
Abstract
This article describes pathogenic concepts and factors, in particular glycolipid abnormalities, that create cell dysfunction and synaptic loss in neurodegenerative diseases. By phenocopying lysosomal storage disorders, such as Gaucher disease and related disorders, age- and dose-dependent changes in glycolipid cell metabolism can lead to Parkinson’s disease and related dementias. Recent results show that perturbation of sphingolipid metabolism can precede or is a part of abnormal protein handling in both genetic and idiopathic Parkinson’s disease and Lewy body dementia. In aging and genetic predisposition with lipid disturbance, α-synuclein’s normal vesicular and synaptic role may be detrimentally shifted toward accommodating and binding such lipids. Specific neuronal glycolipid, protein, and vesicular interactions create potential pathophysiology that is amplified by astroglial and microglial immune mechanisms resulting in neurodegeneration. This perspective provides a new logic for therapeutic interventions that do not focus on protein aggregation, but rather provides a guide to the complex biology and the common sequence of events that lead to age-dependent neurodegenerative disorders.
Similar content being viewed by others

Introduction
It is now evident from genetic variant analysis that, lipid and lipid transport, autophagic-lysosomal and inflammatory pathways carry significant and impactful genetic risk for age-dependent neurodegenerative diseases [1,2,3]. However, it is less clear how their biological interactions lead to the pathology seen in many age-related neurodegenerative disorders, such as Parkinson’s disease (PD), Alzheimer’s disease (AD), and dementias. For example in PD, the first genetic identification of Mendelian genetic human cases involved proteins such as α-synuclein (SNCA), LRRK2, parkin, PINK1, and DJ-1 [4,5,6,7,8,9,10,11,12,13]. Prominently, early discoveries uncovered mutations in the α-synuclein gene in rare families with autosomal dominant PD and the presence of α-synuclein in Lewy bodies in both genetic and sporadic disease [14]. Genome-wide associated studies (GWAS) have also established gene variants associated with lysosomal pathways, lipids, lipoproteins, and inflammation as risk factors for PD, including GBA1, DGKQ, LAMP1, SCARB2/LIMP-2, glycoprotein NMB (GPNMB), and HLA [2, 15]. Critically, it has been shown that the majority of sporadic PD cases (56%) carry at least one putative damaging variant in a lysosomal storage disorder gene, and 21% of patients carry multiple alleles [16]. For these reasons and the pathobiology described in this article, PD clinical trials have begun to try to alleviate the lysosomal dysfunction caused by GBA deficiencies and glycolipid changes in PD patients (ClinicalTrials.gov Identifiers NCT02906020, NCT02941822, NCT02914366).
Glycolipid changes may be causative of the proteinopathy in Parkinson’s disease
Many neurological diseases such as AD, PD, and the ataxias are conventionally linked to the intracellular accumulation and aggregation of proteins. In the case of PD, α-synuclein and the formation of Lewy bodies has been linked to the disease. However, the key scientific and conceptual question is whether the rare familial hereditary α-synuclein protein abnormalities are representing the same sequence of pathological events that are seen in other genetic causes of PD, or the vast majority of sporadic cases. Significant data shows that lipid vesicles, membrane fragments, and cytoskeletal elements are all found within the α-synuclein-coated Lewy body aggregates at the end-stage of disease in post-mortem PD brain [17], perhaps indicating a generalized cellular dysfunction prior to protein deposition in this disease. Relevant to such cellular dysfunction, brain glycosphingolipid substrate levels are elevated in the brain with increasing age, as well as in sporadic PD [18, 19]. Complex glycolipids are located in the plasma and intracellular membranes of mammalian cells and are particularly enriched in the brain where they have essential roles in cell-cell and cell-matrix interactions, as well as in cell adhesion, modulation of membrane receptors, and signal transduction [20]. As discussed in this article, the physiological burden of increased glycolipid levels in neurons in the aging brain and in PD is therefore likely to influence many cellular organelles and pathways, including lipid membranes, vesicle transport, protein:protein and protein:membrane interactions, autophagic clearance, and neuroinflammation.
How does varying lipid abnormalities over time lead to different diseases?
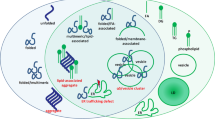
Usually presenting clinically by a very early age, homozygote and biallelic GBA1 mutations in Gaucher disease cause glucocerebrosidase (GCase) loss of function with organ failures of the liver, spleen, and bone [21]. GCase is a lysosomal enzyme involved in the metabolism of glycosphingolipids [22]. The loss of GCase enzyme function resulting from disease-causing GBA1 mutations occurs through destabilization of protein folding or by preventing synthesis of the full-length protein through frameshifts. Gaucher disease patients without primary neurological manifestations typically have some residual GCase activity and are classified as having non-neuronopathic (type 1) Gaucher disease. Acute neuronopathic Gaucher disease (type 2) affects patients in infancy or even prenatally, with severe neurodegeneration and early lethality [21], and as a result of severe or null mutations in GBA1, these patients have little or no GCase activity. Clinically, chronic neuronopathic (type 3) Gaucher disease patients show a degree of neurological involvement but have longer survival (past infancy) [21]. A massive accumulation of GBA-related glycosphingolipids, including glucosylsphingosine and glucosylceramide and related metabolite accumulation are found in the brain in type 2 and type 3 Gaucher disease, with glucosylsphingosine elevations in the brain in chronic neuronopathic Gaucher disease from 22 to 51 times normal, and in acute neuronopathic Gaucher disease from 38 to 694 times normal [23]. Massively increased lipid levels in severe childhood lysosomal storage disease create a different pattern of organ and cellular failure than seen in PD and LBD, where relatively modest but sustained lipid level abnormalities create neurodegeneration later in life. In PD patient carriers of Gaucher disease-linked mutations, haploinsufficiency of GBA1 produces on average a 30–50% reduction of GCase activity, paralleled with increases in glycolipid substrates [24]. Heterozygous mutations in GBA1 are associated with an earlier onset of disease and a faster rate of cognitive decline in PD and LBD [25,26,27,28,29]. How do these glycolipid changes of the same biochemical origin cause such different pathologies? For Gaucher’s disease, the pattern of degeneration parallels several sphingolipidoses lysosomal storage disorders in that as a primary function, the liver responds to the massive metabolic lipid disturbances, and both the liver and spleen become seriously compromised. However, with a chronic lifelong, or acquired by age, relatively lower dose of the same lipid species, the liver and spleen manage such glycolipid metabolic load, whereas in vulnerable neural systems, neurodegenerative changes occur. In PD, the midbrain dopaminergic and other brain and neuronal regions will fail to manage such chronic low-level elevations of glycolipids over time (Fig. 1) [31, 32].

Time and glycosphingolipid dose-dependent phenotypic disease expressions. GBA1 homozygous or compound heterozygous mutations cause Gaucher disease with onset in early life. Such mutations cause major reductions in glucocerebrosidase (GCase) enzymatic activity and massive accumulation of sphingolipid substrates (glucosylceramide and glucosylsphingosine), typically leading to fatal organ failures particularly of the liver and spleen. In the case of Parkinson’s disease (PD), with or without heterozygous GBA1 mutations, disease prevalence is in late life (above age 65) and approaches 1–3% of the population. In PD, GCase activity is typically reduced by ~ 50% in the brain and blood with a moderate elevation of glycosphingolipids. Data from post-mortem human brain tissue and in mouse brain also shows that GCase activity is gradually decreased in the brain during normal aging at levels that mirrors genetic GCase haploinsufficiency. Presumably, this contributes to increased risk in all humans with age for PD
Neurons and cellular systems particularly vulnerable to the Parkinson’s disease pathogenic processes
Whereas organ specificity is relevant, for neurodegenerative diseases, it is more critical to understand individual cellular vulnerability to chronic glycolipid changes that ultimately generate the specific pattern of cell loss in the disease. For example, in PD and LBD, age-dependent neuronal vulnerability and regional synaptic and cellular death follows predictable regional patterns that are defined by complex cell, genetic, and tissue interactions [30, 31, 33]. The initial motor symptoms of PD are largely due to the loss of the dopaminergic midbrain neurons. In the midbrain, the vulnerable A9 and less vulnerable A10 dopaminergic neurons in the substantia nigra have different metabolic and gene expression profiles [30, 31, 34], and we have previously shown that providing A9 neurons with genes that are more highly expressed in A10 neurons provides a cellular resilience against PD-related neurodegeneration in vitro and in vivo [30, 31].
The midbrain dopaminergic neurons, in addition to overall anatomical and normal physiological features, have unique gene and protein expression as well as local tissue milieu and glial interactions that are likely to be involved in their selective vulnerability [31, 33, 35] There is evidence indicating that human dopaminergic neurons in the substantia nigra are subjected to increased oxidative stress during their lifetime as they have increased levels of protein and lipid oxidation products when compared to other regions in the brain [36]. This increased oxidative stress is even more evident in PD. Several markers of oxidative stress are present in the substantia nigra of PD patients, including the reduction of glutathione levels, reduced activity of peroxidase and catalase, and even greater increase of lipid and protein oxidations products [37]. Mitochondrial complex I subunits and activity are decreased in PD brains [38, 39]. Of great significance for understanding age-dependent and common forms of PD, many types of mitochondrial toxicity or metabolic compromise can by itself generate α-synuclein elevations and even aggregated forms of α-synuclein [40].
α-Synuclein’s normal vesicular role in synaptic transmission can be changed by pathological accommodation to lipids
α-Synuclein is a synaptic protein that is involved in synaptic release of vesicles in many neuronal systems. Along with β- and γ- forms of the protein, α-synuclein specifically can influence the vesicular pore opening times of many neurotransmitter systems in the brain, including the dopaminergic systems [41]. This protein is involved in synaptic plasticity, and was originally described to be involved in learning paradigms of animals [42]. Of interest to the discussion in this article, we believe that α-synuclein has evolved also for other than vesicular membrane lipid binding functions, and potentially, when elevated, has a toxic gain of function at high levels that can prevent axonal transport and potentially other normal functions of the protein [43, 44]. Normal rodent α-synuclein carries a threonine residue at position 53, analogous to the human A53T mutation [45], and would be expected to have altered lipid binding capacity compared to wildtype human α-synuclein, similar to the human mutant A53T α-synuclein. The interpretation being that when mutant A53T, for example, is present in humans, α-synuclein may have inadequate lipid binding transport capacity for human longevity, with clearly increased risk of PD with this mutation [5]. Human neurons derived from induced pluripotent stem cells (iPSCs) show evidence of large lipid particles accumulating in the cell body in vitro when wild-type α-synuclein is overexpressed [44]. As we will describe below, new data put into the context of previous studies illustrate that the lipid binding domain of α-synuclein and its potential role in lipid transport, in concert with other proteins, could become part of a toxic cascade that ultimately results in PD-like pathology.
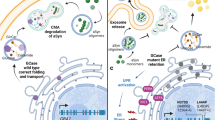
In modeling the genetic predisposition to copy number variant (duplication, triplication) and even mutations in the lipid-binding domains of α-synuclein, AAV-mediated overexpression of α-synuclein in the rat substantia nigra highlights the intrinsic differences between vulnerable and less vulnerable midbrain dopamine neurons and generates a relatively selective or sensitive degeneration of the A9 neurons [43, 46, 47]. α-Synuclein can impair microtubule transport systems, and we have found that overexpression of α-synuclein in the substantia nigra using an AAV system leads to an early blockade of anterograde and retrograde motor proteins along the axons, which is paralleled by neuroinflammation in the target region of the AAV-α-synuclein nigrostriatal neurons, and precedes dopamine neuron degeneration [43]. Findings that α-synuclein overexpression produces such microtubule and transport is widely supported in the literature [48,49,50]. In human neurons derived from iPSCs from PD patients carrying increased dosage of α-synuclein, the resulting α-synuclein oligomerization is associated with mitochondrial axonal transport deficits, reduced ATP levels, and synapse loss [48]. Overexpression of wild-type α-synuclein in human iPSC-derived neurons, is accompanied by massive lipid droplet accumulation [44] and it is perhaps such abnormal α-synuclein-lipid interactions that result in vesicle transport deficits and eventually neuronal degeneration. Significantly, ultrastructural analysis of Lewy pathology in the brain of PD patients shows the presence of lipids, α-synuclein, lysosomes, and mitochondria, and such findings are suggestive of impaired organellar trafficking [17] (Fig. 2a, d).

Lysosomal, lipid, and cellular mechanisms leading to α-synuclein and Lewy body pathology. a Under normal conditions α-synuclein is a natively structured protein with synaptic and vesicular functions. In addition to monomeric α-synuclein, α-synuclein can exist as physiological α-synuclein multimers (tetramers). α-Synuclein is normally transported to intracellular sites, including the synapse, where it participates in synaptic release functions. b Several pathways are involved in the degradation of α-synuclein and maintenance of its steady-state levels, including the ubiquitin-proteasome system (UPS), and c endo-lysosomal pathways. Overexpression of the ubiquitin ligase, Nedd4, can rescue α-synuclein toxicity in animal models through increased degradation of α-synuclein through the endo-lysosomal pathway (see text). c GBA haploinsufficiency, or deficiencies in other lysosomal enzymes, chaperones or transport proteins, can cause dysfunction of the lysosome in Parkinson’s disease (PD) and Lewy body dementia (LBD). Increased expression or function of lysosomal enzymes including GBA and Lamp1 reduce α-synucleinopathy and can rescue midbrain dopamine neuron degeneration in α-synucleinopathy preclinical models. d Damaged mitochondria are degraded through the mitophagy pathway, which can also be disrupted in aging and in familial PD and increases autophagic lysosomal load. e As described in this review, the structural similarity of α-synuclein with apolipoproteins suggests an additional role for α-synuclein in lipid transport. An aberrant accumulation of lipids, including glycosphingolipids in aging and in PD, are associated with abnormal lipidation of α-synuclein, and eventually accumulation of α-synuclein into insoluble high molecular weight forms. Mutations in, overexpression of, and loss of function of α-synuclein, contribute to cellular toxicities and pathology, including perturbation of lipid-vesicle trafficking and axonal transport. Recent advanced microscopic evidence shows that the Lewy body is composed of a large lipid core also including cellular organelles including mitochondria, lipids and protein aggregates. ER (endoplasmic reticulum), Lys (lysosome), Mt (mitochondria), Ub (ubiquitin), Ves (vesicle)
Lysosomal dysfunction in PD highlighted by glucocerebrosidase activity reductions
GCase activity is reduced and glycosphingolipid levels are increased, in the brain of PD patients with and without a GBA1 mutation [18, 51, 52] as well as in the brain in aging [18, 53], and seems to be part of an age-induced phenocopy of the genetic haploinsufficiency of GBA1 seen in patients [53]. The age-induced changes to lysosomal GCase activity and glycosphingolipid homeostasis would be expected to cause lysosomal dysfunction and accumulation of α-synuclein through several interacting mechanisms. Cross-linking studies have identified a physiological conformation of α-synuclein in α-helically folded tetramers [54], which are hypothesized to be in equilibrium with unfolded α-synuclein monomers [55]. α-Synuclein is degraded by different pathways, including proteasomes, macroautophagy, chaperone-mediated autophagy (CMA), and endolysosomes [56]. Several pathways may participate in maintaining a steady state of physiological α-synuclein as well as involved in its degradation, and their combined or sequential failure may lead to its accumulation in the disease. Supporting this, pharmacological GCase inhibition, knockdown of GBA, or GBA mutations, in mice, primary neuronal cultures and in human neurons, causes the accumulation of glucosylceramide and glucosylsphingosine, decreased lysosomal protein degradation, α-synuclein accumulation and altered multimerization, neuroinflammation, and toxicity [57,58,59,60,61,62,63,64,65]. Additional mechanisms of glycosphingolipid toxicity could be caused by leaks from lysosomes, disruption of membrane fluidity, direct interactions with α-synuclein, Ca2+ dysregulation, and neuroinflammation [57, 66]. A moderate accumulation of glycosphingolipids and altered lipid composition in the plasma membrane and membranous organelles in aging and PD would be expected to be associated with multiple pathophysiological consequences and disrupted neuronal function. A reduction of transport of active GCase to the lysosome may also contribute to decreased lysosomal GCase activity in PD. Lysosomal integral membrane protein type-2 (LIMP-2, also known as SCARB-2) binds GCase to allow for trafficking of GCase from the endoplasmic reticulum to lysosomes [67]. Loss of LIMP-2 in mice leads to reduced GCase activity, lipid storage, α-synuclein accumulation, and gliosis in the brain [68], whereas LIMP-2 overexpression or LIMP-2-derived peptides increase endogenous lysosomal GCase and reduce α-synuclein in α-syn-expressing cell lines [68, 69]. Importantly, overexpression of wild-type GBA in neurons, increasing GCase activity through gene therapy, small-molecule chaperones or activators, and reduction of substrate accumulation, prevents α-synucleinopathy of midbrain dopamine neurons in animal models and human neurons [70,71,72,73].
Protein–lipid interactions in aging and neurological diseases
α-Synuclein can bind lipids through its amphipathic N-terminal amino acid domain, and notably, all α-synuclein mutations that drive familial PD (A30P, E46K, G51D, H50Q, A53T) cluster in the lipid binding domain of α-synuclein. α-Synuclein interacts with several types of lipids, including glycosphingolipids, gangliosides, mono and polyunsaturated fatty acids, and acidic phospholipids [57, 74,75,76,77,78,79,80,81]. We have found that aggregated (proteinase-K resistant) α-synuclein increases with GCase inhibition in vivo [58]. In vitro data using human cells indicate that GCase dysfunction leads to lipid changes that destabilize the postulated tetramer configuration of normal α-synuclein [65]. Moreover, reduction of α-synuclein tetramers in mice leads to excess monomers, accumulation of proteinase-K resistant α-synuclein, intraneuronal accumulation of lipid droplets, and dopaminergic degeneration [55]. Our results from experiments evaluating brain aging in mouse and human brain show reduced GCase activity, elevated glycosphingolipids, and the presence of lipid-associated α-synuclein in the aging brain [18, 19, 53] and may be particularly relevant to understanding the cellular changes that precede α-synuclein linked cell toxicity. Interestingly, aging in mice is associated with aberrant lipidation of α-synuclein and phosphorylated Tau, and their accumulation in dopamine-containing Secretogranin II vesicles [19]. These results [19], together with the ability of α-synuclein to bind lipids and the abundance of lipids in Lewy bodies [17, 82], raise the possibilities that α-synuclein could interact with certain types of lipids under pathological conditions promoting its oligomerization and further aggregation (Fig. 2e), and that lipid perturbations are potentially a primary driver of PD and aging pathology. Consequently, α-synuclein aggregation per se may be a consequence of the disease initiation or typical neuronal toxicity caused by other mechanisms, and so attempts to remove oligomeric aggregated α-synuclein as a treatment of the disease may be futile [83].
Potentially important mechanisms involving lipid transport and protein pathophysiology
Appropriate lipid transfer between neurons and glia via lipid transporters such as apolipoproteins is essential for maintaining metabolic integrity of neurons. Elevation of reactive oxygen species and mitochondrial dysfunction in neurons leads to elevated lipid production in neurons and subsequent accumulation of lipid droplets, a lipid storage organelle, in glial cells [84]. Impairment of lipid transport from neurons to glia under conditions of oxidative stress results in neuronal damage and leads to neurodegeneration [85, 86].
Apolipoprotein E (apoE) is the most ubiquitous brain lipoprotein and acts as a scaffold for the formation of lipoprotein particles. ApoE transports and delivers cholesterol and other lipids via binding to cell surface apoE receptors. The e4 allele of apoE (APOE ε4) is the strongest genetic risk factor for sporadic AD and is found in ~ 60% of AD patients. Compared with apoE3, apoE4 is less able to promote cholesterol efflux and forms smaller CNS lipoproteins [87]. From clinical studies, it is clear that APOE ε4 is also linked with increased risk for dementias across the spectrum of Lewy body diseases, including dementia with Lewy bodies, and PD [88,89,90]. Curiously, mouse apoE is more amyloidogenic than the human apoE isoforms, including apoE4 [91]. Potentially, this could indicate an evolutionary pressure for better lipid handling provided by apoE2 and apoE3 isoforms in humans, since the mouse lifespan is so much shorter than humans.
Previous studies in PD experimental models have addressed and shown an involvement of cholesterol metabolism in α-synucleinopathy, and subsequent amelioration of α-synucleinopathy with cholesterol lowering drugs [92]. Clinical and genetic data support an even broader notion of dysregulation of lipid metabolism in PD and LBD [2, 18]. Relevant to the interactions of α-synuclein and lipids is α-synuclein’s structural and functional similarities to apolipoproteins, and α-synuclein has lipid-related functions [42, 74, 75, 78, 93,94,95,96,97,98]. The N-terminal region of α-synuclein contains 11-amino acid repeats which mediate membrane interactions of α-synuclein, characteristic of the 11-amino acid repeats that mediate lipid interactions of apolipoproteins. α-Synuclein can induce membrane curvature and tubulation, and form lipoprotein nanoparticles, similar to other apolipoproteins [93, 97]. In in vitro functional assays, α-synuclein can regulate cholesterol efflux in neuronal cells, which was described by these authors as apolipoprotein-like action [96]. The role of α-synuclein functioning like apolipoproteins is also highlighted by the finding that hippocampal apoE4 pathology is exacerbated in mice that carry human apoE4 with a loss of mouse α-synuclein alleles [95]. Moreover, mice deficient in α-synuclein exhibit an elevation in brain neutral lipid levels [99]. Taken together, these studies highlight a premise for α-synuclein’s function as a lipid-carrying molecule.
Variations in triggering receptor expressed on myeloid cells 2 (TREM2) are also a significant risk factor for developing late onset AD [100]. TREM2 is a lipid sensor and is expressed by microglia in the brain; TREM2 activation is essential for immune function, and microglia expressing TREM2 may sense lipids on damaged neurons and glia [101]. Indeed, the R47H disease-associated mutation impairs TREM2 detection of lipid ligands [101]. ApoE is also a ligand for TREM2 and can bind to apoptotic neuronal cell surfaces and increase TREM2-mediated phagocytosis by microglia [102,103,104], suggesting convergence of ApoE lipoprotein and TREM2 on the same pathway. Recently, ApoE has also been shown to reduce inflammation caused by activated C1q by direct protein-protein interaction, and loss of ApoE function produces oxidized lipids that activate the complement cascade [105].
Multiple lysosomal storage gene dysfunction and disorders create increased risk for Parkinson’s disease
In addition to Gaucher carriers with heterozygote GBA mutations, the increased risk of developing PD is observed in other lysosomal storage diseases with mutations in different enzymes that metabolize lipids in the lysosomes [16, 106, 107] and which are associated with primary accumulation of sphingolipids. α-Synuclein accumulation has also been described in the brain of patients with Sandhoff disease, Fabry’s disease, Krabbe’s disease, and Niemann-Pick disease type C1, as well as in several mouse models carrying mutations in lysosomal storage disorder genes [108,109,110,111,112,113,114]. Therefore, the accumulation of several different lipids could contribute to toxicity in neurons. Further studies will be necessary to determine if the lipids of other lysosomal storage disease than Gaucher’s disease also accumulate in PD and in the aging brain. In summary, it appears that accumulation of glycolipids can cause the entire cascade of events associated with PD, including lysosomal blockade, increased intracellular α-synuclein, and fulminant neuroinflammation leading to synaptic degeneration and death (Fig. 2).
Also relevant to PD risk and lysosomal storage disorders, ATP13A2 is a lysosomal ATPase that acidifies the lysosomal lumen and therefore allows the lysosomal proteases to properly function [115]. Mutations in ATP13A2 are associated with Kufor-Rakeb syndrome and young onset PD [116,117,118], as well as to the lysosomal storage disorder, neuronal ceroid lipofuscinosis. ATP13A2 levels are decreased in dementia with Lewy bodies and PD cases [115, 119]. ATP13A2 depletion has been shown to promote neurodegeneration with endolysosomal abnormalities and reduce the degradation of lysosomal substrates [115, 120,121,122,123]. While ATP13A2 knockout from primary neurons and ATP13A2 PD patients-derived fibroblasts lead to lysosomal dysfunction with subsequent accumulation of α-synuclein and toxicity [115, 121], ATP13A2 knockout mice had lysosomal dysfunction and toxicity without the accumulation of α-synuclein [120]. An explanation for these findings could be that there are compensatory mechanisms in living systems that already present a burden of α-synuclein. Interestingly, heterozygous ATP13A2 knockout mice leads to significant gliosis, which over time would likely be detrimental to function of vulnerable neuronal populations [124].
Dysfunction in mitophagy, cell, and vesicular transport mechanisms in neurodegenerative diseases
Autophagy-dependent removal of damaged mitochondria (mitophagy) (Fig. 2d) is responsible for the selective removal of superfluous and damaged mitochondria under a variety of pathophysiological conditions and is essential to relieve oxidative stress and prevent cytosolic and axonal damage and subsequent cell death. Dysfunction in various steps of the autophagy pathway, for example in autophagosome formation, lysosomal function, autophagic cargo, and autolysosome formation, has been implicated in several neurological and degenerative diseases including PD, AD, polyglutamine diseases, and amyotrophic lateral sclerosis [125]. Abnormalities in mitophagy are described in human sporadic and familial PD patient-derived neurons and fibroblasts (reviewed in [126]). Aging of post-mitotic cells is associated with impaired mitochondrial quality control leading to accumulation of large senescent mitochondria [127]. Of interest, mitochondrial function is compromised in fibroblasts and neurons isolated from PD patients carrying the LRRK2 G2019S mutation [128, 129], and PD patient LRRK2 G2019S mutation fibroblasts show decreased mitophagy by pathways that block the formation of autophagosomes [130].
Relevant for PD and LBD pathology, the targeting of α-synuclein to lysosomes occurs through the fusion with endosomal vesicles (Fig. 2c). Nedd4, an ubiquitin-ligase that has been shown to improve endosomal trafficking of several substrates [131], was shown to ubiquitinate α-synuclein and target it for degradation through the endolysosomal pathway [132]. Consistent with this, the lysosomal inhibitor chloroquine as well as the knockdown of critical endosomal proteins, but not proteasomal inhibitors, prevents the degradation of α-synuclein by Nedd4 [132]. In addition, Nedd4 was found to be increased in dopaminergic neurons containing Lewy bodies [132], suggesting a compensatory increase of this ubiquitin-ligase in conditions of α-synuclein accumulation. In Drosophila and rat models of α-synucleinopathy, Nedd4 overexpression prevents neuronal dysfunction and pathology [133], and NAB2, an activator of the Nedd4 pathway, rescues α-synuclein toxicity in vitro in both yeast and human neuron α-synucleinopathy models [134].
Rab proteins are a family of small GTPase proteins that are important for the vesicular transport from the ER to the Golgi complex but currently they are recognized as having a much broader role in virtually all types of vesicular transport systems (see Fig. 2a), including lysosomes, autophagic and synaptic vesicles [135,136,137]. Rab proteins were described to be involved in many steps of vesicular transport cycle, including formation of vesicles, vesicular motility along cytoskeleton components, and tethering and fusion to target membranes [138]. Lipids are also transported by Rab proteins, including Rab7, Rab5, Rab1, and Rab18 (reviewed in [138]), and Rabs are dependent on lipids for their function by lipid-induced posttranslational modification (attachment of geranyl group at the carboxyl-terminal cysteine residues) [138, 139]. Dysfunction in membrane trafficking and links to Rab proteins has been described for many neurodegenerative disorders, including AD, PD, Huntington’s disease (HD), amyotrophic lateral sclerosis, and Charcot-Marie-Tooth disease [140]. Our work on Rab function in PD first demonstrated that Rab3B, which is enriched in synaptic vesicles and involved in exocytosis and neurotransmitter release, is intrinsically more highly expressed in A10 dopaminergic neurons, which are less vulnerable to degeneration in PD, than in A9 substantia nigra pars compacta neurons [30, 31]. Gene transfer of Rab3B to A9 dopaminergic neurons in rats increased neurotransmitter content, number and size of synaptic vesicles, levels of presynaptic proteins, and improved vesicular transmitter handling and storage capacity of dopaminergic neurons [30]. In a model of retrograde degeneration of midbrain dopaminergic neurons in rodents induced by oxidative stress (which is potentially associated with lipid disruptions and peroxidation of synaptic vesicles), AAV-mediated overexpression of Rab3B improved motor function and protected dopaminergic neurons from degeneration [30]. Moreover, Rab3B overexpression also improves early metabolic changes caused by α-synucleinopathy in rodents (Hallett and Isacson, unpublished data). Elevated expression of a different Rab protein, Rab1a, normalizes α-synuclein levels in human PD neurons with SNCA triplication, potentially through transport to appropriate autophagic systems [141]. Also in the context of PD, Rab modulation of vesicle formation, delivery, tethering, and fusion has been shown to be partially dependent on the kinase activity of LRRK2, a different gene involved in familial PD [142, 143]. Rab proteins could therefore be a pharmacological target for improving vesicular and axonal transport deficits caused by abnormal lipid-protein interactions in PD and LBD.
Pivotal roles of astroglial and microglial cells in the pathophysiology and inflammatory responses seen in age-related neurodegenerative diseases
Abnormal lipid, protein, and metabolic stressors (for example, due to mitochondrial dysfunction or abnormal lipid metabolism) within PD-susceptible tissue and neurons are likely to signal distress to surrounding cells, including microglia and astrocytes [43, 144]. Neurons are in constant communication with glia. In the context of inflammatory and immunological responses, surprisingly it has been shown that both AD related β-amyloid and α-synuclein can participate in anti-microbial functions [145, 146], and so it is conceivable that the neuroimmune responses are retained and executed in the context of sterile inflammation caused by neuronal dysfunction and damage prior to cell death. Interestingly, the toll-like receptors that are involved in responses to bacteria and viruses have receptor agonists that belong to lipid classes [147] and when severe metabolic and lipid disturbances occur within dysfunctional neurons, such molecules, including fatty acids, may be participating in direct activation of neuroinflammation in brain tissue [148]. A causal link between the accumulation of senescent glial cells with cognitive decline and neuronal loss has been described in a mouse model of tauopathy, and genetic elimination of the senescent glia or pharmacological elimination, prevented cognitive decline and pathology [149].
In yeast and human cells, with extreme calcium, redox, vesicle trafficking, or α-synuclein protein load, all organisms die relatively acutely. However, in the age-dependent degeneration seen in PD (for example), the cells in a living tissue context in many cases are able to homeostatically respond to relatively minor changes in these disruptive mechanisms. In the brain, many different types of microglia (in different proinflammatory or phagocytic states) are in second-to-second surveillance of the overall cellular energy status, and what appears to be a large number of cellular / intracellular signals received. Consequently, immune regulation is an important part of cell growth, degeneration, and death in the more complex organisms that have immune systems. This intricate immune regulation can be altered in positive or negative directions to create abnormal neurodegeneration [150,151,152,153]. There is a very significant immunological genetic load that is associated with AD and PD for HLA locus risk and very prominent genetic markers, including the microglial markers that are associated with neurodegeneration.
The role of inflammation in PD has been inferred long ago when patients developed parkinsonism after encephalitis caused by 1918 influenza pandemic, known as von Economo disease [154]. The connection of inflammation and parkinsonism is also observed with other virus types. Experimental infection by H5N1 in mice show that the virus travels from the peripheral to central nervous system causing microglial activation, α-synuclein aggregation, and death of nigral dopaminergic neurons [155]. Traumatic brain injuries are also known to cause profound brain inflammation [156, 157], and in addition to increase in the levels of α-synuclein in the brain [158,159,160], it also increases the risk of PD [160, 161], further supporting the connection between inflammation and PD. Despite the fact that many studies support the role of inflammation in the process of neurodegeneration in PD and in other neurodegenerative diseases as well [144, 145, 162], the attention has been lately channeled into searching the role of disease-mutated proteins in PD, which can give clues about the pathophysiology of the disease but can be misleading if analyzed independently.
Post-mortem analysis of PD brains shows important microglial activation and glial invasion in areas where dopaminergic neurons degenerate [163, 164]. Even though inflammation and glial activation may be regarded as a secondary process [165], several studies in the literature support a more direct participation of inflammation in PD. First, proinflammatory molecules, including TNFα, IL-1β, and IL-6, and several toll-like receptors (TLRs) are elevated in PD brains [166,167,168,169]. Second, injection of PD-related toxins in experimental models, including MPTP and 6-hydroxydopamine, causes pronounced inflammation that leads to degeneration of dopaminergic neurons [169,170,171]. Injection of the TLR4 agonist, lipopolysaccharide (LPS), into the nigrostriatal pathway of rodents expressing normal or elevated levels of α-synuclein is associated with degeneration of dopaminergic neurons in the substantia nigra [172, 173], which is abated in mice that lack α-synuclein. Importantly, LPS injection also induces accumulation of intracellular insoluble aggregated α-synuclein [173]. LPS injection also increases the vulnerability of dopaminergic neurons to rotenone and 6-hydroxydopamine [152, 174] in a mechanism that involves the activation of several proinflammatory cytokines, in particular, IL-1β [152, 175]. In agreement, administration of IL-1 receptor antagonists significantly reduces TNF-α and IFN-γ levels, and attenuates the death of dopaminergic neurons caused by the LPS-induced sensitization at non-toxic doses [152]. In an additional inflammatory model, injection of the TLR3 agonist polyinosinic:polycytidylic acid [poly(I:C)] to the substantia nigra leads to sustained inflammation, increased susceptibility to low doses of 6-hydroxydopamine, and interfered with proteins involved with synaptic transmission and axonal transport [176]. In PD brain, TLR2 expression is elevated and correlates with accumulation of SDS-soluble α-synuclein [168] and TLR9 is also elevated in the brain of patients with PD compared to controls [169]. Substantia nigra dopamine degeneration induced by MPTP in mice is blocked by TLR9 deficiency [177].
In the cell biological lipid framework, knockout of GBA in zebrafish (which are devoid of α-synuclein) causes early microglial activation, reduction in motor activity, decrease of dopaminergic neurons by 30%, and the presence of intraneuronal ubiquitin-positive inclusions [178], supporting a role for inflammation in lipidopathy. Systemic administration of the GCase inhibitor CBE in mice also causes pronounced inflammation and complement activation parallel to α-synuclein accumulation and toxicity [58, 59]. We recently showed that levels of GPNMB, which is associated with astrocytes, microglia, and macrophages, and modulation of neuroinflammatory responses [179,180,181], are increased in the substantia nigra of patients with PD [151]. Remarkably, in experimental models, levels of GPNMB in the brain are elevated following lipid elevations caused by CBE, but are not altered by pure α-synucleinopathy, suggesting a potential for a dominant role of lipid-induced degeneration in PD [151]. Subsequent to our finding of GPNMB elevations in PD, it has also been discovered that GPNMB is elevated in the brain and CSF of patients with sporadic AD, and that GPNMB levels increase with disease progression in distinct AD transgenic mouse models and primarily colocalize with a distinct population of microglial cells located around amyloid plaques [182]. GPNMB elevations have also been shown in the brains, CSF, and plasma of the lysosomal storage disorders’ Gaucher disease and Niemann-Pick type C [183,184,185], further supporting an association of GPNMB in glycolipid dysregulation.
Converging inflammatory mechanisms in neurodegenerative diseases such as PD
In addition to the evident brain degeneration that most people think of as PD, in practically every patient there is significant involvement of the peripheral nervous system [56] and also evidence of inflammation in the gastrointestinal system, and potentially also in other organ systems such as the skin [186]. Clearly, PD genetics and aging is a cell biological problem that affects the whole body and can signal dysfunction and cause inflammatory reactions in regions that do not cause severe symptoms caused by neurodegeneration. In this context, there are a number of inflammatory molecules and cascades associated with the colitis like syndrome that is typically seen in many PD patients [187]. It is also possible to obtain evidence of lipidopathy in epidermal skin cells from PD patients with GBA mutations [188].
GWAS of sporadic cases of multiple neurological diseases have identified risk genes that are associated with the immune system [189]. Moreover, several genes linked with familial neurodegenerative diseases are reported to have roles within the immune system [146, 190,191,192,193], for example, LRRK2 and α-synuclein in PD. The familial PD-associated gene, LRRK2, is expressed in both neurons and immune cells in human brain, and peripheral myeloid cells express LRRK2 at high levels, and the expression of LRRK2 is upregulated by inflammatory signals [194,195,196,197]. Overexpression of α-synuclein in transgenic mice can promote inflammation. Mice overexpressing 5–15 times α-synuclein under a hamster prion promoter have a clear motor phenotype and gliosis but no formation of Lewy body-like inclusions or neuronal degeneration [198], and mice overexpressing α-synuclein under the Thy1 promoter show microglial activation [199]. Interestingly, Thy1-α-synuclein overexpressing mice also express human α-synuclein in the peripheral nervous system as well the brain, and these mice exhibit autonomic dysfunction [200]. Also, while intracerebral injection of fibrillated α-synuclein promotes the accumulation of aggregated α-synuclein in the brain of α-synuclein transgenic and non-transgenic mice [201, 202], a key event is the protracted robust inflammation in the brain of injected mice, even in non-transgenic mice, where the accumulation of aggregated α-synuclein is less clear [203]. This evidence taken together supports that inflammation may in some cases precede the substantial accumulation of pathological α-synuclein inclusions, and that the α-synuclein per se may further induce the inflammatory process. Whereas α-synuclein (and beta and gamma) are normally found primarily on synaptic vesicles, with pathological gain of function, including lipidation, the localization and distribution of α-synuclein can be altered potentially eliciting immune reactions. Supporting a role for α-synuclein in immune responses is the finding that silencing of endogenous α-synuclein in the substantia nigra also induces a neuroinflammatory response and degeneration of dopaminergic neurons [204]. In aging, a disruption of glial function with senescence is sufficient to create an inflammatory condition in hippocampal and other brain regions that may relate to significant functional memory deficits [149]. In summary, many pathological signals, including metabolic, mitochondrial dysfunction, and lipid abnormalities, are likely to contribute to, converge on, and perpetuate the inflammatory process in PD. These findings indicate that inflammatory processes may represent both an early and late event in the pathophysiology of PD. Even though the use of nonsteroidal anti-inflammatory drugs decrease the incidence of PD by 15% [205], the identification of markers to diagnose the disease at initial stages may help establish immune modulatory strategies for the disease.
Concluding remarks
The knowledge and analysis presented in this article provide new opportunities for identifying, measuring, and treating distinct features of age-dependent neurological diseases such as PD and several types of dementia disorders, at both cellular and systemic levels. For PD, the classic pathological sign used to diagnose at post-mortem have changed very little since the original observations of Lewy bodies in the substantia nigra and loss of dopamine neurons. The PD motor syndrome is fully explained by the loss of function and synapses linked to the midbrain dopaminergic system. Nevertheless, beyond the identification of α-synuclein as part of the Lewy body and the genetic risk that rare forms of mutations or multiplication of α-synuclein creates in families, few additional cellular pathological changes have gained diagnostic prominence. A surprising observation from familial genetics has shown that mutations that impair function in GCase in one allele (haploinsufficiency) create PD rather than Gaucher disease which is associated with loss of function in both GBA alleles and an early onset and fatal lipid storage disease throughout many organs in the body. An important insight that explains much of PD pathology is that aging by itself leads to a phenocopy of the GBA deficiency in patients and in mouse models [18, 53]. Genetic data has also put a spotlight on additional lysosomal enzymatic function, where > 50% of PD patients carry a mutation in a lysosomal storage disorder gene [16]. This is consistent with biochemical perturbations of various glycolipid pathways leading to PD pathological changes [58, 108,109,110,111,112,113,114], and electron microscopic evidence shows that the Lewy body is in fact composed of cellular organelles, membranes, and lipids, together with α-synuclein [17]. Lipids and α-synuclein changes occur with age [18, 19, 53] in ways that simulate many of the molecular changes associated with genetic risk factors. Given that for PD research cellular and animal models reveal that lipid and α-synuclein can interact to modify synaptic and vesicular transport, this points to several common pathways and potential pathological gain of α-synuclein function that trigger and sustains the development of PD.
On the new horizon are observations and experiments that reveal how glial and neuron physiological interactions are critical for the disease progression and final pathology. Microglia can both initiate and drive the disease under certain conditions [144, 152, 176]. Moreover, dysfunctional astrocytes can play critical roles in the initiation and progression of the disease. In a global context outside the brain, it is now clear that neurodegenerative disease pathology is present in many peripheral and central nervous system circuitries at the same time, with significant symptoms to patients from each system [56]. Relevant to such systemic manifestations appears to be many immune regulatory changes that are apparent in tissues and cellular systems affected by the disease before and after symptoms. Similar to PD, it is now evident that maladaptive immune responses can drive AD pathobiology. Finally, it is important to obtain diagnostic biomarkers that mirror these mechanisms to treat the actual primary causes of cellular dysfunction, rather than down-stream reactions or end-stage pathology of amyloidogenic proteins.
Abbreviations
- AD:
-
Alzheimer’s disease
- ER:
-
Endoplasmic reticulum
- GCase:
-
Glucocerebrosidase
- GPNMB:
-
Glycoprotein NMB
- IL-1β:
-
Interleukin-1 beta
- IL-6:
-
Interleukin-1 6
- LBD:
-
Lewy body dementia
- LPS:
-
Lipopolysaccharide
- PD:
-
Parkinson’s disease
- TLR:
-
Toll-like receptor
- TNFα:
-
Tumor necrosis factor alpha
- TREM2:
-
Triggering receptor expressed on myeloid cells 2
References
Shi H, Belbin O, Medway C, Brown K, Kalsheker N, Carrasquillo M, Proitsi P, Powell J, Lovestone S, Goate A, et al. Genetic variants influencing human aging from late-onset Alzheimer’s disease (LOAD) genome-wide association studies (GWAS). Neurobiol Aging. 2012;33:1849.e1845–18.
Klemann C, Martens GJM, Sharma M, Martens MB, Isacson O, Gasser T, Visser JE, Poelmans G. Integrated molecular landscape of Parkinson’s disease. NPJ Parkinsons Dis. 2017;3:14.
Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, Kerchner GA, Ayalon G, Bingol B, Sheng M, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet. 2017;49:1511–6.
Kumaran R, Cookson MR. Pathways to parkinsonism redux: convergent pathobiological mechanisms in genetics of Parkinson’s disease. Hum Mol Genet. 2015;24:R32–44.
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7.
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–8.
Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–73.
Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1167–9.
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A. Nussbaum R, et al: alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841.
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–7.
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–8.
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–60.
Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–9.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40.
Billingsley KJ, Bandres-Ciga S, Saez-Atienzar S, Singleton AB. Genetic risk factors in Parkinson’s disease. Cell Tissue Res. 2018;373:9–20.
Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, Heutink P, Shulman JM. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain. 2017;140:3191–203.
Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, Castaño-Díez D, Schweighauser G, Graff-Meyer A, Goldie KN, et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci. 2019;22:1099-1109.
Rocha EM, Smith GA, Park E, Cao H, Brown E, Hallett P, Isacson O. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann Clin Transl Neurol. 2015;2:433–8.
Brekk OR, Moskites A, Isacson O, Hallett PJ. Lipid-dependent deposition of alpha-synuclein and Tau on neuronal Secretogranin II-positive vesicular membranes with age. Sci Rep. 2018;8:15207.
Xu YH, Barnes S, Sun Y, Grabowski GA. Multi-system disorders of glycosphingolipid and ganglioside metabolism. J Lipid Res. 2010;51:1643–75.
Pastores GM, Hughes DA. Gaucher disease. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, LJH B, Stephens K, Amemiya A, editors. GeneReviews((R)). Seattle: University of Washington, Seattle; 1993.
Sidransky E. Gaucher disease: insights from a rare Mendelian disorder. Discov Med. 2012;14:273–81.
Orvisky E, Park JK, LaMarca ME, Ginns EI, Martin BM, Tayebi N, Sidransky E. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: correlation with phenotype and genotype. Mol Genet Metab. 2002;76:262–70.
Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, Mazzoni P, Pauciulo MW, Nichols WC, Gan-Or Z, et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain. 2015;138:2648–58.
Liu G, Boot B, Locascio JJ, Jansen IE, Winder-Rhodes S, Eberly S, Elbaz A, Brice A, Ravina B, van Hilten JJ, et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann Neurol. 2016;80:674–85.
Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, Buchman AS, Larson EB, Crane PK, Kaye JA, et al. GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology. 2012;79:1944–50.
Mata IF, Samii A, Schneer SH, Roberts JW, Griffith A, Leis BC, Schellenberg GD, Sidransky E, Bird TD, Leverenz JB, et al. Glucocerebrosidase gene mutations: a risk factor for Lewy body disorders. Arch Neurol. 2008;65:379–82.
Seto-Salvia N, Pagonabarraga J, Houlden H, Pascual-Sedano B, Dols-Icardo O, Tucci A, Paisan-Ruiz C, Campolongo A, Anton-Aguirre S, Martin I, et al. Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson’s disease course. Mov Disord. 2012;27:393–9.
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361:1651–61.
Chung CY, Koprich JB, Hallett PJ, Isacson O. Functional enhancement and protection of dopaminergic terminals by RAB3B overexpression. Proc Natl Acad Sci U S A. 2009;106:22474–9.
Chung CY, Seo H, Sonntag KC, Brooks A, Lin L, Isacson O. Cell type-specific gene expression of midbrain dopaminergic neurons reveals molecules involved in their vulnerability and protection. Hum Mol Genet. 2005;14:1709–25.
Surmeier DJ, Guzman JN, Sanchez-Padilla J, Schumacker PT. The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in Parkinson’s disease. Neuroscience. 2011;198:221–31.
Greene JG, Dingledine R, Greenamyre JT. Gene expression profiling of rat midbrain dopamine neurons: implications for selective vulnerability in parkinsonism. Neurobiol Dis. 2005;18:19–31.
Brownell AL, Jenkins BG, Elmaleh DR, Deacon TW, Spealman RD, Isacson O. Combined PET/MRS brain studies show dynamic and long-term physiological changes in a primate model of Parkinson disease. Nat Med. 1998;4:1308–12.
Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700.
Floor E, Wetzel MG. Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. J Neurochem. 1998;70:268–75.
Gotz ME, Freyberger A, Riederer P. Oxidative stress: a role in the pathogenesis of Parkinson’s disease. J Neural Transm Suppl. 1990;29:241–9.
Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet. 1989;1:1269.
Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y. Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem Biophys Res Commun. 1989;163:1450–5.
Sherer TB, Kim JH, Betarbet R, Greenamyre JT. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol. 2003;179:9–16.
Logan T, Bendor J, Toupin C, Thorn K, Edwards RH. alpha-Synuclein promotes dilation of the exocytotic fusion pore. Nat Neurosci. 2017;20:681–9.
George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–72.
Chung CY, Koprich JB, Siddiqi H, Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J Neurosci. 2009;29:3365–73.
Fanning S, Haque A, Imberdis T, Baru V, Barrasa MI, Nuber S, Termine D, Ramalingam N, Ho GPH, Noble T, et al. Lipidomic analysis of alpha-synuclein neurotoxicity identifies stearoyl CoA desaturase as a target for parkinson treatment. Mol Cell. 2019;73:1001–1014.e1008.
Rochet JC, Conway KA, Lansbury PT Jr. Inhibition of fibrillization and accumulation of prefibrillar oligomers in mixtures of human and mouse alpha-synuclein. Biochemistry. 2000;39:10619–26.
Ulusoy A, Decressac M, Kirik D, Bjorklund A. Viral vector-mediated overexpression of alpha-synuclein as a progressive model of Parkinson’s disease. Prog Brain Res. 2010;184:89–111.
Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ, Bjorklund A. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci. 2002;22:2780–91.
Prots I, Grosch J, Brazdis RM, Simmnacher K, Veber V, Havlicek S, Hannappel C, Krach F, Krumbiegel M, Schutz O, et al. alpha-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc Natl Acad Sci U S A. 2018;115:7813–8.
Volpicelli-Daley LA. Effects of alpha-synuclein on axonal transport. Neurobiol Dis. 2017;105:321–7.
Ordonez DG, Lee MK, Feany MB. Alpha-synuclein induces mitochondrial dysfunction through spectrin and the actin cytoskeleton. Neuron. 2018;97:108–124.e106.
Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW, Schapira AH. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol. 2012;72:455–63.
Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E, Cooper A, Garner B, Halliday GM. Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain. 2014;137:834–48.
Hallett PJ, Huebecker M, Brekk OR, Moloney EB, Rocha EM, Priestman DA, Platt FM, Isacson O. Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiol Aging. 2018;67:189–200.
Bartels T, Choi JG, Selkoe DJ. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–10.
Nuber S, Rajsombath M, Minakaki G, Winkler J, Muller CP, Ericsson M, Caldarone B, Dettmer U, Selkoe DJ. Abrogating native alpha-synuclein tetramers in mice causes a L-DOPA-responsive motor syndrome closely resembling Parkinson’s disease. Neuron. 2018;100:75–90.e75.
Engelender S, Isacson O. The threshold theory for Parkinson’s disease. Trends Neurosci. 2017;40:4–14.
Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, Toker NJ, Jeon S, Fredriksen K, Mazzulli JR. Reversible conformational conversion of alpha-synuclein into toxic assemblies by glucosylceramide. Neuron. 2018;97:92–107.e110.
Rocha EM, Smith GA, Park E, Cao H, Graham AR, Brown E, McLean JR, Hayes MA, Beagan J, Izen SC, et al. Sustained systemic glucocerebrosidase inhibition induces brain alpha-synuclein aggregation, microglia and complement C1q activation in mice. Antioxid Redox Signal. 2015;23:550–64.
Ginns EI, Mak SK, Ko N, Karlgren J, Akbarian S, Chou VP, Guo Y, Lim A, Samuelsson S, LaMarca ML, et al. Neuroinflammation and alpha-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol Genet Metab. 2014;111:152–62.
Cleeter MW, Chau KY, Gluck C, Mehta A, Hughes DA, Duchen M, Wood NW, Hardy J, Mark Cooper J, Schapira AH. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem Int. 2013;62:1–7.
Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52.
Rockenstein E, Clarke J, Viel C, Panarello N, Treleaven CM, Kim C, Spencer B, Adame A, Park H, Dodge JC, et al. Glucocerebrosidase modulates cognitive and motor activities in murine models of Parkinson’s disease. Hum Mol Genet. 2016;25:2645–60.
Schondorf DC, Aureli M, McAllister FE, Hindley CJ, Mayer F, Schmid B, Sardi SP, Valsecchi M, Hoffmann S, Schwarz LK, et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun. 2014;5:4028.
Xu YH, Sun Y, Ran H, Quinn B, Witte D, Grabowski GA. Accumulation and distribution of alpha-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol Genet Metab. 2011;102:436–47.
Kim S, Yun SP, Lee S, Umanah GE, Bandaru VVR, Yin X, Rhee P, Karuppagounder SS, Kwon SH, Lee H, et al. GBA1 deficiency negatively affects physiological alpha-synuclein tetramers and related multimers. Proc Natl Acad Sci U S A. 2018;115:798–803.
D'Auria L, Reiter C, Ward E, Moyano AL, Marshall MS, Nguyen D, Scesa G, Hauck Z, van Breemen R, Givogri MI, Bongarzone ER. Psychosine enhances the shedding of membrane microvesicles: implications in demyelination in Krabbe’s disease. PLoS One. 2017;12:e0178103.
Reczek D, Schwake M, Schröder J, Hughes H, Blanz J, Jin X, Brondyk W, Van Patten S, Edmunds T, Saftig P. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell. 2007;131:770–83.
Rothaug M, Zunke F, Mazzulli JR, Schweizer M, Altmeppen H, Lüllmann-Rauch R, Kallemeijn WW, Gaspar P, Aerts JM, Glatzel M, et al. LIMP-2 expression is critical for β-glucocerebrosidase activity and α-synuclein clearance. Proc Natl Acad Sci U S A. 2014;111:15573–8.
Zunke F, Andresen L, Wesseler S, Groth J, Arnold P, Rothaug M, Mazzulli JR, Krainc D, Blanz J, Saftig P, et al. Characterization of the complex formed by β -glucocerebrosidase and the lysosomal integral membrane protein type-2. Proc Natl Acad Sci U S A. 2016;113:3791-6.
Rocha EM, Smith GA, Park E, Cao H, Brown E, Hayes MA, Beagan J, McLean JR, Izen SC, Perez-Torres E, et al. Glucocerebrosidase gene therapy prevents alpha-synucleinopathy of midbrain dopamine neurons. Neurobiol Dis. 2015;82:495–503.
Aflaki E, Borger DK, Moaven N, Stubblefield BK, Rogers SA, Patnaik S, Schoenen FJ, Westbroek W, Zheng W, Sullivan P, et al. A new Glucocerebrosidase chaperone reduces alpha-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and parkinsonism. J Neurosci. 2016;36:7441–52.
Sardi SP, Viel C, Clarke J, Treleaven CM, Richards AM, Park H, Olszewski MA, Dodge JC, Marshall J, Makino E, et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc Natl Acad Sci U S A. 2017;114:2699–704.
Mazzulli JR, Zunke F, Tsunemi T, Toker NJ, Jeon S, Burbulla LF, Patnaik S, Sidransky E, Marugan JJ, Sue CM, Krainc D. Activation of beta-glucocerebrosidase reduces pathological alpha-synuclein and restores lysosomal function in Parkinson’s patient midbrain neurons. J Neurosci. 2016;36:7693–706.
Sharon R, Goldberg MS, Bar-Josef I, Betensky RA, Shen J, Selkoe DJ. alpha-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc Natl Acad Sci U S A. 2001;98:9110–5.
Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, Nussbaum RL. Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J Biol Chem. 2002;277:6344–52.
Perrin RJ, Woods WS, Clayton DF, George JM. Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J Biol Chem. 2001;276:41958–62.
Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9.
Jo E, McLaurin J, Yip CM, St George-Hyslop P, Fraser PE. Alpha-synuclein membrane interactions and lipid specificity. J Biol Chem. 2000;275:34328–34.
Kubo S, Nemani VM, Chalkley RJ, Anthony MD, Hattori N, Mizuno Y, Edwards RH, Fortin DL. A combinatorial code for the interaction of alpha-synuclein with membranes. J Biol Chem. 2005;280:31664–72.
Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–96.
Gaspar R, Pallbo J, Weininger U, Linse S, Sparr E. Ganglioside lipids accelerate alpha-synuclein amyloid formation. Biochim Biophys Acta, Proteins Proteomics. 2018;1866:1062-72.
Gai WP, Yuan HX, Li XQ, Power JT, Blumbergs PC, Jensen PH. In situ and in vitro study of colocalization and segregation of alpha-synuclein, ubiquitin, and lipids in Lewy bodies. Exp Neurol. 2000;166:324–33.
Espay AJ, Vizcarra JA, Marsili L, Lang AE, Simon DK, Merola A, Josephs KA, Fasano A, Morgante F, Savica R, et al. Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology. 2019;92:329–37.
Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J, Graham BH, Quintana A, Bellen HJ. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell. 2015;160:177–90.
Liu L, MacKenzie KR, Putluri N, Maletic-Savatic M, Bellen HJ. The glia-neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab. 2017;26:719–737.e716.
Ioannou MS, Jackson J, Sheu SH, Chang CL, Weigel AV, Liu H, Pasolli HA, Xu CS, Pang S, Matthies D, et al. Neuron-astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell. 2019;177:1522-35.e14.
Rebeck GW. The role of APOE on lipid homeostasis and inflammation in normal brains. J Lipid Res. 2017;58:1493–9.
Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, Buchman AS, Larson EB, Crane PK, Kaye JA, et al. APOE epsilon4 increases risk for dementia in pure synucleinopathies. JAMA Neurol. 2013;70:223–8.
Mata IF, Leverenz JB, Weintraub D, Trojanowski JQ, Hurtig HI, Van Deerlin VM, Ritz B, Rausch R, Rhodes SL, Factor SA, et al. APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol. 2014;71:1405–12.
Gao J, Huang X, Park Y, Liu R, Hollenbeck A, Schatzkin A, Mailman RB, Chen H. Apolipoprotein E genotypes and the risk of Parkinson disease. Neurobiol Aging. 2011;32:2106.e2101.
Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM. Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2002;9:305–18.
Koob AO, Ubhi K, Paulsson JF, Kelly J, Rockenstein E, Mante M, Adame A, Masliah E. Lovastatin ameliorates alpha-synuclein accumulation and oxidation in transgenic mouse models of alpha-synucleinopathies. Exp Neurol. 2010;221:267–74.
Varkey J, Isas JM, Mizuno N, Jensen MB, Bhatia VK, Jao CC, Petrlova J, Voss JC, Stamou DG, Steven AC, Langen R. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J Biol Chem. 2010;285:32486–93.
Emamzadeh FN. Role of apolipoproteins and alpha-Synuclein in Parkinson’s disease. J Mol Neurosci. 2017;62:344–55.
Bar R, Boehm-Cagan A, Luz I, Kleper-Wall Y, Michaelson D. The effects of apolipoprotein E genotype, a-synuclein deficiency, and sex on brain synaptic and Alzheimer’s disease-related pathology. Alzheimers Dement. 2018;10:1–11.
Hsiao JT, Halliday GM, Kim WS. alpha-synuclein regulates neuronal cholesterol efflux. Molecules. 2017;22 https://doi.org/10.3390/molecules22101769.
Varkey J, Mizuno N, Hegde BG, Cheng N, Steven AC, Langen R. alpha-Synuclein oligomers with broken helical conformation form lipoprotein nanoparticles. J Biol Chem. 2013;288:17620–30.
Ouberai MM, Wang J, Swann MJ, Galvagnion C, Guilliams T, Dobson CM, Welland ME. alpha-synuclein senses lipid packing defects and induces lateral expansion of lipids leading to membrane remodeling. J Biol Chem. 2013;288:20883–95.
Barcelo-Coblijn G, Golovko MY, Weinhofer I, Berger J, Murphy EJ. Brain neutral lipids mass is increased in alpha-synuclein gene-ablated mice. J Neurochem. 2007;101:132–41.
Yeh FL, Hansen DV, Sheng M. TREM2, microglia, and neurodegenerative diseases. Trends Mol Med. 2017;23:512–33.
Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160:1061–71.
Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, Zheng H, Li X, Rademakers R, Kang SS, Xu H, et al. Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J Biol Chem. 2015;290:26043–50.
Bailey CC, DeVaux LB, Farzan M. The triggering receptor expressed on myeloid cells 2 binds apolipoprotein E. J Biol Chem. 2015;290:26033–42.
Jendresen C, Arskog V, Daws MR, Nilsson LN. The Alzheimer’s disease risk factors apolipoprotein E and TREM2 are linked in a receptor signaling pathway. J Neuroinflammation. 2017;14:59.
Yin C, Ackermann S, Ma Z, Mohanta SK, Zhang C, Li Y, Nietzsche S, Westermann M, Peng L, Hu D, et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med. 2019;25:496–506.
Gan-Or Z, Ozelius LJ, Bar-Shira A, Saunders-Pullman R, Mirelman A, Kornreich R, Gana-Weisz M, Raymond D, Rozenkrantz L, Deik A, et al. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology. 2013;80:1606–10.
Foo JN, Liany H, Bei JX, Yu XQ, Liu J, Au WL, Prakash KM, Tan LC, Tan EK. Rare lysosomal enzyme gene SMPD1 variant (p.R591C) associates with Parkinson’s disease. Neurobiol Aging. 2013;34:2890.e2813–95.
Nelson MP, Tse TE, O'Quinn DB, Percival SM, Jaimes EA, Warnock DG, Shacka JJ. Autophagy-lysosome pathway associated neuropathology and axonal degeneration in the brains of alpha-galactosidase A-deficient mice. Acta Neuropathol Commun. 2014;2:20.
Nelson MP, Boutin M, Tse TE, Lu H, Haley ED, Ouyang X, Zhang J, Auray-Blais C, Shacka JJ. The lysosomal enzyme alpha-galactosidase a is deficient in Parkinson’s disease brain in association with the pathologic accumulation of alpha-synuclein. Neurobiol Dis. 2018;110:68–81.
Suzuki K, Iseki E, Togo T, Yamaguchi A, Katsuse O, Katsuyama K, Kanzaki S, Shiozaki K, Kawanishi C, Yamashita S, et al. Neuronal and glial accumulation of alpha- and beta-synucleins in human lipidoses. Acta Neuropathol. 2007;114:481–9.
Saito Y, Suzuki K, Hulette CM, Murayama S. Aberrant phosphorylation of alpha-synuclein in human Niemann-Pick type C1 disease. J Neuropathol Exp Neurol. 2004;63:323–8.
Shachar T, Lo Bianco C, Recchia A, Wiessner C, Raas-Rothschild A, Futerman AH. Lysosomal storage disorders and Parkinson’s disease: Gaucher disease and beyond. Mov Disord. 2011;26:1593–604.
Smith BR, Santos MB, Marshall MS, Cantuti-Castelvetri L, Lopez-Rosas A, Li G, van Breemen R, Claycomb KI, Gallea JI, Celej MS, et al. Neuronal inclusions of alpha-synuclein contribute to the pathogenesis of Krabbe disease. J Pathol. 2014;232:509–21.
Cachon-Gonzalez MB, Wang SZ, Ziegler R, Cheng SH, Cox TM. Reversibility of neuropathology in Tay-Sachs-related diseases. Hum Mol Genet. 2014;23:730–48.
Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A. 2012;109:9611–6.
Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–91.
Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G, Fabbrini G, Marconi R, Fincati E, Abbruzzese G, et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology. 2007;68:1557–62.
Lees AJ, Singleton AB. Clinical heterogeneity of ATP13A2 linked disease (Kufor-Rakeb) justifies a PARK designation. Neurology. 2007;68:1553–4.
Murphy KE, Cottle L, Gysbers AM, Cooper AA, Halliday GM. ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathol Commun. 2013;1:11.
Kett LR, Stiller B, Bernath MM, Tasset I, Blesa J, Jackson-Lewis V, Chan RB, Zhou B, Di Paolo G, Przedborski S, et al. alpha-synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2. J Neurosci. 2015;35:5724–42.
Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci. 2012;32:4240–6.
Schultheis PJ, Fleming SM, Clippinger AK, Lewis J, Tsunemi T, Giasson B, Dickson DW, Mazzulli JR, Bardgett ME, Haik KL, et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and age-dependent sensorimotor deficits. Hum Mol Genet. 2013;22:2067–82.
Bae EJ, Lee C, Lee HJ, Kim S, Lee SJ. ATP13A2/PARK9 deficiency neither cause lysosomal impairment nor Alter alpha-Synuclein metabolism in SH-SY5Y cells. Exp Neurobiol. 2014;23:365–71.
Rayaprolu S, Seven YB, Howard J, Duffy C, Altshuler M, Moloney C, Giasson BI, Lewis J. Partial loss of ATP13A2 causes selective gliosis independent of robust lipofuscinosis. Mol Cell Neurosci. 2018;92:17–26.
Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Fullgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93:1015–34.
Grunewald A, Kumar KR, Sue CM. New insights into the complex role of mitochondria in Parkinson’s disease. Prog Neurobiol. 2019;177:73-93.
Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem. 2002;269:1996–2002.
Smith GA, Jansson J, Rocha EM, Osborn T, Hallett PJ, Isacson O. Fibroblast biomarkers of sporadic Parkinson’s disease and LRRK2 kinase inhibition. Mol Neurobiol. 2016;53:5161–77.
Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, McLean JR, Carrillo-Reid L, Xie Z, Osborn T, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med. 2012;4:141ra190.
Korecka JA, Thomas R, Christensen DP, Hinrich AJ, Ferrari EJ, Levy SA, Hastings ML, Hallett PJ, Isacson O. Mitochondrial clearance and maturation of autophagosomes are compromised in LRRK2 G2019S familial Parkinson's disease patient fibroblasts. Hum Mol Genet. 2019. https://doi.org/10.1093/hmg/ddz126.
Belgareh-Touze N, Leon S, Erpapazoglou Z, Stawiecka-Mirota M, Urban-Grimal D, Haguenauer-Tsapis R. Versatile role of the yeast ubiquitin ligase Rsp5p in intracellular trafficking. Biochem Soc Trans. 2008;36:791–6.
Tofaris GK, Kim HT, Hourez R, Jung JW, Kim KP, Goldberg AL. Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc Natl Acad Sci U S A. 2011;108:17004–9.
Davies SE, Hallett PJ, Moens T, Smith G, Mangano E, Kim HT, Goldberg AL, Liu JL, Isacson O, Tofaris GK. Enhanced ubiquitin-dependent degradation by Nedd4 protects against alpha-synuclein accumulation and toxicity in animal models of Parkinson’s disease. Neurobiol Dis. 2014;64:79–87.
Chung CY, Khurana V, Auluck PK, Tardiff DF, Mazzulli JR, Soldner F, Baru V, Lou Y, Freyzon Y, Cho S, et al. Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science. 2013;342:983–7.
Luzio JP, Hackmann Y, Dieckmann NM, Griffiths GM. The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harb Perspect Biol. 2014;6:a016840.
Binotti B, Jahn R, Chua JJ. Functions of Rab proteins at presynaptic sites. Cells. 2016;5 https://doi.org/10.3390/cells5010007.
Tanida I. Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal. 2011;14:2201–14.
Bhuin T, Roy JK. Rab proteins: the key regulators of intracellular vesicle transport. Exp Cell Res. 2014;328:1–19.
Calero M, Chen CZ, Zhu W, Winand N, Havas KA, Gilbert PM, Burd CG, Collins RN. Dual prenylation is required for Rab protein localization and function. Mol Biol Cell. 2003;14:1852–67.
Kiral FR, Kohrs FE, Jin EJ, Hiesinger PR. Rab GTPases and membrane trafficking in neurodegeneration. Curr Biol. 2018;28:R471–r486.
Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D. alpha-synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci U S A. 2016;113:1931–6.
Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. 2016;5 https://doi.org/10.7554/eLife.12813.
Steger M, Diez F, Dhekne HS, Lis P, Nirujogi RS, Karayel O, Tonelli F, Martinez TN, Lorentzen E, Pfeffer SR, et al. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife. 2017;6 https://doi.org/10.7554/eLife.31012.
Deleidi M, Isacson O. Viral and inflammatory triggers of neurodegenerative diseases. Sci Transl Med. 2012;4:121ps123.
Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, Lefkowitz A, McColl G, Goldstein LE, Tanzi RE, Moir RD. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. 2016;8:340ra372.
Tomlinson JJ, Shutinoski B, Dong L, Meng F, Elleithy D, Lengacher NA, Nguyen AP, Cron GO, Jiang Q, Roberson ED, et al. Holocranohistochemistry enables the visualization of alpha-synuclein expression in the murine olfactory system and discovery of its systemic anti-microbial effects. J Neural Transm (Vienna). 2017;124:721–38.
Ruysschaert JM, Lonez C. Role of lipid microdomains in TLR-mediated signalling. Biochim Biophys Acta. 1848;2015:1860–7.
Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, Challis C, Schretter CE, Rocha S, Gradinaru V, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. 2016;167:1469–1480.e1412.
Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 2018;562:578-582.
Gonzalez H, Pacheco R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J Neuroinflammation. 2014;11:201.
Moloney EB, Moskites A, Ferrari EJ, Isacson O, Hallett PJ. The glycoprotein GPNMB is selectively elevated in the substantia nigra of Parkinson’s disease patients and increases after lysosomal stress. Neurobiol Dis. 2018;120:1–11.
Koprich JB, Reske-Nielsen C, Mithal P, Isacson O. Neuroinflammation mediated by IL-1beta increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson’s disease. J Neuroinflammation. 2008;5:8.
Presumey J, Bialas AR, Carroll MC. Complement system in neural synapse elimination in development and disease. Adv Immunol. 2017;135:53–79.
Picard F, de Saint-Martin A, Salmon E, Hirsch E, Marescaux C. Postencephalitic stereotyped involuntary movements responsive to L-Dopa. Mov Disord. 1996;11:567–70.
Jang H, Boltz D, Sturm-Ramirez K, Shepherd KR, Jiao Y, Webster R, Smeyne RJ. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci U S A. 2009;106:14063–8.
Feuerstein GZ, Liu T, Barone FC. Cytokines, inflammation, and brain injury: role of tumor necrosis factor-alpha. Cerebrovasc Brain Metab Rev. 1994;6:341–60.
Ghirnikar RS, Lee YL, Eng LF. Inflammation in traumatic brain injury: role of cytokines and chemokines. Neurochem Res. 1998;23:329–40.
Newell KL, Boyer P, Gomez-Tortosa E, Hobbs W, Hedley-Whyte ET, Vonsattel JP, Hyman BT. Alpha-synuclein immunoreactivity is present in axonal swellings in neuroaxonal dystrophy and acute traumatic brain injury. J Neuropathol Exp Neurol. 1999;58:1263–8.
Uryu K, Giasson BI, Longhi L, Martinez D, Murray I, Conte V, Nakamura M, Saatman K, Talbot K, Horiguchi T, et al. Age-dependent synuclein pathology following traumatic brain injury in mice. Exp Neurol. 2003;184:214–24.
Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, Lee VM, Trojanowski JQ, Smith DH. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 2007;208:185–92.
Smith DH, Uryu K, Saatman KE, Trojanowski JQ, McIntosh TK. Protein accumulation in traumatic brain injury. NeuroMolecular Med. 2003;4:59–72.
Weiner HL, Frenkel D. Immunology and immunotherapy of Alzheimer’s disease. Nat Rev Immunol. 2006;6:404–16.
McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285–91.
Banati RB, Daniel SE, Blunt SB. Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov Disord. 1998;13:221–7.
Hurley SD, O'Banion MK, Song DD, Arana FS, Olschowka JA, Haber SN. Microglial response is poorly correlated with neurodegeneration following chronic, low-dose MPTP administration in monkeys. Exp Neurol. 2003;184:659–68.
Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett. 1994;180:147–50.
Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett. 1994;165:208–10.
Dzamko N, Gysbers A, Perera G, Bahar A, Shankar A, Gao J, Fu Y, Halliday GM. Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol. 2017;133:303–19.
Ros-Bernal F, Hunot S, Herrero MT, Parnadeau S, Corvol JC, Lu L, Alvarez-Fischer D, Carrillo-de Sauvage MA, Saurini F, Coussieu C, et al. Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in parkinsonism. Proc Natl Acad Sci U S A. 2011;108:6632–7.
Depino AM, Earl C, Kaczmarczyk E, Ferrari C, Besedovsky H, del Rey A, Pitossi FJ, Oertel WH. Microglial activation with atypical proinflammatory cytokine expression in a rat model of Parkinson’s disease. Eur J Neurosci. 2003;18:2731–42.
Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O. Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur J Neurosci. 2002;15:991–8.
Herrera AJ, Castano A, Venero JL, Cano J, Machado A. The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol Dis. 2000;7:429–47.
Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM. Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J Neurosci. 2008;28:7687–98.
Fan LW, Tien LT, Lin RC, Simpson KL, Rhodes PG, Cai Z. Neonatal exposure to lipopolysaccharide enhances vulnerability of nigrostriatal dopaminergic neurons to rotenone neurotoxicity in later life. Neurobiol Dis. 2011;44:304–16.
Pott Godoy MC, Tarelli R, Ferrari CC, Sarchi MI, Pitossi FJ. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson’s disease. Brain. 2008;131:1880–94.
Deleidi M, Hallett PJ, Koprich JB, Chung CY, Isacson O. The toll-like receptor-3 agonist polyinosinic:polycytidylic acid triggers nigrostriatal dopaminergic degeneration. J Neurosci. 2010;30:16091–101.
Maatouk L, Compagnion AC, Sauvage MC, Bemelmans AP, Leclere-Turbant S, Cirotteau V, Tohme M, Beke A, Trichet M, Bazin V, et al. TLR9 activation via microglial glucocorticoid receptors contributes to degeneration of midbrain dopamine neurons. Nat Commun. 2018;9:2450.
Keatinge M, Bui H, Menke A, Chen YC, Sokol AM, Bai Q, Ellett F, Da Costa M, Burke D, Gegg M, et al. Glucocerebrosidase 1 deficient Danio rerio mirror key pathological aspects of human Gaucher disease and provide evidence of early microglial activation preceding alpha-synuclein-independent neuronal cell death. Hum Mol Genet. 2015;24:6640–52.
Tanaka H, Shimazawa M, Kimura M, Takata M, Tsuruma K, Yamada M, Takahashi H, Hozumi I, Niwa J, Iguchi Y, et al. The potential of GPNMB as novel neuroprotective factor in amyotrophic lateral sclerosis. Sci Rep. 2012;2:573.
Srinivasan K, Friedman BA, Larson JL, Lauffer BE, Goldstein LD, Appling LL, Borneo J, Poon C, Ho T, Cai F, et al. Untangling the brain’s neuroinflammatory and neurodegenerative transcriptional responses. Nat Commun. 2016;7:11295.
Neal ML, Boyle AM, Budge KM, Safadi FF, Richardson JR. The glycoprotein GPNMB attenuates astrocyte inflammatory responses through the CD44 receptor. J Neuroinflammation. 2018;15:73.
Huttenrauch M, Ogorek I, Klafki H, Otto M, Stadelmann C, Weggen S, Wiltfang J, Wirths O. Glycoprotein NMB: a novel Alzheimer’s disease associated marker expressed in a subset of activated microglia. Acta Neuropathol Commun. 2018;6:108.
Kramer G, Wegdam W, Donker-Koopman W, Ottenhoff R, Gaspar P, Verhoek M, Nelson J, Gabriel T, Kallemeijn W, Boot RG, et al. Elevation of glycoprotein nonmetastatic melanoma protein B in type 1 Gaucher disease patients and mouse models. FEBS Open Bio. 2016;6:902–13.
Marques AR, Gabriel TL, Aten J, van Roomen CP, Ottenhoff R, Claessen N, Alfonso P, Irun P, Giraldo P, Aerts JM, van Eijk M. Gpnmb is a potential marker for the visceral pathology in Niemann-Pick type C disease. PLoS One. 2016;11:e0147208.
Zigdon H, Savidor A, Levin Y, Meshcheriakova A, Schiffmann R, Futerman AH. Identification of a biomarker in cerebrospinal fluid for neuronopathic forms of Gaucher disease. PLoS One. 2015;10:e0120194.
Trivedi DK, Sinclair E, Xu Y, Sarkar D, Walton-Doyle C, Liscio C, Banks P, Milne J, Silverdale M, Kunath T, et al. Discovery of volatile biomarkers of Parkinson’s disease from sebum. ACS Cent Sci. 2019;5:599-606.
Devos D, Lebouvier T, Lardeux B, Biraud M, Rouaud T, Pouclet H, Coron E, Bruley des Varannes S, Naveilhan P, Nguyen JM, et al. Colonic inflammation in Parkinson’s disease. Neurobiol Dis. 2013;50:42–8.
Garcia-Sanz P, Orgaz L, Bueno-Gil G, Espadas I, Rodriguez-Traver E, Kulisevsky J, Gutierrez A, Davila JC, Gonzalez-Polo RA, Fuentes JM, et al. N370S-GBA1 mutation causes lysosomal cholesterol accumulation in Parkinson’s disease. Mov Disord. 2017;32:1409–22.
Gan L, Cookson MR, Petrucelli L, La Spada AR. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat Neurosci. 2018;21:1300–9.
Dzamko N, Geczy CL, Halliday GM. Inflammation is genetically implicated in Parkinson’s disease. Neuroscience. 2015;302:89–102.
Brockmann K, Schulte C, Schneiderhan-Marra N, Apel A, Pont-Sunyer C, Vilas D, Ruiz-Martinez J, Langkamp M, Corvol JC, Cormier F, et al. Inflammatory profile discriminates clinical subtypes in LRRK2-associated Parkinson’s disease. Eur J Neurol. 2017;24:427–e426.
Jay TR, von Saucken VE, Landreth GE. TREM2 in neurodegenerative diseases. Mol Neurodegener. 2017;12:56.
Villegas-Llerena C, Phillips A, Garcia-Reitboeck P, Hardy J, Pocock JM. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr Opin Neurobiol. 2016;36:74–81.
Miklossy J, Arai T, Guo JP, Klegeris A, Yu S, McGeer EG, McGeer PL. LRRK2 expression in normal and pathologic human brain and in human cell lines. J Neuropathol Exp Neurol. 2006;65:953–63.
Hakimi M, Selvanantham T, Swinton E, Padmore RF, Tong Y, Kabbach G, Venderova K, Girardin SE, Bulman DE, Scherzer CR, et al. Parkinson’s disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm. 2011;118:795–808.
Thevenet J, Pescini Gobert R, Hooft van Huijsduijnen R, Wiessner C, Sagot YJ. Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS One. 2011;6:e21519.
Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, Korzenik JR, Rioux JD, Daly MJ, Xavier RJ, Podolsky DK. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol. 2010;185:5577–85.
Gomez-Isla T, Irizarry MC, Mariash A, Cheung B, Soto O, Schrump S, Sondel J, Kotilinek L, Day J, Schwarzschild MA, et al. Motor dysfunction and gliosis with preserved dopaminergic markers in human alpha-synuclein A30P transgenic mice. Neurobiol Aging. 2003;24:245–58.
Watson MB, Richter F, Lee SK, Gabby L, Wu J, Masliah E, Effros RB, Chesselet MF. Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp Neurol. 2012;237:318–34.
Hallett PJ, McLean JR, Kartunen A, Langston JW, Isacson O. alpha-synuclein overexpressing transgenic mice show internal organ pathology and autonomic deficits. Neurobiol Dis. 2012;47:258–67.
Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209:975–86.
Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–53.
Sacino AN, Brooks M, McGarvey NH, McKinney AB, Thomas MA, Levites Y, Ran Y, Golde TE, Giasson BI. Induction of CNS alpha-synuclein pathology by fibrillar and non-amyloidogenic recombinant alpha-synuclein. Acta Neuropathol Commun. 2013;1:38.
Benskey MJ, Sellnow RC, Sandoval IM, Sortwell CE, Lipton JW, Manfredsson FP. Silencing alpha synuclein in mature nigral neurons results in rapid Neuroinflammation and subsequent toxicity. Front Mol Neurosci. 2018;11:36.
Gagne JJ, Power MC. Anti-inflammatory drugs and risk of Parkinson disease: a meta-analysis. Neurology. 2010;74:995–1002.
Acknowledgements
Not applicable.
Funding
This research was supported (OI, PH) by NIH/NINDS 1R01NS092667, NIH/NIA R01 AG060195, the Michael J Fox Foundation, Harold and Ronna Cooper Family, The Orchard Foundation, The Consolidated Anti-Aging Foundation. SE was supported while at the Neuroregeneration Institute by a sabbatical fellowship from Technion University, Israel.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
OI drafted and finalized the article with PH. SE contributed to the writing of the review. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Authors’ information
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
All the authors agree with the content of this publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hallett, P.J., Engelender, S. & Isacson, O. Lipid and immune abnormalities causing age-dependent neurodegeneration and Parkinson’s disease. J Neuroinflammation 16, 153 (2019). https://doi.org/10.1186/s12974-019-1532-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-019-1532-2