
Abstract
ApoE is the major lipid and cholesterol carrier in the CNS. There are three major human polymorphisms, apoE2, apoE3, and apoE4, and the genetic expression of APOE4 is one of the most influential risk factors for the development of late-onset Alzheimer's disease (AD). Neuroinflammation has become the third hallmark of AD, together with Amyloid-β plaques and neurofibrillary tangles of hyperphosphorylated aggregated tau protein. This review aims to broadly and extensively describe the differential aspects concerning apoE. Starting from the evolution of apoE to how APOE's single-nucleotide polymorphisms affect its structure, function, and involvement during health and disease. This review reflects on how APOE's polymorphisms impact critical aspects of AD pathology, such as the neuroinflammatory response, particularly the effect of APOE on astrocytic and microglial function and microglial dynamics, synaptic function, amyloid-β load, tau pathology, autophagy, and cell–cell communication. We discuss influential factors affecting AD pathology combined with the APOE genotype, such as sex, age, diet, physical exercise, current therapies and clinical trials in the AD field. The impact of the APOE genotype in other neurodegenerative diseases characterized by overt inflammation, e.g., alpha- synucleinopathies and Parkinson's disease, traumatic brain injury, stroke, amyotrophic lateral sclerosis, and multiple sclerosis, is also addressed. Therefore, this review gathers the most relevant findings related to the APOE genotype up to date and its implications on AD and CNS pathologies to provide a deeper understanding of the knowledge in the APOE field.
Similar content being viewed by others

Background
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by gradual cognitive decline and memory loss. AD represents 60 to 70% of dementia cases and affects more than 50 million people worldwide [1]. Reported deaths from AD increased 148% between 2000 and 2018, and the economic burden worldwide was estimated at $305 billion for 2020 [1, 2].
The two main pathological hallmarks of AD are the accumulation of extracellular amyloid plaques and intraneuronal deposition of hyperphosphorylated microtubule-associated protein tau in the form of neurofibrillary tangles (NFTs). The classical hypothesis regarding AD’s pathophysiology is the amyloid cascade hypothesis postulated by Hardy and Higgins in 1992 [3]. Its premise is that amyloid plaques result from an imbalance between the production and clearance of amyloid-β (Aβ) peptides and that accumulation of Aβ peptides is the leading cause of AD. Aβ peptides are generated by the cleavage of the amyloid precursor protein (APP), a single-pass transmembrane- type-1 integral glycoprotein that can be cleaved and metabolized by α-secretases (non-amyloidogenic pathway) or by β-secretases (amyloidogenic pathway). Under physiological conditions, the Aβ peptide balance between production and clearance is maintained [4].
In the amyloidogenic pathway, APP is cleaved by β-secretase (also known as BACE-1), producing soluble APPβ (sAPPβ) and membrane-bound C-terminal 99 or 89 amino acid fragments, also known as β-CTF. Cleavage of APP CTFs leads to the extracellular secretion of Aβ peptides mainly composed of 39 to 42 amino acid length residues, in which Aβ1-40 and Aβ1-42 are predominant in vivo species. When soluble Aβ is over-produced, or its clearance is impaired, it self-assembles to form the amyloid fibrils constituting amyloid plaques. At certain levels and conformations, Aβ becomes toxic for neurons in many ways, inducing inflammation, oxidative stress, alterations in calcium homeostasis and membrane potentials, and cytoskeleton disruption [5,6,7]. Thus, according to the amyloid cascade hypothesis, Aβ accumulation also drives tau pathology. The physiological role of tau is closely related to the maintenance of neuronal morphology, synaptic plasticity, and axonal transport of organelles by its interaction with α-tubulin and β-tubulin [8,9,10]. Tau is encoded by the MAPT gene. There are six tau isoforms described in mammals, which differ in their microtubule-binding domains. Hyperphosphorylation of tau will negatively affect its binding properties to microtubules and enhance tau self-assembling capacity to form oligomers, fibrils, and eventually NFTs [11].
Multiple findings have called the Amyloid hypothesis into question in the past years. Some of the reasons are the failure of most anti-amyloid-based therapeutics, lack of neuropathological correlations between amyloid plaque density and disease severity, or lack of clinically predictive AD mouse models. Accordingly, new etiological hypotheses and strategies have grown, for instance, a focus on the senescence-associated secretory phenotype in the central nervous system (CNS) cells emerged as a promising therapeutic strategy for multiple age-related conditions [12, 13]. The prion-like propagation hypothesis was proposed for AD, Parkinson’s disease (PD), and Amyotrophic Lateral Sclerosis (ALS) based on the ability of misfolded proteins from neurodegenerative diseases to seed and propagate pathology in a prion-like manner [14]. The cholinergic hypothesis is based on the progressive loss of cholinergic innervations during AD and was the starting point for using cholinesterase inhibitors to reduce AD symptoms [15,16,17]. The inflammatory-infectious hypothesis of AD suggests that dysbiosis of human intestinal microflora is responsible for AD [18]. Further, recent findings point to tau as the main factor leading to the development and progression of AD, even proposing that p-tau can accelerate Aβ cleavage from APP [19, 20]. Despite all these hypotheses, AD’s etiology is still largely unknown, and multiple pathways and targets are still under examination.
AD cases have commonly been categorized into early-onset or late-onset AD cases (EOAD and LOAD). This categorization is based on an arbitrary age cut-off set, corresponding to the age at which clinical symptoms start. It is still applied in current research and clinical trials, as recently reviewed by Reitz and colleagues, to determine potential overlap among these categories [21]. EOAD cases represent between 2 and 10% of all AD cases [22]. Patients develop symptoms before the age of 65 and are mostly related to autosomal dominant genetic mutations in APP, situated on chromosome 21 [23], presenilin1 (PSEN1), on chromosome 14 [24, 25] and Presenilin 2 (PSEN2), on chromosome 1 [26, 27]. Heritability of EOAD ranges between 92–100% [28, 29], so some refer to it as familial EOAD. However, some cases follow a non-mendelian pattern of heritability, perhaps due to new rare variants [30]. LOAD has an estimated heritability rate of 60 to 80% [31]. Most LOAD cases are sporadic and present an undetermined familial pattern of disease. LOAD pathogenesis is characterized by a prolonged asymptomatic preclinical phase in which Aβ alterations begin approximately 10 to 20 years before the onset of the symptoms [32].
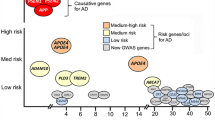
The most influential genetic risk factor associated with LOAD is the expression of polymorphisms in the apolipoprotein E gene (APOE). APOE’s role in increasing AD risk is complex and multifactorial. The latest findings revealed that expressing the APOE2 allele robustly decreases LOAD risk through Aβ-dependent and independent mechanisms [33], whereas the expression of one 4 allele increases threefold the risk of developing AD and expressing two 4 alleles increases risk 9 to 15-fold [34, 35]. The detrimental effects of APOE on AD pathology have been linked to its immunomodulatory functions [36]. APOE can alter the expression profile of astrocytes and microglia, the key players in the central nervous system’s immune response [37, 38]. APOE4 exacerbates Aβ aggregation, tau pathology, neuroinflammation, and neurodegeneration [39], but the mechanisms through which APOE4 exerts its detrimental effects in AD pathology are still under study. Advances in single-cell analysis, transcriptomics, and metabolomic studies have shed some light on the effect of APOE upon multiple aspects of AD pathology. In line with this, genome-wide association studies (GWAS) have identified over 44 risk loci that influence the risk of suffering LOAD. However, there are still plenty of genetic risk factors remaining uncharacterized. These genetic variants are related to multiple pathways, such as endocytosis and intracellular trafficking, APP processing, tau metabolism, lipid and energetic metabolism, synaptic plasticity, inflammation, innate neuronal immunity, glial cell activation, and proteolytic and transcription processes [21, 40,41,42].
Thus, this review aims to extensively describe the influence of APOE and its polymorphisms in different factors influencing AD, bringing special attention to the latest advances in the AD field regarding APOE’s influence in glial activity, synaptic function, cellular processes such as autophagy, and signaling cascades. We also aim to underline the latest information relative to clinical trials in AD, external factors that may influence AD progression, such as diet, sex, and physical exercise. However, the implications of APOE are not limited to AD. The APOE genotype is also related to the severity of other proteinopathies and neurodegenerative diseases characterized by overt neuroinflammation [43]. Hence, we also aim to comment on the influence of the APOE genotype in other neurodegenerative diseases.
Evolutionary aspects of APOE
Apolipoproteins are well-preserved proteins throughout evolution present in aquatic and terrestrial vertebrates. Apolipoproteins belong to the phylogenetically older and larger protein family that include the Pfam family domain PF01442. This domain is also present in some choanoflagellates, thereby dating the emergence of apolipoproteins earlier than the appearance of animals. From this family, a large number of apolipoproteins emerged. APOE orthologs are observed in fish, amphibians, reptiles, and mammals. An analysis of the APOE phylogeny comparing 18 species showed that APOE differed the most in fish and frogs, indicating significant gene changes occurred in the gene since the divergence from fish and amphibians [44]. Interestingly, APOE is absent in birds [45], which additionally lack many of the major proteins involved in cholesterol internalization [46]. This phenomenon is considered an avian-specific loss; since APOE is present in alligators (Gene ID: 109,284,755). The apolipoprotein family has undergone numerous gain and loss of function events in the vertebrate lineage [47]. It has been proposed that the APOE gene evolved from APOC-1 genome duplication, an event dated approximately 400 million years ago in a common ancestor of fish and mammals [48]. Interestingly, the low-density lipoprotein receptor (LDLR) family, to which apoE binds, is much older and appears to have evolved rapidly after the appearance of multicellular organisms [49]. Humans are the only known species in the animal kingdom with multiple functionally polymorphic variants of APOE. Chimpanzees (Pan Troglodytes Verus), our closest evolutionary relatives (divergence approx. 5–7.5 million years ago), appear to have a monomorphic APOE gene which is similar to our human APOE4 [50]. The same happens for Denisovans and Neanderthals, although only a few individual fossils have been sequenced. The APOE2, APOE3, and APOE4 variants result from three of the four haplotypes predicted from combinations of the alleles of the two single-nucleotide polymorphisms rs429358, and rs7412. A fourth and extremely rare haplotype consists of the combination of an arginine at position 112 and a cysteine at position 158, and is defined as APOE1, also known as APOE3r. APOE1 was described in 3 Italian families [51]. It is still unknown what drove the evolution of the allelic variants. Speculations include that APOE3 might result from an adaptation to meat-eating since lipoproteins play essential roles in dietary functions [52]. Comparing APOE from different species also revealed that species with similar diets cluster more closely, indicating that changes in APOE could reflect differing diets of animals [44]. APOE3 and APOE2 are also thought to play significant roles in the evolution of human longevity. Humans have exceptionally long lifespans compared with both our primate brethren and mammalian cousins. This increased lifespan presumably evolved in ancestors carrying APOE4/4, which is associated with increased mortality and morbidity. Some propose that over millennia the longevity and health of “post-reproductive” individuals increase the fitness of their offspring’s offspring, also known as the “grandmother” hypothesis, which could explain why APOE3 is now the dominant allele [53]. Some even argue that APOE3 and APOE2 were mutations that negate an earlier deleterious mutation fixed in the human line [54]. This hypothesis suggests that chimpanzee APOE sequences are most similar to the APOE4 human allele. However, functionally chimpanzee apoE protein resembles that deriving from the expression of the APOE3 allele [50]. A possible explanation for this is that humans faced an evolutionary bottleneck at some point in time, and a detrimental mutation occurred in the APOE4 gene, possibly at position 61, as most mammals, including the great apes, present a threonine molecule at this site [55, 56]. Consequently, the APOE3 and APOE2 genes, based on specific amino acid substitutions, are thought to compensate for the detrimental change in human APOE4. An alternative hypothesis is that APOE4 played a more critical role in immune function in an era when infections were the primary selective pressure on humans. Thus, potentially making APOE4 more prevalent [57]. Supporting this, a study conducted on the forager-horticulturalists Tsimane people suggested that APOE4 could better preserve cognitive function in the case of high parasite burden compared to APOE3 [58]. Either way, both simulations and sequence studies strongly suggest that the prevalence of APOE2 and APOE3 result of natural selection [59, 60].
ApoE structure and isoforms
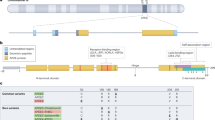
ApoE is a 34 kDa glycoprotein composed of 299 amino acids and belongs to the family of amphiphilic exchangeable lipoproteins. The human APOE gene is located in chromosome 19q13.32, constituted by four exons and three introns [61]. The historical progress in understanding apoE structure was recently documented in the review by Chen and colleagues [62]. From the human allelic variations, APOE derives in six major genotypes, three of them homozygous (APOE2/2, APOE3/3, and APOE4/4) and three heterozygous (APOE2/3, APOE2/4, and APOE3/4). The resultant proteins differ from each other in the amino acids located at positions 112 and 158, resulting in apoE2 (Cys112; Cys158), apoE3 (Cys112; Arg158), and apoE4 (Arg112; Arg158). The human apoE protein contains three main domains, an N-terminal and a C-terminal domain joined by a flexible hinge region. The N- and C-terminal domains contain the receptor-binding and the lipid-binding regions, respectively [55, 63]. The alterations in the amino acid sequences between apoE isoforms are responsible for the differential protein folding of apoE, leading to the differences in the lipid and receptor binding abilities of the different apoE isoforms [64]. Conversely, the mouse apoE gene (m-apoE) is located on chromosome 7 and exists as one isoform. Homology between the promoter regions of human and m-apoE is less than 40%, and m-apoE interactions resemble those of human apoE3 [56, 65]. It is also relevant to note that human and m-apoE differ in functionality in most AD processes, such as Aβ clearance, neuroinflammation, and synaptic integrity [66,67,68]. These disparities justified using the human APOE gene sequence in transgenic pre-clinical AD models.
The differential functional properties that confer apoE its polymorphisms urged determining the population’s allelic frequency of APOE alleles. The global human allele frequencies of APOE2, E3, and E4 are 7%, 79%, and 15% each and result from non-synonymous SNPs located at exon number 4 of the APOE gene [69, 70]. More details related to the global distribution of the APOE allele frequency in humans can be found in various reviews [71,72,73]. SNPs in the surroundings of the APOE gene can also lead to variations in the apoE CSF levels, such as the TOMM40 gene, the APOE promoter, and distal APOE enhancer elements (MEU and BCR) [70]. The case report from Arboleda-Velasquez and colleagues informed about resistance to autosomal dominant AD in a Colombian woman carrying two copies of the Christchurch mutation [74]. The APOE Christchurch mutation presents a Ser-136 variant [R136S] [75]. This mutation strongly decreases apoE-LDLR and apoE-heparin-binding [74, 76].
However, it is interesting to note that heterozygous carriers of the Christchurch mutations seem not to benefit from its protective effects, according to the findings from Hernandez I. and colleagues [77]. Lately, it has been reported the APOE3-Jacksonville mutation, which presents an alteration in the lipid-binding region and has been reported to reduce Aβ aggregation in 5xFAD mice [78]. Thus, further studies regarding the influence of this mutation in the development of AD would result in great value (Fig. 1).

ApoE protein structure and mutations. The human apoE protein is characterized by three main domains, an N-terminal containing the receptor-binding region and a C-terminal domain where the lipid-binding region is located. A flexible hinge region joins N-terminal and C-terminal domains. Mutations at positions 112 and 158 will give rise to the most prevalent apoE isoforms, apoE2, apoE3, and apoE4. Other apoE variants characterized are the Christchurch mutation, which presents a Ser-136 variant [R136S]. ApoE Jacksonville mutation, also called V236E, variant is located at position 236 proximal to the lipid-binding region and reduces apoE self-aggregation. Lastly, the apoE R251G variant induces a single amino acid switch at position 251 and has been initially linked with decreased risk of suffering AD
ApoE production, secretion and lipidation in the CNS
ApoE production and secretion
ApoE production and secretion are highly cell- and tissue-specific, and inducible by various transcription factors and regulatory elements, hormones, cytokines, or lipids [79, 80]. Astrocytes are the major producers of apoE in the CNS, including specialized astrocytes, Bergmann glia, tanycytes, pituicytes, and retinal Müller cells [81]. In addition, microglia, oligodendrocytes, pericytes, the choroid plexus, and neurons under stress situations can also produce apoE to a lesser extent [82,83,84,85]. ApoE is produced following the classical secretory pathway; it is synthesized in the endoplasmic reticulum, post-translationally glycosylated and sialylated in the Golgi network, transported to the plasma membrane, and secreted [79, 86]. Multiple studies have focused on the impact of protein glycosylation in AD pathogenesis [87,88,89], apoE produced by astrocytes is more heavily sialylated and glycosylated, and some studies suggest the existence of tissue-specific apoE glycoforms [79, 90].
ApoE expression and secretion by astrocytes are regulated by the liver X receptor (LXR) and the retinoid X receptor (RXR), DNA-binding factors implicated in cholesterol metabolism. LXR has also been related to the immune response during inflammation and blood–brain barrier (BBB) function [91,92,93]. Brain-derived neurotrophic factor (BDNF) can stimulate LXR to induce APOE expression and astrocytic cholesterol efflux [94]. Recently, Axl receptor tyrosine kinase was also found to regulate APOE in human astrocytes [95]. LXR and RXR also are involved in the transcriptional regulation of APOE [96]. RXR is primarily expressed in hepatocytes, even so, it has been related to brain clean-up after stroke, and RXR agonists have been proposed as a therapeutic approach for AD treatment as are directly targeting LXR and RXR [97,98,99].
Additionally, ATP binding cassette transporter A1 (ABCA1) is transcriptionally regulated by LXR [100]. ABCA1 is one of the main transporters involved in cholesterol efflux regulation through lipoproteins. ABCA1 plays a crucial role in forming high-density lipoproteins (HDL) [101, 102]. It has been demonstrated that the higher tendency of apoE4 to self-aggregate decreases ABCA1 membrane-recycling and therefore leads to lower lipidation of apoE4 particles (Fig. 2) [103].

Roles of distinct APOE isoforms in astrocyte-neuron communication. The liver X receptor (LXR) and retinoid X receptor (RXR) activation regulate APOE isoforms expression in astrocytes. ATP-binding cassette transporter (ABCA1) is responsible for apoE lipidation and secretion to the extracellular space, so as to lipidate apoE already present in the extracellular space. ApoE can be later on recognized by several neuronal lipid receptors including VLDL-receptor (VLDLR), LDLR, Low-density lipoprotein receptor-related protein 1 (LRP1), and heparan sulfate proteoglycans (HSPGs). The distinct apoE isoform lipidation status promotes distinct receptor affinities. The apoE4 isoform presents alterations in its physiological pathways compared to other isoforms, such as low lipidation status, the formation of non-lipidated apoE4 aggregates, and a poor lipid turnover towards astrocytes, which leads to neuronal lipid-droplet accumulation. This promotes mitochondrial damage and reactive oxygen species (ROS) production. Moreover, apoE4 promotes endosome formation and degradation of synaptic receptors like AMPA or NMDA, leading to impaired synaptic function
The brain is the richest body organ in cholesterol, bearing approximately 20% of total body cholesterol, and apoE is the primary responsible for lipid transport and maintenance of cholesterol homeostasis in the brain. ApoE is essential in providing neurons with cholesterol and carrying out excessive cholesterol clearance [104, 105]. ApoE is involved in cell membrane support, synaptic plasticity, signal transduction, proteostasis immunomodulation, and repair after an injury [106,107,108].
ApoE lipidation
To perform its correct functions, apoE must be secreted and lipidated. ApoE lipidation is mediated by cell-surface receptors, ABCA1 and ATP Binding Cassette Subfamily G Member 1 (ABCG1), which moderate cholesterol efflux to apoE [109]. ABCA1 and ABCG1 are expressed in neurons, glial cells, and macrophages [110, 111]. It has been demonstrated that APOE4 promotes greater expression levels of ADP-ribosylation factor 6 (ARF6), a protein involved in ABCA1 recycling, by sequestering ABCA1 in the late endosomes [103]. The receptor-binding abilities, molecular stability, and correct functioning of apoE are conditioned by its lipidation status. Isoform variations of APOE will directly impact its lipidation (apoE4 < apoE3 < apoE2) [112, 113]. In vitro model results pointed out that apoE lipidation prevents its self-aggregation [112]. Thus, due to the critical impact of lipidation on apoE’s physiological and pathological roles, increasing apoE lipidation has been proposed as a target for AD treatment [114].
Lipidated apoE will be internalized through apoE receptors, e.g., low-density lipoprotein receptor-related protein 1 (LRP1) and LDLR [71, 115]. LDLR belongs to the LRP family, seven structurally close-related transmembrane proteins that include LRP1, LRP1b, LRP2, LRP4, LRP8, VLDLR, and LDLR [116]. Previous findings reported that LRP1 preferentially binds to recombinant apoE, lipoprotein particles enriched for apoE, and HDL particles derived from CSF; Whereas LRPs and VLDLR bind all apoE particles, including lipid-free apoE in the case of VLDLR, LDLRs preferentially bind to lipidated apoE particles [117,118,119]. Apolipoprotein E receptor 2 (ApoER2), also known as LRP8, and VLDLR are core components of the Reelin signaling pathway implicated in regulating learning, neuronal migration, dendritic growth, and synaptic plasticity [120,121,122]. VLDLR mRNA expression levels are the highest in the cortex and the cerebellum, particularly in astrocytes, microglia, oligodendrocytes, pyramidal neurons, neuroblasts, glioblasts, and Cajal-Retzius cells, whereas ApoER2 expression in the CNS is related to the brain areas such as the cerebellum, hippocampus, olfactory bulb, neocortex, and cortical neurons, especially in granule cells of the dentate gyrus and the pyramidal cells [123, 124].
Impact of apoE isoforms on receptor and target binding
Allelic variation of APOE will influence the lipid particle size that apoE binds to, the type of lipids and proteoglycans that apoE will interact with, the brain lipid profile, and apoE’s interacting abilities with its receptors. For instance, a study comparing apoE particle size from temporal lobe brain homogenates of AD and control patients by size-exclusion chromatography found that apoE particles in APOE4/4 patients were twice as big as those from APOE3/3 AD patients and APOE3/3 control subjects [125]. However, this study did not include genotype control APOE4/4 subjects, and no comparison was performed between APOE4/4 and APOE3/3 control subjects. The increased particle size in APOE4/4 subjects might be associated with the fact that the APOE4 genotype is related to high blood LDL-cholesterol levels [126].
It has been proposed that only monomeric apoE can bind lipid vesicles and that phospholipids play a role in successful apoE-receptor interaction [127, 128]. ApoE4 peptides present a high lipid-binding affinity related to a lower recycling capacity and increased intracellular cholesterol accumulation [129, 130]. Combining chemical cross-linking with mass spectrometry, Henry N. and colleagues showed that apoE4 adjusts its conformation for its recognition by LDLR [131]. ApoE4 can also bind LRP1 with greater affinity than apoE3 and apoE2 [132]. ApoE2 is characterized by reduced binding affinity to LDLRs, which relates to the high susceptibility of APOE2/2 carriers to develop type III hyperlipoproteinemia [133]. Nonetheless, recent meta-analysis studies have also revealed APOE4 as a hyperlipidemia risk factor [134].
Targeted lipidomic analysis in APOE knock-in mice have evidenced a higher susceptibility of lipidic alterations in the entorhinal cortex (EC) of APOE4 mice [135]. Shotgun lipidomic studies in the parietal lobe of AD brains from APOE2, APOE3 and APOE4 carriers evidenced significantly lower phospholipid levels in APOE4 carriers and marked differences in the brain lipidomes between APOE3/3 and APOE4 carriers [136]. Posterior studies have showed an upregulation of several phospholipid classes in APOE3/3 human brain samples compared to APOE4/4 carriers [137].
Regarding apoE’s lipid-binding abilities, apoE4 presents a binding preference for VLDLs and LDLs, while apoE3 presents a higher affinity towards HDLs [138]. ApoE3 and apoE2 bind HDL with higher affinity than apoE4, whereas apoE4 strongly binds VLDLs [139, 140]. Frieden and colleagues proposed a mechanistic explanation for the functional differences in lipid binding between apoE3 and apoE4 based on their full-length monomeric structure and their domain-domain interactions using molecular dynamics [141].
ApoE can also bind proteoglycans and glycosaminoglycans [142]. ApoE4 binds more strongly to heparan and dermatan-sulfate than apoE2 and apoE3 [143]. However, lipid-free apoE4 tends to bind to heparin to a lower extent than lipid-free apoE3 and apoE2, whereas lipidated apoE isoforms tend to bind to heparin in a similar manner [62, 144]. Both lipidated and non-lipidated apoE bind heparan sulfate proteoglycans (HSPGs) through the N-terminal binding site to clear remnant lipoproteins, particularly in the liver [117, 145,146,147]. The extracellular matrix is very rich in HSPGs, which have been proposed as a therapeutic target in tauopathies due to their tendency to associate with tau aggregates [148]. HSPGs are also related to neuroinflammation and colocalize with the core of amyloid deposits [149, 150]. HSPGs can bind Aβ and facilitate its oligomerization and aggregation [151]. In vitro studies have proposed that the interaction between HSPGs and LRP1 mediates Aβ uptake by primary neurons [152]. Moreover, genetically engineered animal models lacking HSPGs by conditional deletion of the Ext1 gene show lower Aβ deposition and higher clearance [153]. A summary of the identified apoE receptors is reflected in Table 1. The interacting affinities of the apoE isoforms with lipid receptors and its molecular targets are depicted in Tables 2 and 3.
Impact of APOE polymorphisms on CNS cell physiology
ApoE isoforms can affect multiple cellular functions in various cell types of the CNS, such as synaptic function in neurons, lipid homeostasis in astrocytes, and immune responses in microglial cells [178]. ApoE isoforms also affect mitochondrial biogenesis and dynamics [179]. The study by Area-Gomez and colleagues observed alterations in the mitochondrial respiration process among the distinct APOE genotypes, such as upregulation of genes related to oxidative phosphorylation decreases in mitochondrial respiration in the cortex and hippocampus of aged female and male APOE4/4 mice versus APOE3/3 [180]. Inorganic polyphosphate (Poly-P), proposed as an alternative to ATP for cellular energy storage, is involved in mitochondrial function and related to the mitochondrial processes affected in AD, such as calcium homeostasis alteration and acceleration of amyloid fibril formation [181]. ApoE was identified as a Poly-P ligand; however, the differential effect of apoE isoforms and their interaction with Poly-P are yet to be elucidated [182, 183]. APOE4 is closely related to synaptic degeneration, as observed from studies with cerebral organoids from AD patients carrying APOE4, in which APOE4 carriers presented higher levels of cellular apoptosis and decreased synaptic integrity [184]. Studies performing RNA-seq in iPSC neurons and glial cells have reported dramatic switches in the transcriptomic profile when performing isogenic conversion of APOE4 into APOE3 and a reduction in the APOE4-related phenotype [178, 185].
Distribution of apoE in the brain, cerebrospinal fluid, and blood
Over the past years, the potential diagnostic value of knowing apoE levels in biological fluids has gained substantial interest. So as to evaluate the impact of APOE’s allelic isoforms on the fluid concentrations of apoE in the cerebrospinal fluid (CSF) and plasma. As one of the main lipoproteins in the CSF, apoE preferentially binds HDLs and transports cholesterol within the CNS [104, 186]. The impact of the APOE genotype on the transport of HDL particles, cognition and its connection with the development of neurodegenerative diseases has been reviewed by other authors [187, 188].
Studies examining apoE protein levels in the brains of APOE knock-in mouse models have observed a genotype-dependent decrease in apoE levels apoE2 > apoE3 > apoE4 [189,190,191]. In the case of targeted replacement APOE3/4 mice, apoE4 constitutes 30 to 40% of total apoE levels, and newly synthesized apoE4 presents an enhanced degradation and reduced half-life compared to apoE3 [189].
Regarding the expression pattern of apoE in different brain areas, Donald Schmechel’s group analyzed apoE protein distribution in the cerebellum, frontal cortex, and hippocampus of APOE targeted-replacement mice, which revealed significant regional differences but no difference between the APOE genotypes in the apoE regional distribution [192]. The cerebellum was the area with the highest apoE expression, and the hippocampus the lowest. The authors also compared plasma apoE levels between APOE2, APOE3 and APOE4 transgenic mice and found that plasma apoE2 levels were 16-fold higher than the other two isoforms [192]. However, these findings contrast with those in Non-Obese Diabetic (NOD) background (FRGN) humanized liver mice in which they observed that the highest brain apoE levels (endogenous mouse apoE) were found in the hippocampus and the lowest in the thalamus [193]. Furthermore, these authors also found that the brain tissue levels, specifically in the cortex and to some extent in the hippocampus, are lower in mice with humanized APOE4 livers versus those with non-APOE4 livers [193]. Additional studies from APOE targeted replacement mice have shown that mice expressing apoE4 present lower CSF apoE levels than the other isoforms [189, 190, 194].
Plasmatic and CNS apoE do not cross the BBB, thus constituting two independent apoE pools; Peripheral apoE will be primarily produced by hepatocytes, whereas in the CNS, astrocytes will be the major apoE producers [195, 196]. APOE knock-in mice studies evaluating the influence of hepatic apoE on Aβ deposition observed that genetic deletion of hepatic APOE did not influence brain apoE levels but induced plasma lipid profile changes and a decrease in plasmatic apoE levels [197]. Lane-Donovan and colleagues evidenced that genetic restoration of plasmatic apoE into wild-type levels in APOE knock-out mice normalized plasma lipids [107]. Furthermore, while restoring plasmatic apoE levels in the prior study did not rescue synaptic loss, it completely restored learning and memory in mice [107]. Thus, suggesting that both CNS and plasmatic apoE are independent parameters impacting brain health. Interestingly, results derived from humanized-APOE4-liver mice recently evidenced a negative impact of hepatic APOE4 in synaptic integrity, insulin signaling and increased neuroinflammation coupled to lower brain apoE4 expression [193]. Therefore, the potential negative contribution of peripheral apoE4 to the development of pathological brain processes should not be ruled out.
Studies of apoE levels in human CSF and plasma have shown results not always in line with those found in mice. For instance, some human studies found that the APOE genotype does not influence the CSF apoE levels [198, 199]. Nonetheless, the latest findings further point out a difference in the CSF apoE isoform composition in APOE heterozygous subjects [199,200,201]. Nonetheless, neither the total CSF apoE concentration nor its isoforms were related to Aβ status nor the degree of dementia of the AD patients, in agreement with prior studies [199, 200, 202].
A recent publication from Berger and colleagues evaluated the proteomic changes in the CSF of AD Neuroimaging Initiative (ADNI) samples by applying linear regression models adjusted by age, sex, APOE4 copy number, and linear models to adjust by AD clinical status or CSF levels of Aβ or tau. The authors observed that increasing APOE4 copy number was related to decreased CSF levels of C-reactive protein (a classical inflammatory marker), which correlated with cognitive impairment and AD progression [203, 204]. The study by Cruchaga and colleagues examined the correlation between CSF and plasma apoE levels in 641 AD patients and controls, in which they found a very low correlation [205]. However, it was observed that men presented significantly lower plasmatic apoE levels than women [205]. The prior findings correlate with Nielsen and colleagues who demonstrated higher apoE4 levels in females versus males [206]. Nielsen and colleagues also observed an age-dependent increase in the apoE3 isoform levels only in females [206]. Posterior studies from the same group found that, specifically in males, plasma apoE3 levels were negatively correlated to plasma glucose levels which were associated with brain glucose metabolism [207]. Hence, these findings could be related to the higher incidence of AD in women and the link between low plasma apoE levels and an increased risk of suffering dementia [208].
Human studies have identified low plasma apoE values as a significant risk factor for AD and other types of dementia [209]. Low plasma apoE levels described in APOE4 carriers resulted from a specific lowering of the apoE4 isoform [199, 206]. Whether plasma apoE levels are relevant to brain processing mechanisms remains controversial [195]. Nevertheless, a higher ratio of plasma apoE4 to apoE3 isoform levels in cognitively healthy APOE3/4 carriers was associated with reduced glucose metabolism in the hippocampus and gray matter volume reductions in several brain areas [206]. Low plasma apoE levels negatively correlate with cognition and CSF biomarkers [210]. Notably, whereas CSF apoE levels did not appear to be affected by the APOE genotype or AD diagnosis, some studies suggest that amyloidosis is associated with reduced CSF apoE levels in an APOE4 genotype-dependent manner [199,200,201]. Interestingly, the fluid composition of apoE isoforms in APOE heterozygous individuals differs between plasma and CSF, where for example, APOE3/4 subjects exhibit a different apoE4/apoE3 ratio in both compartments [199, 201, 206]. Lastly, it must also be noted that there are significant differences between plasma and CNS apoE turnover rates, supporting the idea that the pathways implicated in apoE metabolism differ between the CNS and the periphery [211].
APOE in the neuroinflammatory response
Neuroinflammation
Inflammation in the CNS, also called neuroinflammation, is a short-term body reaction to a noxious stimulus. Neuroinflammation aims to repair brain damage and restore brain homeostasis. Thus, it has short-term beneficial effects, yet excessive and unrestrained neuroinflammation is deleterious [212, 213]. Multiple factors can trigger neuroinflammation, such as traumatic brain injury, stroke, high-fat diet, exposure to drugs of abuse or aging [214,215,216,217]. Chronic neuroinflammation is a common characteristic of numerous neurodegenerative diseases, including Alzheimer’s disease, in which chronic neuroinflammation has become the third hallmark of its pathogenesis [218]. During neuroinflammation, the abnormal production of pro-inflammatory cytokines will trigger multiple signaling pathways [219, 220]. The two key cell types driving neuroinflammation in the CNS are microglial cells and astrocytes [221].
Microglial cells constitute the brain-resident macrophages of the CNS. They represent 10% of the cell population in the CNS and are the first-line defense against pathogenic agents or brain damage. Microglial activation will vary according to the type of stimulus, the intensity, and context, and under physiological conditions, they perform constant surveillance of the microenvironment in a very dynamic way through a vast array of proteins and receptors defined as the sensome [222, 223]. Some of the physiological roles in which microglia are involved are neurogenesis, synaptic pruning, or synaptic plasticity [224,225,226]. There is a close relationship between microglial morphology and function [227, 228]. Traditionally, microglia were categorized into two different phenotypes, M1 phenotype (classical activation) and M2 phenotype (alternative activation). Nowadays, this dichotomic classification is obsolete, and there is evidence that microglia can display mixed profiles in vivo and may also be regionally heterogeneous [229, 230]. In response to a noxious stimulus, microglia will suffer a series of morphological changes and induce an increase in the expression of surface receptors and the release of pro-inflammatory cytokines. Prolongation of this inflammatory process will compromise microglial cells’ homeostatic functions, including the maintenance of the BBB integrity [231, 232].
When comparing across species, Sullivan and colleagues observed a glial pattern of APOE expression in humans, African green monkeys, and knock-in APOE mice [192]. In humans, APOE isoforms will impact glial activation and migration and synaptic function [233, 234]. APOE4 knock-in mice present increased glial activation after LPS insult, increased production of pro-inflammatory cytokines, and an extensive loss of synaptic proteins compared to APOE3 and APOE2 knock-in mice [235, 236].
Even though astrocytes are the principal producers of apoE in the brain, microglial cells can also act as an apoE source under specific circumstances; for example, a recent publication from William Rebeck’s group evidenced that astrocytes secrete two differentially glycosylated apoE species, whereas microglia secrete a single apoE species while containing two intracellular apoE forms, the three of them glycosylated [237]. Primary cultures of glial cells from APOE knock-in mice studying the microglial and astrocytic response after priming with exogenous TNF-α or LPS showed that TNF-α reduces cellular expression and secretion of apoE in APOE4 astrocytes [237]. In contrast, LPS insult did not produce changes in astrocytic apoE levels. Nonetheless, APOE3 and APOE2 microglia showed increased secretion of apoE after LPS treatment, whereas no effect was observed in APOE4 microglia. Thus, suggesting that APOE4 astrocytes and microglia present dysfunctional responses towards inflammatory insults [237].
However, neuroinflammation is a complex process that will eventually entwine all the cells in the CNS. During neuroinflammation, activated microglia release pro-inflammatory cytokines like tumor necrosis factor-α (TNF-α), interleukin 1-β (IL1-β), IL-6, IL-12, interferon-γ (IFN-γ), and molecule C1 from the complement system (C1q) to their environment [218]. Additionally, innate and adaptative immune responses will also affect neuroinflammation. Immune cells such as T cells, CD4+ cells and B cells can cross the BBB and contribute to the neuroinflammatory process [238]. ApoE can influence immune responses by multiple mechanisms; for instance, in vitro and mouse studies have evidenced the lymphocyte modulatory action of apoE [239, 240]. ApoE also promotes neutrophil activation [241] and mediates antigen presentation on natural-killer cells [242]. Macrophages will also infiltrate through the BBB during neuroinflammation [243, 244], and it has been demonstrated that LPS can repress APOE gene expression in macrophages through the Tpl-2/MEK/ERK pathway [245]. Interestingly, even if the implications of adaptative immunity in AD are still largely unknown, promising small-scale studies suggest the implication of specialized CD8 T cells linked to the AD neurodegenerative process [246]. Therefore, it is vital to determine the molecular mechanisms comprising apoE and immunity in the neuroinflammatory process.
Microglial receptors and their interaction with apoE
As mentioned in the previous section, microglia constantly analyze their surrounding environment through the sensome [223]. Since macrophages and microglial cells share a common origin, they both express macrophage markers, such as F4/80, CD11b, CX3Cr1, Iba1, and CD45 [247,248,249]. Additionally, as the main antigen-presenting cells of the CNS, microglia express in their surface MHC proteins, which allow antigen presentation to CD8 + and CD4 + T cells (cytotoxic and helper T cells) [250]. Whereas MHC I proteins are ubiquitously expressed, microglial expression of MHC II proteins is considered a marker of neurodegeneration and has been related to AD [251, 252]. A study investigating the effects of the human MHC II (HLA) and its relationship with APOE genotype, revealed that the mutation DRB1*13 in human HLA-II was found to be protective against age-related changes in the neural network functioning and presented a similar effect as to carry APOE2 allele [253].
Among the collection of receptors present in microglia, receptors able to recognize stimuli of quite different nature are known as pattern recognition receptors (PRRs), which are located, with some exceptions, on the plasma membrane of microglia. The main PRRs include toll-like receptors (TLRs), inflammasome-forming nucleotide-binding oligomerization domain (nod)-like receptors (NLRs), the receptor for advanced glycation end-products (RAGE), and triggering receptor expressed on myeloid cells 2 (TREM2) [254, 255]. Concerning PPR’s ligands, the microglia field has consistently used the terminology pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs). DAMPS include released or secreted molecules from injured neurons or other cell types [230]. However, the term neurodegeneration-associated molecular patterns (NAMPs) recently rose to describe the danger signals present during brain disease conditions. Likewise, it must be highlighted that while classical DAMPs have traditionally been associated with binding to different PRRs, among other receptors, NAMPS can also activate TREM2 and further drive the acquisition of the disease-associated microglial phenotype (DAM) [256, 257]. However, regardless of these definitions, DAMPS and NAMPS co-exist in the diseased brain, thus giving rise to complex microglia polarization states. Indeed, recent massive transcriptomic analyses have demonstrated that microglia with different transcriptomic profiles co-exist under various disease conditions [258], thus overcoming the old simplistic view that microglia polarize into two opposite polarization states (pro-inflammatory and anti-inflammatory, or tumor-supportive phenotype) [259].
Without a doubt, TREM2 is more than a rising star in the microglia field, whose attention has exponentially increased since it was identified as a risk gene for AD (for reviews, see [255, 260]). In the brain, TREM2 is essentially expressed by microglia [255], and consequently, elucidation of microglia-associated roles of TREM2 and the implications of the APOE genotype under disease conditions is a considerable challenge in the field. Interestingly, upregulated TREM2 mRNA expression levels have been detected in human isolated-microglial cells from AD patients [261, 262]. A remarkable finding was the identification of a rare variant of TREM2 (R47H) as a robust risk gene of AD by two independent studies, followed by the identification of an additional variant (R62H), thus, supporting the significant role of TREM2 in microglia biology associated to neurodegeneration [263, 264]. At that time, Zhang and colleagues evidenced that TREM2 and protein tyrosine kinase binding protein (tyrobp) (also known as DAP12; the adaptor/signaling partner of TREM2) are strongly associated with LOAD pathophysiology. The relevance of the TREM2/DAP12 axis is illustrated by the fact that over 40% of sensome proteins are somewhat related to DAP12 [223]. Typical TREM2 ligands include phosphatidylserine present in apoptotic cells, glycolipids, sphingomyelin, and sulfatide present in damaged myelin. However, other beckoned ligands include apoE and clusterin (CLU/ApoJ), Aβ itself, phosphatidylinositol, and phosphatidylcholine [255, 260]. Besides, it has been demonstrated the ability of galectin-3 (gal-3) to bind to and stimulate TREM2 [265]. TREM2 senses the environment under disease conditions through an extracellular domain that belongs to the immunoglobulin superfamily [266]. TREM2 interacts with two different adaptor proteins upon ligand binding, DAP12 and DAP10. DAP12 activates spleen tyrosine kinase (SYK) and triggers downstream pathways involved in survival, migration, proliferation, activation, and phagocytosis, whereas DAP10 leads to Phosphatidylinositol 3-kinase (PI3K) recruitment and subsequent phosphorylation cascade that promotes Ca2+ mobilization, integrin activation, cytoskeleton rearrangement, mechanistic target of rapamycin (mTOR) and mitogen-activated protein kinase (MAPK) signaling [266].
The significance of TREM2 in governing microglia activation states has been validated thanks to the advent of massive transcriptomic analysis at the single-cell level. Keren-Shaul and colleagues identified a microglia subpopulation displaying a unique molecular signature in 5xFAD mice (a transgenic mouse model of AD) [267]. This microglial phenotype was named “disease-associated microglia” (DAM) and is characterized by downregulation of essential homeostatic genes, including P2ry12, Cx3cr1, and Tmem119, along with upregulation of selective genes including apoE, Itgax Ctsd, Igf1, Lpl, Clec7a, Tyrobp, and Trem2 [267]. The authors identified two clusters of activated microglia in 5xFAD mice that were not present in WT mice. It was suggested that one of the clusters corresponded to an intermediate state (Stage 1 DAM) between homeostatic microglia and DAM (stage 2 DAM) [267]. Mechanistically, the authors concluded that the switch from homeostatic microglia to stage 1 DAM was TREM2-independent, while the shift from transition microglia to DAM was TREM2-dependent [267]. Interestingly, apoE was found to be involved in the TREM2-independent step together with tyrobp [268].
In an independent study, Krasemann and colleagues analyzed single-cell microglial transcripts from animal models of neurodegeneration, including AD, ALS, and MS [269]. This study revealed a strikingly similar transcriptional network across the different disease conditions tested and evidenced, similar to DAM, loss of homeostatic genes (e.g., P2ry12, Tmem119, Gpr34, Csf1r, Hexb, and Mertk), and upregulation of genes such as apoE, Spp1, Itgax, Lgals3, Axl, Clec7a, and Ccl2 [269]. This microglial phenotype was named microglia neurodegenerative phenotype (MGnD) and demonstrated critical roles of both TREM2 and apoE in driving MGnD transcriptional profile [269].
It remains somehow paradoxical that DAM microglial cells are suggested to be neuroprotective, whereas MGnD are neurotoxic [267, 269]. The protective/harmful role of microglia is probably associated with the context, a view notably exemplified by TREM2 signaling. Thus, the absence of TREM2 in AD mice leads to increased neuritic dystrophies associated with Aβ plaques [270, 271]. In contrast, studies in tau transgenic mice have rendered conflicting results regarding the role of TREM2 in tau pathology, including a reduction and exacerbation of the pathology [272, 273]. Another set of studies has highlighted the prominent roles of TREM2 in the seeding/spreading of both Aβ and tau pathologies [274, 275]. In a seeding model based on intracerebral injection of Aβ-rich brain extracts from AD patients or aged APP mice, the absence of TREM2 led to increased seeded amyloid plaques exhibiting low levels of microglia-derived apoE deposition in amyloid plaques in mouse brains [274]. In line with these observations, reduced apoE in amyloid plaques was found in the brains of AD patients carrying different TREM loss-of-function variants [274]. These observations could be associated with reduced microglia clustering to Aβ plaques associated with TREM2 deficiency and the subsequent inability to develop a DAM program [267] and again highlight that microglia may dictate quite polarized directions at the crossroads of Aβ and tau pathologies.
Deeply related to the APOE context, TREM2 signaling is critical for the efficient expression of genes required for lipid metabolism in human microglia, a process dependent on phospholipase CY2 (PLCϒ2) signaling [276, 277]. PLCϒ2 signaling can also act downstream of TLR activation, and it has been proposed that this important node may enable microglia to elicit either a polarization state through TREM2 activation or a pro-inflammatory response through TLR activation [276]. This research group also observed that a TREM2 loss of function leads to aberrant lipid metabolism and enhanced pro-inflammatory response. In parallel, other authors have also observed that TREM2 deletion decreases microglial cell survival and impairs microglial phagocytosis of apoE [276, 278]. Thus, in keeping with a switch from a beneficial to a neurotoxic pro-inflammatory phenotype, both TREM2 and PLCϒ2 are AD risk genes [279]. Regarding ApoE’s interaction with TLRs, it has been described a deleterious effect of the APOE4 genotype in AD through the TLR4-dependent pathway [269], and apoE3 can inhibit macrophage activation driven by TLR4 [280], TLRs recruit TIR domain-containing adaptor proteins such as MyD88, TRIF, TIRAP/TRAM, or TRAM, leading to activation of different signal transduction pathways, including NF-κB and MAP kinases and hence inducing inflammatory cytokine gene expression [281]. Upon ligand binding, it is well known that TLR activation primes microglia, the first step for effective inflammasome activation [282]. This step includes expressing NLRP3 and pro-IL-1β in an NF-κB dependent manner [282]. TREM2 will negatively regulate TLR-induced cytokine production [283]. The interaction between apoE and inflammasome activation has been described in peripheral lavage fluid macrophages, but no interaction has been yet reported between microglial NLRP3 and CNS apoE [284]. However, Wong and colleagues have observed in human AD brains and transgenic mice tissue that 25-hydrocholesterol, a relevant inflammatory mediator produced by microglia, promotes IL-1β mediated neuroinflammation in an apoE isoform-dependent trend (apoE4 > > apoE3/apoE2), and apoE4-expressing microglia produce higher levels of 25-hydrocholesterol [285].
Another receptor family that has gained growing relevance are TAM receptors, including tyro3, axl and mer [286, 287]. These receptors have long been associated with the phagocytic clearance of apoptotic cells [288]. Homeostatic microglia express high gene levels of macrophage efferocytosis receptor c-Mer tyrosine kinase (Mertk) [267]. However, Axl is highly upregulated in the DAM phenotype, including plaque-associated microglia [256]. In vitro models have also shown Axl as a potential regulator of apoE homeostasis in astrocytes [95]. In APP/PS1 mice lacking TAM receptors, transcriptomic response to Aβ plaques was blunted, including a downregulation of genes related to lipid metabolism, including apoE [289]. Overall, this study suggests that microglial TAM receptors recognize and phagocytose Aβ plaques to further promote the formation of dense-core plaques [289]. Hence a prominent role of apoE in the formation of dense-core plaques is plausible in line with the well-accepted function of APOE4 in amyloidogenesis, enhancing fibrillization and compaction [290, 291].
In prior sections of this manuscript, we have addressed some of the relevant functions of the LRP1 receptor and its interaction with apoE. LRP1 is an endocytic and signal transduction-related cell surface receptor. Aside from being expressed in microglia, it is also highly expressed in neurons and to a lesser extent in neuroblasts, radial glia, astrocytes, and oligodendrocyte progenitor cells (OPCs) [292]. In-vitro primary microglial cell studies showed that LRP1 activation suppresses microglia activation through modulating JNK and NF-kB signaling pathways [293, 294]. On the contrary, results derived from spinal cord-derived primary microglial cultures showed that LRP1 plays a significant role in microglial cell activation in response to LPS insult and amplifies neuroinflammatory signaling pathways [295]. It has also been proposed that LRP1 mediates apoE’s effect on microglial inflammation [296]. Similarly, it was recently observed in animal models of AD that brain LRP1 silencing increased the inflammatory response by the TLR4/NFκB/MAPKs signaling pathway [297].
It is worth noting that microglial cells constitute the major source of the complement system component C1q. The complement cascade comprises soluble and membrane-bound proteins and represents the section of the immune system devoted to pathogen and cellular debris recognition and synapse elimination during developmental and adult stages [298, 299]. It has been demonstrated that all apoE polymorphisms inhibit the classical complement cascade by acting in the early activation stages by binding with high affinity to C1q. C1q-apoE complexes formation correlates with cognitive decline and suppression of complement protein C5 ameliorated inflammation [300].
APOE and microglial dynamics
The phenotypic analysis of microglia has become a hot topic in recent past years and highlighted the heterogenicity of microglial cells across the brain.
Recent advances in transcriptomic, bulk-RNA-seq, single-cell RNA-seq, and imaging techniques have underlined the significance of studying microglial dynamics during inflammatory pathologies. Albeit some of these techniques have led to conflicting results, they have allowed relating each microglial phenotype with a particular molecular signature to some extent, a previously reviewed by multiple authors [227, 229, 258, 301, 302]. However, establishing a direct link between microglial phenotypes and their specific functions remains challenging, as evidenced by other authors [229, 303].
The highest microglial heterogenicity has been observed at early postnatal time points, particularly in white-matter brain areas, such as the cerebellum and the Corpus Callosum [301]. Interestingly, some immature sub-populations of microglia are characterized by high gene-expression levels of APOE [304]. A review covering developmental microglial dynamics was performed by Menassa and colleagues [305].
Under non-pathological conditions, the so-called resting state microglia are characterized by highly ramified processes and a small rounded soma, together with the gene expression of homeostatic microglial markers, such as P2ry12, Tmem119, and Sall1 [223, 306,307,308]. Aside from homeostatic microglia, intermediate activation-state microglia, DAM, and MGnD microglial phenotypes described by Keren-Shaul, Krasemann, and colleagues, additional transcriptional microglial phenotypes have been recently identified [267, 269]. For instance, single-cell sequencing from white and grey matter led to the characterization of White-matter microglia (WAM), which are relevant in the AD context because the brains of AD patients present a high white matter loss, which is also related to motor and cognitive dysfunction [309,310,311]. Prior studies have shown that APOE4 can modulate white matter integrity [312]. It has been proposed a link between APOE4 polymorphism and an increase of white matter hyperintensity lesioned areas observed by MRI in AD patients [313, 314]. WAMs present higher expression levels of ubiquitin-specific protease 18 (Usp-18), which contributes to microglial quiescence by negatively regulating the activation of Stat-1 and abrogating IFN signaling [311, 315]. WAMs also rely on TREM2 signaling and are age-dependent. In AD mouse models, WAMs signature is APOE-dependent, whereas in aged brains, WAMs show an APOE-independent signature [311]. As previously mentioned, the study from He and colleagues related to the apoE receptor LRP1 indicated that LRP1 might modulate microglial response via the JNK and NFκB pathways [297]. Since white matter injury is related to myelin and axonal loss after different events, additional studies point out that selective blockade of LRP1 in OPCs could potentially enhance myelin repair [316, 317]. Conversely, in a rat model of subarachnoid hemorrhage, an intraperitoneal dose of an apoE-mimic peptide and LRP1 ligand (COG1410) revealed that microglial LRP1 activation alleviates white matter injury and improves neurological functioning by shifting microglia towards an M2 phenotype through the activation of the Shc/PI3k/Akt signaling pathway [318]. A study from Hammond and Colleagues evaluated the microglial subpopulations from embryonic stages until old age and after the induction of a local demyelinating injury. Among the microglial subpopulations found, the authors performed a comparison between axonal-tract-associated microglia (ATM), DAM, and injury-responsive microglia (IRM) by using canonical correlation analyses. The authors observed that ATM microglia are characterized by upregulated developmental genes, such as Gpnmb and Fabp5, and IRM microglia presents downregulated expression levels of P2ry12 and Cx3cr and upregulated transcription factor Ifi204, interferon-response genes Ifi27l2a and Cxcl10 [267, 308]. Interestingly, ATM, DAM, and IRM microglial groups shared 12 core genes among their transcriptomic structure, including among those genes Spp1, Lpl, and apoE [319]. Sala-Frigerio and colleagues evaluated more than 10 thousand individual microglial cells derived from the cortex and hippocampus of APP mice. The authors observed multiple microglial clusters; among them, transiting-response microglia (TRM) and Activated-response microglia (ARM) transcriptomic profiles were similar. These clusters present an increased expression of apoE and inflammatory markers paired with the downregulation of homeostatic microglial markers. ARMs also present an overexpression of MHC-II genes. TRM express relatively lower apoE and MHC-II markers than ARMs and do not appear to show tissue regeneration genes [308]. They also revealed ARMs as the meeting point for aging processes, sex, and AD risk factors because ARM response was abolished with APOE deletion. Finally, the authors also identified a small microglial sub-cluster corresponding up to 1,2% of the microglial subpopulations called cycling/proliferating microglia (CPM), enriched in genes related to DNA replication, chromatin rearrangement, and the cell cycle, such as Top2a, Mcm2, Tubb5, Cdk1, and Mki67.
Aging is also a significant factor influencing microglial function. Marschallinguer and colleagues performed a study on C57BL/6 J male mice to assess structural differences between young and old microglia (three-month versus twenty-month-old mice) by analyzing their cytoplasmic content through transmission electron microscopy. The authors identified a novel state of microglial cells present in the aging brain called lipid-droplet accumulating microglia (LDAM). LDAM were abundant in the aged hippocampus and rich in neutral lipids, such as tri-, di- and monoacylglycerols and cholesteryl esters. Transcriptional analysis of LDAM revealed a differential expression of genes related to the production of ROS and NO (kl, ppp1cb, jak, cat, rap1b), as well as genes related to phagosome maturation pathways, vesicular transport (rab5b and rab7), and cd22, which negatively regulates microglial phagocytosis. The authors also compared the LDAM transcriptional phenotype to the previously characterized microglial phenotypes, such as aging microglia, DAM, MGnD microglia, and the clusters discovered by Li and colleagues, and Hammond and colleagues. They determined that the LDAM transcriptional phenotype was distinct to those previously characterized, standing out by presenting defective phagocytosis, together with a high production of reactive oxygen and nitrogen species and pro-inflammatory cytokines. The authors also investigated the plausible genetic regulators of microglial lipid droplet formation by CRISPR-Cas9 screening in a microglial mouse-derived BV2 cells, finding slc33a1, snx17, grn, and vps35 as neurodegeneration-related genes and thus pointing to a possible link between LDAM and neurodegeneration. Nonetheless, it is interesting that neither APOE nor TREM2 was regulated in LDAM [320]. Thus, more research must be guided towards the study of microglial heterogenicity, as well as determining how factors such as age, sex, and the APOE genotype impact microglial phenotypes and their function during health and disease. The microglial phenotypes described and the most relevant hallmark genes expressed are summarized in Table 4.
APOE and astrocytic function
Although apoE can be produced by different cell types in the brain, the vast majority of brain apoE is generated by astrocytes [81, 321]. Astrocytes have a supportive role in neuronal function, mature neurons predominantly rely on cholesterol provided by astrocytes, and apoE plays an essential role in transporting cholesterol from astrocytes to neurons [322]. Human iPSC in vitro models have evidenced that apoE protein and mRNA levels are decreased in APOE4 astrocytes compared to APOE3 astrocytes [178]. Astrocyte-derived apoE4 is poorly lipidated compared to apoE3, suggesting that astrocyte-derived apoE3 is more efficient in transporting cholesterol to neurons than apoE4 [323, 324].
Although it remains unclear why apoE4 particles are less lipidated than apoE3, the poor lipidation status of apoE4 is likely linked to impaired cholesterol metabolism in astrocytes. The study by Julia TCW and colleagues showed an increased cholesterol synthesis by APOE4 compared to APOE3 IPSC-derived astrocytes, a finding in line with the previously described increased cholesterol biosynthesis, including increased intracellular cholesterol levels and secretion, by APOE4 IPSC-induced astrocytes [325]. Lin and colleagues also described increased intracellular cholesterol accumulation in APOE4 compared to APOE3 IPSC-derived astrocytes [178]. Of note, increased cholesterol secretion by astrocytes is contradictory to previous findings in APOE knock-in mice, as cholesterol release was observed to be increased in astrocytes from APOE3 knock-in compared to APOE4 knock-in mouse astrocytes [189, 323]. Moreover, genetic expression of APOE4 in iPSC-derived astrocytes will also induce an increase in the intracellular unsaturated fatty acid levels and lipid droplets, which it is possible to restore with choline supplementation [326]
Interestingly, although lipid metabolism regulated by astrocytes in the brain mainly includes lipid transport from astrocytes to neurons, recent studies have described a ‘neuron-astrocyte shuttle’, where toxic fatty acids in neurons are transferred to astrocytes for neutralization, a transfer that also relies on apoE [327,328,329]. Neurons have a limited capacity to store fatty acids in lipid droplets or to catabolize fatty acids [330]. Excessive free fatty acids in the cytosol could lead to lipid peroxidation, toxicity, mitochondrial dysfunction, and neurodegeneration. Therefore, it is highly relevant to control fatty acid metabolism [331,332,333]. In contrast to neurons, astrocytes could store fatty acids in lipid droplets and catabolize the lipids by mitochondrial respiration [327, 334]. The transport of fatty acids from neurons to astrocytes to neutralize or avoid fatty acid-induced toxicity depends on apoE [327]. More specifically, apoE4 has shown to be less efficient in transporting fatty acids to astrocytes [328]. In addition, the same study also described increased lipid droplet accumulation and reduced fatty acid oxidation in astrocytes expressing APOE4, causing not only diminished transfer of toxic fatty acids away from neurons but also reduced capacity of astrocytes to neutralize toxic lipid species [326, 328].
In addition to the role of astrocytes in lipid metabolism in the brain, astrocytes also have a supportive role in brain energy homeostasis. Humanized APOE4 knock-in astrocytes have a reduced mitochondrial capacity compared to APOE3 knock-in astrocytes [328, 335]. In addition, APOE4 astrocytes are linked to altered glucose utilization, e.g., increased glycolytic activity, reduced glucose uptake and oxidation, increased aerobic glycolysis and increased lactate synthesis [336, 337]. APOE4 astrocytes appear to present a shift in glucose metabolism toward a less oxidative tricarboxylic acid cycle and an increased glucose flux towards the pentose phosphate pathway and de novo- biosynthetic routes [336]. Qi and colleagues argued that mitochondrial respiration preferentially relies on glucose metabolism rather than fatty acid β-oxidation, e.g., because of a higher oxygen consumption rate in APOE4 in the presence of glucose substrate [328]. Other authors have reviewed additional mechanisms through which the APOE4 polymorphism impacts glial cell functioning [38]. Further research is required to understand the bioenergetic function in APOE4 astrocytes thoroughly.
APOE in Alzheimer’s disease
APOE, amyloid-β load and AD pathology
In 1993, the link between apoE4 and LOAD was first described by Corder and colleagues and thereafter the number of studies on APOE4, LOAD and Aβ pathology increased tremendously [338]. In general, APOE4 carriers have an increased plaque load and density compared to non-APOE4 carriers [160, 339, 340]. Similarly, mice expressing the human apoE4 isoform have more Aβ deposits and neuritic amyloid plaques than mice expressing other human apoE isoforms [66, 341]. The effect of human APOE on Aβ deposition is isoform-dependent with APOE2, opposite to APOE4, showing the lowest Aβ plaque deposition [66]. A complete lack of APOE expression in AD transgenic mice overexpressing human mutant APP, but not PSEN1, dramatically reduces deposition of Aβ in the brain and lowers both Aβ40 and Aβ42 deposits, highlighting the important role of human APOE in amyloid deposition [341,342,343]. Nonetheless, APOE deletion in a more aggressive AD mouse model containing mutations in both APP and PSEN1, such as APP/PS1 mice, induces an overall increase in plaque size [236]. Interestingly, the prior study also observed a decrease in the Aβ plaque load positive for X-34, an amyloid stain that preferentially binds mature fibrillar plaques.
The effects of APOE deletion in mouse models used to study amyloid pathology are not fully understood and therefore require further investigation.
Compared to endogenous m-apoe expression, all human APOE isoforms reduce plaque deposition and onset, with the strongest reduction induced by expression of the human APOE2 isoform in mice [66, 344]. Besides parenchymal amyloid plaque deposition, APOE4 has also been linked to cerebral amyloid angiopathy (CAA) formation in an AD transgenic mouse model (Tg2576) cross-bred with humanized APOE-knock in mice [344].
More recently, amyloid positivity in the brain detected by positron emission tomography (PET) scan using Pittsburgh compound B (PiB) and reduction in CSF Aβ42 were shown to be predictors of clinical AD [345]. APOE genotype was shown to affect amyloid PET scan results and CSF Aβ levels, with cognitively normal APOE4 carriers evidencing increased PIB+ amyloid burden in the brain and reduced levels of Aβ42 in CSF compared to non-APOE4 carriers [346,347,348]. The strongest effect with age on amyloid burden and Aβ42 CSF levels was seen in APOE4 homozygotes, suggesting a dose-dependent APOE4 effect on amyloid plaque burden prior to clinical AD onset [346].
Although the mechanistic link between APOE4 and plaque development remains poorly understood, a critical role of APOE4 in plaque seeding has been described [290, 349]. Antisense oligonucleotide (ASOs) that suppress APOE expression prior to plaque onset reduced plaque load in humanized APOE4 knock-in / APP/PS1-21 mice compared to non-ASOs treated controls [290]. Interestingly, APOE reduction by ASOs after plaque onset did not affect plaque load in APOE4 / APP/PS21 mice. Additionally, another study also supports a role of APOE4 predominantly in plaque seeding and not in plaque growth [349]. In this study, inducible mouse models were generated overexpressing human APOE3 or APOE4. Overexpressing human apoE4 during the first 6 months of age, prior to plaque onset, strongly increased plaque load, Aβ oligomers and Aβ levels compared to mice not expressing human apoE. Overexpression of human apoE4 from 6 to 9 months of age, thereby starting apoE expression directly after plaque onset, did not influence plaque load and Aβ levels, strongly indicating an apoE4-dependent effect on plaque seeding, but not growth. Human apoE3 is suggested to have a weaker effect on plaque formation as both these two studies did not find a clear effect in the presence of apoE3 [290, 349].
The exact mechanism promoting amyloid plaque formation remains poorly understood, but is most likely caused by either increased Aβ production and/or aggregation, and/or decreased Aβ clearance. Although some conflicting data have been published [350], most studies found that human apoE, in particular apoE4, enhances Aβ fibril formation in vitro [291, 351, 352]. Differences in the preparations of apoE and/or Aβ peptides could be an explanation for the observed differences in in vitro Aβ fibrillization studies. The lipidation status of apoE, especially reduction of apoE lipidation by depleting ABCA1, contributes to increased amyloid deposition in AD transgenic mice [353, 354]. ApoE co-deposits within amyloid plaques [355] and the form of apoE localizing to insoluble Aβ deposits in AD appears to be poorly lipidated [354].
Prior to the onset of amyloid plaques, Aβ accumulates intracellularly in AD vulnerable neurons [356]. Intraneuronal accumulation of Aβ1-42 in late endosomal and lysosomal compartments is enhanced by the presence of apoE4 in mice [357]. The binding of lipidated apoE to ApoER2 receptors could trigger APP and BACE1 internalization, resulting in increased intracellular Aβ generation [358]. ApoE4 binding leads to more enhanced APP and BACE1 endocytosis than apoE3 and apoE2, consequently resulting in a stronger increase in Aβ production by apoE4. More recently, an apoE-isoform dependent effect on APP transcription and Aβ secretion was reported, with apoE4 inducing the strongest increase in APP transcription, followed by apoE3 and lastly apoE2 [359].
Besides increased Aβ generation and aggregation, amyloid pathology is also influenced by disrupted Aβ clearance. Aβ can be cleared from the brain via several clearance pathways, including enzymatic degradation, clearance through the BBB, and cellular internalization and subsequent lysosomal degradation by astrocytes, microglia and neurons [34, 360]. In general, apoE4 is associated with lower Aβ clearance [34, 361]. However, the exact mechanisms of Aβ clearance and the role of apoE in this are complex. One of the mechanisms to clear Aβ from the brain is by proteases, such as neprilysin and insulin-degrading enzyme (IDE) [362, 363]. Both intracellular and extracellular enzymatic clearance of soluble Aβ is enhanced by apoE [364]. Highly lipidated apoE increases Aβ clearance by proteases to the highest extent [364]. Human post-mortem studies have shown reduced neprilysin and IDE expression in APOE4 compared to non-APOE4 carriers, suggesting potential differences among apoE isoforms on proteolytic activity [365, 366].
Aβ can be degraded intracellularly via lysosomal degradation in astrocytes, microglia, and neurons. In all cell types, apoE4 has been linked to a reduced cellular clearance of Aβ [364, 367, 368]. ApoE influences Aβ internalization and degradation in microglia in an isoform-dependent manner with the slowest Aβ uptake and the least efficient Aβ degradation by apoE4 [178, 364]. ApoE was also reported to moderate Aβ cellular degradation by reducing intracellular cholesterol levels, without direct interaction between apoE and Aβ being required [369]. Reducing intracellular cholesterol in microglia results in accelerated endosomal transport of Aβ towards lysosomes [369]. In astrocytes, APOE4 induces impaired Aβ1-42 internalization compared to APOE3 astrocytes [178]. An APOE-dependent mechanism of Aβ clearance in astrocytes was proposed as APOE−/− astrocytes were unable to degrade Aβ [368]. However, more recently, Aβ uptake and clearance by LDLR in astrocytes was shown to also take place in the absence of APOE, pointing towards an APOE-independent Aβ uptake mechanism [370]. An indirect apoE-induced inhibition of Aβ uptake by competing for the same receptors, such as LRP1 receptors, could block Aβ internalization and subsequent degradation [371]. Neurons can also degrade Aβ by cellular internalization and lysosomal degradation [367]. Although the research on neuronal Aβ clearance and apoE remains limited, apoE3 seems to enhance neuronal Aβ uptake, endosomal trafficking and lysosomal degradation more strongly than apoE4 [367].
Another clearance mechanism of Aβ is by the BBB. Both in vitro and in vivo studies showed that apoE could modulate BBB-related Aβ clearance [372,373,374]. ApoE4 impaired Aβ removal via the BBB compared to apoE3 and apoE2. The presence of apoE4 has also been associated with BBB breakdown [375,376,377]. Impairment of the BBB might further dysregulate Aβ clearance from the brain. The precise mechanism of apoE in Aβ clearance by the BBB remains poorly understood. ApoE2, apoE3, apoE2-Aβ and apoE3-Aβ complexes can be cleared by LRP1 and VLDLR at the BBB at a substantial faster rate than apoE4 and apoE4-Aβ complexes [372]. Additionally to BBB clearance of Aβ, apoE also modulates soluble Aβ clearance from interstitial fluid (ISF) in mice in an apoE-isoform dependent manner, shown by an increased half-life of Aβ in ISF in PDAPP/APOE4 compared to PDAPP/APOE3 and PDAPP/APOE2 mice [378]. More research is needed to better understand the role of apoE in Aβ clearance from the ISF.
In sum, APOE4 has been highly associated with changes in amyloid pathology in AD. APOE4 has shown to increase plaque load, in particularly by enhancing plaque seeding rather than growth, and could influence Aβ production and aggregation. At the same time, Aβ clearance has been shown to be least efficient in the presence of an APOE4 genotype. The most crucial process of APOE related to amyloid pathology remains unclear, and additional studies are required to increase the knowledge about the effect of APOE and amyloid pathology in AD.
APOE and Tau pathology
As priorly mentioned in our manuscript, aggregates of hyperphosphorylated tau protein conforming NFTs constitute one of the hallmarks of AD. NFTs correlate with the cognitive deficits observed in AD patients, but whether they drive AD pathology is still under debate [379,380,381].
Initial histopathological evidence established a relation between apoE protein and NFTs in AD brains [382,383,384]. Tau pathology is tightly related to neurodegeneration, and it has long been considered that tau aggregation into NFTs result in a toxic gain of function [385]. Tau pathology originates in the EC and builds out to the hippocampus, temporal and cortical areas, been the cerebellum the least affected area. Even so, the differential vulnerability of brain regions to tau pathology is still unknown [386]. Throughout different studies, APOE4 carriers display a robust amnestic phenotype with a more severe and typical medial and temporal spread of tau, following the stereotypical Braak staging system characterized in AD [387,388,389,390]. On the other hand, APOE4 non-carriers show a non-Braak pattern of tau PET uptake with lower levels in the EC and higher in the neocortex, a finding that is hypothesized to be mediated by Aβ deposition [391,392,393].
There are numerous theories for p-tau spreading through the brain. Various studies point out that it is a process driven by microglia, whereas other authors also suggest tau spreading occurs through neuronal communication pathways [394, 395]. Van der Kant and colleagues reviewed the role of microglia in AD-related tau pathology independently of Aβ, being apoE a key regulator of this phenomenon [396].
On that note, the APOE4 genotype exacerbates tau-mediated pathogenesis, neurotoxicity neuroinflammation, and neurodegeneration in numerous pre-clinical models [390, 397,398,399,400]. The study from Kang Su and colleagues observed that apoE4 potentiates tau neurotoxicity by binding the monoamine transporter 2 (VMAT2), inhibiting neurotransmitter active transport into synaptic vesicles, and eventually leading to Locus Coeruleus (LC) degeneration [400]. In line with the previous findings, in the P301S mouse model of tauopathy, together with knock-in or knockout APOE, APOE4 expressing mice showed the strongest tau-induced neurodegeneration compared to the other two isoforms, whereas APOE knockout mice were protected from this effect [397]. This same research group from David M Holtzman recently showed the potential of reducing APOE4 levels with antisense oligonucleotides in P301S/APOE4 mice, which protected against tau pathology [401]. For a review on how APOE affects both tau pathology and neurodegeneration through its immunomodulatory function, the reader is referred to the review article from Shi and Holtzman [36].
Data from human iPSCs-derived cell types have shown a relation between apoE4 and tau pathology in neurons and glial cells [178, 402, 403]. It has been documented that selective removal of astrocyte-derived apoE4 protects against tau-mediated neurodegeneration [404]. AAV-mediated overexpression of APOE human isoforms in astrocytes revealed that the astrocytic overexpression of APOE4 enhances tau phosphorylation and aggregation, both in vitro and in the P301L mouse tau model [399]. However, AAV-mediated neuronal overexpression in rTg4510 and PS19 mice failed to alter p-tau burden [405]. Additionally, other tauopathy models have addressed an association between the cleaved c-terminal fragment of apoE4 and tau [383, 406].
In terms of brain imaging evidence, studies using the tau PET tracer 18F-AV-1451, also known as 18F-flortaucipir, showed contradictory results, ranging from an association between medial temporal tau and apoE4 independently of Aβ in cognitive performance [407]; to establishing no association between medial temporal tau and apoE4 in cognitively normal individuals, mild cognitive impairment (MCI) or AD patients [408, 409]. The use of the tau tracers 18F-MK6240 and 18F-flortaucipir revealed a relationship between medial temporal tau and apoE4 beyond Aβ [390, 410]. Rodriguez-Vieitez and Nielsen thoroughly reviewed the effects of total tau load in this matter [411]. On top of that, an association between APOE4 and sex has been described in PET studies, with women carrying the allele showing a more widespread tau pathology [412, 413].
Analysis of CSF from AD patients and controls revealed that both apoE3 and apoE4 are positively associated with total tau and phosphorylated tau concentrations [199]. Similar findings have been reported from MCI and AD groups, but only concerning APOE4 carriers, and when comparing tau and apoE levels [414,415,416,417]. Another study showed that in CSF of APOE4-carrying AD patients, tau levels were associated with impaired cortical plasticity, cognitive decline, and astrocyte survival [418]. Overall, correlating apoE and tau levels in CSF samples is complex and not as evident as relating apoE to Aβ levels. Most findings are contradictory and are highly dependent on the disease stage [411]. It is also relevant to consider that measuring CSF tau only provides information on global soluble tau in the entire brain, while tau PET imaging informs of the specific regional distribution of NFTs [409]. In addition, tau PET seems to be a more accurate reflection of disease severity than CSF studies, and concordance between PET and CSF tau levels may rely on the disease stage and which tau fragments are studied [419, 420].
Interestingly, the protective effect of APOE2 in Aβ load has not been reported concerning tau pathology since there are no significant changes in the levels of CSF tau in these carriers, with one exception reporting lower phosphorylated tau levels [421, 422]. Tau PET studies reported similar findings, evidencing that the protective effect of APOE2 on developing AD is primarily linked to the resistance against Aβ deposition [409]. Nevertheless, APOE2 homozygosity has been shown to enhance tau pathology, increase the risk of primary tauopathies, and is more frequent in primary age-related tauopathy as opposed to AD cases [423, 424].
Despite this data, direct interaction between apoE and tau has not been reported in vivo, and it has to be taken into account that apoE is primarily secreted, whereas tau tends to be located in intraneuronal and axonal regions, although evidence has shown that it can also be present in the extracellular space [425]. The mechanism by which the APOE genotype might affect tau deposition is unknown, but the two primary metabolic receptors in charge of mediating clearance of apoE lipoproteins, LDLR and LRP1, have been shown to regulate tau propagation in opposite ways. LDLR overexpression reduces tau pathology via apoE-linked mechanisms, whereas LRP1 knockdown reduces tau propagation [426, 427]. No differential effect has been established in terms of apoE isoforms, but Rauch and colleagues suggest that apoE4 would be the least effective at inhibiting tau spread, since as previously commented, this genotype presents the lowest levels of circulating apoE protein in the brain.
To conclude, there is still much debate about the influence of APOE genotypes, particularly on how APOE4 impacts tau burden. In the context of AD, it is difficult to isolate a direct connection between apoE and tau due to the effect of Aβ, and it is argued that the effect of the APOE4 allele on tau pathology may be secondary to the marked effect of the APOE genotype on Aβ burden.
APOE and synaptic function in AD
Synapses are among the earliest cellular sites affected by AD, and a loss of synaptic terminals appears to be the best correlate to cognitive impairment in AD patients [428,429,430,431]. Reduced levels of pre-and post-synaptic proteins, such as synaptophysin and neurogranin, are found in AD post-mortem brains compared to age-matched controls [432, 433]. Brain regions with a higher active default state, e.g., the hippocampus and EC, seem particularly vulnerable to AD [434]. Aβ oligomers, rather than amyloid plaques, have been shown to cause synaptic toxicity, resulting in synaptic dysfunction, loss of synapses, and disrupted synaptic plasticity, including long-term potentiation (LTP) and long-term depression (LTD) [435,436,437,438]. Oligomeric Aβ localized near plaques is detected at post-synaptic terminals and correlates with spine loss close to amyloid plaques [435]. Aβ1-42 accumulation occurs at synaptic terminals even earlier than amyloid plaques formation in AD transgenic mice suggesting an early effect of Aβ on synaptic pathology [439, 440].
APOE4 is among the LOAD risk genes related to synaptic function impairments in AD [441]. ApoE, as the primary lipid carrier in the brain, will transport lipids, e.g., cholesterol, from astrocytes to neurons. Cholesterol is of high relevance for axonal growth, synapse formation, and spine remodeling; highlighting the role of apoE as a cholesterol transporter in the maintenance of synapses [442,443,444]. Notably, during neuronal stress and injury, apoE expression and cholesterol transport from astrocytes to neurons increase as part of a neuronal repair mechanism [445]. Moreover, astrocytic apoE may modulate neuronal function by altering histone acetylation in an isoform-specific manner (apoE3 > apoE4), and astrocytic-derived microRNAs (miRNAs) may selectively silence genes related to neuronal cholesterol synthesis [446]. Even without neuronal injury, apoE has been associated with changes in neuronal outgrowth and synapse density in normal brain functioning [234, 447]. Prior studies have reported generalized reduced neuronal outgrowth in APOE4 than in APOE3 neurons [447,448,449,450]. Additionally, synaptic density is decreased in neurons expressing human apoE4 [234], perhaps by a reduced spine formation or an enhanced spine removal, as shown in in vitro models [451].
AD is also related to disrupted synaptic plasticity. Multiple studies previously investigated the role of apoE in synaptic plasticity, particularly on LTP. Overall, the APOE4 genotype is associated with decreased LTP compared to APOE3 in mouse and in vitro models [452, 453]. Reduced LTP induction induced by apoE4 is dependent on NMDA receptor functioning [454]. Although most studies support a detrimental effect of apoE4 on LTP, conflicting data have reported increased LTP in young APOE4 mice compared to APOE3 mice [455]. The differences in LTP stimulation protocols, such as high-frequency stimulation, or tetanic stimulation, could potentially explain the different effects of apoE4 observed on LTP [106]. In line with reduced LTP in the presence of apoE4, poor learning and memory outcomes have been described in APOE4 targeted-replacement mice, suggesting that APOE4 impairs synaptic function and also worsens cognitive function [456,457,458].
Whether apoE affects synaptic function by its direct presence at synaptic terminals remains poorly studied. ApoE is present in a subset of synapses, co-localizing to 18–36% of synapses [459]. A recent study observed a synapse-associated localization of apoE in primary neuron cultures, in which both apoE3 and apoE4 co-localize with vGlut1-positive pre-synaptic terminals [460]. ApoE co-localizes with synaptic markers in both healthy controls and AD subjects, pointing towards a not purely pathological role of apoE at synaptic terminals. Interestingly, enhanced accumulation of oligomeric Aβ is observed in synapses positive for apoE [459]. In addition, apoE/Aβ complexes accumulate in human cortical synaptic terminals, especially during AD, and their expression increases in the presence of apoE4, compared to brains from non-APOE4 carriers [461]. Nonetheless, additional research is still needed to address whether ApoE4-induced synaptic dysfunction is dependent or independent of Aβ/APP accumulation.
Although many apoE-related changes in synaptic function have been described in observational studies, the mechanisms by which apoE affects synaptic function remain not fully understood. For instance, in vitro electrophysiological evaluation in vitro of human hiPSCs neurons treated with astrocyte-derived extracellular vesicles (EVs) revealed an enhanced electrophysiological function paired with an anti-apoptotic behavior. Among the cargos found inside the EVs of this study, heat shock proteins, LRP1, alpha-synuclein, and apoE were detected, among others. [462]. However, the previous study did not evaluate the effect of the different apoE isoforms, which would be of great interest in the future. In line with the prior findings related to astrocytic apoE, the Holzman group observed that astrocytic deletion of APOE4 rescues synapse loss [398]. Another study using male embryonic stem cells observed an effect of apoE on synaptogenesis and concluded that apoE stimulates CREB and cFOS signal transduction cascades and isoform-dependent decrease of neuronal signaling (ApoE2 < ApoE3 < ApoE4) [184].
Novel approaches such as synaptometry time of flight (SynTOF), a technique combining mass spectrometry and flow cytometry, have been developed to elucidate cerebral single synaptic events in health and disease. Montine TJ and colleagues have implemented the SynTOF technique in synaptosomes derived from non-human primates, PS/APP mice, and post-mortem tissue from individuals solely with AD neuropathologic change [463]. Some of the most striking findings in this study included the absence of Aβ at human synapses, diverging from prior authors’ findings (e.g. [459]. The authors also observed an increase in the hippocampal synaptic levels of the marker CD47, apoE, and a possible link between low apoE levels in pre-synaptic events and AD resilience [463]. Although one of the possible limitations of this technique is relying on optimal antibody binding, it constitutes a promising tool for future studies.
Impairment of endosome recycling has been described as a potential mechanism causing synaptic dysfunction induced by apoE4 [452, 464]. APOE4 has been associated with significantly reduced endosomal recycling and increased intracellular accumulation of apoE [130, 452]. ApoE4-induced endosome recycling impairment could result in the trapping of apoE, ApoER2 receptors, and glutamatergic receptors involved in synaptic plasticity, such as NMDA and AMPA receptors in endosomes [452]. As a consequence, synaptic receptor trapping in endosomes induces impaired synaptic regulation. Additionally, apoE4-induced intracellular trapping of ApoER2 receptors results in reduced ApoER2 receptor binding by Reelin [465], leading to dysfunctional synaptic plasticity regulation. A recent study discovered the involvement of NHE6, a proton channel present in early endosomes; since NHE6 inhibition rescued impaired Reelin-mediated synaptic plasticity induced by apoE4 [464]. More research is required to define better the role of apoE and its isoforms on synaptic function.
Implications of APOE in autophagy
Endocytosis, phagocytosis, and autophagy are closely related functions involved in the cellular trafficking and recycling system. Briefly, endocytosis is involved in the recycling and internalization of macromolecules from the plasma membrane, autophagy in the removal of intracellular sources and organelles, and phagocytosis in the degradation of extracellular materials. Importantly, autophagy and phagocytosis share many molecular pathways and mediators as LC3, Beclin1, ATG5, or ATG7 in microglia. [466,467,468]. Regarding autophagy, although there are three forms (microautophagy, macroautophagy, and chaperone-mediated autophagy), the term typically refers to macroautophagy. In addition to the well-known response to starvation, autophagy is also involved in different processes as degrading dysfunctional organelles, pathogens, or aggregated proteins [469, 470].
The specific role of autophagy in AD has been extensively investigated in the last years (see review [469]). Already in the early preclinical AD stages, there are alterations in the autophagy-lysosomal network associated with the intracellular accumulation of Aβ [471, 472]. Endo-lysosomal trafficking and autophagy are essential in the formation of Aβ from APP [471], APP degradation and Aβ clearance [466, 473]. Autophagy is an additional mechanism for the clearance of Aβ by microglia [474]. A failure in the complex pathway of cell trafficking and autophagy mediates Aβ accumulation [475]. Furthermore, this alteration influences other elements involved in AD pathophysiology, such as neuroinflammation [476].
Autophagy and neuroinflammation interact at several levels regulating each other bidirectionally. Several cytokines induce or inhibit autophagy; meanwhile, autophagy decreases the secretion of other cytokines [477]. However, this relationship is much more complex in the context of AD; as previously described above, an alteration in autophagy is associated with Aβ accumulation. Therefore it has been observed that autophagy in microglia reduces neuroinflammation [478], but the secretion of pro-inflammatory factors is associated with an increase in autophagy [479, 480]. All this indicates that other factors, such as differential gene expression, must be involved in this complex interplay.
Some LOAD-associated alleles are linked with endocytic/autophagic pathways, being APOE4 the most relevant [481, 482]. ApoE is involved in Aβ internalization by binding to the Aβ12-28 region, being apoE4 much less efficient in this function compared to apoE2 and 3, leading to a higher accumulation of Aβ in both patients and animal models carrying APOE4 [471, 483, 484]. Nevertheless, the specific mechanism by which APOE4 alters autophagy is not yet fully understood, although several publications support that it is one of the main pathological processes regulated by APOE4 in AD (see review [485]).
Concerning APOE4 effects upon glial cell autophagy, Simonovitch and colleagues observed in vitro that APOE4 expression in astrocytes induced lower autophagic flux and a reduced capacity to clear Aβ compared with APOE3-astrocytes [470]. Then, in vivo, they noticed a decrease in autophagy and mitophagy in the hippocampus of APOE4 mice [486]. Also in APOE4 mice, Nuriel and colleagues observed, using transcriptomics, a dysregulation of the endosomal-lysosomal pathway compared to APOE3 mice [487]. Likewise, Pomillo and colleagues presented in vitro and in vivo evidence of microglial autophagy impairment and defective Aβ degradation as AD progresses [488].
Regarding human brain samples, Parcon and colleagues observed a reduction in mRNA transcripts related to autophagy in APOE4 carriers compared with APOE3. They noticed lower mRNA levels of LC3, p62, or LAMP2, all of which are essential components in the autophagic pathway. In the same work, authors observed, using APOE3-expressing astrocytes (T98G), an upregulation on mRNA levels of LC3, p62, and LAMP2 after autophagy induction. This response was not detected in APOE4-T98G cells [489]. Previously, Zhu and colleagues noticed an apoE4-specific increase in the expression of synaptojanin 1 (SYNJ1), a phospholipid regulator which, when induced, caused an increased Aβ clearance and reduced synaptic damage in an AD mice model [490, 491]. Consequently, there is an alteration of autophagy in APOE4-models (in vitro and in vivo) and in human APOE4-carriers. Furthermore, this alteration is part of the vicious circle associated with the progression of the pathology since it involves other elements such as neuroinflammation, cell death, among others.
The complete mechanism that underlies the link between APOE4 and autophagy is not yet fully understood, but some processes have been suggested to be involved, such as the interaction with autophagy regulators (i.e., TFEB and mTOR) or phospholipid dysregulation. Parcon and colleagues demonstrated in vitro and in silico that apoE4 could bind to the CLEAR promoter, blocking or interfering with the CLEAR-binding of the transcription factor EB (TFEB) [489], a master regulator of lysosome function and autophagy [492]. Subsequently, Lima and colleagues found that the interaction with CLEAR was apoE4-specific, as they did not observe interaction with apoE3 nor apoE2 [493]. Reduced levels of TFEB in lymphocytes and monocytes have been detected in patients with AD, so as in mouse models [494, 495]. Therefore, according to these works, APOE4 could reduce the autophagic response through interference with TFEB activation.
mTORC1 is one of the primary regulators of cell growth with multiple pleiotropic functions, such as the repression of autophagy [496]. In AD, the mTORC1 pathway is upregulated, correlating with cognitive decline in AD patients [497, 498]. Although several works have studied the relation between APOE4 and the mTORC1 pathway, to date, there is no consensus on this issue. Even though apoE4 receptors VLDLR and apoER2 activate PI3K/AKT/mTORC1 [499, 500], Yates and colleagues observed that the alteration in the mTORC1 pathway was independent of apoE status in brains of AD-patients [501]. However, Li and colleagues noticed in the hippocampus of APOE4 Wistar rats an activation of the mTORC1 pathway, decreased autophagy, and AD-like alterations [502]. Finally, different studies showed that treatment with rapamycin (inhibitor of mTOR) has a protective effect in the brain of APOE4 transgenic mice and E4FAD mice (5xFAD cross-bred with APOE4 mice) [503,504,505]. These and other previous results have raised interest in rapamycin as a drug of potential interest in AD treatment [506].
The control of autophagy by APOE could involve other mechanisms such as the dysregulation of phospholipids (e.g., phosphoinositol phosphate (PIP) and phosphoinositol biphosphate (PIP2)). PIP and PIP2 mediate autophagy, regulating the mTOR pathway or controlling membrane dynamics [507]. Zhu and colleagues observed an exacerbated reduction in PIP2 in APOE4 carriers’ brains compared to APOE3. Decreased levels of PIP2 were associated with an increase in Synj1 [491], a PIP2-degrading enzyme [507] significantly upregulated in AD brains [508] and associated with reduced internalization and degradation of Aβ [509]. Synj1 appeared upregulated in the neocortex and hippocampus of APOE4/4 mice. These mice have significant memory impairment, abrogated by the genetic knockdown of Synj1 [491]. Cao and colleagues identified the miRNA miR-195 as a candidate in regulating the apoE-synj1-PIP2 pathway. The overexpression of miR-195 restored cognitive deficits and ameliorated amyloid and p-tau pathology in APOE4 individuals [510]. This study offers a new potential therapeutic approach to the regulation of apoE4 and its effect on autophagy in AD.
Altogether, different studies established a clear link between autophagy and APOE4, which may be one of the main cellular processes underlying the detrimental effect of APOE4 in AD. Although the mechanisms involved are not fully understood, different shreds of evidence show the alteration of relevant elements such as mTOR1 or PIP2. Dissecting the components involved in this complex process may offer new and hopeful therapeutic approaches in AD treatment. A summary of the effects of the APOE4 genotype on the AD-related changes can be seen in Table 5.
Connecting APOE with other non-genetic AD-associated risk factors
Age
Aging is considered one of the greatest risk factors implicated in developing neurodegenerative diseases, including AD [519, 520]. Normal cognitive aging is related to neurocognitive changes, such as alterations in processing speed, loss of attention, memory-related deficits, decline in verbal fluency, visuospatial abilities, and executive functioning, whereas some of the structural and functional changes include gray and white matter volume decline [521, 522]. Nonetheless, the exact mechanisms and molecular pathways connecting aging and the development of neurodegenerative diseases are yet unknown. In addition, it is still not determined the connection between the APOE genotype, aging, and the development of neurodegeneration. An age-dependent increase of apoE CNS levels has been documented, and these levels can be affected by the amyloid pathological status [201]. A case–control study comparing the age-specific association between the APOE genotype and AD in the CSF of more than a thousand AD cases reported that the highest impact of APOE4 on AD risk was between the ages of 65 and 70 [523].
Brain aging is related to immunosenescence and low-grade inflammation (inflammaging), to which activated macrophages and monocytes will contribute and promote neuroinflammation, a key event in chronic age-related neurodegenerative diseases [524, 525]. Similarly, systemic inflammation is also a predictor of brain aging [526, 527]. Previous studies have reported how the hypothalamic-derived release of inflammatory mediators controls systemic aging [528]. Quantifying the inflammatory-protein concentration in the CSF and human plasma of AD patients at different AD stages showed a correlation of CSF inflammatory changes with Aβ and tau levels, neurodegeneration, and cognition [529]. The APOE4 genotype is associated with lower C-reactive protein levels, which in turn, correlates with greater APOE-related AD risk [526]. Conversely, the expression of the APOE2 allele has been connected with increased life longevity and protecting against age-induced cognitive decline independently of AD pathology [530].
The study from Zhao and colleagues evaluated in APOE targeted-replacement mice the effect of age, sex, and the APOE genotype in the brain transcriptome and serum metabolome, in which they found a strong impact of age in the brain transcriptome together with brain-specific elevated SerpinA3n levels in APOE4 mice [531]. Serpina3n is a serine protease inhibitor, it is the mouse orthologue of the human alpha-1antichymotrypsin, and an inflammatory protein overexpressed in the AD brain, able to induce tau hyperphosphorylation in neurons [532, 533]. SerpinA3n is also involved in traumatic brain injury (TBI) and multiple sclerosis (MS); during TBI, SerpinA3n deficiency aggravates neuronal apoptosis and cognitive disfunction after hippocampal stab injury, and progressive MS patients present increased SerpinA3n protein levels have [534, 535].
Aging is also associated with chronic glial activation, which correlates with age-related cognitive decline and neurodegeneration [536]. Thus, microglial depletion and repopulation has been proposed as potential therapy to normalize chronic immune activation [537,538,539]. Another example would be the study from Coleman and colleagues in which microglial depletion and repopulation in primary organotypic hippocampal slice cultures normalized persistent pro-inflammatory gene expression [540]. Interestingly, it has been noted that the brain can only perform one whole repopulation event at a time, however, with sufficient time, it can ultimately repopulate after repeated depletion events [541].
Sex
Female sex, together with advanced age and the expression of APOE4 genotype, constitutes one of the most relevant risk factors for the development of AD. In the United States, two-thirds of the diagnosed AD cases correspond to women [542, 543]. This fact is often associated with the higher longevity of women [544]. On the other hand, recent translational imaging studies documented an AD-phenotype characterized by low metabolic activity and high Aβ deposition in perimenopausal and postmenopausal women aged 40–60 years old [545]. This prior study also evidenced an increased Aβ deposition in women carrying the APOE4 genotype. Likewise, it was recently confirmed the sex-specificity between AD biomarkers and CSF apoE, where baseline CSF apoE levels in women were significantly associated with Aβ and tau levels [417], which does not entirely align with results from previous studies [546, 547].
Neuroinflammation is a strong contributor in AD pathophysiology; however, the impact of glial cell sexual dimorphism in the neuroinflammatory response is still yet fairly unknown. Studies characterizing the neuroinflammatory response in male and female C57Bl/6 N mice suggest that type 1 interferons could be one of the potential sex-mediating cytokines that influencing cognition [548].
Microglial sexual dimorphism has been taken under consideration in the context of AD and synaptic function, ischemic stroke, and traumatic brain injury [549,550,551,552]. Of note, microglial responses to environmental challenges occur in a sex- and time-dependent manner. A mouse and human study addressing the microbiome’s influence in prenatal and adult microglia revealed that antibiotic treatment of adult mice led to sexually biased microglial responses [553]. Further on, regarding astrocytic implications in neuroinflammation, sex-mediated differences in the astrocytic response towards an inflammatory stimulus have been described in primary astrocytic cell lines [554, 555]. ApoE can target synapses and impact neuronal activity differently if released by neurons or astrocytes [460]. Live-calcium imaging techniques in vitro cultured neuron models have evidenced that neuron-released apoE induces the highest neuronal firing in the presence of apoE3 and not apoE4, whereas astrocytic-derived apoE induces the strongest neuronal firing with apoE4 [460]. Additionally, it has been reported in APOE4 astrocytes from male but not female APOE-targeted replacement mice higher calcium hyperactivity [556].
The impact of sexual differences in glial physiology have been reviewed by Crespo-Castrillo and colleagues [557]. Further studies taking into account the cellular sexual dimorphism will be of great value to further unravel the interaction between sex and the APOE genotype in neurodegenerative diseases, particularly in AD.
Diet
Dietary habits and lifestyle strongly impact gut microbiota composition, and the gut microbiome is gaining increasing relevance as a modifier of the susceptibility and progression of neurological and neurodegenerative diseases [558,559,560]. Evidence between healthy dietary patterns, their potential anti-neuroinflammatory action, and their influence on age-related cognition has been reviewed by McGrattan and colleagues [561].
Multiple studies have shown that the gut microglioma and the gut-brain axis play a significant role in brain function and impacts neuroinflammation and AD progression [562, 563]. Additionally, some epidemiological studies propose a link between type 2 diabetes mellitus (T2DM) and AD [564, 565].
AD and T2DM share as a common feature the presence of misfolded proteins, Aβ and islet amyloid polypeptide, respectively [566]. T2DM is one of the prime risk factors for AD development, together with the expression of the APOE4 allele. Nonetheless, the precise mechanisms by which the APOE genotype and diabetes contribute to AD risk are still undetermined. EOAD and LOAD are characterized by different metabolic connectivity features [567]. It has been proposed that Insulin influences AD pathology by directly interacting with Aβ peptides [568, 569].
Insulin resistance and impaired glucose metabolism are two extensively reviewed AD features [568]. The association between the APOE genotype polymorphisms and the risk of suffering T2DM and cardiovascular disease have also been reported, pointing at APOE4 as a risk factor for T2DM and cardiovascular diseases [565, 570,571,572]. APOE influences insulin resistance and glucose metabolism in the brain [336, 573, 574]. TaqMan arrays studies in APOE transgenic mice revealed that brains from APOE4 mice are the most deficient profile on glucose uptake and metabolism [575]. APOE isoforms cause differential effects on the functioning of the brain insulin receptor. It has been observed that apoE3 binds with higher affinity to the insulin receptor than apoE4 [576], and a single insulin injection in APOE4-carrying mice induced increased activation of the insulin upstream brain signaling pathways, such as the Akt pathway, coupled to higher phosphorylation levels of the Ser202 tau residue [577]. It has also been reported in primary neuron studies that apoE4 interacts with the insulin receptor, trapping it in the late endosomes and impairing its trafficking, thus causing impaired insulin signaling, insulin-coupled mitochondrial respiration and glycolysis [578].
A high-fat diet has been shown to increase gliosis in APOE3 mice but not in APOE4 mice [579]. Precision nutrition interventional approaches have been suggested for APOE4 carriers [580]. A systematic review of the clinical records from the United States National Alzheimer Coordinating center revealed that diabetes is linked to earlier cognitive decline during aging in APOE2 and APOE3 carriers, but not in APOE4 carriers, in which the effect of diabetes was negligible, perhaps because APOE4 carriers are already more prone to suffer vascular impairment and cognitive decline [581]. The prior study also observed lower AD neuropathological hallmarks in APOE3 carriers [581]. Thus, future studies evaluating the impact of diet and the APOE genotype in AD pathology will be of great value.
Physical exercise
Several robust studies have shown the health benefits of physical activity in cardiopulmonary function, neuroplasticity, cognition, and bone density [582, 583]. Interestingly, related to bone density, it was recently reported the discovery of a specialized bone-cell progenitor that supports immune cell generation in response to physical exercise [584]. Physical activity has also been associated with a lower risk of developing dementia [585,586,587,588,589]. However, these studies are inconclusive to what extent exercise may prevent or postpone AD development [590,591,592,593]. Anthropologists hypothesize that the evolution of increased physical activity 2 million years ago might have been a prerequisite for human cognitive longevity due to the reduction in amyloid pathology and vascular burden of APOE4 [53]. In that context, it is intriguing whether or not the APOE genotype impacts the association between a physically active lifestyle and the development of dementia, particularly in AD.
A wide array of studies evaluated the impact of physical exercise intervention upon multiple aspects of AD pathology. Various meta-analyses have studied the effect of exercise in cognitive function, suggesting that exercise training could delay cognitive function decline in AD-risk and AD patients [594, 595]. Other authors have addressed the impact of physical exercise upon various AD biomarkers, such as hippocampal volume, in which some studies observed volumetric increases in the hippocampus, whereas others failed to do so [596, 597]. Alternative AD biomarkers evaluated regarding the impact of physical exercise are Aβ turnover, cerebral blood flow, neurotrophin release, and inflammation [598,599,600].
Although vascular dementia and AD are considered independent disorders, vascular dysfunction also plays a key role in AD pathogenesis [601,602,603]. Indeed, early exercise intervention in a model of chronic cerebral hypoperfusion led to normalized capillary density and decreased leukocyte rolling in chronic hypoperfused mouse brains, paired with a decrease in hippocampal microglial activation and an improvement of microvessel-covering by astrocytes [604].
Few exercise studies discriminate between APOE genotypes, and among those who do, results are diverse, as reported by recent systematic reviews [605, 606]. Regarding the risk of getting diagnosed with AD, some studies show that the lower risk of developing all-cause dementia and AD among highly active persons is more pronounced in APOE4 non-carriers [586, 587, 607, 608]. In contrast, other studies demonstrate that this association is more valid for people carrying the APOE4 [593], whereas results reported by our research group, and others, do not reveal any impact of the APOE genotype on this relationship [590, 609, 610]. Regarding cognition, some studies find that the association between physical activity and preserved cognition is more noticeable among APOE4 carriers [610,611,612]. Opposingly, others reveal an attenuation or even absence of this relationship in APOE4-carriers [608, 613, 614], while some display no impact of the genotype [609, 615,616,617].
Further, neuroimaging studies indicate that the benefits of physical activity on cortical activation, prefrontal cortex connectivity, and preservation of the hippocampal volume might be limited to APOE4 carriers [618, 619]. Moreover, even among APOE4 carriers, a randomized controlled exercise intervention only improved hippocampal blood flow in those who initially suffered from hypertension [620]. Considering the amyloid pathology, Head and colleagues demonstrated that a more sedentary lifestyle was associated with higher amyloid PET, but only in APOE4-carriers [621]. Their findings align with a recent systematic review, suggesting that the association between physical activity and the amyloid burden is more evident in APOE4 carriers [605]. Still, exercise intervention in patients with mild AD did not affect tau or amyloid biomarkers in CSF, regardless of the apoE status [622], and this lack of impact correlates with another study [623].
The involvement of neuroinflammation in the pathophysiology of AD has caught increasing interest during the past decade. Exercise affects the inflammatory status peripherally, and several studies suggest that exercise may also reduce neuroinflammation [600, 624, 625]. Still, the impact of the APOE genotype in neuroinflammation is not yet fully elucidated. Jensen and colleagues detected a stabilizing effect on plasma IFNγ following exercise only among patients with mild AD who were APOE4 carriers [624]. Experimentally, APOE deficiency compromises the microglial response to amyloid plaques [236], and exercise may prevent the age-related decrease in astrocytic APOE expression, which has been liked to microglial activation, an effect absent in APOE knockout mice [626]. Therefore, there is still a knowledge gap regarding the impact of APOE on the neuroinflammatory effect of exercise. Increased insight in this area could potentially open up new treatment strategies combining exercise interventions with future pharmacological therapies to target specific neuroinflammatory pathological events early in AD patients depending on APOE genotype. A limitation is that nearly all of the investigations made in this field only consider the influence of APOE on dementia risk and do not examine if this genotype affects exercise behavior. A study from the UK biobank that included over 350 000 individuals revealed that the APOE4 variant was the most strongly linked gene to moderate and vigorous physical activity [627]. Likewise, a study comparing over 200 athletes with controls found an implication of the APOE single nucleotide polymorphism (SNP) rs429358 in the modulation of the distinction between power athletes and sedentary individuals [628]. On the other hand, in our previous study that included 10 000 individuals, the APOE4 genotype was equally distributed between groups with different levels of lifestyle physical activity [590]. Further, if participants in a study are aware of their APOE4 genotype and what that implies in terms of long-term risks, they may be more motivated to become more physically active, which may influence their outcomes.
The discrepancy between studies may be attributable to several factors, such as the type of exercise assessment and outcome measurement. For instance, a recent systematic review concluded that most studies with conflicting results used outcomes such as clinical diagnosis, which are not sensitive to preclinical cognitive fluctuations [606]. Timing of exercise intervention or physical activity lifestyle assessment are also relevant parameters to consider since physical activity may need to occur when it is still possible to prevent the pathological process. This prior fact may be significantly important for individuals with genetic predispositions, such as the APOE4 carriers. The study by Krell-Roesch and colleagues is a representative example of when physical activity in midlife seems to preserve memory function, particularly in APOE4-carriers, whereas only non-carriers benefitted from engaging in physical activity later in life [629]. In addition, it is necessary to note that studies with shorter follow-up times (e.g. > 5 years) risk finding associations due to reverse causation since individuals in a prodromal disease stage may engage less in physical activity [586, 590, 630, 631]. Conversely, the association between physical activity and a lower risk of mortality in several diseases ultimately leads to an extended lifespan where neurodegeneration eventually will develop. Therefore, it is crucial to discuss the time point when physical exercise significantly decreases the risks and improves the patient’s life quality. Being physically active may prevent dementia development only during a specific time for individuals with genetic constraints such as APOE4, and effects may be lost with very long follow-up but still be clinically relevant.
Current AD therapies and clinical trials. Targeting APOE4
Over the last few years, many experimental studies have focused on animal models targeting APOE4, showing promising results. These include the increase of apoE levels using bexarotene, apoE mimetics, blocking the apoE-Aβ interaction, APOE silencing, and the use of genome editing to switch from APOE4 to APOE2 or APOE3 isoforms. These advances have been carefully reviewed in Lancet Neurology in 2021 (see review [632]). Nonetheless, despite promising results in animal models, there is a low number of completed or ongoing clinical trials [632].
Two clinical trials have focused on increasing apoE levels by administering bexarotene, an RXR agonist. Oral administration of bexarotene in APP/PS1 mice enhanced Aβ clearance, reduced Aβ plaques area, and improved cognitive deficits [633]. In humans, bexarotene showed poor CNS penetration in healthy subjects and resulted in a modest increase of apoE levels in CSF [634]. In phase 2 clinical trials, bexarotene administration showed a significant reduction in brain amyloid in APOE4 noncarriers, with no changes in APOE4 carriers, so the primary outcome of the clinical trial was negative [635]. Another clinical trial followed a similar approach with probucol (NCT02707458), a non-statin lipid-lowering drug; however, there are still no results published to date [632].
In addition to those mentioned above, two Phase 1 ongoing clinical trials show novel APOE4 therapeutic approaches in patients with AD or MCI (NCT03887741 and NCT03634007). The first trial focuses on the genetic switch of APOE4 to APOE2 through adenoviral vectors (AAV). The trial will assess the safety of the intracisternal administration of the AAV in patients APOE4 homozygotes with AD (NCT03634007). This approach showed promising results in an APOE4 knock-in model, increasing apoE lipidation and decreasing endogenous Aβ [636, 637]. The second clinical trial aims to assess the safety of plasmapheresis from young APOE3 homozygotes into APOE4 homozygotes with MCI (NCT03887741). Both clinical trials are still in the recruitment phase.
The use of immunotherapy (e.g., aducanumab, gantenerumab, lecanemab) is one of the most promising recent advances in treating AD. However, their implementation is not without controversy. The administration of these antibodies potentially induced amyloid-related imaging abnormalities (ARIA), associated with the development of brain hemorrhages. The cause of this adverse response is still unknown. Nevertheless, different works showed an exacerbated ARIA response in APOE4 carriers [638, 639]. Immunotherapy against AD opens another attractive therapeutic alternative, the use of anti-apoE antibodies. To date, studies in animal models have shown that the use of anti-apoE monoclonal antibodies such as HJ6-3 and HAE-4 reduces Aβ and apoE levels in the brain, along with an improvement in cognitive performance [640,641,642,643]. Specifically, the use of the HAE-4 antibody that identifies low-lipidated apoE reduced both parenchymal Aβ plaques and Aβ in the cerebral vasculature without vascular complications associated with Aβ antibodies [643]. These studies evidence the potential interest of this strategy, which could be interesting to test in clinical trials in the coming years.
The knowledge about APOE’s role in AD has increased significantly in recent years. Different animal model studies highlighted the potential therapeutic implications of APOE, but many aspects require further investigation. The advent of new therapeutic approaches such as gene editing or immunotherapy, together with all the knowledge accumulated to date, implies that this is only the beginning of a promising research field.
Implications of APOE in other neurodegenerative diseases
The main hallmark of neurodegeneration will be the loss of neuronal structure and connectivity that can lead to altered movement (ataxia) or dementia [644]. Among the primary mechanisms involved in most neurodegenerative diseases are abnormal protein accumulation, immune system activation, and neuronal death [645]. These three mechanisms are tightly connected, and their interaction determines most of the detrimental events involved in the development of the pathologies.
There is growing evidence that prolonged brain neuroinflammation and cytokine release boosts neurodegeneration and exacerbates Aβ and NFT pathology and the progression of other neurodegenerative diseases [221, 646, 647]. Additional events contributing to neurodegeneration include oxidative stress events, mitochondrial dysfunction, Golgi apparatus fragmentation, disruption of axonal transport, neurotrophins dysfunction, and cell death mechanisms such as apoptosis and necrosis [648, 649].
Despite the vast array of neurodegenerative diseases described in humans, there some similarities among them. Perhaps the most outstanding are the abnormal protein accumulation, neuronal death, and the activation of the innate immune system. Innate immune system activation in the brain is commonly driven by glial cells, especially microglia. The main focus of this review is the role of APOE, and its isoforms, during neuroinflammation observed in neurodegenerative diseases, with particular attention to its role in neuroinflammation. A tremendous scientific effort in the neurodegenerative diseases field is centered on resolving the puzzle of AD pathology. However, other diseases characterized by overt neuroinflammation can also shed light on the impact of the APOE genotype and how the brain works in disease conditions, and due to the similarities before mentioned, apply that knowledge to other research areas.
In this section, we are exploring the importance of the APOE genotype in neurodegenerative diseases other than AD. A common feature of the brain diseases in this section is the abnormal protein accumulation within the neurons or an acute detrimental event. The intraneuronal protein accumulation could lead to a different role of apoE compared to the mechanism involving apoE and the immune system response. For instance, the role of APOE in the neuronal lysosome or autophagy regulation might play a more significant role than the recently described role of APOE in microglial phenotype activity [269, 650, 651]. However, stroke or traumatic brain injury elicits a robust inflammatory response mainly driven by microglial cells. Thus, understanding the dual role of apoE in the regulation of the immune system or the intraneuronal metabolism is a great challenge for the scientific community nowadays.
Altogether, APOE might share common mechanisms of action among neurodegenerative diseases with an abnormal protein accumulation or an acute detrimental event. In the coming section, we will explore in more detail the latest finding on the role of APOE in Parkinson’s disease and other synucleopathies, stroke, TBI, ALS, or MS (Fig. 3).

Mechanisms involved in apoE-related neurodegeneration. The influence of the APOE genotype has been associated with CNS diseases such as Alzheimer’s Disease (AD), Parkinson’s Disease (PD), Traumatic Brain Injury (TBI), Stroke, Amyotrophic Lateral Sclerosis (ALS), and Multiple Sclerosis (MS). In particular, APOE4 promotes synaptic impairment, amyloid-β (Aβ) protein seeding, aggregation, Aβ-plaque formation and defective protein clearance in AD. APOE4 will contribute to additional pathological features characteristic of AD, but also present in other CNS pathologies such as, for instance, impairment of the autophagy/endosomal system, altered microglial reactivity and neuroinflammation, and blood–brain barrier disruption
Parkinson’s disease
Parkinson's Disease (PD) constitutes the second most prevalent neurodegenerative disease. PD characterizes by the intracellular accumulation of proteinaceous aggregates called Lewy Bodies (LBs). LBs constitute rounded intracellular aggregates mainly composed of α-syn, yet, additional proteins and lipids can also be present, conferring these aggregates a highly complex composition, for further details regarding structural, morphological, and biochemical properties of LBs, refer to the review from Fares and colleagues [652]. α-syn is a 140 amino acid protein composed of a C-terminal acidic domain, a central domain highly hydrophobic and involved in aggregation, and an N-terminal domain with lipid-binding similar to apoE’s lipid-binding domain [653]. Remarkably, all six known mutations in the SCNA gene that leads to familial PD are located in the N-terminal domain [654]. This domain possesses hexameric repeats of 11 amino acids, characteristic of apolipoproteins, which confers α-syn lipid-binding abilities [655]. The apoE-cholesterol pathway differentially affects both AD and Lewy Body dementia (LBD), and the relevance of cholesterol in Lewy Body pathology has been recently reviewed [656,657,658]. Firstly, α-syn is known to change its morphology in response to lipid binding; secondly, most of the known PD-related mutations influence cholesterol levels; and lastly, α-syn deficiency leads to increase neuronal lipids in mice [659]. Interaction between α-syn and apoE protein has also been documented. In vitro experiments showed that all isoforms of apoE, but apoE4 in particular, increases α-syn aggregation [660]. In human samples, fragments from apoE protein have been recently discovered to form part of the majority of the LBs [661]. Additionally, CSF lipid vesicles from PD patients, but not control patients, contain both apoE and α-syn, indicating that increased neuronal cholesterol efflux could be a late event in PD [662].
In PD, protein aggregates are primarily found in the dopaminergic neurons of the Substantia Nigra; other areas such as the cortical region, hippocampus, amygdala, olfactory bulb, or gut nervous system, can be affected in diverse subtypes or stages of the disease [663]. Importantly, LBD pathology is not only present in PD, but other related diseases like PD with dementia (PDD), from which is practically indistinguishable when its fully clinically developed, and LBD, that commonly has some degree of concomitant AD [664, 665].
From a genetic perspective, APOE mutations have long been related to a higher risk of developing PD. However, several studies showed discrepancies suggesting that APOE mutations may be more related to a higher risk of worsening cognitive function and not to motor degeneration [666,667,668,669,670,671]. Nonetheless, it has been detected the presence of apoE-positive neuromelanin-containing dopaminergic neurons in substantia nigra of PD patients [662].
Neuroinflammation has long been considered a central player in PD onset and development [672]. However, despite the recent description of microglial phenotypes associated with several neurodegenerative diseases, no microglial phenotype has been associated with PD [673]. Moreover, albeit the APOE relevance for the neurodegenerative phenotypes of microglia, data on the effect of APOE variants in PD-associated microglia are scarce. Recently, Choi and colleagues described a microglial phenotype associated with microglial α-syn clearance [674]. This phenotype has several transcriptomic similarities with other neurodegenerative phenotypes previously described by Krasemann and colleagues [673], such as upregulation of Axl, Lpl, and importantly, apoE. Thus, investigating the role of apoE isoforms in this microglial phenotype could help better understand the role of apoE and microglia in PD onset, dementia development, and α-syn clearance.
Synucleinopathies and amyloidopathies are commonly found simultaneously in the brain of PD and AD patients, hindering the effect of APOE mutations in synucleinopathies' progression. Lately, APOE genetic mutations have been considered a risk factor for developing dementia in all synucleinopathies subtypes, including PD, but not related to PD onset [675]. The study by Dickson and colleagues, including more than 600 hundred PDD and LBD post-mortem samples, showed that severity of LB pathology was associated with APOE4 variant independently of the severity of concomitant AD, yet, the analysis only included cortical regions [676]. Two additional studies recently showed implications of apoE4 in synucleinopathies not related to AD [677, 678]. Goldberg and colleagues propose that APOE4 could be related not only with the severity of LB pathology but also with the extension of the pathology to other areas, in comparison with APOE2 or APOE3 variants where the pathology is more prone to be restricted to the midbrain in mouse models [679]. Prior results could suggest that APOE4 is related to the progression of LB pathology from the classical PD to related LB diseases like PDD or DLB. Overall, even if APOE4 is not considered as a risk factor for developing PD, recent studies evidence that it is risk factor for developing dementia, worsens cognitive function, and can be related to the spreading of LB burden from the midbrain to other areas, perhaps due to distinct roles of APOE isoforms in microglia-associated α-syn clearance.
Other synucleinopathies
In addition to its association with an increased risk of AD, we have previously addressed in this review that APOE4 is also an outstanding genetic risk factor for LBD [680]. Analogously to AD, the APOE2 variant appears protective and delays LBD onset [680]. Furthermore, the presence of apoE4 negatively affects cognitive performance in PD patients and increases the risk of dementia in pure synucleinopathies (with limited AD co-pathology) [666, 681]. There are no studies associating APOE4 with altered risk of suffering oligodendrocytic synucleinopathy multiple system atrophy (MSA), although astrocyte-secreted apoE4 unexpectedly was found to reduce the uptake of α-synuclein in cultures of oligodendrocytic cells [682].
The implications of APOE4 in synucleinopathies are still poorly understood, but recent studies suggest that the APOE gene regulates α-syn brain pathology independently of its effects on AD-related Aβ and tau pathology and that specifically APOE4 exacerbates α-syn pathology [683, 684]. A study led by Paslawski reported that α-synuclein could co-localize with apoE-containing lipoparticles in the CSF, and that apoE CSF levels were higher in patients with early PD compared to controls [662]. Intriguingly, APOE4 has also been associated with higher levels of CSF α-syn in patients in the AD continuum where patients with mild cognitive impairment (MCI) who, after 24 months follow-up fulfilled the clinical criteria for an AD diagnosis, exhibited higher CSF α-syn levels in an APOE4 dose-dependent manner [685]. The same study further reported that higher CSF α-synuclein levels were associated with brain Aβ burden as assessed by PiB-PET in carriers of autosomal-dominant AD mutations that in addition carried the APOE4 gene variant. Indeed, accumulating studies propose α-synuclein to be an active player in not only synucleinopathies but also in AD pathophysiology in which APOE4/apoE4—α-syn interactions may play a still-to-be defined role [686].
Traumatic Brain injury
According to the United States CDC, TBI can be caused by a bump, blow, or jolt inflicted to the head or by a penetrating head injury that will cause a disruption of the normal brain function and will have short- and long-term effects on health [687, 688]. TBI is considered as one of the risk factors for the development of AD and it is associated with an earlier dementia onset [689,690,691]. The recovery rates among people that have suffered TBI differ among individuals, and multiple metanalyses point out a relationship between the APOE4 genotype and increased risk of poor long-term outcomes following TBI [692, 693].
The relationship between APOE and TBI has been extensively studied. RNA-sequencing studies together with bioinformatic analysis have associated TBI with neuroinflammation and neurodegeneration, outlining apoE, Mapt, and Trem2 as differentially expressed genes and astrocytes and microglia as key cells involved [694,695,696]. The study from Giarratana and colleagues documented that PKCε activation and consequent increase in BDNF levels decreased inflammation and microglial activation in APOE4 knock in mice subjected to mild TBI [697].
BBB dysfunction constitutes one of the pathophysiological events present in TBI and has been reported in TBI patients [698]. ApoE is implicated in preserving tight-junctions integrity at the BBB in an isoform-dependent manner, in which studies with APOE4-KI mice have reported higher BBB permeability than in APOE3-KI mice [377, 699]. It has been documented that APOE3 and APOE4 mice react similarly to TBI, albeit APOE3 mice present higher BBB recovery rates than APOE4 mice [700]. Moreover, both APOE4 homozygous and heterozygous individuals are characterized with BBB breakdown in brain areas such as the hippocampus and the medial temporal lobe that may contribute to cognitive decline, and apoE4 accelerates BBB breakdown [375, 701, 702].
Early studies on TBI reported an increase of tau levels in the CSF of TBI patients. Parallelly, an increase of intraneuronal Aβ1-42 accumulation in the terminal ends of dystrophic axons has been observed after TBI. Nonetheless, the specific role of intraneuronal Aβ in TBI is still unknown. A study from Abu Hamdeh S. and colleagues reported increased levels of Amyloid oligomers/protofibrils and Aβ1-42 in the brain tissue of postmortem TBI patients heterozygous for the APOE4 allele and concluded that soluble intermediary Aβ tends to aggregate more rapidly after TBI in the presence of apoE4 [703, 704]. Interestingly, in the study conducted by Montagne and colleagues, authors did not observe a correlation between the BBB breakdown increase in APOE4 carriers and the Aβ or tau CSF levels, yet, prior studies have reported increased phosphorylated tau levels in APOE4 mice after TBI in the CSF and hippocampal region [705, 706].
Repeated TBI is known to initiate cerebrovascular diseases, and can lead to chronic traumatic encephalopathy (CTE). Historically CTE is known as dementia pugilistica, it was first described in boxers in 1928, and closely related to cerebrovascular dysfunction [707]. Neuropathologically, CTE is characterized by NFTs similar to those found in AD and diffuse Aβ plaques, particularly in APOE4 carriers [708,709,710]. A postmortem study of 1275 human brains evaluated the link between the APOE genotype and the development of cerebrovascular disease, concluding that the APOE2 genotype was not protective and significantly increased the risk of suffering hemorrhages when cerebrovascular amyloid angiopathy was present [711]. Therefore, a better understanding of the mechanisms involving TBI and APOE could be of great interest to discover possible disease-modifying biomarkers to halt AD progression.
Stroke
Stroke consists of a disruption in the brain’s blood flow either due to a lack of blood flow (ischemia) or to bleeding (hemorrhage) [712]. The global burden of ischemic strokes is fourfold higher than hemorrhagic [713].
The APOE genotype influences the age onset of stroke and its severity. Notably, the expression of the APOE4 allele is related to delayed recovery of verbal memory function and a reduced EC volume evaluated one year after the ischemic stroke event [714]. APOE4 has been shown to accelerate the risk of developing stroke-associated dementia, possibly related to the implication of APOE4 in BBB dysfunction, as commented in the prior TBI section of this review [715, 716]. Similarly, a meta-analysis carried out by the Swedish Heart and Lung foundation with combined information including the APOE alleles, sex, age of onset of ischemic stroke, stroke severity, and outcome, established a link between the APOE4 allele expression and younger age of stroke onset, so as a link between APOE4 and favorable stoke outcomes, whereas APOE2 allele male carriers were directly related with poor stroke outcomes. Nonetheless, the authors did not observe any association between the APOE alleles and stroke severity [717]. In a Chinese population meta-analysis, the APOE4 genotype could also predict different types of strokes, including ischemic stroke, intracerebral hemorrhage, and subarachnoid hemorrhage [718].
The neuroinflammation-related effects of hypoxic-ischemia in the brain have previously been described, leading to a microglial cell proliferation increase, astrocyte reactivity, and release of pro-inflammatory cytokines [719, 720]. Additionally, a unique subtype of microglial cells in the thalamus of C57BL6J mice subjected to ischemic stroke was recently reported. This microglial subtype is transcriptomically characterized by upregulated apoE, Axl, LpL, CSF1, and Cst7 levels [721]. Related to the neuroinflammatory response during stroke, Tomas Deierborg’s group demonstrated in an in vivo stroke model the deleterious role of gal-3 dependent inflammatory response, in which the lack of gal-3 reduced the infarct area in the stroke-induced model, and intranigral injection of LPS in gal-3 knock-out mice reduced neuronal death compared to gal-3 WT mice [722].
Endothelial cells and astrocytes will also play a significant role after stroke; Jun Xiang and colleagues evaluated how astrocyte-endothelial cell interaction could influence stoke pathogenesis in an oxygen and glucose deprivation-reoxygenation (OGD-R) model and reported that astrocyte-derived apoE activated the PI3k/eNOS pathway in endothelial cells as protection mechanism [723].
Ischemic stroke can induce white and grey matter damage, limits axonal regeneration and remyelination, and might lead to vascular dementia and Alzheimer’s disease [724, 725]. The APOE2 genotype has also been associated with white matter disease in ischemic stroke patients, together with APOE4 [726, 727]. Related to this, Hayden Y. and colleagues aimed to better characterize the interaction between white matter ischemic lesions, the APOE4 genotype, and amyloid pathology in AD using E4FAD mice subjected to a subcortical ischemic stroke. The authors did not observe any effect of the 5xFAD transgene on the stroke lesion, nor in the cortical amyloid deposition between the sham and stroke E4FAD mice, despite that, there was a high microglial reactivity and cortical neuron damage in the stroke-induced E4FAD mice, suggesting that the microglial reactivity induced by the subcortical ischemic lesion may be promoting Aβ clearance. Still, this study was limited to early time points (7 to 14 days post-stroke assessment) and young mice [728].
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a progressive and fatal neurodegenerative disease characterized by deposition of aggregated proteins that will affect motor neurons integrity. Motor-neuron dysfunction will lead to muscle weakness until complete loss of voluntary muscle movement, including breathing, speaking, eating difficulties, and some individuals are also diagnosed with frontotemporal dementia [729, 730]. Although ALS etiopathology is multifactorial, the most prevalent mutations present in ALS are in the proteins Superoxide-dismutase1 (SOD-1) and TAR DNA-binding protein 1 (TDP-43) [731, 732]. Defective energetic metabolism, disruption of iron homeostasis, and neuroinflammation are also components of ALS pathogenesis [733]. ALS-related neuroinflammation comprises reactive microglia and astrocytic cells, together with T-cell and macrophage infiltrations in the CNS, and activation of inflammatory pathways [734,735,736]. As mentioned in previous sections of our manuscript, the microglial APOE-dependent molecular signature identified by Krasemann and colleagues observed as well in ALS models [269].
Additionally, many therapeutic approaches have targeted astrocytes, the main apoE-producing cells, to potentially stop ALS progression [737]. In the early two-thousands, the APOE genotype gained attention as a potential marker for ALS onset and progression, leading to conflicting results. Some studies suggest the APOE2 genotype to be protective, and APOE4 detrimental for ALS onset, whereas others suggest that the APOE genotype does not influence ALS clinical course [738, 739]. More recent studies evaluating the influence of the APOE genotype in brain metabolism during ALS showed that the APOE2 genotype correlates with hypometabolism in the brain motor areas usually affected by frontotemporal dementia, however, no evidence of the implication APOE genotype in ALS development risk has been found so far [740, 741].
Interestingly, alterations several receptors related to apoE have been related to the ALS phenotype. The study from Andres-Benito and colleagues revealed an altered gene expression of ABCA1 and ABC transporters in the cortex of ALS patients [742]. Genetic variants in the LXR are associated with age onset in ALS patients, being the LXR SNP rs2695121 variant associated with a 30% increase in ALS duration, but none of the genetic variants studied were linked to ALS risk [743].
Lipid dysregulation or lipid cacostasis is also one of the pathological hallmarks of ALS, contributing to neurodegeneration [744,745,746]. The study by Abdel-Khalik and colleagues reported a defective cholesterol metabolism in the brains of ALS patients [747]. Lipidomic analysis performed in the motor cortex and spinal cord of animal models of ALS revealed an abnormal lipid droplet accumulation in defective astrocytes and mitochondrial dysfunction [748].
The current advances in experimental techniques could be beneficial for future studies to determine the implications of APOE genotype in the lipid dysregulation mechanisms and the molecular pathways involved in ALS pathology.
Multiple Sclerosis
MS is a neuroinflammatory and demyelinating relapsing–remitting disease that will course with selective cortical neuron damage [749]. The contribution of the APOE polymorphisms in MS onset has long been discussed, and often results have been inconsistent due to lack of power in the studies. For instance, APOE4 is linked to a more rapid progression and severity of MS, however, the meta-analysis performed by Lill CM and colleagues in more than twenty-nine thousand subjects didn’t support a role of the APOE4 and APOE2 polymorphisms on MS susceptibility [750,751,752]. On the other hand, an extensive UK-Biobank study evidenced the potential health risks subjected to the expression of the different APOE alleles. The authors revealed an association between APOE2 with an increased risk of suffering MS among other disease conditions, such as ischemic heart failure and hypercholesterolemia, which correlated with previous findings [753, 754]. This study also evidenced ethnic-derived physiological differences among APOE4-carriers, that should be considered for future studies [753].
Recent studies point to a lipid dysregulation in MS patients and cognitive dysfunction, in which LXRs gained attention as a potential target [755,756,757] since LXR activation ameliorated disease severity in experimental autoimmune encephalomyelitis (EAE) models [758, 759].
BBB dysregulation will also be one of the first cerebrovascular abnormalities observed during MS. A compromised BBB will allow the infiltration of T cells, B cells, and macrophages into the CNS, and has been associated with the development of cognitive impairment [760, 761]. APOE deficiency and APOE4 overexpression in mice can accelerate BBB breakdown by activating the NF- κB and MMP9 pathway [376, 702]. Additionally, an evaluation of the impact of the APOE4 genotype upon cognitive performance in early MS patients revealed that APOE4 homozygosity was associated with lower cognitive performance, but this effect was absent in APOE4 heterozygous or APOE2 carriers [762, 763]
Glial cell activation is also known to contribute to the progression of MS. Schrirmer L. and colleagues reviewed glial cell diversity during MS, where they noted prior studies showing that White matter lesions during MS are more severe than those to gray matter and where astrocytes overexpress LRP1, and to phagocytose myelin at the injured sites [764,765,766]. Nonetheless, the possible interaction between these events and the APOE genotype is yet unknown.
Regarding the microglial response during MS, R. Plemel and colleagues studied microglial response after acute demyelination. The authors labeled microglia and CNS-associated macrophages to differentiate them from brain-infiltrating macrophages, and through single-cell RNA sequencing observed multiple microglial activation states, in which they found genes linked to DAM microglia, so as an apoE upregulation at the lesion sites [767]. Besides, activation of Trem2 in microglia increases the density of oligodendrocyte precursors in demyelinated areas, thus potentially promoting remyelination and axonal integrity [768].
Therefore, there are still many aspects of MS pathology in which the contribution of the APOE genotype could be further addressed, such as dyslipidemia related to MS and the possible implications of APOE, its isoforms, and its interacting receptors.
Conclusions
The APOE genotype dramatically influences many aspects of cell function in the CNS and impacts the progression of AD and other CNS diseases characterized by overt inflammation. Remarkably, new APOE genetic variants are still being discovered and provide more insights into the impact of the APOE genotype in neurodegenerative diseases. For instance, the APOE Jacksonville mutation (V236E), characterized by a mutation in the lipid-binding region, was reported to reduce Aβ aggregation in 5xFAD mice and manifested the relevance of lipid metabolism during AD [78]. Further, the R251G variant evidenced by Rabinovici and colleagues induces a single amino acid switch at position 251 and has been initially associated with decreased risk of suffering AD [769].
Furthermore, the biological significance of altered apoE levels in the CNS or the periphery are still uncertain. For instance, low plasmatic levels of apoE are related to a higher risk of developing dementia [770]. However, it was recently reported that low presynaptic apoE levels could be related to AD resilience [463]. Thus, it would result in significant interest in future studies to determine the influence of the altered levels of apoE isoforms in different compartments as well as which cells acting as a source of apoE, neurons, astrocytes, microglia, pericytes, and the implications of each source.
The APOE genotype will also impact the microenvironment and functionality of the neighboring cells, and the molecular pathways triggered in consequence. Besides, additional genetic and external factors aside from apoE influencing neuroinflammation and neurodegeneration will also contribute to differences in the performance of memory tasks in young versus more age advanced stages. The latter statement refers to the study by Zolakei and colleagues in which they observed slower memory decay in middle-age APOE4 carriers [771]. Additionally, it is relevant to highlight that up to date, it is still unclear how the total concentration of apoE nor its isoforms could be related to the Aβ, p-tau status, the degree of dementia, or affect other biological parameters.
Some points that could not be discussed further in depth in this review are the implications of peripheral versus CNS apoE and the impact of the gut-brain axis in the development of neurodegenerative diseases [772]. In line with this, it must also be noted that the APOE genotype can influence peripheral diseases such as cardiovascular diseases or macular degeneration. Paradoxically, the APOE4 genotype protects against macular degeneration onset, whereas APOE2 carriers presented an earlier disease onset [773]. The implications of microglial cells in degenerative eye diseases such as primary open-angle glaucoma and age-related macular degeneration have been previously described, and animal studies suggest targeting microglial cells as a potential treatment for eye disorders [774]. Ocular disorders present some common hallmarks of neurodegenerative diseases and CNS pathologies extensively discussed by other authors [775], such as the connection between apoE metabolism and retinal inflammation linked to age-related macular degeneration [776]. Therefore, the implications of the APOE genotype in inflammatory eye-related diseases and their connection to the development of Alzheimer’s disease are highly relevant.
So far, it is yet unknown to which extent the APOE alleles are expressed in heterozygotes; and whether the differences in the protein expression levels of each isoform could result from post-secretory or pre-secretory mechanisms influencing apoE levels, related, for instance, to the findings pointing to an inverse correlation found between plasma and CSF levels of apoE4/apoE3 in heterozygote individual studies [201].
The outcome of this extensive literature review supports a neurodegenerative disease-promoting role of apoE4, arguably through a toxic gain of function. Nevertheless, there is still much to investigate and learn from APOE physiology and its implication in the context of neurodegenerative diseases. Current therapeutic strategies include targeting APOE4 as upstream as possible or its conversion to other isoforms, apoE lipidation, anti-apoE4 immunotherapies, antisense oligonucleotide therapies, among others including non-pharmacological approaches, such as diet changes and physical exercise. Techniques such as single-cell sequencing, novel approaches such as synTOF, female and male animal models of APOE, and clinical studies taking into account the APOE genotype will be of great value in puzzling out the role of APOE in neuroinflammation and neurodegeneration.
Availability of data and materials
Not applicable.
Abbreviations
- AAV:
-
Adenoviral vectors
- ABCA1:
-
ATP binding cassette transporter A1
- ABCG1:
-
ATP Binding Cassette Subfamily G Member 1
- AD:
-
Alzheimer's disease
- ADNI:
-
Alzheimer’s Disease Neuroimaging Initiative
- ALS:
-
Amyotrophic Lateral Sclerosis
- apoE:
-
Apolipoprotein E protein
- APOE:
-
Human apolipoprotein E gene
- ApoER2:
-
Apolipoprotein E receptor 2
- APP:
-
Amyloid precursor protein
- ARF6:
-
ADP-ribosylation factor 6
- ARIA:
-
Amyloid-related imaging abnormalities
- ARM:
-
Activated-response microglia
- ASOs:
-
Antisense oligonucleotide
- ATM:
-
Axonal tract-associated microglia
- Aβ:
-
Amyloid beta
- BBB:
-
Blood–brain barrier
- BDNF:
-
Brain-derived neurotrophic factor
- C1q:
-
Molecule C1 from the complement system
- CAA:
-
Cerebral amyloid angiopathy
- CLU/ApoJ:
-
Clusterin
- CNS:
-
Central nervous system
- CPM:
-
Cycling/proliferating microglia
- CSF:
-
Cerebrospinal fluid
- DAM:
-
Disease-associated microglial phenotype
- DAMPs:
-
Danger-associated molecular patterns
- e.g.:
-
For example
- EAE:
-
Experimental autoimmune encephalomyelitis
- EC:
-
Entorhinal cortex
- EOAD:
-
Early-onset Alzheimer's disease
- EVs:
-
Extracellular vesicles
- gal-3:
-
Galectin-3
- GWAS:
-
Genome-wide association studies
- HDL:
-
High-density lipoproteins
- HMGB1:
-
High-mobility group box protein 1
- HSPGs:
-
Heparan sulfate proteoglycans
- IDE:
-
Insulin-degrading enzyme
- IFN-γ:
-
Interferon-γ
- IL1-β:
-
Interleukin 1-β
- IRM:
-
Injury-responsive microglia
- ISF:
-
Interstitial fluid
- LBD:
-
Lewy Body dementia
- LBs:
-
Lewy Bodies
- LC:
-
Locus coeruleus
- LDAM:
-
Lipid-droplet accumulating microglia
- LDLR:
-
Low-density lipoprotein receptor
- LOAD:
-
Late-onset Alzheimer's disease
- LRP1:
-
Low-density lipoprotein receptor-related protein 1
- LRRs:
-
Leucine-rich repeats
- LTD:
-
Long-term depression
- LTP:
-
Long-term potentiation
- LXR:
-
Liver X receptor
- MAPK:
-
Mitogen-activated protein kinase
- m-Apoe :
-
Mouse apolipoprotein E gene
- MCI:
-
Mild cognitive impairment
- Mertk:
-
C-Mer tyrosine kinase
- MGnD:
-
Microglia neurodegenerative phenotype
- MSA:
-
Multiple system atrophy
- mTOR:
-
Rapamycin
- NAMPs:
-
Neurodegeneration-associated molecular patterns
- NFTs:
-
Neurobibrillary tangles
- NHE6:
-
Sodium-hydrogen exchanger 6
- NLRs:
-
Inflammasome-forming nucleotide-binding oligomerization domain (nod)-like receptors
- NSAIDs:
-
Non-steroidal anti-inflammatory drugs
- OPCs:
-
Oligodendrocyte progenitor cells
- PAMPs:
-
Pathogen-associated molecular patterns
- PD:
-
Parkinson’s disease
- PDD:
-
Parkinson's disease with dementia
- PET:
-
Positron emission tomography
- PI3K:
-
Phosphatidylinositol 3-kinase
- PiB:
-
Pittsburgh compound B
- PIP:
-
Phosphoinositol phosphate
- PIP2:
-
Phosphoinositol biphosphate
- PLCϒ2:
-
Phospholipase CY2
- Poly-P:
-
Inorganic polyphosphate
- PRRs:
-
Pattern recognition receptors
- PSEN1:
-
Presenilin1
- PSEN2:
-
Presenilin 2
- RAGE:
-
Receptor for advanced glycation end-products
- RXR:
-
Retinoid X receptor
- sAPPβ:
-
Soluble APPβ
- SNP:
-
Single nucleotide polymorphism
- SYK:
-
Spleen tyrosine kinase
- SYNJ1:
-
Synaptojanin 1
- SynTOF:
-
Synaptometry time of flight
- T2DM:
-
Type 2 diabetes mellitus
- TBI:
-
Traumatic brain injury
- TIR:
-
Toll/IL-1 receptor
- TLRs:
-
Toll-like receptors
- TNF-α:
-
Tumor necrosis factor-α
- TREM2:
-
Receptor expressed on myeloid cells 2
- TRM:
-
Transiting-response microglia
- tyrobp:
-
Protein tyrosine kinase binding protein
- Usp-18:
-
Ubiquitin-specific protease 18
- WAM:
-
White-matter microglia
- α-syn:
-
Alpha-synuclein
References
Gauthier S, Rosa-Neto P, Morais JA, Webster C. World Alzheimer Report 2021: Journey through the diagnosis of dementia. London: Alzheimer’s Disease International; 2021.
Wong W. Economic burden of Alzheimer disease and managed care considerations. Am J Manag Care. 2020;26(8 Suppl):S177–83.
Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–5.
Haass C, et al. Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med. 2012;2(5):a006270.
Harris ME, et al. Direct evidence of oxidative injury produced by the Alzheimer’s beta-amyloid peptide (1–40) in cultured hippocampal neurons. Exp Neurol. 1995;131(2):193–202.
Reiss AB, et al. Amyloid toxicity in Alzheimer’s disease. Rev Neurosci. 2018;29(6):613–27.
Soria Lopez JA, Gonzalez HM, Leger GC. Alzheimer’s disease. Handb Clin Neurol. 2019;167:231–55.
Kosik KS. The molecular and cellular biology of tau. Brain Pathol. 1993;3(1):39–43.
Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med. 2012;2(7):a006247.
Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17(1):5–21.
Alonso AD, et al. Hyperphosphorylation of Tau Associates With Changes in Its Function Beyond Microtubule Stability. Front Cell Neurosci. 2018;12:338.
Saez-Atienzar S, Masliah E. Cellular senescence and Alzheimer disease: the egg and the chicken scenario. Nat Rev Neurosci. 2020;21(8):433–44.
Walton CC, et al. Senescence as an Amyloid Cascade: The Amyloid Senescence Hypothesis. Front Cell Neurosci. 2020;14:129.
Duyckaerts C, Clavaguera F, Potier MC. The prion-like propagation hypothesis in Alzheimer’s and Parkinson’s disease. Curr Opin Neurol. 2019;32(2):266–71.
Hampel H, et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain. 2018;141(7):1917–33.
Hampel H, et al. Revisiting the Cholinergic Hypothesis in Alzheimer’s Disease: Emerging Evidence from Translational and Clinical Research. J Prev Alzheimers Dis. 2019;6(1):2–15.
Haake A, et al. An update on the utility and safety of cholinesterase inhibitors for the treatment of Alzheimer’s disease. Expert Opin Drug Saf. 2020;19(2):147–57.
Sochocka M, et al. The Gut Microbiome Alterations and Inflammation-Driven Pathogenesis of Alzheimer’s Disease-a Critical Review. Mol Neurobiol. 2019;56(3):1841–51.
Arnsten AFT, et al. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimers Dement. 2021;17(1):115–24.
Paspalas CD, et al. The aged rhesus macaque manifests Braak stage III/IV Alzheimer’s-like pathology. Alzheimers Dement. 2018;14(5):680–91.
Reitz C, Rogaeva E, Beecham GW. Late-onset vs nonmendelian early-onset Alzheimer disease: A distinction without a difference? Neurol Genet. 2020;6(5):e512.
Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med. 2016;18(5):421–30.
Goate A, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349(6311):704–6.
Larner AJ. Presenilin-1 mutations in Alzheimer’s disease: an update on genotype-phenotype relationships. J Alzheimers Dis. 2013;37(4):653–9.
Sherrington R, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375(6534):754–60.
Levy-Lahad E, et al. A familial Alzheimer’s disease locus on chromosome 1. Science. 1995;269(5226):970–3.
Rogaev EI, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376(6543):775–8.
Wingo TS, et al. Autosomal recessive causes likely in early-onset Alzheimer disease. Arch Neurol. 2012;69(1):59–64.
Cacace R, Sleegers K, Van Broeckhoven C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 2016;12(6):733–48.
Ayodele T, et al. Early-Onset Alzheimer’s Disease: What Is Missing in Research? Curr Neurol Neurosci Rep. 2021;21(2):4.
Carmona S, Hardy J, Guerreiro R. The genetic landscape of Alzheimer disease. Handb Clin Neurol. 2018;148:395–408.
Troncoso JC, et al. Neuropathology of preclinical and clinical late-onset Alzheimer’s disease. Ann Neurol. 1998;43(5):673–6.
Li Z, et al. APOE2: protective mechanism and therapeutic implications for Alzheimer’s disease. Mol Neurodegener. 2020;15(1):63.
Yamazaki Y, et al. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15(9):501–18.
Sando SB, et al. APOE epsilon 4 lowers age at onset and is a high risk factor for Alzheimer’s disease; a case control study from central Norway. BMC Neurol. 2008;8:9.
Shi Y, Holtzman DM. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol. 2018;18(12):759–72.
Ahmad MH, Fatima M, Mondal AC. Influence of microglia and astrocyte activation in the neuroinflammatory pathogenesis of Alzheimer’s disease: Rational insights for the therapeutic approaches. J Clin Neurosci. 2019;59:6–11.
Fernandez CG, et al. The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Front Aging Neurosci. 2019;11:14.
Tzioras M, et al. Invited Review: APOE at the interface of inflammation, neurodegeneration and pathological protein spread in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2019;45(4):327–46.
Zhang Q, et al. Risk prediction of late-onset Alzheimer’s disease implies an oligogenic architecture. Nat Commun. 2020;11(1):4799.
Hinz FI, Geschwind DH. Molecular Genetics of Neurodegenerative Dementias. Cold Spring Harb Perspect Biol. 2017;9(4):a023705.
Kunkle BW, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414–30.
Robinson JL, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain. 2018;141(7):2181–93.
Kasap M, et al. Apolipoprotein E phylogeny and evolution. Cell Biochem Funct. 2008;26(1):43–50.
Barakat HA, St Clair RW. Characterization of plasma lipoproteins of grain- and cholesterol-fed White Carneau and Show Racer pigeons. J Lipid Res. 1985;26(10):1252–68.
Mello CV, Lovell PV. Avian genomics lends insights into endocrine function in birds. Gen Comp Endocrinol. 2018;256:123–9.
Liu J-Q, et al. Gain and loss events in the evolution of the apolipoprotein family in vertebrata. BMC Evol Biol. 2019;19(1):209.
Huebbe P, Rimbach G. Evolution of human apolipoprotein E (APOE) isoforms: Gene structure, protein function and interaction with dietary factors. Ageing Res Rev. 2017;37:146–61.
Dieckmann M, Dietrich MF, Herz J. Lipoprotein receptors–an evolutionarily ancient multifunctional receptor family. Biol Chem. 2010;391(11):1341–63.
McIntosh AM, et al. The apolipoprotein E (APOE) gene appears functionally monomorphic in chimpanzees (Pan troglodytes). PLoS ONE. 2012;7(10):e47760.
Seripa D, et al. The genetics of the human APOE polymorphism. Rejuvenation Res. 2011;14(5):491–500.
Finch CE, Stanford CB. Meat-adaptive genes and the evolution of slower aging in humans. Q Rev Biol. 2004;79(1):3–50.
Raichlen DA, Alexander GE. Exercise, APOE genotype, and the evolution of the human lifespan. Trends Neurosci. 2014;37(5):247–55.
Enard W. Functional primate genomics–leveraging the medical potential. J Mol Med (Berl). 2012;90(5):471–80.
Weisgraber KH. Apolipoprotein E: structure-function relationships. Adv Protein Chem. 1994;45:249–302.
Raffai RL, et al. Introduction of human apolipoprotein E4 “domain interaction” into mouse apolipoprotein E. Proc Natl Acad Sci U S A. 2001;98(20):11587–91.
van Exel E, et al. Effect of APOE epsilon4 allele on survival and fertility in an adverse environment. PLoS ONE. 2017;12(7):e0179497.
Trumble BC, et al. Apolipoprotein E4 is associated with improved cognitive function in Amazonian forager-horticulturalists with a high parasite burden. FASEB J. 2017;31(4):1508–15.
Fullerton SM, et al. Apolipoprotein E variation at the sequence haplotype level: implications for the origin and maintenance of a major human polymorphism. Am J Hum Genet. 2000;67(4):881–900.
Drenos F, Kirkwood TB. Selection on alleles affecting human longevity and late-life disease: the example of apolipoprotein E. PLoS ONE. 2010;5(4):e10022.
Das HK, et al. Isolation, characterization, and mapping to chromosome 19 of the human apolipoprotein E gene. J Biol Chem. 1985;260(10):6240–7.
Chen Y, et al. Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis. Neuron. 2021;109(2):205–21.
Frieden C, Garai K. Concerning the structure of apoE. Protein Sci. 2013;22(12):1820–5.
Weisgraber KH, Innerarity TL, Mahley RW. Abnormal lipoprotein receptor-binding activity of the human E apoprotein due to cysteine-arginine interchange at a single site. J Biol Chem. 1982;257(5):2518–21.
Maloney B, et al. Important differences between human and mouse APOE gene promoters: limitation of mouse APOE model in studying Alzheimer’s disease. J Neurochem. 2007;103(3):1237–57.
Fagan AM, et al. Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2002;9(3):305–18.
Liao F, et al. Murine versus human apolipoprotein E4: differential facilitation of and co-localization in cerebral amyloid angiopathy and amyloid plaques in APP transgenic mouse models. Acta Neuropathol Commun. 2015;3:70.
Tai LM, et al. EFAD transgenic mice as a human APOE relevant preclinical model of Alzheimer’s disease. J Lipid Res. 2017;58(9):1733–55.
Eisenberg DT, Kuzawa CW, Hayes MG. Worldwide allele frequencies of the human apolipoprotein E gene: climate, local adaptations, and evolutionary history. Am J Phys Anthropol. 2010;143(1):100–11.
Bekris LM, et al. Multiple SNPs within and surrounding the apolipoprotein E gene influence cerebrospinal fluid apolipoprotein E protein levels. J Alzheimers Dis. 2008;13(3):255–66.
Husain MA, Laurent B, Plourde M. APOE and Alzheimer’s Disease: From Lipid Transport to Physiopathology and Therapeutics. Front Neurosci. 2021;15:630502.
Abondio P, et al. The Genetic Variability of APOE in Different Human Populations and Its Implications for Longevity. Genes (Basel). 2019;10(3):222.
Kern S, et al. The distribution of apolipoprotein E genotype over the adult lifespan and in relation to country of birth. Am J Epidemiol. 2015;181(3):214–7.
Arboleda-Velasquez JF, et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019;25(11):1680–3.
Wardell MR, et al. Apolipoprotein E2-Christchurch (136 Arg––Ser). New variant of human apolipoprotein E in a patient with type III hyperlipoproteinemia. J Clin Invest. 1987;80(2):483–90.
Lalazar A, et al. Site-specific mutagenesis of human apolipoprotein E. Receptor binding activity of variants with single amino acid substitutions. J Biol Chem. 1988;263(8):3542–5.
Hernandez I, Gelpi E, Molina-Porcel L, Bernal S, Rodríguez-Santiago B, Dols-Icardo O, Ruiz A, Alcolea D, Boada M, Lleó A, Clarimón J. Heterozygous APOE Christchurch in familial Alzheimer's disease without mutations in other Mendelian genes. Neuropathol Appl Neurobiol. 2021;47(4):579-82. https://doi.org/10.1111/nan.12670.
Liu CC, et al. APOE3-Jacksonville (V236E) variant reduces self-aggregation and risk of dementia. Sci Transl Med. 2021;13(613):eabc9375.
Kockx M, Traini M, Kritharides L. Cell-specific production, secretion, and function of apolipoprotein E. J Mol Med (Berl). 2018;96(5):361–71.
Paik YK, et al. Identification and characterization of transcriptional regulatory regions associated with expression of the human apolipoprotein E gene. J Biol Chem. 1988;263(26):13340–9.
Boyles JK, et al. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest. 1985;76(4):1501–13.
Morikawa M, et al. Production and characterization of astrocyte-derived human apolipoprotein E isoforms from immortalized astrocytes and their interactions with amyloid-beta. Neurobiol Dis. 2005;19(1–2):66–76.
Bruinsma IB, et al. Apolipoprotein E protects cultured pericytes and astrocytes from D-Abeta(1–40)-mediated cell death. Brain Res. 2010;1315:169–80.
Xu Q, et al. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006;26(19):4985–94.
Buttini M, et al. Cellular source of apolipoprotein E4 determines neuronal susceptibility to excitotoxic injury in transgenic mice. Am J Pathol. 2010;177(2):563–9.
Kockx M, et al. Secretion of apolipoprotein E from macrophages occurs via a protein kinase A and calcium-dependent pathway along the microtubule network. Circ Res. 2007;101(6):607–16.
Frenkel-Pinter M, et al. Interplay between protein glycosylation pathways in Alzheimer’s disease. Sci Adv. 2017;3(9):e1601576.
Boix CP, et al. Amyloid precursor protein glycosylation is altered in the brain of patients with Alzheimer’s disease. Alzheimers Res Ther. 2020;12(1):96.
Haukedal H, Freude KK. Implications of Glycosylation in Alzheimer’s Disease. Front Neurosci. 2020;14:625348.
Flowers SA, et al. O-glycosylation on cerebrospinal fluid and plasma apolipoprotein E differs in the lipid-binding domain. Glycobiology. 2020;30(2):74–85.
Schulman IG. Liver X receptors link lipid metabolism and inflammation. FEBS Lett. 2017;591(19):2978–91.
Carbó JM, León TE, Font-Díaz J, De la Rosa JV, Castrillo A, Picard FR, Staudenraus D, Huber M, Cedó L, Escolà-Gil JC, Campos L, Bakiri L, Wagner EF, Caelles C, Stratmann T, Van Ginderachter JA, Valledor AF. Pharmacologic Activation of LXR Alters the Expression Profile of Tumor-Associated Macrophages and the Abundance of Regulatory T Cells in the Tumor Microenvironment. Cancer Res. 2021;81(4):968-85. https://doi.org/10.1158/0008-5472.CAN-19-3360.
Wouters E, et al. Liver X Receptor Alpha Is Important in Maintaining Blood-Brain Barrier Function. Front Immunol. 2019;10:1811.
Spagnuolo MS, et al. Brain-derived neurotrophic factor modulates cholesterol homeostasis and Apolipoprotein E synthesis in human cell models of astrocytes and neurons. J Cell Physiol. 2018;233(9):6925–43.
Zhao W, et al. Axl receptor tyrosine kinase is a regulator of apolipoprotein E. Mol Brain. 2020;13(1):66.
Hong C, Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov. 2014;13(6):433–44.
Wood H. Retinoid X receptor mediates brain clean-up after stroke. Nat Rev Neurol. 2020;16(3):128–9.
Schierle S, Merk D. Therapeutic modulation of retinoid X receptors - SAR and therapeutic potential of RXR ligands and recent patents. Expert Opin Ther Pat. 2019;29(8):605–21.
Fitz NF, et al. Therapeutic targeting of nuclear receptors, liver X and retinoid X receptors, for Alzheimer’s disease. Br J Pharmacol. 2019;176(18):3599–610.
Koldamova R, Lefterov I. Role of LXR and ABCA1 in the pathogenesis of Alzheimer’s disease - implications for a new therapeutic approach. Curr Alzheimer Res. 2007;4(2):171–8.
Jacobo-Albavera L, et al. The Role of the ATP-Binding Cassette A1 (ABCA1) in Human Disease. Int J Mol Sci. 2021;22(4):1593.
Fitz NF, et al. Abca1 deficiency affects Alzheimer’s disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J Neurosci. 2012;32(38):13125–36.
Rawat V, et al. ApoE4 Alters ABCA1 Membrane Trafficking in Astrocytes. J Neurosci. 2019;39(48):9611–22.
Mahley RW. Central Nervous System Lipoproteins: ApoE and Regulation of Cholesterol Metabolism. Arterioscler Thromb Vasc Biol. 2016;36(7):1305–15.
Nieweg K, Schaller H, Pfrieger FW. Marked differences in cholesterol synthesis between neurons and glial cells from postnatal rats. J Neurochem. 2009;109(1):125–34.
Kim J, et al. Apolipoprotein E in synaptic plasticity and Alzheimer’s disease: potential cellular and molecular mechanisms. Mol Cells. 2014;37(11):767–76.
Lane-Donovan C, et al. Genetic Restoration of Plasma ApoE Improves Cognition and Partially Restores Synaptic Defects in ApoE-Deficient Mice. J Neurosci. 2016;36(39):10141–50.
Tensaouti Y, Yu TS, Kernie SG. Apolipoprotein E regulates the maturation of injury-induced adult-born hippocampal neurons following traumatic brain injury. PLoS ONE. 2020;15(3):e0229240.
Vance JE, Hayashi H. Formation and function of apolipoprotein E-containing lipoproteins in the nervous system. Biochim Biophys Acta. 2010;1801(8):806–18.
Karasinska JM, et al. ABCA1 influences neuroinflammation and neuronal death. Neurobiol Dis. 2013;54:445–55.
Tarr PT, Edwards PA. ABCG1 and ABCG4 are coexpressed in neurons and astrocytes of the CNS and regulate cholesterol homeostasis through SREBP-2. J Lipid Res. 2008;49(1):169–82.
Hubin E, et al. Apolipoprotein E associated with reconstituted high-density lipoprotein-like particles is protected from aggregation. FEBS Lett. 2019;593(11):1144–53.
Morrow JA, et al. Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry. 2000;39(38):11657–66.
Lanfranco MF, Ng CA, Rebeck GW. ApoE Lipidation as a Therapeutic Target in Alzheimer’s Disease. Int J Mol Sci. 2020;21(17):6336.
Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(3):a006312.
May P, et al. The LDL receptor-related protein (LRP) family: an old family of proteins with new physiological functions. Ann Med. 2007;39(3):219–28.
Zhao N, et al. Apolipoprotein E, Receptors, and Modulation of Alzheimer’s Disease. Biol Psychiatry. 2018;83(4):347–57.
Ruiz J, et al. The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J Lipid Res. 2005;46(8):1721–31.
Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10(5):333–44.
Dlugosz P, Nimpf J. The Reelin Receptors Apolipoprotein E receptor 2 (ApoER2) and VLDL Receptor. Int J Mol Sci. 2018;19(10):3090.
Jossin Y. Reelin Functions, Mechanisms of Action and Signaling Pathways During Brain Development and Maturation. Biomolecules. 2020;10(6):964.
D’Arcangelo G, et al. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24(2):471–9.
Reddy SS, et al. Similarities and differences in structure, expression, and functions of VLDLR and ApoER2. Mol Neurodegener. 2011;6:30.
Leon M, et al. Therapeutic approach targeting apolipoprotein E binding region and low-density lipoprotein receptor for Alzheimer’s disease. Neuroimmunol Neuroinflammation. 2018;5:30.
Nelson TJ, Sen A. Apolipoprotein E particle size is increased in Alzheimer’s disease. Alzheimers Dement (Amst). 2019;11:10–8.
Garcia AR, et al. APOE4 is associated with elevated blood lipids and lower levels of innate immune biomarkers in a tropical Amerindian subsistence population. Elife. 2021;10:e68231.
Prakashchand DD, Mondal J. Conformational Reorganization of Apolipoprotein E Triggered by Phospholipid Assembly. J Phys Chem B. 2021;125(20):5285–95.
Garai K, Baban B, Frieden C. Dissociation of apolipoprotein E oligomers to monomer is required for high-affinity binding to phospholipid vesicles. Biochemistry. 2011;50(13):2550–8.
Hsieh YH, Chou CY. Structural and functional characterization of human apolipoprotein E 72–166 peptides in both aqueous and lipid environments. J Biomed Sci. 2011;18:4.
Heeren J, et al. Impaired recycling of apolipoprotein E4 is associated with intracellular cholesterol accumulation. J Biol Chem. 2004;279(53):55483–92.
Henry N, et al. Lipidated apolipoprotein E4 structure and its receptor binding mechanism determined by a combined cross-linking coupled to mass spectrometry and molecular dynamics approach. PLoS Comput Biol. 2018;14(6):e1006165.
Cooper JM, et al. Regulation of tau internalization, degradation, and seeding by LRP1 reveals multiple pathways for tau catabolism. J Biol Chem. 2021;296:100715.
Khalil YA, et al. APOE gene variants in primary dyslipidemia. Atherosclerosis. 2021;328:11–22.
Zhao XN, et al. Association between apolipoprotein gene polymorphisms and hyperlipidemia: a meta-analysis. BMC Genom Data. 2021;22(1):14.
Miranda AM, et al. Effects of APOE4 allelic dosage on lipidomic signatures in the entorhinal cortex of aged mice. Transl Psychiatry. 2022;12(1):129.
Lefterov I, et al. APOE2 orchestrated differences in transcriptomic and lipidomic profiles of postmortem AD brain. Alzheimers Res Ther. 2019;11(1):113.
Fitz NF, et al. Phospholipids of APOE lipoproteins activate microglia in an isoform-specific manner in preclinical models of Alzheimer’s disease. Nat Commun. 2021;12(1):3416.
Dong LM, Weisgraber KH. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J Biol Chem. 1996;271(32):19053–7.
Nguyen D, et al. Molecular basis for the differences in lipid and lipoprotein binding properties of human apolipoproteins E3 and E4. Biochemistry. 2010;49(51):10881–9.
Li H, et al. Molecular mechanisms responsible for the differential effects of apoE3 and apoE4 on plasma lipoprotein-cholesterol levels. Arterioscler Thromb Vasc Biol. 2013;33(4):687–93.
Frieden C, Wang H, Ho CMW. A mechanism for lipid binding to apoE and the role of intrinsically disordered regions coupled to domain-domain interactions. Proc Natl Acad Sci U S A. 2017;114(24):6292–7.
Shuvaev VV, Laffont I, Siest G. Kinetics of apolipoprotein E isoforms-binding to the major glycosaminoglycans of the extracellular matrix. FEBS Lett. 1999;459(3):353–7.
Yamauchi Y, et al. Role of the N- and C-terminal domains in binding of apolipoprotein E isoforms to heparan sulfate and dermatan sulfate: a surface plasmon resonance study. Biochemistry. 2008;47(25):6702–10.
Futamura M, et al. Two-step mechanism of binding of apolipoprotein E to heparin: implications for the kinetics of apolipoprotein E-heparan sulfate proteoglycan complex formation on cell surfaces. J Biol Chem. 2005;280(7):5414–22.
Ji ZS, et al. Role of heparan sulfate proteoglycans in the binding and uptake of apolipoprotein E-enriched remnant lipoproteins by cultured cells. J Biol Chem. 1993;268(14):10160–7.
Saito H, et al. Characterization of the heparin binding sites in human apolipoprotein E. J Biol Chem. 2003;278(17):14782–7.
Mahley RW, Ji ZS. Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res. 1999;40(1):1–16.
Alavi Naini SM, Soussi-Yanicostas N. Heparan Sulfate as a Therapeutic Target in Tauopathies: Insights From Zebrafish. Front Cell Dev Biol. 2018;6:163.
Sandwall E, et al. Heparan sulfate mediates amyloid-beta internalization and cytotoxicity. Glycobiology. 2010;20(5):533–41.
Zhang X, Wang B, Li JP. Implications of heparan sulfate and heparanase in neuroinflammation. Matrix Biol. 2014;35:174–81.
Brunden KR, et al. pH-dependent binding of synthetic beta-amyloid peptides to glycosaminoglycans. J Neurochem. 1993;61(6):2147–54.
Kanekiyo T, et al. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J Neurosci. 2011;31(5):1644–51.
Liu CC, et al. Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-beta clearance and aggregation in Alzheimer’s disease. Sci Transl Med. 2016;8(332):332ra44.
Zhang Y, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929–47.
Motoi Y, et al. Neuronal localization of a novel mosaic apolipoprotein E receptor, LR11, in rat and human brain. Brain Res. 1999;833(2):209–15.
Yajima R, et al. ApoE-isoform-dependent cellular uptake of amyloid-beta is mediated by lipoprotein receptor LR11/SorLA. Biochem Biophys Res Commun. 2015;456(1):482–8.
Zhao S, et al. Selective expression of LDLR and VLDLR in myelinating oligodendrocytes. Dev Dyn. 2007;236(9):2708–12.
Rapp A, Gmeiner B, Huttinger M. Implication of apoE isoforms in cholesterol metabolism by primary rat hippocampal neurons and astrocytes. Biochimie. 2006;88(5):473–83.
May P, et al. Neuronal LRP1 functionally associates with postsynaptic proteins and is required for normal motor function in mice. Mol Cell Biol. 2004;24(20):8872–83.
Rebeck GW, et al. Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron. 1993;11(4):575–80.
Wicher G, et al. Low density lipoprotein receptor-related protein-2/megalin is expressed in oligodendrocytes in the mouse spinal cord white matter. J Neurosci Res. 2006;83(5):864–73.
Bento-Abreu A, et al. Megalin is a receptor for albumin in astrocytes and is required for the synthesis of the neurotrophic factor oleic acid. J Neurochem. 2008;106(3):1149–59.
Alvira-Botero X, et al. Megalin interacts with APP and the intracellular adapter protein FE65 in neurons. Mol Cell Neurosci. 2010;45(3):306–15.
Zhang H, et al. A Role of Low-Density Lipoprotein Receptor-Related Protein 4 (LRP4) in Astrocytic Abeta Clearance. J Neurosci. 2020;40(28):5347–61.
Mosca TJ, et al. Presynaptic LRP4 promotes synapse number and function of excitatory CNS neurons. Elife. 2017;6:e27347.
Halleskog C, et al. WNT signaling in activated microglia is proinflammatory. Glia. 2011;59(1):119–31.
Guerit S, et al. Astrocyte-derived Wnt growth factors are required for endothelial blood-brain barrier maintenance. Prog Neurobiol. 2021;199:101937.
Huang Y, et al. Lrp5/6 are required for cerebellar development and for suppressing TH expression in Purkinje cells via beta-catenin. Mol Brain. 2016;9:7.
Chow HM, et al. Low-Density Lipoprotein Receptor-Related Protein 6 Cell Surface Availability Regulates Fuel Metabolism in Astrocytes. Adv Sci (Weinh). 2021;8(16):e2004993.
Liu CC, et al. Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer’s disease. Neuron. 2014;84(1):63–77.
Ulland TK, et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell. 2017;170(4):649-663 e13.
Ito H, Hamerman JA. TREM-2, triggering receptor expressed on myeloid cell-2, negatively regulates TLR responses in dendritic cells. Eur J Immunol. 2012;42(1):176–85.
Rosciszewski G, et al. Toll-Like Receptor 4 (TLR4) and Triggering Receptor Expressed on Myeloid Cells-2 (TREM-2) Activation Balance Astrocyte Polarization into a Proinflammatory Phenotype. Mol Neurobiol. 2018;55(5):3875–88.
Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173(6):3916–24.
Tang SC, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104(34):13798–803.
Yamauchi K, et al. Characterization of apolipoprotein E-containing lipoproteins in cerebrospinal fluid: effect of phenotype on the distribution of apolipoprotein E. Clin Chem. 1999;45(9):1431–8.
Mahley RW, Huang Y, Rall SC Jr. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia). Questions, quandaries, and paradoxes. J Lipid Res. 1999;40(11):1933–49.
Lin YT, et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron. 2018;98(6):1141-1154 e7.
Yin J, et al. Effect of ApoE isoforms on mitochondria in Alzheimer disease. Neurology. 2020;94(23):e2404–11.
Area-Gomez E, et al. APOE4 is Associated with Differential Regional Vulnerability to Bioenergetic Deficits in Aged APOE Mice. Sci Rep. 2020;10(1):4277.
Cremers CM, et al. Polyphosphate: A Conserved Modifier of Amyloidogenic Processes. Mol Cell. 2016;63(5):768–80.
Borden EA, et al. Is there a link between inorganic polyphosphate (polyP), mitochondria, and neurodegeneration? Pharmacol Res. 2021;163:105211.
Smith SA, et al. Novel Polyphosphate-Binding Proteins in Human Plasma and Platelet Releasates. Blood. 2011;118(21):986–986.
Huang YA, et al. Differential Signaling Mediated by ApoE2, ApoE3, and ApoE4 in Human Neurons Parallels Alzheimer’s Disease Risk. J Neurosci. 2019;39(37):7408–27.
Zhao J, et al. APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids. Nat Commun. 2020;11(1):5540.
Vitali C, Wellington CL, Calabresi L. HDL and cholesterol handling in the brain. Cardiovasc Res. 2014;103(3):405–13.
Hottman DA, et al. HDL and cognition in neurodegenerative disorders. Neurobiol Dis. 2014;72 Pt A:22–36.
Mahley RW. Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J Mol Med (Berl). 2016;94(7):739–46.
Riddell DR, et al. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J Neurosci. 2008;28(45):11445–53.
Sullivan PM, et al. Reduced levels of human apoE4 protein in an animal model of cognitive impairment. Neurobiol Aging. 2011;32(5):791–801.
Ramaswamy G, et al. Effect of domain interaction on apolipoprotein E levels in mouse brain. J Neurosci. 2005;25(46):10658–63.
Sullivan PM, et al. Marked regional differences of brain human apolipoprotein E expression in targeted replacement mice. Neuroscience. 2004;124(4):725–33.
Giannisis A, Patra K, Edlund AK, Nieto LA, Benedicto-Gras J, Moussaud S, de la Rosa A, Twohig D, Bengtsson T, Fu Y, Bu G, Bial G, Foquet L, Hammarstedt C, Strom S, Kannisto K, Raber J, Ellis E, Nielsen HM. Brain integrity is altered by hepatic APOE ε4 in humanized-liver mice. Mol Psychiatry. 2022. https://doi.org/10.1038/s41380-022-01548-0.
Ulrich JD, et al. In vivo measurement of apolipoprotein E from the brain interstitial fluid using microdialysis. Mol Neurodegener. 2013;8:13.
Linton MF, et al. Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J Clin Invest. 1991;88(1):270–81.
Getz GS, Reardon CA. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J Lipid Res. 2009;50(Suppl):S156–61.
Huynh TV, et al. Lack of hepatic apoE does not influence early Abeta deposition: observations from a new APOE knock-in model. Mol Neurodegener. 2019;14(1):37.
Wahrle SE, et al. Apolipoprotein E levels in cerebrospinal fluid and the effects of ABCA1 polymorphisms. Mol Neurodegener. 2007;2:7.
Martinez-Morillo E, et al. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer’s disease patients and controls. Acta Neuropathol. 2014;127(5):633–43.
Minta K, et al. Quantification of total apolipoprotein E and its isoforms in cerebrospinal fluid from patients with neurodegenerative diseases. Alzheimers Res Ther. 2020;12(1):19.
Baker-Nigh AT, et al. Human Central Nervous System (CNS) ApoE Isoforms Are Increased by Age, Differentially Altered by Amyloidosis, and Relative Amounts Reversed in the CNS Compared with Plasma. J Biol Chem. 2016;291(53):27204–18.
Simon R, et al. Total ApoE and ApoE4 isoform assays in an Alzheimer’s disease case-control study by targeted mass spectrometry (n=669): a pilot assay for methionine-containing proteotypic peptides. Mol Cell Proteomics. 2012;11(11):1389–403.
Fernandes A, et al. C-reactive protein as a predictor of mild cognitive impairment conversion into Alzheimer’s disease dementia. Exp Gerontol. 2020;138:111004.
Berger M, et al. The INTUIT Study: Investigating Neuroinflammation Underlying Postoperative Cognitive Dysfunction. J Am Geriatr Soc. 2019;67(4):794–8.
Cruchaga C, et al. Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer’s disease. Hum Mol Genet. 2012;21(20):4558–71.
Nielsen HM, et al. Peripheral apoE isoform levels in cognitively normal APOE epsilon3/epsilon4 individuals are associated with regional gray matter volume and cerebral glucose metabolism. Alzheimers Res Ther. 2017;9(1):5.
Edlund AK, et al. Plasma Apolipoprotein E3 and Glucose Levels Are Associated in APOE varepsilon3/varepsilon4 Carriers. J Alzheimers Dis. 2021;81(1):339–54.
Rasmussen KL, et al. Plasma apolipoprotein E levels and risk of dementia: A Mendelian randomization study of 106,562 individuals. Alzheimers Dement. 2018;14(1):71–80.
Rasmussen KL, et al. APOE and dementia - resequencing and genotyping in 105,597 individuals. Alzheimers Dement. 2020;16(12):1624–37.
Patra K, et al. Plasma Apolipoprotein E Monomer and Dimer Profile and Relevance to Alzheimer’s Disease. J Alzheimers Dis. 2019;71(4):1217–31.
Wildsmith KR, et al. In vivo human apolipoprotein E isoform fractional turnover rates in the CNS. PLoS ONE. 2012;7(6):e38013.
DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139(Suppl 2):136–53.
Yong HYF, et al. The benefits of neuroinflammation for the repair of the injured central nervous system. Cell Mol Immunol. 2019;16(6):540–6.
Schimmel SJ, Acosta S, Lozano D. Neuroinflammation in traumatic brain injury: A chronic response to an acute injury. Brain Circ. 2017;3(3):135–42.
Jayaraj RL, et al. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation. 2019;16(1):142.
Jones JD. Potential of Glial Cell Modulators in the Management of Substance Use Disorders. CNS Drugs. 2020;34(7):697–722.
Perkins AE, et al. Assessment of neuroinflammation in the aging hippocampus using large-molecule microdialysis: Sex differences and role of purinergic receptors. Brain Behav Immun. 2021;91:546–55.
Guzman-Martinez L, et al. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front Pharmacol. 2019;10:1008.
Shabab T, et al. Neuroinflammation pathways: a general review. Int J Neurosci. 2017;127(7):624–33.
Wang WY, et al. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med. 2015;3(10):136.
Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. 2020;9(1):42.
Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–8.
Hickman SE, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16(12):1896–905.
Diaz-Aparicio I, et al. Microglia Actively Remodel Adult Hippocampal Neurogenesis through the Phagocytosis Secretome. J Neurosci. 2020;40(7):1453–82.
Paolicelli RC, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–8.
Bar E, Barak B. Microglia roles in synaptic plasticity and myelination in homeostatic conditions and neurodevelopmental disorders. Glia. 2019;67(11):2125–41.
Gomez-Nicola D, Perry VH. Microglial dynamics and role in the healthy and diseased brain: a paradigm of functional plasticity. Neuroscientist. 2015;21(2):169–84.
Zanier ER, et al. Shape descriptors of the “never resting” microglia in three different acute brain injury models in mice. Intensive Care Med Exp. 2015;3(1):39.
Tan YL, Yuan Y, Tian L. Microglial regional heterogeneity and its role in the brain. Mol Psychiatry. 2020;25(2):351–67.
Garcia-Revilla J, et al. Reformulating Pro-Oxidant Microglia in Neurodegeneration. J Clin Med. 2019;8(10):1719.
Ronaldson PT, Davis TP. Regulation of blood-brain barrier integrity by microglia in health and disease: A therapeutic opportunity. J Cereb Blood Flow Metab. 2020;40(1_suppl):S6–24.
Smith JA, et al. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. 2012;87(1):10–20.
Cudaback E, et al. Apolipoprotein E isoform-dependent microglia migration. FASEB J. 2011;25(6):2082–91.
Dumanis SB, et al. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci. 2009;29(48):15317–22.
Zhu Y, et al. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia. 2012;60(4):559–69.
Ulrich JD, et al. ApoE facilitates the microglial response to amyloid plaque pathology. J Exp Med. 2018;215(4):1047–58.
Lanfranco MF, et al. Expression and secretion of apoE isoforms in astrocytes and microglia during inflammation. Glia. 2021;69(6):1478–93.
Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. 2012;12(9):623–35.
Laskowitz DT, et al. Altered immune responses in apolipoprotein E-deficient mice. J Lipid Res. 2000;41(4):613–20.
Tenger C, Zhou X. Apolipoprotein E modulates immune activation by acting on the antigen-presenting cell. Immunology. 2003;109(3):392–7.
Terkeltaub RA, et al. Apolipoprotein (apo) E inhibits the capacity of monosodium urate crystals to stimulate neutrophils. Characterization of intraarticular apo E and demonstration of apo E binding to urate crystals in vivo. J Clin Invest. 1991;87(1):20–6.
van den Elzen P, et al. Apolipoprotein-mediated pathways of lipid antigen presentation. Nature. 2005;437(7060):906–10.
Winkler B, et al. Brain inflammation triggers macrophage invasion across the blood-brain barrier in Drosophila during pupal stages. Sci Adv. 2021;7(44):eabh0050.
Takata F, et al. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front Cell Neurosci. 2021;15:661838.
Gafencu AV, et al. Inflammatory signaling pathways regulating ApoE gene expression in macrophages. J Biol Chem. 2007;282(30):21776–85.
Gate D, et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature. 2020;577(7790):399–404.
Butovsky O, Weiner HL. Microglial signatures and their role in health and disease. Nat Rev Neurosci. 2018;19(10):622–35.
Lee E, et al. Distinct Features of Brain-Resident Macrophages: Microglia and Non-Parenchymal Brain Macrophages. Mol Cells. 2021;44(5):281–91.
Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2018;18(4):225–42.
Schetters STT, et al. Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front Immunol. 2017;8:1905.
Hopperton KE, et al. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: a systematic review. Mol Psychiatry. 2018;23(2):177–98.
Mathys H, et al. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017;21(2):366–80.
James LM, et al. The effects of human leukocyte antigen DRB1*13 and apolipoprotein E on age-related variability of synchronous neural interactions in healthy women. EBioMedicine. 2018;35:288–94.
Kigerl KA, et al. Pattern recognition receptors and central nervous system repair. Exp Neurol. 2014;258:5–16.
Ulland TK, Colonna M. TREM2 - a key player in microglial biology and Alzheimer disease. Nat Rev Neurol. 2018;14(11):667–75.
Deczkowska A, et al. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell. 2018;173(5):1073–81.
Deczkowska A, Amit I, Schwartz M. Microglial immune checkpoint mechanisms. Nat Neurosci. 2018;21(6):779–86.
Stratoulias V, et al. Microglial subtypes: diversity within the microglial community. EMBO J. 2019;38(17):e101997.
Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19(8):987–91.
Gratuze M, Leyns CEG, Holtzman DM. New insights into the role of TREM2 in Alzheimer’s disease. Mol Neurodegener. 2018;13(1):66.
Celarain N, et al. TREM2 upregulation correlates with 5-hydroxymethycytosine enrichment in Alzheimer’s disease hippocampus. Clin Epigenetics. 2016;8:37.
Gosselin D, et al. An environment-dependent transcriptional network specifies human microglia identity. Science. 2017;356(6344):eaal3222.
Guerreiro R, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–27.
Jonsson T, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368(2):107–16.
Boza-Serrano A, et al. Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. 2019;138(2):251–73.
Jay TR, von Saucken VE, Landreth GE. TREM2 in Neurodegenerative Diseases. Mol Neurodegener. 2017;12(1):56.
Keren-Shaul H, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell. 2017;169(7):1276-1290 e17.
Audrain M, Haure-Mirande JV, Mleczko J, Wang M, Griffin JK, St George-Hyslop PH, Fraser P, Zhang B, Gandy S, Ehrlich ME. Reactive or transgenic increase in microglial TYROBP reveals a TREM2-independent TYROBP-APOE link in wild-type and Alzheimer's-related mice. Alzheimers Dement. 2021;17(2):149-63. https://doi.org/10.1002/alz.12256.
Krasemann S, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity. 2017;47(3):566-581 e9.
Zhong L, et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med. 2017;214(3):597–607.
Yuan P, et al. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron. 2016;90(4):724–39.
Bemiller SM, et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol Neurodegener. 2017;12(1):74.
Leyns CEG, et al. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci U S A. 2017;114(43):11524–9.
Parhizkar S, et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci. 2019;22(2):191–204.
Leyns CEG, et al. TREM2 function impedes tau seeding in neuritic plaques. Nat Neurosci. 2019;22(8):1217–22.
Andreone BJ, et al. Alzheimer’s-associated PLCgamma2 is a signaling node required for both TREM2 function and the inflammatory response in human microglia. Nat Neurosci. 2020;23(8):927–38.
Nugent AA, et al. TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron. 2020;105(5):837-854 e9.
McQuade A, et al. Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat Commun. 2020;11(1):5370.
Magno L, et al. Alzheimer’s disease phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res Ther. 2019;11(1):16.
Zhu Y, Kodvawala A, Hui DY. Apolipoprotein E inhibits toll-like receptor (TLR)-3- and TLR-4-mediated macrophage activation through distinct mechanisms. Biochem J. 2010;428(1):47–54.
Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461.
Shen X, et al. Caspases orchestrate microglia instrumental functions. Prog Neurobiol. 2018;171:50–71.
Zhou Y, et al. TLR4 Targeting as a Promising Therapeutic Strategy for Alzheimer Disease Treatment. Front Neurosci. 2020;14:602508.
Gordon EM, et al. Apolipoprotein E is a concentration-dependent pulmonary danger signal that activates the NLRP3 inflammasome and IL-1beta secretion by bronchoalveolar fluid macrophages from asthmatic subjects. J Allergy Clin Immunol. 2019;144(2):426-441 e3.
Wong MY, et al. 25-Hydroxycholesterol amplifies microglial IL-1beta production in an apoE isoform-dependent manner. J Neuroinflammation. 2020;17(1):192.
Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science. 2001;293(5528):306–11.
Fourgeaud L, et al. TAM receptors regulate multiple features of microglial physiology. Nature. 2016;532(7598):240–4.
Zagorska A, et al. Diversification of TAM receptor tyrosine kinase function. Nat Immunol. 2014;15(10):920–8.
Huang Y, et al. Microglia use TAM receptors to detect and engulf amyloid beta plaques. Nat Immunol. 2021;22(5):586–94.
Huynh TV, et al. Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of beta-amyloidosis. Neuron. 2017;96(5):1013-1023 e4.
Ma J, et al. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature. 1994;372(6501):92–4.
Auderset L, Cullen CL, Young KM. Low Density Lipoprotein-Receptor Related Protein 1 Is Differentially Expressed by Neuronal and Glial Populations in the Developing and Mature Mouse Central Nervous System. PLoS ONE. 2016;11(6):e0155878.
Yang L, et al. LRP1 modulates the microglial immune response via regulation of JNK and NF-kappaB signaling pathways. J Neuroinflammation. 2016;13(1):304.
Chuang TY, et al. LRP1 expression in microglia is protective during CNS autoimmunity. Acta Neuropathol Commun. 2016;4(1):68.
Brifault C, et al. LRP1 deficiency in microglia blocks neuro-inflammation in the spinal dorsal horn and neuropathic pain processing. Glia. 2019;67(6):1210–24.
Pocivavsek A, et al. Microglial low-density lipoprotein receptor-related protein 1 modulates c-Jun N-terminal kinase activation. J Neuroimmunol. 2009;214(1–2):25–32.
He Y, et al. Silencing of LRP1 Exacerbates Inflammatory Response Via TLR4/NF-kappaB/MAPKs Signaling Pathways in APP/PS1 Transgenic Mice. Mol Neurobiol. 2020;57(9):3727–43.
Schafer DP, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705.
Crehan H, Hardy J, Pocock J. Microglia, Alzheimer’s disease, and complement. Int J Alzheimers Dis. 2012;2012:983640.
Yin C, et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med. 2019;25(3):496–506.
Masuda T, et al. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020;30(5):1271–81.
Bachiller S, et al. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front Cell Neurosci. 2018;12:488.
De Schepper S, Crowley G, Hong S. Understanding microglial diversity and implications for neuronal function in health and disease. Dev Neurobiol. 2021;81(5):507-23. https://doi.org/10.1002/dneu.22777.
Masuda T, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019;566(7744):388–92.
Menassa DA, Gomez-Nicola D. Microglial Dynamics During Human Brain Development. Front Immunol. 2018;9:1014.
Davis BM, et al. Characterizing microglia activation: a spatial statistics approach to maximize information extraction. Sci Rep. 2017;7(1):1576.
Dubbelaar ML, et al. The Kaleidoscope of Microglial Phenotypes. Front Immunol. 2018;9:1753.
Sala Frigerio C, et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Abeta Plaques. Cell Rep. 2019;27(4):1293-1306 e6.
Mito R, et al. Fibre-specific white matter reductions in Alzheimer’s disease and mild cognitive impairment. Brain. 2018;141(3):888–902.
Ji F, et al. White matter microstructural abnormalities and default network degeneration are associated with early memory deficit in Alzheimer’s disease continuum. Sci Rep. 2019;9(1):4749.
Safaiyan S, et al. White matter aging drives microglial diversity. Neuron. 2021;109(7):1100-1117.e10.
Heise V, et al. The APOE varepsilon4 allele modulates brain white matter integrity in healthy adults. Mol Psychiatry. 2011;16(9):908–16.
Mirza SS, et al. APOE epsilon4, white matter hyperintensities, and cognition in Alzheimer and Lewy body dementia. Neurology. 2019;93(19):e1807–19.
Sudre CH, et al. APOE epsilon4 status is associated with white matter hyperintensities volume accumulation rate independent of AD diagnosis. Neurobiol Aging. 2017;53:67–75.
Goldmann T, et al. USP18 lack in microglia causes destructive interferonopathy of the mouse brain. EMBO J. 2015;34(12):1612–29.
Auderset L, et al. Low-Density Lipoprotein Receptor-Related Protein 1 (LRP1) Is a Negative Regulator of Oligodendrocyte Progenitor Cell Differentiation in the Adult Mouse Brain. Front Cell Dev Biol. 2020;8:564351.
Hase Y, et al. White matter degeneration in vascular and other ageing-related dementias. J Neurochem. 2018;144(5):617–33.
Peng J, et al. LRP1 activation attenuates white matter injury by modulating microglial polarization through Shc1/PI3K/Akt pathway after subarachnoid hemorrhage in rats. Redox Biol. 2019;21: 101121.
Hammond TR, et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity. 2019;50(1):253-271 e6.
Marschallinger J, et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci. 2020;23(2):194–208.
Pitas RE, et al. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B, E(LDL) receptors in the brain. J Biol Chem. 1987;262(29):14352–60.
Zhang J, Liu Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell. 2015;6(4):254–64.
Gong JS, et al. Apolipoprotein E (ApoE) isoform-dependent lipid release from astrocytes prepared from human ApoE3 and ApoE4 knock-in mice. J Biol Chem. 2002;277(33):29919–26.
Zhao J, et al. APOE epsilon4/epsilon4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum Mol Genet. 2017;26(14):2690–700.
Tcw J, et al. Cholesterol and matrisome pathways dysregulated in astrocytes and microglia. Cell. 2022;185(13):2213-2233 e25.
Sienski G, et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci Transl Med. 2021;13(583):eaaz4564.
Ioannou MS, et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell. 2019;177(6):1522-1535 e14.
Qi G, et al. ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism. Cell Rep. 2021;34(1):108572.
Liu L, et al. The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab. 2017;26(5):719-737 e6.
Schonfeld P, Reiser G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J Cereb Blood Flow Metab. 2013;33(10):1493–9.
Schonfeld P, Reiser G. Brain energy metabolism spurns fatty acids as fuel due to their inherent mitotoxicity and potential capacity to unleash neurodegeneration. Neurochem Int. 2017;109:68–77.
Nguyen TB, et al. DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev Cell. 2017;42(1):9-21 e5.
Sultana R, Perluigi M, Butterfield DA. Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer disease brain. Free Radic Biol Med. 2013;62:157–69.
Bailey AP, et al. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell. 2015;163(2):340–53.
Schmukler E, et al. Altered mitochondrial dynamics and function in APOE4-expressing astrocytes. Cell Death Dis. 2020;11(7):578.
Williams HC, et al. APOE alters glucose flux through central carbon pathways in astrocytes. Neurobiol Dis. 2020;136:104742.
Farmer BC, et al. APOEpsilon4 lowers energy expenditure in females and impairs glucose oxidation by increasing flux through aerobic glycolysis. Mol Neurodegener. 2021;16(1):62.
Corder EH, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–3.
Tiraboschi P, et al. Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology. 2004;62(11):1977–83.
Schmechel DE, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(20):9649–53.
Holtzman DM, et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2000;97(6):2892–7.
Bales KR, et al. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 1999;96(26):15233–8.
Bales KR, et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997;17(3):263–4.
Fryer JD, et al. Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci. 2005;25(11):2803–10.
Hansson O, et al. CSF biomarkers of Alzheimer’s disease concord with amyloid-beta PET and predict clinical progression: A study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018;14(11):1470–81.
Morris JC, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67(1):122–31.
Reiman EM, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106(16):6820–5.
Sunderland T, et al. Cerebrospinal fluid beta-amyloid1-42 and tau in control subjects at risk for Alzheimer’s disease: the effect of APOE epsilon4 allele. Biol Psychiatry. 2004;56(9):670–6.
Liu CC, et al. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron. 2017;96(5):1024-1032 e3.
Wood SJ, Chan W, Wetzel R. Seeding of A beta fibril formation is inhibited by all three isotypes of apolipoprotein E. Biochemistry. 1996;35(38):12623–8.
Wisniewski T, et al. Acceleration of Alzheimer’s fibril formation by apolipoprotein E in vitro. Am J Pathol. 1994;145(5):1030–5.
Castano EM, et al. Fibrillogenesis in Alzheimer’s disease of amyloid beta peptides and apolipoprotein E. Biochem J. 1995;306(Pt 2):599–604.
Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem. 2005;280(52):43224–35.
Wahrle SE, et al. Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem. 2005;280(52):43236–42.
Namba Y, et al. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991;541(1):163–6.
Gouras GK, et al. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156(1):15–20.
Zhao W, et al. Human APOE genotype affects intraneuronal Abeta1-42 accumulation in a lentiviral gene transfer model. Hum Mol Genet. 2014;23(5):1365–75.
He X, et al. Apolipoprotein receptor 2 and X11 alpha/beta mediate apolipoprotein E-induced endocytosis of amyloid-beta precursor protein and beta-secretase, leading to amyloid-beta production. J Neurosci. 2007;27(15):4052–60.
Huang YA, et al. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell. 2017;168(3):427-441 e21.
Tarasoff-Conway JM, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11(8):457–70.
Kanekiyo T, Xu H, Bu G. ApoE and Abeta in Alzheimer’s disease: accidental encounters or partners? Neuron. 2014;81(4):740–54.
Iwata N, et al. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6(2):143–50.
Qiu WQ, et al. Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem. 1998;273(49):32730–8.
Jiang Q, et al. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58(5):681–93.
Miners JS, et al. Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy. J Neuropathol Exp Neurol. 2006;65(10):1012–21.
Cook DG, et al. Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer’s disease is associated with the apolipoprotein E-epsilon4 allele. Am J Pathol. 2003;162(1):313–9.
Li J, et al. Differential regulation of amyloid-beta endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J Biol Chem. 2012;287(53):44593–601.
Koistinaho M, et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med. 2004;10(7):719–26.
Lee CY, et al. Apolipoprotein E promotes beta-amyloid trafficking and degradation by modulating microglial cholesterol levels. J Biol Chem. 2012;287(3):2032–44.
Basak JM, et al. Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Abeta uptake and degradation by astrocytes. J Biol Chem. 2012;287(17):13959–71.
Verghese PB, et al. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc Natl Acad Sci U S A. 2013;110(19):E1807–16.
Deane R, et al. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118(12):4002–13.
Robert J, et al. Clearance of beta-amyloid is facilitated by apolipoprotein E and circulating high-density lipoproteins in bioengineered human vessels. Elife. 2017;6:e29595.
Ma Q, et al. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-beta42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol Neurodegener. 2018;13(1):57.
Montagne A, et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 2020;581(7806):71–6.
Bell RD, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485(7399):512–6.
Nishitsuji K, et al. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J Biol Chem. 2011;286(20):17536–42.
Castellano JM, et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med. 2011;3(89):89ra57.
Braak H, et al. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70(11):960–9.
Scholl M, et al. PET Imaging of Tau Deposition in the Aging Human Brain. Neuron. 2016;89(5):971–82.
Bejanin A, et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain. 2017;140(12):3286–300.
Huang Y, et al. Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci U S A. 2001;98(15):8838–43.
Brecht WJ, et al. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci. 2004;24(10):2527–34.
Rohn TT, et al. Identification of an amino-terminal fragment of apolipoprotein E4 that localizes to neurofibrillary tangles of the Alzheimer’s disease brain. Brain Res. 2012;1475:106–15.
Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4:13.
Hu W, et al. Expression of Tau Pathology-Related Proteins in Different Brain Regions: A Molecular Basis of Tau Pathogenesis. Front Aging Neurosci. 2017;9:311.
Vogel JW, et al. Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat Med. 2021;27(5):871–81.
Sanchez JS, et al. The cortical origin and initial spread of medial temporal tauopathy in Alzheimer’s disease assessed with positron emission tomography. Sci Transl Med. 2021;13(577):eabc0655.
Braak H, et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112(4):389–404.
Baek MS, et al. Effect of APOE epsilon4 genotype on amyloid-beta and tau accumulation in Alzheimer’s disease. Alzheimers Res Ther. 2020;12(1):140.
Yu L, et al. Disentangling the effects of age and APOE on neuropathology and late life cognitive decline. Neurobiol Aging. 2014;35(4):819–26.
Mattson MP, Arumugam TV. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab. 2018;27(6):1176–99.
Whitwell JL, et al. [(18) F]AV-1451 clustering of entorhinal and cortical uptake in Alzheimer’s disease. Ann Neurol. 2018;83(2):248–57.
Vogel JW, et al. Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat Commun. 2020;11(1):2612.
Shi Y, et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med. 2019;216(11):2546–61.
van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid-beta-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci. 2020;21(1):21–35.
Shi Y, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549(7673):523–7.
Wang C, et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron. 2021;109(10):1657-1674.e7.
Jablonski AM, et al. Astrocytic expression of the Alzheimer’s disease risk allele, ApoEepsilon4, potentiates neuronal tau pathology in multiple preclinical models. Sci Rep. 2021;11(1):3438.
Kang SS, et al. ApoE4 inhibition of VMAT2 in the locus coeruleus exacerbates Tau pathology in Alzheimer’s disease. Acta Neuropathol. 2021;142(1):139–58.
Litvinchuk A, et al. Apolipoprotein E4 Reduction with Antisense Oligonucleotides Decreases Neurodegeneration in a Tauopathy Model. Ann Neurol. 2021;89(5):952–66.
Wang C, et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med. 2018;24(5):647–57.
Wadhwani AR, et al. Neuronal apolipoprotein E4 increases cell death and phosphorylated tau release in alzheimer disease. Ann Neurol. 2019;85(5):726–39.
Wang C, et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron. 2021;109(10):1657-1674 e7.
Koller EJ, et al. Intracerebral Expression of AAV-APOE4 Is Not Sufficient to Alter Tau Burden in Two Distinct Models of Tauopathy. Mol Neurobiol. 2020;57(4):1986–2001.
Rohn TT, et al. Immunolocalization of an amino-terminal fragment of apolipoprotein E in the Pick’s disease brain. PLoS ONE. 2013;8(12):e80180.
Weigand AJ, et al. APOE interacts with tau PET to influence memory independently of amyloid PET in older adults without dementia. Alzheimers Dement. 2021;17(1):61–9.
Johnson KA, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol. 2016;79(1):110–9.
Salvado G, et al. Differential associations of APOE-epsilon2 and APOE-epsilon4 alleles with PET-measured amyloid-beta and tau deposition in older individuals without dementia. Eur J Nucl Med Mol Imaging. 2021;48(7):2212–24.
Therriault J, et al. Association of Apolipoprotein E epsilon4 With Medial Temporal Tau Independent of Amyloid-beta. JAMA Neurol. 2020;77(4):470–9.
Rodriguez-Vieitez E, Nielsen HM. Associations Between APOE Variants, Tau and alpha-Synuclein. Adv Exp Med Biol. 2019;1184:177–86.
Ramanan VK, et al. Association of Apolipoprotein E varepsilon4, Educational Level, and Sex With Tau Deposition and Tau-Mediated Metabolic Dysfunction in Older Adults. JAMA Netw Open. 2019;2(10):e1913909.
Yan S, et al. Association of sex and APOE epsilon4 with brain tau deposition and atrophy in older adults with Alzheimer’s disease. Theranostics. 2020;10(23):10563–72.
Risacher SL, et al. The role of apolipoprotein E (APOE) genotype in early mild cognitive impairment (E-MCI). Front Aging Neurosci. 2013;5:11.
Deming Y, et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol. 2017;133(5):839–56.
Wattmo C, Blennow K, Hansson O. Cerebro-spinal fluid biomarker levels: phosphorylated tau (T) and total tau (N) as markers for rate of progression in Alzheimer’s disease. BMC Neurol. 2020;20(1):10.
Liu Y, et al. The Associations of Cerebrospinal Fluid ApoE and Biomarkers of Alzheimer’s Disease: Exploring Interactions With Sex. Front Neurosci. 2021;15:633576.
Koch G, et al. CSF tau is associated with impaired cortical plasticity, cognitive decline and astrocyte survival only in APOE4-positive Alzheimer’s disease. Sci Rep. 2017;7(1):13728.
Leuzy A, et al. Longitudinal tau and metabolic PET imaging in relation to novel CSF tau measures in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2019;46(5):1152–63.
Wolters EE, et al. Regional [(18)F]flortaucipir PET is more closely associated with disease severity than CSF p-tau in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2020;47(12):2866–78.
Chiang GC, et al. Hippocampal atrophy rates and CSF biomarkers in elderly APOE2 normal subjects. Neurology. 2010;75(22):1976–81.
Grothe MJ, et al. Multimodal characterization of older APOE2 carriers reveals selective reduction of amyloid load. Neurology. 2017;88(6):569–76.
Robinson AC, et al. Influence of APOE genotype in primary age-related tauopathy. Acta Neuropathol Commun. 2020;8(1):215.
Zhao N, et al. APOE epsilon2 is associated with increased tau pathology in primary tauopathy. Nat Commun. 2018;9(1):4388.
Yamada K, et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. 2011;31(37):13110–7.
Rauch JN, et al. LRP1 is a master regulator of tau uptake and spread. Nature. 2020;580(7803):381–5.
Shi Y, et al. Overexpressing low-density lipoprotein receptor reduces tau-associated neurodegeneration in relation to apoE-linked mechanisms. Neuron. 2021;109(15):2413-2426.e7.
DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27(5):457–64.
Terry RD, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572–80.
Scheff SW, et al. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging. 2006;27(10):1372–84.
Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298(5594):789–91.
Davidsson P, Blennow K. Neurochemical dissection of synaptic pathology in Alzheimer’s disease. Int Psychogeriatr. 1998;10(1):11–23.
Reddy PH, et al. Differential loss of synaptic proteins in Alzheimer’s disease: implications for synaptic dysfunction. J Alzheimers Dis. 2005;7(2):103–17 (discussion 173-80).
Buckner RL, et al. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25(34):7709–17.
Koffie RM, et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009;106(10):4012–7.
Sheng M, Sabatini BL, Sudhof TC. Synapses and Alzheimer’s disease. Cold Spring Harb Perspect Biol. 2012;4(5):a005777.
Lambert MP, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95(11):6448–53.
Tu S, et al. Oligomeric Abeta-induced synaptic dysfunction in Alzheimer’s disease. Mol Neurodegener. 2014;9:48.
Klementieva O, et al. Pre-plaque conformational changes in Alzheimer’s disease-linked Abeta and APP. Nat Commun. 2017;8:14726.
Takahashi RH, et al. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161(5):1869–79.
Perdigao C, et al. Intracellular Trafficking Mechanisms of Synaptic Dysfunction in Alzheimer’s Disease. Front Cell Neurosci. 2020;14:72.
Mauch DH, et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294(5545):1354–7.
Pfrieger FW. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol Life Sci. 2003;60(6):1158–71.
Liu CC, et al. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106–18.
Mahley RW, Huang Y. Apolipoprotein e sets the stage: response to injury triggers neuropathology. Neuron. 2012;76(5):871–85.
Li X, et al. Astrocytic ApoE reprograms neuronal cholesterol metabolism and histone-acetylation-mediated memory. Neuron. 2021;109(6):957-970 e8.
Nathan BP, et al. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science. 1994;264(5160):850–2.
Nathan BP, et al. Apolipoprotein E4 inhibits, and apolipoprotein E3 promotes neurite outgrowth in cultured adult mouse cortical neurons through the low-density lipoprotein receptor-related protein. Brain Res. 2002;928(1–2):96–105.
Teter B, et al. Human apolipoprotein E isoform-specific differences in neuronal sprouting in organotypic hippocampal culture. J Neurochem. 1999;73(6):2613–6.
Wang C, et al. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiol Dis. 2005;18(2):390–8.
Nwabuisi-Heath E, et al. ApoE4 delays dendritic spine formation during neuron development and accelerates loss of mature spines in vitro. ASN Neuro. 2014;6(1):e00134.
Chen Y, et al. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci U S A. 2010;107(26):12011–6.
Trommer BL, et al. ApoE isoform affects LTP in human targeted replacement mice. NeuroReport. 2004;15(17):2655–8.
Korwek KM, et al. ApoE isoform-dependent changes in hippocampal synaptic function. Mol Neurodegener. 2009;4:21.
Kitamura HW, et al. Age-dependent enhancement of hippocampal long-term potentiation in knock-in mice expressing human apolipoprotein E4 instead of mouse apolipoprotein E. Neurosci Lett. 2004;369(3):173–8.
Rodriguez GA, et al. Young APOE4 targeted replacement mice exhibit poor spatial learning and memory, with reduced dendritic spine density in the medial entorhinal cortex. Learn Mem. 2013;20(5):256–66.
Andrews-Zwilling Y, et al. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci. 2010;30(41):13707–17.
Bour A, et al. Middle-aged human apoE4 targeted-replacement mice show retention deficits on a wide range of spatial memory tasks. Behav Brain Res. 2008;193(2):174–82.
Koffie RM, et al. Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-beta. Brain. 2012;135(Pt 7):2155–68.
Konings SC, et al. Astrocytic and Neuronal Apolipoprotein E Isoforms Differentially Affect Neuronal Excitability. Front Neurosci. 2021;15:734001.
Bilousova T, et al. Apolipoprotein E/Amyloid-beta Complex Accumulates in Alzheimer Disease Cortical Synapses via Apolipoprotein E Receptors and Is Enhanced by APOE4. Am J Pathol. 2019;189(8):1621–36.
Chun C, et al. Astrocyte-derived extracellular vesicles enhance the survival and electrophysiological function of human cortical neurons in vitro. Biomaterials. 2021;271:120700.
Phongpreecha T, et al. Single-synapse analyses of Alzheimer’s disease implicate pathologic tau, DJ1, CD47, and ApoE. Sci Adv. 2021;7(51):eabk0473.
Xian X, et al. Reversal of ApoE4-induced recycling block as a novel prevention approach for Alzheimer’s disease. Elife. 2018;7:e40048.
Weeber EJ, et al. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J Biol Chem. 2002;277(42):39944–52.
Gao S, et al. Genetic variation within endolysosomal system is associated with late-onset Alzheimer’s disease. Brain. 2018;141(9):2711–20.
Bonilla DL, et al. Autophagy Regulates Phagocytosis by Modulating the Expression of Scavenger Receptors. Immunity. 2013;39(3):537–47.
Fu R, et al. Phagocytosis of Microglia in the Central Nervous System Diseases. Mol Neurobiol. 2014;49(3):1422–34.
Di Meco A, et al. Autophagy Dysfunction in Alzheimer’s Disease: Mechanistic Insights and New Therapeutic Opportunities. Biol Psychiat. 2020;87(9):797–807.
Simonovitch S, et al. Impaired Autophagy in APOE4 Astrocytes. J Alzheimers Dis. 2016;51(3):915–27.
Van Acker ZP, Bretou M, Annaert W. Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: impact of genetic risk factors. Molecular Neurodegener. 2019;14(1):20.
Avrahami L, et al. Inhibition of Glycogen Synthase Kinase-3 Ameliorates β-Amyloid Pathology and Restores Lysosomal Acidification and Mammalian Target of Rapamycin Activity in the Alzheimer Disease Mouse Model. J Biol Chem. 2013;288(2):1295–306.
Xiao Q, et al. Neuronal-Targeted TFEB Accelerates Lysosomal Degradation of APP, Reducing A Generation and Amyloid Plaque Pathogenesis. J Neurosci. 2015;35(35):12137–51.
Cho M-H, et al. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 2014;10(10):1761–75.
Nixon RA. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: inseparable partners in a multifactorial disease. FASEB J. 2017;31(7):2729–43.
Francois A, et al. Involvement of interleukin-1beta in the autophagic process of microglia: relevance to Alzheimer’s disease. J Neuroinflammation. 2013;10:151.
Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med. 2017;6(1):1.
Wu A-G, et al. Targeting microglial autophagic degradation in NLRP3 inflammasome-mediated neurodegenerative diseases. Ageing Res Rev. 2021;65:101202.
Delgado MA, et al. Toll-like receptors control autophagy. EMBO J. 2008;27(7):1110–21.
King BC, et al. Outside in: Roles of complement in autophagy. Br J Pharmacol. 2020;178(14):2786–801.
Cataldo AM, et al. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157(1):277–86.
Lambert J-C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–8.
Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120(23):4081–91.
Kuszczyk MA, et al. Blocking the Interaction between Apolipoprotein E and Aβ Reduces Intraneuronal Accumulation of Aβ and Inhibits Synaptic Degeneration. Am J Pathol. 2013;182(5):1750–68.
Schmukler E, Michaelson DM, Pinkas-Kramarski R. The Interplay Between Apolipoprotein E4 and the Autophagic–Endocytic–Lysosomal Axis. Mol Neurobiol. 2018;55(8):6863–80.
Simonovitch S, et al. The Effects of APOE4 on Mitochondrial Dynamics and Proteins in vivo. J Alzheimers Dis. 2019;70(3):861–75.
Nuriel T, et al. The Endosomal-Lysosomal Pathway Is Dysregulated by APOE4 Expression in Vivo. Front Neurosci. 2017;11:702.
Pomilio C, et al. Microglial autophagy is impaired by prolonged exposure to beta-amyloid peptides: evidence from experimental models and Alzheimer’s disease patients. Geroscience. 2020;42(2):613–32.
Parcon PA, et al. Apolipoprotein E4 inhibits autophagy gene products through direct, specific binding to CLEAR motifs. Alzheimers Dement. 2018;14(2):230–42.
McIntire LBJ, et al. Reduction of Synaptojanin 1 Ameliorates Synaptic and Behavioral Impairments in a Mouse Model of Alzheimer’s Disease. J Neurosci. 2012;32(44):15271–6.
Zhu L, et al. Phospholipid dysregulation contributes to ApoE4-associated cognitive deficits in Alzheimer’s disease pathogenesis. Proc Natl Acad Sci. 2015;112(38):11965–70.
Cortes CJ, La Spada AR. TFEB dysregulation as a driver of autophagy dysfunction in neurodegenerative disease: Molecular mechanisms, cellular processes, and emerging therapeutic opportunities. Neurobiol Dis. 2019;122:83–93.
Lima D, et al. Electrochemical detection of specific interactions between apolipoprotein E isoforms and DNA sequences related to Alzheimer’s disease. Bioelectrochemistry. 2020;133:107447.
Tiribuzi R, et al. miR128 up-regulation correlates with impaired amyloid β(1–42) degradation in monocytes from patients with sporadic Alzheimer’s disease. Neurobiol Aging. 2014;35(2):345–56.
Reddy K, et al. Dysregulation of Nutrient Sensing and CLEARance in Presenilin Deficiency. Cell Rep. 2016;14(9):2166–79.
Sabatini DM. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc Natl Acad Sci. 2017;114(45):11818–25.
Caccamo A, et al. Molecular Interplay between Mammalian Target of Rapamycin (mTOR), Amyloid-β, and Tau. J Biol Chem. 2010;285(17):13107–20.
Shafei MA, Harris M, Conway ME. Divergent Metabolic Regulation of Autophagy and mTORC1—Early Events in Alzheimer’s Disease? Front Aging Neurosci. 2017;9:173.
Herz J, Chen Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat Rev Neurosci. 2006;7(11):850–9.
Rebeck GW, et al. Mol Neurodegener. 2006;1(1).
Yates SC, et al. Dysfunction of the mTOR pathway is a risk factor for Alzheimer’s disease. Acta Neuropathol Commun. 2013;1(1):3.
Li W, et al. Proteomes analysis reveals the involvement of autophagy in AD-like neuropathology induced by noise exposure and ApoE4. Environmental Research. 2019;176:108537.
Lin A-L, Butterfield DA, Richardson A. mTOR: Alzheimer’s disease prevention for APOE4 carriers. Oncotarget. 2016;7(29):44873–4.
Lin A-L, et al. Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer’s disease. J Cereb Blood Flow Metab. 2016;37(1):217–26.
Lin A-L, et al. APOE genotype-dependent pharmacogenetic responses to rapamycin for preventing Alzheimer’s disease. Neurobiol Dis. 2020;139:104834.
Kaeberlein M, Galvan V. Rapamycin and Alzheimer’s disease: Time for a clinical trial? Sci Transl Med. 2019;11(476):eaar4289.
Dallrmi C, Devereaux KA, De Paolo G. The Role of Lipids in the Control of Autophagy. Curr Biol. 2013;23(1):R33–45.
Ando K, et al. The lipid phosphatase Synaptojanin 1 undergoes a significant alteration in expression and solubility and is associated with brain lesions in Alzheimer’s disease. Acta Neuropathol Commun. 2020;8(1):79.
Zhu L, et al. Reduction of Synaptojanin 1 Accelerates Aβ Clearance and Attenuates Cognitive Deterioration in an Alzheimer Mouse Model. J Biol Chem. 2013;288(44):32050–63.
Cao J, Huang M, Guo L, Zhu L, Hou J, Zhang L, Pero A, Ng S, El Gaamouch F, Elder G, Sano M, Goate A, Tcw J, Haroutunian V, Zhang B, Cai D. MicroRNA-195 rescues ApoE4-induced cognitive deficits and lysosomal defects in Alzheimer's disease pathogenesis. Mol Psychiatry. 2021;26(9):4687-701. https://doi.org/10.1038/s41380-020-0824-3.
Bales KR, et al. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J Neurosci. 2009;29(21):6771–9.
Youmans KL, et al. APOE4-specific changes in Abeta accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem. 2012;287(50):41774–86.
Greenberg SM, et al. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol. 1995;38(2):254–9.
Nelson PT, et al. APOE-epsilon2 and APOE-epsilon4 correlate with increased amyloid accumulation in cerebral vasculature. J Neuropathol Exp Neurol. 2013;72(7):708–15.
Tesseur I, et al. Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. Am J Pathol. 2000;156(3):951–64.
Zhou M, et al. APOE4 Induces Site-Specific Tau Phosphorylation Through Calpain-CDK5 Signaling Pathway in EFAD-Tg Mice. Curr Alzheimer Res. 2016;13(9):1048–55.
Ljungberg MC, et al. Truncated apoE forms tangle-like structures in a neuronal cell line. NeuroReport. 2002;13(6):867–70.
Nuriel T, et al. The Endosomal-Lysosomal Pathway Is Dysregulated by APOE4 Expression in Vivo. Front Neurosci. 2017;11:702.
Hou Y, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15(10):565–81.
Li Z, et al. Aging and age-related diseases: from mechanisms to therapeutic strategies. Biogerontology. 2021;22(2):165–87.
Harada CN, Natelson Love MC, Triebel KL. Normal cognitive aging. Clin Geriatr Med. 2013;29(4):737–52.
Fjell AM, Walhovd KB. Structural brain changes in aging: courses, causes and cognitive consequences. Rev Neurosci. 2010;21(3):187–221.
Saddiki H, et al. Age and the association between apolipoprotein E genotype and Alzheimer disease: A cerebrospinal fluid biomarker-based case-control study. PLoS Med. 2020;17(8):e1003289.
Di Benedetto S, et al. Contribution of neuroinflammation and immunity to brain aging and the mitigating effects of physical and cognitive interventions. Neurosci Biobehav Rev. 2017;75:114–28.
Otani K, Shichita T. Cerebral sterile inflammation in neurodegenerative diseases. Inflamm Regen. 2020;40(1):28.
Corlier F, et al. Systemic inflammation as a predictor of brain aging: Contributions of physical activity, metabolic risk, and genetic risk. Neuroimage. 2018;172:118–29.
Walker KA. Inflammation and neurodegeneration: chronicity matters. Aging (Albany NY). 2018;11(1):3–4.
Zhang G, et al. Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature. 2013;497(7448):211–6.
Cullen NC, et al. Accelerated inflammatory aging in Alzheimer’s disease and its relation to amyloid, tau, and cognition. Sci Rep. 2021;11(1):1965.
Shinohara M, et al. APOE2 is associated with longevity independent of Alzheimer’s disease. Elife. 2020;9:e62199.
Zhao N, et al. Alzheimer’s Risk Factors Age, APOE Genotype, and Sex Drive Distinct Molecular Pathways. Neuron. 2020;106(5):727-742 e6.
Horvath AJ, et al. The murine orthologue of human antichymotrypsin: a structural paradigm for clade A3 serpins. J Biol Chem. 2005;280(52):43168–78.
Padmanabhan J, et al. Alpha1-antichymotrypsin, an inflammatory protein overexpressed in Alzheimer’s disease brain, induces tau phosphorylation in neurons. Brain. 2006;129(Pt 11):3020–34.
Fissolo N, et al. CSF SERPINA3 Levels Are Elevated in Patients With Progressive MS. Neurol Neuroimmunol Neuroinflamm. 2021;8(2):e941.
Wang ZM, et al. SerpinA3N deficiency deteriorates impairments of learning and memory in mice following hippocampal stab injury. Cell Death Discov. 2020;6:88.
Angelova DM, Brown DR. Microglia and the aging brain: are senescent microglia the key to neurodegeneration? J Neurochem. 2019;151(6):676–88.
Koellhoffer EC, McCullough LD, Ritzel RM. Old Maids: Aging and Its Impact on Microglia Function. Int J Mol Sci. 2017;18(4):769.
Niraula A, Sheridan JF, Godbout JP. Microglia Priming with Aging and Stress. Neuropsychopharmacology. 2017;42(1):318–33.
Rice RA, et al. Microglial repopulation resolves inflammation and promotes brain recovery after injury. Glia. 2017;65(6):931–44.
Coleman LG Jr, Zou J, Crews FT. Microglial depletion and repopulation in brain slice culture normalizes sensitized proinflammatory signaling. J Neuroinflammation. 2020;17(1):27.
Najafi AR, et al. A limited capacity for microglial repopulation in the adult brain. Glia. 2018;66(11):2385–96.
Rajan KB, et al. Population estimate of people with clinical Alzheimer’s disease and mild cognitive impairment in the United States (2020–2060). Alzheimers Dement. 2021;17(12):1966–75.
Beam CR, et al. Differences Between Women and Men in Incidence Rates of Dementia and Alzheimer’s Disease. J Alzheimers Dis. 2018;64(4):1077–83.
Mielke MM, Vemuri P, Rocca WA. Clinical epidemiology of Alzheimer’s disease: assessing sex and gender differences. Clin Epidemiol. 2014;6:37–48.
Mosconi L, et al. Menopause impacts human brain structure, connectivity, energy metabolism, and amyloid-beta deposition. Sci Rep. 2021;11(1):10867.
Hohman TJ, et al. Sex-Specific Association of Apolipoprotein E With Cerebrospinal Fluid Levels of Tau. JAMA Neurol. 2018;75(8):989–98.
Li G, et al. Cerebrospinal fluid biomarkers for Alzheimer’s and vascular disease vary by age, gender, and APOE genotype in cognitively normal adults. Alzheimers Res Ther. 2017;9(1):48.
Posillico CK, Garcia-Hernandez RE, Tronson NC. Sex differences and similarities in the neuroimmune response to central administration of poly I:C. J Neuroinflammation. 2021;18(1):193.
Fisher DW, Bennett DA, Dong H. Sexual dimorphism in predisposition to Alzheimer’s disease. Neurobiol Aging. 2018;70:308–24.
Qi S, et al. X chromosome escapee genes are involved in ischemic sexual dimorphism through epigenetic modification of inflammatory signals. J Neuroinflammation. 2021;18(1):70.
Han J, et al. Uncovering sex differences of rodent microglia. J Neuroinflammation. 2021;18(1):74.
Kalimon OJ, Sullivan PG. Sex Differences in Mitochondrial Function Following a Controlled Cortical Impact Traumatic Brain Injury in Rodents. Front Mol Neurosci. 2021;14:753946.
Thion MS, et al. Microbiome Influences Prenatal and Adult Microglia in a Sex-Specific Manner. Cell. 2018;172(3):500-516 e16.
Chistyakov DV, et al. Sex-Mediated Differences in LPS Induced Alterations of TNFalpha, IL-10 Expression, and Prostaglandin Synthesis in Primary Astrocytes. Int J Mol Sci. 2018;19(9):2793.
Santos-Galindo M, et al. Sex differences in the inflammatory response of primary astrocytes to lipopolysaccharide. Biol Sex Differ. 2011;2:7.
Larramona-Arcas R, et al. Sex-dependent calcium hyperactivity due to lysosomal-related dysfunction in astrocytes from APOE4 versus APOE3 gene targeted replacement mice. Mol Neurodegener. 2020;15(1):35.
Crespo-Castrillo A, Arevalo MA. Microglial and Astrocytic Function in Physiological and Pathological Conditions: Estrogenic Modulation. Int J Mol Sci. 2020;21(9):3219.
Leeming ER, et al. Effect of Diet on the Gut Microbiota: Rethinking Intervention Duration. Nutrients. 2019;11(12):2862.
Fang P, et al. The Microbiome as a Modifier of Neurodegenerative Disease Risk. Cell Host Microbe. 2020;28(2):201–22.
Cryan JF, et al. The gut microbiome in neurological disorders. Lancet Neurol. 2020;19(2):179–94.
McGrattan AM, et al. Diet and Inflammation in Cognitive Ageing and Alzheimer’s Disease. Curr Nutr Rep. 2019;8(2):53–65.
Megur A, et al. The Microbiota-Gut-Brain Axis and Alzheimer’s Disease: Neuroinflammation Is to Blame? Nutrients. 2020;13(1):37.
Ma Q, et al. Impact of microbiota on central nervous system and neurological diseases: the gut-brain axis. J Neuroinflammation. 2019;16(1):53.
Carlsson CM. Type 2 diabetes mellitus, dyslipidemia, and Alzheimer’s disease. J Alzheimers Dis. 2010;20(3):711–22.
Chatterjee S, Mudher A. Alzheimer’s Disease and Type 2 Diabetes: A Critical Assessment of the Shared Pathological Traits. Front Neurosci. 2018;12:383.
DeToma AS, et al. Misfolded proteins in Alzheimer’s disease and type II diabetes. Chem Soc Rev. 2012;41(2):608–21.
Chung J, et al. Glucose Metabolic Brain Networks in Early-Onset vs. Late-Onset Alzheimer’s Disease. Front Aging Neurosci. 2016;8:159.
Arnold SE, et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol. 2018;14(3):168–81.
Kellar D, Craft S. Brain insulin resistance in Alzheimer’s disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol. 2020;19(9):758–66.
El-Lebedy D, Raslan HM, Mohammed AM. Apolipoprotein E gene polymorphism and risk of type 2 diabetes and cardiovascular disease. Cardiovasc Diabetol. 2016;15:12.
Zhen J, et al. Association of ApoE Genetic Polymorphism and Type 2 Diabetes with Cognition in Non-Demented Aging Chinese Adults: A Community Based Cross-Sectional Study. Aging Dis. 2018;9(3):346–57.
Luo JQ, et al. The Associations between Apolipoprotein E Gene Epsilon2/Epsilon3/Epsilon4 Polymorphisms and the Risk of Coronary Artery Disease in Patients with Type 2 Diabetes Mellitus. Front Physiol. 2017;8:1031.
Rhea EM, Raber J, Banks WA. ApoE and cerebral insulin: Trafficking, receptors, and resistance. Neurobiol Dis. 2020;137:104755.
Fallaize R, et al. APOE genotype influences insulin resistance, apolipoprotein CII and CIII according to plasma fatty acid profile in the Metabolic Syndrome. Sci Rep. 2017;7(1):6274.
Wu L, Zhang X, Zhao L. Human ApoE Isoforms Differentially Modulate Brain Glucose and Ketone Body Metabolism: Implications for Alzheimer’s Disease Risk Reduction and Early Intervention. J Neurosci. 2018;38(30):6665–81.
Chan ES, et al. Differential interaction of Apolipoprotein-E isoforms with insulin receptors modulates brain insulin signaling in mutant human amyloid precursor protein transgenic mice. Sci Rep. 2015;5:13842.
Traversy MT, et al. Altered cerebral insulin response in transgenic mice expressing the epsilon-4 allele of the human apolipoprotein E gene. Psychoneuroendocrinology. 2017;77:203–10.
Zhao N, et al. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron. 2017;96(1):115-129 e5.
Jones NS, Watson KQ, Rebeck GW. High-fat diet increases gliosis and immediate early gene expression in APOE3 mice, but not APOE4 mice. J Neuroinflammation. 2021;18(1):214.
Norwitz NG, et al. Precision Nutrition for Alzheimer’s Prevention in ApoE4 Carriers. Nutrients. 2021;13(4):1362.
Shinohara M, et al. Interaction between APOE genotype and diabetes in cognitive decline. Alzheimers Dement (Amst). 2020;12(1):e12006.
Li X, et al. Effects of Exercise Training on Cardiopulmonary Function and Quality of Life in Elderly Patients with Pulmonary Fibrosis: A Meta-Analysis. Int J Environ Res Public Health. 2021;18(14):7643.
Benedetti MG, et al. The Effectiveness of Physical Exercise on Bone Density in Osteoporotic Patients. Biomed Res Int. 2018;2018:4840531.
Sacma M, Geiger H. Exercise generates immune cells in bone. Nature. 2021;591(7850):371–2.
Blondell SJ, Hammersley-Mather R, Veerman JL. Does physical activity prevent cognitive decline and dementia?: A systematic review and meta-analysis of longitudinal studies. BMC Public Health. 2014;14:510.
Tan ZS, et al. Physical Activity, Brain Volume, and Dementia Risk: The Framingham Study. J Gerontol A Biol Sci Med Sci. 2017;72(6):789–95.
Tolppanen AM, et al. Leisure-time physical activity from mid- to late life, body mass index, and risk of dementia. Alzheimers Dement. 2015;11(4):434-443 e6.
Nyberg J, et al. Cardiovascular and cognitive fitness at age 18 and risk of early-onset dementia. Brain. 2014;137(Pt 5):1514–23.
Hotting K, Roder B. Beneficial effects of physical exercise on neuroplasticity and cognition. Neurosci Biobehav Rev. 2013;37(9 Pt B):2243–57.
Hansson O, et al. Midlife physical activity is associated with lower incidence of vascular dementia but not Alzheimer’s disease. Alzheimers Res Ther. 2019;11(1):87.
Najar J, et al. Cognitive and physical activity and dementia: A 44-year longitudinal population study of women. Neurology. 2019;92(12):e1322–30.
Ravaglia G, et al. Physical activity and dementia risk in the elderly: findings from a prospective Italian study. Neurology. 2008;70(19 Pt 2):1786–94.
Rovio S, et al. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurol. 2005;4(11):705–11.
Panza GA, et al. Can Exercise Improve Cognitive Symptoms of Alzheimer’s Disease? J Am Geriatr Soc. 2018;66(3):487–95.
Du Z, et al. Physical activity can improve cognition in patients with Alzheimer’s disease: a systematic review and meta-analysis of randomized controlled trials. Clin Interv Aging. 2018;13:1593–603.
Frederiksen KS, et al. Effects of Physical Exercise on Alzheimer’s Disease Biomarkers: A Systematic Review of Intervention Studies. J Alzheimers Dis. 2018;61(1):359–72.
Firth J, et al. Effect of aerobic exercise on hippocampal volume in humans: A systematic review and meta-analysis. Neuroimage. 2018;166:230–8.
De la Rosa A, et al. Physical exercise in the prevention and treatment of Alzheimer’s disease. J Sport Health Sci. 2020;9(5):394–404.
Stranahan AM, Martin B, Maudsley S. Anti-inflammatory effects of physical activity in relationship to improved cognitive status in humans and mouse models of Alzheimer’s disease. Curr Alzheimer Res. 2012;9(1):86–92.
Mee-Inta O, Zhao ZW, Kuo YM. Physical Exercise Inhibits Inflammation and Microglial Activation. Cells. 2019;8(7):691.
Emrani S, et al. Alzheimer’s/Vascular Spectrum Dementia: Classification in Addition to Diagnosis. J Alzheimers Dis. 2020;73(1):63–71.
Kalaria R. Similarities between Alzheimer’s disease and vascular dementia. J Neurol Sci. 2002;203–204:29–34.
Bandyopadhyay S. Role of Neuron and Glia in Alzheimer’s Disease and Associated Vascular Dysfunction. Front Aging Neurosci. 2021;13:653334.
Leardini-Tristao M, et al. Physical exercise promotes astrocyte coverage of microvessels in a model of chronic cerebral hypoperfusion. J Neuroinflammation. 2020;17(1):117.
de Frutos-Lucas J, et al. Does APOE genotype moderate the relationship between physical activity, brain health and dementia risk? A systematic review. Ageing Res Rev. 2020;64:101173.
Tokgoz S, Claassen J. Exercise as Potential Therapeutic Target to Modulate Alzheimer’s Disease Pathology in APOE epsilon4 Carriers: A Systematic Review. Cardiol Ther. 2021;10(1):67–88.
Podewils LJ, et al. Physical activity, APOE genotype, and dementia risk: findings from the Cardiovascular Health Cognition Study. Am J Epidemiol. 2005;161(7):639–51.
Fenesi B, et al. Physical Exercise Moderates the Relationship of Apolipoprotein E (APOE) Genotype and Dementia Risk: A Population-Based Study. J Alzheimers Dis. 2017;56(1):297–303.
Buchman AS, et al. Total daily physical activity and the risk of AD and cognitive decline in older adults. Neurology. 2012;78(17):1323–9.
Jensen CS, et al. Patients with Alzheimer’s disease who carry the APOE epsilon4 allele benefit more from physical exercise. Alzheimers Dement (N Y). 2019;5:99–106.
Colovati MES, et al. Interaction between physical exercise and APOE gene polymorphism on cognitive function in older people. Braz J Med Biol Res. 2020;54(2):e10098.
Deeny SP, et al. Exercise, APOE, and working memory: MEG and behavioral evidence for benefit of exercise in epsilon4 carriers. Biol Psychol. 2008;78(2):179–87.
Stern Y, et al. Effect of aerobic exercise on cognition in younger adults: A randomized clinical trial. Neurology. 2019;92(9):e905–16.
Fernandez-Matarrubia M, et al. An Active Lifestyle Is Associated with Better Cognitive Function Over Time in APOE varepsilon4 Non-Carriers. J Alzheimers Dis. 2021;79(3):1257–68.
Rodriguez FS, et al. APOE e4-genotype and lifestyle interaction on cognitive performance: Results of the LIFE-Adult-study. Health Psychol. 2018;37(2):194–205.
Lyall DM, et al. Assessing for interaction between APOE epsilon4, sex, and lifestyle on cognitive abilities. Neurology. 2019;92(23):e2691–8.
Stringa N, et al. Physical Activity as Moderator of the Association Between APOE and Cognitive Decline in Older Adults: Results from Three Longitudinal Cohort Studies. J Gerontol A Biol Sci Med Sci. 2020;75(10):1880–6.
Kerestes R, et al. Multimodal evaluation of the amygdala’s functional connectivity. Neuroimage. 2017;148:219–29.
Smith JC, et al. Physical activity reduces hippocampal atrophy in elders at genetic risk for Alzheimer’s disease. Front Aging Neurosci. 2014;6:61.
Kaufman CS, et al. Aerobic exercise improves hippocampal blood flow for hypertensive Apolipoprotein E4 carriers. J Cereb Blood Flow Metab. 2021;41(8):2026–37.
Head D, et al. Exercise Engagement as a Moderator of the Effects of APOE Genotype on Amyloid Deposition. Arch Neurol. 2012;69(5):636–43.
Steen Jensen C, et al. Cerebrospinal Fluid Amyloid Beta and Tau Concentrations Are Not Modulated by 16 Weeks of Moderate- to High-Intensity Physical Exercise in Patients with Alzheimer Disease. Dement Geriatr Cogn Disord. 2016;42(3–4):146–58.
Stojanovic M, et al. Physical Exercise and Longitudinal Trajectories in Alzheimer Disease Biomarkers and Cognitive Functioning. Alzheimer Dis Assoc Disord. 2020;34(3):212–9.
Jensen CS, et al. Exercise as a potential modulator of inflammation in patients with Alzheimer’s disease measured in cerebrospinal fluid and plasma. Exp Gerontol. 2019;121:91–8.
Kohman RA, et al. Exercise reduces activation of microglia isolated from hippocampus and brain of aged mice. J Neuroinflammation. 2013;10:114.
Soto I, et al. APOE Stabilization by Exercise Prevents Aging Neurovascular Dysfunction and Complement Induction. PLoS Biol. 2015;13(10):e1002279.
Klimentidis YC, et al. Genome-wide association study of habitual physical activity in over 377,000 UK Biobank participants identifies multiple variants including CADM2 and APOE. Int J Obes (Lond). 2018;42(6):1161–76.
Wojciechowicz B, Laguette MN, Sawczuk M, Humińska-Lisowska K, Maciejewska-Skrendo A, Ficek K, Michałowska-Sawczyn M, Leońska-Duniec A, Kaczmarczyk M, Chycki J, Trybek G, September AV, Cięszczyk P. Are KIF6 and APOE polymorphisms associated with power and endurance athletes? Eur J Sport Sci. 2021;21(9):1283-9. https://doi.org/10.1080/17461391.2020.1817983.
Krell-Roesch J, et al. Association of non-exercise physical activity in mid- and late-life with cognitive trajectories and the impact of APOE epsilon4 genotype status: the Mayo Clinic Study of Aging. Eur J Ageing. 2019;16(4):491–502.
Sabia S, et al. Physical activity, cognitive decline, and risk of dementia: 28 year follow-up of Whitehall II cohort study. BMJ. 2017;357:j2709.
de Bruijn RF, et al. The association between physical activity and dementia in an elderly population: the Rotterdam Study. Eur J Epidemiol. 2013;28(3):277–83.
Serrano-Pozo A, Das S, Hyman BT. APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021;20(1):68–80.
Cramer PE, et al. ApoE-Directed Therapeutics Rapidly Clear -Amyloid and Reverse Deficits in AD Mouse Models. Science. 2012;335(6075):1503–6.
Ghosal K, et al. A randomized controlled study to evaluate the effect of bexarotene on amyloid-β and apolipoprotein E metabolism in healthy subjects. Alzheimer’s & Dementia: Translational Research & Clinical Interventions. 2016;2(2):110–20.
Cummings JL, et al. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene in moderate Alzheimer’s disease. Alzheimer’s Research & Therapy. 2016;8(1):4.
Hu J, et al. Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Abeta metabolism in apoE4-targeted replacement mice. Mol Neurodegener. 2015;10:6.
Rosenberg JB, et al. AAVrh. 10-Mediated APOE2 Central Nervous System Gene Therapy for APOE4-Associated Alzheimer’s Disease. Human Gene Therapy Clin Dev. 2018;29(1):24–47.
Sperling R, et al. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012;11(3):241–9.
VandeVrede L, et al. Symptomatic amyloid-related imaging abnormalities in an APOE ε4/ε4 patient treated with aducanumab. Alzheimers Dement. 2020;12(1):e12101.
Liao F, et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J Clin Investig. 2018;128(5):2144–55.
Kim J, et al. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Aβ amyloidosis. J Exp Med. 2012;209(12):2149–56.
Liao F, et al. Anti-ApoE Antibody Given after Plaque Onset Decreases A Accumulation and Improves Brain Function in a Mouse Model of A Amyloidosis. J Neurosci. 2014;34(21):7281–92.
Xiong M, et al. APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Sci Transl Med. 2021;13(581):eabd7522.
Przedborski S, Vila M, Jackson-Lewis V. Neurodegeneration: what is it and where are we? J Clin Invest. 2003;111(1):3–10.
Gupta R, et al. Post-translational modifications: Regulators of neurodegenerative proteinopathies. Ageing Res Rev. 2021;68:101336.
Minter MR, Taylor JM, Crack PJ. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J Neurochem. 2016;136(3):457–74.
Garwood CJ, et al. Astrocytes are important mediators of Abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011;2:e167.
Jellinger KA. Basic mechanisms of neurodegeneration: a critical update. J Cell Mol Med. 2010;14(3):457–87.
Jellinger KA. Cell death mechanisms in neurodegeneration. J Cell Mol Med. 2001;5(1):1–17.
Van Acker ZP, Bretou M, Annaert W. Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: impact of genetic risk factors. Mol Neurodegener. 2019;14(1):20.
Nguyen AT, et al. APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol. 2020;140(4):477–93.
Fares MB, Jagannath S, Lashuel HA. Reverse engineering Lewy bodies: how far have we come and how far can we go? Nat Rev Neurosci. 2021;22(2):111–31.
Villar-Piqué A, Lopes da Fonseca T, Outeiro TF. Structure function and toxicity of alpha-synuclein: the Bermuda triangle in synucleinopathies. J Neurochem. 2016;1391:240–55.
Ruf VC, et al. Different Effects of α-Synuclein Mutants on Lipid Binding and Aggregation Detected by Single Molecule Fluorescence Spectroscopy and ThT Fluorescence-Based Measurements. ACS Chem Neurosci. 2019;10(3):1649–59.
Hsiao JT, Halliday GM, Kim WS. α-Synuclein Regulates Neuronal Cholesterol Efflux. Molecules. 2017;22(10):1769.
García-Sanz P, Aerts JMFG, Moratalla R. The Role of Cholesterol in α-Synuclein and Lewy Body Pathology in GBA1 Parkinson’s Disease. Mov Disord. 2021;36(5):1070–85.
Hallett PJ, Engelender S, Isacson O. Lipid and immune abnormalities causing age-dependent neurodegeneration and Parkinson’s disease. J Neuroinflammation. 2019;16(1):153.
Borroni B, et al. APOE genotype and cholesterol levels in lewy body dementia and Alzheimer disease: investigating genotype-phenotype effect on disease risk. Am J Geriatr Psychiatry. 2006;14(12):1022–31.
Barceló-Coblijn G, et al. Brain neutral lipids mass is increased in alpha-synuclein gene-ablated mice. J Neurochem. 2007;101(1):132–41.
Emamzadeh FN, et al. Effects of different isoforms of apoE on aggregation of the α-synuclein protein implicated in Parkinson’s disease. Neurosci Lett. 2016;618:146–51.
Rohn TT, Mack JM. Apolipoprotein E Fragmentation within Lewy Bodies of the Human Parkinson’s Disease Brain. Int J Neurodegener Dis. 2018;1(1):002.
Paslawski W, et al. alpha-synuclein-lipoprotein interactions and elevated ApoE level in cerebrospinal fluid from Parkinson’s disease patients. Proc Natl Acad Sci U S A. 2019;116(30):15226–35.
Braak H, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211.
Sezgin M, et al. Parkinson’s Disease Dementia and Lewy Body Disease. Semin Neurol. 2019;39(2):274–82.
Outeiro TF, et al. Dementia with Lewy bodies: an update and outlook. Mol Neurodegener. 2019;14(1):5.
Mata IF, et al. APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol. 2014;71(11):1405–12.
Federoff M, et al. A large study reveals no association between APOE and Parkinson’s disease. Neurobiol Dis. 2012;46(2):389–92.
Eerola J, et al. Apolipoprotein E (APOE), PARKIN and catechol-O-methyltransferase (COMT) genes and susceptibility to sporadic Parkinson’s disease in Finland. Neurosci Lett. 2002;330(3):296–8.
Harhangi BS, et al. APOE and the risk of PD with or without dementia in a population-based study. Neurology. 2000;54(6):1272–6.
Zareparsi S, et al. Age at onset of Parkinson disease and apolipoprotein E genotypes. Am J Med Genet. 2002;107(2):156–61.
Tan MMX, et al. Genome-Wide Association Studies of Cognitive and Motor Progression in Parkinson’s Disease. Mov Disord. 2021;36(2):424–33.
Poewe W, et al. Parkinson disease. Nat Rev Dis Primers. 2017;3(1):17013.
Krasemann S, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity. 2017;47(3):566-581.e9.
Choi I, et al. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun. 2020;11(1):1386.
Pang S, et al. Meta-Analysis of the Relationship between the. Parkinsons Dis. 2018;2018:9497147.
Dickson DW, et al. ε4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology. 2018;91(12):e1182–95.
Zhao N, et al. APOE4 exacerbates α-synuclein pathology and related toxicity independent of amyloid. Sci Transl Med. 2020;12(529):eaay1809.
Davis AA, Inman CE, Wargel ZM, Dube U, Freeberg BM, Galluppi A, Haines JN, Dhavale DD, Miller R, Choudhury FA, Sullivan PM, Cruchaga C, Perlmutter JS, Ulrich JD, Benitez BA, Kotzbauer PT, Holtzman DM. APOEgenotype regulates pathology and disease progression in synucleinopathy. Sci Transl Med. 2020;12(529):eaay3069. https://doi.org/10.1126/scitranslmed.aay3069.
Goldberg TE, Huey ED, Devanand DP. Association of APOE e2 genotype with Alzheimer’s and non-Alzheimer’s neurodegenerative pathologies. Nat Commun. 2020;11(1):4727.
Berge G, et al. Apolipoprotein E epsilon2 genotype delays onset of dementia with Lewy bodies in a Norwegian cohort. J Neurol Neurosurg Psychiatry. 2014;85(11):1227–31.
Tsuang D, et al. APOE epsilon4 increases risk for dementia in pure synucleinopathies. JAMA Neurol. 2013;70(2):223–8.
Ogaki K, et al. Multiple system atrophy and apolipoprotein E. Mov Disord. 2018;33(4):647–50.
Davis AA, et al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci Transl Med. 2020;12(529):eaay3069.
Zhao N, et al. APOE4 exacerbates alpha-synuclein pathology and related toxicity independent of amyloid. Sci Transl Med. 2020;12(529):eaay1809.
Twohig D, et al. The relevance of cerebrospinal fluid alpha-synuclein levels to sporadic and familial Alzheimer’s disease. Acta Neuropathol Commun. 2018;6(1):130.
Twohig D, Nielsen HM. alpha-synuclein in the pathophysiology of Alzheimer’s disease. Mol Neurodegener. 2019;14(1):23.
Dixon KJ. Pathophysiology of Traumatic Brain Injury. Phys Med Rehabil Clin N Am. 2017;28(2):215–25.
Faul M, Coronado V. Epidemiology of traumatic brain injury. Handb Clin Neurol. 2015;127:3–13.
Dams-O’Connor K, et al. Traumatic brain injury as a risk factor for Alzheimer’s disease: current knowledge and future directions. Neurodegener Dis Manag. 2016;6(5):417–29.
Schaffert J, et al. Traumatic brain injury history is associated with an earlier age of dementia onset in autopsy-confirmed Alzheimer’s disease. Neuropsychology. 2018;32(4):410–6.
Grasset L, et al. Association of traumatic brain injury with dementia and memory decline in older adults in the United States. Alzheimers Dement. 2020;16(6):853–61.
Zhou W, et al. Meta-analysis of APOE4 allele and outcome after traumatic brain injury. J Neurotrauma. 2008;25(4):279–90.
McFadyen CA, et al. Apolipoprotein E4 Polymorphism and Outcomes from Traumatic Brain Injury: A Living Systematic Review and Meta-Analysis. J Neurotrauma. 2021;38(8):1124–36.
Karve IP, Taylor JM, Crack PJ. The contribution of astrocytes and microglia to traumatic brain injury. Br J Pharmacol. 2016;173(4):692–702.
Muza P, et al. APOE Genotype Specific Effects on the Early Neurodegenerative Sequelae Following Chronic Repeated Mild Traumatic Brain Injury. Neuroscience. 2019;404:297–313.
Zhao J, et al. Identification of target genes in neuroinflammation and neurodegeneration after traumatic brain injury in rats. PeerJ. 2019;7:e8324.
Giarratana AO, et al. APOE4 genetic polymorphism results in impaired recovery in a repeated mild traumatic brain injury model and treatment with Bryostatin-1 improves outcomes. Sci Rep. 2020;10(1):19919.
Wu Y, et al. Blood-Brain Barrier Dysfunction in Mild Traumatic Brain Injury: Evidence From Preclinical Murine Models. Front Physiol. 2020;11:1030.
Lochhead JJ, et al. Structure, Function, and Regulation of the Blood-Brain Barrier Tight Junction in Central Nervous System Disorders. Front Physiol. 2020;11:914.
Main BS, et al. Apolipoprotein E4 impairs spontaneous blood brain barrier repair following traumatic brain injury. Mol Neurodegener. 2018;13(1):17.
Montagne A, et al. APOE4 accelerates advanced-stage vascular and neurodegenerative disorder in old Alzheimer’s mice via cyclophilin A independently of amyloid-β. Nature Aging. 2021;1(6):506–20.
Teng Z, et al. ApoE Influences the Blood-Brain Barrier Through the NF-kappaB/MMP-9 Pathway After Traumatic Brain Injury. Sci Rep. 2017;7(1):6649.
Abu Hamdeh S, et al. Rapid amyloid-beta oligomer and protofibril accumulation in traumatic brain injury. Brain Pathol. 2018;28(4):451–62.
Stein TD, et al. Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathol. 2015;130(1):21–34.
Cao J, et al. ApoE4-associated phospholipid dysregulation contributes to development of Tau hyper-phosphorylation after traumatic brain injury. Sci Rep. 2017;7(1):11372.
Franz G, et al. Amyloid beta 1–42 and tau in cerebrospinal fluid after severe traumatic brain injury. Neurology. 2003;60(9):1457–61.
Ramos-Cejudo J, et al. Traumatic Brain Injury and Alzheimer’s Disease: The Cerebrovascular Link. EBioMedicine. 2018;28:21–30.
Ling H, Hardy J, Zetterberg H. Neurological consequences of traumatic brain injuries in sports. Mol Cell Neurosci. 2015;66(Pt B):114–22.
Roberts GW, Allsop D, Bruton C. The occult aftermath of boxing. J Neurol Neurosurg Psychiatry. 1990;53(5):373–8.
DeKosky ST, et al. Association of increased cortical soluble abeta42 levels with diffuse plaques after severe brain injury in humans. Arch Neurol. 2007;64(4):541–4.
Goldberg TE, Huey ED, Devanand DP. Associations of APOE e2 genotype with cerebrovascular pathology: a postmortem study of 1275 brains. J Neurol Neurosurg Psychiatry. 2020:jnnp-2020-323746. https://doi.org/10.1136/jnnp-2020-323746.
Amarenco P, et al. Classification of stroke subtypes. Cerebrovasc Dis. 2009;27(5):493–501.
Kalaria RN, Akinyemi R, Ihara M. Stroke injury, cognitive impairment and vascular dementia. Biochim Biophys Acta. 2016;1862(5):915–25.
Werden E, et al. APOE varepsilon4 Carriers Show Delayed Recovery of Verbal Memory and Smaller Entorhinal Volume in the First Year After Ischemic Stroke. J Alzheimers Dis. 2019;71(1):245–59.
Montagne A, Nation DA, Zlokovic BV. APOE4 Accelerates Development of Dementia After Stroke: Is There a Role for Cerebrovascular Dysfunction? Stroke. 2020;51(3):699–700.
Pendlebury ST, et al. APOE-epsilon4 Genotype and Dementia Before and After Transient Ischemic Attack and Stroke: Population-Based Cohort Study. Stroke. 2020;51(3):751–8.
Lagging C, et al. APOE epsilon4 is associated with younger age at ischemic stroke onset but not with stroke outcome. Neurology. 2019;93(19):849–53.
Chen C, Hu Z. ApoE Polymorphisms and the Risk of Different Subtypes of Stroke in the Chinese Population: A Comprehensive Meta-Analysis. Cerebrovasc Dis. 2016;41(3–4):119–38.
Qiu L, et al. Less neurogenesis and inflammation in the immature than in the juvenile brain after cerebral hypoxia-ischemia. J Cereb Blood Flow Metab. 2007;27(4):785–94.
Buscemi L, et al. Spatio-temporal overview of neuroinflammation in an experimental mouse stroke model. Sci Rep. 2019;9(1):507.
Cao Z, et al. Unique Subtype of Microglia in Degenerative Thalamus After Cortical Stroke. Stroke. 2021;52(2):687–98.
Cheng X, et al. Galectin-3 causes enteric neuronal loss in mice after left sided permanent middle cerebral artery occlusion, a model of stroke. Sci Rep. 2016;6:32893.
Xiang J, et al. Melatonin-induced ApoE expression in mouse astrocytes protects endothelial cells from OGD-R induced injuries. Transl Psychiatry. 2020;10(1):181.
Wang Y, et al. White matter injury in ischemic stroke. Prog Neurobiol. 2016;141:45–60.
Vijayan M, Reddy PH. Stroke, Vascular Dementia, and Alzheimer’s Disease: Molecular Links. J Alzheimers Dis. 2016;54(2):427–43.
Lemmens R, et al. Association of apolipoprotein E epsilon2 with white matter disease but not with microbleeds. Stroke. 2007;38(4):1185–8.
Wang R, et al. Effects of vascular risk factors and APOE epsilon4 on white matter integrity and cognitive decline. Neurology. 2015;84(11):1128–35.
Hayden EY, et al. Modeling Mixed Vascular and Alzheimer’s Dementia Using Focal Subcortical Ischemic Stroke in Human ApoE4-TR:5XFAD Transgenic Mice. Transl Stroke Res. 2020;11(5):1064–76.
van den Bos MAJ, et al. Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques. Int J Mol Sci. 2019;20(11):2818.
Restivo DA, et al. ALS dysphagia pathophysiology: differential botulinum toxin response. Neurology. 2013;80(7):616–20.
Vaughan SK, et al. The ALS-inducing factors, TDP43(A315T) and SOD1(G93A), directly affect and sensitize sensory neurons to stress. Sci Rep. 2018;8(1):16582.
Jeon GS, et al. Pathological Modification of TDP-43 in Amyotrophic Lateral Sclerosis with SOD1 Mutations. Mol Neurobiol. 2019;56(3):2007–21.
Cheng Y, Chen Y, Shang H. Aberrations of biochemical indicators in amyotrophic lateral sclerosis: a systematic review and meta-analysis. Transl Neurodegener. 2021;10(1):3.
Rizzo F, et al. Cellular therapy to target neuroinflammation in amyotrophic lateral sclerosis. Cell Mol Life Sci. 2014;71(6):999–1015.
Liu J, Wang F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front Immunol. 2017;8:1005.
Dutta K, et al. Mitigation of ALS Pathology by Neuron-Specific Inhibition of Nuclear Factor Kappa B Signaling. J Neurosci. 2020;40(26):5137–54.
Izrael M, Slutsky SG, Revel M. Rising Stars: Astrocytes as a Therapeutic Target for ALS Disease. Front Neurosci. 2020;14:824.
Jawaid A, et al. Does apolipoprotein E genotype modify the clinical expression of ALS? Eur J Neurol. 2011;18(4):618–24.
Li YJ, et al. Apolipoprotein E is associated with age at onset of amyotrophic lateral sclerosis. Neurogenetics. 2004;5(4):209–13.
Canosa A, et al. Correlation between Apolipoprotein E genotype and brain metabolism in amyotrophic lateral sclerosis. Eur J Neurol. 2019;26(2):306–12.
Govone F, et al. Lack of association between APOE gene polymorphisms and amyotrophic lateral sclerosis: a comprehensive meta-analysis. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(7–8):551–6.
Andres-Benito P, et al. Amyotrophic lateral sclerosis, gene deregulation in the anterior horn of the spinal cord and frontal cortex area 8: implications in frontotemporal lobar degeneration. Aging (Albany NY). 2017;9(3):823–51.
Mouzat K, et al. Liver X Receptor Genes Variants Modulate ALS Phenotype. Mol Neurobiol. 2018;55(3):1959–65.
Ingre C, et al. Lipids, apolipoproteins, and prognosis of amyotrophic lateral sclerosis. Neurology. 2020;94(17):e1835–44.
Gonzalez De Aguilar JL. Lipid Biomarkers for Amyotrophic Lateral Sclerosis. Front Neurol. 2019;10:284.
Dodge JC, et al. Neutral Lipid Cacostasis Contributes to Disease Pathogenesis in Amyotrophic Lateral Sclerosis. J Neurosci. 2020;40(47):9137–47.
Abdel-Khalik J, et al. Defective cholesterol metabolism in amyotrophic lateral sclerosis. J Lipid Res. 2017;58(1):267–78.
Chaves-Filho AB, et al. Alterations in lipid metabolism of spinal cord linked to amyotrophic lateral sclerosis. Sci Rep. 2019;9(1):11642.
Schirmer L, et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature. 2019;573(7772):75–82.
Lill CM, et al. Closing the case of APOE in multiple sclerosis: no association with disease risk in over 29 000 subjects. J Med Genet. 2012;49(9):558–62.
Fazekas F, et al. Apolipoprotein E epsilon 4 is associated with rapid progression of multiple sclerosis. Neurology. 2001;57(5):853–7.
Tamam Y, et al. Association of apolipoprotein E genotypes with prognosis in multiple sclerosis. Eur Rev Med Pharmacol Sci. 2011;15(10):1122–30.
Lumsden AL, et al. Apolipoprotein E (APOE) genotype-associated disease risks: a phenome-wide, registry-based, case-control study utilising the UK Biobank. EBioMedicine. 2020;59:102954.
Carlin C, et al. Involvement of apolipoprotein E in multiple sclerosis: absence of remyelination associated with possession of the APOE epsilon2 allele. J Neuropathol Exp Neurol. 2000;59(5):361–7.
Radikova Z, et al. Lipoprotein profiling in early multiple sclerosis patients: effect of chronic inflammation? Lipids Health Dis. 2020;19(1):49.
Noori H, et al. The correlation between dyslipidemia and cognitive impairment in multiple sclerosis patients. Mult Scler Relat Disord. 2019;36:101415.
Mailleux J, et al. Active liver X receptor signaling in phagocytes in multiple sclerosis lesions. Mult Scler. 2018;24(3):279–89.
Hindinger C, et al. Liver X receptor activation decreases the severity of experimental autoimmune encephalomyelitis. J Neurosci Res. 2006;84(6):1225–34.
Pineda-Torra I, et al. Disrupted Lipid Metabolism in Multiple Sclerosis: A Role for Liver X Receptors? Front Endocrinol (Lausanne). 2021;12:639757.
Minagar A, Alexander JS. Blood-brain barrier disruption in multiple sclerosis. Mult Scler. 2003;9(6):540–9.
Geng J, et al. Blood-Brain Barrier Disruption Induced Cognitive Impairment Is Associated With Increase of Inflammatory Cytokine. Front Aging Neurosci. 2018;10:129.
Engel S, Graetz C, Salmen A, Muthuraman M, Toenges G, Ambrosius B, Bayas A, Berthele A, Heesen C, Klotz L, Kümpfel T, Linker RA, Meuth SG, Paul F, Stangel M, Tackenberg B, Then Bergh F, Tumani H, Weber F, Wildemann B, Zettl UK, Antony G, Bittner S, Groppa S, Hemmer B, Wiendl H, Gold R, Zipp F, Lill CM, Luessi F; German Competence Network of Multiple Sclerosis. Is APOE ε4 associated with cognitive performance in early MS? Neurol Neuroimmunol Neuroinflamm. 2020;7(4):e728. https://doi.org/10.1212/NXI.0000000000000728.
Parmenter BA, et al. Cognitive impairment in patients with multiple sclerosis: association with the APOE gene and promoter polymorphisms. Mult Scler. 2007;13(1):25–32.
Prins M, et al. Pathological differences between white and grey matter multiple sclerosis lesions. Ann N Y Acad Sci. 2015;1351:99–113.
Schirmer L, et al. Diversity and Function of Glial Cell Types in Multiple Sclerosis. Trends Immunol. 2021;42(3):228–47.
Ponath G, et al. Myelin phagocytosis by astrocytes after myelin damage promotes lesion pathology. Brain. 2017;140(2):399–413.
Plemel JR, et al. Microglia response following acute demyelination is heterogeneous and limits infiltrating macrophage dispersion. Sci Adv. 2020;6(3):eaay6324.
Cignarella F, et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 2020;140(4):513–34.
Le Guen Y, et al. Association of Rare APOE Missense Variants V236E and R251G With Risk of Alzheimer Disease. JAMA Neurol. 2022;79(7):652–63.
Rasmussen KL, et al. Plasma levels of apolipoprotein E and risk of dementia in the general population. Ann Neurol. 2015;77(2):301–11.
Zokaei N, et al. Sex and APOE: A memory advantage in male APOE epsilon4 carriers in midlife. Cortex. 2017;88:98–105.
Dinan TG, Cryan JF. Gut instincts: microbiota as a key regulator of brain development, ageing and neurodegeneration. J Physiol. 2017;595(2):489–503.
Baird PN, et al. The epsilon2 and epsilon4 alleles of the apolipoprotein gene are associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2004;45(5):1311–5.
Wang SK, Cepko CL. Targeting Microglia to Treat Degenerative Eye Diseases. Front Immunol. 2022;13:843558.
Marchesi N, et al. Ocular Neurodegenerative Diseases: Interconnection between Retina and Cortical Areas. Cells. 2021;10(9):2394.
Hu ML, Quinn J, Xue K. Interactions between Apolipoprotein E Metabolism and Retinal Inflammation in Age-Related Macular Degeneration. Life (Basel). 2021;11(7):635.
Acknowledgements
Not applicable.
Funding
Open access funding provided by Lund University. This work was supported by The Strategic Research Area MultiPark (Multidisciplinary Research in neurodegenerative diseases) at Lund University, the Swedish Alzheimer Foundation, the Swedish Brain Foundation, Crafoord Foundation, Swedish Dementia Association, G&J Kock Foundation, Olle Engkvist Foundation, the Swedish Medical Research Council, the Swedish Parkinson Foundation, the A.E. Berger Foundation, the Thurings Foundation, and the Swedish mental health foundation to T.D., M.S, A.B-S. The Royal Physiographic Society of Lund grant to R.F–C. SYNDEGEN, Marie Curie grant n°721802 to G.K.G and S.K. We also thank the Spanish Ministerio de Ciencia e Innovación/FEDER/UE/PID2021-124096OB-I00; the Spanish Junta de Andalucia/FEDER/EU P18-RT-1372 and the Spanish FEDER I + D + i-USE US-1264806 to J.L.V.
Author information
Authors and Affiliations
Contributions
Design and conceptualization by R.F–C and T.D. R.F–C, S.K, J.F-R, J.G-R, L.C-F, M.S, I.M, A.B-S, J.L.V, H.N, G.K.G contributed in writing the manuscript. R.F–C and S.K edited and formatted the manuscript. J. F-R, H.N, G.K.G and T.D revised, supervised, and corrected the manuscript. J.G-R created the figures and figure legends. All the authors have approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fernández-Calle, R., Konings, S.C., Frontiñán-Rubio, J. et al. APOE in the bullseye of neurodegenerative diseases: impact of the APOE genotype in Alzheimer’s disease pathology and brain diseases. Mol Neurodegeneration 17, 62 (2022). https://doi.org/10.1186/s13024-022-00566-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-022-00566-4