
Abstract
Pancreatic ductal adenocarcinoma (PDA) is a lethal disease with limited response to cytotoxic chemoradiotherapy, as well as newer immunotherapies. The PDA tumor microenvironment contains infiltrating immune cells including cytotoxic T cells; however, there is an overall immunosuppressive milieu. Hypoxia is a known element of the solid tumor microenvironment and may promote tumor survival. Through various mechanisms including, but not limited to, those mediated by HIF-1α, hypoxia also leads to increased tumor proliferation and metabolic changes. Furthermore, epithelial to mesenchymal transition is promoted through several pathways, including NOTCH and c-MET, regulated by hypoxia. Hypoxia-promoted changes also contribute to the immunosuppressive phenotype seen in many different cell types within the microenvironment and thereby may inhibit an effective immune system response to PDA. Pancreatic stellate cells (PSCs) and myofibroblasts appear to contribute to the recruitment of myeloid derived suppressor cells (MDSCs) and B cells in PDA via cytokines increased due to hypoxia. PSCs also increase collagen secretion in response to HIF-1α, which promotes a fibrotic stroma that alters T cell homing and migration. In hypoxic environments, B cells contribute to cytotoxic T cell exhaustion and produce chemokines to attract more immunosuppressive regulatory T cells. MDSCs inhibit T cell metabolism by hoarding key amino acids, modulate T cell homing by cleaving L-selectin, and prevent T cell activation by increasing PD-L1 expression. Immunosuppressive M2 phenotype macrophages promote T cell anergy via increased nitric oxide (NO) and decreased arginine in hypoxia. Increased numbers of regulatory T cells are seen in hypoxia which prevent effector T cell activation through cytokine production and increased CTLA-4. Effective immunotherapy for pancreatic adenocarcinoma and other solid tumors will need to help counteract the immunosuppressive nature of hypoxia-induced changes in the tumor microenvironment. Promising studies will look at combination therapies involving checkpoint inhibitors, chemokine inhibitors, and possible targeting of hypoxia. While no model is perfect, assuring that models incorporate the effects of hypoxia on cancer cells, stromal cells, and effector immune cells will be crucial in developing successful therapies.
Similar content being viewed by others

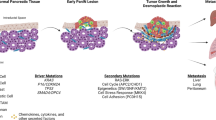

Background
Pancreatic ductal adenocarcinoma (PDA) is projected to be the second highest cause of death from cancer in the United States within the next 10 years [1, 2]. The lethality of the disease is in part due to lack of effective screening resulting in later stage diagnoses, as well as poor response to standard therapies including surgery, systemic chemotherapy, and external beam radiation [3,4,5,6]. Immunotherapy has heralded a new era in oncologic treatment that may ultimately improve outcomes, while having fewer toxic side effects than systemic chemotherapy. The overarching goal of immunotherapy is to enhance the body’s immune response to tumor cells. The strategy of blocking immune checkpoints to potentiate immune-mediated tumor cell killing has been successful in several tumors such as melanoma and certain phenotypes of lung cancer, but has not been successful in many other solid tumors such as PDA [7,8,9].
The reason for the effectiveness of immunotherapy in some tumors more than others has been a subject of intense focus. Initially, this was thought to be due to a paucity of immune cells infiltrating PDA tumors, however many studies have since shown there is a variable but substantial population of tumor-infiltrating lymphocytes (TIL) in PDA [10,11,12]. Another theory was that PDA was not as immunogenic as other tumors, but several neoepitopes have been identified as recognizable by T cells [13]. PDA in particular has a robust tumor microenvironment composed of myofibroblasts and immune cells that often outnumber carcinoma cells [12]. The interactions among these cells are undoubtedly a major driving factor of immunotherapy resistance in PDA, but hypoxia has an underlying influence that is not yet fully understood.
The tumor microenvironments of many solid tumors are known to be hypoxic [14,15,16]. In PDA, there is a decrease in tissue partial oxygen pressure in tumors, with median pO2 0–5.3 mmHg (0-0.7%) compared to nearby normal pancreas at pO2 24.3–92.7 mmHg (3.2–12.3%) [17]. For reference, normal pO2 is 160 mmHg (21.1%) in air and 100 mmHg (13.2%) in arterial blood [18]. Further studies have shown that this hypoxia is heterogeneous throughout the tumor and not static [17, 19, 20]. Many reviews have summarized in general the pro-survival and pro-metastatic changes that a tumor undergoes in a hypoxic environment [21,22,23,24,25]. Additionally, hypoxia also induces changes in the other cells in the tumor microenvironment that encourage immunosuppression, which may play a role in diminishing the efficacy of immunotherapy in PDA.
Signaling pathways in response to hypoxia
A large number of downstream effects of hypoxia are mediated by a transcription factor called hypoxia inducible factor (HIF) [23]. Three variants of the alpha subunit of HIF have been discovered, with HIF-1α being the most commonly studied. Based on current knowledge, HIF-3α primarily acts to promote or inhibit other HIF complexes [21]. The HIF variants are constitutively expressed proteins. Primary regulation is achieved by hydroxylation of a proline in normoxic conditions by a prolyl hydroxylase unique to each HIFα variant [26]. The hydroxyl group tags the molecule for degradation via von Hippel-Lindau protein (vHL). In hypoxic conditions, the iron atom in prolyl hydroxylase stays reduced and the enzyme is unable to add the hydroxyl group to the HIFα unit [26]. This allows HIF-1α to bind to the HIF-1β molecule and translocate to the nucleus where it acts as a transcription factor on many promoter sequences.
Post-translational modifications, such as phosphorylation and acetylation, of the different variants influence binding abilities and therefore transcriptional effects [21]. Additionally, owing to their different prolyl hydroxylases, HIF-1α and HIF-2α accumulate at different oxygen levels. HIF-2α tends to accumulate at higher oxygen levels (2–5%), whereas HIF-1α does not accumulate until lower oxygen levels (0–2%) [26]. HIF-1α mRNA also degrades very quickly even in hypoxic conditions making its effects shorter lived [27]. While HIF-2α has been studied less than HIF-1α, there have been data to show that HIF-2α uniquely promotes chronic pancreatitis in mouse models, as well as mucinous cyst neoplasms in the presence of an oncogenic KRAS mutation [28].
Hypoxia promotes tumorigenesis in carcinoma cells
Tumor cells in PDA have many advantages under hypoxic conditions. KRAS mutations, which are seen in around 95% of PDA tumors, work to alter the cell metabolism to function in hypoxic environments [29]. Glycolysis becomes the primary means of obtaining energy via downstream effects of HIF-1α and persists even if normoxic conditions are restored—a phenomenon known as the Warburg hypothesis [29, 30]. To increase the glucose supply, HIF-1α mediates increased transcription of GLUT1 and GLUT3 transporters, as well as increased production of pyruvate and lactate dehydrogenase [31, 32]. The lactate produced from glycolysis is further used as energy for surrounding cells and impairs T cell cytokine production [33]. Other metabolic enzymes that are upregulated by PDA in hypoxia include carbonic anhydrase and indoleamine 2,3 dioxygenase (IDO), which also impair immune cell function through the creation of acidic and tryptophan-depleted environments, respectively [32, 34,35,36]. An additional source of energy for PDA cells is autophagy, which in tumors with loss of p53 has been correlated with increased tumor progression [37,38,39]. Hypoxia has been shown to increase autophagy via HIF-1α, which promotes survival of PDA tumor cells, particularly those that are undifferentiated [40,41,42].
Mechanisms that promote terminal cell differentiation are inhibited via interaction with HIF-1α and NOTCH signaling in PDA and other tumor types [43,44,45]. This is thought to promote cancer cell “stemness” in hypoxic niches in the PDA microenvironment [46, 47]. The quiescent cell is also less affected by systemic chemotherapy and radiotherapy that acts on rapidly dividing cells, therefore promoting tumor recurrence [48,49,50]. Additional mechanisms of cell cycle regulation in tumors involve the differential expression of HIF-1α and HIF-2α. As mentioned earlier, extreme hypoxia promotes HIF-1α stabilization and actually decreases cellular proliferation by halting the cell cycle via c-MYC in some tumors [51]. Molecular inhibitors of c-MYC halt cell cycle progress in PDA and may also block hypoxic signaling [52,53,54]. Evidence supports that, conversely, HIF-2α promotes proliferation with cells entering the cell cycle via stabilization of MYC and increased DNA repair enzymes [25, 27].
Another benefit of a hypoxic environment for PDA is an increase in cell migration. There are numerous pathways activated by HIF-1α that contribute to the epithelial to mesenchymal transition (EMT) [55,56,57]. EMT involves loss of normal cell-to-cell adhesion molecules seen in terminally differentiated epithelial cells and promotes molecules used for cell movement and angioinvasion typically seen in less differentiated cells. Well documented molecules that are increased in hypoxic PDA include matrix metalloproteinases (MMPs) via up regulated fascin and QSOX1 that subsequently decrease the immediately surrounding extracellular matrix (ECM) to allow tumor cell movement [58, 59]. Cadherins are cell adhesion molecules involved in maintaining epithelial tissue architecture, with increased N-cadherin and loss of E-cadherin expression associated with greater invasive potential in cancer. Through HIF-1α-mediated NF-κB pathways, hypoxia increases N-cadherin to allow transendothelial migration into blood vessels [60]. The transcription factors snail and slug, also HIF-1α promoted, are expressed in many pancreatic cancer cell lines and act to decrease E-cadherin seen in normal epithelial cell-to-cell adhesion [61]. Twist is another transcription factor that prevents E-cadherin formation as well as increased vascular endothelial growth factor (VEGF)-A, but is primarily up regulated in PDA after HIF-2α stabilization [61,62,63]. Hedgehog signaling in PDA, which is potentiated by hypoxia, also down regulates E-cadherin and up regulates vimentin, which promotes invadopodia formation and angioinvasion [64].
Tumor cell survival in hypoxia also requires changes to avoid internal mechanisms of apoptosis and increase resistance to chemotherapy. In PDA cell lines, HIF-2α up regulates survivin production, which provides resistance to apoptosis by tumor necrosis factor (TNF) related apoptosis inducing ligand (TRAIL) [65]. Hypoglycemia-mediated apoptosis is also prevented in PDA by hypoxia-induced up regulation of asparagine synthetase and subsequent prevention of c-jun-NH2 terminal kinase/phospho-stress-activated protein kinase activation, which is also a method for cisplatin resistance [66]. Many pancreatic cell lines down-regulate expression of BNIP3, which is a gene involved in hypoxia-mediated cell-induced apoptosis [67, 68]. Additionally, loss of BNIP3 expression in hypoxia has been associated with resistance to gemcitabine and 5-fluorouracil [69]. Gemcitabine resistance is also increased in hypoxia via the PI3K/Akt/NF-κB pathways that increases anti-apoptotic proteins such as Bcl-XL, FLIP, and cIAP [4, 46, 70]. Hypoxia also promotes resistance to radiotherapy through decreased production of DNA free radicals and increased DNA repair enzymes as described above [46, 71].
Hypoxia induces several changes on the cell surface of tumors to promote cell survival. While not yet shown in PDA, hypoxia in prostate cancer encourages tumor cells to shed their major histocompatibility complex (MHC) class I molecules via decreased nitric oxide (NO) and increased matrix metalloproteinases (MMPs) [72]. There are increased soluble levels of MHC class I chains A and B, suggesting that PDA may use this mechanism to avoid recognition by adaptive and innate immune mechanisms [73]. Additionally, human leukocyte antigen G (HLA-G) is a component of MHC class I expressed in a minority of PDA tumors that induces immunosuppression by interacting with receptors on antigen presenting cells [74]. The up regulation of HLA-G transcription is also mediated by HIF-1α, however some other types of tumors actually have decreased HLA-G expression in hypoxia [35]. Hypoxia also promotes increased programmed death ligand-1 (PD-L1) cell surface expression in a variety of solid tumors via the PTEN/PI3K pathway through HIF-1α [72]. Increased PD-L1 expression prevents effector T cells from initiating apoptosis of cancer cells and can actually lead to anergy or apoptosis of the T cells [72]. A minority of human PDA samples with baseline PD-L1 upregulation were also seen to have downregulation of MHC class I [75]. Hypoxia also stimulated CD47 expression in PDA, which is a co-stimulatory molecule that blocks pro-phagocytic signals in myeloid derived suppressor cells (MDSCs) and macrophages [76, 77]. Tumor cells also increase CD39 and CD73 in response to hypoxia, which promotes extracellular adenosine accumulation and can lead to T cell apoptosis [78, 79].
The effects of hypoxia on the tumor microenvironment
As discussed above, carcinoma cells themselves respond to hypoxia in self-promoting ways and encourage a continued hypoxic and acidic environment within the tumor stroma. The resulting landscape causes non-carcinoma tumor cells to shift towards an overall tumor-supportive and immunosuppressive milieu. The resulting cell phenotypes in the microenvironment have a direct influence on effector T cell function and resulting ineffectiveness of immunotherapies.
Pancreatic stellate cells and fibroblasts
Major contributors of the tumor microenvironment are activated pancreatic stellate cells (PSCs) and myofibroblasts. PSCs are identified as having large vitamin A droplets in their inactivated state, which they lose when they become differentiated in response to pancreatic injury or inflammation [80]. Cytokines shown to activate PSCs include TGF-β, TNF-α, IL-1, and IL-6; however, some suggest they are capable of autocrine signaling [81, 82]. The role and origin of PSCs are still not fully elucidated; however, they produce ECM molecules such as alpha smooth muscle actin (aSMA), type I collagen, fibronectin, and periostin that lead to pancreatic fibrosis [81, 83]. There are differences between PSCs and fibroblasts such as cell shape, amount of different ECM molecules produced, and scavenger receptors, but often they are grouped together in discussions [82]. In pancreatic intraductal neoplasms (PanIN), which are precursor lesions to PDA, fibroblasts show a CD34+ aSMA− phenotype, whereas in PDA, these are reversed to CD34− aSMA+, demonstrating an increase in aSMA production as the lesion progresses from non-invasive to invasive [84]. Activated fibroblasts in PDA can often be identified by the serine protease fibroblast activating protein (FAP) expression, although this is also seen on some tumor cells, and has been associated with increased desmoplasia and worse prognosis [85, 86].
PSCs have a significant role as potentiators of immunosuppression in PDA. Increased type I collagen density produced in fibrosis interferes with chemokines used in T cell homing causing them to become “trapped” away from tumor cells. Fibroblasts also produce increased CXCL12 which is another method of inhibiting T cell homing [84, 87]. Fibronectin deposition in the ECM encourages more rapid migration of tumor cells [86]. Periostin increases fibroblast growth factor (FGF) 2 which promotes macrophage differentiation into the M2 phenotype as well as encourages PDA proliferation [88]. Cytokines produced by PSCs also exert significant influence on the tumor-infiltrating immune cells. The most impactful of this is secretion of IL-6 and M-CSF which recruits MDSCs [89, 90]. They also secrete IL-1 and TGF-β which work to continually activate PSCs to continue forming a fibrotic environment in an autocrine fashion [80].
In response to hypoxia, tumor cells produce the sonic hedgehog ligand which acts in a paracrine manner on myofibroblasts by binding to the Patched-1 receptor. The resulting downstream effects of this interaction include increased desmoplasia with production of aSMA, type I collagen, fibronectin, and periostin [91, 92]. Like in tumor cells, hypoxia up regulates carbonic anhydrase and GLUT1 and GLUT3 transporters in PSCs to further contribute to the immunosuppressive microenvironment. Additionally, hypoxia promotes connective tissue growth factor production by PSCs, which helps inhibit apoptosis in tumor cells [93]. VEGF production from PSCs is also increased in hypoxic conditions, which works in a paracrine fashion to encourage PSC migration [91, 94, 95].
Plasma or B cells
Responsible for the humoral immune response, B cells were once thought to reside primarily in lymphoid tissue, but studies have recently shown that they also infiltrate the tumor microenvironment [96]. CXCL13, a primary chemokine for B cell migration, is expressed by fibroblasts in the PDA stroma [97]. While not as extensively studied as other immune cell populations, there have been conflicting data about the role of B cells in the anti-tumor response. It has been elucidated that B cells, much like the rest of tumor-infiltrating immune cells, exist on a spectrum of activation that can encourage or inhibit T cell responses [96, 98]. One study reported that IL-35-producing B cells stimulate pancreatic neoplasia development starting from PanIN in both human and mouse models with KRAS mutations [97]. Another study looking at B cell distribution in human PDA demonstrated that B cells retained in tertiary lymphoid tissue gave a survival benefit, which was not seen when B cells were infiltrating into the tumor stroma [99]. Additionally, there are increased levels of B cell activating factor (BAFF) expressed by B cells infiltrating PDA with a correlation with increased EMT-related gene expression in tumor cells [100]. In other solid tumors, B cells have been found to increase tumor invasiveness through secretion of IL-8 [101].
B cells can produce a variety of cytokines and chemokines that have been implicated in immunosuppression. Regulatory B cells (Bregs) secrete IL-10 and TGF-β that induce Treg differentiation via Stat3 as well as M2 macrophage development in different murine cancer models [96, 98, 102]. In other solid tumors, B cells have been implicated in programming myeloid derived suppressor cells (MDSCs) to increase their immunosuppressive activity [98]. Bregs have also been shown to express PD-L1 on their cell surfaces, which can directly inhibit T cells [98]. In PDA models, B cell deficiency has shown to decrease the desmoplastic reaction of tumors [103]. B cells grown in a PDA environment were also shown to encourage Th2 differentiation of CD4 + T cells in a manner dependent on Bruton’s tyrosine kinase [103].
While not yet shown in PDA, it has been shown in other tumors that HIF-1α induces CXCR4 and HIF-2α induces CXCL12 production by B cells [104, 105]. This recruits MDSCs and regulatory T cells (Tregs) to the tumor environment. Interestingly, HIF-1α knockout in the pancreas of mouse PDA showed a large increase in the number of B cells in the tumor microenvironment [106]. The mechanism for this was thought to be increased levels of B cell attractant chemokines such as CXCL13 in the HIF-1α knockout mice PDA models, suggesting that hypoxia helps decrease B cell tumor infiltration [106]. Depletion of B cells via anti-CD20 antibody in the HIF-1α knockout PDA model allowed T cell infiltration into the tumor, but did not change Treg percentage [106].
Myeloid derived suppressor cells
MDSCs are a progenitor cell type derived from the bone marrow that has recently received increasing attention. These cells can give rise to macrophages, dendritic cells, and granulocytes, among others, and can have a major influence in the immunophenotype of the tumor microenvironment despite their usual low numbers [107]. MDSCs normally circulate in the bloodstream and are drawn to areas of inflammation and ischemia through chemokine molecules such as CXCL12 produced by fibroblasts, as well as growth factors such as granulocyte–macrophage colony stimulating factor (GM-CSF) from tumor cells in PDA [108,109,110]. Both peripheral blood, bone marrow, and PDA tumors in humans showed MDSC accumulation compared to healthy controls [111, 112]. MDSCs secrete immunosuppressive cytokines such as IL-6, IL-10, and TGF-β, which promote Treg differentiation and inhibit co-stimulatory molecules on antigen presenting cells (APCs) [107, 113]. Macrophage differentiation is also heavily influenced by MDSCs via the production of IL-4, IL-10, and IL-13, which promote an immunosuppressive M2 phenotype via STAT6 [107, 114].
Additional ways that MDSCs exert inhibitory influences on effector T cells is through cell metabolism, although this has primarily been demonstrated in other solid tumors. Via STAT3 and NF-κB pathway activation, MDSCs decrease essential amino acids for T cells, such as tryptophan in breast cancer [115]. l-arginine is depleted via cleavage by arginase-1 and cysteine is accumulated in the cytoplasm due increase uptake and lack of exporter on MDSCs [113, 116]. Metabolites also accumulate to inhibit T cells such as adenosine via up regulation of CD39 and CD73 that cleave ADP and AMP, respectively [79, 107, 117]. MDSCs also nitrate tyrosine residues on T cell receptors that prevent them from accurately recognizing antigens. This occurs via peroxynitrate generation from NO and reactive oxygen species (ROS). In murine PDA models specifically, production of pancreatic adenocarcinoma upregulated factor (PAUF) by tumor cells resulted in increased levels of arginase, NO, and ROS produced by MDSCs [118]. Finally, MDSCs cleave L-selectin due to constitutive expression of ADAM17 on their cell surface, which impairs T cell homing [119].
Lung cancer models have shown increased CD39 and CD73 production by MDSCs in hypoxia [120]. In mouse models of liver tumors, hypoxia increased PD-L1 expression on MDSCs, which had an immunosuppressive effect on T cells [121]. Subsequent blockage of this by PD-L1 inhibition led to decreased IL-10, IL-6, and TGF-β production [121]. Additionally, MDSCs have been shown to increase in number and remain undifferentiated in hepatocellular carcinoma in hypoxic conditions via up regulation of CCL26 and CD391L [122, 123]. Interestingly, HIF-1α stabilization in MDSCs in the lymphoma environment, but not the spleen, supported differentiation into macrophages with increased arginase and NO synthetase levels [124].
Macrophages
Macrophages are a primary component of the innate immune response, and there has been significant interest in the role of tumor-associated macrophages (TAMs) in the recent years. Macrophages may differentiate from cells in the tumor microenvironment or be recruited via CCL2, CCL5, and CXCL12 [108, 125]. As with most cells in the immune microenvironment, macrophages exist on a spectrum from immunostimulatory to immunosuppressive, which is thought to be adaptive to situations like chronic infections [126]. The milieu of cells in the tumor microenvironment produces cytokines such as IL-4, IL-10, IL-13, and M-CSF that encourage M2 or immunosuppressive phenotype differentiation [127]. Many studies have looked at models for macrophages in a variety of tumors and all have shown that increased TAMs led to decreased survival through various mechanisms [128,129,130,131].
Looking at immunosuppressive mechanisms in PDA specifically, macrophages isolated from human PDA have been shown to induce EMT related changes in various cell lines for both M1 and M2 macrophages [132]. Macrophages in PDA also secrete FAP, a serine proteinase, which encourages fibroblasts in the tumor environment to promote tumor angiogenesis and metastasis [87, 133]. Additionally, macrophages are thought to induce PDA cells to produce cytidine deaminase, which metabolizes gemcitabine to promote resistance [134].
M2 macrophages tend to be found in more hypoxic regions of PDA, whereas M1 macrophages tend to be in normoxic regions farther from cancer cells [135, 136]. This spatial arrangement is thought to be due to mechanisms related to IL-6, TGF-β, and M-CSF, as well as semaphorin 3A/neuropilin-1 [137,138,139]. After migration, semaphorin is then down regulated by HIF-1α, which helps retain M2 macrophages in the hypoxic areas [137, 140]. Interestingly, in bacterial infections, HIF-1α also increases production of IFN-γ which is pro-inflammatory and promotes a more M1 type phenotype, although this was not studied in PDA directly [141]. Secretion of TGF-β and IL-10 from tumor cells and the surrounding environment then promotes macrophage switching to M2 phenotype [108].
TAMs drive significant metabolic changes that influence the microenvironment. In response to hypoxia in a breast cancer model, HIF-1α acts quickly to increase NO via inducible nitric oxide synthetase (iNOS) in macrophages which causes T cell suppression [142]. HIF-2α acts more slowly via increased arginase which decreases the arginine required for NO synthesis and thus counteracts some of the action of HIF-1α; however, the lack of arginine causes more long-term anergy of T cells [142, 143]. Surprisingly, high quantities of NO produced by macrophages in response to HIF-1α can actually lead to tumor suppression and death in early stages [108]. IL-4 can counteract this by up regulating HIF-2α to increase arginase as well as non-HIF mechanisms to increase arginase via NF-κB [142]. In addition to NO regulation, other metabolic pathways such as IDO, which leads to tryptophan depletion, are up regulated in hypoxic macrophages in hepatocellular carcinoma [144].
There are several other proposed mechanisms through which macrophages having an immunosuppressive influence in a hypoxic environment. In a study looking at mouse models of breast, colon, and liver tumors, knockout of HIF-1α and HIF-2α decreased tumor growth, but only HIF-2α knockout lead to decreased expression of macrophage colony-stimulating factor receptor (M-CSFR) and CXCR4 on tumor-infiltrating macrophages [145]. Increased HIF-1α stabilization has also been shown to correlate with increased PD-L1 expression on macrophage cell surfaces [145]. Hypoxia also causes macrophages to produce MMP-7 which can cleave Fas ligand from neighboring cells and protect them from cell-mediated killing [140, 146]. Additionally, increased MMP-2 and MMP-9 have been seen in other solid tumors in the setting of increased tumor cell invasion [136].
Dendritic cells
Dendritic cells (DCs) are hematopoetic in origin and their main functions are phagocytosis and antigen presentation. Based on the presence or absence of co-stimulatory molecules, DCs can induce pro-inflammatory responses or immune tolerance in other immune cell populations [147]. DCs initially exist in their immature forms, and after exposure to different environmental stimuli, can become more immunostimulatory or immunosuppressive along a spectrum of myeloid forms, which stimulate Th1 response, and plasmacytoid forms, which stimulate a Th2 response [108, 148]. Tumors such as PDA benefit from immature DCs and prevent maturation via production of VEGF, IL-10, IL-6, and GM-CSF, among others [149, 150]. IL-8 also influences DC migration in colon cancer models [151]. Dendritic cells have CXCR1 and CXCR2 on their cell surfaces that binds to IL-8, but the amount of IL-8 did not affect the expression of MHC class II or co-stimulatory molecules such as CD80 and CD86 [151]. Prolonged exposure to IL-8 caused internalization of CXCR1 and CXCR2 prevented further migration towards IL-8 producing tumor cells [151].
In PDA, increased levels of circulating myeloid DCs can be predictive of longer survival after surgical resection [152, 153]. In addition to decreased co-stimulatory molecules such as CD40 and CD80 that prevent T cell activation, DCs in PDA produce a variety of chemokines and cytokines that help support an immunosuppressive environment [148]. DCs can produce CCL22 which recruits immunosuppressive Tregs in response to IL1a and TGF-β [154, 155]. Looking at miRNA in PDA, studies have shown that exosomes from tumor cells can alter cell surface expression of toll-like receptor (TLR) 4 [156]. PAUF is a ligand for TLR4, which can induce the production of pro-inflammatory TNF-α and IL-12 by DCs [156, 157]. Additionally, Smad4, a transcription factor that mediates TGF-β transduction, is repressed in DCs in the PDA environment through miRNA which prevents their antigen presentation and differentiation [158].
There are conflicting reports on the effects of hypoxia on DCs. In some reports, hypoxia increases the ability of DCs to interact with cytotoxic T cells [159]. Hypoxia has been shown to decrease circulating plasmacytoid DCs with corresponding increase in TNF-α and IL-6, although increase in CXCL12 could signify tissue migration [160]. One experiment demonstrated that DCs without HIF-1α had less CD278 on their cell surface, and the T cells in co-culture produced less granzyme B mRNA [161]. This has been shown to be due to the PI3K/Akt pathways [162]. It is not certain if DCs have decreased migration and phagocytic capabilities in a hypoxic environment [163, 164]. There does seem to be increased osteopontin secreted by hypoxic DCs in a breast mouse model that encourage tumor migration [165]. Hypoxia encourages the Th2 phenotype via increase in CD44 as well as adenosine receptor A2b although this has not been verified in PDA specifically [166, 167]. Additionally, PD-L1 expression on DC membranes is increased due to HIF-1α in hypoxia [121, 145]. Some immature DCs with prolonged exposure to hypoxia can actually be induced to undergo apoptosis via up regulation of BNIP3 and BAX [168].
Helper and regulatory T cells
CD4+ T cells have several different subtypes that result from terminal differentiation of naive progenitor cells, although there is some overlap between these classifications. The most commonly researched in PDA are T helper (Th) 1, Th2, Treg, and Th17 [169]. Th1 cells are pro-inflammatory and express IFN-γ to promote APCs and prime CD8+ cells, while Th2 cells may encourage tumor growth through IL-5 production, although they do recruit eosinophils through IL-4 and IL-13 [170]. Tregs are identified by FOXP3 gene expression and are predominately immunosuppressive through TGF-β production and repression of effector T cell proliferation [170]. Less clear is the role of Th17 cells that have been studied in autoimmune disease and may be associated with prolonged survival in some cancers [171, 172]. In addition to differing roles, CD4+ subtypes are also located differently in the PDA microenvironment. While the percentage of CD4+ cells that were Th1 remained stable throughout the tumor, tumor periphery, and healthy pancreas, Th2 and Treg cells were more likely to be in the central tumor, whereas Th17 cells were more likely to be in the healthy pancreas [169]. Another study showed that Tregs tend to accumulate early in the malignant process, even in PanIN mouse models, but primarily remain in peritumoral lymph nodes [173].
Due to poor response of PDA to immunomodulating therapies such as checkpoint inhibition, a significant focus has been placed on the immunosuppressive Tregs. CCL5 is produced by PDA which encourages migration of Tregs into the tumor microenvironment due to their CCR5 expression [174]. Treg migration into PDA is also encouraged by L1CAM expression on tumor cells [175]. Tregs in PDA express CTLA-4, which competes for co-stimulatory ligands CD80 and CD86 and prevents CD28 binding necessary for effector T cell activation [173]. Many Tregs also express CD25, which acts as an IL-2 receptor and contributes to FOXP3 expression in some Tregs [173, 176]. Overall, increased ratios of Treg:effector T cells is associated with tumor progression and worse outcome [12].
Hypoxia influences immunosuppression as well as subtype differentiation of CD4+ cells. Activation of the T cell receptor actually increases HIF-1α downstream effects via PI3K/mTOR and protein kinase C mechanisms through stabilization of HIF-1α even in the absence of hypoxia [140, 177]. HIF-1α promotes differentiation into FOXP3+ cells via increased gene transcription [178]. If sufficient TGF-β is also present, then these cells become Tregs, but combined IL-6 and HIF-1α instead promotes Th17 cells [179, 180]. Addition of IL-6 actually counteracts the effects of hypoxia on Treg proliferation with IL-1 having a more moderate counteractive effect [177, 179]. These mechanisms are thought to be mediated by STAT3, which also decreases IFN-γ production and decreases Th1 phenotype markers [177, 180]. Interestingly, HIF-1α can also bind to the FOXP3 protein to promote its degradation which can also promote Th17 and other pro-inflammatory phenotypes [178]. HIF-2α, however, does not promotes FOXP3 transcription in murine models of inflammation [179]. Treg numbers are further increased in hypoxia due to CCL28 production by tumor cells that increases Treg chemotaxis [181]. Hypoxia does not appear to affect levels of co-stimulatory molecules such as CD23 and CTLA-4 in CD4 + cells [179].
Effector T cells
Effector or CD8+ T cells are the primary cytotoxic agents of the adaptive immune systems. Most of the immunosuppressive mechanisms discussed above involve preventing effector T cells from undergoing the steps required to induce apoptosis in tumor cells: [1] migration of a naive CD8+ T cell into the area of the tumor, [2] antigen-presentation to the CD8+ cell between the T cell receptor (TCR) and MHC class I molecule with appropriate co-stimulation (via cytokines from CD4+ cells or CD28 and CD80/B7-1 or CD86/B7-2 on the APC), [3] clonal expansion of CD8+ cells, [4] recognition of the antigen again on the tumor cell, and [5] initiation of cytotoxic process with production and release of perforin, granzyme, and granulysin which trigger the caspase cascade in tumor cells [182, 183]. Alternatively, CD8+ cells have Fas ligand/CD95L on their cell surface that can bind Fas on target cells, which then promotes procaspases in the target cell, but this is rarely expressed in tumor cells [182]. The above describes more common protein-recognizing αβ CD8+ cells; however, there is a separate population of γδ CD8+ cells that recognize lipid antigens and do not require the same antigen presentation steps to become activated. Indeed, γδ CD8+ cells are being studied as well in immunotherapy for PDA [184].
Among the more common αβ CD8+ cells, there have been both stimulatory and inhibitory effects of hypoxia. HIF-1α is essential to the expression of the co-stimulatory molecule CD137 on effector T cells in several solid tumors, although this has not been confirmed in PDA [185]. Also, granzyme B production is increased in hypoxia, leading to increased lytic capacity of T cells in more mildly hypoxic compared to atmospheric oxygen [186,187,188]. Negative effects include decreased effector T cell migration into the tumor in hypoxia. Poorly formed vasculature forms in the hypoxic PDA environment, and in combination with IL-10 down regulating integrins such as αLβ2 on the vascular endothelium, T cell extravasation is diminished [140, 189]. HIF-1α also decreases production of the pro-inflammatory cytokines IL-2 and IFN-γ by CD8+ cells, even when stabilized under normoxic conditions [177, 186]. ROS resulting from hypoxia can have immunosuppressive and even lethal effects on T cells. Superoxide is an ROS produced in mitochondria from activation of STAT3 and NADPH, which can then activate the caspase cascade and cause T cell apoptosis [188]. Reactive nitrogen species such as peroxynitrite, are generated from ROS, and can prevent the binding of molecules to T cell receptors through nitration of receptor amino acids [190].
More recent studies have also been examining the changes in metabolism between naive, effector, and memory CD8+ cell populations. Upon activation by binding at the TCR and the appropriate co-receptors, CD8+ cells preferentially use glycolysis similar to APCs after binding of TLRs or other pathogen-receptor-recognition pathways [191, 192]. The focus on glycolysis is thought to be maintained in part due to HIF-1α stabilization even in the absence of hypoxia [193]. However, when glucose becomes scarce in the tumor microenvironment due to uptake by cancer cells, CD8+ cells switch to oxidative phosphorylation and have increased PD-1 expression [193]. The hypoxic tumor environment prevents the successful transition to oxidative phosphorylation, which can lead to decreased proliferation and increased LAG3 expression in melanoma mouse models [193]. Multiple studies have shown that in hypoxic and glucose deficient states, CD8+ cells switch to fatty acid oxidation via PPAR-α pathway signaling, and that this transition is necessary to prevent T cell exhaustion in this environment [192, 193]. Interestingly, many are studying PPAR antagonists in solid tumors to disrupt similar pathways in cancer cells, but may have negative effects on the increase of fatty acid oxidation in T cells [194].
Future directions
Remarkable progress has been made in the field of immunotherapy. Particularly in melanoma and lung cancers, patients enjoy longer survival, and in rare cases, complete remission in response to immunotherapy [195, 196]. Immunotherapy for pancreatic cancer has not yet shown the same degree of success, but many have chronicled the progress thus far in detailed reviews [197,198,199,200]. Single-agent immunotherapy clinical trials in human PDA have particularly been ineffective, thought in part to be due to decreased PD-1/PD-L1 expression compared to other tumors, but many ongoing studies are examining combinations, including with cytotoxic chemotherapy and radiation [75, 201]. Novel therapies including DC vaccines, chimeric antigen receptor T cells, and miRNA inhibitors are being developed, to name a few [202]. It is likely that effective treatment will take a combination of these therapies and that the heterogeneity of PDA will prevent a one-size-fits-all treatment model.
During the testing of new therapies it is important to replicate the conditions inside the human tumor as much as possible. Hypoxia, as described above, has a significant influence on the immune response to PDA, yet most new therapies are tested in cell cultures in atmospheric oxygen environments. Particularly with T cells, atmospheric oxygen compared to physiologic oxygen can cause lower intracellular NO and decreased CD69, which can cause increased T cell proliferation [203]. Further studies also need to be done regarding regulation of T cell metabolism in hypoxia, including promotion of fatty acid oxidation in glucose and oxygen poor environments. Mouse models, while they do have hypoxic tumor environments, also have limitations. Both genetically engineered and xenograft PDA mouse models do not have as robust a T cell infiltrate as human tumors, despite having more circulating lymphocytes [12, 84, 204]. Other differences between mice and humans include regulators of iNOS in macrophages, induction of Th1 responses in T cells in response to IFN-α, and expression of T cell co-stimulatory molecules such as CD28 [204]. Trends lately have been to “de-sterilize” laboratory animal environments to help their immune systems better reflect those in humans, but barriers remain [205, 206]. Newer organoid models have also been developed that enable a 3-D structure, heterogeneity, and interactions between cell types [207]. Advantages of the organoid model include a longer lifespan than a tumor slice culture model and the ability to expand and use in xenografts unlike a tumor slice culture model [208].
Targeting of the hypoxic environment in solid tumors has also been attempted (Table 1). The most basic of these ideas is to create a tumor microenvironment with increased oxygen or ROS to sensitive the cancer cells to radiation and other therapies, however others argue that anti-oxidants such as N-acetylcysteine that decrease ROS may actually decrease ROS and therefore downstream effects of EMT and immunosuppression [209]. Other methods include pro-drugs that become activated in hypoxia or drugs that hone in on HIF-1α active cells are being developed [20, 210, 211]. Evofosfamide (TH-302), a mustard-based derivative, is a cytotoxic pro-drug that is converted to an active metabolite in hypoxic conditions [211]. Evofosfamide has been used to decrease resistance to radiotherapy in pancreatic cancer, but a recent clinical trial for non-small cell lung cancer using this drug in combination with tarloxotonib, a hypoxia-activated tyrosine kinase inhibitor, was stopped early due to futility [211, 212]. Another drug developed over a decade ago is POP33, a fusion protein that consists of a transduction domain to deliver the drug into cells, a HIF-1α dependent stabilization domain, and a cleaved caspase pro-enzyme [210]. While this showed promise in a mouse model of PDA, there has yet to be a successful human application. While no direct HIF inhibitors have been used in clinical trials for pancreatic adenocarcinoma, therapies that target heat shock protein (HSP) 90 have shown to also lead to HIF degradation and are currently being tested [213, 214].
Major limitations of targeting HIF with inhibitors are the rapid degradation of the molecule, as well as the highly conserved nature of the transcription pathways and potential for negative systemic effects [215, 216]. Indeed several clinical trials have examined targeting STAT3, Notch, PI3K, and Hh pathways in PDA without strongly favorable results [217,218,219,220,221,222]. Several therapies have been developed to target the more downstream effects of hypoxia, however. Countering Warburg metabolism is one strategy, as is encouraging fatty acid oxidation in T cells. CD73 upregulation and subsequent accumulation of immunosuppressive adenosine has been targeted via anti-CD73 antibodies, as well as anti-A2A adenosine receptor inhibition, which is present on T cells [78, 120]. Preventing hypoxic upregulation of MMP-9, used in cancer cell and MDSC migration, via zoledronic acid is also being studied as combination therapy in PDA [223, 224]. Cytokine and chemokines that are upregulated in hypoxia have also been targeted. Immunosuppressive TGF-β and IL-6 are the targets of several clinical trials in PDA [225,226,227,228]. Increased PD-L1 expression is seen on on carcinoma cells in PDA as well as MDSCs and macrophages; this has been targeted with both anti-PD-L1 antibodies and inhibition of pyruvate kinase M2, another molecule that binds in the PD-L1 promoter [229]. Many clinical trials treating PDA that are actively recruiting involve PD-L1 inhibition (Table 1). The CXCR4-CXCL12 axis as well as the CCR5 chemokine with its multiple receptors are also being targeted in PDA [84, 230]. It is possible that combining more developed immunotherapy such as checkpoint and/or chemokine inhibitors with hypoxia targeting may finally overcome the severe immunosuppressive milieu in PDA.
Conclusions
Hypoxia exists in PDA in a heterogeneous manner, and the complex immunosuppressive environment in PDA is exacerbated in hypoxic conditions. Immunotherapy in PDA is not yet successful, likely due to the numerous immunosuppressive pathways upregulated in hypoxia. Tumor heterogeneity will prevent a one-size fits all approach for traditional chemoradiotherapies as well as immunotherapies, but it is important to test in conditions that most resemble the hypoxic human tumor microenvironment.
Abbreviations
- PDA:
-
Pancreatic ductal adenocarcinoma
- TIL:
-
tumor-infiltrating lymphocytes
- HIF:
-
hypoxia inducible factor
- vHL:
-
von Hippel-Lindau protein
- IDO:
-
indoleamine 2,3 dioxygenase
- EMT:
-
epithelial to mesenchymal transition
- MMPs:
-
matrix metalloproteinases
- ECM:
-
extracellular matrix
- VEGF:
-
vascular endothelial growth factor
- TNF:
-
tumor necrosis factor
- TRAIL:
-
TNF related apoptosis inducing ligand
- MHC:
-
major histocompatibility complex
- NO:
-
nitric oxide
- HLA-G:
-
human leukocyte antigen G
- PD-L1:
-
programmed death ligand 1
- MDSCs:
-
myeloid derived suppressor cells
- PSCs:
-
pancreatic stellate cells
- aSMA:
-
alpha smooth muscle actin
- PanIN:
-
pancreatic intraductal neoplasms
- FAP:
-
fibroblast activating protein
- BAFF:
-
B cell activating factor
- Bregs:
-
Regulatory B cells
- Tregs:
-
Regulatory T cells
- GM-CSF:
-
granulocyte–macrophage colony stimulating factor
- APCs:
-
antigen presenting cells
- PAUF:
-
pancreatic adenocarcinoma upregulated factor
- TAMs:
-
tumor-associated macrophages
- iNOS:
-
inducible nitric oxide synthetase
- M-CSFR:
-
macrophage colony-stimulating factor receptor
- DCs:
-
dendritic cells
- TLR:
-
toll-like receptor
- TCR:
-
T cell receptor
- HSP:
-
heat shock protein
- LAG3:
-
lymphocyte activation gene 3
References
Siegel RL, Miller KD, Jemal A (2018) Cancer statistics, 2018. CA Cancer J Clin 68(1):7–30
Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM (2014) Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 74(11):2913–2921
Binenbaum Y, Na’ara S, Gil Z (2015) Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist Updat 23:55–68
Yokoi K, Fidler IJ (2004) Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin Cancer Res 10(7):2299–2306
Sheahan AV, Biankin AV, Parish CR, Khachigian LM (2018) Targeted therapies in the management of locally advanced and metastatic pancreatic cancer: a systematic review. Oncotarget. 9(30):21613–21627
Hidalgo M, Cascinu S, Kleeff J, Labianca R, Löhr J, Neoptolemos J et al (2015) Addressing the challenges of pancreatic cancer: future directions for improving outcomes. Pancreatology. 15(1):8–18
McCormick KA, Coveler AL, Rossi GR, Vahanian NN, Link C, Chiorean EG (2016) Pancreatic cancer: update on immunotherapies and algenpantucel-L. Hum Vaccin Immunother 12(3):563–575
Ahn DH, Ramanathan RK, Bekaii-Saab T (2018) Emerging Therapies and Future Directions in Targeting the Tumor Stroma and Immune System in the Treatment of Pancreatic Adenocarcinoma. Cancers (Basel). 10(6):193
Pillarisetty VG (2014) The pancreatic cancer microenvironment: an immunologic battleground. Oncoimmunology 3(8):e950171
Fukunaga A, Miyamoto M, Cho Y, Murakami S, Kawarada Y, Oshikiri T et al (2004) CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas 28(1):26
Carstens JL, Sampaio P, Yang D, Barua S, Wang H, Rao A et al (2017) Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun. 8:15095
Shibuya KC, Goel VK, Xiong W, Sham JG, Pollack SM, Leahy AM et al (2014) Pancreatic ductal adenocarcinoma contains an effector and regulatory immune cell infiltrate that is altered by multimodal neoadjuvant treatment. PLoS ONE 9(5):e96565
Bailey P, Chang DK, Forget M, Lucas FAS, Alvarez HA, Haymaker C et al (2016) Exploiting the neoantigen landscape for immunotherapy of pancreatic ductal adenocarcinoma. Sci Rep. 6:35848
Barsoum IB, Smallwood CA, Siemens DR, Graham CH (2014) A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 74(3):665–674
Doktorova H, Hrabeta J, Khalil MA, Eckschlager T (2015) Hypoxia-induced chemoresistance in cancer cells: the role of not only HIF-1. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 159(2):166–177
Moreno Roig E, Yaromina A, Houben R, Groot AJ, Dubois L, Vooijs M (2018) Prognostic Role of Hypoxia-Inducible Factor-2α Tumor Cell Expression in Cancer Patients: a Meta-Analysis. Front Oncol 8:224
Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM, et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys 2000 Nov 01,;48(4):919-922
Carreau A, Hafny-Rahbi BE, Matejuk A, Grillon C, Kieda C (2011) Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med 15(6):1239–1253
Lohse I, Lourenco C, Ibrahimov E, Pintilie M, Tsao M, Hedley DW (2014) Assessment of hypoxia in the stroma of patient-derived pancreatic tumor xenografts. Cancers (Basel). 6(1):459–471
Conway JRW, Warren SC, Herrmann D, Murphy KJ, Cazet AS, Vennin C et al (2018) Intravital imaging to monitor therapeutic response in moving hypoxic regions resistant to PI3K pathway targeting in pancreatic cancer. Cell Rep. 23(11):3312–3326
Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer 2008 Mar;8(3):180-192
López-Lázaro M (2008) The warburg effect: why and how do cancer cells activate glycolysis in the presence of oxygen? Anticancer Agents Med Chem 8(3):305–312
Pouysségur J, Dayan F, Mazure NM (2006) Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 441(7092):437–443
Wilson WR, Hay MP (2011) Targeting hypoxia in cancer therapy. Nat Rev Cancer 11(6):393–410
Semenza GL (2012) Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci 33(4):207–214
Jokilehto T, Jaakkola PM (2010) The role of HIF prolyl hydroxylases in tumour growth. J Cell Mol Med 14(4):758–770
Keith B, Johnson RS, Simon MC (2011) HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 12(1):9–22
Criscimanna A, Duan L, Rhodes JA, Fendrich V, Wickline E, Hartman DJ et al (2013) PanIN-specific regulation of Wnt signaling by HIF2α during early pancreatic tumorigenesis. Cancer Res. 73(15):4781–4790
White E (2013) Exploiting the bad eating habits of Ras-driven cancers. Genes Dev 27(19):2065–2071
Guillaumond F, Leca J, Olivares O, Lavaut M, Vidal N, Berthezène P et al (2013) Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci USA 110(10):3919–3924
Guillaumond F, Iovanna JL, Vasseur S (2014) Pancreatic tumor cell metabolism: focus on glycolysis and its connected metabolic pathways. Arch Biochem Biophys. 545:69–73
Gunda V, Kumar S, Dasgupta A, Singh PK (2018) Hypoxia-induced metabolomic alterations in pancreatic cancer cells. Methods Mol Biol 1742:95–105
Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M et al (2007) Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109(9):3812–3819
Pastorek J, Pastorekova S (2015) Hypoxia-induced carbonic anhydrase IX as a target for cancer therapy: from biology to clinical use. Semin Cancer Biol 31:52–64
Li Y, Patel SP, Roszik J, Qin Y (2018) Hypoxia-Driven immunosuppressive metabolites in the tumor microenvironment: new approaches for combinational immunotherapy. Front Immunol 9:1591
Peng Y, Zhang J, Liang W, Tu M, Lu Z, Wei J et al (2014) Elevation of MMP-9 and IDO induced by pancreatic cancer cells mediates natural killer cell dysfunction. BMC Cancer 14:738
Iacobuzio-Donahue CA, Herman JM (2014) Autophagy, p53, and pancreatic cancer. N Engl J Med 370(14):1352–1353
Yang M, Wang H, Hou Y, Tung H, Chiu T, Shan Y (2015) Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol Cancer 14:179
Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M et al (2015) Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524(7565):361–365
Zhu H, Wang D, Zhang L, Xie X, Wu Y, Liu Y et al (2014) Upregulation of autophagy by hypoxia-inducible factor-1α promotes EMT and metastatic ability of CD133 + pancreatic cancer stem-like cells during intermittent hypoxia. Oncol Rep 32(3):935–942
Zhu H, Wang D, Liu Y, Su Z, Zhang L, Chen F et al (2013) Role of the Hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133 + pancreatic cancer stem-like cells. Cancer Cell Int 13(1):119
Rausch V, Liu L, Apel A, Rettig T, Gladkich J, Labsch S et al (2012) Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J Pathol 227(3):325–335
Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J et al (2005) Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell 9(5):617–628
McGinn O, Gupta VK, Dauer P, Arora N, Sharma N, Nomura A et al (2017) Inhibition of hypoxic response decreases stemness and reduces tumorigenic signaling due to impaired assembly of HIF1 transcription complex in pancreatic cancer. Sci Rep 7(1):7872
Yeung TM, Gandhi SC, Bodmer WF (2011) Hypoxia and lineage specification of cell line-derived colorectal cancer stem cells. Proc Natl Acad Sci USA 108(11):4382–4387
Muz B, de la Puente P, Azab F, Azab AK (2015) The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl) 3:83–92
Al-Assar O, Demiciorglu F, Lunardi S, Gaspar-Carvalho MM, McKenna WG, Muschel RM et al (2014) Contextual regulation of pancreatic cancer stem cell phenotype and radioresistance by pancreatic stellate cells. Radiother Oncol 111(2):243–251
Yang M, Wang H, Hou Y, Tung H, Chiu T, Shan Y (2015) Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol Cancer 14:179
Fitzgerald TL, McCubrey JA (2014) Pancreatic cancer stem cells: association with cell surface markers, prognosis, resistance, metastasis and treatment. Adv Biol Regul 56:45–50
Nomura A, Dauer P, Gupta V, McGinn O, Arora N, Majumdar K et al (2016) Microenvironment mediated alterations to metabolic pathways confer increased chemo-resistance in CD133 + tumor initiating cells. Oncotarget 7(35):56324–56337
Keith B, Johnson RS, Simon MC (2011) HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer 12(1):9–22
Liu X, Xiao X, Shou Q, Yan J, Chen L, Fu H et al (2016) Bufalin inhibits pancreatic cancer by inducing cell cycle arrest via the c-Myc/NF-κB pathway. J Ethnopharmacol 193:538–545
Zhang M, Fan H, Li S (2015) Inhibition of c-Myc by 10058-F4 induces growth arrest and chemosensitivity in pancreatic ductal adenocarcinoma. Biomed Pharmacother 73:123–128
Chien W, Lee DH, Zheng Y, Wuensche P, Alvarez R, Wen DL et al (2014) Growth inhibition of pancreatic cancer cells by histone deacetylase inhibitor belinostat through suppression of multiple pathways including HIF, NFkB, and mTOR signaling in vitro and in vivo. Mol Carcinog 53(9):722–735
Jung H, Fattet L, Yang J (2015) Molecular pathways: linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin Cancer Res 21(5):962–968
Marie-Egyptienne DT, Lohse I, Hill RP (2013) Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: potential role of hypoxia. Cancer Lett 341(1):63–72
Sui H, Zhu L, Deng W, Li Q (2014) Epithelial-mesenchymal transition and drug resistance: role, molecular mechanisms, and therapeutic strategies. Oncol Res Treat 37(10):584–589
Shi C, Fan Y, Liu B, Lou W (2013) HIF1 contributes to hypoxia-induced pancreatic cancer cells invasion via promoting QSOX1 expression. Cell Physiol Biochem 32(3):561–568
Zhao X, Gao S, Ren H, Sun W, Zhang H, Sun J et al (2014) Hypoxia-inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-bundling protein fascin. Cancer Res 74(9):2455–2464
Cheng Z, Sun B, Wang S, Gao Y, Zhang Y, Zhou H et al (2011) Nuclear factor-κB-dependent epithelial to mesenchymal transition induced by HIF-1α activation in pancreatic cancer cells under hypoxic conditions. PLoS ONE 6(8):e23752
Hotz B, Arndt M, Dullat S, Bhargava S, Buhr H, Hotz HG (2007) Epithelial to mesenchymal transition: expression of the regulators snail, slug, and twist in pancreatic cancer. Clin Cancer Res 13(16):4769–4776
Yang J, Zhang X, Zhang Y, Zhu D, Zhang L, Li Y et al (2016) HIF-2α promotes epithelial-mesenchymal transition through regulating Twist2 binding to the promoter of E-cadherin in pancreatic cancer. J Exp Clin Cancer Res 35:26
Liu A, Huang C, Cai X, Xu J, Yang D (2016) Twist promotes angiogenesis in pancreatic cancer by targeting miR-497/VEGFA axis. Oncotarget 7(18):25801–25814
Lei J, Ma J, Ma Q, Li X, Liu H, Xu Q et al (2013) Hedgehog signaling regulates hypoxia induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand-independent manner. Mol Cancer 12:66
Harashima N, Takenaga K, Akimoto M, Harada M (2017) HIF-2α dictates the susceptibility of pancreatic cancer cells to TRAIL by regulating survivin expression. Oncotarget 8(26):42887–42900
Cui H, Darmanin S, Natsuisaka M, Kondo T, Asaka M, Shindoh M et al (2007) Enhanced expression of asparagine synthetase under glucose-deprived conditions protects pancreatic cancer cells from apoptosis induced by glucose deprivation and cisplatin. Cancer Res 67(7):3345–3355
Abe T, Toyota M, Suzuki H, Murai M, Akino K, Ueno M et al (2005) Upregulation of BNIP3 by 5-aza-2′-deoxycytidine sensitizes pancreatic cancer cells to hypoxia-mediated cell death. J Gastroenterol 40(5):504–510
Okami J, Simeone DM, Logsdon CD (2004) Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res 64(15):5338–5346
Erkan M, Kleeff J, Esposito I, Giese T, Ketterer K, Büchler MW et al (2005) Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene 24(27):4421–4432
Chand S, O’Hayer K, Blanco FF, Winter JM, Brody JR (2016) The landscape of pancreatic cancer therapeutic resistance mechanisms. Int J Biol Sci 12(3):273–282
Schwartz DL, Bankson JA, Lemos R, Lai SY, Thittai AK, He Y et al (2010) Radiosensitization and stromal imaging response correlates for the HIF-1 inhibitor PX-478 given with or without chemotherapy in pancreatic cancer. Mol Cancer Ther 9(7):2057–2067
Siemens DR, Hu N, Sheikhi AK, Chung E, Frederiksen LJ, Pross H et al (2008) Hypoxia increases tumor cell shedding of MHC class I chain-related molecule: role of nitric oxide. Cancer Res 68(12):4746–4753
Märten A, von Lilienfeld-Toal M, Büchler MW, Schmidt J (2006) Soluble MIC is elevated in the serum of patients with pancreatic carcinoma diminishing gammadelta T cell cytotoxicity. Int J Cancer 119(10):2359–2365
Zhou L, Niu Z, Liang Z, Zhou W, You L, Wang M et al (2015) HLA-G impairs host immune response and predicts poor prognosis in pancreatic cancer. Am J Transl Res 7(10):2036–2044
Birnbaum DJ, Finetti P, Lopresti A, Gilabert M, Poizat F, Turrini O et al (2016) Prognostic value of PDL1 expression in pancreatic cancer. Oncotarget 7(44):71198–71210
Michaels AD, Newhook TE, Adair SJ, Morioka S, Goudreau BJ, Nagdas S et al (2018) CD47 Blockade as an Adjuvant Immunotherapy for Resectable Pancreatic Cancer. Clin Cancer Res 24(6):1415–1425
Soto-Pantoja DR, Terabe M, Ghosh A, Ridnour LA, DeGraff WG, Wink DA et al (2014) CD47 in the tumor microenvironment limits cooperation between antitumor T-cell immunity and radiotherapy. Cancer Res 74(23):6771–6783
Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S et al (2015) Adenosine receptor 2A blockade increases the efficacy of anti-PD-1 through enhanced antitumor T-cell responses. Cancer Immunol Res 3(5):506–517
Antonioli L, Pacher P, Vizi ES, Haskó G (2013) CD39 and CD73 in immunity and inflammation. Trends Mol Med. 19(6):355–367
Masamune A, Shimosegawa T (2015) Pancreatic stellate cells: a dynamic player of the intercellular communication in pancreatic cancer. Clin Res Hepatol Gastroenterol 39(Suppl 1):98
Masamune A, Shimosegawa T (2013) Pancreatic stellate cells–multi-functional cells in the pancreas. Pancreatology 13(2):102–105
Erkan M, Adler G, Apte MV, Bachem MG, Buchholz M, Detlefsen S et al (2012) StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut 61(2):172–178
Bynigeri RR, Jakkampudi A, Jangala R, Subramanyam C, Sasikala M, Rao GV et al (2017) Pancreatic stellate cell: Pandora’s box for pancreatic disease biology. World J Gastroenterol 23(3):382–405
Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS et al (2013) Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA 110(50):20212
Shi M, Yu D, Chen Y, Zhao C, Zhang J, Liu Q et al (2012) Expression of fibroblast activation protein in human pancreatic adenocarcinoma and its clinicopathological significance. World J Gastroenterol 18(8):840–846
Lee H, Mullins SR, Franco-Barraza J, Valianou M, Cukierman E, Cheng JD (2011) FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer 11:245
Jiang G, Xu W, Du J, Zhang K, Zhang Q, Wang X et al (2016) The application of the fibroblast activation protein α-targeted immunotherapy strategy. Oncotarget 7(22):33472–33482
Keklikoglou I, Kadioglu E, Bissinger S, Langlois B, Bellotti A, Orend G et al (2018) Periostin limits tumor response to VEGFA inhibition. Cell Rep 22(10):2530–2540
Masamune A, Watanabe T, Kikuta K, Shimosegawa T (2009) Roles of pancreatic stellate cells in pancreatic inflammation and fibrosis. Clin Gastroenterol Hepatol 7(11 Suppl):48
Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS et al (2013) Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res 73(10):3007–3018
Erkan M, Reiser-Erkan C, Michalski CW, Deucker S, Sauliunaite D, Streit S et al (2009) Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 11(5):497–508
Spivak-Kroizman TR, Hostetter G, Posner R, Aziz M, Hu C, Demeure MJ et al (2013) Hypoxia triggers hedgehog-mediated tumor-stromal interactions in pancreatic cancer. Cancer Res 73(11):3235–3247
Bennewith KL, Huang X, Ham CM, Graves EE, Erler JT, Kambham N et al (2009) The role of tumor cell-derived connective tissue growth factor (CTGF/CCN2) in pancreatic tumor growth. Cancer Res 69(3):775–784
Masamune A, Kikuta K, Watanabe T, Satoh K, Hirota M, Shimosegawa T (2008) Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am J Physiol Gastrointest Liver Physiol 295(4):709
Moir JAG, Mann J, White SA (2015) The role of pancreatic stellate cells in pancreatic cancer. Surg Oncol 24(3):232–238
Nelson BH (2010) CD20 + B cells: the other tumor-infiltrating lymphocytes. J Immunol 185(9):4977–4982
Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB et al (2016) IL35-producing b cells promote the development of pancreatic neoplasia. Cancer Discov 6(3):247–255
Schwartz M, Zhang Y, Rosenblatt JD (2016) B cell regulation of the anti-tumor response and role in carcinogenesis. J Immunother Cancer 4:40
Castino GF, Cortese N, Capretti G, Serio S, Di Caro G, Mineri R et al (2016) Spatial distribution of B cells predicts prognosis in human pancreatic adenocarcinoma. Oncoimmunology 5(4):e1085147
Koizumi M, Hiasa Y, Kumagi T, Yamanishi H, Azemoto N, Kobata T et al (2013) Increased B cell-activating factor promotes tumor invasion and metastasis in human pancreatic cancer. PLoS ONE 8(8):e71367
Reis ST, Leite KRM, Piovesan LF, Pontes-Junior J, Viana NI, Abe DK et al (2012) Increased expression of MMP-9 and IL-8 are correlated with poor prognosis of Bladder Cancer. BMC Urol 12:18
Tadmor T, Zhang Y, Cho H, Podack ER, Rosenblatt JD (2011) The absence of B lymphocytes reduces the number and function of T-regulatory cells and enhances the anti-tumor response in a murine tumor model. Cancer Immunol Immunother 60(5):609–619
Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B et al (2016) Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov 6(3):270–285
Martin SK, Diamond P, Williams SA, To LB, Peet DJ, Fujii N et al (2010) Hypoxia-inducible factor-2 is a novel regulator of aberrant CXCL12 expression in multiple myeloma plasma cells. Haematologica 95(5):776–784
Piovan E, Tosello V, Indraccolo S, Masiero M, Persano L, Esposito G et al (2007) Differential regulation of hypoxia-induced CXCR4 triggering during B-cell development and lymphomagenesis. Cancer Res 67(18):8605–8614
Lee KE, Spata M, Bayne LJ, Buza EL, Durham AC, Allman D et al (2016) Hif1a deletion reveals pro-neoplastic function of B cells in pancreatic neoplasia. Cancer Discov 6(3):256–269
Ostrand-Rosenberg S (2010) Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother 59(10):1593–1600
Casazza A, Di Conza G, Wenes M, Finisguerra V, Deschoemaeker S, Mazzone M (2014) Tumor stroma: a complexity dictated by the hypoxic tumor microenvironment. Oncogene 33(14):1743–1754
Xu Q, Wang Z, Chen X, Duan W, Lei J, Zong L et al (2015) Stromal-derived factor-1α/CXCL12-CXCR4 chemotactic pathway promotes perineural invasion in pancreatic cancer. Oncotarget 6(7):4717–4732
Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ et al (2012) Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 21(6):822–835
Porembka MR, Mitchem JB, Belt BA, Hsieh C, Lee H, Herndon J et al (2012) Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol Immunother 61(9):1373–1385
Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM et al (2014) Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 63(11):1769–1781
Gabrilovich DI, Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9(3):162–174
Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S (2007) Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol 179(2):977–983
Yu J, Du W, Yan F, Wang Y, Li H, Cao S et al (2013) Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol 190(7):3783–3797
Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S (2010) Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res 70(1):68–77
Leone RD, Emens LA (2018) Targeting adenosine for cancer immunotherapy. J Immunother Cancer 6(1):57
Song J, Lee J, Kim J, Jo S, Kim YJ, Baek JE et al (2016) Pancreatic adenocarcinoma up-regulated factor (PAUF) enhances the accumulation and functional activity of myeloid-derived suppressor cells (MDSCs) in pancreatic cancer. Oncotarget 7(32):51840
Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S (2009) Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4 + and CD8 + T cells. J Immunol 183(2):937–944
Li J, Wang L, Chen X, Li L, Li Y, Ping Y et al (2017) CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 6(6):e1320011
Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P et al (2014) PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med 211(5):781–790
Chiu DK, Xu IM, Lai RK, Tse AP, Wei LL, Koh H et al (2016) Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 64(3):797–813
Chiu DK, Tse AP, Xu IM, Cui JD, Lai RK, Li LL et al (2017) Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nature Communications 8:517
Corzo CA, Condamine T, Lu L, Cotter MJ, Youn J, Cheng P et al (2010) HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med 207(11):2439–2453
Cui R, Yue W, Lattime EC, Stein MN, Xu Q, Tan X (2016) Targeting tumor-associated macrophages to combat pancreatic cancer. Oncotarget 7(31):50735–50754
Chang Y, Hsu T, Lin H, Chio C, Chiu AW, Chen N et al (2004) Modulation of macrophage differentiation and activation by decoy receptor 3. J Leukoc Biol 75(3):486–494
Gajewski TF, Schreiber H, Fu Y (2013) Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14(10):1014–1022
Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K et al (2014) The cellular and molecular origin of tumor-associated macrophages. Science 344(6186):921–925
Lao L, Fan S, Song E (2017) Tumor associated macrophages as therapeutic targets for breast cancer. Adv Exp Med Biol 1026:331–370
Zhong X, Chen B, Yang Z (2018) The role of tumor-associated macrophages in colorectal carcinoma progression. Cell Physiol Biochem 45(1):356–365
Krishnan V, Schaar B, Tallapragada S, Dorigo O (2018) Tumor associated macrophages in gynecologic cancers. Gynecol Oncol 149(1):205–213
Helm O, Held-Feindt J, Grage-Griebenow E, Reiling N, Ungefroren H, Vogel I et al (2014) Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis. Int J Cancer 135(4):843–861
Arnold JN, Magiera L, Kraman M, Fearon DT (2014) Tumoral immune suppression by macrophages expressing fibroblast activation protein-α and heme oxygenase-1. Cancer Immunol Res 2(2):121–126
Weizman N, Krelin Y, Shabtay-Orbach A, Amit M, Binenbaum Y, Wong RJ et al (2014) Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 33(29):3812–3819
Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y et al (2013) Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer 108(4):914–923
Henze A, Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Invest 2016 Oct 03,;126(10):3672-3679
Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M et al (2013) Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 24(6):695–709
Zhang J, Cao J, Ma S, Dong R, Meng W, Ying M et al (2014) Tumor hypoxia enhances Non-Small Cell Lung Cancer metastasis by selectively promoting macrophage M2 polarization through the activation of ERK signaling. Oncotarget 5(20):9664–9677
Guo X, Xue H, Shao Q, Wang J, Guo X, Chen X et al (2016) Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSFR. Oncotarget 7(49):80521–80542
Chouaib S, Noman MZ, Kosmatopoulos K, Curran MA (2017) Hypoxic stress: obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 36(4):439
Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL et al (2005) HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest 115(7):1806–1815
Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG et al (2010) Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res 70(19):7465–7475
Rodriguez PC, Quiceno DG, Ochoa AC (2007) l-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 109(4):1568–1573
Ye L, Chen W, Bai X, Xu X, Zhang Q, Xia X et al (2016) Hypoxia-Induced Epithelial-to-Mesenchymal Transition in Hepatocellular Carcinoma Induces an Immunosuppressive Tumor Microenvironment to Promote Metastasis. Cancer Res 76(4):818–830
Barsoum IB, Smallwood CA, Siemens DR, Graham CH (2014) A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res 74(3):665–674
Fingleton B, Vargo-Gogola T, Crawford HC, Matrisian LM (2001) Matrilysin [MMP-7] expression selects for cells with reduced sensitivity to apoptosis. Neoplasia 3(6):459–468
Fainaru O, Almog N, Yung CW, Nakai K, Montoya-Zavala M, Abdollahi A et al (2010) Tumor growth and angiogenesis are dependent on the presence of immature dendritic cells. FASEB J 24(5):1411–1418
Bellone G, Carbone A, Smirne C, Scirelli T, Buffolino A, Novarino A, et al. Cooperative induction of a tolerogenic dendritic cell phenotype by cytokines secreted by pancreatic carcinoma cells. J Immunol 2006 Sep 01,;177(5):3448-3460
Gabrilovich D (2004) Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol 4(12):941–952
Bharadwaj U, Li M, Zhang R, Chen C, Yao Q (2007) Elevated interleukin-6 and G-CSF in human pancreatic cancer cell conditioned medium suppress dendritic cell differentiation and activation. Cancer Res 67(11):5479–5488
Alfaro C, Suárez N, Martínez-Forero I, Palazón A, Rouzaut A, Solano S et al (2011) Carcinoma-derived interleukin-8 disorients dendritic cell migration without impairing T-cell stimulation. PLoS ONE 6(3):e17922
Yamamoto T, Yanagimoto H, Satoi S, Toyokawa H, Yamao J, Kim S et al (2012) Circulating myeloid dendritic cells as prognostic factors in patients with pancreatic cancer who have undergone surgical resection. J Surg Res 173(2):299–308
Hirooka S, Yanagimoto H, Satoi S, Yamamoto T, Toyokawa H, Yamaki S et al (2011) The role of circulating dendritic cells in patients with unresectable pancreatic cancer. Anticancer Res 31(11):3827–3834
Wiedemann GM, Knott MML, Vetter VK, Rapp M, Haubner S, Fesseler J et al (2016) Cancer cell-derived IL-1α induces CCL22 and the recruitment of regulatory T cells. Oncoimmunology 5(9):e1175794
Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E et al (2005) Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4 + CD25 + regulatory T cell proliferation. J Exp Med 202(7):919–929
Zhou M, Chen J, Zhou L, Chen W, Ding G, Cao L (2014) Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell Immunol 292(1–2):65–69
Kang TH, Kim YS, Kim S, Yang B, Lee J, Lee H et al (2015) Pancreatic adenocarcinoma upregulated factor serves as adjuvant by activating dendritic cells through stimulation of TLR4. Oncotarget 6(29):27751–27762
Du J, Wang J, Tan G, Cai Z, Zhang L, Tang B et al (2012) Aberrant elevated microRNA-146a in dendritic cells (DC) induced by human pancreatic cancer cell line BxPC-3-conditioned medium inhibits DC maturation and activation. Med Oncol 29(4):2814–2823
Jantsch J, Chakravortty D, Turza N, Prechtel AT, Buchholz B, Gerlach RG et al (2008) Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol 180(7):4697–4705
Yilmaz A, Ratka J, Rohm I, Pistulli R, Goebel B, Asadi Y et al (2016) Decrease in circulating plasmacytoid dendritic cells during short-term systemic normobaric hypoxia. Eur J Clin Invest 46(2):115–122
Wobben R, Hüsecken Y, Lodewick C, Gibbert K, Fandrey J, Winning S (2013) Role of hypoxia inducible factor-1α for interferon synthesis in mouse dendritic cells. Biol Chem 394(4):495–505
Filippi I, Morena E, Aldinucci C, Carraro F, Sozzani S, Naldini A (2014) Short-term hypoxia enhances the migratory capability of dendritic cell through HIF-1α and PI3K/Akt pathway. J Cell Physiol 229(12):2067–2076
Ogino T, Onishi H, Suzuki H, Morisaki T, Tanaka M, Katano M (2012) Inclusive estimation of complex antigen presentation functions of monocyte-derived dendritic cells differentiated under normoxia and hypoxia conditions. Cancer Immunol Immunother 61(3):409–424
Köhler T, Reizis B, Johnson RS, Weighardt H, Förster I (2012) Influence of hypoxia-inducible factor 1α on dendritic cell differentiation and migration. Eur J Immunol 42(5):1226–1236
Yang M, Ma C, Liu S, Sun J, Shao Q, Gao W et al (2009) Hypoxia skews dendritic cells to a T helper type 2-stimulating phenotype and promotes tumour cell migration by dendritic cell-derived osteopontin. Immunology 128(1 Suppl):237
Yang M, Liu Y, Ren G, Shao Q, Gao W, Sun J et al (2015) Increased expression of surface CD44 in hypoxia-DCs skews helper T cells toward a Th2 polarization. Sci Rep 5:13674
Yang M, Ma C, Liu S, Shao Q, Gao W, Song B et al (2010) HIF-dependent induction of adenosine receptor A2b skews human dendritic cells to a Th2-stimulating phenotype under hypoxia. Immunol Cell Biol 88(2):165–171
Naldini A, Morena E, Pucci A, Miglietta D, Riboldi E, Sozzani S et al (2012) Hypoxia affects dendritic cell survival: role of the hypoxia-inducible factor-1α and lipopolysaccharide. J Cell Physiol 227(2):587–595
Amedei A, Niccolai E, Benagiano M, Della Bella C, Cianchi F, Bechi P et al (2013) Ex vivo analysis of pancreatic cancer-infiltrating T lymphocytes reveals that ENO-specific Tregs accumulate in tumor tissue and inhibit Th1/Th17 effector cell functions. Cancer Immunol Immunother 62(7):1249–1260
Kim H, Cantor H (2014) CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol Res 2(2):91–98
Punt S, Langenhoff JM, Putter H, Fleuren GJ, Gorter A, Jordanova ES (2015) The correlations between IL-17 vs Th17 cells and cancer patient survival: a systematic review. Oncoimmunology 4(2):e984547
Liao Y, Wang B, Huang Z, Shi M, Yu X, Zheng L et al (2013) Increased circulating Th17 cells after transarterial chemoembolization correlate with improved survival in stage III hepatocellular carcinoma: a prospective study. PLoS ONE 8(4):e60444
Bengsch F, Knoblock DM, Liu A, McAllister F, Beatty GL (2017) CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol Immunother 66(12):1609–1617
Tan MCB, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE et al (2009) Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol 182(3):1746–1755
Grage-Griebenow E, Jerg E, Gorys A, Wicklein D, Wesch D, Freitag-Wolf S et al (2014) L1CAM promotes enrichment of immunosuppressive T cells in human pancreatic cancer correlating with malignant progression. Mol Oncol 8(5):982–997
Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ et al (2012) CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med 4(134):134ra62
Gnanaprakasam JNR, Sherman JW, Wang R (2017) MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev 35:63–70
Hsu T, Lai M (2018) Hypoxia-inducible factor 1α plays a predominantly negative role in regulatory T cell functions. J Leukoc Biol 104(5):911–918
Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P et al (2012) Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci USA 109(41):2784
Dang EV, Barbi J, Yang H, Jinasena D, Yu H, Zheng Y et al (2011) Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146(5):772–784
Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang L et al (2011) Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 475(7355):226–230
Bakshi RK, Cox MA, Zajac AJ. Cytotoxic T Lymphocytes. Encyclopedia of Medical Immunology: Springer: New York; 2014. p. 332–2
Hoyer S, Prommersberger S, Pfeiffer IA, Schuler-Thurner B, Schuler G, Dörrie J et al (2014) Concurrent interaction of DCs with CD4(+) and CD8(+) T cells improves secondary CTL expansion: it takes three to tango. Eur J Immunol 44(12):3543–3559
Oberg H, Peipp M, Kellner C, Sebens S, Krause S, Petrick D et al (2014) Novel bispecific antibodies increase γδ T-cell cytotoxicity against pancreatic cancer cells. Cancer Res 74(5):1349–1360
Palazón A, Martínez-Forero I, Teijeira A, Morales-Kastresana A, Alfaro C, Sanmamed MF et al (2012) The HIF-1α hypoxia response in tumor-infiltrating T lymphocytes induces functional CD137 (4-1BB) for immunotherapy. Cancer Discov 2(7):608–623
Caldwell CC, Kojima H, Lukashev D, Armstrong J, Farber M, Apasov SG et al (2001) Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J Immunol 167(11):6140–6149
Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E et al (2013) Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol 14(11):1173–1182
Hildeman DA, Mitchell T, Teague TK, Henson P, Day BJ, Kappler J et al (1999) Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity 10(6):735–744
Takeichi T, Mocevicius P, Deduchovas O, Salnikova O, Castro-Santa E, Büchler MW et al (2012) αL β2 integrin is indispensable for CD8 + T-cell recruitment in experimental pancreatic and hepatocellular cancer. Int J Cancer 130(9):2067–2076
Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L et al (2007) Altered recognition of antigen is a mechanism of CD8 + T cell tolerance in cancer. Nat Med 13(7):828–835
Nicoli F, Paul S, Appay V (2018) Harnessing the induction of CD8 + T-Cell responses through metabolic regulation by pathogen-recognition-receptor triggering in antigen presenting cells. Front Immunol 9:2372
Nicoli F, Papagno L, Frere JJ, Cabral-Piccin MP, Clave E, Gostick E et al (2018) Naïve CD8 + T-Cells engage a versatile metabolic program upon activation in humans and differ energetically from memory CD8 + T-cells. Front Immunol 9:2736
Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A et al (2017) Enhancing CD8 + T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 32(3):391
Gou Q, Gong X, Jin J, Shi J, Hou Y (2017) Peroxisome proliferator-activated receptors (PPARs) are potential drug targets for cancer therapy. Oncotarget 8(36):60704–60709
Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R et al (2016) Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA 315(15):1600–1609
Shroff GS, de Groot PM, Papadimitrakopoulou VA, Truong MT, Carter BW (2018) Targeted therapy and immunotherapy in the treatment of non-small cell lung cancer. Radiol Clin North Am 56(3):485–495
Bauer C, Kühnemuth B, Duewell P, Ormanns S, Gress T, Schnurr M (2016) Prevailing over T cell exhaustion: New developments in the immunotherapy of pancreatic cancer. Cancer Lett 381(1):259–268
Kotteas E, Saif MW, Syrigos K (2016) Immunotherapy for pancreatic cancer. J Cancer Res Clin Oncol 142(8):1795–1805
Kunk PR, Bauer TW, Slingluff CL, Rahma OE (2016) From bench to bedside a comprehensive review of pancreatic cancer immunotherapy. J Immunother Cancer 4:14
Foley K, Kim V, Jaffee E, Zheng L (2016) Current progress in immunotherapy for pancreatic cancer. Cancer Lett 381(1):244–251
Zheng L (2017) PD-L1 expression in pancreatic cancer. J Natl Cancer Inst. 109:6
Chiorean EG, Coveler AL (2015) Pancreatic cancer: optimizing treatment options, new, and emerging targeted therapies. Drug Des Devel Ther 9:3529
Atkuri KR, Herzenberg LA, Niemi A, Cowan T, Herzenberg LA (2007) Importance of culturing primary lymphocytes at physiological oxygen levels. Proc Natl Acad Sci USA 104(11):4547–4552
Mestas J, Hughes CCW (2004) Of mice and not men: differences between mouse and human immunology. J Immunol 172(5):2731–2738
Tao L, Reese TA (2017) Making Mouse Models That Reflect Human Immune Responses. Trends Immunol. 38(3):181–193
Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA et al (2016) Recapitulating adult human immune traits in laboratory mice by normalizing environment. Nature 532(7600):512
Boj SF, Hwang C, Baker LA, Chio IIC, Engle DD, Corbo V et al (2015) Organoid models of human and mouse ductal pancreatic cancer. Cell 160(1–2):324–338
Tsai S, McOlash L, Palen K, Johnson B, Duris C, Yang Q et al (2018) Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 18(1):335
Shimojo Y, Akimoto M, Hisanaga T, Tanaka T, Tajima Y, Honma Y et al (2013) Attenuation of reactive oxygen species by antioxidants suppresses hypoxia-induced epithelial-mesenchymal transition and metastasis of pancreatic cancer cells. Clin Exp Metastasis 30(2):143–154
Kizaka-Kondoh S, Itasaka S, Zeng L, Tanaka S, Zhao T, Takahashi Y et al (2009) Selective killing of hypoxia-inducible factor-1-active cells improves survival in a mouse model of invasive and metastatic pancreatic cancer. Clin Cancer Res 15(10):3433–3441
Hajj C, Russell J, Hart CP, Goodman KA, Lowery MA, Haimovitz-Friedman A et al (2017) A combination of radiation and the hypoxia-activated prodrug evofosfamide (TH-302) is efficacious against a human orthotopic pancreatic tumor model. Transl Oncol 10(5):760–765
Salem A, Asselin M, Reymen B, Jackson A, Lambin P, West CML et al (2018) Targeting hypoxia to improve non-small cell lung cancer outcome. J Natl Cancer Inst 110:1
Belalcazar A, Shaib WL, Farren MR, Zhang C, Chen Z, Yang L et al (2017) Inhibiting heat shock protein 90 and the ubiquitin-proteasome pathway impairs metabolic homeostasis and leads to cell death in human pancreatic cancer cells. Cancer 123(24):4924–4933
Bobrov E, Skobeleva N, Restifo D, Beglyarova N, Cai KQ, Handorf E et al (2017) Targeted delivery of chemotherapy using HSP90 inhibitor drug conjugates is highly active against pancreatic cancer models. Oncotarget 8(3):4399–4409
Wigerup C, Påhlman S, Bexell D (2016) Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther 08(164):152–169
Li Y, Patel SP, Roszik J, Qin Y (2018) Hypoxia-driven immunosuppressive metabolites in the tumor microenvironment: new approaches for combinational immunotherapy. Front Immunol 9:1591
Mosquera C, Maglic D, Zervos EE (2016) Molecular targeted therapy for pancreatic adenocarcinoma: a review of completed and ongoing late phase clinical trials. Cancer Genet 209(12):567–581
De Jesus-Acosta A, Laheru D, Maitra A, Arcaroli J, Rudek MA, Dasari A et al (2014) A phase II study of the gamma secretase inhibitor RO4929097 in patients with previously treated metastatic pancreatic adenocarcinoma. Invest New Drugs 32(4):739–745
O’Neil BH, Scott AJ, Ma WW, Cohen SJ, Leichman L, Aisner DL et al (2015) A phase II/III randomized study to compare the efficacy and safety of rigosertib plus gemcitabine versus gemcitabine alone in patients with previously untreated metastatic pancreatic cancer. Ann Oncol 26(9):1923–1929
Ma WW, Messersmith WA, Dy GK, Weekes CD, Whitworth A, Ren C et al (2012) Phase I study of Rigosertib, an inhibitor of the phosphatidylinositol 3-kinase and Polo-like kinase 1 pathways, combined with gemcitabine in patients with solid tumors and pancreatic cancer. Clin Cancer Res 18(7):2048–2055
Kim EJ, Sahai V, Abel EV, Griffith KA, Greenson JK, Takebe N et al (2014) Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin Cancer Res 20(23):5937–5945
Ko AH, LoConte N, Tempero MA, Walker EJ, Kate Kelley R, Lewis S et al (2016) A phase i study of FOLFIRINOX Plus IPI-926, a hedgehog pathway inhibitor, for advanced pancreatic adenocarcinoma. Pancreas 45(3):370–375
Vitellius C, Fizanne L, Menager-Tabourel E, Nader J, Baize N, Laly M et al (2018) The combination of everolimus and zoledronic acid increase the efficacy of gemcitabine in a mouse model of pancreatic adenocarcinoma. Oncotarget 9(46):28069–28082
Sanford DE, Porembka MR, Panni RZ, Mitchem JB, Belt BA, Plambeck-Suess SM et al (2013) A study of zoledronic acid as neo-adjuvant, perioperative therapy in patients with resectable pancreatic ductal adenocarcinoma. J Cancer Ther 4(3):797–803
Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S et al (2018) IL-6 and PD-L1 antibody blockade combination therapy reduces tumor progression in murine models of pancreatic cancer. Gut 67(2):320
Goumas FA, Holmer R, Egberts J, Gontarewicz A, Heneweer C, Geisen U et al (2015) Inhibition of IL-6 signaling significantly reduces primary tumor growth and recurrencies in orthotopic xenograft models of pancreatic cancer. International Journal of Cancer 137(5):1035–1046
Pu N, Zhao G, Gao S, Cui Y, Xu Y, Lv Y et al (2018) Neutralizing TGF-β promotes anti-tumor immunity of dendritic cells against pancreatic cancer by regulating T lymphocytes. Central-European Journal of Immunology 43(2):123
Soares KC, Rucki AA, Kim V, Foley K, Solt S, Wolfgang CL et al (2015) TGF-β blockade depletes T regulatory cells from metastatic pancreatic tumors in a vaccine dependent manner. Oncotarget 6(40):43005
Palsson-McDermott EM, Dyck L, Zasłona Z, Menon D, McGettrick AF, Mills KHG et al (2017) Pyruvate kinase M2 is required for the expression of the immune checkpoint PD-L1 in immune cells and tumors. Front Immunol 8:1300
Tan MCB, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE et al (2009) Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol 182(3):1746–1755
Authors’ contributions
SKD compiled the references and wrote the manuscript. KMS and KPL edited the manuscript. VGP edited the manuscript and provided concept formation. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to acknowledge the support of Tumor Immunology and Microenvironment group at the University of Washington.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
None.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Daniel, S.K., Sullivan, K.M., Labadie, K.P. et al. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma. Clin Trans Med 8, 10 (2019). https://doi.org/10.1186/s40169-019-0226-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40169-019-0226-9