
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a highly devastating disease with poor prognosis and rising incidence. Late detection and a particularly aggressive biology are the major challenges which determine therapeutic failure. In this review, we present the current status and the recent advances in PDAC treatment together with the biological and immunological hallmarks of this cancer entity. On this basis, we discuss new concepts combining distinct treatment modalities in order to improve therapeutic efficacy and clinical outcome – with a specific focus on protocols involving radio(chemo)therapeutic approaches.
Similar content being viewed by others


Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most prevalent neoplastic disease of the pancreas accounting for more than 90% of all pancreatic malignancies [1]. To date, PDAC is the fourth most frequent cause of cancer-related deaths worldwide with a 5-year overall survival of less than 8% [2]. The incidence of PDAC is expected to rise further in the future, and projections indicate a more than two-fold increase in the number of cases within the next ten years, both in terms of new diagnoses as well as in terms of PDAC-related deaths in the U.S. as well as in European countries ([3, 4], www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/pancreatic-cancer#heading-Zero). A particular reason for this – apart from the general aging of our society – is the evident implication of obesity and type 2 diabetes, two emerging public health challenges, in PDAC etiology [5,6,7]. Life style habits, including alcohol and tobacco abuse, which are well-known to increase the risk for several other types of cancer, such as lung cancer and squamous cell carcinomas of the head and neck region [8,9,10], also appear to be involved in PDAC development [11,12,13,14,15]. Finally, for a subgroup of approximately 5-6% of all PDAC patients, genetic predispositions, such as germline mutations in the genes BRCA1/2, ATM, MLH1, TP53, or CDKN2A, represent further risk factors [16,17,18].
Current treatment standards and recent advances in PDAC chemo- and/or radiotherapy
Efficacy and outcome of PDAC treatment are largely determined by the stage of disease at the time of diagnosis. Surgical resection followed by adjuvant chemotherapy is the only possibly curative therapy available, yet only 10-20% of PDAC patients present with resectable PDAC stages, while the residual 80-90% show locally advanced, non-resectable stages or – in the majority – distant metastases [19, 20]. Systemic chemotherapy is commonly employed as first-line treatment in patients with non-resectable or borderline-resectable tumors. This encompasses nucleoside analogues, including gemcitabine and capecitabine, or the pyrimidine analogue 5-fluorouracil (5-FU) in monotherapy settings or in combination with other treatment modalities, such as radiotherapy, respectively [20,21,22]. FOLFIRINOX, a poly-chemotherapeutic regimen composed of folinic acid, 5-FU, irinotecan, and oxaliplatin, has been reported to nearly double median survival in the metastasized stage as compared to gemcitabine alone [23], and the combination of gemcitabine and a nanoparticle albumin-bound paclitaxel (nab-paclitaxel) has also been shown to significantly improve overall survival [24]. However, these protocols are associated with relevantly higher toxicity, thus often preventing their application in elderly patients and/or patients with poor performance status, but overall quality of life was reported to increase [25].
Radio(chemo)therapy has been rather infrequently adopted for the treatment of PDAC, since the majority of patients suffer from disseminated stages in which local treatment procedures are of secondary importance [26]. Nevertheless, neoadjuvant radiotherapy has the potential to improve PDAC resectability in locally advanced or primarily inoperable/borderline-operable patients, and its beneficial effects on local tumor control are well documented [27, 28]. Compared to other cancer entities, PDAC tumors exhibit a rather high degree of radioresistance – a characteristic which is currently addressed by combining PDAC radiotherapy with radiosensitizing agents, including gemcitabine, capecitabine, or 5-FU, respectively [28, 29]. According to the guidelines of the National Comprehensive Cancer Network (NCCN), the use of radio(chemo)therapy is recommended for PDAC patients with borderline-resectable tumors, and several regimens involving capecitabine, gemcitabine, or 5-FU have been clinically implemented [29, 30]. The advances of modern external beam radiation techniques, including image-guided radiation therapy (IGRT), stereotactic body radiation therapy (SBRT), and ablative radiation therapy, as well as the combination with novel chemotherapeutic protocols have clearly widened the spectrum of radiotherapeutic options [27, 31, 32].
Expecting increased toxicities when combining more aggressive treatment approaches, sequential application is currently being evaluated in the randomized phase III CONKO-007 trial for PDAC patients with borderline-resectable, non-metastatic disease (NCT01827553). Preliminary results from an interims analysis document a promising outcome with higher rates of resectability, confirming previous phase II findings [27, 30, 33]. As the performance of systemic therapies gradually improves, local tumor control moves back into the focus of interest, both with respect to symptom control as well as with respect to quality of life. In consequence, the importance of local radiotherapy for the treatment of PDAC patients is constantly growing. SBRT is a highly conformal radiation technique which is employed to deliver high doses in a small number of fractions. Due to its steep dose gradients around the target volume, SBRT efficiently spares adjacent organs at risk resulting in relevantly lower toxicity. In several studies, SBRT achieved significant improvements in pain control paralleled by increased local tumor control [34]. Hence, SBRT can be seen as an effective and safe therapeutic option, and its use in multimodality treatment concepts and/or in palliative settings is considered more and more frequently.
In several other cancer entities, e.g. in melanoma and lung cancer, the implementation of immunotherapeutic approaches, specifically immune checkpoint inhibition, has proven compelling success [35,36,37,38]. Yet, at least so far, treatment efficacy in PDAC has been rather limited [35, 39], and checkpoint inhibition has only received approval for the small subset of PDAC tumors with high microsatellite instability (1-2% of all cases) [40, 41]. This may be due to the strongly immunosuppressive, desmoplastic PDAC microenvironment, the relatively low mutational burden (resulting in a low number of neo-antigens), as well as other biological and/or immunological hallmarks of PDAC which are discussed in this review [42].
Biological and immunological hallmarks of PDAC
Tumor plasticity and heterogeneity
The pancreas contains cells of exocrine (acinar), epithelial (ductal), and endocrine (α, β, δ, ε) origin among which acinar cells are well known for their high degree of plasticity. This plasticity is considered to drive pancreas homeostasis and regeneration, as – in contrast to other organs of the gastrointestinal tract – the pancreas seems to lack a defined stem cell compartment [43]. In a process called acinar-to-ductal metaplasia (ADM), acinar cells transdifferentiate to more epithelial (ductal-like) phenotypes when experiencing certain macro- and microenvironmental stimuli, e.g. tissue damage, inflammatory, or stress conditions [44, 45]. During ADM, acinar cells acquire ‘progenitor cell-like’ characteristics which render them more susceptible to pro-oncogenic hits, such as activating mutations in the proto-oncogene KRAS, eventually transforming them into pancreatic intra-epithelial neoplasias (PanINs). This transformation is generally considered as the initial step in PDAC development followed by sequential progression involving genetic hits in several tumor suppressor genes [46] (Fig. 1).

Multi-step PDAC carcinogenesis. Modified from [47].
In order to examine the mutational and transcriptional landscape of PDAC, a number of next generation sequencing approaches were initiated in the last years [48,49,50,51]. In conjunction, these studies showed that the gene encoding the proto-oncogenic GTPase KRAS as well as several tumor suppressor genes, including tumor suppressor protein 53 (TP53), cyclin-dependent kinase inhibitor 2A (CDKN2A), and mothers against decapentaplegic homologue 4 (SMAD4), exhibit the most frequent alterations and/or mutations in PDAC [49]. For instance, KRAS was not only found to be mutated in most PDAC tumors (> 90%), its mutant alleles were additionally amplified in a subgroup of samples, resulting in acceleration of their tumor-promoting potential [52]. Furthermore, RAC-beta serine/threonine-protein kinase (AKT2) is frequently overexpressed, and the activity of its upstream regulator phosphoinositide 3-kinase (PI3K) is often elevated in PDAC leading to increased tumor cell survival [53, 54]. Apart from these key mutations, several more uncommon alterations, such as germline mutations in DNA damage repair genes (e.g. breast cancer early onset genes 1/2 (BRCA1/2), partner and localizer of BRCA2 (PALB2), and ataxia telangiectasia mutated protein serine/threonine kinase ATM), or somatic mutations in DNA mismatch repair regulator genes leading to increased microsatellite instability have been found in certain subsets of patients [55]. Of note, the transcriptomic landscape of PDAC is not entirely governed by genetic alterations. Integrated epigenetic regulatory circuits comprising chromatin-based mechanisms, such as DNA methylation and histone post-translational modification, as well as regulation by non-coding RNAs are also largely distorted in PDAC. In this regard, key tumor suppressor genes have been described to be repressed, and oncogenes upregulated due to epigenetic alterations [56]. Furthermore, epigenetic (re-)programing is fundamentally linked to tumor progression and metastasis formation [57, 58], and the epigenetic landscapes of human PDAC subtypes differ substantially [59].
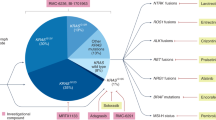
PDAC is a highly heterogenic disease, and various attempts have been undertaken to define distinct subtypes with the aim of stratifying patients towards personalized treatment strategies [49, 50, 60,61,62]. Currently available transcriptome-based classifications were extracted via unsupervised clustering methods and differ in the numbers of subtypes identified. Nevertheless, all share common subtypes, including a classical/canonical subtype hallmarked by epithelial-like gene expression, and a quasi-mesenchymal/basal-like subtype characterized by a more mesenchymal gene expression pattern and poorer prognosis (Fig. 2). These subtypes meanwhile can be stratified by immunohistochemistry using hepatocyte nuclear factor 1A (HNF1A) and cytokeratin-81 (KRT81) as markers [64]. Furthermore, subtypes related to exocrine pancreas function have been described as well as subtypes with expression signatures of immune cell-related genes [50, 61, 62]. Although to date there is still no consensus classification which would be the prerequisite for clinical application, retrospective as well as prospective analyses have shown that subtype-based stratification has the potential for genomics-driven precision medicine [64, 65]. The PDAC subtypes obviously stem from inter-tumoral heterogeneity. Yet, intra-tumoral heterogeneity needs to be considered as well, and tumor cell plasticity might render these classifications dynamic, especially upon therapeutic intervention.

Molecular classifications of PDAC. Modified from [63].
Desmoplastic, hypoxic, immunosuppressive microenvironment
A crucial hallmark of PDAC is the existence of extensive desmoplastic stroma which can constitute up to 90% of the tumor volume and is commonly considered to originate from cancer-associated fibroblasts (CAFs) [42] (Fig. 3). Distinct subtypes of CAFs with either myofibroblastic or inflammatory phenotypes have been identified [67, 68], and the major source of CAFs appear to be pancreatic stellate cells which upon activation, e.g. by injury or chronic inflammation, start depositing huge amounts of extra cellular matrix, including laminins, fibronectins, collagens, and hyaluronan [69,70,71,72]. Interestingly, expression of focal adhesion kinase 1 (FAK1) in PDAC cells has recently been reported to be decisive for this process as pharmacological targeting of FAK1 interfered with the formation of desmoplasia, thus offering a potential target for therapeutic intervention [73]. Hypoxia is another key feature of the PDAC microenvironment, and it is closely interlinked with desmoplasia. It originates from desmoplasia-associated hypovascularization and vice versa favors desmoplastic progression by activating pancreatic stellate cells [74,75,76]. PDAC hypoxia and desmoplasia, which are observed in clinical samples as well as in genetically engineered mouse models, seem to represent barriers to T cell infiltration – intriguingly both for effector as well as regulatory T cells – and T cell activation [77,78,79]. Moreover, hypoxia and desmoplasia are accompanied by a strong accumulation of myeloid cells [80, 81]. Macrophages that are recruited adopt an immunosuppressive, pro-angiogenic M2-like state, block CD4+ T cell entry into the PDAC microenvironment, support PDAC progression, and thus are a marker of negative clinical prognosis [76, 82, 83]. Systemic frequencies of monocytes and granulocytes are elevated in PDAC patients, and due to their pathological activation and immunosuppressive function they are classified as monocytic or polymorphonuclear myeloid-derived suppressor cells (MDSCs), respectively. Both populations are potent suppressors of T cell function and inhibit anti-tumor immune responses [84, 85]. Recently, the CXCL-1/CXCR2-axis has been shown to be crucially involved in intra-tumoral recruitment of MDSCs, suppressing CD8+ T cell infiltration and function as well as compromising responsiveness to immunotherapy [86]. Apart from these innate immune cell subpopulations, immunosuppressive T and B cell subpopulations, including regulatory T cells, γδ T cells, and regulatory B cells, have been described in the PDAC microenvironment. They do not only block activation but also infiltration of effector T cells resulting in low intra-tumoral CD8+ T cell frequencies [87,88,89]. These effector T cells appear to be antigen-experienced, but tumor antigen recognition and/or T cell activation seem to be disturbed [90]. However, the intra-tumoral T cell repertoire shows enrichment in distinct T cell receptors, suggesting that in principle PDAC tumors are sites of local T cell expansion [91].

PDAC desmoplasia. Modified from [66].
On the cytokine level, the PDAC microenvironment represents a comparable degree of complexity. Nevertheless, the dominating cytokines seem to be transforming growth factor beta (TGF-β), interleukin (IL-) 6, IL-8, IL-10, IL-35, granulocyte macrophage colony-stimulating factor (GM-CSF), CC-chemokine ligand 2 (CCL-2), CXC-chemokine ligand 1 (CXCL-1), and CXCL-13. In complex networks they orchestrate the recruitment and education of innate and adaptive immune cells as well as their crosstalk with tumor cells, CAFs, and other cells in the PDAC microenvironment, culminating in the desmoplastic, immunosuppressive milieu that has been described above [92,93,94].
Metastasis formation
Another feature of PDAC is its early progression to metastatic disease [1]. In advanced stages, patients show invasion of the (retro)peritoneum, the liver, and other gastrointestinal organs, as well as – in some cases – the vascular and/or the nervous system [95]. The key drivers of PDAC metastasis formation are still poorly understood, especially since the genetic composition of most metastases is closely resembling the one of the corresponding primary tumors [96,97,98]. Nevertheless, metastasis formation appears to be a clonal process, since primary PDAC tumors are composed of different subclones with individual metastatic potential, and most of the metastases show high levels of clonality, indicating that they initially evolved from one or only a few disseminated tumor cells [96, 98]. Mechanistic studies with genetically traceable mouse models identified a crucial involvement of epithelial-to-mesenchymal transition (EMT) explaining also why the quasi-mesenchymal PDAC subtype as characterized by stronger expression of mesenchymal genes may be associated with poorer prognosis due to accelerated metastasis formation [61, 62, 99] (Fig. 4). EMT so far has been considered to be orchestrated by a complex network of transcription factors which repress epithelial gene expression and/or induce mesenchymal gene expression, including twist-related protein 1 and 2 (TWIST1/2), snail family zinc finger protein SNAI1 and 2 (SNAI1/2), zinc finger E-box-binding homeobox 1 and 2 (ZEB1/2), and paired mesoderm homeobox protein 1 (PRRX1a/b) [100, 101]. Especially the EMT activator ZEB1 has been assigned a central role for tumor cell plasticity and metastasis formation in murine PDAC models [102]. miRNAs, particularly miR-10, miR-21 and members of the miR-200 family, constitute another regulatory level of EMT and are closely interlinked with the EMT transcription factors via diverse feedback and feedforward circuits [103, 104]. Recently, a novel, partial program of EMT has been described which is driven by post-translational internalization of epithelial proteins resulting in cluster-like rather than single-cell dissemination [105].

PDAC epithelial-mesenchymal transition and metastasis formation.
Several parameters of the tumor micro- and macroenvironment are known to influence EMT regulation. Amongst those, hypoxia, inflammation, and metabolic stress appear to be of special importance [100]. Interestingly, high blood glucose concentration, a crucial characteristic of diabetes, has also been shown to facilitate EMT and metastasis formation [7], thus linking a documented risk factor to a relevant tumorbiological process. In order to colonize foreign tissues circulating PDAC cells must undergo a reverse form of EMT (MET) and re-acquire the epithelial state [106, 107]. Morphologically and mechanistically, MET displays many features of EMT in an inverse manner. However, the details of this process as well as its master regulators are still being investigated.
EMT/MET phenomena seem to be crucial elements in the process of metastasis formation, yet gene expression profiling and epigenomic comparisons between primary tumor cells and metastatic cells also disclosed an involvement of other mechanisms, such as rewiring of the carbohydrate metabolism, e.g. in the oxidative branch of the pentose phosphate pathway, as well as shifts in energy consumption [58, 108, 109]. Further studies revealed a (re-)activation of embryonic programs and/or elevated expression levels of cancer stem cell markers, including forkhead box protein A1 (FOXA1), aldehyde dehydrogenase 1 (ALDH1), ATP-binding cassette sub-family G member 2 (ABCG2), and hepatocyte growth factor receptor (c-Met), in metastatic PDAC cells, suggesting a close relationship between retrograde developmental transition, cancer cell stemness and biological features of metastasis formation [57, 110]. Finally, the primary tumor appears to condition the future target organ of metastasis by releasing soluble factors and/or exosomes, thus generating a pre-metastatic niche – even in the status of a premalignant lesion [111]. Key players in this regard have been identified to be tissue inhibitor of metalloproteinases 1 (TIMP-1) and macrophage migration inhibitory factor (MIF) [112, 113].
Therapy resistance
A signature hallmark of PDAC is its high degree of resistance against virtually any kind of therapy [114,115,116]. Accordingly, overcoming treatment resistance will be essential in order to improve the overall prognosis of PDAC.
The therapeutic success of current first-line chemotherapy involving cytidine analogues, the poly-chemotherapeutic protocol FOLFIRINOX, or gemcitabine plus nab-paclitaxel, respectively, is strongly limited by intrinsic and/or acquired chemoresistance, and the underlying mechanisms are only poorly understood [21, 115]. Several predictive biomarkers have been identified, e.g. increased expression of ribonucleotide reductase catalytic subunits M1/2 (RRM1/2), an enzyme catalyzing the reduction of ribonucleotides, or human equilibrative nucleoside transporter 1 (hENT1), a transmembrane protein which imports nucleosides into the cytosol [117, 118]. In preclinical studies, it was observed that elevated expression levels of RRM1 indeed mediate resistance of PDAC cells to gemcitabine [117,118,119], yet no association between RRM1 expression and OS was detected in clinical analyses [120]. Similar examples are given by integrin-linked kinase (ILK) [121] and hypoxia-inducible, pro-apoptotic factor BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) [122]. Furthermore, cells of the microenvironment limit the efficacy of gemcitabine treatment. Recent data show that CAFs contribute to gemcitabine failure by metabolizing gemcitabine to the active metabolite 2′,2′-difluorodeoxycytidine-5′-triphosphate (dFdCTP). However, since dFdCTP cannot cross cell membranes, this process scavenges gemcitabine and reduces the effective concentration of the active metabolite in the tumor cells [123]. In case of FOLFIRINOX treatment, increased expression of thymidylate synthase (TS) and the 5-FU-catabolizing enzyme dihydropyrimidine dehydrogenase (DPD) were shown to contribute to therapy resistance, both in preclinical models and in retrospective clinical analyses [119, 124]. However, despite all these efforts, biomarker-based, individualized chemotherapy protocols are far from being clinical standard. This is predominantly due to a lack of prospective validation studies, let alone randomized controlled trials.
PDAC tumors also exhibit a high degree of radioresistance often resulting in tumor progression even during therapy [125]. As in case of chemoresistance, the responsible mechanisms appear to be multifactorial. From a biophysical point of view, the hypoxic PDAC microenvironment reduces the biological effectiveness of photon irradiation by 2-3 fold as compared to well-oxygenated tissues and, thus, attenuates its therapeutic efficacy [126, 127]. Additionally, several studies revealed an overexpression of key regulators of the DNA damage response, e.g. RAD51, in PDAC which contribute to accelerated repair of radiation-induced DNA damage [128, 129]. Other studies provided evidence for an implication of Integrin- or SMAD signaling in PDAC radioresistance [130,131,132]. Finally, increased recruitment of monocytes upon irradiation stimulating tumor cell proliferation and neovascularization in response to therapy have been discussed [133]. In order to counteract PDAC radioresistance, several approaches focused on adjusting radiotherapeutic protocols. As such, radiotherapy meanwhile is frequently combined with concomitant chemotherapy (radiochemotherapy), using gemcitabine, 5-FU, or capecitabine as radiosensitizing agents [134, 135]. Additionally, stereotactic irradiation regimens with higher single doses, including SBRT and ablative body radiotherapy, are increasingly being employed aiming at the delivery of higher biologically active doses to the tumor [26, 31, 136]. However, therapeutic success is still rather limited, and future attempts should evaluate the clinical potential of biologically and/or immunologically optimized radiochemotherapy strategies.
Novel approaches of mechanism-based, molecularly targeted therapies
Biologically targeted therapies (1,363 words)
Since less than 20% of all PDAC patients exhibit surgically resectable disease at time of presentation, systemic chemotherapy is currently the most frequently applied treatment option [21]. Albeit the development of novel poly-chemotherapy protocols, the overall prognosis, and survival rate of PDAC patients still remain poor. Hence, there is a strong demand for novel, biologically motivated treatment strategies with higher specificity for PDAC-relevant, tumor-driving targets. The genomic landscape of PDAC is dominated by a handful of signature genes which are affected by aberrations and mutations at high frequencies: KRAS, CDKN2A, TP53, and SMAD4 [49, 51]. All of these genes are still basically considered to be undruggable, although agents targeting mutant TP53 have been developed, and attempts to pharmacologically manipulate RAS function are constantly increasing [137, 138]. So far, substances targeting downstream effectors of these major PDAC drivers or other regulators which are also frequently altered, including BRAF, ERK, PI3K/AKT, and mTOR, are in the focus of investigation.
The mitogen-activated protein kinase (MAPK) signaling cascade offers promising perspectives in this regard, because PDAC cells are known to depend on MAPK signaling, both in terms of progression and metastasis formation [139, 140]. The most apical possibility to interfere with MAPK signaling is targeting the epidermal growth factor receptor (EGFR). However, a phase III trial evaluating the efficacy of anti-EGFR treatment with cetuximab in addition to gemcitabine-based chemotherapy showed no significant improvement in clinical outcome [141]. Recent data attributed this to a compensatory activation of Integrin β1 signaling [142]. Downstream of EGFR, KRAS constitutes a near-perfect target for PDAC treatment as revealed by preclinical RNA interference experiments [143]. However, clinical RNA interference is challenging, and no reliable KRAS inhibitors have been described so far [144]. Nevertheless, pharmacological disruption of the interaction between KRAS and phosphodiesterase PDEδ was shown to efficiently suppress PDAC progression in vitro and in vivo [145]. The only targeting approach for MAPK signaling that has entered the clinical routine thus far is the combination of gemcitabine and the EGFR-specific tyrosine kinase inhibitor erlotinib [146]. Although EGFR is considered to be its only target, erlotinib was reported to be similarly effective in tumors with wildtype or hyperactive mutants of KRAS, respectively [147]. This implies that either inhibition of tyrosine kinases other than EGFR or feedback regulatory mechanisms between hyperactivated KRAS and EGFR may be involved, respectively [148,149,150,151]. Sunitinib, a tyrosine kinase inhibitor that does not target EGFR, failed to show similar performance when combined with gemcitabine [152], and preclinical data support the notion that indeed inhibition of gemcitabine-induced MAPK signaling by erlotinib accounts for the observed clinical benefits [153]. Several other inhibitors of MAPK signaling, including inhibitors of EGFR, MEK, ERK, and corresponding protein phosphatases, have shown convincing performance in preclinical studies [154,155,156], but their potential for clinical implementation remains to be examined, as for instance in ACCEPT, a randomized phase II trial combining gemcitabine with the EGFR inhibitor afatinib (NCT01728818).
Single-drug treatments – most likely – will not be sufficient to improve the therapeutic outcome of PDAC [157]. Instead, dual or even multiple targeting strategies appear to be required in order to achieve significant advances. One example is the concomitant inhibition of MAPK and PI3K/AKT signaling. Preclinical data revealed that inhibition of MAPK signaling results in potent compensatory activation of PI3K/AKT signaling and vice versa, each being of importance for PDAC progression [158, 159]. Indeed, concomitant inhibition of MAPK and PI3K/AKT signaling did interfere with tumor progression to significantly greater extent than the single-drug treatments in preclinical PDAC models [158, 160]. However, other studies reported only modest effects of combined MAPK and PI3K/AKT inhibition [161,162,163], and clinical trialing of this combination failed [164]. One potential explanation could be that inhibitors of different target specificities were employed. A more detailed characterization of the target spectrum of these inhibitors would clarify this and could also help to find new targets for mechanism-based therapies. In this regard, upstream and/or transcriptional regulators of PI3K expression, such as transducin beta-like 1 (TBL1), may also be of interest as studies in genetic mouse models have identified them as crucial checkpoints in PDAC development and progression [165]. Nevertheless, if this mechanism can be exploited therapeutically remains unclear [166].
The mammalian target of rapamycin (mTOR) pathway is best known for its functions in cell survival, proliferation, motility, and evasion of apoptosis [167]. In several preclinical studies, mTOR inhibitors revealed promising results [168,169,170,171], but it was also reported that inhibition of mTOR stimulates feedback activation mechanisms involving MEK/ERK or AKT signaling, respectively, further emphasizing the need for combinatorial treatment regimens [172,173,174,175,176]. Not surprisingly, multi-pathway inhibition regimens are commonly associated with higher levels of toxicity [177]. This toxicity often interferes with clinical implementation. Nevertheless, clinical trials evaluating mTOR inhibition as monotherapy in PDAC altogether failed [178,179,180], and combined modality approaches of mTOR inhibition in conjunction with capecitabine revealed only limited improvements as compared to capecitabine alone [181]. These findings raise the question whether mTOR inhibitors, despite their successful clinical implementation for the treatment of neuroendocrine pancreatic tumors, may at all represent a therapeutic alternative for the treatment of PDAC [182], or whether such approaches have been inadequately tested in the clinic.
PDAC is commonly considered a hypovascularized tumor [183], but relevant expression of vascular endothelial growth factor A (VEGF-A) has been observed [184]. Therefore, the VEGF-A-specific antibody bevacizumab was tested in combination with gemcitabine in a randomized phase III trial with locally advanced PDAC but failed to show improved outcome [185]. A possible explanation could be the expression of other VEGF isoforms. However, complementary phase III trials which evaluated the VEGF receptor tyrosine kinase inhibitor axitinib in combination with gemcitabine, or the combination of bevacizumab, gemcitabine, and erlotinib, respectively, also failed [186, 187]. In summary, these results render therapeutic targeting of angiogenesis a questionable approach for the treatment of PDAC [188].
A subset of PDAC tumors (approximately 15% of all cases) is characterized by mutations in genes that are related to the DNA damage response [54]. Amongst those, PDAC tumors carrying mutations in BRCA1/2 genes are of highest interest as they are supposed to be defective in homologous recombination DNA damage repair [189]. Accordingly, patients with BRCA1/2-mutated tumors were reported to benefit significantly more from platinum-based chemotherapy than patients with BRCA1/2 wildtype tumors [190, 191]. For BRCA1/2-deficient tumors, the inhibition of Poly-(ADP-ribose)-polymerase (PARP) may be promising, since this enzyme shares an axis of synthetic lethality with BRCA1/2 [192]. Initial trials examining the therapeutic potential of PARP inhibitors in patients with BRCA1/2-deficient PDAC reported promising results [193,194,195,196]. Currently, the randomized phase III POLO trial is evaluating PARP inhibition in patients who received first-line platinum-based chemotherapy, and results are awaited in 2019 (NCT02184195). Beyond BRCA1/2, mutations in other genes of the DNA damage response, including ATM, may select for PARP inhibitor sensitivity [197].
In addition to the described genetic alterations, PDAC tumors display relevant changes in epigenetic modifications, including DNA methylation, histone post-translational modification, nucleosome remodeling, and regulation by non-coding RNAs [56]. In contrast to genetic alterations, epigenetic modifications are in principle reversible, and it is plausible to assume that pharmacological interference with epigenetic mechanisms underlying PDAC pathology and progression could open new therapeutic perspectives [198]. Preclinical results of epigenetic therapies have so far been promising, PDAC cell plasticity could be reduced, and resistance against standard chemotherapy was attenuated. However, in mono-agent settings, epigenetic therapeutics did not provide any measurable benefits, demanding for combined modality settings, e.g. in conjunction with chemotherapy or in form of multi-agent combinations, such as combined inhibition of bromodomain and extra-terminal motif (BET) proteins and histone deacetylases (HDACs) [199]. Currently, various phase I/II trials are ongoing which will determine the clinical perspectives of such approaches. Despite all efforts, individualized, mechanism-based treatment strategies for PDAC are still far from being clinical standard [200].
Therapeutic targeting of hypoxia and metastasis formation appears to be very attractive in the PDAC context, since hypoxia is a principal determinant of therapy resistance and metastasis formation, and metastases are the major cause of death [20, 74]. Regardless of all preclinical efforts [201], however, no therapeutic strategy could so far be established. Sort of alternatively, efforts to (re-)activate the immune system in order to detect and combat macro- and micro-metastases have been undertaken and will be discussed in the following.
Immunotherapy
Immunotherapy implementing immune checkpoint inhibitors has revolutionized cancer treatment in the last years [202]. Therapeutic antibodies targeting cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) or the axis of programmed cell death protein 1 (PD-1) and its corresponding ligand PD-L1 have shown compelling results in several different cancer types, including metastasized melanoma and lung cancer [36, 203]. Hence, immune checkpoint inhibition was also tested in PDAC [35, 39], but compared to melanoma and lung cancer, considerably smaller numbers of patients (approximately 2%) exhibited clinical benefits [40, 204]. Consistently, the responding tumors showed high levels of microsatellite instability, providing a mechanistic explanation as well as a potential future stratification marker, since microsatellite instability is known to increase the number of tumor-associated neo-antigens [205].
A major determinant of the immunotherapeutic success are tumor-specific T cells and their (re-)activation. Although their numbers have been described to be rather low in PDAC patients [90], recent data suggest that the tumor-reactive T-cell repertoire is similar to the one found in melanoma where T cell-based therapies meanwhile have relevant therapeutic impact [91]. Further studies showed that neo-antigen quality rather than quantity, and strong intra-tumoral CD8+ T cell infiltration are associated with prolonged survival, indicating that the stimulation of anti-tumor T cell responses can indeed be a promising strategy for the treatment of PDAC [60, 206, 207]. Along these lines, different vaccination strategies employing various kinds of antigens have already been tested [208,209,210]. The Algenpantucel-L vaccine consisting of irradiated, allogeneic pancreatic tumor cells stably expressing alpha-1,3-galactosyltransferase 2 (A3GALT2), a glycosylating enzyme that mainly targets lipids and extracellular proteins, turned out to be the most promising candidate for a PDAC-targeting vaccine [209]. However, this vaccine failed to improve treatment efficacy when being tested in a randomized phase III trial combined with the standard of care [211]. Other antigens that were examined include peptides derived from human telomerase 1 (TERT1) and GVAX, a vaccine comprised of autologous or allogeneic tumor cells expressing the dendritic cell-stimulating cytokine GM-CSF [212, 213]. Unfortunately, none of these vaccines achieved convincing clinical results. In principle, common PDAC driver mutations, such as KRASG12D, can harbor tumor-specific, T cell epitopes [214]. An ongoing phase II trial first predicts such neo-antigens using exome-sequencing of tumor biopsies, followed by production of personalized dendritic cell vaccines loaded with the respective epitopes (NCT03300843) [215]. Whether this strategy turns out to be successful needs to be awaited. Overall, several vaccination approaches could successfully elicit measurable anti-tumor T cell responses, yet so far none of these strategies resulted in clear clinical benefits [216].
Antigen-independent immunostimulatory therapies aim at the activation of antigen-presenting cells. Diverse receptor-ligand-axes have been explored in this regard. As such, treatment with agonistic anti-CD40 antibodies is well known to activate antigen-presenting cells and to polarize macrophages towards the pro-inflammatory M1-like state [217, 218]. However, clinical evaluation of this strategy in PDAC patients disclosed only short-term responses, and no long-term anti-tumor immunity was observed [219]. Nevertheless, CD40 stimulation in combination with chemotherapy and immune checkpoint blockade is currently under clinical investigation in a phase I/II trial (NCT03214250). Complementary approaches to achieve activation of antigen-presenting cells involve ligand-dependent stimulation of pattern recognition receptors (PRRs) [220]. Indeed, agonists of toll-like receptors (TLRs), RIG-I-like helicases (RLHs), and the stimulator of interferon genes (STING) revealed encouraging results in preclinical PDAC models [221,222,223], but their clinical potential remains to be elucidated.
Bypassing the in situ steps of T cell priming by antigen-presenting cells, adoptive transfer of T cells carrying chimeric antigen receptors (CARs) has proven powerful clinical performance in B-cell malignancies [224]. CAR T cells recognize specific cancer cell surface antigens through a single-chain variable fragment (scFv) whose ligation stimulates T cell activation via the intracellular domains of the CAR construct, resulting in efficient T cell-mediated killing of the target cell [225]. PDAC exhibits several tumor-specific antigens, such as carcinoembryonic antigen (CEA), mesothelin (MSLN), and mucin 1 (MUC1), which are promising determinants for CAR T cell therapy [226, 227]. However, for solid cancer entities, intra-tumoral recruitment and trafficking of CAR T cells as well as the commonly observed immunosuppressive tumor microenvironment appear to be major challenges. Intelligent combinations, thus, are needed in order to overcome these obstacles.
A cardinal feature of the immunosuppressive PDAC microenvironment is its massive stromal content and the excessive deposition of extracellular matrix, including hyaluronan [72]. Early phase clinical trials combining recombinant human hyaluronidase 20 (rHuPH20) with gemcitabine and nab-paclitaxel revealed promising results, particularly in those patients whose tumors were characterized by high levels of hyaluronan [228]. Reporting of the HALO-109-301 phase III trial (NCT02715804) is awaited in order to fully assess the clinical performance of this approach [229]. Inhibition of FAK1, a tyrosine kinase involved in the process of CAF generation, constitutes another approach to interfere with stromal function in PDAC, and pharmacological FAK1 inhibition eventually rendered preclinical PDAC model systems more susceptible to T cell immunotherapy and immune checkpoint inhibition [73]. Other studies showed that genetic ablation or inhibition of FAK1 also increases PDAC responsiveness to gemcitabine and nab-paclitaxel [230, 231]. In rather strong contrast, genetic deletion of stromal myofibroblasts in PDAC mouse models led to disease exacerbation and diminished animal survival due to enhanced regulatory T cell-mediated immunosuppression, clearly calling for caution when targeting components of PDAC stroma [78].
On a cellular level, massive infiltration by myeloid cells, such as MDSCs, and resulting exclusion of CD8+ T cells are major hallmarks of the immunosuppressive PDAC microenvironment [86, 232]. Several myeloid cell-targeting approaches have been investigated in recent years in order to overcome these mechanisms of immunosuppression [82, 233, 234]. Chemokine receptor 2 (CCR2), for instance, is known to contribute to the infiltration of pancreatic tumors by monocytes and macrophages, and this is associated with reduced patient survival and poor outcome [235]. Strikingly, the combination of CCR2 blockade and gemcitabine/nab-paclitaxel chemotherapy showed promising results in phase I trials [85, 236]. However, the follow-up phase Ib/II trial (NCT02732938) was discontinued due to strategic considerations, and instead phase I/II trials with combined modality approaches of CCR2 blockade in conjunction with pre-operative SBRT and immune checkpoint inhibition were recently initiated (NCT03778879, NCT03767582). Another target that regulates the function of macrophages and MDSCs in PDAC is M-CSF. Preclinical data suggest that M-CSF blockade can indeed reprogram macrophages and thus, synergize with immune checkpoint inhibition, but the clinical potential of this strategy remains to be examined [237].
In summary, (re-)activating anti-PDAC immunity in order to improve the overall clinical outcome appears clearly more challenging than extrapolated experiences from other cancer entities have suggested. Probably the most promising strategies would incorporate combinations of different immunotherapeutic approaches and/or combinations with other (classical) treatment modalities, such as chemotherapy and/or radiotherapy [238].
Combined modality treatment approaches encompassing radio(chemo)therapy
In order to improve the efficacy and the outcome of clinical PDAC treatment, it will be inevitable to develop novel treatment strategies which combine different therapeutic modalities aiming at achieving synergism [239]. The rationale for such approaches is to outcompete therapy resistance, but their development remains challenging as combined modality treatments are frequently associated with higher toxicity levels [240]. We already discussed several combined modality attempts involving different chemotherapeutics, either with each other or with novel, molecularly targeted inhibitors. At this point, we want to concentrate on combinatorial approaches involving radiotherapy (Fig. 5).

Combined modality perspectives for the treatment of PDAC.
Radiotherapy has rather infrequently been used for the treatment of PDAC. Nevertheless, there have been approaches to improve the efficacy of radiotherapy in PDAC. One obvious strategy is to combine radiotherapy with radiosensitizing agents which either can be classical chemotherapeutic drugs, such as gemcitabine or 5-FU, or – as has been reported more recently – molecularly designed inhibitors that target specific proteins and/or structures involved in PDAC radioresistance [28, 125]. The MAPK pathway is a very attractive target [140], and preclinical data derived from different PDAC mouse models showed that interference with MAPK signaling by cetuximab treatment can indeed increase the efficacy of radiochemotherapy [241, 242]. Encouraged by these observations, several clinical trials were initiated, yet with only modest results [243,244,245,246]. The major reason was the persistently high rate of distant failure due to metastasis formation, rather than poor local control [244, 246].
Pharmacological intervention with the PI3K/AKT and the mTOR pathway has also been examined with regards to its radiosensitizing potential. Several preclinical studies obtained basically positive results [247,248,249,250,251,252,253]. However, due to very unfavorable pharmaceutical properties of the employed substances, e.g. elevated toxicity levels and crossover inhibition, none of these approaches have entered the clinic thus far.
A very direct approach of radiosensitization is the employment of molecularly designed drugs which target components of the DNA damage response, specifically the upstream kinases ATM, ATR, CHK1/2, and DNA-PK [254,255,256]. Several of these inhibitors displayed convincing synergism with ionizing irradiation or DNA-damaging chemotherapy in preclinical PDAC model systems [257,258,259,260,261,262,263], but the transferability into the clinic remains to be investigated – particularly in view of local control versus distant failure. PARP is another example for a DNA damage response regulator that can be targeted by highly refined inhibitors, and preclinical data suggest that PARP inhibition indeed can radiosensitize PDAC cells [264]. However, since PARP is known to share synthetic lethality with BRCA1/2 [192], PARP inhibition may turn out to be only effective in BRCA1/2 deficient tumors [265]. This is a general lesson that has been learned in the era of molecularly targeted therapy: Molecularly designed therapy requires upfront molecular diagnostics and proper patient stratification, since otherwise promising agents are prone to fail if they are trialed in the wrong subgroups of patients.
Apart from its potential to induce tumor cell death, radiotherapy is known to recondition the tumor microenvironment and to stimulate systemic anti-tumor immune responses – a phenomenon summarized as abscopal effects of radiotherapy [266,267,268]. However, in the monotherapy setting, radiation is often not sufficient to break the immunosuppressive milieu of established tumors, and combinations with immunostimulating agents are required. As an example, radiotherapy plus GM-CSF, a potent stimulator of antigen-presenting cell maturation, produced objective abscopal responses in a subset of patients with different metastatic tumors [269], and a recent case report showed similar effects in a patient with metastatic pancreatic cancer [270]. In preclinical model systems, PDAC tumors have been reported to regress convincingly upon immunotherapeutic targeting of CCL2 or PD-L1 in combination with radiotherapy via a reduction of intra-tumoral immunosuppressive myeloid cells and enhanced recruitment of tumor-specific T cells [133, 271], and the clinical performance of this approach will be investigated (NCT03778879, NCT03767582). Similarly, radiotherapy has been described to reprogram tumor-infiltrating macrophages towards an M1-like phenotype and to favor intra-tumoral recruitment of adoptively transferred T cells in a mouse model of neuroendocrine pancreatic cancer [272]. These observations were confirmed by pilot data from patients with advanced PDAC stages undergoing neoadjuvant irradiation prior to tumor resection revealing 3- to 5-fold increases in intra-epithelial CD4+ and CD8+ T cells as compared to non-irradiated control patients [272, 273]. If these findings may also be transferred to combinations with PDAC-specific CAR T cells remains to be examined. On a mechanistic level, cytosolic DNA-sensing upon irradiation-induced DNA damage and type I interferon signaling appear to be involved in the immunostimulating effects of radiotherapy [274, 275]. Accordingly, artificial activation of cytosolic DNA sensors, such as STING, was shown to increase the efficacy of radiotherapy by enhancing CD8+ T cell responses – at least in preclinical PDAC models [276].
From clinical experiences with other cancer entities it is becoming increasingly evident that the combination of radiotherapy and immunotherapy requires very careful considerations regarding timing, dosing, and treatment sequence in order to achieve the best outcome [266]. This may be of particular interest for PDAC with its highly challenging immunosuppressive microenvironment. In brief, higher single doses of radiotherapy, e.g. SBRT or ablative protocols, applied in neoadjuvant settings appear to be beneficial, and immunotherapy needs to be started before or with the first irradiation fraction, respectively [266]. However, the optimal treatment regimen and the best combination of agents for PDAC remain unclear as well as the impact of additional chemotherapy and other factors, such as type II diabetes and/or obesity. A pilot study addressing some of these combinatorial issues added radiotherapy to CD40-dependent immunostimulation plus anti-CTLA-4/anti-PD-1-mediated immune checkpoint blockade in genetically engineered PDAC mouse models and utilized machine learning algorithms to extract signature patterns for each therapeutic component [277]. Along these lines, more in depth-analyses are needed in order to fully exploit the synergism between radiotherapy and immunotherapy. Nevertheless, several clinical phase I/II trials combining radiotherapy with different immunotherapeutic approaches have been initiated for advanced PDAC, and first results are awaited [278] (NCT02648282, NCT03161379, NCT03767582, NCT03563248).
Conclusions
PDAC represents a cancer entity of extraordinarily high malignancy, particularly poor prognosis, and constantly increasing patient numbers. Its aggressive biology and the fact that most patients present in advanced or disseminated stages of disease render the development of novel PDAC treatment strategies one of the superordinate challenges in current oncological research. Results of the last 20 years have led to the establishment of a detailed multi-step model of PDAC development and progression. Although this has unquestionably reformed our understanding of PDAC as a disease, none of these findings could be successfully translated into a therapeutic breakthrough so far. It is becoming increasingly evident that the clinical performance of single-agent therapies lags behind the original expectations, and instead intelligent combinations appear to be required. In this regard, radiotherapeutic protocols, and particularly modern radiation techniques with high conformality and steep dose gradients, represent attractive partners both for biologically motivated as well as for immunotherapeutic strategies. Importantly, however, this will require in-depth optimization of timing, dosing, and treatment sequences, as well as careful upfront patient stratification. Otherwise per se promising combinations run the risk of failing prematurely.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- 5-FU:
-
5-Fluorouracil
- A3GALT2 :
-
Alpha-1,3-galactosyltransferase 2
- ABCG2 :
-
ATP-binding cassette sub-family G member 2
- ADM:
-
Acinar-to-ductal metaplasia
- AKT :
-
RAC-beta serine/threonine-protein kinase
- ALDH1 :
-
Aldehyde dehydrogenase 1
- ATM :
-
Ataxia telangiectasia mutated protein serine/threonine kinase
- ATR :
-
ATM- and Rad3-related kinase
- BET :
-
Bromodomain and extra-terminal motif
- BNIP3 :
-
BCL2/adenovirus E1B 19 kDa protein-interacting protein 3
- BRAF :
-
v-Raf murine sarcoma viral oncogene homolog B
- BRCA1/2 :
-
Breast cancer early onset 1/2
- CAF:
-
Cancer-associated fibroblast
- CAR T cell:
-
Chimeric antigen receptor T cell
- CCL-2 :
-
CC-chemokine ligand 2
- CD:
-
Cluster of differentiation
- CDKN2A :
-
Cyclin-dependent kinase inhibitor 2A
- CEA :
-
Carcinoembryonic antigen
- CHK1/2 :
-
Checkpoint kinase 1/2
- c-Met :
-
Hepatocyte growth factor receptor
- CTLA-4 :
-
Cytotoxic T lymphocyte-associated protein 4
- CXCL-1 :
-
CXC-chemokine ligand 1
- CXCR2 :
-
CXC-chemokine receptor 2
- dFdCTP:
-
2′,2′-difluorodeoxycytidine-5′-triphosphate
- DNA-PK :
-
DNA-dependent protein kinase
- DPD :
-
Dihydropyrimidine dehydrogenase
- EGFR :
-
Epidermal growth factor receptor
- EMT:
-
Epithelial-to-mesenchymal transition
- ERK :
-
Extracellular signal-regulated kinase
- FAK1 :
-
Focal adhesion kinase 1
- FOLFIRINOX:
-
Poly-chemotherapeutic regimen composed of folinic acid, 5-FU, irinotecan, and oxaliplatin
- FOXA1 :
-
Forkhead box protein A1
- GM-CSF :
-
Granulocyte macrophage stimulating factor
- HDAC :
-
Histone deacetylases
- hENT1 :
-
Human equilibrative nucleoside transporter 1
- HNF1A :
-
Hepatocyte nuclear factor 1A
- IGRT:
-
Image-guided radiotherapy
- IL :
-
Interleukin
- ILK :
-
Integrin-linked kinase
- KRAS :
-
Proto-oncogene from Kirsten rat sarcoma virus
- KRT81 :
-
Cytokeratin-81
- MAPK :
-
Mitogen-activated protein kinase
- M-CSF :
-
Macrophage colony stimulating factor
- MDSC:
-
Myeloid-derived suppressor cell
- MEK :
-
Mitogen-activated protein kinase kinase
- MET:
-
Mesenchymal-to-epithelial transition
- MIF :
-
Macrophage migration inhibitory factor
- MSLN :
-
Mesothelin
- mTOR :
-
Mammalian target of rapamycin
- MUC1 :
-
Mucin I
- NCCN:
-
National Comprehensive Cancer Network
- OS:
-
Overall survival
- PALB2 :
-
Partner and localizer of BRCA2
- PAnIN:
-
Pancreatic intra-epithelial neoplasias
- PARP1/2 :
-
Poly-(ADP-ribose)-polymerase 1/2
- PD-1 :
-
Programmed cell death 1
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PDEδ :
-
Photoreceptor cGMP phosphodiesterase δ subunit
- PD-L1 :
-
Programmed cell death ligand 1
- PI3K :
-
Phosphoinositide 3-kinase
- PRR :
-
Pattern recognition receptor
- PRXX1a/b :
-
Paired mesoderm homeobox protein 1a/b
- rHuPH20:
-
Recombinant human hyaluronidase 20
- RIG-I :
-
Retinoic acid inducible gene I
- RLH :
-
RIG-I-like helicases
- RRM1/2 :
-
Ribonucleotide reductase catalytic subunits M1/2
- SBRT:
-
Stereotactic body radiotherapy
- scFv:
-
single-chain variable fragment
- SMAD4 :
-
Mothers against decapentaplegic homologue 4
- SNAI1/2 :
-
Snail family zinc finger protein 1/2
- STING :
-
Stimulator of interferon genes
- TBL1 :
-
Transducin beta-like 1
- TERT1 :
-
Telomerase reverse transcriptase 1
- TGF-β :
-
Transforming growth factor β
- TIMP-1 :
-
Tissue inhibitor of metalloproteinases 1
- TLR :
-
Toll-like receptor
- TP53 :
-
Tumor protein 53
- TS :
-
Thymidylate synthase
- TWIST1/2 :
-
Twist-related proteins 1/2
- VEGF-A :
-
Vascular endothelial growth factor A
- ZEB1/2 :
-
Zinc finger E-box-binding homeobox 1/2
References
Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, Neale RE, Tempero M, Tuveson DA, Hruban RH, et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30.
Quante AS, Ming C, Rottmann M, Engel J, Boeck S, Heinemann V, Westphalen CB, Strauch K. Projections of cancer incidence and cancer-related deaths in Germany by 2020 and 2030. Cancer Med. 2016;5(9):2649–56.
Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–21.
Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348(17):1625–38.
Font-Burgada J, Sun B, Karin M. Obesity and Cancer: The Oil that Feeds the Flame. Cell Metab. 2016;23(1):48–62.
Rahn S, Zimmermann V, Viol F, Knaack H, Stemmer K, Peters L, Lenk L, Ungefroren H, Saur D, Schafer H, et al. Diabetes as risk factor for pancreatic cancer: Hyperglycemia promotes epithelial-mesenchymal-transition and stem cell properties in pancreatic ductal epithelial cells. Cancer Lett. 2018;415:129–50.
Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91(14):1194–210.
Blot WJ, McLaughlin JK, Winn DM, Austin DF, Greenberg RS, Preston-Martin S, Bernstein L, Schoenberg JB, Stemhagen A, Fraumeni JF Jr. Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res. 1988;48(11):3282–7.
Blot WJ. Alcohol and cancer. Cancer Res. 1992;52(7 Suppl):2119s–23s.
Gapstur SM, Jacobs EJ, Deka A, McCullough ML, Patel AV, Thun MJ. Association of alcohol intake with pancreatic cancer mortality in never smokers. Arch Intern Med. 2011;171(5):444–51.
Pelucchi C, Galeone C, Polesel J, Manzari M, Zucchetto A, Talamini R, Franceschi S, Negri E, La Vecchia C. Smoking and body mass index and survival in pancreatic cancer patients. Pancreas. 2014;43(1):47–52.
Olson SH, Chou JF, Ludwig E, O'Reilly E, Allen PJ, Jarnagin WR, Bayuga S, Simon J, Gonen M, Reisacher WR, et al. Allergies, obesity, other risk factors and survival from pancreatic cancer. Int J Cancer. 2010;127(10):2412–9.
Delitto D, Zhang D, Han S, Black BS, Knowlton AE, Vlada AC, Sarosi GA, Behrns KE, Thomas RM, Lu X, et al. Nicotine Reduces Survival via Augmentation of Paracrine HGF-MET Signaling in the Pancreatic Cancer Microenvironment. Clin Cancer Res. 2016;22(7):1787–99.
Zhang S, Wang C, Huang H, Jiang Q, Zhao D, Tian Y, Ma J, Yuan W, Sun Y, Che X, et al. Effects of alcohol drinking and smoking on pancreatic ductal adenocarcinoma mortality: A retrospective cohort study consisting of 1783 patients. Sci Rep. 2017;7(1):9572.
Petersen GM, Amundadottir L, Fuchs CS, Kraft P, Stolzenberg-Solomon RZ, Jacobs KB, Arslan AA, Bueno-de-Mesquita HB, Gallinger S, Gross M, et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat Genet. 2010;42(3):224–8.
Pihlak R, Valle JW, McNamara MG. Germline mutations in pancreatic cancer and potential new therapeutic options. Oncotarget. 2017;8(42):73240–57.
Hu C, Hart SN, Polley EC, Gnanaolivu R, Shimelis H, Lee KY, Lilyquist J, Na J, Moore R, Antwi SO, et al. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA. 2018;319(23):2401–9.
Gillen S, Schuster T. Meyer Zum Buschenfelde C, Friess H, Kleeff J: Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010;7(4):e1000267.
Werner J, Combs SE, Springfeld C, Hartwig W, Hackert T, Buchler MW. Advanced-stage pancreatic cancer: therapy options. Nat Rev Clin Oncol. 2013;10(6):323–33.
Manji GA, Olive KP, Saenger YM, Oberstein P. Current and Emerging Therapies in Metastatic Pancreatic Cancer. Clin Cancer Res. 2017;23(7):1670–8.
Teague A, Lim KH, Wang-Gillam A. Advanced pancreatic adenocarcinoma: a review of current treatment strategies and developing therapies. Ther Adv Med Oncol. 2015;7(2):68–84.
Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardiere C, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–25.
Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–703.
Gourgou-Bourgade S, Bascoul-Mollevi C, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Boige V, et al. Impact of FOLFIRINOX compared with gemcitabine on quality of life in patients with metastatic pancreatic cancer: results from the PRODIGE 4/ACCORD 11 randomized trial. J Clin Oncol. 2013;31(1):23–9.
Hall WA, Goodman KA. Radiation therapy for pancreatic adenocarcinoma, a treatment option that must be considered in the management of a devastating malignancy. Radiat Oncol. 2019;14(1):114.
Roeder F. Neoadjuvant radiotherapeutic strategies in pancreatic cancer. World J Gastrointest Oncol. 2016;8(2):186–97.
Brunner TB, Scott-Brown M. The role of radiotherapy in multimodal treatment of pancreatic carcinoma. Radiat Oncol. 2010;5:64.
Mukherjee S, Hurt CN, Bridgewater J, Falk S, Cummins S, Wasan H, Crosby T, Jephcott C, Roy R, Radhakrishna G, et al. Gemcitabine-based or capecitabine-based chemoradiotherapy for locally advanced pancreatic cancer (SCALOP): a multicentre, randomised, phase 2 trial. Lancet Oncol. 2013;14(4):317–26.
Murphy JE, Wo JY, Ryan DP, Jiang W, Yeap BY, Drapek LC, Blaszkowsky LS, Kwak EL, Allen JN, Clark JW, et al. Total Neoadjuvant Therapy With FOLFIRINOX Followed by Individualized Chemoradiotherapy for Borderline Resectable Pancreatic Adenocarcinoma: A Phase 2 Clinical Trial. JAMA Oncol. 2018;4(7):963–9.
Reyngold M, Parikh P, Crane CH. Ablative radiation therapy for locally advanced pancreatic cancer: techniques and results. Radiat Oncol. 2019;14(1):95.
Boldrini L, Cusumano D, Cellini F, Azario L, Mattiucci GC, Valentini V. Online adaptive magnetic resonance guided radiotherapy for pancreatic cancer: state of the art, pearls and pitfalls. Radiat Oncol. 2019;14(1):71.
Nanda RH, El-Rayes B, Maithel SK, Landry J. Neoadjuvant modified FOLFIRINOX and chemoradiation therapy for locally advanced pancreatic cancer improves resectability. J Surg Oncol. 2015;111(8):1028–34.
Buwenge M, Macchia G, Arcelli A, Frakulli R, Fuccio L, Guerri S, Grassi E, Cammelli S, Cellini F, Morganti AG. Stereotactic radiotherapy of pancreatic cancer: a systematic review on pain relief. J Pain Res. 2018;11:2169–78.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–65.
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373(17):1627–39.
Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020–30.
Larkin J, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373(13):1270–1.
Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33(8):828–33.
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–13.
Lemery S, Keegan P, Pazdur R. First FDA Approval Agnostic of Cancer Site - When a Biomarker Defines the Indication. N Engl J Med. 2017;377(15):1409–12.
Dougan SK. The Pancreatic Cancer Microenvironment. Cancer J. 2017;23(6):321–5.
Puri S, Folias AE, Hebrok M. Plasticity and dedifferentiation within the pancreas: development, homeostasis, and disease. Cell Stem Cell. 2015;16(1):18–31.
Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JP, Pan FC, Akiyama H, Wright CV, Jensen K, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22(6):737–50.
Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, Vasile E, DePinho RA, Jacks T. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009;16(5):379–89.
Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, Hruban RH, Maitra A, Kinzler K, Vogelstein B, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142(4):730–3 e739.
Morris JP, Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer. 2010;10(10):683–95.
Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744.
Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501.
Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531(7592):47–52.
Cancer Genome Atlas Research Network. Electronic address aadhe, Cancer Genome Atlas Research N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017;32(2):185–203 e113.
Mueller S, Engleitner T, Maresch R, Zukowska M, Lange S, Kaltenbacher T, Konukiewitz B, Ollinger R, Zwiebel M, Strong A, et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature. 2018;554(7690):62–8.
Altomare DA, Tanno S, De Rienzo A, Klein-Szanto AJ, Tanno S, Skele KL, Hoffman JP, Testa JR. Frequent activation of AKT2 kinase in human pancreatic carcinomas. J Cell Biochem. 2002;87(4):470–6.
Ying H, Dey P, Yao W, Kimmelman AC, Draetta GF, Maitra A, DePinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016;30(4):355–85.
Hu ZI, Shia J, Stadler ZK, Varghese AM, Capanu M, Salo-Mullen E, Lowery MA, Diaz LA Jr, Mandelker D, Yu KH, et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin Cancer Res. 2018;24(6):1326–36.
Iguchi E, Safgren SL, Marks DL, Olson RL, Fernandez-Zapico ME. Pancreatic Cancer, A Mis-interpreter of the Epigenetic Language. Yale J Biol Med. 2016;89(4):575–90.
Roe JS, Hwang CI, Somerville TDD, Milazzo JP, Lee EJ, Da Silva B, Maiorino L, Tiriac H, Young CM, Miyabayashi K, et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell. 2017;170(5):875–88 e820.
McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, Word AE, Carrer A, Salz TH, Natsume S, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet. 2017;49(3):367–76.
Lomberk G, Blum Y, Nicolle R, Nair A, Gaonkar KS, Marisa L, Mathison A, Sun Z, Yan H, Elarouci N, et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat Commun. 2018;9(1):1978.
Connor AA, Denroche RE, Jang GH, Timms L, Kalimuthu SN, Selander I, McPherson T, Wilson GW, Chan-Seng-Yue MA, Borozan I, et al. Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017;3(6):774–83.
Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015;47(10):1168–78.
Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17(4):500–3.
Veenstra VL, Garcia-Garijo A, van Laarhoven HW, Bijlsma MF. Extracellular Influences: Molecular Subclasses and the Microenvironment in Pancreatic Cancer. Cancers (Basel). 2018;10(2). https://doi.org/10.3390/cancers10020034.
Muckenhuber A, Berger AK, Schlitter AM, Steiger K, Konukiewitz B, Trumpp A, Eils R, Werner J, Friess H, Esposito I, et al. Pancreatic Ductal Adenocarcinoma Subtyping Using the Biomarkers Hepatocyte Nuclear Factor-1A and Cytokeratin-81 Correlates with Outcome and Treatment Response. Clin Cancer Res. 2018;24(2):351–9.
Aung KL, Fischer SE, Denroche RE, Jang GH, Dodd A, Creighton S, Southwood B, Liang SB, Chadwick D, Zhang A, et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin Cancer Res. 2018;24(6):1344–54.
Nielsen MF, Mortensen MB, Detlefsen S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J Gastroenterol. 2016;22(9):2678–700.
Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, Tuveson DA. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019;9(2):282–301.
Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA, Lee EJ, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214(3):579–96.
Melstrom LG, Salazar MD, Diamond DJ. The pancreatic cancer microenvironment: A true double agent. J Surg Oncol. 2017;116(1):7–15.
Masamune A, Watanabe T, Kikuta K, Shimosegawa T. Roles of pancreatic stellate cells in pancreatic inflammation and fibrosis. Clin Gastroenterol Hepatol. 2009;7(11 Suppl):S48–54.
Apte MV, Haber PS, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut. 1999;44(4):534–41.
Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62(1):112–20.
Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22(8):851–60.
Erkan M, Kurtoglu M, Kleeff J. The role of hypoxia in pancreatic cancer: a potential therapeutic target? Expert Rev Gastroenterol Hepatol. 2016;10(3):301–16.
Heinemann V, Reni M, Ychou M, Richel DJ, Macarulla T, Ducreux M. Tumour-stroma interactions in pancreatic ductal adenocarcinoma: rationale and current evidence for new therapeutic strategies. Cancer Treat Rev. 2014;40(1):118–28.
Li N, Li Y, Li Z, Huang C, Yang Y, Lang M, Cao J, Jiang W, Xu Y, Dong J, et al. Hypoxia Inducible Factor 1 (HIF-1) Recruits Macrophage to Activate Pancreatic Stellate Cells in Pancreatic Ductal Adenocarcinoma. Int J Mol Sci. 2016;17(6). https://doi.org/10.3390/ijms17060799.
Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, Marshall JF, Chin-Aleong J, Chelala C, Gribben JG, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145(5):1121–32.
Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–34.
Daniel SK, Sullivan KM, Labadie KP, Pillarisetty VG. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma. Clin Transl Med. 2019;8(1):10.
Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67(19):9518–27.
Mahajan UM, Langhoff E, Goni E, Costello E, Greenhalf W, Halloran C, Ormanns S, Kruger S, Boeck S, Ribback S, et al. Immune Cell and Stromal Signature Associated With Progression-Free Survival of Patients With Resected Pancreatic Ductal Adenocarcinoma. Gastroenterology. 2018;155(5):1625–39 e1622.
Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128–41.
Hu H, Hang JJ, Han T, Zhuo M, Jiao F, Wang LW. The M2 phenotype of tumor-associated macrophages in the stroma confers a poor prognosis in pancreatic cancer. Tumour Biol. 2016;37(7):8657–64.
Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150.
Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17(5):651–62.
Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, Richman LP, Lin JH, Sun YH, Rech AJ, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity. 2018;49(1):178–193.e177.
Daley D, Zambirinis CP, Seifert L, Akkad N, Mohan N, Werba G, Barilla R, Torres-Hernandez A, Hundeyin M, VRK M, et al. gammadelta T Cells Support Pancreatic Oncogenesis by Restraining alphabeta T Cell Activation. Cell. 2016;166(6):1485–99 e1415.
Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, Gorjestani S, Liudahl SM, Truitt M, Olson P, et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2016;6(3):270–85.
Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, Bar-Sagi D. IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer Discov. 2016;6(3):247–55.
Stromnes IM, Hulbert A, Pierce RH, Greenberg PD, Hingorani SR. T-cell Localization, Activation, and Clonal Expansion in Human Pancreatic Ductal Adenocarcinoma. Cancer Immunol Res. 2017;5(11):978–91.
Poschke I, Faryna M, Bergmann F, Flossdorf M, Lauenstein C, Hermes J, Hinz U, Hank T, Ehrenberg R, Volkmar M, et al. Identification of a tumor-reactive T-cell repertoire in the immune infiltrate of patients with resectable pancreatic ductal adenocarcinoma. Oncoimmunology. 2016;5(12):e1240859.
Johnson BA 3rd, Yarchoan M, Lee V, Laheru DA, Jaffee EM. Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin Cancer Res. 2017;23(7):1656–69.
Yako YY, Kruger D, Smith M, Brand M. Cytokines as Biomarkers of Pancreatic Ductal Adenocarcinoma: A Systematic Review. PLoS One. 2016;11(5):e0154016.
Roshani R, McCarthy F, Hagemann T. Inflammatory cytokines in human pancreatic cancer. Cancer Lett. 2014;345(2):157–63.
Poruk KE, Firpo MA, Adler DG, Mulvihill SJ. Screening for pancreatic cancer: why, how, and who? Ann Surg. 2013;257(1):17–26.
Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467(7319):1114–7.
Makohon-Moore AP, Zhang M, Reiter JG, Bozic I, Allen B, Kundu D, Chatterjee K, Wong F, Jiao Y, Kohutek ZA, et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat Genet. 2017;49(3):358–66.
Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467(7319):1109–13.
Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1-2):349–61.
Wang S, Huang S, Sun YL. Epithelial-Mesenchymal Transition in Pancreatic Cancer: A Review. Biomed Res Int. 2017;2017:2646148.
Takano S, Reichert M, Bakir B, Das KK, Nishida T, Miyazaki M, Heeg S, Collins MA, Marchand B, Hicks PD, et al. Prrx1 isoform switching regulates pancreatic cancer invasion and metastatic colonization. Genes Dev. 2016;30(2):233–47.
Krebs AM, Mitschke J, Lasierra Losada M, Schmalhofer O, Boerries M, Busch H, Boettcher M, Mougiakakos D, Reichardt W, Bronsert P, et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol. 2017;19(5):518–29.
Giovannetti E, van der Borden CL, Frampton AE, Ali A, Firuzi O, Peters GJ. Never let it go: Stopping key mechanisms underlying metastasis to fight pancreatic cancer. Semin Cancer Biol. 2017;44:43–59.
Mees ST, Mardin WA, Wendel C, Baeumer N, Willscher E, Senninger N, Schleicher C, Colombo-Benkmann M, Haier J. EP300--a miRNA-regulated metastasis suppressor gene in ductal adenocarcinomas of the pancreas. Int J Cancer. 2010;126(1):114–24.
Aiello NM, Maddipati R, Norgard RJ, Balli D, Li J, Yuan S, Yamazoe T, Black T, Sahmoud A, Furth EE, et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev Cell. 2018;45(6):681–95 e684.
Yao D, Dai C, Peng S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol Cancer Res. 2011;9(12):1608–20.
Reichert M, Bakir B, Moreira L, Pitarresi JR, Feldmann K, Simon L, Suzuki K, Maddipati R, Rhim AD, Schlitter AM, et al. Regulation of Epithelial Plasticity Determines Metastatic Organotropism in Pancreatic Cancer. Dev Cell. 2018;45(6):696–711.e698.
Stratford JK, Bentrem DJ, Anderson JM, Fan C, Volmar KA, Marron JS, Routh ED, Caskey LS, Samuel JC, Der CJ, et al. A six-gene signature predicts survival of patients with localized pancreatic ductal adenocarcinoma. PLoS Med. 2010;7(7):e1000307.
Chaika NV, Yu F, Purohit V, Mehla K, Lazenby AJ, DiMaio D, Anderson JM, Yeh JJ, Johnson KR, Hollingsworth MA, et al. Differential expression of metabolic genes in tumor and stromal components of primary and metastatic loci in pancreatic adenocarcinoma. PLoS One. 2012;7(3):e32996.
Abel EV, Simeone DM. Biology and clinical applications of pancreatic cancer stem cells. Gastroenterology. 2013;144(6):1241–8.
Kruger A. Premetastatic niche formation in the liver: emerging mechanisms and mouse models. J Mol Med (Berl). 2015;93(11):1193–201.
Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, Becker A, Hoshino A, Mark MT, Molina H, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17(6):816–26.
Grunwald B, Harant V, Schaten S, Fruhschutz M, Spallek R, Hochst B, Stutzer K, Berchtold S, Erkan M, Prokopchuk O, et al. Pancreatic Premalignant Lesions Secrete Tissue Inhibitor of Metalloproteinases-1, Which Activates Hepatic Stellate Cells Via CD63 Signaling to Create a Premetastatic Niche in the Liver. Gastroenterology. 2016;151(5):1011–24 e1017.
Amrutkar M, Gladhaug IP. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers (Basel). 2017;9(11). https://doi.org/10.3390/cancers9110157.
Grasso C, Jansen G, Giovannetti E. Drug resistance in pancreatic cancer: Impact of altered energy metabolism. Crit Rev Oncol Hematol. 2017;114:139–52.
Morrison AH, Byrne KT, Vonderheide RH. Immunotherapy and Prevention of Pancreatic Cancer. Trends Cancer. 2018;4(6):418–28.
Nakano Y, Tanno S, Koizumi K, Nishikawa T, Nakamura K, Minoguchi M, Izawa T, Mizukami Y, Okumura T, Kohgo Y. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br J Cancer. 2007;96(3):457–63.
Nakahira S, Nakamori S, Tsujie M, Takahashi Y, Okami J, Yoshioka S, Yamasaki M, Marubashi S, Takemasa I, Miyamoto A, et al. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int J Cancer. 2007;120(6):1355–63.
Kurata N, Fujita H, Ohuchida K, Mizumoto K, Mahawithitwong P, Sakai H, Onimaru M, Manabe T, Ohtsuka T, Tanaka M. Predicting the chemosensitivity of pancreatic cancer cells by quantifying the expression levels of genes associated with the metabolism of gemcitabine and 5-fluorouracil. Int J Oncol. 2011;39(2):473–82.
Valsecchi ME, Holdbrook T, Leiby BE, Pequignot E, Littman SJ, Yeo CJ, Brody JR, Witkiewicz AK. Is there a role for the quantification of RRM1 and ERCC1 expression in pancreatic ductal adenocarcinoma? BMC Cancer. 2012;12:104.
Duxbury MS, Ito H, Benoit E, Waseem T, Ashley SW, Whang EE. RNA interference demonstrates a novel role for integrin-linked kinase as a determinant of pancreatic adenocarcinoma cell gemcitabine chemoresistance. Clin Cancer Res. 2005;11(9):3433–8.
Erkan M, Kleeff J, Esposito I, Giese T, Ketterer K, Buchler MW, Giese NA, Friess H. Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene. 2005;24(27):4421–32.
Hessmann E, Patzak MS, Klein L, Chen N, Kari V, Ramu I, Bapiro TE, Frese KK, Gopinathan A, Richards FM, et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut. 2018;67(3):497–507.
Caparello C, Meijer LL, Garajova I, Falcone A, Le Large TY, Funel N, Kazemier G, Peters GJ, Vasile E, Giovannetti E. FOLFIRINOX and translational studies: Towards personalized therapy in pancreatic cancer. World J Gastroenterol. 2016;22(31):6987–7005.
Huguet F, Mukherjee S, Javle M. Locally advanced pancreatic cancer: the role of definitive chemoradiotherapy. Clin Oncol (R Coll Radiol). 2014;26(9):560–8.
Quintiliani M. Modification of radiation sensitivity: the oxygen effect. Int J Radiat Oncol Biol Phys. 1979;5(7):1069–76.
Matsumoto S, Kishimoto S, Saito K, Takakusagi Y, Munasinghe JP, Devasahayam N, Hart CP, Gillies RJ, Mitchell JB, Krishna MC. Metabolic and Physiologic Imaging Biomarkers of the Tumor Microenvironment Predict Treatment Outcome with Radiation or a Hypoxia-Activated Prodrug in Mice. Cancer Res. 2018;78(14):3783–92.
Maacke H, Jost K, Opitz S, Miska S, Yuan Y, Hasselbach L, Luttges J, Kalthoff H, Sturzbecher HW. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene. 2000;19(23):2791–5.
Mathews LA, Cabarcas SM, Hurt EM, Zhang X, Jaffee EM, Farrar WL. Increased expression of DNA repair genes in invasive human pancreatic cancer cells. Pancreas. 2011;40(5):730–9.
Wang F, Xia X, Yang C, Shen J, Mai J, Kim HC, Kirui D, Kang Y, Fleming JB, Koay EJ, et al. SMAD4 Gene Mutation Renders Pancreatic Cancer Resistance to Radiotherapy through Promotion of Autophagy. Clin Cancer Res. 2018;24(13):3176–85.
Cordes N, Frick S, Brunner TB, Pilarsky C, Grutzmann R, Sipos B, Kloppel G, McKenna WG, Bernhard EJ. Human pancreatic tumor cells are sensitized to ionizing radiation by knockdown of caveolin-1. Oncogene. 2007;26(48):6851–62.
Hehlgans S, Eke I, Storch K, Haase M, Baretton GB, Cordes N. Caveolin-1 mediated radioresistance of 3D grown pancreatic cancer cells. Radiother Oncol. 2009;92(3):362–70.
Kalbasi A, Komar C, Tooker GM, Liu M, Lee JW, Gladney WL, Ben-Josef E, Beatty GL. Tumor-Derived CCL2 Mediates Resistance to Radiotherapy in Pancreatic Ductal Adenocarcinoma. Clin Cancer Res. 2017;23(1):137–48.
Murphy JD, Adusumilli S, Griffith KA, Ray ME, Zalupski MM, Lawrence TS, Ben-Josef E. Full-dose gemcitabine and concurrent radiotherapy for unresectable pancreatic cancer. Int J Radiat Oncol Biol Phys. 2007;68(3):801–8.
Loehrer PJ Sr, Feng Y, Cardenes H, Wagner L, Brell JM, Cella D, Flynn P, Ramanathan RK, Crane CH, Alberts SR, et al. Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: an Eastern Cooperative Oncology Group trial. J Clin Oncol. 2011;29(31):4105–12.
Chang DT, Schellenberg D, Shen J, Kim J, Goodman KA, Fisher GA, Ford JM, Desser T, Quon A, Koong AC. Stereotactic radiotherapy for unresectable adenocarcinoma of the pancreas. Cancer. 2009;115(3):665–72.
Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell. 2017;170(6):1062–78.
Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014;13(11):828–51.
Agbunag C, Bar-Sagi D. Oncogenic K-ras drives cell cycle progression and phenotypic conversion of primary pancreatic duct epithelial cells. Cancer Res. 2004;64(16):5659–63.
Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer. 2014;111(5):817–22.
Philip PA, Benedetti J, Corless CL, Wong R, O'Reilly EM, Flynn PJ, Rowland KM, Atkins JN, Mirtsching BC, Rivkin SE, et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J Clin Oncol. 2010;28(22):3605–10.
Kim YJ, Jung K, Baek DS, Hong SS, Kim YS. Co-targeting of EGF receptor and neuropilin-1 overcomes cetuximab resistance in pancreatic ductal adenocarcinoma with integrin beta1-driven Src-Akt bypass signaling. Oncogene. 2017;36(18):2543–52.
Fleming JB, Shen GL, Holloway SE, Davis M, Brekken RA. Molecular consequences of silencing mutant K-ras in pancreatic cancer cells: justification for K-ras-directed therapy. Mol Cancer Res. 2005;3(7):413–23.
Baines AT, Xu D, Der CJ. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem. 2011;3(14):1787–808.
Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens PI, et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497(7451):638–42.
Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960–6.
Boeck S, Jung A, Laubender RP, Neumann J, Egg R, Goritschan C, Ormanns S, Haas M, Modest DP, Kirchner T, et al. KRAS mutation status is not predictive for objective response to anti-EGFR treatment with erlotinib in patients with advanced pancreatic cancer. J Gastroenterol. 2013;48(4):544–8.
Conradt L, Godl K, Schaab C, Tebbe A, Eser S, Diersch S, Michalski CW, Kleeff J, Schnieke A, Schmid RM, et al. Disclosure of erlotinib as a multikinase inhibitor in pancreatic ductal adenocarcinoma. Neoplasia. 2011;13(11):1026–34.
Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE, Carpenter ES, Halbrook CJ, Hall JC, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012;22(3):304–17.
Schneeweis C, Wirth M, Saur D, Reichert M, Schneider G. Oncogenic KRAS and the EGFR loop in pancreatic carcinogenesis-A connection to licensing nodes. Small GTPases. 2018;9(6):457–64.
Diersch S, Wirth M, Schneeweis C, Jors S, Geisler F, Siveke JT, Rad R, Schmid RM, Saur D, Rustgi AK, et al. Kras(G12D) induces EGFR-MYC cross signaling in murine primary pancreatic ductal epithelial cells. Oncogene. 2016;35(29):3880–6.
Bergmann L, Maute L, Heil G, Russel J, Weidmann E, Koberle D, Fuxius S, Weigang-Kohler K, Aulitzky WE, Wormann B, et al. A prospective randomised phase-II trial with gemcitabine versus gemcitabine plus sunitinib in advanced pancreatic cancer: a study of the CESAR Central European Society for Anticancer Drug Research-EWIV. Eur J Cancer. 2015;51(1):27–36.
Miyabayashi K, Ijichi H, Mohri D, Tada M, Yamamoto K, Asaoka Y, Ikenoue T, Tateishi K, Nakai Y, Isayama H, et al. Erlotinib prolongs survival in pancreatic cancer by blocking gemcitabine-induced MAPK signals. Cancer Res. 2013;73(7):2221–34.
Vaseva AV, Blake DR, Gilbert TSK, Ng S, Hostetter G, Azam SH, Ozkan-Dagliyan I, Gautam P, Bryant KL, Pearce KH, et al. KRAS Suppression-Induced Degradation of MYC Is Antagonized by a MEK5-ERK5 Compensatory Mechanism. Cancer Cell. 2018;34(5):807–22 e807.
Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, Aziz M, Kassner M, Bryant KL, Pierobon M, et al. Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer Cell. 2016;29(1):75–89.
Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, Gorgulu K, Dantes Z, Wormann SM, Diakopoulos KN, et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med. 2018;24(7):954–60.
Zeitouni D, Pylayeva-Gupta Y, Der CJ, Bryant KL. KRAS Mutant Pancreatic Cancer: No Lone Path to an Effective Treatment. Cancers (Basel). 2016;8(4). https://doi.org/10.3390/cancers8040045.
Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM, Charles RP, Rabinovich BA, Hann B, Dankort D, et al. A central role for RAF-->MEK-->ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2012;2(8):685–93.
Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, Hieber M, Arbeiter A, Klein S, Kong B, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23(3):406–20.
Alagesan B, Contino G, Guimaraes AR, Corcoran RB, Deshpande V, Wojtkiewicz GR, Hezel AF, Wong KK, Loda M, Weissleder R, et al. Combined MEK and PI3K inhibition in a mouse model of pancreatic cancer. Clin Cancer Res. 2015;21(2):396–404.
Ischenko I, Petrenko O, Hayman MJ. A MEK/PI3K/HDAC inhibitor combination therapy for KRAS mutant pancreatic cancer cells. Oncotarget. 2015;6(18):15814–27.
Junttila MR, Devasthali V, Cheng JH, Castillo J, Metcalfe C, Clermont AC, Otter DD, Chan E, Bou-Reslan H, Cao T, et al. Modeling targeted inhibition of MEK and PI3 kinase in human pancreatic cancer. Mol Cancer Ther. 2015;14(1):40–7.
Ciuffreda L, Del Curatolo A, Falcone I, Conciatori F, Bazzichetto C, Cognetti F, Corbo V, Scarpa A, Milella M. Lack of growth inhibitory synergism with combined MAPK/PI3K inhibition in preclinical models of pancreatic cancer. Ann Oncol. 2017;28(11):2896–8.
Chung V, McDonough S, Philip PA, Cardin D, Wang-Gillam A, Hui L, Tejani MA, Seery TE, Dy IA, Al Baghdadi T, et al. Effect of Selumetinib and MK-2206 vs Oxaliplatin and Fluorouracil in Patients With Metastatic Pancreatic Cancer After Prior Therapy: SWOG S1115 Study Randomized Clinical Trial. JAMA Oncol. 2017;3(4):516–22.
Stoy C, Sundaram A, Rios Garcia M, Wang X, Seibert O, Zota A, Wendler S, Mannle D, Hinz U, Sticht C, et al. Transcriptional co-factor Transducin beta-like (TBL) 1 acts as a checkpoint in pancreatic cancer malignancy. EMBO Mol Med. 2015;7(8):1048–62.
Fiskus W, Sharma S, Saha S, Shah B, Devaraj SG, Sun B, Horrigan S, Leveque C, Zu Y, Iyer S, et al. Pre-clinical efficacy of combined therapy with novel beta-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia. 2015;29(6):1267–78.
Populo H, Lopes JM, Soares P. The mTOR signalling pathway in human cancer. Int J Mol Sci. 2012;13(2):1886–918.
Wei F, Zhang Y, Geng L, Zhang P, Wang G, Liu Y. mTOR inhibition induces EGFR feedback activation in association with its resistance to human pancreatic cancer. Int J Mol Sci. 2015;16(2):3267–82.
Utomo WK, Narayanan V, Biermann K, van Eijck CH, Bruno MJ, Peppelenbosch MP, Braat H. mTOR is a promising therapeutical target in a subpopulation of pancreatic adenocarcinoma. Cancer Lett. 2014;346(2):309–17.
Lou HZ, Weng XC, Pan HM, Pan Q, Sun P, Liu LL, Chen B. The novel mTORC1/2 dual inhibitor INK-128 suppresses survival and proliferation of primary and transformed human pancreatic cancer cells. Biochem Biophys Res Commun. 2014;450(2):973–8.
Morran DC, Wu J, Jamieson NB, Mrowinska A, Kalna G, Karim SA, Au AY, Scarlett CJ, Chang DK, Pajak MZ, et al. Targeting mTOR dependency in pancreatic cancer. Gut. 2014;63(9):1481–9.
Soares HP, Ni Y, Kisfalvi K, Sinnett-Smith J, Rozengurt E. Different patterns of Akt and ERK feedback activation in response to rapamycin, active-site mTOR inhibitors and metformin in pancreatic cancer cells. PLoS One. 2013;8(2):e57289.
Soares HP, Ming M, Mellon M, Young SH, Han L, Sinnet-Smith J, Rozengurt E. Dual PI3K/mTOR Inhibitors Induce Rapid Overactivation of the MEK/ERK Pathway in Human Pancreatic Cancer Cells through Suppression of mTORC2. Mol Cancer Ther. 2015;14(4):1014–23.
O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66(3):1500–8.
Hassan Z, Schneeweis C, Wirth M, Veltkamp C, Dantes Z, Feuerecker B, Ceyhan GO, Knauer SK, Weichert W, Schmid RM, et al. MTOR inhibitor-based combination therapies for pancreatic cancer. Br J Cancer. 2018;118(3):366–77.
Driscoll DR, Karim SA, Sano M, Gay DM, Jacob W, Yu J, Mizukami Y, Gopinathan A, Jodrell DI, Evans TR, et al. mTORC2 Signaling Drives the Development and Progression of Pancreatic Cancer. Cancer Res. 2016;76(23):6911–23.
Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, Gunn S, Smetzer L, Mays TA, Kaiser B, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18(8):2316–25.
Wolpin BM, Hezel AF, Abrams T, Blaszkowsky LS, Meyerhardt JA, Chan JA, Enzinger PC, Allen B, Clark JW, Ryan DP, et al. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J Clin Oncol. 2009;27(2):193–8.
Javle MM, Shroff RT, Xiong H, Varadhachary GA, Fogelman D, Reddy SA, Davis D, Zhang Y, Wolff RA, Abbruzzese JL. Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: results of two phase II studies. BMC Cancer. 2010;10:368.
Iriana S, Ahmed S, Gong J, Annamalai AA, Tuli R, Hendifar AE. Targeting mTOR in Pancreatic Ductal Adenocarcinoma. Front Oncol. 2016;6:99.
Kordes S, Klumpen HJ, Weterman MJ, Schellens JH, Richel DJ, Wilmink JW. Phase II study of capecitabine and the oral mTOR inhibitor everolimus in patients with advanced pancreatic cancer. Cancer Chemother Pharmacol. 2015;75(6):1135–41.
Chan J, Kulke M. Targeting the mTOR signaling pathway in neuroendocrine tumors. Curr Treat Options Oncol. 2014;15(3):365–79.
Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–61.
Itakura J, Ishiwata T, Friess H, Fujii H, Matsumoto Y, Buchler MW, Korc M. Enhanced expression of vascular endothelial growth factor in human pancreatic cancer correlates with local disease progression. Clin Cancer Res. 1997;3(8):1309–16.
Kindler HL, Niedzwiecki D, Hollis D, Sutherland S, Schrag D, Hurwitz H, Innocenti F, Mulcahy MF, O'Reilly E, Wozniak TF, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J Clin Oncol. 2010;28(22):3617–22.
Kindler HL, Ioka T, Richel DJ, Bennouna J, Letourneau R, Okusaka T, Funakoshi A, Furuse J, Park YS, Ohkawa S, et al. Axitinib plus gemcitabine versus placebo plus gemcitabine in patients with advanced pancreatic adenocarcinoma: a double-blind randomised phase 3 study. Lancet Oncol. 2011;12(3):256–62.
Van Cutsem E, Vervenne WL, Bennouna J, Humblet Y, Gill S, Van Laethem JL, Verslype C, Scheithauer W, Shang A, Cosaert J, et al. Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J Clin Oncol. 2009;27(13):2231–7.
Craven KE, Gore J, Korc M. Overview of pre-clinical and clinical studies targeting angiogenesis in pancreatic ductal adenocarcinoma. Cancer Lett. 2016;381(1):201–10.
Powell SN, Kachnic LA. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene. 2003;22(37):5784–91.
Golan T, Kanji ZS, Epelbaum R, Devaud N, Dagan E, Holter S, Aderka D, Paluch-Shimon S, Kaufman B, Gershoni-Baruch R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer. 2014;111(6):1132–8.
Blair AB, Groot VP, Gemenetzis G, Wei J, Cameron JL, Weiss MJ, Goggins M, Wolfgang CL, Yu J, He J. BRCA1/BRCA2 Germline Mutation Carriers and Sporadic Pancreatic Ductal Adenocarcinoma. J Am Coll Surg. 2018;226(4):630–7 e631.
Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;5(4):387–93.
Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244–50.
Bendell J, O'Reilly EM, Middleton MR, Chau I, Hochster H, Fielding A, Burke W, Burris H 3rd. Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann Oncol. 2015;26(4):804–11.
Lowery MA, Kelsen DP, Capanu M, Smith SC, Lee JW, Stadler ZK, Moore MJ, Kindler HL, Golan T, Segal A, et al. Phase II trial of veliparib in patients with previously treated BRCA-mutated pancreas ductal adenocarcinoma. Eur J Cancer. 2018;89:19–26.
O'Reilly EM, Lee JW, Lowery MA, Capanu M, Stadler ZK, Moore MJ, Dhani N, Kindler HL, Estrella H, Maynard H, et al. Phase 1 trial evaluating cisplatin, gemcitabine, and veliparib in 2 patient cohorts: Germline BRCA mutation carriers and wild-type BRCA pancreatic ductal adenocarcinoma. Cancer. 2018;124(7):1374–82.
Perkhofer L, Schmitt A, Romero Carrasco MC, Ihle M, Hampp S, Ruess DA, Hessmann E, Russell R, Lechel A, Azoitei N, et al. ATM Deficiency Generating Genomic Instability Sensitizes Pancreatic Ductal Adenocarcinoma Cells to Therapy-Induced DNA Damage. Cancer Res. 2017;77(20):5576–90.
Hessmann E, Johnsen SA, Siveke JT, Ellenrieder V. Epigenetic treatment of pancreatic cancer: is there a therapeutic perspective on the horizon? Gut. 2017;66(1):168–79.
Mazur PK, Herner A, Mello SS, Wirth M, Hausmann S, Sanchez-Rivera FJ, Lofgren SM, Kuschma T, Hahn SA, Vangala D, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21(10):1163–71.
Krantz BA, O'Reilly EM. Biomarker-Based Therapy in Pancreatic Ductal Adenocarcinoma: An Emerging Reality? Clin Cancer Res. 2018;24(10):2241–50.
McDonald PC, Chafe SC, Brown WS, Saberi S, Swayampakula M, Venkateswaran G, Nemirovsky O, Gillespie JA, Karasinska JM, Kalloger SE, et al. Regulation of pH by Carbonic Anhydrase 9 Mediates Survival of Pancreatic Cancer Cells With Activated KRAS in Response to Hypoxia. Gastroenterology. 2019. https://doi.org/10.1053/j.gastro.2019.05.004.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64.
Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23.
Sahin IH, Askan G, Hu ZI, O'Reilly EM. Immunotherapy in pancreatic ductal adenocarcinoma: an emerging entity? Ann Oncol. 2017;28(12):2950–61.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8.
Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, Herbst B, Askan G, Bhanot U, Senbabaoglu Y, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551(7681):512–6.
Bauer C, Kuhnemuth B, Duewell P, Ormanns S, Gress T, Schnurr M. Prevailing over T cell exhaustion: New developments in the immunotherapy of pancreatic cancer. Cancer Lett. 2016;381(1):259–68.
Gjertsen MK, Buanes T, Rosseland AR, Bakka A, Gladhaug I, Soreide O, Eriksen JA, Moller M, Baksaas I, Lothe RA, et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma. Int J Cancer. 2001;92(3):441–50.
Hardacre JM, Mulcahy M, Small W, Talamonti M, Obel J, Krishnamurthi S, Rocha-Lima CS, Safran H, Lenz HJ, Chiorean EG. Addition of algenpantucel-L immunotherapy to standard adjuvant therapy for pancreatic cancer: a phase 2 study. J Gastrointest Surg. 2013;17(1):94–100 discussion p. 100-101.
Yan J, Pankhong P, Shin TH, Obeng-Adjei N, Morrow MP, Walters JN, Khan AS, Sardesai NY, Weiner DB. Highly optimized DNA vaccine targeting human telomerase reverse transcriptase stimulates potent antitumor immunity. Cancer Immunol Res. 2013;1(3):179–89.
NewLink Genetics Announces Results from Phase 3 IMPRESS Trial of Algenpantucel-L for Patients with Resected Pancreatic Cancer [https://globenewswire.com/news-release/2016/05/09/837878/0/en/NewLink-Genetics-Announces-Results-from-Phase-3-IMPRESS-Trial-of-Algenpantucel-L-for-Patients-with-Resected-Pancreatic-Cancer.html]. Accessed 30 July.
Middleton G, Silcocks P, Cox T, Valle J, Wadsley J, Propper D, Coxon F, Ross P, Madhusudan S, Roques T, et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): an open-label, randomised, phase 3 trial. Lancet Oncol. 2014;15(8):829–40.
Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T, Springett G, Morse M, Zeh H, Cohen D, Fine RL, et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol. 2015;33(12):1325–33.
Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, Gartner JJ, Zheng Z, Li YF, Ray S, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350(6266):1387–90.
Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med. 2016;375(23):2255–62.
Bauer C, Dauer M, Saraj S, Schnurr M, Bauernfeind F, Sterzik A, Junkmann J, Jakl V, Kiefl R, Oduncu F, et al. Dendritic cell-based vaccination of patients with advanced pancreatic carcinoma: results of a pilot study. Cancer Immunol Immunother. 2011;60(8):1097–107.
Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–95.
Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229(1):152–72.
Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–6.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–20.
Jacobs C, Duewell P, Heckelsmiller K, Wei J, Bauernfeind F, Ellermeier J, Kisser U, Bauer CA, Dauer M, Eigler A, et al. An ISCOM vaccine combined with a TLR9 agonist breaks immune evasion mediated by regulatory T cells in an orthotopic model of pancreatic carcinoma. Int J Cancer. 2011;128(4):897–907.
Duewell P, Steger A, Lohr H, Bourhis H, Hoelz H, Kirchleitner SV, Stieg MR, Grassmann S, Kobold S, Siveke JT, et al. RIG-I-like helicases induce immunogenic cell death of pancreatic cancer cells and sensitize tumors toward killing by CD8(+) T cells. Cell Death Differ. 2014;21(12):1825–37.
Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11(7):1018–30.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018;378(5):439–48.
Benmebarek MR, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int J Mol Sci. 2019;20(6). https://doi.org/10.3390/ijms20061283.
Posey AD Jr, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, Stone JD, Madsen TD, Schreiber K, Haines KM, et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity. 2016;44(6):1444–54.
Akce M, Zaidi MY, Waller EK, El-Rayes BF, Lesinski GB. The Potential of CAR T Cell Therapy in Pancreatic Cancer. Front Immunol. 2018;9:2166.
Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS, Braiteh F, Ritch PS, Zalupski MM, Bahary N, et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J Clin Oncol. 2018;36(4):359–66.
Doherty GJ, Tempero M, Corrie PG. HALO-109-301: a Phase III trial of PEGPH20 (with gemcitabine and nab-paclitaxel) in hyaluronic acid-high stage IV pancreatic cancer. Future Oncol. 2018;14(1):13–22.
Kanteti R, Mirzapoiazova T, Riehm JJ, Dhanasingh I, Mambetsariev B, Wang J, Kulkarni P, Kaushik G, Seshacharyulu P, Ponnusamy MP, et al. Focal adhesion kinase a potential therapeutic target for pancreatic cancer and malignant pleural mesothelioma. Cancer Biol Ther. 2018;19(4):316–27.
Begum A, Ewachiw T, Jung C, Huang A, Norberg KJ, Marchionni L, McMillan R, Penchev V, Rajeshkumar NV, Maitra A, et al. The extracellular matrix and focal adhesion kinase signaling regulate cancer stem cell function in pancreatic ductal adenocarcinoma. PLoS One. 2017;12(7):e0180181.
Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348(6230):74–80.
Albeituni SH, Ding C, Yan J. Hampering immune suppressors: therapeutic targeting of myeloid-derived suppressor cells in cancer. Cancer J. 2013;19(6):490–501.
Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, Greenberg PD, Hingorani SR. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014;63(11):1769–81.
Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, Mitchem JB, Plambeck-Suess SM, Worley LA, Goetz BD, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. 2013;19(13):3404–15.
Linehan D, Noel MS, Hezel AF, Wang-Gillam A, Eskens F, Sleijfer S, Desar IME, Erdkamp F, Wilmink J, Diehl J, et al. Overall survival in a trial of orally administered CCR2 inhibitor CCX872 in locally advanced/metastatic pancreatic cancer: Correlation with blood monocyte counts. J Clin Oncol. 2018;36(5_suppl):92.
Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–69.
Schnurr M, Duewell P, Bauer C, Rothenfusser S, Lauber K, Endres S, Kobold S. Strategies to relieve immunosuppression in pancreatic cancer. Immunotherapy. 2015;7(4):363–76.
Vokes EE. Combined modality therapy of solid tumours. Lancet. 1997;349(Suppl 2):SII4–6.
Niyazi M, Maihoefer C, Krause M, Rodel C, Budach W, Belka C. Radiotherapy and “new” drugs-new side effects? Radiat Oncol. 2011;6:177.
Buchsbaum DJ, Bonner JA, Grizzle WE, Stackhouse MA, Carpenter M, Hicklin DJ, Bohlen P, Raisch KP. Treatment of pancreatic cancer xenografts with Erbitux (IMC-C225) anti-EGFR antibody, gemcitabine, and radiation. Int J Radiat Oncol Biol Phys. 2002;54(4):1180–93.
Morgan MA, Parsels LA, Kollar LE, Normolle DP, Maybaum J, Lawrence TS. The combination of epidermal growth factor receptor inhibitors with gemcitabine and radiation in pancreatic cancer. Clin Cancer Res. 2008;14(16):5142–9.
Demols A, Mahin C, Maréchal R, Delaunoit T, Borbath I, Hendlisz A, Jacquy C, Mitine C, Laethem JV. Cetuximab plus chemoradiation combined therapy for locally advanced inoperable pancreatic adenocarcima: A phase I study. J Clin Oncol. 2008;26(15_suppl):4629.
Munter M, Timke C, Abdollahi A, Friess H, Jaeger D, Heeger S, Buchler M, Debus J, Huber P, Krempien R. Final results of a phase II trial [PARC-Study ISRCTN56652283] for patients with primary inoperable locally advanced pancreatic cancer combining intensity modulated radiotherapy (IMRT) with cetuximab and gemcitabine. J Clin Oncol. 2008;26(15_suppl):4613.
Arnoletti JP, Frolov A, Eloubeidi M, Keene K, Posey J, Wood T, Greeno E, Jhala N, Varadarajulu S, Russo S, et al. A phase I study evaluating the role of the anti-epidermal growth factor receptor (EGFR) antibody cetuximab as a radiosensitizer with chemoradiation for locally advanced pancreatic cancer. Cancer Chemother Pharmacol. 2011;67(4):891–7.
Rembielak AI, Jain P, Jackson AS, Green MM, Santorelli GR, Whitfield GA, Crellin A, Garcia-Alonso A, Radhakrishna G, Cullen J, et al. Phase II Trial of Cetuximab and Conformal Radiotherapy Only in Locally Advanced Pancreatic Cancer with Concurrent Tissue Sampling Feasibility Study. Transl Oncol. 2014;7(1):55–64.
Hayman TJ, Wahba A, Rath BH, Bae H, Kramp T, Shankavaram UT, Camphausen K, Tofilon PJ. The ATP-competitive mTOR inhibitor INK128 enhances in vitro and in vivo radiosensitivity of pancreatic carcinoma cells. Clin Cancer Res. 2014;20(1):110–9.
Dai ZJ, Gao J, Kang HF, Ma YG, Ma XB, Lu WF, Lin S, Ma HB, Wang XJ, Wu WY. Targeted inhibition of mammalian target of rapamycin (mTOR) enhances radiosensitivity in pancreatic carcinoma cells. Drug Des Devel Ther. 2013;7:149–59.
Williams TM, Flecha AR, Keller P, Ram A, Karnak D, Galban S, Galban CJ, Ross BD, Lawrence TS, Rehemtulla A, et al. Cotargeting MAPK and PI3K signaling with concurrent radiotherapy as a strategy for the treatment of pancreatic cancer. Mol Cancer Ther. 2012;11(5):1193–202.
Park JH, Jung KH, Kim SJ, Fang Z, Yan HH, Son MK, Kim J, Kang YW, Lee JE, Han B, et al. Radiosensitization of the PI3K inhibitor HS-173 through reduction of DNA damage repair in pancreatic cancer. Oncotarget. 2017;8(68):112893–906.
Yang L, Yang G, Ding Y, Dai Y, Xu S, Guo Q, Xie A, Hu G. Inhibition of PI3K/AKT Signaling Pathway Radiosensitizes Pancreatic Cancer Cells with ARID1A Deficiency in Vitro. J Cancer. 2018;9(5):890–900.
Chang L, Graham PH, Hao J, Ni J, Bucci J, Cozzi PJ, Kearsley JH, Li Y. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014;5:e1437.
Durrant DE, Das A, Dyer S, Tavallai S, Dent P, Kukreja RC. Targeted Inhibition of Phosphoinositide 3-Kinase/Mammalian Target of Rapamycin Sensitizes Pancreatic Cancer Cells to Doxorubicin without Exacerbating Cardiac Toxicity. Mol Pharmacol. 2015;88(3):512–23.
Lomax ME, Folkes LK, O'Neill P. Biological consequences of radiation-induced DNA damage: relevance to radiotherapy. Clin Oncol (R Coll Radiol). 2013;25(10):578–85.
O'Connor MJ. Targeting the DNA Damage Response in Cancer. Mol Cell. 2015;60(4):547–60.
Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481(7381):287–94.
Prevo R, Fokas E, Reaper PM, Charlton PA, Pollard JR, McKenna WG, Muschel RJ, Brunner TB. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol Ther. 2012;13(11):1072–81.
Fokas E, Prevo R, Pollard JR, Reaper PM, Charlton PA, Cornelissen B, Vallis KA, Hammond EM, Olcina MM, Gillies McKenna W, et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012;3:e441.
Wallez Y, Dunlop CR, Johnson TI, Koh SB, Fornari C, Yates JWT, Bernaldo de Quiros Fernandez S, Lau A, Richards FM, Jodrell DI. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol Cancer Ther. 2018;17(8):1670–82.
Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC, Arumugarajah S, Hylander-Gans L, Morosini D, Simeone DM, et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010;70(12):4972–81.
Venkatesha VA, Parsels LA, Parsels JD, Zhao L, Zabludoff SD, Simeone DM, Maybaum J, Lawrence TS, Morgan MA. Sensitization of pancreatic cancer stem cells to gemcitabine by Chk1 inhibition. Neoplasia. 2012;14(6):519–25.
Engelke CG, Parsels LA, Qian Y, Zhang Q, Karnak D, Robertson JR, Tanska DM, Wei D, Davis MA, Parsels JD, et al. Sensitization of pancreatic cancer to chemoradiation by the Chk1 inhibitor MK8776. Clin Cancer Res. 2013;19(16):4412–21.
Al-Ejeh F, Pajic M, Shi W, Kalimutho M, Miranda M, Nagrial AM, Chou A, Biankin AV, Grimmond SM, Australian Pancreatic Cancer Genome I, et al. Gemcitabine and CHK1 inhibition potentiate EGFR-directed radioimmunotherapy against pancreatic ductal adenocarcinoma. Clin Cancer Res. 2014;20(12):3187–97.
Tuli R, Surmak AJ, Reyes J, Armour M, Hacker-Prietz A, Wong J, DeWeese TL, Herman JM. Radiosensitization of Pancreatic Cancer Cells In Vitro and In Vivo through Poly (ADP-ribose) Polymerase Inhibition with ABT-888. Transl Oncol. 2014. https://doi.org/10.1016/j.tranon.2014.04.003.
Rajan A, Carter CA, Kelly RJ, Gutierrez M, Kummar S, Szabo E, Yancey MA, Ji J, Mannargudi B, Woo S, et al. A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clin Cancer Res. 2012;18(8):2344–51.
Brix N, Tiefenthaller A, Anders H, Belka C, Lauber K. Abscopal, immunological effects of radiotherapy: Narrowing the gap between clinical and preclinical experiences. Immunol Rev. 2017;280(1):249–79.
Filatenkov A, Baker J, Mueller AM, Kenkel J, Ahn GO, Dutt S, Zhang N, Kohrt H, Jensen K, Dejbakhsh-Jones S, et al. Ablative Tumor Radiation Can Change the Tumor Immune Cell Microenvironment to Induce Durable Complete Remissions. Clin Cancer Res. 2015;21(16):3727–39.
Lennon S, Oweida A, Milner D, Phan AV, Bhatia S, Van Court B, Darragh L, Mueller AC, Raben D, Martinez-Torrecuadrada JL, et al. Pancreatic Tumor Microenvironment Modulation by EphB4-ephrinB2 Inhibition and Radiation Combination. Clin Cancer Res. 2019;25(11):3352-65.
Golden EB, Chhabra A, Chachoua A, Adams S, Donach M, Fenton-Kerimian M, Friedman K, Ponzo F, Babb JS, Goldberg J, et al. Local radiotherapy and granulocyte-macrophage colony-stimulating factor to generate abscopal responses in patients with metastatic solid tumours: a proof-of-principle trial. Lancet Oncol. 2015;16(7):795–803.
Shi F, Wang X, Teng F, Kong L, Yu J. Abscopal effect of metastatic pancreatic cancer after local radiotherapy and granulocyte-macrophage colony-stimulating factor therapy. Cancer Biol Ther. 2017;18(3):137–41.
Azad A, Yin Lim S, D'Costa Z, Jones K, Diana A, Sansom OJ, Kruger P, Liu S, McKenna WG, Dushek O, et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol Med. 2017;9(2):167–80.
Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, Pfirschke C, Voss RH, Timke C, Umansky L, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24(5):589–602.
Timke C, Winnenthal HS, Klug F, Roeder FF, Bonertz A, Reissfelder C, Rochet N, Koch M, Tjaden C, Buechler MW, et al. Randomized controlled phase I/II study to investigate immune stimulatory effects by low dose radiotherapy in primarily operable pancreatic cancer. BMC Cancer. 2011;11:134.
Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41(5):843–52.
Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, Demaria S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8:15618.
Baird JR, Friedman D, Cottam B, Dubensky TW Jr, Kanne DB, Bambina S, Bahjat K, Crittenden MR, Gough MJ. Radiotherapy Combined with Novel STING-Targeting Oligonucleotides Results in Regression of Established Tumors. Cancer Res. 2016;76(1):50–61.
Rech AJ, Dada H, Kotzin JJ, Henao-Mejia J, Minn AJ, Twyman-Saint Victor C, Vonderheide RH. Radiotherapy and CD40 Activation Separately Augment Immunity to Checkpoint Blockade in Cancer. Cancer Res. 2018;78(15):4282–91.
Lee V, Rodriguez C, Shupe E-M, Chen L, Parkinson R, Durham JN, Sugar E, Wilt C, McIntyre KR, Hacker-Prietz A, et al. Phase II study of GM-CSF secreting allogeneic pancreatic cancer vaccine (GVAX) with PD-1 blockade antibody and stereotactic body radiation therapy (SBRT) for locally advanced pancreas cancer (LAPC). J Clin Oncol. 2017;35(15_suppl):TPS4154.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (CRC1321 projects P13, P14 and P16 to GS, KL, MS, and JM, and SCHN664/6-1 to MS), the international doctoral program ‘i-Target: Immunotargeting of cancer’ funded by the Elite Network of Bavaria (to MS and KL), the Bundesministerium fuer Bildung und Forschung (BMBF 01EK1511A to JM), and the European Union (EU-FP-7: EPC-TM to JM, Marie-Sklodowska-Curie ‘Training Network for the Immunotherapy of Cancer (IMMUTRAIN)’ to MS).
The funding body did not have any influence on conception and content of this review manuscript.
Author information
Authors and Affiliations
Contributions
MO, PM, and KL: conception and writing. KL: illustration. All listed authors were involved in drafting and revising of the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Orth, M., Metzger, P., Gerum, S. et al. Pancreatic ductal adenocarcinoma: biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat Oncol 14, 141 (2019). https://doi.org/10.1186/s13014-019-1345-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13014-019-1345-6