immune tolerance of tumor cells, such as regulatory T cells (Treg cells) [2, 3], myeloid-derived suppressor cells (MDSCs) [4], dendritic cells (DCs) [5], and M2 macrophages [6, 7].The second mechanism of immunosuppression involves induction of the expression of immunosuppressive molecules or their receptors, including programmed death-ligand 1/programmed death-1 (PD-L1/PD-1), galectin-9/TIM-3, IDO1, LAG-3, and CTLA4, which are known as the immune checkpoints and can inhibit the activation of effector T lymphocytes, ultimately leading to tumor immune escape. Thus, blocking these immune checkpoints has become an important direction of immunotherapy in recent years to eliminate the immune suppression and restore immune system function. Among these immune checkpoint blockers, PD-L1/PD-L1 antagonists account for the largest proportion of drugs approved by the FDA in recent years and are currently in clinical trials.
PD-1, also known as cluster of differentiation 279 (CD279), has attracted a substantial amount of attention in the field of cancer research in recent years. PD-1 was originally cloned from a drug-treated mouse hybridoma and hematopoietic progenitor cell lines in an apoptotic state by subtractive hybridization in 1992, and was considered to be mainly involved in the process of cell apoptosis, from which its name is derived [8]. Human PD-1 is encoded by the PDCD1 gene, located on chromosome 2q37, which is a type I transmembrane protein composed of 288 amino acid residues, belonging to the immunoglobulin CD28 family. PD-1 is expressed in a wide range of immune cells, including peripherally activated T cells, B cells, monocytes, natural killer (NK) cells, and certain DCs. Weaker PD-1 expression has also been detected on the surface of immature T cells and B cells located in the thymus and bone marrow during specific developmental stages [9, 10]. When binding to its ligand, PD-1 can activate intracellular signaling pathways and inhibit the activation of immune cells, thereby reducing the secretion of antibodies and cytokines by immune cells to even exhaust the immune cell and thus maintain immune system homeostasis. PD-L1 (B7-H1 or CD274) was the first ligand of PD-1 discovered [11], which belongs to the B7 family and is located on human chromosome 9 p24.2. Its amino acid structure is similar to that of PD-1. PD-L1 is widely expressed. In addition to lymphocytes, PD-L1 is also widely expressed in non-blood cells such as in lung, vascular endothelium, reticular fibroblasts, non-parenchymal liver cells, mesenchymal stem cells, islet cells, astrocytes, neuronal cells, and keratinocytes [9, 12, 13]. In addition, PD-L1 also shows abnormally high expression in tumor cells, which is considered the main factor responsible for promoting the ability of tumor immune escape [14,15,16,17].
However, the therapeutic effect of a PD-1/PD-L1 antagonist against solid tumors is currently not satisfactory. In PD-L1-positive metastatic melanoma or lung cancer, the effective rate of anti-PD-L1 antagonists is only 40–50%. In colorectal cancer, although the PD-L1-positive rate is 40–50%, anti-PD-1 or anti-PD-L1 drugs show very low efficacy [18]. This poor treatment response, in addition to the high variation of genetic mutations among individuals, may also be related to the complex microenvironment of tumors. The role of the tumor microenvironment in tumor growth and metastasis has long been recognized. Recent studies have also shown that many cytokines and tumor-derived exosomes in the tumor microenvironment can induce the expression of PD-L1 and promote tumor immune escape. This review provides a summary of recent research progress toward understanding the molecular mechanism of PD-L1/PD-1 in tumor immune escape, and the regulation of PD-1 and PD-L1 in the tumor microenvironment. This research progress and indication of remaining questions can help to better understand the tumor immune escape mechanism toward developing more effective immunotherapies for cancer patients.
Tumor microenvironment
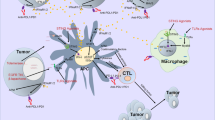
A tumor is not simply a cell mass composed of malignant cells but is actually composed of a large number of non-transformed cells recruited by malignant cells, eventually forming a complex structure composed of both malignant cells and non-transformed cells, and their interaction forms the tumor microenvironment [19,20,21,22,23,24]. The tumor microenvironment consists mainly of vasculature, extracellular matrix (ECM) [25, 26], and other non-malignant cells surrounding the tumor, as well as a complex signaling molecule network that sustains the internal connections of the microenvironment, including growth factors, cytokines, chemokines, and exosomes [27, 28] (Fig. 1). In recent years, with the development of biological technology, different types of cells were identified in the microenvironment, including stromal cells, fibroblasts, fat cells, vascular endothelial cells, and immune cells such as T lymphocytes, B lymphocytes, NK cells, tumor-associated macrophages, and so on [27]. Most of these cells can secrete cytokines and play a role in promoting or inhibiting tumors. Among them, mesenchymal cells and fibroblasts can secrete growth factors such as hepatocyte growth factor, fibroblast growth factor, vascular endothelial growth factor (VEGF), metal secretory proteins MMP2 and CXCL12, and chemokines in the tumor microenvironment. These cytokines not only promote the growth and survival of malignant tumor cells but also their invasion and migration [29, 30]. Vascular endothelial cells produce blood vessels that supply oxygen to tumor cells and carry away metabolic waste. However, the blood vessels generated inside the tumor are incomplete and have weak function; thus, new blood vessels need to be generated constantly, which further complicates the internal blood vessel network [31]. The adipose tissue is an important component of the tumor microenvironment, which can provide a hypoxic and inflammatory microenvironment for tumors [32]. In particular, interleukin (IL)-6, leptin, and adiponectin secreted by fat cells play an important role in tumor growth [33].

Cell growth factors and cytokines secreted in the tumor microenvironment. Abbreviations: CAFS, cancer-associated fibroblasts; Mφ, macrophage; Breg: B regulatory cell; T cells, T lymphocytes; NK, natural killer cell; DC, dendritic cells; TAN, tumor-associated neutrophil; MDSC, myeloid-derived suppressor cell; TAAC, tumor-associated adipose cells; VEC, vascular endothelial cell; ECM, extracellular matrix
Immune cells are the most important defensive weapon of the human body. The immune system is composed of various immune cells to protect against invading or infectious pathogens and to eliminate damaged or cancerous cells [34, 35]. The immune cells located in the tumor microenvironment comprise T cells, Tregs, B regulatory cells (Bregs), NK cells, DCs, MDSCs, and macrophages, among others. Tumor-infiltrating T lymphocytes are important effector cells in the immune system and can be divided into CD4+ T cells (helper T cells) and CD8+ T cells (cytotoxic T cells). These cells can secrete anti-tumor cytokines such as interferon (IFN)-γ, tumor necrosis factor (TNF)-α, IL-17, and IL-2. Tregs and Bregs are immunosuppressive cells in the immune system that secrete IL-10, IL-35, and transforming growth factor (TGF)-β to suppress the immune response of T lymphocytes, and thus prevent damage from the excessive activation of T cells. NK cells account for approximately 10% of the peripheral lymphocytes and are widely distributed in the peripheral blood, lymph nodes, spleen, and bone marrow, but can also migrate to an inflammatory site under the induction of chemokines. The main role of NK cells is to exert cytotoxicity. After activation, NK cells can secrete IFN-γ, TNF-α, and granulocyte macrophage-colony-stimulating factor (GM-CSF) to exert anti-tumor effects [36]. DCs can express immune co-stimulatory molecules as well as initial inflammatory factors to promote Th1 and cytotoxicity, including IL-1, IL-12, and IL-23 [37]. MDSCs are also a class of immunosuppressive cells that can up-regulate the immunosuppressive products nitric oxide synthase and reactive oxygen species to inhibit the activity of immune cytotoxic T cells [38]. Neutrophils are mainly abundant in the peripheral blood, and can produce a large number of proteases and growth factors such as MMP9 and VEGF, contributing to promoting the growth and metastasis of tumor cells [39]. Macrophages can be divided into M1 type and M2 type macrophages: M1-type macrophages have anti-tumor characteristics and secrete pro-inflammatory factors such as IL-6, IL-8, IL-1β, IFN-γ, and TNF-α, whereas M2-type macrophages have tumor-promoting properties and secrete anti-inflammatory factors such as IL-10, IL-4, epithelial growth factor (EGF), TGF-β, and IL-19, thereby playing an important role in promoting tumor cell proliferation and migration [40, 41].
The ECM consists of the basement membrane and intercellular mass, which is an important barrier for tumor metastasis. The ECM contains a large number of growth factors, cytokines, and various metalloproteinases secreted by tumor cells and other cells in the microenvironment, along with various acidic substances produced by tumor metabolism to maintain the weak acidic environment of tumors, which could induce the tumor epithelial-mesenchymal transition (EMT) and facilitate a low-oxygen environment. Indeed, owing to the rapid cell growth of tumors and the insufficiency of tumor angiogenesis, a hypoxic environment is a common characteristic of the tumor microenvironment. In addition, exosomes carrying non-coding RNAs are another important component of the tumor microenvironment, which contribute to the growth and migration of tumor cells [42, 43].
Since the PD-L1/PD-1 signaling pathway is one of the important pathways of tumor immune escape, regulating the expression level of PD-L1 could help to manipulate these components of the tumor microenvironment through inhibition of the activation of T cells so as to eliminate the immune surveillance of the tumor microenvironment and prevent tumors from achieving immune evasion.
Effect of the PD-L1/PD-1 signaling pathway in immune cells
PD-1 is a specific receptor of PD-L1. After binding, PD-1 can inhibit the activation of lymphocytes, reduce the secretion of lymphocyte cytokines, and thus promote the apoptosis of lymphocytes. PD-1 protein is a type I transmembrane protein with a molecular weight of 55–60 kDa, composed of an extracellular IgV-like domain, hydrophobic transmembrane region, and intracellular region. The C-terminal and N-terminal amino acid residues of the intracellular domain of PD-1 have two independent phosphorylation sites: the immunoreceptor tyrosine-based inhibitory motif (ITIM) and the immunoreceptors tyrosine-based switch motif (ITSM) [44]. The ITSM is an important structural site for PD-1 to exert its biological function: when PD-1 binds to PD-L1, the ITSM region will be phosphorylated to activate a series of intracellular signaling pathways and achieve efficient immune inhibition. However, the specific mechanism of PD-1 activation differs between T and B lymphocytes.
T cells
The classical model of the T cell-mediated immune response during pathogen invasion involves the regulation of two signals. The first signal is the combination of the pathogen-derived peptide antigen and the major histocompatibility complex on the surface of an antigen-presenting cell (APC), which transmits signals to T cells that are identified by antigen-specific receptors. However, the first signal presented by the APC is not sufficient to activate the T cell immune response, and a second signal is required so that the immunostimulatory molecule ligand expressed by the APC interacts with the T cell receptor (TCR). Through this interaction, the signal of activation or inhibition is transmitted to T cells to regulate the immune response. Therefore, the immune co-stimulating molecule in the second signal is the key molecule of the T cell immune response, which plays an important role in T cell activation, tolerance, and apoptosis [45]. PD-L1/PD-1 can inhibit the TCR-mediated activation of T cells. The specific mechanism is as follows. When the TCR signal is activated, the ligand-bound PD-1 intracellular tyrosine is phosphorylated and then activated. PD-1 then recruits SHP-1 and SHP-2 to the C-terminal ITSM, which can dephosphorylate the TCR activation signals CD-3ζ and ZAP70, leading to downstream PI3K/AKT signaling inhibition. PI3K/AKT inactivation downregulates the expression of the cell survival gene Bcl-xl and promotes T lymphocyte apoptosis, while simultaneously inhibiting the secretion of cytokines by T lymphocytes [46]. This mechanism differs from the activation of AKT by CTLA4, in which PD-1 binds to the p85α subunit and p110σ subunit of PI3K in the TCR molecular cluster to activate early T cells, thereby inhibiting the phosphorylation of PI3K. PD-1 has also been suggested to activate PTEN, thereby inhibiting TCR-mediated PI3K/AKT activation [47, 48]. In addition, PD-1 can inhibit activation of the RAS-ERK1/2 signaling pathway to consequently inhibit the proliferation of T lymphocytes; alternatively, PD-1 can inhibit the activation of PKCδ and reduce the secretion level of IL-2 by T cells [49, 50] (Fig. 2b).

PD-1 signaling in B cells and T cells. a In B cells, upon PD-1 activation, SHP-2 is recruited to the C-terminal of PD-1 and dephosphorylates downstream members of the BCR pathway (e.g., SyK, Igα/β), thereby disrupting the normal BCR response as well as inhibiting PLCγ2, ERK, and PI3K signaling. This PD-1 activation consequently reduces the stability of the immunological synapse as well as B cell cycle arrest and causes disorder of Ca2+ mobilization. b In T cells, when PD-1 combines with PD-L1, SHP-1/2 are recruited to the C-terminal of PD-1 immediately and dephosphorylate key signal transducers, including the ZAP70, CD3δ, and PI3K pathways, thus suppressing TCR-mediated cell proliferation and cytokine production
B cells
PD-1 can also inhibit the activation of B cells. To understand the mechanism, researchers constructed a chimeric molecule composed of the extracellular region of the IgG Fc IIB receptor and the intracellular region of PD-1. After expressing this molecule in B lymphocyte strains, they found that PD-1 prevented B cell receptor (BCR)-mediated growth inhibition, and also inhibited the intracellular transport of Ca2+ and the tyrosine phosphorylation of some effector molecules [51]. These effects occurred because when the ligand-bound PD-1 is linked to BCR, the two tyrosines on the PD-1 ITSM are phosphorylated so that SHP-2 molecules are recruited to the C-terminus of PD-1 and phosphorylated. Phosphorylated SHP-2 can then dephosphorylate BCR signaling molecules such as SyK and Igα/β, leading to dephosphorylation of the downstream molecules PI3K, PLCγ2, and ERK, resulting in acute Ca2+ disorder and long-term growth stagnation. In addition, the expression of PD-1 can weaken the immune response of B lymphocytes to antigens [52]. Activation of PD-1 also inhibits the secretion of antibodies by B cells in the presence of type 2 antigen stimulation [53, 54] (Fig. 2a).
Therefore, many factors in the tumor microenvironment can induce PD-L1 expression and promote immune escape, which will be discussed in detail in the following section.
Regulation network of PD-1/PD-L1 in the tumor microenvironment
Regulation of PD-1
As an immunosuppressive molecule, PD-1 can inhibit the activation of T lymphocytes and promote T lymphocytes apoptosis. PD-1 is also affected by the tumor environment (Fig. 3). The expression of PD-1 is strictly regulated in T lymphocytes, with low or even undetectable expression in naïve T lymphocytes, but is rapidly induced when the TCR signal is activated [55]. TGF-β plays an important role in in this process. When TGF-β is blocked, the TCR activation-induced PD-1 can be inhibited significantly [56]. Moreover, when stimulated by an antigen (e.g., anti-CD3), CD4+ T lymphocytes, CD8+ T cells, NK T cells, and monocytes are all induced to express PD-1 on the cell membrane [57]. This may be related to the body’s own protective mechanism, which can inhibit the excessive activation of immune cells. Upon TCR activation, the influx of calcium ions into cells will activate NFATc1, when then translocates to the nucleus where it can bind to the 5′-terminal region of the PD-1 promoter, thereby increasing the transcription level of the PD-1 gene [58]. In addition, IL-12 and IL-6 can also induce PD-1 upon TCR activation by changing the chromatin structure of the PD-1 gene and enhancing the transcription of PD-1 by activating STAT3/STAT4. However, this effect cannot be achieved by IL-6 or IL-12 alone [59], and requires the proximal cis-element CRC of the PD-1 promoter, along with the transcription factors FOXO1 and NF-κB [60]. Other factors in the tumor microenvironment can also regulate the expression of PD-1. IL-7, IL-15, and IL-21 can induce PD-1 expression in peripheral T lymphocytes, although up-regulated PD-1 does not affect the expansion and survival of T cells by these cytokines, but rather inhibits the secretion of cytokines in T lymphocytes [61]. In macrophages, IFN-α can also regulate the expression of PD-1 by activating the JAK/STAT signaling pathway, which can form the P48/STAT1/STAT2 complex that binds to the ISRE binding site on the PD-1 promoter, thereby enhancing PD-1 transcription [62]. In addition, in mouse T lymphocytes, IFN-α can also act synergistically with TCR signals to regulate the expression of PD-1, and generates strong inhibition feedback signals for T lymphocyte-mediated immune responses [63]. In addition, the inflammatory factors TNF-α and IL-6 can also neutralize the growth inhibition of PD-1 on T lymphocytes in osteoarthritis, which is mainly achieved by inducing the secretion of soluble PD-1 to interfere with the interaction of PD-1 and PD-L1 [64]. To date, few studies have focused on the regulation of PD-1, and thus the detailed regulation mechanism is not very clear. With the development and deepening of immunotherapy, it is believed that more extensive and in-depth research will be conducted on the regulation mechanism of PD-1 in the future.

The regulation network of PD-1 in the tumor microenvironment
Regulation of PD-L1
In addition to being widely expressed on the surface of T lymphocytes, B lymphocytes, DCs, and macrophages, high expression of PD-L1 is also found on the surface of many tumor cells, causing T cell exhaustion and immune tolerance, leading to immune escape [65]. As summarized above, a variety of cytokines and exosomes in the tumor microenvironment [66,67,68] can induce the expression of PD-L1, enhance the PD-L1/PD-1 signal to inhibit CTL activation in the tumor microenvironment, and thereby promote tumor escape. The mechanism of induction of PD-L1 by these cytokines and exosomes is schematically summarized in Figs. 4 and 5. Here, we focus on some of the key players of PD-L1 regulation: IFN-γ, TNF-a, cell growth factors, hypoxia, and exosomes.

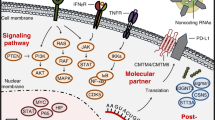
Epigenetic modification of PD-L1 by the tumor microenvironment. a IFN-γ can regulate the translation of PD-L1 mRNA via upregulating miR-155 or downregulating miR-513, and EGF can enhance the mRNA stability via the RAS-ERK1/2-TPP pathway. b EGF can reduce PD-L1 degradation via upregulating B3NT3 or downregulating GSK3β; alternatively, EGF can enhance PD-L1 protein stability via the PTEN/PI3K/mTOR /S6K1 pathway

Transcription regulation network of PD-L1 in the tumor microenvironment
IFN-γ
IFN is a bioactive glycoprotein that is secreted by cells when they are infected by viruses, with antiviral, antibacterial, antitumor, and immune regulation functions [69]. IFN-γ belongs to the type II IFN family, which is mainly secreted by CD8+ T lymphocytes, NK cells, and macrophages. When two IFN-γ molecules interact with IFNGR1/IFNGR2, a biologically active tetrameric complex can be formed, which plays an important role in both innate and adaptive immunity [70]. IFN-γ has long been considered an anti-inflammatory cytokine that plays an important role in antiviral and bacterial infections as well as anti-tumor adjuvant therapy [71]. However, in recent years, accumulating evidence has indicated that IFN-γ does not show complete anti-tumor efficacy, and in some cases, can also actually promote tumor growth and resist tumor immunological monitoring [72]. For example, adjuvant treatment of IFN-γ to melanoma patients had to be terminated prematurely because these patients had a worse outcome than those who did not receive the treatment [73]. In another trial, the total survival time of patients with advanced ovarian cancer that received a combined regimen with IFN-γ and carboplatin/paclitaxel was significantly shorter than that of patients treated with carboplatin/paclitaxel alone [74]. These results demonstrated that IFN-γ therapy not only is ineffective at inhibiting the tumor but also promotes the growth of the tumor in certain conditions. The poorer prognosis of patients under IFN-γ regimens suggests that IFN-γ may have a positive regulating effect on certain tumor-promoting factors. With increased research focus on immune checkpoints in recent years, IFN-γ was shown to induce the expression of PD-L1 and thus promote the escape of tumor cells from the body’s immune surveillance by protecting tumor cells from specific T lymphocytes, and ultimately promoting tumor progression [75]. When IFN-γ binds to its receptor, the dimer formed changes the conformation of the receptor, and the allosteric complex narrows the distance between JAK1/JAK2 and IFNGR1/2, prompting the autophosphorylation and activation of JAK2 [76].
IFN-γ has multiple ways of inducing the expression of PD-L1, which is related to the type of tumor. In gastric cancer, IFN-γ induces PD-L1 expression via the JAK2/STAT1/IFR-1 signaling pathway [77, 78]. However, in lung cancer, IFN-γ induces PD-L1 expression through the JAK/STAT3 and PI3K-AKT signaling pathways, leading to immune escape [79]. Similar results have been obtained in dermal fibroblasts. In addition, the phosphorylation of MAPK and PI3K in dermal fibroblasts was found to result in the release of NF-κB, which enters the nucleus and directly binds to the PD-L1 promoter to activate its transcription [80]. However, in myeloma cells, the expression of PD-L1 induced by IFN-γ was shown to mainly occur through the MEK/ERK signaling pathway, with only a weak effect of the JAK-STAT signaling pathway and no influence of the PI3K/AKT and NF-κB signaling pathways. In addition, the inducible expression of PD-L1 by IFN-γ also relies on Toll-like receptor (TLR) signaling, as inhibiting the expression of MyD88 and TRAF6 will directly block the expression of PD-L1 induced by IFN-γ [81]. Furthermore, IFN-γ can modulate the expression of PD-L1 via microRNA (miRNA) regulation. PD-L1 is the target gene of miRNA-155 and miRNA-513, and IFN-γ can inhibit the expression of miRNA-513 to relieve the translation inhibition of PD-L1, thereby enhancing the expression of PD-L1 protein [82]. However, IFN-γ can also increase the expression level of miRNA-155, which may contribute to a feedback inhibition loop of PD-L1 expression induced by IFN-γ [83].
Overall, these findings demonstrate that the effect of IFN-γ on a tumor is complex. However, further study of the regulation mechanism of PD-L1 expression by IFN-γ will be helpful to understand the promotion of tumorigenesis by IFN-γ, and to find a new method to reduce the side effects (i.e., tumor-promoting effects) in the treatment of tumors with IFN-γ therapy.
TNF-α
TNF was originally identified as a direct contributor to tumor hemorrhagic necrosis. However, subsequent studies indicated that TNF-α is an important inflammatory factor in addition to its effects of killing tumor cells. TNF-α is produced by kinase-activated macrophages, which bind to the receptor of specific homologous trimers on the cell membrane. By activating the caspase protease, JNK, and transcription factor NF-κB signaling pathways, TNF-α can induce inflammation, and promote cell growth, differentiation, and apoptosis. Recent studies have found that TNF-α also induces the expression of PD-L1. In a mouse model, the expression of PD-L1 increased during the maturation of monocytes to macrophages, and reached its peak when monocytes differentiated into macrophages. Moreover, these effects were found to be independent of IFN-γ expression. Although an abundance of IFN-γ and TNF-α will be produced during the differentiation of monocytes into macrophages, in vitro experiments confirmed that certain cytokines would be produced when tumor cells are co-cultured with monocytes, which could promote the secretion of TNF-α by monocytes along with the expression of PD-L1 on the surface of monocytes [84]. The expression of PD-L1 is deficient in the monocytes of patients with systemic lupus erythematosus, but can be reconstituted by the exogenous addition of TNF-α [85]. In addition, HIV infection often leads to increased expression of the co-immunosuppressive molecule PD-L1, mainly due to the role of Tat protein. In DCs, induction of Tat protein on PD-L1 was found to be closely related to TNF-α [86]. TNF-α can up-regulate the mRNA and protein levels of PD-L1, which is mainly completed by activating the NF-κB and ERK1/2 signaling pathways [87]. In addition, TNF-α can also modulate the expression of PD-L1 through miRNA-155 [83]. As an inflammatory cytokine, TNF-α plays an important role in activating inflammatory cells, killing pathogens, participating in tissue repair, and inducing angiogenesis and connective tissue formation. However, recent studies have shown that TNF-α can also induce the expression of the immunosuppressive molecule PD-L1 to promote immune escape. Therefore, the role of TNF-α in tumors is still unclear and needs further study.
ILs
ILs are types of cytokines produced by various cells that play an important role in the maturation, activation, proliferation, and immunomodulation of immune cells. In addition, ILs are involved in various physiological and pathological responses. For example, IL-27 is a key immunosuppressive cytokine that can induce the expression of PD-L1 in initial T cells, which is a STAT1-dependent event. In vivo experiments showed that T cells expressing a TCR transgene and IL-27-induced PD-L1 could inhibit the differentiation of Th17 cells and avoid the development of severe autoimmune encephalomyelitis [88]. Other than T cells, induced expression of PD-L1 by IL-27 can be found in ovarian cancer cells, prostate cancer cells, and non-small cell lung cancer cells [89]. As pro-inflammatory cytokines, IL-6 and IL-17 also mediate the expression of PD-L1 in the tumor microenvironment. Epithelial growth factor receptor (EGFR) regulates the expression of PD-L1 and cell proliferation through the IL6/JAK/STAT3 signaling pathway [90]. Moreover, high expression of PD-L1 and low expression of NKG2D can enhance the radiotherapy tolerance of non-small cell lung cells, which is mainly achieved through the IL-6/MEK/ERK signaling pathway [91]. Further, inhibition of the IL-6 signal pathway can reduce the expression level of PD-L1 and enhance the sensitivity of cancer cells to the cytotoxicity of NK cells [92]. Although IL-10 itself does not directly induce the expression of PD-L1, after inhibiting the expression of IL-10 in the environment, the expression level of PD-L1 also decreases. This suggests that IL-10 can induce the expression of PD-L1 via certain factors present in cells [93]. IL-12 is best known for its important role in the activation of NK cells and T lymphocytes but has also been shown to induce PD-L1 expression. This inducing effect depends on activation of the NF-κB signal, which may be related to the direct induction of IFN-γ by IL-12 [94]. Similar to TNF-α, IL-17 can also induce the expression of PD-L1 through the synergistic effect of the NF-κB and ERK1/2 signaling pathways [87]. In human pluripotent interstitial cells, IL-25 can inhibit the immune response of Th17 cells via the IL-25/STAT3/PD-L1 complex [95].
Cell growth factors
EGF is a small-molecule active polypeptide that is widely distributed in the human body, consisting of 53 amino acids. EGF can combine with its receptor EGFR and activate EGFR signaling pathways to promote cell growth. Moreover, the EGFR signaling pathway plays an important role in tumor cell migration and proliferation, and mutations of EGFR can be found in many types of tumor cells. The mutated EGFR no longer needs to be combined with EGF to have a continuous activation effect, leading to malignant proliferation and metastasis of the tumor. Recent studies have shown that EGFR signals can not only regulate the growth and invasion of tumors but also play an important role in their immune escape. In various cancers such as lung cancer, breast cancer, head and neck cancer, esophageal cancer, and salivary adenoid cystic carcinoma, EGF was found to up-regulate the expression of PD-L1. However, the specific mechanism by which EGF induces PD-L1 differs according to the cancer type [96,97,98,99,100]. In lung cancer and head and neck cancer, EGFR can increase the expression of PD-L1 via activating the JAK/STAT1 signaling pathway [98, 101]. In addition, PI3K/AKT and MEK/ERK are also important pathways for inducing the expression of PD-L1 in lung cancer cells with EGFR mutation [102]. MYC is an important transcription factor in tumors and is also involved in the regulation of PD-L1 by EGFR. In the EGFR-derived PD-L1 signaling pathway, inhibiting the expression of MYC can significantly reduce the expression of PD-L1 [100, 103]. In T-ALL cells, we found that MYC can be directly combined with the PD-L1 promoter to upregulate the expression of PD-L1 [104], suggesting that the EGFR pathway could up-regulate the expression of the MYC transcription factor and promote its nuclear translocation to ultimately up-regulate the expression of PD-L1. EGF not only induces the transcription of PD-L1 but also affects its protein stability and biological function. EGFR-RAS is a classical intracellular signal pathway, and RAS has been shown to regulate the mRNA stability of PD-L1. In lung cancer and colorectal cancer, RAS can enhance the stability of PD-L1 mRNA via the phosphorylation of TPP by MEK signaling, as TPP can negatively regulate the stability of PD-L1 mRNA. EGFR-RAS can also increase the protein level of PD-L1 and enhance the immune tolerance of the tumor [105]. In tumor cells, a decrease in the PTEN copy number is often accompanied by an increase in the EGFR copy number [106], and the decreased PTEN leads to activation of the PI3K/AKT signaling pathway. The activated PI3K/AKT signal can in turn stabilize the protein level of PD-L1 through mTOR/S6K1 and promote immune escape of the tumor [107]. In addition, EGF can inhibit the protein level of GSK3β in cells, while GSK3β can phosphorylate PD-L1, making it more susceptible to ubiquitination and degradation to ultimately increase the stability of PD-L1 protein. The glycosylation level of the PD-L1 protein itself also directly affects the degradation of GSK3β [97]. B3GNT3 is one of the proteins that glycosylate PD-L1. EGF can upregulate the expression of B3GNT3 to promote the glycosylation of PD-L1, which will also enhance the stability of PD-L1 protein and promote its biological function [108]. Therefore, blocking the EGFR signaling pathway, in addition to inhibiting tumor growth, can also inhibit tumor immune escape, which is undoubtedly a benefit for EGFR blockage therapy.
The TGF-β family of polypeptide signaling molecules plays an important role in regulating cell growth and differentiation. Similar to its dual role in both promoting and inhibiting cancer, TGF-β is a double-edged sword with respect to tumor immune escape. In hepatocellular carcinoma and lung cancer, TGF-β can induce the expression of PD-L1 on the surface of DCs, which depends on the activation of STAT3 [109, 110]. However, in systemic lupus erythematosus, TNF-α can induce the expression of PD-L1 in mononuclear cells, while TGF-β has the opposite effect. This is consistent with a previous report showing that TGF-β could inhibit the expression of proximal epithelial cell PD-L1 in renal tubules, although the exact mechanism is unclear [111]. TGF-β signaling pathways mainly comprise the classic TGF-β/Smads signaling pathways and the non-classical TGF-β/EGFR signaling pathways, and recent studies have shown that the influence of TGF-β on PD-L1 is almost always dependent on the latter, while the role of the former pathway in regulating the expression of PD-L1 has not been reported to date.
In addition, GM-CSF can also activate the expression of PD-L1 in neutrophils, which promotes the inhibition of their T cell immune activation and achieves immunosuppression via the JAK2/STAT3 signal pathway [112, 113].
Hypoxia
During the occurrence and development of tumors, especially solid tumors, it is impossible to rapidly establish new blood vessels in the tumor microenvironment to match the rapid proliferation rate of tumor cells. Furthermore, the newly established endovascularization results in abnormalities in structure and function. Ultimately, the metabolism of tumor cells results in a tumor microenvironment characterized by a decrease of oxygen content, lack of nutrients, and accumulation of a large number of acidic substances, and these conditions are extremely detrimental to the growth of tumor cells [114,115,116,117,118]. However, in an anoxic microenvironment, tumor cells can change their metabolic mode to conduct metabolic reprogramming via changing the expression of glycolytic-related proteins such as GLUT1, GLUT3, PKM2, and LDHA to ultimately increase the uptake of glucose and the production of energy [119,120,121,122]. Moreover, the invasion and metastasis of tumor cells are promoted by altering the expression of E-cadherin, N-cadherin, Snail, and vimentin, which are all EMT markers [123, 124]. Tumor immune escape is also an important strategy for tumor cells to survive in this hypoxic microenvironment.
Hypoxia-inducible factors (HIFs) are the most important proteins for cell-induced expression in hypoxic environments. Under the action of normal oxygen, proline residues in the conserved region of HIF subunits are hydroxylated by proline hydroxylase, and hydroxylated HIF is identified and ubiquitinated by the VHL E3 ubiquitination ligase so that the ubiquitinated HIFs are rapidly degraded by the proteasome [125]. However, under hypoxic conditions, the prolinyl hydroxylase of HIF protein is inhibited, which stabilizes the protein level of HIFα, resulting in induction of the expression of its target genes to induce tumor immune escape [126]. HIF induces tumor immune escape mainly by inducing the expression of PD-L1. Under hypoxia, the expression of PD-L1 has been shown to be elevated in T lymphocytes, DCs, MDSCs, and macrophages, as well as in prostate cancer cells, breast cancer cells, and colorectal cancer cells [127, 128]. Hypoxia can increase the expression of PD-L1 and suppress the killing effect of CTL on tumor cells. The induced expression of PD-L1 by hypoxia mainly occurs at the transcriptional level. The hypoxia-induced expression of PD-L1 is achieved through HIF-1α rather than HIF-2α in breast cancer and prostate cancer [128], and similar results were obtained in MDSCs. Immunoprecipitation and luciferase reporter gene experiments confirmed that HIF-1α could activate the transcription of PD-L1 via binding to the HRE site on the PD-L1 promoter [127]. However, in clear cell renal cell carcinoma, the induced expression of PD-L1 mainly occurs through HIF-2α rather than HIF-1α [129]. This difference may be related to the cell types, because over 90% of patients with clear cell renal cell carcinoma will present with pure homozygous inactivation of VHL protein, which is the E3 ubiquitination ligase of the HIF ligase subunit. This inactivation leads to high levels of HIF protein accumulation in cells [130]. In addition, other studies have suggested indirect regulation of PD-L1 by the host HIF, because the expression levels of GLUT1 and PD-L1 show a strong correlation in a hypoxic state. However, the specific mechanism of action remains to be elucidated [131].
Exosomes
Almost all cells release exosomes, which are extracellular vesicles with a diameter of 40–150 nm. The outer part of exosomes is a lipid bilayer structure, along with proteins, DNA, and RNA from exosome-derived cells [42]. Exosomes were previously considered to represent a form of cell waste; however, recent studies have shown that exosomes can be transmitted as signal molecules to other cells to alter their function. Exosomes have also been shown to play important roles in tumor cell growth, migration, and angiogenesis [43]. More recent studies have also shown that exosomes play an important role in immunosuppression. In chronic lymphocytic leukemia, tumor-derived exosomes can induce the tumor immunosuppressive reaction in monocytes. Monocyte activation mainly occurs through the non-coding small RNA Yh4 in exosomes through the TLR7 signaling pathway in monocytes, which promotes the secretion of cytokines by monocytes and ultimately induces the expression of PD-L1 [132]. Tumor-derived exosomes can also promote the polarization of monocytes to M2 macrophages and the expression of PD-L1 in M2-polarized macrophages through STAT3 phosphorylation, further enhancing the immunosuppressive effects [133]; however, it is not yet clear whether exosomes also activate STAT3 through TLR7 in this case. In addition, the same study showed that tumor-derived exosomes containing PD-L1 protein have strong immunosuppressive effects [134]. Since research on exosomes is still in a preliminary stage, new functions of exosomes are expected to be discovered in the future.
Non-coding RNA
The expression of PD-L1 in tumor cells is not only affected by the various cytokines and exosomes in the tumor microenvironment but also by various intracellular regulatory signals, including those derived from non-coding RNA (ncRNAs) [135], which transfer between cancer cells and the tumor microenvironment via exosomes and gap junctions [136]. Indeed, ncRNAs have been demonstrated to play an important role in tumor growth, metabolism, and migration, as well as in regulating the expression of PD-L1 and mediating immune escape (Fig. 6). Long non-coding RNA (lncRNA) is an endogenous RNA molecule with a length greater than 200 nt and does not encode proteins [137,138,139,140,141,142]. LncRNAs represent the majority of the sequences transcribed in the human genome, and are more abundant than protein-coding genes and small-molecule RNAs (such as miRNA), and also show more diverse and extensive patterns in regulating gene expression [143,144,145,146,147]. The lncRNA NKX2–1-AS1 shares some overlapping regions with NKX2–1, but there expression levels are not correlated. Instead, NKX2–1-AS1 can interact with NKX2–1 protein and interfere with its binding to the PD-L1 promoter, thereby negatively regulating the expression of PD-L1 [148]. However, there have been few studies on PD-L1 regulation by lncRNAs, and it is believed that there will be more reports on the role of lncRNAs in immune escape in the near future. MiRNAs are single-stranded small RNA molecules with a size of about 22 nt. They are generated by Dicer enzyme processing single-stranded RNA precursors with a hairpin structure of about 70–90 nt, and approximately 30–50% protein-coding genes are currently known to be regulated by miRNAs [149,150,151]. Mature miRNAs regulate genes in two ways: one is to bind to the target gene mRNA and promote its degradation, and the other is to inhibit the translation of target mRNA [152]. In recent years, a role of miRNAs in the regulation of PD-L1 has also been discovered. The regulatory effect of miRNA on PD-L1 occurs not only by directly binding to the PD-L1 mRNA, but also by indirectly regulating the expression of PD-L1. As the earliest miRNA identified to regulate PD-L1, miR-513 plays an important role in the IFN-induced expression of PD-L1. In cholangiocarcinoma cells, it can directly target the 3′-untranslated region (UTR) of PD-L1 and inhibit the translation of PD-L1. Overexpression of miR-513 in the induction environment can block the induction effect of IFN-γ on PD-L1 [82, 153]. In human dermal lymphoid endothelial cells and dermal fibroblasts, miR-155 induced by IFN-γ and TNF-α can also target the 3′-UTR of PD-L1 and inhibit the translation level of PD-L1 [83]. Therefore, miR-513 and miR-155 can be considered an IFNγ-induced feedback regulation loop of PD-L1. In non-small-cell lung cancer, miR-34a/b/c induced by wild-type P53 can bind to the 3′-UTR of PD-L1, promote the degradation of PD-L1 mRNA, and thereby inhibit the immune escape of non-small-cell lung cancer. In mouse models, tail intravenous injection of miR-34a contained in liposomes effectively inhibited tumor growth [154]. In addition, miR-200 can also target PD-L1 and regulate the expression of PD-L1 in non-small-cell lung cancer [155]. Several miRNAs have been shown to directly regulate the expression of PD-L1, including miR-142-5p, miR-93, and miR106b in pancreatic cancer [156, 157]; miR-138-5p in colorectal cancer [158]; miR-217 in laryngeal cancer [159]; miR-17-5p in melanoma [160]; miR-200b, miR-152, and miR-570 in gastric cancer [161, 162]; miR-15a, miR-193a, and miR-16 in malignant pleural mesothelioma [163]; miR-497-5p in clear cell renal cell carcinoma [164]; and miR-140, miR-142, miR-340, and miR-383 in cervical cancer [165]. Another way to regulate PD-L1 is through other members of the PD-L1 signaling pathway. In colorectal cancer, miR-20, miR-21, and miR130b can inhibit the expression level of PTEN, which is an upstream inhibitor of PD-L1. Therefore, these miRNAs can indirectly promote the expression of PD-L1 by inhibiting PTEN [166]. Moreover, miR-18a has similar functions. In cervical cancer, miR-18a can target PTEN, WNK2, and SOX6, leading to activation of the PI3K/AKT, MEK/ERK, and Wnt/β-catenin signaling pathways, which can promote the expression of PD-L1 [165]. In non-small cell lung cancer, miR-197 can also regulate PD-L1 by targeting CKS1B/STAT3 signaling [167].

Role of non-coding RNAs in regulating PD-L1
Furthermore, recent studies have shown that circulating RNAs (circRNAs) can also regulate the expression of PD-L1. CircRNAs are a special class of non-coding RNA molecules with a closed loop structure, which is not affected by RNA exonuclease, making them more stable and less prone to degradation than other RNAs [168,169,170,171]. CircRNA molecules are rich in miRNA-binding sites, and can thus act as an miRNA sponge in cells to relieve the inhibition of miRNA on the target gene and increase its expression level. In colorectal cancer, the circRNA molecule Hsa_circ_0020397 was shown to inhibit the activity of miR-138 via an RNA sponge effect, thus promoting the expression of PD-L1 [172]. This was the first report that a circRNA can regulate the expression of PD-L1, suggesting that circRNAs might also play an important role in tumor immune escape.
Conclusions and prospects
Immune escape mediated by the PD-L1/PD-1 signaling pathway has emerged as a hot topic in anti-tumor research and in the field of cancer translational medicine. However, targeted PD-L1/PD-1 immunotherapy has not achieved the desired effects in the treatment of various types of cancers, especially for solid tumors, with a low response rate overall. One reason for this inconsistent and poor response may be related to individual differences among patients as well as tumor heterogeneity within a single patient, given that the expression of PD-L1 varies in different tumors. In general, antagonistic drugs designed to block PD-L1/PD-1 show good effects for patients with high expression of PD-L1 or PD-1, whereas the therapeutic efficacy is poor for patients with low PD-L1/PD-1 expression, and the treatment could bring about serious side effects. Some patients that were treated with PD-L1/PD-1 antagonistic drugs actually showed rapid growth of the tumor [173]. A follow-up investigation into 131 patients that were treated with PD-1 antagonists found that 12 patients (9%) experienced deterioration, and those aged 65 and older had a higher rate of deterioration [174]. At present, there is no uniform standard for the preoperative detection of PD-L1/PD-1, making it difficult to achieve individualized treatment for a given patient with respect to the most suitable drug and dosage. In addition, the mechanism of tumor formation is very complex, and the tumor microenvironment plays an important role in the regulation of PD-L1. A variety of cytokines secreted in the tumor microenvironment can cause immunosuppression by inducing PD-L1 expression. This review summarized the main regulation mechanisms of PD-1 and PD-L1 by cytokines, exosomes, and ncRNAs in the tumor microenvironment, demonstrating the wide variation of co-molecules and pathways involved. Specifically, IFNγ can induce the expression of PD-L1 through the JAK/STAT and PI3K-AKT signaling pathways to promote tumor immune escape [79]. TNF-α can directly increase the mRNA and protein levels of PD-L1, thus promoting immune escape. IL-6 and IL-17 can also regulate PD-L1 expression by means of the JAK/STAT3 and NF-κB signaling pathways, respectively. Some growth factors such as EGF, TGF-β, and GM-CSF can also induce the expression of PD-L1 and promote the occurrence of immunosuppression. Interestingly, most of these cytokines with PD-L1-inducing abilities are inflammatory factors.
Although the effects of inflammation on tumor growth and metastasis have long been recognized, recent research has revealed that inflammatory factors can also promote the immune escape of tumors. This review further points to the potential importance of the inflammatory microenvironment for tumor immune escape. Thus, inhibiting the occurrence of inflammation in the tumor microenvironment, or inhibiting the secretion of certain inflammatory factors, may have a desirable effect for tumor treatment [175]. A study with a melanoma mouse model showed that treatment with both PD-1 inhibitors and TNF-α inhibitors had a better therapeutic effect than treatment with PD-1 inhibitors alone [176]. This indicated that the high expression of inflammatory factors in the tumor microenvironment may be one of the important factors for the poor efficacy of immunotherapy. Thus, a combination of anti-inflammatory drugs and PD-L1/PD-1 inhibitors might lead to a better treatment outcome for cancer patients.
At present, most of the drugs developed to inhibit PD-L1/PD-1 signaling pathways are antibodies; however, the latest research shows that metformin may also be effective for suppressing PD-L1/PD-1 signaling pathways. Metformin can activate AMPK [177], which can directly phosphorylate PD-L1, thus inhibiting the PD-L1 glycosylation level, resulting in PD-L1 degradation and inhibiting immune escape [178]. However, there is still much to discover about the tumor microenvironment, and the regulation network of PD-L1 by cytokines remains to be uncovered in detail. Therefore, further study of the tumor microenvironment, especially the role of inflammatory cytokines, may provide new ideas for immunotherapy and a new development direction for the effective use of PD-L1/PD-1 inhibitors in cancer treatment.
Abbreviations
- APC:
-
Antigen-presenting cell
- BCR:
-
B cell receptor
- Bregs:
-
B regulatory cells
- CTL:
-
Cytolytic T lymphocytes
- CTLA4:
-
Cytotoxic T-lymphocyte-associated protein 4
- CXCL12:
-
C-X-C motif chemokine 12
- DC:
-
Dentritic cell
- ECM:
-
Extracellular matrix
- EGF:
-
Epithelial growth factor
- EMT:
-
Epithelial-mesenchymal transition
- GM-CSF:
-
Granulocyte macrophage-colony-stimulating factor
- HIFs:
-
Hypoxia-inducible factors
- HIV:
-
Human immunodeficiency virus
- IDO1:
-
Indoleamine-pyrrole 2,3-dioxygenase
- IFN:
-
Interferon
- IFNGR:
-
Interferon gamma receptor
- IL:
-
Interleukin
- ITIM:
-
Immunoreceptor tyrosine-based inhibitory motif
- ITSM:
-
Immunoreceptors tyrosine-based switch motif
- LAG-3:
-
Lymphocyte-activation gene 3
- MDSC:
-
Myeloid-derived suppressor cell
- MMP2:
-
Matrix metalloproteinase-2
- NK:
-
Natural killer
- PD-1:
-
Programmed death-1
- PD-L1:
-
Programmed death ligand-1
- TCR:
-
T cell receptor
- TGF:
-
Transforming growth factor
- Th1:
-
Type I T helper cell
- TIM-3:
-
T-cell immunoglobulin and mucin-domain containing-3
- TNF:
-
Tumor necrosis factor
- Tregs:
-
Regulatory T cells
- VEGF:
-
Vascular endothelial growth factor
- VHL:
-
Von Hippel–Lindau tumor suppressor
References
Jiang XJ, Wang J, Deng XY, Li XL, Li XY, Zeng ZY, et al. Immunotherapy targeted to immune checkpoint: a revolutionary breakthrough in cancer therapy. Prog Biochem Biophys. 2018;45(11):1178–86.
Jiang X, Shapiro DJ. The immune system and inflammation in breast cancer. Mol Cell Endocrinol. 2014;382(1):673–82.
Wang YA, Li XL, Mo YZ, Fan CM, Tang L, Xiong F, et al. Effects of tumor metabolic microenvironment on regulatory T cells. Mol Cancer. 2018;17(1):168.
Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. 2013;190(7):3783–97.
Benencia F, Muccioli M, Alnaeeli M. Perspectives on reprograming cancer-associated dendritic cells for anti-tumor therapies. Front Oncol. 2014;4:72.
Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). 2014;6(3):1670–90.
Liao Q, Zeng Z, Guo X, Li X, Wei F, Zhang W, et al. LPLUNC1 suppresses IL-6-induced nasopharyngeal carcinoma cell proliferation via inhibiting the Stat3 activation. Oncogene. 2014;33(16):2098–109.
Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11(11):3887–95.
Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704.
Nishimura H, Agata Y, Kawasaki A, Sato M, Imamura S, Minato N, et al. Developmentally regulated expression of the PD-1 protein on the surface of double-negative (CD4-CD8-) thymocytes. Int Immunol. 1996;8(5):773–80.
Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5(12):1365–9.
Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol. 2002;169(10):5538–45.
Zhong X, Tumang JR, Gao W, Bai C, Rothstein TL. PD-L2 expression extends beyond dendritic cells/macrophages to B1 cells enriched for V(H)11/V(H)12 and phosphatidylcholine binding. Eur J Immunol. 2007;37(9):2405–10.
Qin T, Zeng YD, Qin G, Xu F, Lu JB, Fang WF, et al. High PD-L1 expression was associated with poor prognosis in 870 Chinese patients with breast cancer. Oncotarget. 2015;6(32):33972–81.
Lin YM, Sung WW, Hsieh MJ, Tsai SC, Lai HW, Yang SM, et al. High PD-L1 expression correlates with metastasis and poor prognosis in Oral squamous cell carcinoma. PLoS One. 2015;10(11):e0142656.
Zhou Y, Miao J, Wu H, Tang H, Kuang J, Zhou X, et al. PD-1 and PD-L1 expression in 132 recurrent nasopharyngeal carcinoma: the correlation with anemia and outcomes. Oncotarget. 2017;8(31):51210–23.
Tang Y, He Y, Shi L, Yang L, Wang J, Lian Y, et al. Co-expression of AFAP1-AS1 and PD-1 predicts poor prognosis in nasopharyngeal carcinoma. Oncotarget. 2017;8(24):39001–11.
Sznol M. Blockade of the B7-H1/PD-1 pathway as a basis for combination anticancer therapy. Cancer J. 2014;20(4):290–5.
Yan Q, Zeng Z, Gong Z, Zhang W, Li X, He B, et al. EBV-miR-BART10-3p facilitates epithelial-mesenchymal transition and promotes metastasis of nasopharyngeal carcinoma by targeting BTRC. Oncotarget. 2015;6(39):41766–82.
Xiao K, Yu Z, Li X, Li X, Tang K, Tu C, et al. Genome-wide analysis of Epstein-Barr virus (EBV) integration and strain in C666-1 and Raji cells. J Cancer. 2016;7(2):214–24.
Song Y, Li X, Zeng Z, Li Q, Gong Z, Liao Q, et al. Epstein-Barr virus encoded miR-BART11 promotes inflammation-induced carcinogenesis by targeting FOXP1. Oncotarget. 2016;7(24):36783–99.
He B, Li W, Wu Y, Wei F, Gong Z, Bo H, et al. Epstein-Barr virus-encoded miR-BART6-3p inhibits cancer cell metastasis and invasion by targeting long non-coding RNA LOC553103. Cell Death Dis. 2016;7(9):e2353.
Tu C, Zeng Z, Qi P, Li X, Yu Z, Guo C, et al. Genome-Wide Analysis of 18 Epstein-Barr Viruses Isolated from Primary Nasopharyngeal Carcinoma Biopsy Specimens. J Virol. 2017;91(17):e00301-17.
Fan C, Tang Y, Wang J, Xiong F, Guo C, Wang Y, et al. The emerging role of Epstein-Barr virus encoded microRNAs in nasopharyngeal carcinoma. J Cancer. 2018;9(16):2852–64.
Wei F, Tang L, He Y, Wu Y, Shi L, Xiong F, et al. BPIFB1 (LPLUNC1) inhibits radioresistance in nasopharyngeal carcinoma by inhibiting VTN expression. Cell Death Dis. 2018;9(4):432.
Wei F, Wu Y, Tang L, He Y, Shi L, Xiong F, et al. BPIFB1 (LPLUNC1) inhibits migration and invasion of nasopharyngeal carcinoma by interacting with VTN and VIM. Br J Cancer. 2018;118(2):233–47.
Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–37.
Wang JP, Tang YY, Fan CM, Guo C, Zhou YH, Li Z, et al. The role of exosomal non-coding RNAs in cancer metastasis. Oncotarget. 2017;9(15):12487–502.
Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, et al. Role of tumor microenvironment in tumorigenesis. J Cancer. 2017;8(5):761–73.
Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–22.
Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86(3):353–64.
Gilbert CA, Slingerland JM. Cytokines, obesity, and cancer: new insights on mechanisms linking obesity to cancer risk and progression. Annu Rev Med. 2013;64:45–57.
Zhang Q, Sun LJ, Yang ZG, Zhang GM, Huo RC. Influence of adipocytokines in periprostatic adipose tissue on prostate cancer aggressiveness. Cytokine. 2016;85:148–56.
Tu C, Zeng Z, Qi P, Li X, Guo C, Xiong F, et al. Identification of genomic alterations in nasopharyngeal carcinoma and nasopharyngeal carcinoma-derived Epstein-Barr virus by whole genome sequencing. Carcinogenesis. 2018;39(12):1517-28.
Yi M, Cai J, Li J, Chen S, Zeng Z, Peng Q, et al. Rediscovery of NF-kappaB signaling in nasopharyngeal carcinoma: how genetic defects of NF-kappaB pathway interplay with EBV in driving oncogenesis? J Cell Physiol. 2018;233(8):5537–49.
Aktas ON, Ozturk AB, Erman B, Erus S, Tanju S, Dilege S. Role of natural killer cells in lung cancer. J Cancer Res Clin Oncol. 2018;144(6):997–1003.
Butt AQ, Mills KH. Immunosuppressive networks and checkpoints controlling antitumor immunity and their blockade in the development of cancer immunotherapeutics and vaccines. Oncogene. 2014;33(38):4623–31.
Lu T, Ramakrishnan R, Altiok S, Youn JI, Cheng P, Celis E, et al. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest. 2011;121(10):4015–29.
Ocana A, Nieto-Jimenez C, Pandiella A, Templeton AJ. Neutrophils in cancer: prognostic role and therapeutic strategies. Mol Cancer. 2017;16(1):137.
Jackaman C, Tomay F, Duong L, Abdol Razak NB, Pixley FJ, Metharom P, et al. Aging and cancer: the role of macrophages and neutrophils. Ageing Res Rev. 2017;36:105–16.
Tevis KM, Cecchi RJ, Colson YL, Grinstaff MW. Mimicking the tumor microenvironment to regulate macrophage phenotype and assessing chemotherapeutic efficacy in embedded cancer cell/macrophage spheroid models. Acta Biomater. 2017;50:271–9.
Kalluri R. The biology and function of exosomes in cancer. J Clin Invest. 2016;126(4):1208–15.
Shao C, Yang F, Miao S, Liu W, Wang C, Shu Y, et al. Role of hypoxia-induced exosomes in tumor biology. Mol Cancer. 2018;17(1):120.
Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Lorenz M, et al. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity. 2004;20(3):337–47.
Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–48.
Hofmeyer KA, Jeon H, Zang X. The PD-1/PD-L1 (B7-H1) pathway in chronic infection-induced cytotoxic T lymphocyte exhaustion. J Biomed Biotechnol. 2011;2011:451694.
Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209(6):1201–17.
Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol. 2013;33(16):3091–8.
Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal. 2012;5(230):ra46.
Wartewig T, Kurgyis Z, Keppler S, Pechloff K, Hameister E, Ollinger R, et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature. 2017;552(7683):121–5.
Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A. 2001;98(24):13866–71.
Nicholas KJ, Zern EK, Barnett L, Smith RM, Lorey SL, Copeland CA, et al. B cell responses to HIV antigen are a potent correlate of viremia in HIV-1 infection and improve with PD-1 blockade. PLoS One. 2013;8(12):e84185.
Haas KM. Programmed cell death 1 suppresses B-1b cell expansion and long-lived IgG production in response to T cell-independent type 2 antigens. J Immunol. 2011;187(10):5183–95.
Xia Y, Jeffrey Medeiros L, Young KH. Signaling pathway and dysregulation of PD1 and its ligands in lymphoid malignancies. Biochim Biophys Acta. 2016;1865(1):58–71.
Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8(5):765–72.
Rekik R, Belhadj Hmida N, Ben Hmid A, Zamali I, Kammoun N, Ben AM. PD-1 induction through TCR activation is partially regulated by endogenous TGF-beta. Cell Mol Immunol. 2015;12(5):648–9.
Keir ME, Francisco LM, Sharpe AH. PD-1 and its ligands in T-cell immunity. Curr Opin Immunol. 2007;19(3):309–14.
Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 regulates PD-1 expression upon T cell activation. J Immunol. 2008;181(7):4832–9.
Austin JW, Lu P, Majumder P, Ahmed R, Boss JM. STAT3, STAT4, NFATc1, and CTCF regulate PD-1 through multiple novel regulatory regions in murine T cells. J Immunol. 2014;192(10):4876–86.
Bally AP, Tang Y, Lee JT, Barwick BG, Martinez R, Evavold BD, et al. Conserved region C functions to regulate PD-1 expression and subsequent CD8 T cell memory. J Immunol. 2017;198(1):205–17.
Kinter AL, Godbout EJ, McNally JP, Sereti I, Roby GA, O'Shea MA, et al. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J Immunol. 2008;181(10):6738–46.
Cho HY, Lee SW, Seo SK, Choi IW, Choi I, Lee SW. Interferon-sensitive response element (ISRE) is mainly responsible for IFN-alpha-induced upregulation of programmed death-1 (PD-1) in macrophages. Biochim Biophys Acta. 2008;1779(12):811–9.
Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, et al. IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol. 2011;186(5):2772–9.
Bommarito D, Hall C, Taams LS, Corrigall VM. Inflammatory cytokines compromise programmed cell death-1 (PD-1)-mediated T cell suppression in inflammatory arthritis through up-regulation of soluble PD-1. Clin Exp Immunol. 2017;188(3):455–66.
Deng X, Xiong F, Li X, Xiang B, Li Z, Wu X, et al. Application of atomic force microscopy in cancer research. J Nanobiotechnology. 2018;16(1):102.
Yang Y, Liao Q, Wei F, Li X, Zhang W, Fan S, et al. LPLUNC1 inhibits nasopharyngeal carcinoma cell growth via down-regulation of the MAP kinase and cyclin D1/E2F pathways. PLoS One. 2013;8(5):e62869.
Zhang W, Fan S, Zou G, Shi L, Zeng Z, Ma J, et al. Lactotransferrin could be a novel independent molecular prognosticator of nasopharyngeal carcinoma. Tumour Biol. 2015;36(2):675–83.
Zhou Y, Liao Q, Li X, Wang H, Wei F, Chen J, et al. HYOU1, regulated by LPLUNC1, is up-regulated in nasopharyngeal carcinoma and associated with poor prognosis. J Cancer. 2016;7(4):367–76.
Spaccarelli N, Rook AH. The use of interferons in the treatment of cutaneous T-cell lymphoma. Dermatol Clin. 2015;33(4):731–45.
Martinez R, de Villavicencio-Diaz TN, Sanchez A, Ramos Y, Ferro JN, Gonzalez LG, et al. Comparative proteomic analysis of growth hormone secretagogue A233 treatment of murine macrophage cells J774A.2 indicates it has a role in antiviral innate response. Biochem Biophys Rep. 2016;5:379–87.
Alshaker HA, Matalka KZ. IFN-gamma, IL-17 and TGF-beta involvement in shaping the tumor microenvironment: the significance of modulating such cytokines in treating malignant solid tumors. Cancer Cell Int. 2011;11:33.
Mandai M, Hamanishi J, Abiko K, Matsumura N, Baba T, Konishi I. Dual faces of IFNgamma in Cancer progression: a role of PD-L1 induction in the determination of pro- and antitumor immunity. Clin Cancer Res. 2016;22(10):2329–34.
Meyskens FL Jr, Kopecky KJ, Taylor CW, Noyes RD, Tuthill RJ, Hersh EM, et al. Randomized trial of adjuvant human interferon gamma versus observation in high-risk cutaneous melanoma: a southwest oncology group study. J Natl Cancer Inst. 1995;87(22):1710–3.
Alberts DS, Marth C, Alvarez RD, Johnson G, Bidzinski M, Kardatzke DR, et al. Randomized phase 3 trial of interferon gamma-1b plus standard carboplatin/paclitaxel versus carboplatin/paclitaxel alone for first-line treatment of advanced ovarian and primary peritoneal carcinomas: results from a prospectively designed analysis of progression-free survival. Gynecol Oncol. 2008;109(2):174–81.
Bo H, Fan L, Li J, Liu Z, Zhang S, Shi L, et al. High Expression of lncRNA AFAP1-AS1 Promotes the Progression of Colon Cancer and Predicts Poor Prognosis. J Cancer. 2018;9(24):4677-83.
Grayfer L, Belosevic M. Molecular characterization, expression and functional analysis of goldfish (Carassius aurutus L.) interferon gamma. Dev Comp Immunol. 2009;33(2):235–46.
Mimura K, Teh JL, Okayama H, Shiraishi K, Kua LF, Koh V, et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 2018;109(1):43–53.
Moon JW, Kong SK, Kim BS, Kim HJ, Lim H, Noh K, et al. IFNgamma induces PD-L1 overexpression by JAK2/STAT1/IRF-1 signaling in EBV-positive gastric carcinoma. Sci Rep. 2017;7(1):17810.
Zhang X, Zeng Y, Qu Q, Zhu J, Liu Z, Ning W, et al. PD-L1 induced by IFN-gamma from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int J Clin Oncol. 2017;22(6):1026–33.
Lee SK, Seo SH, Kim BS, Kim CD, Lee JH, Kang JS, et al. IFN-gamma regulates the expression of B7-H1 in dermal fibroblast cells. J Dermatol Sci. 2005;40(2):95–103.
Liu J, Hamrouni A, Wolowiec D, Coiteux V, Kuliczkowski K, Hetuin D, et al. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood. 2007;110(1):296–304.
Gong AY, Zhou R, Hu G, Li X, Splinter PL, O'Hara SP, et al. MicroRNA-513 regulates B7-H1 translation and is involved in IFN-gamma-induced B7-H1 expression in cholangiocytes. J Immunol. 2009;182(3):1325–33.
Yee D, Shah KM, Coles MC, Sharp TV, Lagos D. MicroRNA-155 induction via TNF-alpha and IFN-gamma suppresses expression of programmed death ligand-1 (PD-L1) in human primary cells. J Biol Chem. 2017;292(50):20683–93.
Hartley G, Regan D, Guth A, Dow S. Regulation of PD-L1 expression on murine tumor-associated monocytes and macrophages by locally produced TNF-alpha. Cancer Immunol Immunother. 2017;66(4):523–35.
Ou JN, Wiedeman AE, Stevens AM. TNF-alpha and TGF-beta counter-regulate PD-L1 expression on monocytes in systemic lupus erythematosus. Sci Rep. 2012;2:295.
Planes R, BenMohamed L, Leghmari K, Delobel P, Izopet J, Bahraoui E. HIV-1 tat protein induces PD-L1 (B7-H1) expression on dendritic cells through tumor necrosis factor alpha- and toll-like receptor 4-mediated mechanisms. J Virol. 2014;88(12):6672–89.
Wang X, Yang L, Huang F, Zhang Q, Liu S, Ma L, et al. Inflammatory cytokines IL-17 and TNF-alpha up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol Lett. 2017;184:7–14.
Hirahara K, Ghoreschi K, Yang XP, Takahashi H, Laurence A, Vahedi G, et al. Interleukin-27 priming of T cells controls IL-17 production in trans via induction of the ligand PD-L1. Immunity. 2012;36(6):1017–30.
Carbotti G, Barisione G, Airoldi I, Mezzanzanica D, Bagnoli M, Ferrero S, et al. IL-27 induces the expression of IDO and PD-L1 in human cancer cells. Oncotarget. 2015;6(41):43267–80.
Zhang N, Zeng Y, Du W, Zhu J, Shen D, Liu Z, et al. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int J Oncol. 2016;49(4):1360–8.
Shen MJ, Xu LJ, Yang L, Tsai Y, Keng PC, Chen Y, et al. Radiation alters PD-L1/NKG2D ligand levels in lung cancer cells and leads to immune escape from NK cell cytotoxicity via IL-6-MEK/Erk signaling pathway. Oncotarget. 2017;8(46):80506–20.
Xu L, Chen X, Shen M, Yang DR, Fang L, Weng G, et al. Inhibition of IL-6-JAK/Stat3 signaling in castration-resistant prostate cancer cells enhances the NK cell-mediated cytotoxicity via alteration of PD-L1/NKG2D ligand levels. Mol Oncol. 2018;12(3):269–86.
Wang X, Ni S, Chen Q, Ma L, Jiao Z, Wang C, et al. Bladder cancer cells induce immunosuppression of T cells by supporting PD-L1 expression in tumour macrophages partially through interleukin 10. Cell Biol Int. 2017;41(2):177–86.
Xiong HY, Ma TT, Wu BT, Lin Y, Tu ZG. IL-12 regulates B7-H1 expression in ovarian cancer-associated macrophages by effects on NF-kappaB signalling. Asian Pac J Cancer Prev. 2014;15(14):5767–72.
Wang WB, Yen ML, Liu KJ, Hsu PJ, Lin MH, Chen PM, et al. Interleukin-25 mediates transcriptional control of PD-L1 via STAT3 in multipotent human mesenchymal stromal cells (hMSCs) to suppress Th17 responses. Stem Cell Reports. 2015;5(3):392–404.
Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3(12):1355–63.
Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632.
Concha-Benavente F, Srivastava RM, Trivedi S, Lei Y, Chandran U, Seethala RR, et al. Identification of the cell-intrinsic and -extrinsic pathways downstream of EGFR and IFNgamma that induce PD-L1 expression in head and neck Cancer. Cancer Res. 2016;76(5):1031–43.
Zhang W, Pang Q, Yan C, Wang Q, Yang J, Yu S, et al. Induction of PD-L1 expression by epidermal growth factor receptor-mediated signaling in esophageal squamous cell carcinoma. Onco Targets Ther. 2017;10:763–71.
Wang Y, Hu J, Wang Y, Ye W, Zhang X, Ju H, et al. EGFR activation induced Snail-dependent EMT and myc-dependent PD-L1 in human salivary adenoid cystic carcinoma cells. Cell Cycle. 2018;17(12):1457-70.
Cheng CC, Lin HC, Tsai KJ, Chiang YW, Lim KH, Chen CG, et al. Epidermal growth factor induces STAT1 expression to exacerbate the IFNr-mediated PD-L1 axis in epidermal growth factor receptor-positive cancers. Mol Carcinog. 2018;57(11):1588-98.
Ota K, Azuma K, Kawahara A, Hattori S, Iwama E, Tanizaki J, et al. Induction of PD-L1 expression by the EML4-ALK Oncoprotein and downstream signaling pathways in non-small cell lung Cancer. Clin Cancer Res. 2015;21(17):4014–21.
Wang J, Jia Y, Zhao S, Zhang X, Wang X, Han X, et al. BIN1 reverses PD-L1-mediated immune escape by inactivating the c-MYC and EGFR/MAPK signaling pathways in non-small cell lung cancer. Oncogene. 2017;36(45):6235–43.
Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352(6282):227–31.
Coelho MA, de Carne TS, Rana S, Zecchin D, Moore C, Molina-Arcas M, et al. Oncogenic RAS signaling promotes tumor Immunoresistance by stabilizing PD-L1 mRNA. Immunity. 2017;47(6):1083–99 e6.
Freeman SS, Allen SW, Ganti R, Wu J, Ma J, Su X, et al. Copy number gains in EGFR and copy number losses in PTEN are common events in osteosarcoma tumors. Cancer. 2008;113(6):1453–61.
Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13(1):84–8.
Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, et al. Eradication of triple-negative breast Cancer cells by targeting glycosylated PD-L1. Cancer Cell. 2018;33(2):187–201 e10.
Ni XY, Sui HX, Liu Y, Ke SZ, Wang YN, Gao FG. TGF-beta of lung cancer microenvironment upregulates B7H1 and GITRL expression in dendritic cells and is associated with regulatory T cell generation. Oncol Rep. 2012;28(2):615–21.
Song S, Yuan P, Wu H, Chen J, Fu J, Li P, et al. Dendritic cells with an increased PD-L1 by TGF-beta induce T cell anergy for the cytotoxicity of hepatocellular carcinoma cells. Int Immunopharmacol. 2014;20(1):117–23.
Starke A, Wuthrich RP, Waeckerle-Men Y. TGF-beta treatment modulates PD-L1 and CD40 expression in proximal renal tubular epithelial cells and enhances CD8 cytotoxic T-cell responses. Nephron Exp Nephrol. 2007;107(1):e22–9.
Thorn M, Guha P, Cunetta M, Espat NJ, Miller G, Junghans RP, et al. Tumor-associated GM-CSF overexpression induces immunoinhibitory molecules via STAT3 in myeloid-suppressor cells infiltrating liver metastases. Cancer Gene Ther. 2016;23(6):188–98.
Wang TT, Zhao YL, Peng LS, Chen N, Chen W, Lv YP, et al. Tumour-activated neutrophils in gastric cancer foster immune suppression and disease progression through GM-CSF-PD-L1 pathway. Gut. 2017;66(11):1900–11.
Li DW, Dong P, Wang F, Chen XW, Xu CZ, Zhou L. Hypoxia induced multidrug resistance of laryngeal cancer cells via hypoxia-inducible factor-1alpha. Asian Pac J Cancer Prev. 2013;14(8):4853–8.
Borsi E, Terragna C, Brioli A, Tacchetti P, Martello M, Cavo M. Therapeutic targeting of hypoxia and hypoxia-inducible factor 1 alpha in multiple myeloma. Transl Res. 2015;165(6):641–50.
Yi M, Li J, Chen S, Cai J, Ban Y, Peng Q, et al. Emerging role of lipid metabolism alterations in Cancer stem cells. J Exp Clin Cancer Res. 2018;37(1):118.
Tang L, Wei F, Wu Y, He Y, Shi L, Xiong F, et al. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J Exp Clin Cancer Res. 2018;37(1):87.
Zhang Y, Xia M, Jin K, Wang S, Wei H, Fan C, et al. Function of the c-met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol Cancer. 2018;17(1):45.
Semenza GL. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin Cancer Biol. 2009;19(1):12–6.
Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20(1):51–6.
Liang F, Li Q, Li X, Li Z, Gong Z, Deng H, et al. TSC22D2 interacts with PKM2 and inhibits cell growth in colorectal cancer. Int J Oncol. 2016;49(3):1046–56.
Li Q, Chen P, Zeng Z, Liang F, Song Y, Xiong F, et al. Yeast two-hybrid screening identified WDR77 as a novel interacting partner of TSC22D2. Tumour Biol. 2016;37(9):12503–12.
Azab AK, Hu J, Quang P, Azab F, Pitsillides C, Awwad R, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood. 2012;119(24):5782–94.
Zhao X, Gao S, Ren H, Sun W, Zhang H, Sun J, et al. Hypoxia-inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-bundling protein fascin. Cancer Res. 2014;74(9):2455–64.
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–5.
Wong W, Goehring AS, Kapiloff MS, Langeberg LK, Scott JD. mAKAP compartmentalizes oxygen-dependent control of HIF-1alpha. Sci Signal. 2008;1(51):ra18.
Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211(5):781–90.
Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014;74(3):665–74.
Ruf M, Moch H, Schraml P. PD-L1 expression is regulated by hypoxia inducible factor in clear cell renal cell carcinoma. Int J Cancer. 2016;139(2):396–403.
Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013;45(8):860–7.
Koh YW, Han JH, Park SY, Yoon DH, Suh C, Huh J. GLUT1 as a prognostic factor for classical Hodgkin's lymphoma: correlation with PD-L1 and PD-L2 expression. J Pathol Transl Med. 2017;51(2):152–8.
Haderk F, Schulz R, Iskar M, Cid LL, Worst T, Willmund KV, et al. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci Immunol. 2017;2(13):eaah5509.
Gabrusiewicz K, Li X, Wei J, Hashimoto Y, Marisetty AL, Ott M, et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology. 2018;7(4):e1412909.
Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560(7718):382–6.
Gong Z, Zhang S, Zeng Z, Wu H, Yang Q, Xiong F, et al. LOC401317, a p53-regulated long non-coding RNA, inhibits cell proliferation and induces apoptosis in the nasopharyngeal carcinoma cell line HNE2. PLoS One. 2014;9(11):e110674.
Patel JS, Hu M, Sinha G, Walker ND, Sherman LS, Gallagher A, et al. Non-coding RNA as mediators in microenvironment-breast cancer cell communication. Cancer Lett. 2016;380(1):289–95.
Fan C, Wang J, Tang Y, Wang Y, Xiong F, Zhang S, et al. Long non-coding RNA LOC284454 promotes migration and invasion of nasopharyngeal carcinoma via modulating the rho/Rac signaling pathway. Carcinogenesis. 2018. Epub ahead of print. https://doi.org/10.1093/carcin/bgy143.
Lian Y, Xiong F, Yang L, Bo H, Gong Z, Wang Y, et al. Long noncoding RNA AFAP1-AS1 acts AS a competing endogenous RNA of miR-423-5p to facilitate nasopharyngeal carcinoma metastasis through regulating the rho/Rac pathway. J Exp Clin Cancer Res. 2018;37(1):253.
Tang Y, He Y, Zhang P, Wang J, Fan C, Yang L, et al. LncRNAs regulate the cytoskeleton and related rho/ROCK signaling in cancer metastasis. Mol Cancer. 2018;17(1):77.
He Y, Jing Y, Wei F, Tang Y, Yang L, Luo J, et al. Long non-coding RNA PVT1 predicts poor prognosis and induces radioresistance by regulating DNA repair and cell apoptosis in nasopharyngeal carcinoma. Cell Death Dis. 2018;9(2):235.
Yang L, Tang Y, Xiong F, He Y, Wei F, Zhang S, et al. LncRNAs regulate cancer metastasis via binding to functional proteins. Oncotarget. 2017;9(1):1426–43.
Wang Y, Mo Y, Yang X, Zhou R, Wu Z, He Y, et al. Long non-coding RNA AFAP1-AS1 is a novel biomarker in various cancers: a systematic review and meta-analysis based on the literature and GEO datasets. Oncotarget. 2017;8(60):102346–60.
Geisler S, Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol. 2013;14(11):699–712.
Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43(6):904–14.
Tang Y, Wang J, Lian Y, Fan C, Zhang P, Wu Y, et al. Linking long non-coding RNAs and SWI/SNF complexes to chromatin remodeling in cancer. Mol Cancer. 2017;16(1):42.
Fan C, Tang Y, Wang J, Xiong F, Guo C, Wang Y, et al. Role of long non-coding RNAs in glucose metabolism in cancer. Mol Cancer. 2017;16(1):130.
Yu J, Liu Y, Guo C, Zhang S, Gong Z, Tang Y, et al. Upregulated long non-coding RNA LINC00152 expression is associated with progression and poor prognosis of tongue squamous cell carcinoma. J Cancer. 2017;8(4):523–30.
Kathuria H, Millien G, McNally L, Gower AC, Tagne JB, Cao Y, et al. NKX2-1-AS1 negatively regulates CD274/PD-L1, cell-cell interaction genes, and limits human lung carcinoma cell migration. Sci Rep. 2018;8(1):14418.
Smith B, Agarwal P, Bhowmick NA. MicroRNA applications for prostate, ovarian and breast cancer in the era of precision medicine. Endocr Relat Cancer. 2017;24(5):R157–R72.
Gong Z, Yang Q, Zeng Z, Zhang W, Li X, Zu X, et al. An integrative transcriptomic analysis reveals p53 regulated miRNA, mRNA, and lncRNA networks in nasopharyngeal carcinoma. Tumour Biol. 2016;37(3):3683–95.
Xu K, Xiong W, Zhou M, Wang H, Yang J, Li X, et al. Integrating ChIP-sequencing and digital gene expression profiling to identify BRD7 downstream genes and construct their regulating network. Mol Cell Biochem. 2016;411(1–2):57–71.
Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nat Struct Mol Biol. 2012;19(6):586–93.
Gong AY, Zhou R, Hu G, Liu J, Sosnowska D, Drescher KM, et al. Cryptosporidium parvum induces B7-H1 expression in cholangiocytes by down-regulating microRNA-513. J Infect Dis. 2010;201(1):160–9.
Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, et al. PDL1 Regulation by p53 via miR-34. J Natl Cancer Inst. 2015;108(1):djv303.
Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5:5241.
Jia L, Xi Q, Wang H, Zhang Z, Liu H, Cheng Y, et al. miR-142-5p regulates tumor cell PD-L1 expression and enhances anti-tumor immunity. Biochem Biophys Res Commun. 2017;488(2):425–31.
Cioffi M, Trabulo SM, Vallespinos M, Raj D, Kheir TB, Lin ML, et al. The miR-25-93-106b cluster regulates tumor metastasis and immune evasion via modulation of CXCL12 and PD-L1. Oncotarget. 2017;8(13):21609–25.
Zhao L, Yu H, Yi S, Peng X, Su P, Xiao Z, et al. The tumor suppressor miR-138-5p targets PD-L1 in colorectal cancer. Oncotarget. 2016;7(29):45370–84.
Miao S, Mao X, Zhao S, Song K, Xiang C, Lv Y, et al. miR-217 inhibits laryngeal cancer metastasis by repressing AEG-1 and PD-L1 expression. Oncotarget. 2017;8(37):62143–53.
Audrito V, Serra S, Stingi A, Orso F, Gaudino F, Bologna C, et al. PD-L1 up-regulation in melanoma increases disease aggressiveness and is mediated through miR-17-5p. Oncotarget. 2017;8(9):15894–911.
Xie G, Li W, Li R, Wu K, Zhao E, Zhang Y, et al. Helicobacter pylori promote B7-H1 expression by suppressing miR-152 and miR-200b in gastric Cancer cells. PLoS One. 2017;12(1):e0168822.
Wang W, Li F, Mao Y, Zhou H, Sun J, Li R, et al. A miR-570 binding site polymorphism in the B7-H1 gene is associated with the risk of gastric adenocarcinoma. Hum Genet. 2013;132(6):641–8.
Kao SC, Cheng YY, Williams M, Kirschner MB, Madore J, Lum T, et al. Tumor suppressor microRNAs contribute to the regulation of PD-L1 expression in malignant pleural mesothelioma. J Thorac Oncol. 2017;12(9):1421–33.
Qu F, Ye J, Pan X, Wang J, Gan S, Chu C, et al. MicroRNA-497-5p down-regulation increases PD-L1 expression in clear cell renal cell carcinoma. J Drug Target. 2019;27(1):67–74.
Dong P, Xiong Y, Yu J, Chen L, Tao T, Yi S, et al. Control of PD-L1 expression by miR-140/142/340/383 and oncogenic activation of the OCT4-miR-18a pathway in cervical cancer. Oncogene. 2018;37(39):5257–68.
Zhu J, Chen L, Zou L, Yang P, Wu R, Mao Y, et al. MiR-20b, −21, and -130b inhibit PTEN expression resulting in B7-H1 over-expression in advanced colorectal cancer. Hum Immunol. 2014;75(4):348–53.
Fujita Y, Yagishita S, Hagiwara K, Yoshioka Y, Kosaka N, Takeshita F, et al. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Mol Ther. 2015;23(4):717–27.
Zhong Y, Du Y, Yang X, Mo Y, Fan C, Xiong F, et al. Circular RNAs function as ceRNAs to regulate and control human cancer progression. Mol Cancer. 2018;17(1):79.
Zhou R, Wu Y, Wang W, Su W, Liu Y, Wang Y, et al. Circular RNAs (circRNAs) in cancer. Cancer Lett. 2018;425:134–42.
He R, Liu P, Xie X, Zhou Y, Liao Q, Xiong W, et al. circGFRA1 and GFRA1 act as ceRNAs in triple negative breast cancer by regulating miR-34a. J Exp Clin Cancer Res. 2017;36(1):145.
Wang Y, Mo Y, Gong Z, Yang X, Yang M, Zhang S, et al. Circular RNAs in human cancer. Mol Cancer. 2017;16(1):25.
Zhang XL, Xu LL, Wang F. Hsa_circ_0020397 regulates colorectal cancer cell viability, apoptosis and invasion by promoting the expression of the miR-138 targets TERT and PD-L1. Cell Biol Int. 2017;41(9):1056–64.
Berger KN, Pu JJ. PD-1 pathway and its clinical application: a 20year journey after discovery of the complete human PD-1 gene. Gene. 2018;638:20–5.
Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, et al. Hyperprogressive disease is a new pattern of progression in Cancer patients treated by anti-PD-1/PD-L1. Clin Cancer Res. 2017;23(8):1920–8.
Wei F, Wu YF, Tang L, Xiong F, Guo C, Li XY, et al. Trend analysis of cancer incidence and mortality in China. Sci China-Life Sci. 2017;60(11):1271–5.
Bertrand F, Montfort A, Marcheteau E, Imbert C, Gilhodes J, Filleron T, et al. TNFalpha blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat Commun. 2017;8(1):2256.
Yi Y, Chen D, Ao J, Sun S, Wu M, Li X, et al. Metformin promotes AMP-activated protein kinase-independent suppression of DeltaNp63alpha protein expression and inhibits Cancer cell viability. J Biol Chem. 2017;292(13):5253–61.
Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell. 2018;71(4):606–20 e7.
Acknowledgments
Not applicable.
Funding
This study was supported by grants from the National Natural Science Foundation of China (81572787, 81672683, 81672993, 81772928, 81702907, 81772901, 31741049, 21806186 and 81872278), the Overseas Expertise Introduction Project for Discipline Innovation (111 Project, No. 111–2-12) and the Natural Science Foundation of Hunan Province (2016JC2035, 2017SK2015, 2018JJ3704, 2018JJ3815, 2018SK21210 and 2018SK21211).
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
XJ, JW, XD, FX, JG, BX, XW, JM, MZ, XL, YL, GL and WX collected the related paper and drafted the manuscript. CG, and ZZ participated in the design of the review and draft the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jiang, X., Wang, J., Deng, X. et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer 18, 10 (2019). https://doi.org/10.1186/s12943-018-0928-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-018-0928-4