
Abstract
The establishment, remodeling and maintenance of tissular architecture during animal development, and even across juvenile to adult life, are deeply regulated by a delicate interplay of extracellular signals, cell membrane receptors and intracellular signal messengers. It is well known that cell adhesion molecules (cell-cell and cell-extracellular matrix) play a critical role in these processes. Particularly, adherens junctions (AJs) mediated by E-cadherin and catenins determine cell-cell contact survival and epithelia function. Consequently, this review seeks to encompass the complex and prolific knowledge about E-cadherin roles during physiological and pathological states, particularly focusing on the influence exerted by the thyroid hormone (TH).
Similar content being viewed by others

Background
Since the early 70s, prolific and significant research and hypotheses subscribe to the theory that unicellular to multicellular transition has occurred more than once during evolution through different genetic mechanisms for each kingdom in the tree of life. Particularly, cadherin and integrin cell adhesion molecules played a crucial role in metazoan transition [78, 171].
In addition, the interpretation of the cellular role of cadherin has changed over time, from a static membrane protein involved in cell-cell adhesion mechanic events to a membrane receptor involved in very dynamic intracellular signal transductions. These processes would be mediated not only from cell-cell junction platforms, but also from single receptors [29, 189, 216].
In different physiological contexts during development and adulthood, this adhesion is highly plastic, suffering remodeling due to numerous and complex signaling cascades finely coordinated in time and space. Thus, in pathological states such as cancer, the configuration of this adhesion is altered by genetic and epigenetic changes, resulting in modifications in the signaling pathways, loss of inhibition by contact, cell migration, and altered stromal interactions. A key group of these cell-cell adhesion molecules is the superfamily of cadherins, whose prototypical member is E-cadherin. This molecule was found for the first time in epithelial tissues [177, 215] and has been characterized as a potent suppressor of invasion and metastasis in studies dating back to 1990 (reviewed by [199]). E-cadherin plays a key role in determining cell polarity and differentiation, and thereby in the establisment and maintainance of tissue homeostasis. It is also important during development and during the whole life cycle of pluricelular organisms, mainly metazoans.
In this work, four major aspects bring E-cadherin into focus. First, epithelia are composed of cell phenotypes which exhibit the maximum polarity and whose epithelial identity is primarily specified by E-cadherin. Second, E-cadherin is involved in numerous processes such as, cell phenotype selection and migration, and morphogenetic movements occurring during metazoan development [26, 192, 193, 196]. Third, epithelia are usually located in direct contact with mutagenic and/or carcinogenic agents, so in men, 85–90 % of the cases can be attributed to epithelial cancers. Lastly, the complexity of in vivo animal studies has partly concealed the full spectrum of E-cadherin functions. Owing to the latter, the work-goal focuses on E-cadherin control mediated by thyroid hormones in a scarcely studied physio-pathological scenario.
Review
E-cadherin function during metazoan development
Cadherins are a huge family of Ca2+-dependent cell surface adhesion glycoproteins, which are differentially expressed in tissues and ontogeny among broad groups of organisms, from unicellular choanoflagellates to invertebrates and all vertebrates classes. Therefore, cadherin evolution has special relevance for understanding metazoan origins [2, 62, 66, 77, 78, 80, 140, 143, 171, 192, 196].
Classical cadherins, like E(epithelial)-cadherin, bind to β-catenin via the cytoplasmic domain (CD) and promote cell-cell adhesion [174] or are rapidly degraded [32, 76, 86]. As development progresses, cadherins establish and maintain AJs through adaptor proteins to the cytoskeleton [63, 64, 129, 141]. These complex structural units are called cadhesomes [216], and act integrating signals from extracellular and intracellular environments [19, 62, 85–87, 138]. In vertebrates, E-cadherin mRNA and protein are maternally expressed, beginning their zygotic expression from 2-cell embryo or gastrulation [8, 9, 34, 91, 114, 191]. Although E-cadherin is principally expressed in the epidermal ectoderm [191], it has also been found in derivatives of neuroectoderm, mesoderm and endoderm [39, 85, 87]. In some species, embrionary E-cadherin is necessary for 8-cell stage compaction, for trophectoderm expansion [7, 108], and for maintaining epiboly integrity [115]. Later during development, E-cadherin contributes to the morphogenesis of endodermal and neuroectodermal derivates [83], and digestive tract, kidney and skin remodeling [85–87, 179, 197]. In invertebrates, a vertebrate E-cadherin homolog is critical for cell rearrangement [198] and packing geometry [35].
Some studies suggest that three modern cadherin families —lefftyrins, coherins, and hedglings— were present in the last common ancestor of choanoflagellates and metazoans, and they may have evolved to diverse metazoan signaling and adhesion gene families [140]. Several cadherin-analogous functions have been hypothesized in non metazoan unicellular lineages in spite of the fact that cadherins are undocumented in choanoflagellates. These organisms express members of key cell signaling and adhesion protein families that were previously thought to be exclusively found in animals [97, 98]. By studying cell differentiation and development in some choanoflagellates, it may be possible to characterize some ancestral functions (bacterial prey adhesive capture, attachment to environmental substrates, gamete recognition and hormonal signaling) of proteins that regulate animal development [46, 47, 98]. Interesting, numerous studies show interaction processes between bacteria or yeast and metazoan cadherins promoting host invasion [17, 36]. In addition, other studies inform that no receptors or ligands were identified from the nuclear hormone receptor, WNT and TGF-β signaling pathways [98]. As we will explain later, the absence of nuclear hormone receptors and ligands, and molecules involved in Wnt signaling pathways suggest combined features and roles in a single molecule-type: the metazoan cadherins.
Taken together, these data and our results on TH-dependent E-cadherin control [56, 85], support the idea that classical cadherins could have emerged in the premetazoan to metazoan transition to respond to signaling mediated by nuclear hormone receptors and cytoskeleton connexions. Indeed, the development of complex multicellular organisms requires a genetic program regulated by nuclear hormone receptor signaling and dynamic cytoskeleton reorganizations.
E-cadherin structure, function and dysfunction
In their mature state, most cadherins have three segments: the extracellular (EC), trans-membrane(TM) and cytoplasmic (CD) domains (Fig. 1). The EC domain is mainly involved in homophilic recognition and Ca2+-dependent adhesion mechanisms, and it is constituted by a variable amount of cadherin type-repeats (ECn). Each one has a β-sandwich Ig-like folding that contains conserved Ca2+-coordinating regions [15]. Each ECn is enumerated from the outermost N-terminal EC1 to the closest to membrane EC1 + n. The CD domain exhibits the highest variability among the cadherin subfamilies [66, 77] and binds to different intracellular proteins giving both functional connection to the cytoskeleton [15] and diversity [66, 140].

Structure and regulation of E-cadherin gene and protein. Scheme represents human chromosome-16q22.1 cadherin cluster (CDH1/E-cadherin and CDH3/P-cadherin locus), and a regulation model both CDH1 locus and protein. CDH1 locus has 16 exons (black bars), cis-regulatory elements (DHSs, vertical arrows), transcription and translation start sites (small horizontal arrows), several enhancer sequences (green boxes and arrows) ─alternative intron 2-independent gene activation in late embryogenesis (alt), specific expression in (brain) or endoderm (enh), or ectoderm/tissues (tse1-4)─, downregulated sequences (red arrows) ─E-boxes and brain-specific silencer (sil)─, and yolk sac-specific elements outside of intron 2. Locus control region (LCR) could activate or downregulate gene activity (purple arrow). CpG methylation and E-box-bounded specific repressors (Snail1, slug, E47, δEF1/ZEB1 SIP1/ZEB2) control the promoter, as well as poly-ADP ribosylation and repressor inhibition (miR-200). MIR and MaLR regulatory-associated repetitive elements (light blue arrow) were bioinformatically found in introns 2 and 3, and are involved in exonization and increased novo intronic transcription. New transcripts have been revealed from intron 2-transcription and exon 11 skipping (red cross). In Drosophila melanogaster, CDH1-mRNA translation is suppressed by poly-ribosylation of HnRNP attached to E-cadherin 5’UTR. Human CDH1 pro-protein harbors topological domains: signal peptide (S), pro-peptide (PRO), extracellular with cadherin repeats EC1-EC5 domains, transmembrane (TM, with proximal CH2 and distal CH3), cytoplasmic domain (CD); binding sites of delta1-catenin and presenilin-1 (PS1), p120-ctn, β-catenin (βCTN), calcium; ubiquitination and short intracellular half-life sites rich in proline (P), glutamic acid (E), serine (S) and threonine (T) (PEST); motif highly conserved (Leu-Ser-Ser-Leu); acidic residues cluster of endocytic signal (DEE 602–604). Cleavage sites by proteases (scissors): metalloprotease (MMP), gamma-secretase/PS1 (presenilin-1) and caspase 3. Inhibitory or stimulatory phosphorylation sites for casein kinase-1 (CK1) (red) and CK2, GSK3β and PDK1 respectively. N-glycosylation at Asn-483 is essential for expression, folding and trafficking. Ligth blue horizontal lines indicate protein binding domains to E-cadherin.
E-cadherin function control via EC
The mature classical E-cadherin is a single-pass transmembrane glycoprotein with five ectodomains (EC1–5), each made up of ~110 amino acid residues. Their NH2-terminal ectodomain mediates adhesive binding to cadherins present on the surfaces of neighboring cells. Their four interdomain junctions are characterized by three calcium binding sites ─DXD, DRE, and DXNDNAPXF sequence motifs─ [207]. Cadherin function requires calcium to rigidify the EC domain [27, 112, 146] and to stabilize the protein [156, 159] avoiding proteolysis [79, 190], and allowing its proper localization [57, 156, 159].
E-cadherin mediates principally in cell-cell homophilic interactions through self-recognition of the conserved histidine-alanine-valine (HAV) sequence within EC1 [14] (Fig. 1), and a strand-swapping interface. In this surface, a Tryptophan in position 2 (W2/Trp2) inserts into a hydrophobic pocket from another cis (from the same cell surface) or trans (from an adjacent cell) cadherin [100, 111, 182]. Although another cis dimerization has been postulated [15], several models disagree on the roles and numbers of the inner EC domains involved in cadherin homophilic interaction [31, 66, 112, 160].
Cleavage of the ~130 amino acids prodomain of immature cadherin represents the switch from a non adhesive to the functional form [67]. Even though the specific E-cadherin endoprotease has not been identified yet [145], the E-cadherin proprotein contains a furin-cleavable motif [SSPGLRRQKR] (Fig. 1) [73, 136], and sequence specificity for other mammalian convertases [161]. In contrast, mature E-cadherins are inactivated by cleavage in the EC domain mediated by matrix metalloproteinases [37] and others proteases [89, 153], in a process known as ectodomain shedding, thus promoting the invasion [44, 122, 187] (Fig. 1).
In addition, it has also been reported that E-cadherins can adhere heterophilically to integrins αEβ7 and α2β1 [30, 176, 205], killer-cell lectin-like receptors G1 [173], and numerous infectious agent proteins that target E-cadherins as an entry receptor [5, 36, 132].
E-cadherin function control via CD
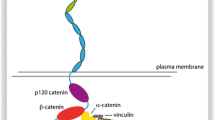
In active functionally adhesion complexes E-cadherin associates with intracellular components forming AJs. E-cadherin CD interacts with several cytoplasmic proteins, being the catenins the best understood: β-catenin or γ-catenin, α-catenin, and p120-catenin (p120-ctn) [41, 128, 133, 134, 165]. A homolog of β-catenin, γ-catenin/plakoglobin [152], can substitute it under some circumstances [38]. When they are fully incorporated in complexes with cadherins, these three catenins associate with a stoichiometry of one of each catenin per cadherin molecule [90, 147]. β-catenin binds directly to the distal ~72 amino acids of the E-cadherin CD through a 30-amino acid “core”, anchoring indirectly to α-catenin (Fig. 1) [76]. P120-ctn binds independently to the ~29 amino acid membrane-proximal region of the cadherin CD [165, 199] (Fig. 1). α-Catenin associates with the actin cytoskeleton [167, 214] and numerous adaptor proteins to strengthen cell-cell adhesion [1, 199].
Several cleavage fragments of E-cadherin CD and disassembling of their adhesive complexes have been reported. During apoptosis or calcium influx, presenilin 1 (PS1)/γ-secretase cleaves between human E-cadherin residues Leu731 and Arg732 [127] and caspase-3 cleaves on site 747-DTRD-750 and releases a 25-kDa fragment [93, 183]. In addition, during tumoral progression calpain mediates E-cadherin proteolysis [168] (Fig. 1).
It has been proposed that while β-catenin and plakoglobin facilitate indirect interactions between classic cadherins and the actin cytoskeleton at AJs in vivo, p120-ctn subfamily members induce the lateral (cis) clustering of cadherins [209], and the tethering of signaling or regulatory entities, such as kinases and phosphatases [118]. P120-ctn also stabilizes cadherins at the cell membrane by modulating cadherin membrane trafficking (endocytosis-recycling) and degradation [21, 139, 211]. Catenins are modified by kinases and/or phosphatases that are enriched at cell-cell contacts [6, 42, 52, 118, 203]. E-cadherins, in turn, are normally maintained in a tyrosine-dephosphorylated state through the action of phosphatases that are crucial for stabilizing AJs [51, 135]. In some cell contexts, phosphatase inhibition promotes release of E-cadherins and β-catenins from cell-cell contacts, enabling cytoplasmic catenins to relocate and function in the nucleus and promoting E-cadherin proteolysis [86]. Like phosphatase inhibition, receptor tyrosine kinase stimulation disassembles cell contacts mediated by cadherin─β-catenin─α-catenin [18, 203]. In addition, p120-ctn is an important modulator of RhoGTPase activities, such as RhoA, Rac1 and Cdc42 [3, 4, 142] and gene transcription [43, 95, 149, 162, 180]. On the other hand, p120-ctn interacts with tubulin influencing microtubule stability and dynamics, and thereby affects cell motility and directional migration independently of the cadherin adhesion system [81, 170] (Fig. 2).

Signaling pathways involved in cell-cell adhesion mediated by E-cadherin in physiological and pathological conditions. a Shortly after their synthesis, cadherins associate with β-catenin and phosphorylation on the RER avoid degradation of uncomplexed cadherins and the pro-region cleavage by furin proteases in the trans-Golgi network. When the cis E-cadherin surface pool increases, pro-domain cleavage induces dimerization, trans homophilic bonding and E-cadherin adhesive activity. Delivery of newly synthesized E-cadherin to the basolateral cell surface should be mediated by Rab 11, golgin-97, Sec5, 6, Protein Associated with Lin Seven 1 (PALS), aquaporin 3 (AQP3), p120-ctn, and possibly β-catenin, via localization at the centrosome. b AJs-disassembly by dysfunction of E-cadherin─catenin complexes (CCC) releases catenins that accumulate in the cytoplasm. β-catenins are then sequestered and phosphorylated by the adenomatous polyposis coli (APC)–axin–glycogen synthase kinase 3β (GSK-3β) complex, inducing their ubiquitination by the E3 ubiquitin-ligase βTrCP subunit for proteosomal degradation. However, if at the same time the Wnt signaling pathway is activated, GSK-3β is repressed and β-catenins are no longer phosphorylated and are translocated to the nucleus where their bind TCF/LEF1 transcription factors and modulate gene expression involved in cell proliferation and migration. Cytoplasmic p120-ctns detached from AJs, in turn, activate Rac1 and Cdc42 through Vav2 (Rho-GEF) and represse Rho, promoting filopodia and lamellipodia projections. PI3K is recruited to the membrane by intact E-cadherin adhesion junctions, where it generates PIP3. This activates Tiam1 (Rho-GEF) and subsequently Rac1 and Cdc42, sequestring the GTPase-activating protein (IQGAP1), avoiding IQGAP-binding to β-catenins, and displacing α-catenins from the CCCs, thereby disrupting the CCC-anchoring to the cytoskeleton. Thus, while the activation of Cdc42 and Rac1 induces the formation of filopodia and lamellipodia respectively, Rho induces the formation of actin stress fibers. Cytoplasmic p120-ctn also can translocate to the nucleus to associate with Kaiso and modulate gene expression
As for α-catenin, it modulates the actin-cytoskeleton organization because it can occur either in a monomeric or a homodimeric form [13, 50, 65]. Upon the application of force on epithelia, the conformational change of monomeric αE-catenin uncovers the vinculin binding site, allowing vinculin to bind, and recruit additional F-actin to the cadherin-catenin complexes [126, 214]. Additionally, ZO-1, spectrin, vinculin [199] and Eplin [1] assemble F-actin to E-cadherin-catenin complexes. In parallel, the non-junctional cytosolic homodimeric αE-catenin pool inhibits the Arp2/3 complex, reducing membrane dynamics by preventing F-actin branch-formation [13].
Dynamics of E-cadherin-mediated cell-cell adhesion
Establishment of adherens junctions
Epithelial AJs are built on a foundation of homophilic contacts between (E or P)-cadherin clusters on the surface of adjacent epithelial cells [62, 117]. Cadherins alone are not sufficient for AJ formation; rather, cooperation between nectins and cadherins is required [166, 188] (Fig. 3). Appearently, the trans-interacting nectin inhibits non-trans-interacting E-cadherin endocytosis through afadin, Rap1, and p120-ctn, thereby further non-trans-interacting E-cadherin accumulates in the nectin-based cell-cell adhesion sites for AJ formation [74]. Once AJs have been established through intracellular partner binding to the cytoskeleton, E-cadherin contacts modulate actin filament organization at the underlying cortex [10, 50, 155] and microtubules network [130, 131].

Dynamics of E-cadherin-mediated cell-cell adhesion Epithelial AJs are constructed on a foundation of homophilic contacts between E-cadherin clusters. Previous contacts between nectins inhibit non-trans-interacting E-cadherin endocytosis through afadin, Rap1, and p120-ctn, and increase their concentration at cell-cell adhesion sites. Immediately after E-cadherin trans-interaction, the junction complexes trigger activation of the phosphatidylinositol-3-kinase (PI3K)–Akt–protein kinase B pathway. Phosphatidylinositol-(3,4,5)-triphosphate (PIP3) is generated, and guanine nucleotide exchange factors are recruited to the membrane, activating Rac1 or Cdc42 and reducing Rho activation, which stimulates membrane and actin dynamics adjacent to the initial site of contact, increasing the probability of additional E-cadherin engagements. Alpha-catenin homodimerizes and is released from the cadherin-catenin complexes to bind at and antagonize with Arp2/3, facilitating the belt formation of unbranched actin filaments. While vinculin, afadin and alpha-actinin link with actin cytoskeleton, β-catenin and p120-ctn also link with tubulin cytoskeleton to route both vesicles of newly synthesized E-cadherin-catenin and E-cadherin-recycling-endosomes to the cell-cell contact sites. Down-stream of Rac and Cdc42, IQGAP1 binds β-catenin, which could localize in membrane ruffles and control cadherin internalization via SNX-1 preventing E-cadherin lysosomal degradation and recycling of E-cadherin back to the cell surface for AJ maintenance. P120-ctn binding covers cadherin juxtamembrane domain inhibiting RhoA locally and adaptor complex-binding that recruits cadherins into a coated pit. Thus, E-cadherin becomes withheld in plasma membrane junction domains. Meanwhile, PtdIns(3,4,5)P3 accumulation in the membrane signals for the formation and expansion of the baso-lateral surface, while Rac1 promotes cell polarity and lumen formation, cell cycle arrest of confluent epithelial cells, and survival of polarized epithelial cells. In parallel, E-cadherin downregulates ligand-dependent receptor tyrosine kinase activation, such as EGFR stabilizing cell-cell contacts. Insert: traffic of E-cadherin vesicles via p120-ctn or β-catenin coupled to kinesin for delivering to newly forming or remodeling junctions
It is known that catenins, including β-catenins, α-catenins, and p120-catenins, bind to immature E-cadherins while traveling through the endoplasmic reticulum and Golgi apparatus [40, 71]. Following synthesis in the rough endoplasmic reticulum and phosphorylation of the cytoplasmic domain, the E-cadherin pro-region is cleaved by furin proteases in the trans-Golgi network, an event that is mandatory for the mature cadherin to function in adhesion [59, 75, 116] (Fig. 2). Via golgin-97 dependent tubulovesicular carriers, E-cadherins leave the Golgi complex [124], and subsequently fuse with an intermediate recycling endosome, in route to the basolateral plasma membrane. This pathway for E-cadherins trafficking is tightly integrated with other proteins involved in epithelial polarity, such as Sec5, 6, and 15 [107, 193], Rab 11 [123], PALS (Protein Associated with Lin Seven 1), and aquaporin 3 (AQP3) [137], and it determines lumen formation [49] (Fig. 2).
The function and regulation of E-cadherins post-translational modifications in vivo have remained poorly defined. While the phosphorylations to eight serine residue cluster within a region that binds β-catenin modulate the β-catenin affinity and strengthen cell–cell adhesion [181], the E-cadherin cytoplasmic O-glycosylation (O-GlcNAc) blocks its cell surface transport, reducing intercellular adhesion [218]. In addition, E-cadherins have four consensus sites Asn-X-Ser/Thr for N-glycosylation in EC4 and EC5 domains [102], most of them conserved among species (Fig. 1). It has been established that, while the N-glycans at Asn 633/483 are essential for E-cadherin folding, trafficking, and proper expression [217], their modification with complex N-glycans weakens AJs [121, 157] (Fig. 2).
Signaling and maintainance of adherens cell-cell junctions
Chemical [3, 85, 86, 149] and mechanical [110, 214] signals from cell-cell junctions can be transduced through E-cadherin-CD via β-, α- and p120-catenin to the cytoplasm or nucleus. Mechanical stimulation generated by tumor cell proliferation leads to β-catenin phosphorylation via Src kinase at the site of its interaction with E-cadherin, increasing β-catenin nuclear localization, and upregulating the oncogenes Myc and Twist1 [204]. In physiological state, the formation of junctional complexes triggers activation of the phosphatidylinositol-3-kinase (PI3K)–Akt–protein kinase B pathway [151] because the PI3K-p85 subunit associates with AJs through direct binding to β-catenin [206] (Fig. 3). After recruitment of PI3K, phosphatidylinositol-(3,4,5)-triphosphate (PIP3) is generated, and guanine nucleotide exchange factors, that contain PIP3-binding pleckstrin homology domains, are recruited to the membrane and activate Rac1 [105, 210] or Cdc42 [94]. This stimulates membrane actin dynamics adjacent to the initial contact site, increasing the probability of additional E-cadherin engagements. α-Catenins, in turn, homodimerizes and are released from the cadherin─catenin complexes to bind at actin and antagonize Arp2/3 function, inhibiting actin branching and facilitating belt formation of unbranched actin filaments. Simultaneously, other actin-binding proteins such as vinculin, afadin and α-actinin link with actin cytoskeleton, and β-catenin and p120-ctn link with tubulin cytoskeleton to route vesicles of E-cadherin─catenin to the cell-cell contact sites. Meanwhile, PtdIns(3,4,5)P3 accumulation in the membrane signals for the formation and expansion of the basolateral surface, whereas Rac1 promotes polarity orientation and lumen formation. In tissue and cell-type specific contexts, Rac1 also triggers the activation of downstream signaling effectors, promoting cell cycle arrest of confluent epithelial cells, and survival of polarized epithelial cells [169]. Therefore, cadherins function in tissue morphogenesis by controlling both cell-cell adhesion and cell signaling. In this way, cadherins are involved in determining cell shape, position, migration [33, 68, 92, 125, 186], polarity [16, 200] and proliferation [96, 172], as well as tissular folding [201, 202]. In adherens junctions, E-cadherins and catenins determine cell-cell contact survival and epithelia function [11].
AJs are very dynamic structures that undergo constant remodeling. This can be low-scale remodelling involving replacement of individual or groups of molecules within the adhesive clusters without disrupting steady-state intercellular adhesions. It can also be large-scale junctional rearrangements that accompany breakdown and reformation of cell-cell contacts [82].
Growth factors (GFs) as specific regulators of E-cadherin─catenin traffic
GFs are responsible for crosstalk between cell proliferation, migration, and adhesion. GFs bind to their specific receptor, cause cell-cell dissociation coupled to E-cadherin endocytosis and recycling back to the cell surface by several mechanisms [22, 23, 148] (Fig. 3). Co-regulation of cadherins and GF signaling is prominent in epithelial to mesenchymal transitions (EMT) and tumorigenesis [20, 88]. E-cadherins, in turn, modulate signal transduction by interacting with receptor tyrosine kinases, including the epidermal growth factor receptor (EGFR) [29, 163]. Among them, thyroid hormones (THs) modulate energy metabolism, having a great influence on growth and development by independent mechanisms [54]. An increasing number of studies show the THs action in different parallel signaling pathways via membrane receptors, cytoplasmic partners and thyroid hormone receptors (TRs) [72, 106]. TRs heterodimerize with retinoid X receptors (RXRs) and bind to T3 response elements (TRE) located within the genomic regions of target genes [212]. In the absence of T3, TRs interact with co-repressor proteins to inhibit TH-regulated target gene transcription (Fig. 4). Following T3 binding, co-repressors are displaced and co-activator proteins are recruited to the ligand-bound TR complex, so as to facilitate T3-dependent activation of the target genes. Complexity increases because the diploid organisms have THRA and THRB genes that encode the TRα and TRβ isoforms respectively, which are ubiquitously expressed [109, 175, 212, 213]. Moreover, depending on species, tissue or experimental systems, there are predominant TR cell isoforms, and each gene can generate different proteins using different promoters and/or alternative splicing [109, 175, 213]. In addition to functions mediated by TRs, THs also exert rapid non-genomic actions that are initiated at the cell membrane. For example, the αvβ3-integrin binds to THs, activating the mitogen-activated protein kinase (MAPK) cascade [45, 119] for modulating the membrane ion channels, Na+/K+ exchanger and Ca2+ATPase, as well as the actin cytoskeletal components [99] (Fig. 4).

T3-actions from genomic and non-genomic effects on cell adhesion and differentiation during vertebrate development. THs modulate energy metabolism, growth and development by independent mechanisms. While thyroid calorigenesis is influenced predominantly via nuclear receptors, many of the TH effects over development are thought to be mediated via cytosolic and membrane partners. E-cadherin trans-interaction triggers activation of the phosphatidylinositol-3-kinase (PI3K)–Akt–protein kinase B pathway bound to β-catenin, generating phosphatidylinositol-(3,4,5)-triphosphate (PIP3), recruitment of guanine nucleotide exchange factors, activation of Rac1 or Cdc42 and Akt, and reduction of Rho activation. In addition, TRα or TRβ forms a cytoplasmic complex with the p85 subunit of PI3K, inducing protein kinaseB/Akt nuclear translocation and inhibition of the Wnt/β-catenin pathway through its interaction and consequent sequestration of β-catenin. The process results in down-regulation of cell proliferation. Simultaneously, TH binding to TRs causes heterodimerization with retinoid X receptors (RXRs), binding to T3 response elements located within the genomic regions and shooting target gene transcription. In the absence of T3, TRs interact with co-repressor proteins to inhibit target gene transcription. Following T3 binding, co-repressors are displaced and co-activator proteins are recruited to the ligand-bound TR complex, facilitating T3-dependent activation of the target genes. Besides the TR-mediated functions, THs also exert rapid non-genomic actions that are initiated at the cell membrane. Integrin αvβ3 is a specific membrane receptor for THs, which mediate activation of the mitogen-activated protein kinase (MAPK) intracellular cascade. TH-dependent MAPK activation subsequently results in modulation of the membrane potential by regulation of ion channels, activation of the Na+/K+ exchanger and Ca2+ATPase, or regulation of actin cytoskeletal components anchored at the cell membrane. TH-activated MAPK, in turn, can rapidly translocate to the nucleus inducing serine phosphorylation of TRs, thereby resulting in the induction of angiogenesis or tumor cell proliferation. Nuclear targets for phosphorylated TRs include the transcription factors p53, STAT1a and STAT3
Among genomic mechanisms, it is known that transcriptional repression of E-cadherins is mediated either by promoter CpG hypermethylation [60] or activation of repressors binded at E-boxes or brain-specific silencers, such as Snail and Slug [194], Twist [208] and E12/E47 (E2A gene product) [154]. In contrast, the miR-200 family directly targets repressors ZEB1 and ZEB2, promoting E-cadherin expression upregulation [103, 104] (Fig. 1). In hormonal signaling routes, it is known that the activated androgen receptor binds to promoter and also represses E-cadherin gene expression, promoting metastasis by EMT [120].
Conversely, our results suggest that T3-TRs promote E-cadherin, β- and α-catenin gene expression in vivo and EMT inhibition [56, 85]. E-cadherin locus up-regulation can be mediated by an enhancer element at intron 1 [12, 25, 69, 70]. In addition, Stemler Stemmler et al. [185] reveal a complex mechanism of gene regulation at mouse E-cadherin locus intron 2. While in differentiated epithelia intron 2 sequences are required both to initiate transcriptional activation and additionally to maintain E-cadherin expression during embryogenesis, the level of intron 2-dependent E-cadherin expression is relative to the tissues and developmental timing. Thus, early embryogenesis requires intron 2 for the onset of expression, but at later stages, a second mechanism initiates E-cadherin expression independently of intron 2, although for high-level expression the support of the intron 2 enhancer elements is still required. The onset of the second wave of expression was detected in the surface ectoderm differentiate to form skin, and in the gut endoderm (around E12.5). A locus control region (LCR), in turn, might influence gene activity for proper activation and downregulation, sited upstream of cadherin clusters [184, 185] with a vital role for the large intron 2 (Fig. 1). Striking, cadherin superfamily genes display a higher average total intron number and significantly longer introns than other genes and across the entire vertebrate lineage [144]. Particularly, the human genome has an uncommon high frequency of MIR and MaLR regulatory-associated repetitive elements at 5’-located introns, concomitant with increased de novo intronic transcription. Therefore, these intronic-specific sites may constitute targets of cadherin superfamily expression regulation, both in homeostasis and illness.
Thus, searching for some of those physiological needs to sustain epithelial life, we have analyzed cell adhesion molecule response to 3,5,3’-triiodothyronine (T3), detecting morphometric evidences of gene upregulation exerted by T3 on E-cadherin, β- and α-catenin expression in different epithelial cell types of the metamorphosing anuran foregut [85]. Coincidentally, mouse β-catenin gene upregulation and transrepression by TH-TR [61, 158], as well as the impact of TH signaling in development, homeostasis and cancer susceptibility of mouse intestine [178] have been reported. T3-TRα1 binds directly to β-catenin gene-intron 1 specific TRE in the intestine, increasing its expression in an epithelial cell-autonomous way [158] (Fig. 5). This is parallel to positive regulation of proliferation-controlling genes, such as type D cyclins and c-myc, which are known targets of the Wnt/β-catenin pathway [158], synergizing Wnt pathway and inducing crypt cell proliferation and promoting tumorigenesis [179]. In contrast, CTNNB1 transrepression is mediated by binding of the TRβ-RXR complexes on TREs located in the human promoter between −807 and −772 (Fig. 5) [61]. Therefore, liganded TRβ acts as a tumor suppressor via inhibition of the expression of a potent tumor promoter, the CTNNB1 gene.

T3-control model of E-cadherin─β-catenin complex on gastrointestinal epithelial cells. THs and RTs function in cell proliferation, differentiation and apoptosis is not homogenous, because it depends strongly on physio-pathological context; that is, the cell-type, ontogeny (progenitor or differentiated cell) and health (normal or tumoral cell). However, it is possible to postulate that, while T3 induces epithelial basal cell proliferation via EGF-EGFR and cAMP-PKA signaling, T3 activates transcription of E-cadherin, β- and α-catenin genes in epithelial cells programmed to differentiate on pre-adult gut epithelia cells and inhibiting their EGF-EGFR dependent proliferative signal, as well as inhibiting their TH-integrin αvβ3 dependent migratory signals. In addition, because E-cadherin increases β-catenin sequestration at the plasma membrane, it then promotes cell differentiation by diminishing the β-catenin/TCF complex pool. At the same time, TSH-TSHR (receptor) signaling via cAMP stabilizes the assembly and retention of E-cadherin at the cell surface. TRα1 binds to β-catenin gene-intron 1-TRE (TRE-int1) in the intestine, increasing its expression via TH-binding. In parallel, TRα1 positively regulates the proliferation-controlling genes such as type D cyclins and c-myc, which are known targets of the Wnt/β-catenin. Increase of β-catenin/Tcf4, in turn, reduces the TRα1 transcriptional activity on its target genes. On the other hand, CTNNB1 transrepression is mediated by binding of the TRβ-RXR complexes to promoter TREs
Even though TH signaling controls the proliferation of the intestinal epithelial progenitors in both amphibians and mammals, it has been suggested that TH control on the Wnt/β-catenin pathway does not appear to play a central role in amphibians [24, 178]. However, our in vivo experiments contradict this hypothesis, since we have detected early upregulation (24 hs) of E-cadherin, β- and α-catenin genes in the Xenopus laevis gut [56]. During metamorphic climax, larval cell apoptosis coexists with pre-adult (juvenile) cell proliferation and differentiation, and with cell-cell junction assembly─disassembly, that require a complex signal network to control tissular homeostasis. We found that T3 modulates epithelial adhesive potential during gut remodeling in X. laevis development, early and directly activating E-cadherin, β-catenin and α-catenin genes, and downstream, modulating small GTPases and other proteins involved in adhesive epithelial properties. Using INSECT2.0 web server to predict the occurrence of Cis-Regulatory Modules (CRMs) [150], we found putative TERs in X. laevis E-cadherin, β-catenin and α-catenin genes, but not in the p120-ctn gene [56]. Among evaluated small GTPases, gastrointestinal Rac1-mRNA levels significantly increased at 24 hs T3-treatment correlated with heightened “lamellipodia” or membrane protrusions. In contrast, at 5 days post T3-treatment RhoA-mRNA levels decrease while the important Rap1-mRNA increasing suggests that membrane Rap1-dependent E-cadherin recycling occurs at metamorphic climax end. Cdc42 and Arp2 actin nucleation protein became constant both during T3-induced and natural metamorphosis [56].
THs exert profound effects on tissues. Among them, cell-type dependent proliferation and differentiation both in mammalian skin [101, 113, 164, 195] and gut [158, 178], as in anuran kidney [84], skin [86] and gut [55, 56, 85]. These effects are regulated by phosphorilation levels of several partner proteins [86] and modulation of gen expression [56, 85]. Among the non-genomic effects of thyroid hormones, it appears that T3 activates PKA to in turn induce β-catenin nuclear translocation by phosphorylation at Ser675 site, thereby β-catenin modulates cyclin-D1 gene transcription and induces cell proliferation (Fig. 5) [53]. In contrast, TH causes astrocyte differentiation through both initial PKA activity and later by phospho-MAP kinase (p-MAPK or p-ERK) [58]. Postbirth, Schwann cell E-cadherin expression is highly regulated through cAMP-PKA activation for maintaining structural integrity [39]. Thus, E-cadherin can negatively regulate, in an adhesion dependent manner, the ligand-dependent activation of divergent classes of RTKs, by inhibiting their ligand-dependent activation in association with a decrease in receptor mobility and in ligand-binding affinity [163] (Fig. 5).
Therefore, the function of THs and their TRs in cell proliferation, differentiation and apoptosis is not homogenous, because it depends strongly on the physio-pathological context; that is, the cell-type, ontogeny (progenitor or differentiated cell) and health (normal or tumoral cell). Although TH-dependent processes are highly coordinated, in turn, the local requirements cannot be governed by global mechanisms, such as an alteration of the thyroid gland function and variations in plasma TH concentrations. Instead, they require tissue-specific regulation mediated by deiodinases. Thus, in mammals, T3 induces type 2 deiodinase (D2) and E-cadherin expression, which sequesters β-catenin and reduces both β-catenin/TCF complex and type 3 deiodinase (D3) activation levels. Consequently, the local active T3-level increases and promotes cell differentiation and reduces its oncogenic effects in intestinal cells (Fig. 5) [48]. Recently, this hypothesis has been again supported by Catalano and coworkers [28], who have found that increased intracellular TH concentration through D3 depletion induces cell differentiation and sharply mitigates tumor formation.
In this context, it is possible to postulate that during organ remodeling T3 induces epithelial stem cell proliferation via EGF-EGFR and cAMP-PKA signaling, and in parallel, T3 leads to repression of this via in a subgroup of these stem cells, and simultaneously increases E-cadherin, β- and α-catenin transcription via TH-RT to differentiate them to pre-adult (juvenile) gut epithelia cells (Fig. 5). In addition, the erichment of junctional E-cadherins sequestrates β-catenins, reducing their β-catenin nuclear translocalization and D3 disponibility, thereby strengthening cell differentiation.
Conclusions
Unquestionably, E-cadherin is deeply involved in establishing cell polarity and differentiation, and thereby in the establishment and maintenance of tissue homeostasis during the development and the entire life of pluricellular organisms, mainly metazoans. Therefore, this transmembrane receptor is a potent suppressor of tumoral invasion and metastasis. Thus, E-cadherin must continuously react both at extra- and intracellular signals, some of which are classical, and very well known.
This review summarizes findings supporting the central role of thyroid hormones in controlling the availability and functionality of E-cadherin, through membrane, cytoplasmic and gene expression activities that regulate cell proliferation, differentiation, migration, and thereby epithelial homeostasis.
Abbreviations
- AJ:
-
Adherens junctions
- AQP3:
-
Aquaporin 3
- CD:
-
Cytoplasmic domain
- D2:
-
Type 2 deiodinase
- D3:
-
Type 3 deiodinase
- EC:
-
Extracellular domain
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial to mesenchymal transitions
- GF:
-
Growth factor
- HAV:
-
Histidine-alanine-valine
- MAPK:
-
Mitogen-activated protein kinase
- O-GlcNAc:
-
O-glycosylation
- p120-ctn:
-
p120-catenin
- PALS:
-
Protein Associated with Lin Seven 1
- PI3K:
-
Phosphatidylinositol-3-kinase
- PIP3:
-
Phosphatidylinositol-(3,4,5)-triphosphate
- PS1:
-
Presenilin 1
- RT:
-
Thyroid hormone receptor
- RTK:
-
Membrane-receptor tyrosine kinase
- RXR:
-
Retinoid X receptor
- T3:
-
3,5,3’-triiodothyronine
- TH:
-
Thyroid hormone
- TM:
-
Trans-membrane domain
- TRE:
-
T3 response element
References
Abe K, Takeichi M. EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc Natl Acad Sci USA. 2008;105:13–9.
Abedin M, King N. The premetazoan ancestry of cadherins. Science. 2008;319:946–8.
Anastasiadis PZ, Moon SY, Thoreson MA, Mariner DJ, Crawford HC, Zheng Y, Reynolds AB. Inhibition of RhoA by p120catenin. Nat Cell Biol. 2000;2:637–44.
Anastasiadis PZ. p120-ctn: A nexus for contextual signaling via Rho GTPases. Biochim Biophys Acta. 2007;1773:34–46.
Anderton JM, Rajam G, Romero-Steiner S, Summer S, Kowalczyk AP, Carlone GM, Sampson JS, Ades EW. E-cadherin is a receptor for the common protein pneumococcal surface adhesin A (PsaA) of Streptococcus pneumoniae. Microb Pathog. 2007;42:225–36.
Andl CD, Rustgi AK. No one-way street: cross-talk between E-cadherin and receptor tyrosine kinase (RTK) signaling: a mechanism to regulate RTK activity. Cancer Biol Ther. 2005;4:28–31.
Ao A, Erickson RP. Injection of antisense RNA specific for E-cadherin demonstrates that E-cadherin facilitates compaction, the first differentiative step of the mammalian embryo. Antisense Res Dev. 1992;2:153–63.
Babb SG, Barnett J, Doedens AL, Cobb N, Liu Q, Sorkin BC, Yelick PC, Raymond PA, Marrs JA. Zebrafish E-cadherin: expression during early embryogenesis and regulation during brain development. Dev Dyn. 2001;221:231–7.
Babb SG, Marrs JA. E-cadherin regulates cell movements and tissue formation in early zebrafish embryos. Dev Dyn. 2004;230:263–77.
Baum B, Perrimon N. Spatial control of the actin cytoskeleton in Drosophila epithelial cells. Nat Cell Biol. 2001;3:883–90.
Baum B, Georgiou M. Dynamics of adherens junctions in epithelial establishment, maintenance, and remodelling. J Cell Biol. 2011;192:907–17.
Behrens J, Löwrick O, Klein-Hitpass L, Birchmeier W. The E-cadherin promoter: functional analysis of a G.C-rich region and an epithelial cell-specific palindromic regulatory element. Proc Natl Acad Sci U S A. 1991;88:11495–9.
Benjamin JM, Kwiatkowski AV, Yang C, Korobova F, Pokutta S, Svitkina T, Weis WI, Nelson WJ. Alpha E-catenin regulates actin dynamics independently of cadherin-mediated cell-cell adhesion. J Cell Biol. 2010;189:339–52.
Blaschuk OW, Sullivan R, David S, Pouliot Y. Identification of a cadherin cell adhesion recognition sequence. Dev Biol. 1990;139:227–9.
Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science. 2002;296:1308–13.
Bosveld F, Bonnet I, Guirao B, Tlili S, Wang Z, Petitalot A, Marchand R, Bardet P-L, Marcq P, Graner F. Mechanical control ofmorphogenesis by Fat/Dachsous/Four-jointed planar cell polarity pathway. Science. 2012;336:724–7.
Boyle EC, Finlay BB. Bacterial pathogenesis: Exploiting cellular adherence. Curr Opin Cell Biol. 2003;15:633–9.
Brembeck FH, Schwarz-Romond T, Bakkers J, Wilhelm S, Hammerschmidt M, Birchmeier W. Essential role of BCL9-2 in the switch between beta-catenin’s adhesive and transcriptional functions. Genes Dev. 2004;18:2225–30.
Brembeck FH, Rosario M, Birchmeier W. Balancing cell adhesion and Wnt signaling, the key role of beta-catenin. Curr Opin Genet Dev. 2006;16:51–9.
Bremm A, Walch A, Fuchs M, Mages J, Duyster J, Keller G, Hermannstadter C, Becker KF, Rauser S, Langer R, von Weyhern CH, Hofler H, Luber B. Enhanced activation of epidermal growth factor receptor caused by tumor-derived E-cadherin mutations. Cancer Res. 2008;68:707–14.
Bryant DM, Stow JL. The ins and outs of E-cadherin trafficking. Trends Cell Biol. 2004;14:427–34.
Bryant DM, Wylie FG, Stow JL. Regulation of endocytosis, nuclear translocation, and signaling of fibroblast growth factor receptor 1 by E-cadherin. Mol Biol Cell. 2005;16:14–23.
Bryant DM, Kerr MC, Hammond LA, Joseph SR, Mostov KE, Teasdale RD, Stow JL. EGF induces macropinocytosis and SNX1-modulated recycling of E-cadherin. J Cell Sci. 2007;120:1818–28.
Buchholz DR, Heimeier RA, Biswajit D, Washington T, Shi Y-B. Pairing morphology with gene expression in thyroid hormone-induced intestinal remodeling and identification of a core set of TH-induced genes across tadpole tissues. Dev Biol. 2007;303:576–90.
Bussemakers MJ, Giroldi LA, van Bokhoven A, Schalken JA. Transcriptional regulation of the human E-cadherin gene in human prostate cancer cell lines: characterization of the human E-cadherin gene promoter. Biochem Biophys Res Commun. 1994;203:1284–90.
Cai D, Chen SC, Prasad M, He L, Wang X, Choesmel-Cadamuro V, Sawyer JK, Danuser G, Montell DJ. Mechanical Feedback through E-Cadherin Promotes Direction Sensing during Collective Cell Migration. Cell. 2014;157:1146–59.
Cailliez F, Lavery R. Cadherin mechanics and complexation: the importance of calcium binding. Biophys J. 2005;89:3895–903.
Catalano V, Dentice M, Ambrosio R, Luongo C, Carollo R, Benfante A, Todaro M, Stassi G, Salvatore D. Activated thyroid hormone promotes differentiation and chemotherapeutic sensitization of colorectal cancer stem cells by regulating Wnt and BMP4 Signaling. Cancer Res. 2016;76:1237–44.
Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4:118–32.
Cepek KL, Shaw SK, Parker CM, Russell GJ, Morrow JS, Rimm DL, Brenner MB. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature. 1994;372:190–3.
Chappuis-Flament S, Wong E, Hicks LD, Kay CM, Gumbiner BM. Multiple cadherin extracellular repeats mediate homophilic binding and adhesion. J Cell Biol. 2001;154:231–43.
Chen YH, Stewart DB, Nelson WJ. Coupling assembly of the E-cadherin/b-catenin complex to efficient endoplasmic reticulum exit and basal-lateral membrane targeting of E-cadherin in polarized MDCK cells. J Cell Biol. 1999;144:687–99.
Chihara D, Nance J. An E-cadherin-mediated hitchhiking mechanism for C. elegans germ cell internalization during gastrulation. Development. 2012;139:2547–56.
Choi YS, Gumbiner B. Expression of cell adhesion molecule E-cadherin in Xenopus embryos begins at gastrulation and predominates in the ectoderm. J Cell Biol. 1989;108:2449–58.
Classen AK, Anderson KI, Marois E, Eaton S. Hexagonal Packing of Drosophila wing epithelial cells by the planar cell polarity pathway. Dev Cell. 2005;9:805–17.
Costa AM, Leite M, Seruca R, Figueiredo C. Adherens junctions as targets of microorganisms: A focus on Helicobacter pylori. FEBS Lett. 2013;587:259–65.
Covington MD, Burghardt RC, Parrish AR. Ischemia-induced cleavage of cadherins in NRK cells requires MT1-MMP (MMP-14). Am J Physiol-Renal Physiol. 2006;290:F43–51.
Cowin P, Kapprel H-P, Franke WW, Tamkun J, Hynes RO. Plakoglobin: A protein common to different kinds of intercellular adhering junctions. Cell. 1986;46:1063–73.
Crawford AT, Desai D, Gokina P, Basak S, Kim HA. E-cadherin expression in postnatal Schwann cells is regulated by the cAMP-dependent protein kinase A pathway. Glia. 2008;56:1637–47.
Curtis MW, Johnson KR, Wheelock MJ. E-cadherin/catenin complexes are formed co-translationally in the endoplasmic reticulum/Golgi compartments. Cell Commun Adhes. 2008;15:365–78.
Daniel JM, Reynolds AB. The tyrosine kinase substrate p120cas binds directly to E-cadherin but not to the adenomatous polyposis coli protein or a-catenin. Mol Cell Biol. 1995;15:4819–24.
Daniel JM, Reynolds AB. Tyrosine phosphorylation and cadherin/catenin function. BioEssays. 1997;19:883–91.
Daniel JM, Reynolds AB. The catenin p120(ctn) interacts with Kaiso, a novel BTB/POZ domain zinc finger transcription factor. Mol Cell Biol. 1999;19:3614–23.
Davies G, Jiang WG, Mason MD. Matrilysin mediates extracellular cleavage of E-cadherin from prostate cancer cells: a key mechanism in hepatocyte growth factor/scatter factor-induced cell-cell dissociation and in vitro invasion. Clin Cancer Res. 2001;7:3289–97.
Davis PJ, Leonard JL, Davis FB. Mechanisms of nongenomic actions of thyroid hormone. Front Neuroendocrinol. 2008;29:211–8.
Dayel MJ, Alegado RA, Fairclough SR, Levin TC, Nichols SA, McDonald K, King N. Cell differentiation and morphogenesis in the colony-forming choanoflagellate Salpingoeca rosetta. Dev Biol. 2011;357:73–82.
Dayel MJ, King N. Prey capture and phagocytosis in the choanoflagellate Salpingoeca rosetta. PLoS ONE. 2014;9:e95577.
Dentice M, Luongo C, Ambrosio R, Sibilio A, Casillo A, Iaccarino A, Troncone G, Fenzi G, Larsen PR, Salvatore D. β-Catenin regulates deiodinase levels and thyroid hormone signaling in colon cancer cells. Gastroenterol. 2012;143:1037–47.
Desclozeaux M, Venturato J, Wylie FG, Kay JG, Joseph SR, Le HT, Stow JL. Active Rab11 and functional recycling endosome are required for E-cadherin trafficking and lumen formation during epithelial morphogenesis. Am J Physiol Cell Physiol. 2008;295:C545–556.
Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI. Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell. 2005;123:903–15.
Duchesne C, Charland S, Asselin C, Nahmias C, Rivard N. Negative regulation of β-catenin signaling by tyrosine phosphatase SHP-1 in intestinal epithelial cells. J Biol Chem. 2003;278:14274–83.
Erez N, Bershadsky A, Geiger B. Signaling from adherens-type junctions. Eur J Cell Biol. 2005;84:235–44.
Fanti M, Singh S, Ledda-Columbano GM, Columbano A, Monga SP. Tri-iodothyronine induces hepatocyte proliferation by protein kinase A-dependent β-catenin activation in rodents. Hepatology. 2014;59:2309–20.
Fisher DA, Hoath S, Lakshmanan J. The thyroid hormone effects on growth and development may be mediated by growth factors. Endocrinol Exp. 1982;16:259–71.
Galetto CD, Izaguirre MF, Bessone V, Casco VH. Isolation and nucleotide analysis sequence of the of Rhinella arenarum β-catenin: An mRNA and protein expression study during the larval stages of the digestive tract development. Gene. 2012;511:256–64.
Galetto CD. Rol de la adhesión mediada por cadherina E. PhD Thesis on Biological Sciences, School of Biochemistry and Biological Sciences of the Litoral National University, Argentina. 2016.
Gamboa-Dominguez A, Dominguez-Fonseca C, Chavarri-Guerra Y, Vargas R, Reyes-Gutierrez E, Green D, Quintanilla-Martinez L, Luber B, Busch R, Becker KF, Becker I, Höfler H, Fend F. E-cadherin expression in sporadic gastric cancer from Mexico: exon 8 and 9 deletions are infrequent events associated with poor survival. Hum Pathol. 2005;36:29–35.
Ghosh M, Gharami K, Paul S, Das S. Thyroid hormone-induced morphological differentiation and maturation of astrocytes involves activation of protein kinase A and ERK signalling pathway. Eur J Neurosci. 2005;22:1609–17.
Gooding JM, Yap KL, Ikura M. The cadherin-catenin complex as a focal point of cell adhesion and signalling: new insights from three-dimensional structures. Bioessays. 2004;26:497–511.
Grady WM, Willis J, Guilford PJ, Dunbier AK, Toro TT, Lynch H, Wiesner G, Ferguson K, Eng C, Park JG, Kim SJ, Markowitz S. Methylation of the CDH1 promoter as the second genetic hit in hereditary diffuse gastric cancer. Nat Genet. 2000;26:16–7.
Guigon CJ, Kim DW, Zhu X, Zhao L, Cheng SY. Tumor suppressor action of liganded thyroid hormone receptor beta by direct repression of beta-catenin gene expression. Endocrinology. 2010;151:5528–36.
Gumbiner BM. Regulation of cadherin adhesive activity. J Cell Biol. 2000;148:399–404.
Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622–34.
Harris TJ, Tepass U. Adherens junctions: from molecules to morphogenesis. Nat Rev Mol Cell Biol. 2010;11:502–14.
Hartsock A, Nelson WJ. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–9.
Hasenahuer MA, Casco VH, Izaguirre MF. New footprints into metazoan C-1 family cadherin evolution. Onl J Bioinform. 2013;14:76–95.
Häussinger D, Ahrens T, Aberle T, Engel J, Stetefeld J, Grzesiek S. Proteolytic E-cadherin activation followed by solution NMR and X-ray crystallography. EMBO J. 2004;23:1699–708.
Hayashi T, Carthew R. Surface mechanics mediate pattern formation in the developing retina. Nature. 2004;431:647–52.
Hennig G, Behrens J, Truss M, Frisch S, Reichmann E, Birchmeier W. Progression of carcinoma cells is associated with alterations in chromatin structure and factor binding at the E-cadherin promoter in vivo. Oncogene. 1995;11:475–84.
Hennig G, Löwrick O, Birchmeier W, Behrens J. Mechanisms identified in the transcriptional control of epithelial gene expression. J Biol Chem. 1996;271:595–602.
Hinck L, Näthke IS, Papkoff J, Nelson WJ. Dynamics of cadherin/catenin complex formation: novel protein interactions and pathways of complex assembly. J Cell Biol. 1994;125:1327–40.
Hiroi Y, Kim HH, Ying H, Furuya F, Huang Z, Simoncini T, Noma K, Ueki K, Nguyen NH, Scanlan TS, Moskowitz MA, Cheng SY, Liao JK. Rapid nongenomic actions of thyroid hormone. Proc Natl Acad Sci U S A. 2006;103:14104–9.
Hoshino H, Konda Y, Takeuchi T. Co-expression of the proprotein-processing endoprotease furin and its substrate transforming growth factor ßl and the differentiation of rat hepatocytes. FEBS Lett. 1997;419:9–12.
Hoshino T, Sakisaka T, Baba T, Yamada T, Kimura T, Takai Y. Regulation of E-cadherin endocytosis by nectin through afadin, Rap1, and p120ctn. J Biol Chem. 2005;280:24095–103.
Huber AH, Weis WI. The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell. 2001;105:391–402.
Huber AH, Stewart DB, Laurents DV, Nelson WJ, Weis WI. The cadherin cytoplasmic domain is unstructured in the absence of beta-catenin. A possible mechanism for regulating cadherin turnover. J Biol Chem. 2001;276:12301–9.
Hulpiau P, van Roy F. Molecular evolution of the cadherin superfamily. Int J Biochem Cell Biol. 2009;41:349–69.
Hulpiau P, van Roy F. New insights into the evolution of metazoan cadherins. Mol Biol Evol. 2011;28:647–57.
Hyafil F, Morello D, Babinet C, Jacob F. A cell surface glycoprotein involved in the compaction of embryonal carcinoma cells and cleavage stage embryos. Cell. 1980;21:927–34.
Hynes RO, Zhao Q. The evolution of cell adhesion. J Cell Biol. 2000;150:F89–96.
Ichii T, Takeichi M. p120-catenin regulates microtubule dynamics and cell migration in a cadherin independent manner. Genes Cells. 2007;12:827–39.
Ivanov AI, Nusrat A, Parkos CA. Endocytosis of the apical junctional complex: mechanisms and possible roles in regulation of epithelial barriers. Bioessays. 2005;27:356–65.
Izaguirre MF, Adur JF, Soler AP, Casco VH. Alterations induced by E-cadherin and beta-catenin antibodies during the development of Bufo arenarum (Anura-Bufonidae). Histol Histopathol. 2001;16:1097–106.
Izaguirre MF, García-Sancho MN, Miranda LA, Tomas J, Casco VH. Expression of cell adhesion molecules in the normal and T3 blocked development of the tadpole’s kidney of Bufo arenarum (Amphibian, Anuran, Bufonidae). Brazilian J Biol. 2008;68:561–9.
Izaguirre MF, Casco VH. T3 regulates E-cadherin, and β- and α-catenin expression in the stomach during the metamorphosis of the toad Rhinella arenarum. Biotech Histochem (USA). 2010;85:305–23.
Izaguirre MF, Larrea D, Adur JF, Diaz-Zamboni JE, Vicente N, Galetto CD, Casco VH. E-cadherin role in epithelial architecture maintenance. Cell Commun Adhes. 2010;17:1–12.
Izaguirre MF, Casco VH. Adhesión Intercelular en el Desarrollo de Vertebrados. Su Rol en el Desarrollo Embrionario y Larval de Anfibios. In: Editorial Académica Española. Saarbrücken: Germany, Press; 2011. p. 189.
Janda E, Nevolo M, Lehmann K, Downward J, Beug H, Grieco M. Raf plus TGFbeta-dependent EMT is initiated by endocytosis and lysosomal degradation of E-cadherin. Oncogene. 2006;25:7117–30.
Johnson SK, Ramani VC, Hennings L, Haun RS. Kallikrein 7 enhances pancreatic cancer cell invasion by shedding E-cadherin. Cancer. 2007;109:1811–20.
Jou TS, Stewart DB, Stappert J, Nelson WJ, Marrs JA. Genetic and biochemical dissection of protein linkages in the cadherin─catenin complex. Proc Natl Acad Sci U S A. 1995;92:5067–71.
Kane DA, McFarland KN, Warga RM. Mutations in half baked/E-cadherin block cell behaviors that are necessary for teleost epiboly. Development. 2005;132:1105–16.
Kardash E, Reichman-Fried M, Maître JL, Boldajipour B, Papusheva E, Messerschmidt EM, Heisenberg CP, Raz E. A role for Rho GTPases and cell–cell adhesion in single-cell motility in vivo. Nat Cell Biol. 2010;12:47–53.
Keller SH, Nigam SK. Biochemical processing of E-cadherin under cellular stress. Biochem Biophys Res Commun. 2003;307:215–23.
Kim SH, Li Z, Sacks DB. E-cadherin-mediated cell-cell attachment activates Cdc42. J Biol Chem. 2000;275:36999–7005.
Kim SW, Park JI, Spring CM, Sater AK, Ji H, Otchere AA, Daniel JM, McCrea PD. Non-canonical Wnt signals are modulated by the Kaiso transcriptional repressor and p120-catenin. Nat Cell Biol. 2004;6:1212–20.
Kim NG, Koh E, Chen X, Gumbiner BM. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc Natl Acad Sci U S A. 2011;108:11930–5.
King N, Hittinger CT, Carroll SB. Evolution of key cells ignaling and adhesion protein families predates animal origins. Science. 2003;301:361–3.
King N, Westbrook MJ, Young SL, Kuo A, Abedin M, Chapman J, Fairclough S, Hellsten U, Isogai Y, Letunic I, Marr M, Pincus D, Putnam N, Rokas A, Wright KJ, Zuzow R, Dirks W, Good M, Goodstein D, Lemons D, Li W, Lyons JB, Morris A, Nichols S, Richter DJ, Salamov A, Sequencing JGI, Bork P, Lim WA, Manning G, Miller WT, McGinnis W, Shapiro H, Tjian R, Grigoriev IV, Rokhsar D. The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature. 2008;451:783–8.
Klein I, Ojamaa K. Thyroid hormone and the cardiovascular system. N Engl J Med. 2001;344:501–9.
Koch AW, Manzur KL, Shan W. Structure-based models of cadherin-mediated cell adhesion: the evolution continues. Cell Mol Life Sci. 2004;61:1884–95.
Komuves LG, Hanley K, Jiang Y, Elias PM, Williams ML, Feingold KR. Ligands and activators of nuclear hormone receptors regulate epidermal differentiation during fetal rat skin development. J Invest Dermatol. 1998;111:429–33.
Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–64.
Korpal M, Kang Y. The emerging role of miR-200 family of microRNAs in epithelial-mesenchymal transition and cancer metastasis. RNA Biol. 2008;5:115–9.
Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–4.
Kovacs EM, Ali RG, McCormack AJ, Yap AS. E-cadherin homophilic ligation directly signals through Rac and phosphatidylinositol 3-kinase to regulate adhesive contacts. J Biol Chem. 2002;277:6708–18.
Kress E, Samarut J, Plateroti M. Thyroid hormones and the control of cell proliferation or cell differentiation: paradox or duality? Mol Cell Endocrinol. 2009;313:36–49.
Langevin J, Morgan MJ, Sibarita JB, Aresta S, Murthy M, Schwarz T, Camonis J, Bellaiche Y. Drosophila exocyst components Sec5, Sec6, and Sec15 regulate DE-Cadherin trafficking from recycling endosomes to the plasma membrane. Dev Cell. 2005;9:355–76.
Larue L, Ohsugi M, Hirchenhain J, Kemler R. E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc Natl Acad Sci U S A. 1994;91:8263–7.
Lazar MA. Thyroid hormone action: a binding contract. J Clin Invest. 2003;112:497–9.
le Duc Q, Shi Q, Blonk I, Sonnenberg A, Wang N, Leckband D, de Rooij J. Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II–dependent manner. J Cell Biol. 2010;189:1107–15.
Leckband D, Sivasankar S. Mechanism of homophilic cadherin adhesion. Curr Opin Cell Biol. 2000;12:587–92.
Leckband D, Prakasam A. Mechanism and dynamics of cadherin adhesion. Annu Rev Biomed Eng. 2006;8:259–87.
Leonhardt JM, Heymann WR. Thyroid disease and the skin. Dermatol Clin. 2002;20:473–81.
Levi G, Gumbiner B, Thiery JP. The distribution of E-cadherin during Xenopus laevis development. Development. 1991;111:159–69.
Levine E, Lee CH, Kintner C, Gumbiner BM. Selective disruption of E-cadherin function in early Xenopus embryos by a dominant negative mutant. Development. 1994;120:901–9.
Lickert H, Bauer A, Kemler R, Stappert J. Casein kinase II phosphorylation of E-cadherin increases E-cadherin/β-catenin interaction and strengthens cell–cell adhesion. J Biol Chem. 2000;275:5090–5.
Lien WH, Stepniak E, Vasioukhin V. Dissecting the role of cadherin–catenin proteins in mammalian epidermis. Proc Natl Acad Sci U S A. 2008;105:15225–6.
Lilien J, Balsamo J. The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta-catenin. Curr Opin Cell Biol. 2005;17:459–65.
Lin HY, Shih A, Davis FB, Davis PJ. Thyroid hormone promotes the phosphorylation of STAT3 and potentiates the action of epidermal growth factor in cultured cells. Biochem J. 1999;338:427–32.
Liu YN, Liu Y, Lee HJ, Hsu YH, Chen JH. Activated androgen receptor downregulates E-Cadherin gene expression and promotes tumor metastasis. Mol Cell Biol. 2008;28:7096–108.
Liwosz A, Lei T, Kukuruzinska MA. N-glycosylation affects the molecular organization and stability of E-cadherin junctions. J Biol Chem. 2006;281:23138–49.
Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol. 1997;139:1861–72.
Lock JG, Stow JL. Rab11 in recycling endosomes regulates the sorting and basolateral transport of E-cadherin. Mol Biol Cell. 2005;16:1744–55.
Lock JG, Hammond LA, Houghton F, Gleeson PA, Stow JL. E-cadherin transport from the trans-Golgi network in tubulovesicular carriers is selectively regulated by golgin-97. Traffic. 2005;6:1142–56.
Lorthongpanich C, Doris TPY, Limviphuvadh V, Knowles BB, Solter D. Developmental fate and lineage commitment of singled mouse blastomeres. Development. 2012;139:3722–31.
Maiden SL, Hardin J. The secret life of α-catenin: moonlighting in morphogenesis. J Cell Biol. 2011;195:543–52.
Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 2002;21:1948–56.
McCrea PD, Gumbiner BM. Purification of a 92-kDa cytoplasmic protein tightly associated with the cell-cell adhesion molecule Ecadherin (uvomorulin). Characterization and extractability of the protein complex from the cell cytostructure. J Biol Chem. 1991;266:4514–20.
McCrea PD, Gu D. The catenin family at a glance. J Cell Sci. 2010;123:637–42.
Meng W, Mushika Y, Ichii T, Takeichi M. Anchorage of microtubule minus ends to adherens junctions regulates epithelial cell-cell contacts. Cell. 2008;135:948–59.
Meng W, Takeichi M. Adherens junction: Molecular architecture and regulation. Cold Spring Harbor Perspect Biol. 2009;1:a002899.
Mengaud J, Lecuit M, Lebrun M, Nato F, Mazie JC, Cossart P. Antibodies to the leucine-rich repeat region of internalin block entry of Listeria monocytogenes into cells expressing E-cadherin. Infect Immun. 1996;64:5430–3.
Nagafuchi A, Takeichi M. Cell binding function of E-cadherin is regulated by the cytoplasmic domain. EMBO J. 1988;7:3679–84.
Nagafuchi A, Takeichi M. Transmembrane control of cadherin-mediated cell adhesion: a 94 kDa protein functionally associated with a specific region of the cytoplasmic domain of E-cadherin. Cell Regul. 1989;1:37–44.
Nakamura Y, Patrushev N, Inomata H, Mehta D, Urao N, Kim HW, Razvi M, Kini V, Mahadev K, Goldstein BJ, McKinney R, Fukai T, Ushio-Fukai M. Role of protein tyrosine phosphatase 1B in vascular endothelial growth factor signaling and cell-cell adhesions in endothelial cells. Circ Res. 2008;102:1182–91.
Nakayama K. Furin: A mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem J. 1997;327:625–35.
Nejsum LN, Nelson WJ. A molecular mechanism directly linking E-cadherin adhesion to initiation of epithelial cell surface polarity. J Cell Biol. 2007;178:323–35.
Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–7.
Nelson WJ. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem Soc Trans. 2008;36:149–55.
Nichols SA, Roberts BW, Richter DJ, Fairclough SR, King N. Origin of metazoan cadherin diversity and the antiquity of the classical cadherin/β-catenin complex. Proc Natl Acad Sci U S A. 2012;109:13046–51.
Niessen CM, Leckband D, Yap AS. Tissue organization by cadherin adhesion molecules: dynamic molecular and cellular mechanisms of morphogenetic regulation. Physiol Rev. 2011;91:691–731.
Noren NK, Liu BP, Burridge K, Kreft B. p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J Cell Biol. 2000;150:567–80.
Oda H, Takeichi M. Evolution: Structural and functional diversity of cadherin at the adherens junction. J Cell Biol. 2011;193:1137–46.
Oliveira P, Sanges R, Huntsman D, Stupka E, Oliveira C. Characterization of the intronic portion of cadherin superfamily members, common cancer orchestrators. Eur J Hum Genet. 2012;20:878–83.
Ozawa M, Kemler R. Correct proteolytic cleavage is required for the cell adhesive function of uvomorulin. J Cell Biol. 1990;111:1645–50.
Ozawa M, Engel J, Kemler R. Single amino acid substitutions in one Ca2+ binding site of uvomorulin abolish the adhesive function. Cell. 1990;63:1033–8.
Ozawa M, Kemler R. Molecular organization of the uvomorulin-catenin complex. J Cell Biol. 1992;116:989–96.
Palacios F, Price L, Schweitzer J, Collard JG, D’Souza-Schorey C. An essential role for ARF6-regulated membrane traffic in adherens junction turnover and epithelial cell migration. EMBO J. 2001;20:4973–86.
Park JI, Kim SW, Lyons JP, Ji H, Nguyen TT, Cho K, Barton MC, Deroo T, Vleminckx K, Moon RT, McCrea PD. Kaiso/p120catenin and TCF/beta-catenin complexes coordinately regulate canonical Wnt gene targets. Dev Cell. 2005;8:843–54.
Parra RG, Rohr CO, Koile D, Perez-Castro C, Yankilevich P. INSECT 2.0: a web-server for genome-wide cis-regulatory modules prediction. Bioinformatics. 2016;32:1229–31.
Pece S, Chiariello M, Murga C, Gutkind JS. Activation of the protein kinase Akt/PKB by the formation of E-cadherin-mediated cell-cell junctions. Evidence for the association of phosphatidylinositol 3-kinase with the E-cadherin adhesion complex. J Biol Chem. 1999;274:19347–51.
Peifer M, McCrea PD, Green KJ, Wieschaus E, Gumbiner BM. The vertebrate adhesive junction proteins b-catenin and plakoglobin and the Drosophila segment polarity gene armadillo form a multigene family with similar properties. J Cell Biol. 1992;118:681–91.
Peralta Soler A, Russo J, Russo IH, Knudsen KA. Soluble fragment of P-cadherin adhesion protein found in human milk. J Cell Biochem. 2002;85:180–4.
Perez-Moreno MA, Locascio A, Rodrigo I, Dhondt G, Portillo F, Nieto MA, Cano A. A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem. 2001;276:27424–31.
Perez-Moreno M, Jamora C, Fuchs E. Sticky business: orchestrating cellular signals at adherens junctions. Cell. 2003;112:535–48.
Pertz O, Bozic D, Koch AW, Fauser C, Brancaccio A, Engel J. A new crystal structure, Ca2+ dependence and mutational analysis reveal molecular details of E-cadherin homoassociation. EMBO J. 1999;18:1738–47.
Pinho SS, Seruca R, Gärtner F, Yamaguchi Y, Gu J, Taniguchi N, Reis CA. Modulation of E-cadherin function and dysfunction by N-glycosylation. Cell Mol Life Sci. 2011;68:1011–20.
Plateroti M, Kress E, Mori JI, Samarut J. Thyroid hormone receptor alpha1 directly controls transcription of the beta-catenin gene in intestinal epithelial cells. Mol Cell Biol. 2006;26:3204–14.
Pokutta S, Herrenknecht K, Kemler R, Engel J. Conformational changes of the recombinant extracellular domain of E-cadherin upon calcium binding. Eur J Biochem. 1994;223:1019–26.
Pokutta S, Weis WI. Structure and mechanism of cadherins and catenins in cell-cell contacts. Annu Rev Cell Dev Biol. 2007;23:237–61.
Posthausa H, Duboisb CM, Lapriseb M-H, Grondinb F, Sutera MM, Müllera E. Proprotein cleavage of E-cadherin by furin in baculovirus over-expression system: potential role of other convertases in mammalian cells. Febs Lett. 1998;438:306–10.
Prokhortchouk A, Hendrich B, Jorgensen H, Ruzov A, Wilm M, Georgiev G, Bird A, Prokhortchouk E. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 2001;15:1613–8.
Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. 2004;23:1739–48.
Radoja N, Stojadinovic O, Waseem A, Tomic-Canic M, Milisavljevic V, Teebor S, Blumenberg M. Thyroid hormones and gamma interferon specifically increase K15 keratin gene transcription. Mol Cell Biol. 2004;24:3168–79.
Reynolds AB, Daniel J, McCrea PD, Wheelock MJ, Wu J, Zhang Z. Identification of a new catenin: the tyrosine kinase substrate p120cas associates with E-cadherin complexes. Mol Cell Biol. 1994;14:8333–42.
Rikitake Y, Takai Y. Interactions of the cell adhesion molecule nectin with transmembrane and peripheral membrane proteins for pleiotropic functions. Cell Mol Life Sci. 2008;65:253–63.
Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS. Alpha 1(E)-catenin is an actin binding and -bundling protein mediating the attachment of F actin to the membrane adhesion complex. Proc Natl Acad Sci U S A. 1995;92:8813–7.
Rios-Doria J, Day KC, Kuefer R, Rashid MG, Chinnaiyan AM, Rubin MA, Day ML. The role of calpain in the proteolytic cleavage of Ecadherin in prostate and mammary epithelial cells. J Biol Chem. 2003;278:30469–77.
Rivard N. Phosphatidylinositol 3-kinase: a key regulator in adherens junction formation and function. Front Biosci. 2009;14:510–22.
Roczniak-Ferguson A, Reynolds AB. Regulation of p120-catenin nucleocytoplasmic shuttling activity. J Cell Sci. 2003;116:4201–12.
Rokas A. The origins of multicellularity and the early history of the genetic toolkit for animal development. Annu Rev Genet. 2008;42:235–51.
Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, Camargo FD. Yap1 acts downstream of α-catenin to control epidermal proliferation. Cell. 2011;144:782–95.
Schwartzkopff S, Gründemann C, Schweier O, Rosshart S, Karjalainen KE, Becker KF, Pircher H. Tumor-associated E-cadherin mutations affect binding to the killer cell lectin-like receptor G1 in humans. J Immunol. 2007;179:1022–9.
Shapiro L, Weis WI. Structure and biochemistry of cadherins and catenins. Cold Spring Harbor Perspect Biol. 2009;1:a003053.
Shi YB, Ishizuya-Oka A. Thyroid hormone regulation of apoptotic tissue remodeling: implications from molecular analysis of amphibian metamorphosis. Prog Nucleic Acid Res Mol Biol. 2001;65:53–100.
Shiraishi K, Tsuzaka K, Yoshimoto K, Kumazawa C, Nozaki K, Abe T, Tsubota K, Takeuchi T. Critical role of the fifth domain of E-cadherin for heterophilic adhesion with alpha E beta 7, but not for homophilic adhesion. J Immunol. 2005;175:1014–21.
Shirayoshi Y, Hatta K, Hosoda M, Tsunasawa S, Sakiyama F, Takeichi M. Cadherin cell adhesion molecules with distinct specificities share a common structure. EMBO J. 1986;5:2485–8.
Sirakov M, Plateroti M. The thyroid hormones and their nuclear receptors in the gut: from developmental biology to cancer. Biochim Biophys Acta. 2011;1812:938–46.
Sirakov M, Skah S, Lone IN, Nadjar J, Angelov D, Plateroti M. Multi-level interactions between the nuclear receptor TRα1 and the WNT effectors β-catenin/Tcf4 in the intestinal epithelium. PLoS ONE. 2012;7:e34162.
Spring CM, Kelly KF, O’Kelly I, Graham M, Crawford HC, Daniel JM. The catenin p120(ctn) inhibits Kaiso-mediated transcriptional repression of the beta-catenin/TCF target gene matrilysin. Exp Cell Res. 2012;305:253–65.
Stappert J, Kemler R. A short core region of E-cadherin is essential for catenin binding and is highly phosphorylated. Cell Adhes Commun. 1994;2:319–27.
Steinberg MS, McNutt PM. Cadherins and their connections: adhesion junctions have broader functions. Curr Opin Cell Biol. 1999;11:554–60.
Steinhusen U, Weiske J, Badock V, Tauber R, Bommert K, Huber O. Cleavage and shedding of E-cadherin after induction of apoptosis. J Biol Chem. 2001;276:4972–80.
Stemmler MP, Hecht A, Kinzel B, Kemler R. Analysis of regulatory elements of E-cadherin with reporter gene constructs in transgenic mouse embryos. Dev Dyn. 2003;227:238–45.
Stemmler MP, Hecht A, Kemler R. E-cadherin intron 2 contains cis-regulatory elements essential for gene expression. Development. 2005;132:965–76.
Stephenson RO, Yamanaka Y, Rossant J. Disorganized epithelial polarity and excess trophectoderm cell fate in preimplantation embryos lacking E-cadherin. Development. 2010;137:3383–91.
Symowicz J, Adley BP, Gleason KJ, Johnson JJ, Ghosh S, Fishman DA, Hudson LG, Stack MS. Engagement of collagen-binding integrins promotes matrix metalloproteinase-9-dependent E-cadherin ectodomain shedding in ovarian carcinoma cells. Cancer Res. 2007;67:2030–9.
Takahashi K, Nakanishi H, Miyahara M, Mandai K, Satoh K, Satoh A, Nishioka H, Aoki J, Nomoto A, Mizoguchi A, Takai Y. Nectin/PRR: an immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with Afadin, a PDZ domain-containing protein. J Cell Biol. 1999;145:539–49.
Takeichi M. Cadherins: a molecular family important in selective cell-cell adhesion. Annu Rev Biochem. 1990;59:237–52.
Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;251:1451–5.
Takeichi M. Morphogenetic roles of classical cadherins. Curr Opin Cell Biol. 1995;7:619–27.
Takeichi M, Nakagawa S, Aono S, Usui T, Uemura T. Patterning of cell assemblies regulated by adhesion receptors of the cadherin superfamily. Philos Trans R Soc Lond B. 2000;355:885–90.
Tanos B, Rodriguez-Boulan E. The epithelial polarity program: machineries involved and their hijacking by cancer. Oncogene. 2008;27:6939–57.
Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–42.
Tomic-Canic M, Day D, Samuels HH, Freedberg IM, Blumenberg M. Novel regulation of keratin gene expression by thyroid hormone and retinoid receptors. J Biol Chem. 1996;271:1416–23.
Tucker RP, Adams JC. Adhesion networks of cnidarians: a postgenomic view. Int Rev Cell Mol Biol. 2014;308:323–77.
Tunggal JA, Helfrich I, Schmitz A, Schwarz H, Günzel D, Fromm M, Kemler R, Krieg T, Niessen CM. E-cadherin is essential for in vivo epidermal barrier function by regulating tight junctions. EMBO J. 2005;24:1146–56.
Uemura T, Oda H, Kraut R, Hayashi S, Kotaoka Y, Takeichi M. Zygotic Drosophila E-cadherin expression is required for processes of dynamic epithelial cell rearrangement in the Drosophila embryo. Genes Dev. 1996;10:659–71.
van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 2008;65:3756–88.
Wang Y, Kaiser MS, Larson JD, Nasevicius A, Clark KJ, Wadman SA, Roberg-Perez SE, Ekker SC, Hackett PB, McGrail M. Moesin1 and VE-cadherin are required in endothelial cells during in vivo tubulogenesis. Development. 2010;137:3119–28.
Wang Y-C, Khan Z, Kaschube M, Wieschaus EF. Differential positioning of adherens junctions is associated with initiation of epithelial folding. Nature. 2012;484:390–3.
Wang Y-C, Khan Z, Wieschaus EF. Distinct Rap1 activity states control the extent of epithelial invagination via α-catenin. Dev Cell. 2013;25:299–309.
Wheelock MJ, Johnson KR. Cadherin-mediated cellular signaling. Curr Opin Cell Biol. 2003;15:509–14.
Whitehead J, Vignjevic D, Fütterer C, Beaurepaire E, Robine S, Farge E. Mechanical factors activate beta-catenin-dependent oncogene expression in APC mouse colon. HFSP J. 2008;2:286–94.
Whittard JD, Craig SE, Mould AP, Koch A, Pertz O, Engel J, Humphries MJ. E-cadherin is a ligand for integrin alpha2beta1. Matrix Biol. 2002;21:525–32.
Woodfield RJ, Hodgkin MN, Akhtar N, Morse MA, Fuller KJ, Saqib K, Thompson NT, Wakelam MJ. The p85 subunit of phosphoinositide 3-kinase is associated with beta-catenin in the cadherin-based adhesion complex. Biochem J. 2001;360:335–44.
Yagi T, Takeichi M. Cadherin superfamily genes: functions, genomic organization, and neurologic diversity. Genes Dev. 2000;14:1169–80.
Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–39.
Yap AS, Niessen CM, Gumbiner BM. The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn. J Cell Biol. 1998;141:779–89.
Yap AS, Kovacs EM. Direct cadherin-activated cell signaling: a view from the plasma membrane. J Cell Biol. 2003;160:11–6.
Yap AS, Crampton MS, Hardin J. Making and breaking contacts: the cellular biology of cadherin regulation. Curr Opin Cell Biol. 2007;19:508–14.
Yen PM. Physiological and molecular basis of thyroid hormone action. Physiol Rev. 2001;81:1097–142.
Yen PM, Ando S, Feng X, Liu Y, Maruvada P, Xia X. Thyroid hormone action at the cellular, genomic and target gene levels. Mol Cell Endocrinol. 2006;246:121–7.
Yonemura S, Wada Y, Watanabe T, Nagafuchi A, Shibata M. Alpha-catenin as a tension transducer that induces adherens junction development. Nat Cell Biol. 2010;12:533–42.
Yoshida C, Takeichi M. Teratocarcinoma cell adhesion: identificacion of a cellsurface protein involved in calcium-dependent cell aggregation. Cell. 1982;28:217–24.
Zaidel-Bar R. Cadherin adhesome at a glance. J Cell Sci. 2013;126:373–8.
Zhou F, Su J, Fu L, Yang Y, Zhang L, Wang L, Zhao H, Zhang D, Li Z, Zha X. Unglycosylation at Asn-633 made extracellular domain of E-cadherin folded incorrectly and arrested in endoplasmic reticulum, then sequentially degraded by ERAD. Glycoconj J. 2008;25:727–40.
Zhu W, Leber B, Andrews DW. Cytoplasmic O-glycosylation prevents cell surface transport of E-cadherin during apoptosis. EMBO J. 2001;20:5999–6007.
Acknowledgements
We are grateful to Professor Diana Mónica Waigandt for her help with the revision of the English version of the manuscript for publication and to Dr. Alejandro Peralta Soler for his critical reading and valuable suggestions to improve the manuscript.
Funding
This work was supported by grants from: National Agency for Scientific and Technological Promotion-MINCYT, Argentina and UNER: PICTO-2009-209 (to Victor Hugo Casco). SCYTFRH-UNER 6019–1 (to Victor Hugo Casco); SCYTFRH-UNER 6067–1 (to Victor Hugo Casco); SCYTFRH-UNER 6088–1 (to María Fernanda Izaguirre).
Availability of data and materials
Not applicable.
Authors’ contributions
IMF and CVH devised and wrote the manuscript. Both authors have read and accepted the final version of the manuscript.
Authors’ information
María Fernanda Izaguirre is BA in Biochemistry and PhD in Biological Sciences Victor Hugo Casco is BA and PhD in Biological Sciences. Both are Professors of Molecular and Cell Biology and carry out research on the function of cell-cell adhesion molecules during development and adult life of animals.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethical approval and consent to participate
The research protocol was approved by the local Research Institutional Ethics Committee of Entre Ríos National University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Izaguirre, M.F., Casco, V.H. E-cadherin roles in animal biology: A perspective on thyroid hormone-influence. Cell Commun Signal 14, 27 (2016). https://doi.org/10.1186/s12964-016-0150-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-016-0150-1