
Abstract
The Alzheimer’s disease (AD) afflicted brain is neuropathologically defined by extracellular amyloid-β (Aβ) plaques and intraneuronal neurofibrillary tangles composed of hyperphosphorylated tau protein. However, accumulating evidence suggests that the presynaptic protein α-synuclein (αSyn), mainly associated with synucleinopathies like Parkinson’s disease (PD), dementia with Lewy bodies (DLB) and multiple system atrophy (MSA), is involved in the pathophysiology of AD. Lewy-related pathology (LRP), primarily comprised of αSyn, is present in a majority of autopsied AD brains, and higher levels of αSyn in the cerebrospinal fluid (CSF) of patients with mild cognitive impairment (MCI) and AD have been linked to cognitive decline. Recent studies also suggest that the asymptomatic accumulation of Aβ plaques is associated with higher CSF αSyn levels in subjects at risk of sporadic AD and in individuals carrying autosomal dominant AD mutations. Experimental evidence has further linked αSyn mainly to tau hyperphosphorylation, but also to the pathological actions of Aβ and the APOEε4 allele, the latter being a major genetic risk factor for both AD and DLB. In this review, we provide a summary of the current evidence proposing an involvement of αSyn either as an active or passive player in the pathophysiological ensemble of AD, and furthermore describe in detail the current knowledge of αSyn structure and inferred function.
Similar content being viewed by others

Background
Alzheimer’s disease (AD) is currently untreatable, the leading cause of dementia and the most prevalent neurodegenerative disorder worldwide [1,2,3]. The disease afflicts tens of millions of patients and burdens scores of caregivers [4]. The vast majority of AD cases arise sporadically either in the form of late-onset AD in individuals ≥65 years old (~90% of patients), or as early-onset AD when patients become AD symptomatic between ~45-65 years old (~6-16% of patients)[1, 5]. A small group of patients (~1%) have early-onset autosomal dominant AD (ADAD) caused by mutations in either the PSEN1, PSEN2 or APP genes. The gradual cognitive decline and eventual dementia due to AD is thought to relate to the increased deposition of insoluble protein aggregates in the form of extracellular senile plaques enriched with aggregated amyloid-β peptide (Aβ), and intracellular neurofibrillary tangles (NFTs) primarily comprised of hyperphosphorylated microtubule-associated protein tau (tau). However, there is significant clinical and pathological heterogeneity between AD patients at all stages of AD pathogenesis. Advances in functional neuroimaging have made ante-mortem identification of aggregated forms of Aβ and tau possible, and along with fluid biomarkers, specifically cerebrospinal fluid (CSF) protein levels of Aβ residues 1-42 (Aβ1-42), total tau (t-tau) and phosphorylated tau (pThr181) (p-tau), offer a high degree of sensitivity and specificity for detecting pre-clinical, prodromal and probable AD [6,7,8,9].
Amongst the repertoire of genetic and lifestyle related risk factors associated with sporadic AD [10,11,12], the most significant risk factors are a family history of sporadic AD, increasing age (≥65 years old) and the presence of the epsilon-4 (ε4) allele of the APOE gene (APOEε4) [1]. After the age of 65 the risk for developing AD doubles every five years eventually effecting >25% of people ≥85 years old [13], while individuals who are APOEε4 heterozygous or homozygous have a 3-15-fold increased risk of AD, respectively [14,15,16]. Moreover, it has been recently shown that the increased risk associated with the APOEε4 allele and AD symptom onset is more pronounced in early onset sporadic AD patients and females. A non-proportional clinical decline was found in both sexes, however, this began at age 75 in females compared to age 80 in males [17].
Interestingly, the APOE gene variants not only affect the risk of AD but also the closely related disorder dementia with Lewy bodies (DLB). Similar to the case with AD, the APOEε4 variant significantly increases the risk of DLB, whereas the APOEε2 variant appears to be protective against both disorders [16]. Dementia with Lewy bodies, as the name implies, is characterized by the occurrence of intracellular Lewy bodies and Lewy neurites which contain high concentrations of aggregated α-synuclein (αSyn). These patients frequently exhibit AD pathology, including Aβ plaques and to some degree tau pathology [18] which ante-mortem can be reflected in an AD-like CSF AD biomarker profile [19,20,21]. The implications of co-occurring AD pathology in DLB are currently unknown. Importantly, co-morbid αSyn pathology has been reported in more than half of all autopsy-confirmed AD cases [22,23,24], and more than 90% of patients with ADAD due to mutations in the PSEN1 gene exhibit αSyn pathology specifically in the amygdala [25]. Studies from our own laboratory and those of others have provided results supporting the notion that αSyn may be involved in the earliest stages of AD development, reflected in altered CSF αSyn levels and associations with brain Aβ-plaque deposition in both sporadic AD and ADAD cases [26, 27]. Importantly, our recent study described a novel link between the APOEε4 risk allele, CSF αSyn levels and Aβ deposition at the very early stages of AD development [27]. Therefore, we speculate that αSyn indeed contributes to the pathophysiology of AD as an underappreciated member of the pathological AD ensemble. In the current report we summarize the evidence provided by clinical and experimental studies linking αSyn to the pathophysiology of AD.
Alzheimer’s disease pathophysiology and diagnosis
Alzheimer’s disease is an untreatable neurodegenerative disorder clinically defined by gradual cognitive decline with impairments in executive function, language, praxis, and visual processing that eventually lead to dementia. Pathologically the symptoms can be correlated to the loss of synapses, neurons, neuropil and an overall reduction of grey matter [28, 29]. The bulk of AD cases arise sporadically after the age of 65 years while roughly 10% [30] of AD diagnoses occur before the age of 65 years (early-onset AD). Many AD patients first present with subjective memory complaints then progress, at variable rates, to mild cognitive impairment (MCI) before ultimately being diagnosed with probable AD [31,32,33]. A small subpopulation (<1%) of early-onset AD patients harbor highly penetrant autosomal dominant mutations in the APP gene which encodes the amyloid precursor protein (APP), or within the PSEN1 and PSEN2 genes encoding two independent catalytic subunits of the APP protease γ-secretase, specifically presenilin-1 (PS1) and presenilin-2 (PS2) [34]. Consequently, the direct link between APP, APP processing and AD formed the basis of the widely accepted amyloid cascade hypothesis which postulates that a protracted asymptomatic disease phase with well-defined spatiotemporal accumulations of pathological Aβ plaques in the temporal lobe, frontal lobe and limbic system precedes widespread neocortical and limbic NFTs, symptom onset, cognitive decline and dementia [34]. Therapeutics designed to reduce Aβ aggregate load or inhibit either β- or γ-secretase have exhibited some promising results in animal models, but have not translated successfully into clinical trials, exemplified by well-publicized failures of costly Phase-II and –III clinical trials. Subsequently, the amyloid cascade hypothesis has become target of a fierce debate [35,36,37]. As a result, many proponents of the amyloid cascade hypothesis have now shifted their focus upstream to examine the toxicity of soluble mono- and multimeric forms of Aβ and/or tau due to a considerable body of evidence supporting this theory [38, 39].
An accurate clinical diagnosis of probable AD is problematic due to the considerable symptomatic heterogeneity between dementia patients [40]. As a result, a definite diagnosis of AD is still only possible upon post-mortem examination [41]. With the aim to improve diagnostics, a concerted push to define robust biomarker-based diagnostic frameworks for pre-clinical and probable AD is currently underway [42, 43]. For example, ante-mortem diagnosis of probable AD may be greatly improved by advances in neuroimaging and CSF biomarker analysis [6]. Structural imaging using magnetic resonance imaging (MRI) combined with functional PET radiotracers such as glucose analogues, Aβ plaque and NFT-specific ligands, have provided seminal insights into the molecular and physical changes occurring from the early to advanced stages of AD [44]. Accumulations of Aβ aggregates can begin years to decades prior to the onset of symptoms but appear to plateau once individuals become symptomatic [6]. The proposed spread of Aβ pathology tends to follow a spatiotemporal pattern beginning in the neocortex and subsequently spreading to the allocortex, basal ganglia, midbrain, and finally appearing in the pons and cerebellum [45]. Tau pathology also begins to develop during the pre-clinical phase of AD but much later relative to Aβ plaques. The occurrence of NFT continues to increase once patients are symptomatic and correlate well with AD symptom onset and cognitive decline [46]. Generally, AD NFT pathology is believed to start in the transentorhinal region and propagate to the entorhinal region, hippocampus and temporal neocortex, superior temporal neocortex, and eventually the entirety of the neocortex [45].
With brain PET imaging techniques for tau and Aβ plaque mainly still under development and/or refinement, the most utilized and informative AD biomarkers are found primarily in the CSF, and to a lesser extent in blood [47]. In CSF, the established AD biomarker panel supporting a diagnosis of probable AD are reductions in Aβ1-42 or the Aβ1-42/Aβ1-40 ratio, and increased levels of total tau and phosphorylated (pThr181) tau. This combination of CSF biomarkers is also sensitive enough to detect asymptomatic pre-clinical AD patients and can predict the decline from MCI to AD [48]. Increased levels of CSF total tau and phosphorylated tau in particular show a robust positive correlation to cognitive decline, a finding also verified by tau PET imaging [49].
Post-mortem observations of α-synuclein in the AD brain
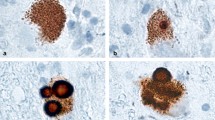
In addition to what we know as classical AD brain pathology including Aβ and tau lesions, numerous studies have also documented the occurrence of co-morbid αSyn or Lewy-related pathology (LRP) in more than 50% of autopsy-confirmed AD brains [22,23,24], please see Table 1 for an overview of the literature. Initially, Ueda and coworkers identified and described a non-Aβ peptide fragment, which we now know to be the non-amyloid-β component (NAC) region of αSyn [54], enriched in Aβ plaques isolated from a single patient. These results were later confirmed in a larger cohort of autopsy-confirmed AD cases [56]. Upon closer examination it indeed has turned out that a substantial proportion of AD patients examined at autopsy exhibited LRP in the neocortex [52, 57], limbic system [22,23,24, 61,62,63, 65], and to a lesser extent in the substantia nigra [23, 65]. Co-occurring LRP may relate to visual hallucinations [63], extrapyramidal symptoms [51, 53, 72] and a shorter more aggressive disease course [59]. Although fewer post-mortem studies have investigated the occurrence of LRP in ADAD cohorts, two studies have shown that individuals with PSEN1 mutations have increased, yet heterogeneous limbic LRP [22, 25]. In a small, but notable ADAD kindred study, three related family members carrying the S170F PSEN1 mutation had widespread limbic, neocortical and brainstem LRP, dementia onset during the third decade of life, severe symptoms and death by the fourth to fifth decade of life [64].
Interestingly, the co-occurrence of LRP in AD is frequently associated with the immunohistochemical co-localization of αSyn and tau pathology [22, 24, 61, 63], and to a lesser extent αSyn and Aβ pathology [62, 65]. Co-immunoprecipitation experiments have shown that αSyn monomers, dimers, trimers and pentamers bind to monomeric Aβ in post-mortem samples from AD and DLB brains [67]. Importantly, two reports from Larson, Lesné and co-workers demonstrated significantly increased levels of intracellular soluble αSyn monomers and oligomers in the inferior temporal lobe of AD brains without any detectable LRP [69, 73]. Specifically, when compared to brain samples from cognitively healthy controls, post-mortem AD brains exhibited increased 17-, 28-, 35- and 56-kDa, but not 72-kDa αSyn species, which correlated to AD-related cognitive decline and reduced synapsin expression within the temporal lobe. Thus, an increased presence of potentially toxic forms of soluble intracellular αSyn, direct interactions between oligomeric αSyn and monomeric Aβ, and the frequent co-localization of αSyn with NFTs and Aβ plaques in brain regions susceptible to AD neurodegeneration may explain the more severe clinical symptoms often found in AD patients with co-occurring LRP. More importantly, the results provided by Larson and colleagues [69, 73] suggest that soluble αSyn and the local concentrations thereof may play an important role in AD regardless of LRP. It is also pertinent to note that LRP [74] and AD-type pathology [75] have been documented in cognitively healthy individuals upon autopsy. For instance, 24% of the autopsied normal control cases from the University of Kentucky Alzheimer’s Disease Center exhibited LRP [74]. This has led to the speculation that these subjects harboring asymptomatic pathology are in a presymptomatic disease stage [74], whilst others argue that insoluble aggregates are inherently inert and/or protective [76]. Yet others have postulated that protein toxicity is context-dependent [77].
Cerebrospinal fluid α-synuclein levels in synucleinopathies versus AD
Whether accumulations of LRP or altered local concentrations of αSyn in the brain parenchyma can be monitored in CSF, similar to the situation with Aβ and tau [49, 78], remains to be established. Accumulating evidence from our own laboratory and those of others have shown that patients with synucleinopathies, mainly Parkinson’s disease (PD) and DLB, frequently exhibit reduced levels of CSF αSyn [19, 79,80,81], for an overview of the literature please see Table 2. Interestingly, reduced CSF αSyn levels do not appear to be associated with clinical or imaging measures of PD severity [95]. Although a substantial amount of results now propose a synucleinopathy-specific reduction of CSF αSyn, consensus on a CSF profile of reduced αSyn in synucleinopathy patients is still lacking mainly due to the high variability between the various αSyn quantification assays, differences in the species of αSyn being quantified and inter-laboratory variability [96]. Nevertheless, several studies including our own have shown that lower CSF αSyn could distinguish PD from AD patients [81, 97]. In contrast to the reported lower CSF αSyn levels in patients with PD and DLB, numerous studies have documented unaltered or slightly increased CSF αSyn levels in patients with MCI and AD [27, 80,81,82, 86, 90, 91, 93, 94]. Higher CSF αSyn levels were also shown to be positively associated with disease progression from sporadic MCI to AD [27, 92] and negatively correlated to cognitive function (MMSE scores)[27, 86, 93]. Our own study in addition exhibited an effect of the APOEε4 variant in MCI patients that progressed to fulfill the diagnostic criteria for AD over a 24 month period. In these patients we documented a dose-response relationship between the APOEε4 allele and CSF αSyn levels where APOEε4 homozygous carriers exhibited the highest CSF αSyn concentrations [26]. Furthermore, in a cross-sectional sample of participants carrying ADAD mutations from the Dominantly Inherited Alzheimer Network (DIAN) we found that CSF αSyn levels were significantly correlated to the estimated years from disease onset [27]. We also found that specifically in asymptomatic (CDR<0.5) DIAN participants who carried ADAD mutations and who also were APOEε4 positive, higher CSF αSyn levels significantly correlated to PiB-PET Aβ plaque burden in several of the ‘early Aβ-accumulating brain areas’. Interestingly, the direction of this relationship between CSF αSyn levels and brain Aβ plaque burden was positive in DIAN participants that were asymptomatic, however, the direction changed to negative when symptoms began to surface (CDR>0.5). Although we found significant associations between CSF αSyn levels and PiB-PET Aβ burden, we, rather unexpectedly, failed to record any significant correlations between CSF αSyn and Aβ1-42 [27]. A relationship between brain Aβ load and CSF αSyn levels was previously proposed by Vergallo and co-authors who examined cognitively normal individuals with subjective memory complaints stratified by AD biomarkers [26]. The same study further confirmed the by us and others previously reported strong correlation between CSF levels of αSyn and both t-tau and p-tau [19, 81, 98]. The biological relevance of the consistently reported correlation between αSyn and tau in CSF is not known. However, both αSyn and tau appear to be secreted by similar exosome-mediated release mechanisms [99, 100] and both proteins can also be endocytosed via the heparin sulfate proteoglycans (HSPGs) pathway [101]. Hence, the extracellular levels of αSyn and tau may be regulated by similar mechanisms reflected in a strong consistent correlation between the CSF concentrations of these proteins in both control subjects and AD patients. Together these results suggest an association between biological processes reflected in altered CSF αSyn levels and the asymptomatic development of AD pathology, which in turn may be under the influence of APOEε4.
α-synuclein structure
α-synuclein is a small 140-residue (14-kDa, pKa= 4.7) protein translated from 5 exons of the SNCA gene located on the long arm of chromosome four at position 21 (4q21.3-q22) [102]. The primary structure of αSyn (Fig. 1a) is subdivided into three regions: an amphipathic N-terminal (amino acids (a.a.) 1-60), a hydrophobic core region known as the NAC (a.a. 61-95), and an unstructured acidic C-terminal (a.a. 96-140). Although the overall secondary structure of αSyn has yet to be determined there is a consensus that the N-terminal and NAC can together fold into α-helices (Fig. 1b). Multiple in vitro experiments have shown that the N-terminal and NAC of αSyn can form a single α-helix (a.a. 3-92) when bound to membranes of low curvature [106,107,108,109] or two amphipathic anti-parallel α-helices (a.a. 3-37 and a.a. 45-92) when bound to lipid membranes of high curvature [103, 110, 111] (Fig. 1b). Within the N-terminal and NAC are seven highly conserved imperfect 11-residue repeats based on the XKTKEGVXXXX motif which form the structural backbone of the α-helices [112] and may also stabilize the native conformations of αSyn [113, 114] (Fig. 1a). The NAC appears to be essential in the aggregation of αSyn due to its ability to adopt β-sheet structures (Fig. 1c) [115] and has also been shown elicit seeding-competent amyloids [116, 117]. Introducing hydrophilic point mutations within the NAC significantly reduces the oligomerization and fibrillation potential of αSyn, further suggesting that the NAC plays a central role in the aggregation of αSyn [116]. These 11-residue repeats are also found in other lipophilic proteins such as apolipoproteins which also form α-helices that interact with membranes, however the a.a. sequence is unique to the synucleins alone [118]. A noteworthy report found that the loss of two repeat motifs led to the formation of β-sheet enriched oligomers and amyloid fibrils, while adding two additional repeats inhibited the formation of both β-sheet enriched oligomers and amyloid fibrils (Fig. 1c). These findings thus highlight the structural and physiological importance of the highly conserved features of αSyn [119]. Importantly, all known autosomal dominant point mutations that cause PD reside within these repeat motifs, specifically A30P[120], E46K[121], A53T[122], G51D[123], and H50Q[124] (Fig. 1a). The location and severity of these pathogenic mutations shows the important relationship between the structure and function of αSyn (Fig. 1b-c).

The structures of αSyn. a Primary structure of αSyn. N-terminal residues are green, the non-amyloid component (NAC) residues are blue, C-terminal residues are grey and disease associated point mutations are red. b Molecular model of a crystal structure of micelle bound human αSyn, PDB 1XQ8[103]. Color scheme is same as in a. c Molecular model of a cryo-electron microscopy structure of four human αSyn proteins (residues 1-121) in a fibril, PDB 6H6B[104]. Color scheme is same as in a. Molecular models created using Deep View Swiss PDB Viewer [105]
The C-terminal of αSyn (a.a. 96-140) is inherently acidic and remains disordered even as the N-terminal adopts secondary structures [125]. There is considerable debate regarding the inherent role of the C-terminal [126] with studies suggesting that it: i) interacts with the N-terminal or NAC to stabilize the native unfolded structure thus preventing fibrillation [127], ii) interacts with the N-terminal or NAC to form compacted monomeric structures which resist the propensity to aggregate at low temperatures [128, 129], iii) binds to calmodulin [130, 131], iv) binds to Fe3+ during αSyn-catalyzed Fe3+→ Fe2+ reduction [132], or iv) binds to both calcium and synaptic vesicles thereby influencing synaptic vesicle clustering, αSyn aggregation kinetics and/or dopamine-induced cytotoxicity [133].
Many reports have confirmed the widely held assumption that αSyn primarily exists as an unfolded protein in dynamic equilibrium with smaller pools of membrane-bound αSyn, however, others have advanced the theory that αSyn exists in pools of soluble helical multimers [134]. The role and nature of αSyn multimers are unclear. Burré and colleagues reported that αSyn multimers assemble when bound to vesicles engaged in SNARE dependent exocytosis [135], while Dettmer and co-authors instead postulated that membrane-free αSyn multimers, specifically αSyn tetramers, form the largest steady-state pool of intracellular αSyn [134]. The latter group has also reported that in human brain lysates, human neuronal cultures, mouse models, and in mouse/human/goat cell lines expressing familial PD αSyn mutations (A53T or E46K), a decrease in the tetramer:monomer ratio correlated to increased neurotoxicity. This led the authors to conclude that αSyn tetramers and low molecular weight multimers disassemble into less-stable higher-energy monomers in order to bind to membranes, however, when monomers are in excess they are also potentially neurotoxic [136,137,138,139,140].
Thus, αSyn appears to exist in a dynamic physical equilibrium fluctuating between potentially numerous metastable homogenous or heterogeneous secondary, tertiary and quaternary structures involving numerous binding partners and physiological processes. To improve our understanding of whether specific species of αSyn lose or gain functions that may promote or intermingle with AD pathophysiological processes more detailed studies of αSyn in the context of AD are needed.
α-synuclein expression and function
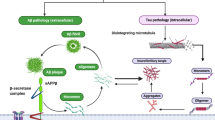
Although abundantly expressed in the kidneys and blood cells, αSyn is predominantly expressed within the presynaptic terminals of neurons [141,142,143] and can also be found in the neuronal nucleus where it appears to co-localize with the nuclear membrane [144]. Expression of αSyn can mainly be found in the cerebral cortex, cerebellum, striatum, thalamus, hippocampus and olfactory bulb [141, 145]. The factors regulating the transcription of αSyn remain largely unknown, however, SNCA has shown an affinity for zinc-finger motifs and responds accordingly to the transcription factors GATA-binding factor 2 (GATA2) [146] and zinc finger and SCAN domain-containing protein 21 (ZSCAN21, [147]. Similar to tau, it has been suggested that αSyn is secreted within exosomes in a calcium-dependent manner [100]. It was further proposed that αSyn can be taken up by cells via HSPGs similar to what has been reported for tau and Aβ [101](Fig. 2). Interestingly, APOE was shown to reduce cellular uptake of Aβ by competing for HSPGs [148] and specifically the APOE4 isoform limited the cellular uptake of αSyn in oligodendrocytes [149]. Several mechanisms have been reported to orchestrate the degradation of αSyn including autophagy and catabolism mediated by the ubiquitin-proteasome system [150]. Also, extracellular proteases like neurosin, also referred to as kallikrein 6, have been shown to degrade αSyn [151, 152]. Notably, the lentivirus-facilitated expression of neurosin in a mouse model of Lewy body disease enhanced clearance of αSyn and reduced αSyn pathology [153]. Hence an interplay between various molecules including APOE may determine the distribution of αSyn between the extracellular and intracellular pools. The balance between these two pools in turn may determine the physiological roles of αSyn in both compartments.

Known (solid lines) and hypothetical (dotted lines) mechanisms shuffling αSyn between the intra-and extracellular compartments with implications for the development of AD pathological lesions. The top left-hand side and middle of the figure depicts physiological processes taking place in the healthy neuron A, the middle of the figure (underneath the healthy neuron A) is the extra-neuronal space with an astrocyte. The bottom half and right-hand side of the figure depicts two independent neurons in pathophysiological conditions, neurons B and C respectively
Although the native role of αSyn is unclear it has been strongly linked to various pre-synaptic processes. Initially αSyn was suggested to play a role in learning and plasticity after George and colleagues reported upregulation of αSyn RNA in the zebra finch song control circuit during a period of song-acquisition-induced synaptic re-arrangement [154].
The first in vivo forays into Syn mouse models showed that αSyn knock-out (KO)[155, 156], β-Syn KO [157], α/β-Syn double-KO [157], and γ-Syn KO [158] animals were all viable, fertile, displayed normal brain development, morphology, synaptic ultrastructure, they had no behavioral abnormalities, and lacked any distinguishable phenotype [155,156,157,158]. However, αSyn-KO, and α/β-Syn double-KO mice exhibited reduced brain dopamine levels compared to wild-type (wt) animals and markedly different responses to electrophysiological interrogation were documented in three different studies. One study reported no differences [157], another study found disinhibition of synaptic depression following paired-pulses [155], and yet another noted prolonged recovery times after a train of stimuli pointing to αSyn having a roll in vesicle trafficking or refilling [148]. Studies using rat primary dopaminergic cell models showed that overexpression of αSyn via viral injection into the substantia nigra of mature animals reduced dopamine release and reuptake by 80-90% [159]. Therefore, one could speculate that αSyn and β-Syn may functionally compensate for one another, and that αSyn may be involved in the long-term synaptic regulation and or maintenance of dopaminergic pathways. The α/β/γ-Syn triple-KO animals were viable but had lower survival rates and age-dependent neuronal dysfunction in the hippocampus and retina [160]. This was not due to a loss of synapses, but rather due to changes in synaptic protein composition and alterations of axonal and synaptic architecture, thereby augmenting the fidelity of the signal. The electrophysiological differences between synuclein null and wt animals were shown to start at a young age but could be rescued by both human and murine αSyn expression [160], again indicating that αSyn has a presynaptic role. Mice harboring a deletion of the presynaptic SNARE complex chaperone cysteine string protein-α (CSPα) had significant neurodegeneration, shortened survival (<4 months), reduced levels SNAP-25, and hindered SNARE-complex assembly [161]. Remarkably, transgenic expression of αSyn completely rescued the neurodegeneration in CSPα-/- animals, while loss of endogenous αSyn exacerbated the neurodegeneration [161], indicating that αSyn has a compensatory, neuroprotective role within the presynaptic SNARE machinery.
Further support for the notion that αSyn has an important role within the presynaptic machinery, specifically in regards to vesicle dynamics, also came from rat primary cell culture studies where αSyn was shown to exclusively co-localize with the transmembrane synaptic vesicle protein synaptophysin [162]. Ultrastructural analysis further revealed that knock-down of αSyn resulted in a significant reduction in the distal/reserve pool of synaptic vesicles [162]. Single molecule fluorescence studies recently have also determined that 70-αSyn units bind per presynaptic vesicle, which is very similar to the ratio of synaptobrevins per vesicle, further indicating αSyn’s involvement in presynaptic vesicle trafficking and/or remodeling. A more detailed investigation regarding the potential interactions between αSyn and synaptic vesicles was conducted using transgenic mice overexpressing human αSyn tagged with GFP, and found that the transgenic neuronal cultures had augmented synaptic architecture, reduced pre-synaptic protein levels and altered release kinetics [163]. Specifically, the primary transgenic neuronal cultures exhibited a reduction in the density of pre-synaptic vesicles and a more heterogeneous distribution of synaptic vesicle sizes with more unusually large (>50nM) vesicles. The same study further showed reduced levels of VAMP2, piccolo, synapsin-1 and amphiphysin paralleled by a reduced frequency of spontaneous neuronal activity [163]. Importantly, these changes were also correlated to increasing levels of proteinase K-resistant αSyn and phosphorylated αSyn (pS129), thereby providing a potential link between synaptic dysfunction and pathological αSyn alterations. Using the same model system it was later confirmed that overexpression of αSyn lead to a specific reduction in in size of the recycling pool of synaptic vesicles and hindered intersynaptic vesicle trafficking compared to wt cultures [164]. Moreover, αSyn-KO cultures showed the opposite effect, as they had a larger recycling pool and increased intersynaptic trafficking of synaptic vesicles. Interestingly, further experiments using transcriptionally encoded human αSyn dimers overexpressed in dissociated αSyn-KO hippocampal neurons resulted in an increased clustering of synaptic vesicles, retarded vesicle motility and augmented recycling kinetics compared to wt neurons [165]. The increased ability of αSyn multimers to cluster synaptic vesicles was significantly reduced by the introduction of seven hydrophilic lysine residues which were thought to disrupt the membrane associated αSyn α-helix, but somewhat surprisingly, there was no difference in synaptic vesicle binding compared to wt αSyn [165]. These findings were further verified and expanded upon by Nemani et al. using murine hippocampal neurons overexpressing human αSyn. They found that compared to wt neurons, αSyn overexpression reduced the size of the reserve- and ready releasable pools of synaptic vesicles, inhibited post-endocytosis vesicle clustering and attenuated SNARE mediated exocytosis [166].
The dynamics of the presynaptic exocytotic/endocytotic fusion pore are also potentially mediated in part by αSyn. Logan et al. showed that overexpressing human αSyn in WT mice neurons retarded the closure of the fusion pore, reduced the number of exocytotic events, but also increased the kinetics of exocytosis which may be the neurons attempt to compensate for the loss in exocytosis frequency [167]. Moreover, when the same authors used wt mice chromaffin cells expressing the A35T αSyn mutation dilation, rather than closure, of the fusion pore was impaired [167, 168].
We find it rather odd that a protein so highly conserved, synaptically enriched, and potentially toxic is at the same time non-essential, and non-specific in its function. We speculate that the transient nature of αSyn’s inherent structures and the large variation in murine models, cell lines and the use of various non-standardized peptides and proteins contribute to the previously reported variation in outcome [169]. To improve our understanding of αSyn during normal and pathological conditions such as in AD, concerted efforts using both in vitro and in vivo models, preferably in a translational manner, and techniques that readily can capture the various molecular species of αSyn are highly warranted.
Experimental evidence supporting a role for α-synuclein in AD pathophysiology
To improve the understanding of potential molecular pathways through which αSyn may be connected to the pathophysiology of AD and neurodegeneration at large, various studies have employed transgenic AD mouse lines in which αSyn was either manipulated, reduced or eliminated. The results of these various studies are however not fully consistent and consensus-building conclusions remain to be drawn. We speculate that at least some of these inconsistencies depend on inter-model variabilities. For example, Spencer and colleagues reported that APP Tg/α-syn KO animals exhibited reduced cholinergic and hippocampal neurodegeneration, less behavioral deficits and increased expression of Rab proteins [170]. A similar APP/αSyn-KO mouse model in the hands of Khan and co-workers exhibited enhanced memory performance but significantly aggravated Aβ plaque pathology. The same authors also showed that elimination of αSyn prevented premature death in the APP mice paralleled by a remarkable rescue of behavioral parameters and prevention of pathological neuronal cell cycle re-entry (CCR). The APP/αSyn-KO mice further exhibited reduced levels of extracellular Aβ oligomers and less αSyn oligomers. In addition, Khan et al. also overexpressed human wt-αSyn in mutant APP mice (APP/αSyn) and found that the behavioral and pathological phenotypes had reversed compared to the APP/αSyn-KO mice. Although the APP/αSyn mice had a significantly reduced Aβ plaque burden, they had exacerbated behavioral deficits, more deleterious neuronal cell cycle re-entry, and increased levels of Aβ oligomers, αSyn oligomers and tau pathology [171]. The expression of APP in the two APP/αSyn-KO mouse models used by Spenser and Khan respectively, was driven by either the mouse Thy1 or the PGDFβ promoter [170, 171]. We speculate that the variability in the reported Aβ pathology between these mouse models may be directly linked to the regulation of APP expression in these mice.
Another remarkable study showed that cortical shRNA-induced reduction of SNCA expression in rats with complete T10-T11 spinal cord transections, resulted in a significant increase in neuronal survival, decreased apoptosis, and stimulated motor and sensory recovery in the hindquarters. In contrast upregulation of SNCA expression exacerbated neuronal death and impeded recovery [172]. The neuroprotective mechanism conferred by down-regulating or KO of αSyn is unknown but may be due to an enhanced ability to sequester toxins in vesicles [173], or through reducing potentially pathologic post-translational modifications of αSyn [174]. Further, the reduction of αSyn in αSyn-KO/KD models could regulate important hippocampal glutamatergic mGluR5 and NMDA receptors [175], and has the potential to regulate various members of the Rab GTPase family which are key intracellular membrane trafficking proteins [175, 176].
Identification of potential mechanisms specifically linking αSyn to AD pathophysiology, in addition to the prominent roles αSyn appears to play in orchestrating synucleinopathies like PD, DLB and MSA, would offer crucial insights into disease-specific pathogenic processes and hopefully allow for the development for more potent interventions. A multitude of studies have already shown molecular interactions between αSyn and tau which may have implications for various neurodegenerative diseases [177]. For example, the C-terminal of αSyn can directly interact with the microtubule binding domain of tau [178], and αSyn seeds have been shown to induce intracellular aggregation of tau [179]. Interactions between tau and αSyn could potentially also be mediated by the serine/threonine kinase glycogen synthase kinase 3-β (GSK3β)[180]. The GSK3β participates in multiple cellular signaling pathways related to development, proliferation, viability, immunity and apoptosis. The GSK3β can orchestrate pathological tau-phosphorylation at over two-dozen disease-associated sites [181,182,183,184]. Other reports have suggested that GSK3β activity could be increased by Aβ, inhibited acetylcholine synthesis [185], and could trigger apoptosis [183]. Hence, the active role GSK3β plays in AD pathophysiology appears to be established. Importantly, several lines of evidence propose an association between GSK3β and αSyn. Kozikowski and co-authors used human neuroblastoma SH-SY5Y cells stably expressing αSyn to screen novel GSK3β inhibitors, and found that the selective inhibition of GSK3β not only prevented tau phosphorylation but also reduced αSyn protein levels [186]. Later reports from different groups confirmed the findings of Kozikowski and colleagues and further demonstrated that αSyn can regulate the activity, but not the expression of GSK3β[187]. Interestingly, the αSyn-NAC region can directly mediate the activity of GSK3β by inducing GSK3β autophosphorylation leading to GSK3β-mediated tau-hyperphosphorylation [180]. However, this was not αSyn specific as β-synuclein was also shown to mediate the same signaling cascade [180].
Whether extracellular levels of αSyn would be of relevance to the activity of GSK3β and its’ effects on tau protein was recently investigated in vitro by Gassowska and colleagues using the PC12 cell line. They found that exogenously added αSyn increased the phosphorylation of tau at the Ser-396 site which could be prevented by the GSK3β inhibitor SB216763[188]. The same study further showed that αSyn both increased the activity of GSK3β and the protein levels thereof [188]. Whether alterations in both extracellular versus intracellular levels of αSyn specifically could promote the actions of GSK3β is an important question, highlighted by the previously described inverse alterations in CSF αSyn levels in patients with synucleinopathies compared to AD patients. Importantly, the processes governing the distribution of αSyn between the extracellular and intracellular pools may be a crucial factor in pathological downstream processes such as tau phosphorylation. As mentioned previously, αSyn can be degraded extracellularly by proteases like neurosin [151, 152] but also intracellularly by lysosomes through the chaperone-mediated autophagy pathway [189]. The latter was also confirmed to occur in vivo during normal and pathological conditions [190]. Similar to tau, αSyn can be endocytosed via the HSPG pathway [101]. This observation offers an opportunity to better understand what biological processes may underlie the increased disease risk of the APOEε4 allele for both AD and DLB [16], as APOE may be involved in the distribution of αSyn between the intracellular and extracellular space. For example, APOE was shown to compete with Aβ for cellular uptake in vitro via the HSPG pathway thereby limiting Aβ uptake [148], and APOE4 can also specifically reduce αSyn uptake in primary human oligodendrocytes [149]. With APOE as an important lipid-carrier in both the CNS and the periphery it is tempting to speculate that the APOE allele-associated alterations in lipid profiles may be linked to various Aβ, tau and αSyn pathological processes due to their ability to interact with free lipids and membranes [191, 192]. Interestingly, an age-dependent reduction in glucocerebrosidase (GCase) activity in mice was found to be associated with lipid-dependent changes in neuronal vesicular membrane compartments which accumulated lipid-stabilized αSyn and phosphorylated tau [193]. The authors specifically commented on the loss of GCase activity which under normal conditions results in the conversion of glucosylceramide into ceramide and glucose [194], and its association with lipid homeostasis in PD and DLB. They further reported a significant correlation between GCase activity and αSyn aggregation in the human PD brain which prompted them to propose that age-associated lipid changes may be related to αSyn and tau pathology [193]. We speculate that an age-related lipid dyshomeostasis could be further aggravated by APOEε4 which down-stream could translate into altered intra- versus extracellular levels of αSyn. Consequently we could hypothesize that in prodromal AD patients [27] the link between APOEε4 and higher CSF αSyn levels may be attributed to higher extracellular levels of αSyn due to a shift in the αSyn intra- versus extracellular pools possibly driven by either lipid-related changes or hampered cellular αSyn uptake in the presence of the APOE4 isoform. Further evidence linking APOE to αSyn were previously exhibited from transgenic mouse models expressing mutant human A30P αSyn (αSyn-A30P)[195]. The αSyn-A30P transgenic mice exhibited behavioral dysfunction, spinal cord astrocytosis, spinal cord motor neuron degeneration, increased levels of both insoluble αSyn and Aβ, and a substantial (4-fold) increase in APOE [195]. This strong effect that mutant αSyn had on APOE led the authors to cross the A30P transgenic mice with wt ApoE-KO mice (A30P/ApoE-KO). The resulting A30P-ApoE-KO mice still had neurodegeneration, but had increased survival rates, delayed onset of behavioral symptoms, reduced neurodegeneration, and significant reductions of Aβ aggregates [195]. Remarkably, knocking out APOE also increased the levels of monomeric αSyn, while reducing levels of oligomers and other larger αSyn aggregates.
Hence, APOEε4, which has been implicated in AD pathophysiology and pathogenesis by processes mainly linked to Aβ but also associated with Aβ-independent pathways [15], may be the missing link connecting Aβ to tau via actions of αSyn. This notion is supported by evidence showing an effect of αSyn on Aβ itself. For example, in 2006 Mandal and colleagues used nuclear magnetic resonance spectroscopy to investigate the interactions between membrane bound αSyn with Aβ1-40 or Aβ1-42 and found that both forms of Aβ interacted with bound αSyn [196]. Notably, αSyn enhanced the toxic oligomerization of Aβ1-42 leading to oligomeric Aβ1-42 mediated cleavage of αSyn and liberation of the NAC, therefore providing a plausible mechanism explaining why the NAC co-aggregates in Aβ plaques in vivo (Fig. 2). In addition, in vitro and in vivo models have shown that αSyn can interact and catalyze the oligomerization of tau [197], and form neurotoxic cation pores with Aβ [67].
Further evidence of a link between αSyn and Aβ pathology was offered from studies using mice expressing wt human αSyn (hαSyn) and human APP (hAPP) (hαSyn/hAPP). The hαSyn/hAPP model recapitulated many of the clinical symptoms and pathology of AD-LBV. Compared to animals expressing only hαSyn or hAPP, the hαSyn/hAPP animals had marked cholinergic neuron loss, intraneuronal LRP, florid Aβ plaques, exhibited marked motor dysfunction and had significant learning and memory deficits [198]. Animals expressing all three AD pathologies (Aβ, tau and αSyn) were developed by crossing 3xTg-AD mice (APPKM670/671NL, tauP301L, PS1M146V) with αSynA35T, and subsequently named AD-LBV mice. The 3xTg-AD mice readily recapitulated Aβ and tau pathology, and after breeding with αSynA35T mice the resulting AD-LBV offspring exhibited accelerated cognitive deterioration, and enhanced accumulations of Aβ, αSyn, and tau pathology compared to 3xTg-AD or αSynA53T animals [199]. The AD-LBV mice exhibited increased levels of insoluble αSyn, phosphorylated αSyn (pS129), Aβ1-42, and tau at younger ages compared to the 3xTg-AD or αSyn-A53T animals [199].
A direct involvement of αSyn in the production of Aβ (Fig. 2) in humans could be suggested by results from Winslow and colleagues who described a novel molecular interaction between presenilin 1 (PS1), an important player in the proteolytic processing of the APP to yield Aβ [200], and αSyn [201]. Complexes of αSyn and PS1 where increased in AD and DLB patients with associated PSEN1 mutations [201]. Another study showed that PSEN1 mutations associated with AD and DLB aggravated phosphorylation and accumulation of αSyn [202].
Together the described results, although varying, from numerous studies of patient samples, animal and cell models propose that αSyn may play a leading role in a number of mechanisms and processes linked to AD. However, the heterogeneity in these processes have so far precluded the identification of exact mechanisms to be replicated in both in vivo and in vitro models. Such mechanisms could not only deepen our knowledge regarding the etiology of AD but also the pathogenesis of various synucleinopathies.
Relevance of proposed α-synuclein propagation to pathophysiology of AD
As aforementioned, the neuropathological lesions of not only AD but also PD/DLB and amyotrophic lateral sclerosis (ALS) are known to spread through the brain in a rather specific pattern, which also serves as a basis for the neuropathological staging of disease severity [203]. The spreading of pathology was previously proposed to result from intra- and intercellular propagation of pathological seeds of tau, Aβ, αSyn and TDP-43 following a prion-like mechanism [204]. Specifically, the term ‘prion-like’ refers to unknown mechanisms in which toxic misfolded monomers or multimers have the innate ability to directly transmit disease to endogenous proteins by initiating an autocatalytic cycle of pathological misfolding and aggregation [203]. The most widely cited studies in support of the prion-like spread of LRP comes from post-mortem studies of two PD patient cohorts who received robust long-term intrastriatal grafts of human fetal midbrain tissue [205,206,207,208]. These studies showed that 1-5% of the grafted dopaminergic neurons exhibited detectable LRP 12-22 years post-transplantation (n=2 patients [205], n=1 patient [206], n=3 patients [207]), which increased to 11-12% after 24 years (n=1 patient [208]). A prion-like and αSyn-supported spread of AD pathology is potentially plausible as; i) it is widely known that in cell-free, cell and animal models, αSyn fibrils are readily able to catalyze the fibrillation of monomeric αSyn into aggregates of varying toxicity, ii) as mentioned herein, αSyn can also seed the aggregation of Aβ and tau, and iii) αSyn has the ability to spread to adjacent cells via secretion in exosomes [209], uptake via endocytosis [210] and cell-surface receptors like HSPGs, or even direct neuron-to-neuron [211, 212] or astrocyte-to-neuron [213] transfer via tunneling nanotubes (Fig. 2). Numerous aspects of a potential prion-like spread of αSyn in AD need to be considered including whether αSyn expression and secretion is altered and whether this is specific to certain brain areas. Importantly, the spatiotemporal spread of αSyn lesions in PD/DLB is nearly inverted to the spread of Aβ and tau pathology as observed in AD [203]. Hence the importance of specific neuronal populations and circuits to the potential spread of pathological seeds may differ between synucleinopathies like PD/DLB and AD. This notion can exemplified by the occurrence of LRP specifically in the amygdala of PSEN1 mutation carriers [25]. Although the amygdala is heavily regulated by an inhibitory network of cells, it primarily responds to glutamate resulting in the secretion of neurotransmitters including acetylcholine, dopamine, serotonin and norepinephrine [214]. Furthermore, local concentrations of molecules like APOE and neurosin may alter the extracellular concentrations of αSyn, and thereby possibly influence the downstream spread of αSyn and AD pathogenesis. Detailed studies are needed in order to elucidate why the amygdala in particular is vulnerable to αSyn pathology.
Concluding remarks
Numerous clinical studies have by now documented the presence of αSyn pathology in patients with AD, however, little attention has been devoted to αSyn as a potential target for AD disease-modification, prevention or cure. The accumulated data from human and murine cell cultures clearly suggests an involvement of αSyn in the GSK3β-mediated phosphorylation of tau which goes hand-in-hand with the strong correlations found between CSF levels of αSyn and tau in multiple neurodegenerative disorders including AD. Importantly, in vivo models also outline possible mechanisms in which αSyn-GSK3β activity could contribute to a pathological feedback loop in AD; Aβ could increase GSK3β activity which in turn can induce tau phosphorylation and αSyn production (and heterotrimeric complexes), all of which (Aβ, tau, and αSyn) can seed the aggregation of one another, leading to potentially more Aβ production, GSK3β activation (via more Aβ and αSyn), tau phosphorylation, cellular dysfunction and apoptosis. Importantly, recent data implies that the AD and DLB genetic risk factor APOEε4 may be linked to processes connecting αSyn to the core AD pathological hallmarks, Aβ plaques and NFTs. Future studies facilitated by emerging techniques to image αSyn in the brain tissue ante-mortem will increase our understanding of the distribution of αSyn, αSyn pathology and its relevance to the pathogenesis and pathophysiology of AD.
References
(USA) AsA: 2018 Alzheimer’s disease facts and figures includes a special report on the financial and personal benefits of early diagnosis. 2018.
Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2.
Savica R, Grossardt BR, Bower JH, Boeve BF, Ahlskog JE, Rocca WA. Incidence of dementia with Lewy bodies and Parkinson disease dementia. JAMA Neurol. 2013;70:1396–402.
Schulz R, Beach SR. Caregiving as a risk factor for mortality: The caregiver health effects study. JAMA. 1999;282:2215–9.
Wattmo C, Wallin ÅK. Early- versus late-onset Alzheimer’s disease in clinical practice: cognitive and global outcomes over 3 years. Alzheimers Res Ther. 2017;9:70.
Jack CR, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. The Lancet Neurology. 2013;12:207–16.
Herukka SK, Simonsen AH, Andreasen N, Baldeiras I, Bjerke M, Blennow K, Engelborghs S, Frisoni GB, Gabryelewicz T, Galluzzi S, et al. Recommendations for cerebrospinal fluid Alzheimer’s disease biomarkers in the diagnostic evaluation of mild cognitive impairment. Alzheimers Dement. 2017;13:285–95.
Simonsen AH, Herukka SK, Andreasen N, Baldeiras I, Bjerke M, Blennow K, Engelborghs S, Frisoni GB, Gabryelewicz T, Galluzzi S, et al. Recommendations for CSF AD biomarkers in the diagnostic evaluation of dementia. Alzheimers Dement. 2017;13:274–84.
Frisoni GB, Boccardi M, Barkhof F, Blennow K, Cappa S, Chiotis K, Démonet J-F, Garibotto V, Giannakopoulos P, Gietl A, et al. Strategic roadmap for an early diagnosis of Alzheimer's disease based on biomarkers. The Lancet Neurology. 2017;16:661–76.
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452–8.
Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer's disease prevalence. The Lancet Neurology. 2011;10:819–28.
Baumgart M, Snyder HM, Carrillo MC, Fazio S, Kim H, Johns H. Summary of the evidence on modifiable risk factors for cognitive decline and dementia: A population-based perspective. Alzheimers Dement. 2015;11:718–26.
Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 2013;80:1778–83.
Corder E, Saunders A, Strittmatter W, Schmechel D, Gaskell P, Small G, Roses A, Haines J, Pericak-Vance M. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–3.
Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–18.
Berge G, Sando SB, Rongve A, Aarsland D, White LR. Apolipoprotein E epsilon2 genotype delays onset of dementia with Lewy bodies in a Norwegian cohort. J Neurol Neurosurg Psychiatry. 2014;85:1227–31.
Liu L, Caselli RJ. Age stratification corrects bias in estimated hazard of APOE genotype for Alzheimer's disease. Alzheimers Dement (N Y). 2018;4:602–8.
Donaghy PC, McKeith IG. The clinical characteristics of dementia with Lewy bodies and a consideration of prodromal diagnosis. Alzheimers Res Ther. 2014;6:46.
Wennstrom M, Londos E, Minthon L, Nielsen HM. Altered CSF orexin and alpha-synuclein levels in dementia patients. J Alzheimers Dis. 2012;29:125–32.
Nielsen HM, Hall S, Surova Y, Nagga K, Nilsson C, Londos E, Minthon L, Hansson O, Wennstrom M. Low levels of soluble NG2 in cerebrospinal fluid from patients with dementia with Lewy bodies. J Alzheimers Dis. 2014;40:343–50.
Paterson RW, Slattery CF, Poole T, Nicholas JM, Magdalinou NK, Toombs J, Chapman MD, Lunn MP, Heslegrave AJ, Foiani MS, et al. Cerebrospinal fluid in the differential diagnosis of Alzheimer's disease: clinical utility of an extended panel of biomarkers in a specialist cognitive clinic. Alzheimers Res Ther. 2018;10:32.
Lippa CF, Fujiwara H, Mann DM, Giasson B, Baba M, Schmidt ML, Nee LE, O'Connell B, Pollen DA, St George-Hyslop P, et al. Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer's disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol. 1998;153:1365–70.
Hamilton RL. Lewy bodies in Alzheimer's disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000;10:378–84.
Arai Y, Yamazaki M, Mori O, Muramatsu H, Asano G, Katayama Y. α-Synuclein-positive structures in cases with sporadic Alzheimer’s disease: morphology and its relationship to tau aggregation. Brain Res. 2001;888:287–96.
Leverenz JB, Fishel MA, Peskind ER, Montine TJ, Nochlin D, Steinbart E, Raskind MA, Schellenberg GD, Bird TD, Tsuang D. Lewy body pathology in familial Alzheimer disease: evidence for disease- and mutation-specific pathologic phenotype. Arch Neurol. 2006;63:370–6.
Vergallo A, Bun RS, Toschi N, Baldacci F, Zetterberg H, Blennow K, Cavedo E, Lamari F, Habert MO, Dubois B, et al. Association of cerebrospinal fluid alpha-synuclein with total and phospho-tau181 protein concentrations and brain amyloid load in cognitively normal subjective memory complainers stratified by Alzheimer's disease biomarkers. Alzheimers Dement. 2018;14:1623–31.
Twohig D, Rodriguez-Vieitez E, Sando SB, Berge G, Lauridsen C, Moller I, Grontvedt GR, Brathen G, Patra K, Bu G, et al. The relevance of cerebrospinal fluid alpha-synuclein levels to sporadic and familial Alzheimer's disease. Acta Neuropathol Commun. 2018;6:130.
Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991;1:213–6.
Dubois B, Feldman HH, Jacova C, Cummings JL, DeKosky ST, Barberger-Gateau P, Delacourte A, Frisoni G, Fox NC, Galasko D, et al. Revising the definition of Alzheimer's disease: a new lexicon. The Lancet Neurology. 2010;9:1118–27.
Zhu XC, Tan L, Wang HF, Jiang T, Cao L, Wang C, Wang J, Tan CC, Meng XF, Yu JT. Rate of early onset Alzheimer’s disease: a systematic review and meta-analysis. Ann Transl Med. 2015;3:38.
Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR Jr, Kaye J, Montine TJ, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer's & dementia : the journal of the Alzheimer’s Association. 2011;7:280–92.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–9.
Dubois B, Hampel H, Feldman HH, Scheltens P, Aisen P, Andrieu S, Bakardjian H, Benali H, Bertram L, Blennow K, et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016;12:292–323.
Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–5.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608.
The amyloid cascade hypothesis has misled the pharmaceutical industry. Biochem Soc Trans 2011, 39:920-923.
Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015;18:794–9.
Choi ML, Gandhi S. Crucial role of protein oligomerization in the pathogenesis of Alzheimer's and Parkinson's diseases. FEBS J. 2018;285:3631–44.
Cline EN, Bicca MA, Viola KL, Klein WL. The Amyloid-beta Oligomer Hypothesis: Beginning of the Third Decade. J Alzheimers Dis. 2018;64:S567–610.
Lam B, Masellis M, Freedman M, Stuss DT, Black SE. Clinical, imaging, and pathological heterogeneity of the Alzheimer's disease syndrome. Alzheimers Res Ther. 2013;5(1).
Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118:5–36.
Galasko D, Golde TE. Biomarkers for Alzheimer’s disease in plasma, serum and blood - conceptual and practical problems. Alzheimers Res Ther. 2013;5:10.
Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535–62.
Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Green RC, Harvey D, Jack CR, Jagust W, Liu E, et al. The Alzheimer’s Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement. 2013;9:e111–94.
Thal DR, Attems J, Ewers M. Spreading of amyloid, tau, and microvascular pathology in Alzheimer's disease: findings from neuropathological and neuroimaging studies. J Alzheimers Dis. 2014;42(Suppl 4):S421–9.
Bejanin A, Schonhaut DR, La Joie R, Kramer JH, Baker SL, Sosa N, Ayakta N, Cantwell A, Janabi M, Lauriola M, et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer's disease. Brain. 2017;140:3286–300.
Blennow K. A Review of Fluid Biomarkers for Alzheimer's Disease: Moving from CSF to Blood. Neurol Ther. 2017;6:15–24.
Lashley T, Schott JM, Weston P, Murray CE, Wellington H, Keshavan A, Foti SC, Foiani M, Toombs J, Rohrer JD, et al. Molecular biomarkers of Alzheimer's disease: progress and prospects. Dis Model Mech. 2018;11.
Saint-Aubert L, Lemoine L, Chiotis K, Leuzy A, Rodriguez-Vieitez E, Nordberg A. Tau PET imaging: present and future directions. Mol Neurodegener. 2017;12:19.
Bergeron C, Pollanen M. Lewy bodies in Alzheimer disease--one or two diseases? Alzheimer Dis Assoc Disord. 1989;3:197–204.
Hansen L, Salmon D, Galasko D, Masliah E, Katzman R, DeTeresa R, Thal L, Pay MM, Hofstetter R, Klauber M, et al. The Lewy body variant of Alzheimer's disease: a clinical and pathologic entity. Neurology. 1990;40:1–8.
Hansen LA, Masliah E, Galasko D, Terry RD. Plaque-only Alzheimer disease is usually the lewy body variant, and vice versa. J Neuropathol Exp Neurol. 1993;52:648–54.
Forstl H, Burns A, Luthert P, Cairns N, Levy R. The Lewy-body variant of Alzheimer's disease. Clinical and pathological findings. Br J Psychiatry. 1993;162:385–92.
Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:11282–6.
Hansen LA, Galasko D, Samuel W, Xia Y, Chen X, Saitoh T. Apolipoprotein-E ε-4 is associated with increased neurofibrillary pathology in the Lewy body variant of Alzheimer's disease. Neurosci Lett. 1994;182:63–5.
Masliah E, Iwai A, Mallory M, Ueda K, Saitoh T. Altered presynaptic protein NACP is associated with plaque formation and neurodegeneration in Alzheimer's disease. Am J Pathol. 1996;148:201–10.
Iwai A, Masliah E, Sundsmo MP, DeTeresa R, Mallory M, Salmon DP, Saitoh T. The synaptic protein NACP is abnormally expressed during the progression of Alzheimer's disease. Brain Res. 1996;720:230–4.
Lippa CF, Fujiwara H, Mann DMA, Giasson B, Baba M, Schmidt ML, Nee LE, O'Connell B, Pollen DA, St. George-Hyslop P, et al. Lewy Bodies Contain Altered α-Synuclein in Brains of Many Familial Alzheimer's Disease Patients with Mutations in Presenilin and Amyloid Precursor Protein Genes. Am J Pathol. 1998;153:1365–70.
Olichney JM, Galasko D, Salmon DP, Hofstetter CR, Hansen LA, Katzman R, Thal LJ. Cognitive decline is faster in Lewy body variant than in Alzheimer's disease. Neurology. 1998;51:351–7.
Lippa CF, Schmidt ML, Lee VM, Trojanowski JQ. Antibodies to alpha-synuclein detect Lewy bodies in many Down's syndrome brains with Alzheimer's disease. Ann Neurol. 1999;45:353–7.
Marui W, Iseki E, Ueda K, Kosaka K. Occurrence of human alpha-synuclein immunoreactive neurons with neurofibrillary tangle formation in the limbic areas of patients with Alzheimer's disease. J Neurol Sci. 2000;174:81–4.
Wirths O, Weickert S, Majtenyi K, Havas L, Kahle PJ, Okochi M, Haass C, Multhaup G, Beyreuther K, Bayer TA. Lewy body variant of Alzheimer's disease: alpha-synuclein in dystrophic neurites of A beta plaques. Neuroreport. 2000;11:3737–41.
Iseki E. Dementia with Lewy bodies: reclassification of pathological subtypes and boundary with Parkinson's disease or Alzheimer's disease. Neuropathology. 2004;24:72–8.
Snider BJ, Norton J, Coats MA, Chakraverty S, Hou CE, Jervis R, Lendon CL, Goate AM, McKeel DW Jr, Morris JC. Novel presenilin 1 mutation (S170F) causing Alzheimer disease with Lewy bodies in the third decade of life. Arch Neurol. 2005;62:1821–30.
Mikolaenko I, Pletnikova O, Kawas CH, O'Brien R, Resnick SM, Crain B, Troncoso JC. Alpha-synuclein lesions in normal aging, Parkinson disease, and Alzheimer disease: evidence from the Baltimore Longitudinal Study of Aging (BLSA). J Neuropathol Exp Neurol. 2005;64:156–62.
Uchikado H, Lin WL, DeLucia MW, Dickson DW. Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol. 2006;65:685–97.
Tsigelny IF, Crews L, Desplats P, Shaked GM, Sharikov Y, Mizuno H, Spencer B, Rockenstein E, Trejo M, Platoshyn O, et al. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer's and Parkinson's diseases. PLoS One. 2008;3:e3135.
Spies PE, Melis RJ, Sjogren MJ, Rikkert MG, Verbeek MM. Cerebrospinal fluid alpha-synuclein does not discriminate between dementia disorders. J Alzheimers Dis. 2009;16:363–9.
Larson ME, Sherman MA, Greimel S, Kuskowski M, Schneider JA, Bennett DA, Lesne SE. Soluble alpha-synuclein is a novel modulator of Alzheimer's disease pathophysiology. J Neurosci. 2012;32:10253–66.
Toledo JB, Gopal P, Raible K, Irwin DJ, Brettschneider J, Sedor S, Waits K, Boluda S, Grossman M, Van Deerlin VM, et al. Pathological alpha-synuclein distribution in subjects with coincident Alzheimer's and Lewy body pathology. Acta Neuropathol. 2016;131:393–409.
Thomas AJ, Mahin-Babaei F, Saidi M, Lett D, Taylor JP, Walker L, Attems J. Improving the identification of dementia with Lewy bodies in the context of an Alzheimer's-type dementia. Alzheimers Res Ther. 2018;10:27.
Portet F, Scarmeas N, Cosentino S, Helzner EP, Stern Y. Extrapyramidal signs before and after diagnosis of incident Alzheimer disease in a prospective population study. Arch Neurol. 2009;66:1120–6.
Larson ME, Greimel SJ, Amar F, LaCroix M, Boyle G, Sherman MA, Schley H, Miel C, Schneider JA, Kayed R, et al. Selective lowering of synapsins induced by oligomeric alpha-synuclein exacerbates memory deficits. Proc Natl Acad Sci U S A. 2017;114:E4648–57.
Markesbery WR, Jicha GA, Liu H, Schmitt FA. Lewy body pathology in normal elderly subjects. J Neuropathol Exp Neurol. 2009;68:816–22.
Zolochevska O, Taglialatela G. Non-Demented Individuals with Alzheimer's Disease Neuropathology: Resistance to Cognitive Decline May Reveal New Treatment Strategies. Curr Pharm Des. 2016;22:4063–8.
Espay AJ, Vizcarra JA, Marsili L, Lang AE, Simon DK, Merola A, Josephs KA, Fasano A, Morgante F, Savica R, et al. Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology. 2019;92:329–37.
Peng C, Gathagan RJ, Lee VM. Distinct alpha-Synuclein strains and implications for heterogeneity among alpha-Synucleinopathies. Neurobiol Dis. 2018;109:209–18.
Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–9.
Tokuda T, Salem SA, Allsop D, Mizuno T, Nakagawa M, Qureshi MM, Locascio JJ, Schlossmacher MG, El-Agnaf OM. Decreased alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson's disease. Biochem Biophys Res Commun. 2006;349:162–6.
Tateno F, Sakakibara R, Kawai T, Kishi M, Murano T. Alpha-synuclein in the cerebrospinal fluid differentiates synucleinopathies (Parkinson Disease, dementia with Lewy bodies, multiple system atrophy) from Alzheimer disease. Alzheimer Dis Assoc Disord. 2012;26:213–6.
Wennstrom M, Surova Y, Hall S, Nilsson C, Minthon L, Bostrom F, Hansson O, Nielsen HM. Low CSF levels of both alpha-synuclein and the alpha-synuclein cleaving enzyme neurosin in patients with synucleinopathy. PLoS One. 2013;8:e53250.
Mollenhauer B, Cullen V, Kahn I, Krastins B, Outeiro TF, Pepivani I, Ng J, Schulz-Schaeffer W, Kretzschmar HA, McLean PJ, et al. Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp Neurol. 2008;213:315–25.
Ohrfelt A, Grognet P, Andreasen N, Wallin A, Vanmechelen E, Blennow K, Zetterberg H. Cerebrospinal fluid alpha-synuclein in neurodegenerative disorders-a marker of synapse loss? Neurosci Lett. 2009;450:332–5.
Kasuga K, Tokutake T, Ishikawa A, Uchiyama T, Tokuda T, Onodera O, Nishizawa M, Ikeuchi T. Differential levels of alpha-synuclein, beta-amyloid42 and tau in CSF between patients with dementia with Lewy bodies and Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2010;81:608–10.
Reesink FE, Lemstra AW, van Dijk KD, Berendse HW, van de Berg WD, Klein M, Blankenstein MA, Scheltens P, Verbeek MM, van der Flier WM. CSF alpha-synuclein does not discriminate dementia with Lewy bodies from Alzheimer's disease. J Alzheimers Dis. 2010;22:87–95.
Korff A, Liu C, Ginghina C, Shi M, Zhang J. Alzheimer's Disease Neuroimaging I: alpha-Synuclein in cerebrospinal fluid of Alzheimer's disease and mild cognitive impairment. J Alzheimers Dis. 2013;36:679–88.
Kapaki E, Paraskevas GP, Emmanouilidou E, Vekrellis K. The diagnostic value of CSF alpha-synuclein in the differential diagnosis of dementia with Lewy bodies vs. normal subjects and patients with Alzheimer's disease. PLoS One. 2013;8:e81654.
Toledo JB, Korff A, Shaw LM, Trojanowski JQ, Zhang J. CSF alpha-synuclein improves diagnostic and prognostic performance of CSF tau and Abeta in Alzheimer's disease. Acta Neuropathol. 2013;126:683–97.
Hansson O, Hall S, Ohrfelt A, Zetterberg H, Blennow K, Minthon L, Nagga K, Londos E, Varghese S, Majbour NK, et al. Levels of cerebrospinal fluid alpha-synuclein oligomers are increased in Parkinson's disease with dementia and dementia with Lewy bodies compared to Alzheimer's disease. Alzheimers Res Ther. 2014;6:25.
Slaets S, Vanmechelen E, Le Bastard N, Decraemer H, Vandijck M, Martin JJ, De Deyn PP, Engelborghs S. Increased CSF alpha-synuclein levels in Alzheimer's disease: correlation with tau levels. Alzheimers Dement. 2014;10:S290–8.
Wang ZY, Han ZM, Liu QF, Tang W, Ye K, Yao YY. Use of CSF alpha-synuclein in the differential diagnosis between Alzheimer's disease and other neurodegenerative disorders. Int Psychogeriatr. 2015;27:1429–38.
Berge G, Sando SB, Albrektsen G, Lauridsen C, Moller I, Grontvedt GR, Brathen G, White LR. Alpha-synuclein measured in cerebrospinal fluid from patients with Alzheimer's disease, mild cognitive impairment, or healthy controls: a two year follow-up study. BMC Neurol. 2016;16:180.
Majbour NK, Chiasserini D, Vaikath NN, Eusebi P, Tokuda T, van de Berg W, Parnetti L, Calabresi P, El-Agnaf OM. Increased levels of CSF total but not oligomeric or phosphorylated forms of alpha-synuclein in patients diagnosed with probable Alzheimer's disease. Sci Rep. 2017;7:40263.
Shi M, Tang L, Toledo JB, Ginghina C, Wang H, Aro P, Jensen PH, Weintraub D, Chen-Plotkin AS, Irwin DJ, et al. Cerebrospinal fluid alpha-synuclein contributes to the differential diagnosis of Alzheimer's disease. Alzheimers Dement. 2018;14:1052–62.
van Dijk KD, Bidinosti M, Weiss A, Raijmakers P, Berendse HW, van de Berg WD. Reduced alpha-synuclein levels in cerebrospinal fluid in Parkinson's disease are unrelated to clinical and imaging measures of disease severity. Eur J Neurol. 2014;21:388–94.
Kruse N, Persson S, Alcolea D, Bahl JM, Baldeiras I, Capello E, Chiasserini D, Bocchio Chiavetto L, Emersic A, Engelborghs S, et al. Validation of a quantitative cerebrospinal fluid alpha-synuclein assay in a European-wide interlaboratory study. Neurobiol Aging. 2015;36:2587–96.
Hong Z, Shi M, Chung KA, Quinn JF, Peskind ER, Galasko D, Jankovic J, Zabetian CP, Leverenz JB, Baird G, et al. DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson's disease. Brain. 2010;133:713–26.
Wang H, Stewart T, Toledo JB, Ginghina C, Tang L, Atik A, Aro P, Shaw LM, Trojanowski JQ, Galasko DR, et al. A Longitudinal Study of Total and Phosphorylated alpha-Synuclein with Other Biomarkers in Cerebrospinal Fluid of Alzheimer's Disease and Mild Cognitive Impairment. J Alzheimers Dis. 2018;61:1541–53.
Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, Jackson B, McKee AC, Alvarez VE, Lee NC, Hall GF. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. 2012;287:3842–9.
Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–51.
Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013;110:E3138–47.
Siddiqui IJ, Pervaiz N, Abbasi AA, Bisaglia M, Mammi S, Bubacco L, Vilar M, Kruger R, Zarranz JJ, Silke AC, et al. The Parkinson Disease gene SNCA: Evolutionary and structural insights with pathological implication. Sci Rep. 2016;6:–24475.
Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem. 2005;280:9595–603.
Guerrero-Ferreira R, Taylor NM, Mona D, Ringler P, Lauer ME, Riek R, Britschgi M, Stahlberg H. Cryo-EM structure of alpha-synuclein fibrils. Elife. 2018;7.
Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–23.
Ferreon AC, Gambin Y, Lemke EA, Deniz AA. Interplay of alpha-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc Natl Acad Sci U S A. 2009;106:5645–50.
Trexler AJ, Rhoades E. Alpha-synuclein binds large unilamellar vesicles as an extended helix. Biochemistry. 2009;48:2304–6.
Trexler AJ, Rhoades E. Function and dysfunction of alpha-synuclein: probing conformational changes and aggregation by single molecule fluorescence. Mol Neurobiol. 2013;47:622–31.
Georgieva ER, Ramlall TF, Borbat PP, Freed JH, Eliezer D. The lipid-binding domain of wild type and mutant alpha-synuclein: compactness and interconversion between the broken and extended helix forms. J Biol Chem. 2010;285:28261–74.
Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9.
Zhu M, Li J, Fink AL. The association of alpha-synuclein with membranes affects bilayer structure, stability, and fibril formation. J Biol Chem. 2003;278:40186–97.
Bendor JT, Logan TP, Edwards RH. The function of alpha-synuclein. Neuron. 2013;79:1044–66.
Sode K, Ochiai S, Kobayashi N, Usuzaka E. Effect of reparation of repeat sequences in the human alpha-synuclein on fibrillation ability. Int J Biol Sci. 2006;3:1–7.
Dettmer U, Newman AJ, von Saucken VE, Bartels T, Selkoe D. KTKEGV repeat motifs are key mediators of normal alpha-synuclein tetramerization: Their mutation causes excess monomers and neurotoxicity. Proc Natl Acad Sci U S A. 2015;112:9596–601.
Li HT, Du HN, Tang L, Hu J, Hu HY. Structural transformation and aggregation of human alpha-synuclein in trifluoroethanol: non-amyloid component sequence is essential and beta-sheet formation is prerequisite to aggregation. Biopolymers. 2002;64:221–6.
Giasson BI, Murray IV, Trojanowski JQ, Lee VM. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem. 2001;276:2380–6.
Luk KC, Song C, O'Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106:20051–6.
George JM. The synucleins. Genome Biol. 2002;3:Reviews3002.
Kessler JC, Rochet JC, Lansbury PT, Jr.: The N-terminal repeat domain of alpha-synuclein inhibits beta-sheet and amyloid fibril formation. Biochemistry 2003, 42:672-678.
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–8.
Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–73.
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–7.
Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N, Pieri L, Madiona K, Durr A, Melki R, et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol. 2013;73:459–71.
Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov Disord. 2013;28:811–3.
Kim TD, Paik SR, Yang CH. Structural and functional implications of C-terminal regions of alpha-synuclein. Biochemistry. 2002;41:13782–90.
Eliezer D. The mysterious C-terminal tail of alpha-synuclein: nanobody's guess. J Mol Biol. 2013;425:2393–6.
Hong DP, Xiong W, Chang JY, Jiang C. The role of the C-terminus of human alpha-synuclein: intra-disulfide bonds between the C-terminus and other regions stabilize non-fibrillar monomeric isomers. FEBS Lett. 2011;585:561–6.
Bertoncini CW, Jung YS, Fernandez CO, Hoyer W, Griesinger C, Jovin TM, Zweckstetter M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc Natl Acad Sci U S A. 2005;102:1430–5.
Dedmon MM, Lindorff-Larsen K, Christodoulou J, Vendruscolo M, Dobson CM. Mapping long-range interactions in alpha-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J Am Chem Soc. 2005;127:476–7.
Martinez J, Moeller I, Erdjument-Bromage H, Tempst P, Lauring B. Parkinson's disease-associated alpha-synuclein is a calmodulin substrate. J Biol Chem. 2003;278:17379–87.
Bertini I, Gupta YK, Luchinat C, Parigi G, Peana M, Sgheri L, Yuan J. Paramagnetism-based NMR restraints provide maximum allowed probabilities for the different conformations of partially independent protein domains. J Am Chem Soc. 2007;129:12786–94.
McDowall JS, Ntai I, Hake J, Whitley PR, Mason JM, Pudney CR, Brown DR. Steady-State Kinetics of α-Synuclein Ferrireductase Activity Identifies the Catalytically Competent Species. Biochemistry. 2017;56:2497–505.
Lautenschlager J, Stephens AD, Fusco G, Strohl F, Curry N, Zacharopoulou M, Michel CH, Laine R, Nespovitaya N, Fantham M, et al. C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat Commun. 2018;9:712.
Dettmer U. Rationally Designed Variants of alpha-Synuclein Illuminate Its in vivo Structural Properties in Health and Disease. Front Neurosci. 2018;12:623.
Burre J, Sharma M, Sudhof TC. alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A. 2014;111:E4274–83.
Bartels T, Choi JG, Selkoe DJ. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–10.
Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, et al. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A. 2011;108:17797–802.
Dettmer U, Newman AJ, Luth ES, Bartels T, Selkoe D. In vivo cross-linking reveals principally oligomeric forms of alpha-synuclein and beta-synuclein in neurons and non-neural cells. J Biol Chem. 2013;288:6371–85.
Dettmer U, Newman AJ, Soldner F, Luth ES, Kim NC, von Saucken VE, Sanderson JB, Jaenisch R, Bartels T, Selkoe D. Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun. 2015;6:7314.
Selkoe D, Dettmer U, Luth E, Kim N, Newman A, Bartels T. Defining the native state of alpha-synuclein. Neurodegener Dis. 2014;13:114–7.
Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–75.
Maroteaux L, Scheller RH. The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res Mol Brain Res. 1991;11:335–43.
Liu S, Ninan I, Antonova I, Battaglia F, Trinchese F, Narasanna A, Kolodilov N, Dauer W, Hawkins RD, Arancio O. alpha-Synuclein produces a long-lasting increase in neurotransmitter release. EMBO J. 2004;23:4506–16.
Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–15.
Nakajo S, Shioda S, Nakai Y, Nakaya K. Localization of phosphoneuroprotein 14 (PNP 14) and its mRNA expression in rat brain determined by immunocytochemistry and in situ hybridization. Brain Res Mol Brain Res. 1994;27:81–6.
Scherzer CR, Grass JA, Liao Z, Pepivani I, Zheng B, Eklund AC, Ney PA, Ng J, McGoldrick M, Mollenhauer B, et al: GATA transcription factors directly regulate the Parkinson's disease-linked gene α-synuclein. 2008.
Clough RL, Dermentzaki G, Stefanis L: Functional dissection of the α-synuclein promoter: transcriptional regulation by ZSCAN21 and ZNF219. 2009, 110:1479-1490.
Fu Y, Zhao J, Atagi Y, Nielsen HM, Liu CC, Zheng H, Shinohara M, Kanekiyo T, Bu G. Apolipoprotein E lipoprotein particles inhibit amyloid-beta uptake through cell surface heparan sulphate proteoglycan. Mol Neurodegener. 2016;11:37.
Ogaki K, Martens YA, Heckman MG, Koga S, Labbe C, Lorenzo-Betancor O, Wernick AI, Walton RL, Soto AI, Vargas ER, et al. Multiple system atrophy and apolipoprotein E. Mov Disord. 2018.
Kim C, Lee SJ. Controlling the mass action of alpha-synuclein in Parkinson's disease. J Neurochem. 2008;107:303–16.
Kasai T, Tokuda T, Yamaguchi N, Watanabe Y, Kametani F, Nakagawa M, Mizuno T. Cleavage of normal and pathological forms of alpha-synuclein by neurosin in vitro. Neurosci Lett. 2008;436:52–6.
Tatebe H, Watanabe Y, Kasai T, Mizuno T, Nakagawa M, Tanaka M, Tokuda T. Extracellular neurosin degrades alpha-synuclein in cultured cells. Neurosci Res. 2010;67:341–6.
Spencer B, Michael S, Shen J, Kosberg K, Rockenstein E, Patrick C, Adame A, Masliah E. Lentivirus mediated delivery of neurosin promotes clearance of wild-type alpha-synuclein and reduces the pathology in an alpha-synuclein model of LBD. Mol Ther. 2013;21:31–41.
George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–72.
Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–52.
Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R, et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J Neurosci. 2002;22:8797–807.
Chandra S, Fornai F, Kwon HB, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, Castillo PE, Sudhof TC. Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc Natl Acad Sci U S A. 2004;101:14966–71.
Ninkina N, Papachroni K, Robertson DC, Schmidt O, Delaney L, O'Neill F, Court F, Rosenthal A, Fleetwood-Walker SM, Davies AM, Buchman VL. Neurons expressing the highest levels of gamma-synuclein are unaffected by targeted inactivation of the gene. Mol Cell Biol. 2003;23:8233–45.
Lundblad M, Decressac M, Mattsson B, Bjorklund A. Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc Natl Acad Sci U S A. 2012;109:3213–9.
Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, Makani S, Tian N, Castillo PE, Buchman VL, Chandra SS. alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A. 2010;107:19573–8.
Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–96.
Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–20.
Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A Pathologic Cascade Leading to Synaptic Dysfunction in -Synuclein-Induced Neurodegeneration. J Neurosci. 2010;30:8083–95.
Scott D, Roy S. alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J Neurosci. 2012;32:10129–35.
Wang L, Das U, Scott DA, Tang Y, McLean PJ, Roy S: α-Synuclein Multimers Cluster Synaptic Vesicles and Attenuate Recycling. 2014, 24.
Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79.
Logan T, Bendor J, Toupin C, Thorn K, Edwards RH. α-Synuclein promotes dilation of the exocytotic fusion pore. Nat Neurosci. 2017;20:681–9.
Logan T, Bendor J, Toupin C, Thorn K, Edwards RH. α-Synuclein promotes dilation of the exocytotic fusion pore. Nat Neurosci. 2017.
Sacino AN, Brooks M, Thomas MA, McKinney AB, McGarvey NH, Rutherford NJ, Ceballos-Diaz C, Robertson J, Golde TE, Giasson BI. Amyloidogenic alpha-synuclein seeds do not invariably induce rapid, widespread pathology in mice. Acta Neuropathol. 2014;127:645–65.
Spencer B, Desplats PA, Overk CR, Valera-Martin E, Rissman RA, Wu C, Mante M, Adame A, Florio J, Rockenstein E, Masliah E. Reducing Endogenous α-Synuclein Mitigates the Degeneration of Selective Neuronal Populations in an Alzheimer's Disease Transgenic Mouse Model. J Neurosci. 2016;36:7971–84.
Khan SS, LaCroix M, Boyle G, Sherman MA, Brown JL, Amar F, Aldaco J, Lee MK, Bloom GS, Lesne SE. Bidirectional modulation of Alzheimer phenotype by alpha-synuclein in mice and primary neurons. Acta Neuropathol. 2018;136:589–605.
Wang YC, Feng GY, Xia QJ, Hu Y, Xu Y, Xiong LL, Chen ZW, Wang HP, Wang TH, Zhou X. Knockdown of alpha-synuclein in cerebral cortex improves neural behavior associated with apoptotic inhibition and neurotrophin expression in spinal cord transected rats. Apoptosis. 2016;21:404–20.
Drolet RE, Behrouz B, Lookingland KJ, Goudreau JL. Mice lacking alpha-synuclein have an attenuated loss of striatal dopamine following prolonged chronic MPTP administration. Neurotoxicology. 2004;25:761–9.
Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290:985–9.
Spencer B, Desplats PA, Overk CR, Valera-Martin E, Rissman RA, Wu C, Mante M, Adame A, Florio J, Rockenstein E, Masliah E. Reducing Endogenous alpha-Synuclein Mitigates the Degeneration of Selective Neuronal Populations in an Alzheimer's Disease Transgenic Mouse Model. J Neurosci. 2016;36:7971–84.
Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev. 2011;91:119–49.
Moussaud S, Jones DR, Moussaud-Lamodiere EL, Delenclos M, Ross OA, McLean PJ. Alpha-synuclein and tau: teammates in neurodegeneration? Mol Neurodegener. 2014;9:43.
Oikawa T, Nonaka T, Terada M, Tamaoka A, Hisanaga S, Hasegawa M. alpha-Synuclein Fibrils Exhibit Gain of Toxic Function, Promoting Tau Aggregation and Inhibiting Microtubule Assembly. J Biol Chem. 2016;291:15046–56.
Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by alpha-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci. 2011;31:7604–18.
Kawakami F, Suzuki M, Shimada N, Kagiya G, Ohta E, Tamura K, Maruyama H, Ichikawa T. Stimulatory effect of alpha-synuclein on the tau-phosphorylation by GSK-3beta. FEBS J. 2011;278:4895–904.
Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–8.
Latimer DA, Gallo JM, Lovestone S, Miller CC, Reynolds CH, Marquardt B, Stabel S, Woodgett JR, Anderton BH. Stimulation of MAP kinase by v-raf transformation of fibroblasts fails to induce hyperphosphorylation of transfected tau. FEBS Lett. 1995;365:42–6.
Takashima A, Noguchi K, Michel G, Mercken M, Hoshi M, Ishiguro K, Imahori K. Exposure of rat hippocampal neurons to amyloid beta peptide (25-35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3 beta. Neurosci Lett. 1996;203:33–6.
Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin ML, Yardin C, Terro F. Tau protein kinases: involvement in Alzheimer's disease. Ageing Res Rev. 2013;12:289–309.
Hoshi M, Takashima A, Noguchi K, Murayama M, Sato M, Kondo S, Saitoh Y, Ishiguro K, Hoshino T, Imahori K. Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3beta in brain. Proc Natl Acad Sci U S A. 1996;93:2719–23.
Kozikowski AP, Gaisina IN, Petukhov PA, Sridhar J, King LT, Blond SY, Duka T, Rusnak M, Sidhu A. Highly potent and specific GSK-3beta inhibitors that block tau phosphorylation and decrease alpha-synuclein protein expression in a cellular model of Parkinson's disease. ChemMedChem. 2006;1:256–66.
Duka T, Duka V, Joyce JN, Sidhu A. Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson’s disease models. FASEB J. 2009;23:2820–30.
Gassowska M, Czapski GA, Pajak B, Cieslik M, Lenkiewicz AM, Adamczyk A. Extracellular alpha-synuclein leads to microtubule destabilization via GSK-3beta-dependent Tau phosphorylation in PC12 cells. PLoS One. 2014;9:e94259.
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–5.
Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, Di Monte DA. Lysosomal degradation of alpha-synuclein in vivo. J Biol Chem. 2010;285:13621–9.
Avdulov NA, Chochina SV, Igbavboa U, Warden CS, Vassiliev AV, Wood WG. Lipid binding to amyloid beta-peptide aggregates: preferential binding of cholesterol as compared with phosphatidylcholine and fatty acids. J Neurochem. 1997;69:1746–52.
Wilson DM, Binder LI. Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer's disease. Am J Pathol. 1997;150:2181–95.
Brekk OR, Moskites A, Isacson O, Hallett PJ. Lipid-dependent deposition of alpha-synuclein and Tau on neuronal Secretogranin II-positive vesicular membranes with age. Sci Rep. 2018;8:15207.
Brady RO, Kanfer J, Shapiro D. The Metabolism of Glucocerebrosides. I. Purification and Properties of a Glucocerebroside-Cleaving Enzyme from Spleen Tissue. J Biol Chem. 1965;240:39–43.
Gallardo G, Schluter OM, Sudhof TC. A molecular pathway of neurodegeneration linking alpha-synuclein to ApoE and Abeta peptides. Nat Neurosci. 2008;11:301–8.
Mandal PK, Pettegrew JW, Masliah E, Hamilton RL, Mandal R. Interaction between Abeta peptide and alpha synuclein: molecular mechanisms in overlapping pathology of Alzheimer's and Parkinson's in dementia with Lewy body disease. Neurochem Res. 2006;31:1153–62.
Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300:636–40.
Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, Mucke L. beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer's disease and Parkinson's disease. Proc Natl Acad Sci U S A. 2001;98:12245–50.
Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic Interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. 2010;30:7281–9.
Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med. 2012;2:a006270.
Winslow AR, Moussaud S, Zhu L, Post KL, Dickson DW, Berezovska O, McLean PJ. Convergence of pathology in dementia with Lewy bodies and Alzheimer's disease: a role for the novel interaction of alpha-synuclein and presenilin 1 in disease. Brain. 2014;137:1958–70.
Kaneko H, Kakita A, Kasuga K, Nozaki H, Ishikawa A, Miyashita A, Kuwano R, Ito G, Iwatsubo T, Takahashi H, et al. Enhanced accumulation of phosphorylated alpha-synuclein and elevated beta-amyloid 42/40 ratio caused by expression of the presenilin-1 deltaT440 mutant associated with familial Lewy body disease and variant Alzheimer's disease. J Neurosci. 2007;27:13092–7.
Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51.
Goedert M. NEURODEGENERATION. Alzheimer's and Parkinson's diseases: The prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science. 2015;349:1255555.
Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Bjorklund A, et al. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–3.
Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med. 2008;14:504–6.
Kurowska Z, Englund E, Widner H, Lindvall O, Li JY, Brundin P. Signs of degeneration in 12-22-year old grafts of mesencephalic dopamine neurons in patients with Parkinson's disease. J Park Dis. 2011;1:83–92.
Li W, Englund E, Widner H, Mattsson B, van Westen D, Latt J, Rehncrona S, Brundin P, Bjorklund A, Lindvall O, Li JY. Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc Natl Acad Sci U S A. 2016;113:6544–9.
Ngolab J, Trinh I, Rockenstein E, Mante M, Florio J, Trejo M, Masliah D, Adame A, Masliah E, Rissman RA. Brain-derived exosomes from dementia with Lewy bodies propagate alpha-synuclein pathology. Acta Neuropathol Commun. 2017;5:46.
Rodriguez L, Marano MM, Tandon A. Import and Export of Misfolded alpha-Synuclein. Front Neurosci. 2018;12:344.
Abounit S, Bousset L, Loria F, Zhu S, de Chaumont F, Pieri L, Olivo-Marin JC, Melki R, Zurzolo C. Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016;35:2120–38.
Dieriks BV, Park TI, Fourie C, Faull RL, Dragunow M, Curtis MA. alpha-synuclein transfer through tunneling nanotubes occurs in SH-SY5Y cells and primary brain pericytes from Parkinson's disease patients. Sci Rep. 2017;7:42984.
Rostami J, Holmqvist S, Lindstrom V, Sigvardson J, Westermark GT, Ingelsson M, Bergstrom J, Roybon L, Erlandsson A. Human Astrocytes Transfer Aggregated Alpha-Synuclein via Tunneling Nanotubes. J Neurosci. 2017;37:11835–53.
LeDoux J. The amygdala. Curr Biol. 2007;17:R868–74.
Acknowledgements
The authors would like to thank the funding organizations (Alzheimer’s Association (2015-NIRG-339824), Stiftelsen Olle Engkvist Byggmästare (184-333), Demensförbundet and Marcus Borgströms Stiftelse) for their support of this work.
Funding
This review was supported by grants from the Alzheimer’s Association (2015-NIRG-339824, HMN), Stiftelsen Olle Engkvist Byggmästare (184-333, HMN). Demensförbundet (DT, HMN) and Marcus Borgströms Stiftelse (HMN).
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
DT and HMN critically reviewed the literature and drafted the manuscript. Both authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Twohig, D., Nielsen, H.M. α-synuclein in the pathophysiology of Alzheimer’s disease. Mol Neurodegeneration 14, 23 (2019). https://doi.org/10.1186/s13024-019-0320-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-019-0320-x