
Abstract
Glioma is the most aggressive and malignant type of primary brain tumor, comprises the majority of central nervous system deaths, and is categorized into different subgroups according to its histological characteristics, including astrocytomas, oligodendrogliomas, glioblastoma multiforme (GBM), and mixed tumors. The forkhead box (FOX) transcription factors comprise a collection of proteins that play various roles in numerous complex molecular cascades and have been discovered to be differentially expressed in distinct glioma subtypes. FOXM1 and FOXOs have been recognized as crucial transcription factors in tumor cells, including glioma cells. Accumulating data indicates that FOXM1 acts as an oncogene in various types of cancers, and a significant part of studies has investigated its function in glioma. Although recent studies considered FOXO subgroups as tumor suppressors, there are pieces of evidence that they may have an oncogenic role. This review will discuss the subtle functions of FOXOs and FOXM1 in gliomas, dissecting their regulatory network with other proteins, microRNAs and their role in glioma progression, including stem cell differentiation and therapy resistance/sensitivity, alongside highlighting recent pharmacological progress for modulating their expression.
Similar content being viewed by others
Introduction
Glioma, a nervous system (CNS) tumor with a high recurrence rate, is responsible for 81% of adults' most primary invasive brain tumors and 30% of CNS malignancies [1]. Gliomas are classified as astrocytoma, oligodendrogliomas, ependymomas, or oligoastrocytoma based on the malignancy intensity and their histologic origination oligodendrocytic and astrocytic components of the CNS [2]. World Health Organization (WHO) categorized glioma into four grades (grades I to IV). Gliomas with WHO grades I and II are classified as low-grade gliomas (LGG). In contrast, those with grades III and IV are classified as high-grade gliomas (HGG)[3]. HGG includes several tumors consisting of glioblastoma multiforme (GBM), anaplastic oligodendroglioma (OA), and anaplastic astrocytoma (AA) [4]. The median survival time of HGGs after conventional treatments is approximately 2 to 5 years for anaplastic glioma [5] and less than 15 months for glioblastoma [6, 7]. LGGs include oligo-astrocytomas or mixed gliomas, astrocytomas, and oligodendrogliomas with an average survival rate of 7 years and eventually progress to HGGs [8]. Conventional treatments were limited to chemotherapy and radiotherapy in the past; however, despite the recent development of novel treatments such as molecular targeted therapy, stem cell therapy, immunotherapy, gene therapy, and genomic corrections, the survival rate of patients has not improved significantly in clinical settings, majorly because of low brain-blood-barrier (BBB) permeability and occurrence of the resistance to treatment [9]. Therefore, a subtle understanding of the molecular mechanisms involved in glioma progression, therapy resistance, and glioma stem cell-induced differentiation is needed for developing the efficacy of available treatments [10].
Transcription factors (TFs) play critical roles in the transcriptional processes that control gene expression; dysregulation of muted TFs is prevalent in glioma and can lead to the development of tumor-related characteristics. Various expressed TFs and their downstream targets in glioma could be utilized for therapeutic goals [11]. FOX proteins are a broad group of transcription factors that play key roles in a variety of cellular mechanisms, including cellular growth, cell differentiation, proliferation, and cell cycle control. FOX proteins are classified according to a DNA binding motif consisting of 80 to 100 amino acids, known as the FKH domain or the fork head box [12, 13]. Thus, they are categorized into 19 subtypes according to similarities in the FKH domain; despite the fact that the FOX proteins have highly similar DNA binding domains, they have diverse tissue-specific transcriptional regulation and regulatory mechanisms that allow them to perform their specialized tasks [14, 15].
FOXO is a member of the FOX family, including four subtypes (FOXO1, FOXO3, FOXO4, and FOXO6). Growth factors which are essential for stimulation of the phosphatidylinositol 3-kinase–protein kinase B (PI3K-AKT), regulate FOXO function and phosphorylation of Akt, resulting in activation of FOXO. Moreover, FOXOs are involved in various physiological and pathological mechanisms, including cell cycle arrest, apoptosis, stem cell differentiation, and oxidative stress [16, 17]. The controversial and complex regulatory functions of FOXOs in tumorigenesis have been documented. Despite their well-known tumor-suppressing properties, FOXOs can potentially induce cancer in some circumstances [18]. For instance, FOXO1s downregulation is linked to poor prognosis and decreased survival rate in myeloid leukemia (AML), soft tissue sarcoma, and breast cancer [19,20,21]. In contrast, the deactivation of FOXO1 in gastric cancer contributes to better outcomes, while its activation in B-cell lymphomas was shown to be associated with cancer progression [22, 23].
FOXM1 is associated with several human carcinomas, and alterations in FOXM1 signaling are correlated with carcinogenesis and oncogenesis in gliomas, prostate, lung, colorectal, breast, and hepatocellular cancers [24, 25]. In malignant glioma, abnormal FOXM1 expression has been discovered to be a prevalent molecular change. Furthermore, increased FOXM1 expression has been linked to radioresistance and poor prognosis in GBM patients [26]. In glioma, FOXM1 interacts with critical signaling pathways and molecules, including MELK, STAT proteins, Wnt/β-Catenin, growth factors, and non-coding RNAs [26,27,28,29].
Several exclusive reviews have emphasized the role of FoxM1 and FoxOs in ovarian cancer [30] and hepatocellular carcinoma [31], respectively. However, a study to do so in gliomas is missing. Therefore, we aimed to conduct this review to fill the missing gaps and shed more light on the role of these transcription factors in the pathogenesis of gliomas.
Methods
First, we searched PubMed on 14 May 2023 to estimate the number of published articles regarding forkhead box transcription factors in glioma using the following terms: ([Name of FOX protein]) AND (Glioma OR Glioblastoma OR Astrocytoma OR Ependymoma OR Oligodendroglioma OR Oligoastrocytoma). According to our initial assessment, the most frequently studied FOX proteins were FOXP3, followed by FOXM1, FOXO3(a), and FOXO1 (Fig. 1). However, as a subtype of regulatory T cells are also termed FOXP3+ cells, the number of studies that evaluated FOXP3 function was relatively few, leading us only to review the function of FOXM1 and FOXOs transcription factors in glioma deeply. Our inclusion criteria were original studies that evaluated these proteins' biological, prognostic, or pharmacological function in gliomas. Exclusion criteria were review articles, case reports or series, letters, editorials, consensus statements, conference abstracts, and retracted articles. The flow chart of the study selection is shown in Fig. 2.

Results of preliminary search of PubMed database

Flowchart of study selection
FoxM1
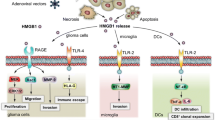
FOXM1 (forkhead box protein M1, also known as HNF-3, HFH-11, or Trident) is a transcription factor whose overexpression was implicated in the carcinogenesis of diverse tumors, especially glioma [32, 33]. This molecule is regulated at different stages of gene expression, including (a) transcriptional level (mostly cis-activated via interaction of different molecules with its binding sites and promotor), (b) post-transcriptional level (notably by non-coding RNAs: including miRNAs, lncRNAs, and circRNAs), (c) post-translational level (via mechanisms such as phosphorylation, ubiquitination, and de-ubiquitination), and (d) direct interaction of protein/RNAs with FOXM1 protein [32]. Due to this variety of targets for controlling FOXM1 expression, its inhibition seems to be a promising strategy in cancer [32]. In higher-grade gliomas, including anaplastic astrocytoma and glioblastoma, the expression of FOXM1 is significantly elevated, resulting in tumor recurrence [34,35,36,37]. In glioma tumor-initiating cells (TICs), FOXM1 is a critical factor implicated in the proliferation and self-renewal of cancer cells [38]. In this section, we will discuss the importance of FOXM1 in glioma progression, alongside mentioning its upstream and downstream regulators (Fig. 3).

A summary of FOXM1 regulation in gliomas. Activation of growth factors and tyrosine kinases can subsequently promote FoxM1 translocation to the nucleus via inducing its translocation. In the nucleus, FOXM1 can transcriptionally regulate the expression of various targets by biding to their promoter, while several transcription factors (e.g., HSF1, FGFR1, HIF-1a, and HMGA2) can transactivate FOXM1. In addition, several miRNAs that target 3’ UTR of FOXM1 mRNA are downregulated in glioma cell lines. All of these processes lead to cell proliferation, migration, invasion, angiogenesis, as well as resistance to chemoradiotherapy
FOXM1 interplay with crucial signaling pathways and molecules in glioma
PI3K/AKT signaling pathway
Phosphatidylinositol 3-kinase (PI3K)/Akt, as an overactivated signaling axis, is known for contributing to the progression of malignant gliomas (GBMs) [39]. It is well established that activation of Akt can directly affect FOXM1 in solid cancers by forming a positive loop in a reciprocal manner [40]. Zhang et al. have shown that increased Akt expression can provoke FOXM1 activity. Their results demonstrated that MYB-related protein B (B-MYB/MYBL2) and FOXM1, both transcriptional factors, are co-expressed together. Their expression is strongly correlated with poor clinical outcomes and grades of gliomas. In addition, decreases in their expression can suppress glioma progression by inducing apoptosis, delay of cells in the G2 phase, and inhibiting migration, invasion, and EMT [41]. These results align with a previous study by Wang et al. which showed that binding chemokine CXCL12 to its receptor CXCR4 could significantly induce FOXM1 expression via the PI3K/AKT pathway [42]. Due to this, using a dual inhibitor of histone deacetylases (HDAC) and PI3K, such as CUDC-907, can suppress the expression and transcriptional activity of FOXM1 in high-grade gliomas, leading to radiosensitization [43].
MELK
Maternal embryonic leucine zipper kinase (MELK) belongs to a group of serine/threonine kinases that physiologically modulates organogenesis during the embryonic period; however, its overexpression leads to the progression of many cancers, including GBM, majorly via activating transcription factors such as FOXM1 [44]. In more detail, the activation of FOXM1 by MELK in GSCs is mediated by another kinase named PLK1. Therefore, targeting the complex composed of these proteins can be considered a desirable target in GBM [28]. The importance of the MELK/FOXM1 complex even gets bolder when EZH2, as an emerging therapeutic molecule in brain tumors [45], was confirmed to be a target of this complex in GBM spheres [46]. The MELK/FOXM1 axis has received more attention in recent years due to its significance in high-grade gliomas, and newer investigations have uncovered other upstream regulators involved in chromatin remodeling, such as SAT1 in the regulation of MELK and EZH2 [47].
STAT proteins
Similar to FOX proteins, STATs (signal transducers and activators of transcription) are a group of transcription factors that are mainly localized in the cytoplasm of cells; however, upon phosphorylation are translocated to the nucleus and affect target genes’ expression following the activation of cytokines (e.g., CXCR4) or growth factors (EGFR)[48, 49]. In GBM cells, FOXM1 is correlated with STAT3 levels, and inhibition of FOXM1 can prevent growth factor- and cytokine-induced STAT3 activation [50]. Schonberg et al. have found that ferritin, which stores and regulates iron ions, is preferentially expressed in GBM stem cells and associated with poor survival. They further noticed the expression of ferritin has the highest correlation with STAT3. Since FOXM1 correlates strongly with STAT3 levels, both of them can be targeted by ferritin knockdown [51, 52]. In addition, the interaction between FOXM1 and STAT3 is necessary for GBM cells’ resistance to radiation and DNA damage, which will be the point of our focus in the next parts [26]. Moreover, not only STAT3 but also STAT1 can control FOXM1 expression in different glioma cell lines (U87, A172, U251, and T98), influencing other signaling pathways implicated in inflammation, such as NF-κB [53].
Wnt/β-catenin signaling pathway
Wnt pathway is an evolutionary conserved pathway required for embryonic differentiation and development, and recent studies frequently addressed the consequences of its dysregulation in glioma tumorigenesis. The Canonical Wnt pathway is also referred to as the Wnt/β-Catenin pathway since it leads to the accumulation of β-Catenin in the nucleus affecting a crucial transcription factor named TCF4 responsible for Wnt target genes expression [54]. A study by Zhang et al. turned out FoxM1 acts as a downstream for canonical Wnt pathway in glioma and is required for β-catenin activation by its translocating to the nucleus, leading to self-renewal and tumorgenicity of GBM-initiating cells (GICs) [55]. More importantly, the expression of the previously mentioned protein transcription factor STAT3 is mediated by FoxM1 via enhancing β-catenin/TCF4 binding to the STAT3 gene promoter [50].
Growth factors
Some studies have mentioned the positive impact of FOXM1 on the expression of growth factors, including vascular endothelial growth factor (VEGF) [56] and epidermal growth factor receptor(EGFR) [57] in high-grade gliomas, all necessary for the growth and proliferation of GSCs. While FOXM1 can target growth factors expression, the receptor of growth factors such as fibroblast growth factor receptor 1 (FGFR1) has been reported to regulate the expression of FOXM1 in GBM stem cells, leading to increased expression of EMT genes, resistance to ionizing radiation, and GBM relapse after chemo-radiotherapy [58].
m6A modification pathway
One of the most common and frequent RNA modifications observed in eukaryotes is the m6A modification, also known as N6-methyladenosine modification. In RNA molecules, it includes attaching a methyl group to the nitrogen atom at the sixth position of the adenosine base [59]. ALKBH5 is an m6A demethylase that plays a critical role in regulating m6A modification. By removing the m6A mark from RNA molecules, ALKBH5 influences RNA stability and metabolism, consequently influencing gene expression and various biological processes in cancers [60]. In GBM stem cells (GSCs), a significantly elevated expression of ALKBH5 has been observed, which is essential for stem cell self-renewal. ALKBH5 could maintain FOXM1 mRNA stability by demethylating its nascent transcripts in GSCs, leading to tumor growth [61]. Thus, selective ALKBH5 inhibitors such as Ena15 and Ena21 are promising strategies against glioma progression as they could decrease tumor growth in different GBM cell lines [62].
Hedgehog signaling pathway
The hedgehog signaling system is a multidimensional molecular signaling network in animals, including humans, that plays a crucial part in embryonic development, tissue maintenance as well as cancer by controlling cell differentiation and proliferation. When hedgehog proteins attach to a receptor known as Patched, they activate another protein named Smoothened. This sets off a chain of intracellular events that activate transcription factors known as GLI proteins. GLI proteins regulate the expression of target genes in the pathway as well as other downstream signaling pathways [63]. There is evidence that GLI1 and FOXM1 are co-expressed in GBM cells. In more detail, it has been shown that FOXM1, via promoting transcription of a nuclear importer protein named IPO7, increases the nuclear localization of GLI1 proteins. The FOXM1/IPO7/GLI1 axis contributes to the proliferation, migration, and invasion of GBM cells [64]. GLI1 has a prominent role in the malignant transformation of immortalized human astrocytes [65]. These data show a positive feedback loop exists between GLI1 and FOXM1 transcription factors in different subtypes of gliomas [64, 66].
Other regulators and the role of non-coding RNAs
Alongside signaling pathways and molecules discussed above, various studies have indicated the relationship between FoxM1 and other molecules involved in glioma progression. As seen in Table 1, FoxM1 can regulate (upstream regulating) or be regulated (as downstream target) by a variety of molecules. Among upstream regulators of FoxM1, the prominent role of non-coding RNAs is worth mentioning. Though these classes of RNAs are not encoded into proteins, they play a significant role in epigenetic regulation of other proteins at different stages of gene expression [67]. Due to the ability of circular RNAs and long non-coding RNAs to act as a sponge for shared microRNAs at the post-transcriptional level, they are also referred to as competing endogenous RNAs (ceRNAs). In addition, the mechanism of action by which microRNAs exert their role is mostly through targeting the 3′ UTR of mRNAs [63]. Since FOXM1 acts as an oncogene in cancers and glioma is not an exception, those miRNAs whose target is FOXM1 are usually downregulated, leading to its overexpression and exacerbating glioma’s malignancy. On the other hand, the downregulation of these miRNAs is affected by oncogenic lncRNAs and circRNAs as well, which are upregulated (Table 1).
FOXM1 and treatment opportunities in glioma
Radiotherapy
While radiotherapy combined with other treatments such as chemotherapy is considered a conventional treatment after surgical resection in high-grade gliomas, failure in treatment is frequently seen due to radioresistant exhibited by tumor cells, particularly glioma stem cells (GSCs). Various molecular pathways are involved in the radioresistance of gliomas; on top of them, there are AKT, Wnt/β-catenin, and STAT3 [104]. Surprisingly, FOXM1 is a downstream target affected by them. For this reason, FOXM1 can be considered a promising target for overcoming radiotherapy resistance in gliomas [105], as activation of the abovementioned oncogenic signaling pathways and proteins subsequently leads to the aberrant activation of this protein and radioresistance. One mechanism by which FOXM1 contributes to radiation resistance is its DNA repair capability. Cells undergoing radiation often overexpress FOXM1 to prevent further DNA damage [106]. Since FOXM1 is involved in cell cycle regulation and DNA repair, it plays a significant role in driving transcriptional response against radiation in high-grade gliomas [105]. Not only FOXM1 but also its targets also have been shown to be implicated in the radioresistance of gliomas. The previously mentioned MYBL2, as a downstream protein upregulated by AKT/FOXM1 axis, can be used as radiosensitivity biomarker for diagnosing patients with no response to radiotherapy [41]. Similarly, the expression of both STAT3 and FOXM1 was shown to be concurrent following radiation treatment in high-grade gliomas [26]. The studies emphasizing the role of FOXM1 in radioresistance in glioma have been summarized in Table 1.
Chemotherapy
The most commonly used chemotherapy regimens against high-grade gliomas are temozolomide (TMZ), bevacizumab, nitrosourea agents (e.g., carmustine), and platinum-based agents (e.g., cisplatin, carboplatin, and oxaliplatin). However, resistance to these drugs is commonly seen [107]. Like many other transcription factors, FOXM1 is strongly associated with the processes related to DNA repair, making glioma cells resistant to chemotherapy as well. Therefore, lowering FOXM1 has been shown to be associated with temozolomide (TMZ) sensitivity in GBM cell lines following the downregulation of DNA-repair-responsible genes such as Rad51 and RFC5 [95, 96]. Various FOXM1 inhibitors have been found in gliomas with chemosensitizing effects on in vivo and in vitro models (Table 2). For example, previous studies have found that FOXM1 can serve as a general target for proteasome inhibitors (PIs) in different cancer cell lines [108]. Bortezomib is a PI that has shown TMZ-sensitizing properties via inhibiting FOXM1 in both cellular and pre-clinical models for the treatment of high-grade gliomas [109]. However, since this agent cannot pass through the blood–brain barrier (BBB), early clinical trials generally have been accompanied by unsatisfactory outcomes, and newer generations of PIs, such as marizomib, were more successful [110]. Takei et al. have shown that GBM patients with low expression of FOXM1 had better overall survival compared to those with high levels of FOXM1 after neoadjuvant therapy with Bortezomib. Therefore, FOXM1 can be used as a biomarker for evaluating response treatment in GBM patients [111]. The treatments which target FOXM1 in glioma have been summarized in Table 2.
Immunotherapy
Immune checkpoint blockade, cytokine therapy, dendritic cell vaccines, viral therapy, and CAR-T therapy all have been tried as immunotherapeutic approaches against gliomas, and among them, immune checkpoint inhibitors and CAR-T therapy have shown promising therapeutic values in clinical trials [112]. A recent clinical trial has highlighted the efficacy of early treatment with a vaccine-based immunotherapy approach using glioma oncoantigens (GOAs) containing FOXM1 before starting chemotherapy or radiotherapy to prevent possible chemo-radio resistance [113]. It has been shown that chimeric antigen receptor (CAR) T cells with costimulatory MyD88 and CD40 (MC) endo-domains have a higher levels of FOXM1, indicating that stimulation of FOXM1 in CAR-T cells might improve the results of immunotherapy [114].
FOXO family
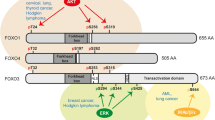
The "O" subfamily of forkhead box transcription factors consists of four members, including FOXO1 (FKHR), FOXO3 (FKHRL1), FOXO4 (AFX), and FOXO6, generally considered tumor suppressors via inducing apoptosis and inhibiting proliferation. While FOXO proteins are majorly silenced following PI3K/AKT and Ras/MEK-ERK(MAPK) pathways overactivity, they can be activated by oxidative stress regulators such as JNK (c-Jun N-terminal kinase) and MST1 (Mammalian Ste20-like kinase). Moreover, varieties of tumoral processes, including invasiveness, angiogenesis, metastasis, and drug response/resistance, are dependent on their deregulation [126]. This standpoint has been revisited against previous thoughts regarding the tumor-suppressive effects of FOXOs. Multiple theories have been proposed to justify this controversy. Depending on the context, the stage in which tumor cells are plays an essential role in the consequences of FOXOs’ transcriptional output. The impact of epigenetics, concurrent signaling pathways, and spatial localization of cells in tumor spheroids were shown as different factors responsible for metastasis-promoting outputs of FOXOs expression [127]. On the one hand, the interplay between PI3K/AKT pathway and FOXOs [126], and on the other hand, the interaction with WNT/β-catenin and TGF-β pathway is supposed to be an essential factor in forming a balance between the anti-tumor and tumor-promoting activity of FOXOs [127].
FOXO1
Post-transcriptional modifications (e.g., phosphorylation, ubiquitination, acetylation, and deacetylation) of FOXO1 were shown to play a substantial role in regulating cell proliferation, apoptosis, autophagy, and oxidative stress [128]. The controversial role of FOXO1 in tumorigenesis is also seen in gliomas. A recent study by Chen et al. has indicated the anti-tumor capacities of FOXO1 in GBMs favor prolonged cell survival and decreased migration, invasion, cell adhesion (EMT), and drug resistance to chemotherapeutic agents such as TMZ, BCNU, or cisplatin [129]. However, these findings are in contrast with their previous study that showed both nuclear and cytoplasmic FOXO1 expression is increased in astrocytomas and GBM cells, associated with poor survival [130]. Likewise, in a recent study with a small sample size by Huang et al. immune-cytoplasmic-staining scores of FOXO1a helped distinguish low-grade-gliomas from non-neoplastic lesions but did not correlate significantly with WHO grades [131]. Later, further research conducted on TCGA-LGG and GTEx brain databases showed that low-grade gliomas have a significantly upregulated FOXO1 expression. A nomogram containing this gene alongside other autophagy-related genes (e.g., GRID2, MYC, PTK6, IKBKE, BIRC5, and TP73) could predict the survival of patients with excellent accuracy (AUC: 0.81–0.90) [132]. Another study has revealed that in both GBM and lower-grade gliomas undergoing hypoxia (higher expression of HIF-1α,) the expression of FOXO1 is also elevated [133]. However, more experimental studies than bioinformatic studies are required to confirm these results. Due to this duality, for each study reviewed here, the tumor-suppressive or tumor-supportive features of FOXOs will be highlighted (Fig. 5 and Table 3).
Akt and FOXO1 in glioma
Given that the PI3K/AKT/mTOR pathway is overactivated in 90 percent of GBMs and is closely related to FOXOs activity, controlling its expression can serve as an indirect approach for targeting FOXOs as well [39, 134]. However, it clearly has been established that inhibition of other signaling pathways and oncogenes should be taken into account for attaining therapeutic response. Tumor suppressor p53 has an old reputation for maintaining radiation response in different cancers [135]. Therefore, its intact activity in GBM stem cells was shown to be necessary for an adequate response to combined treatment with PI3K/mTOR inhibitors and ionizing gamma radiation followed by loss of stemness markers (e. g., SOX2, nestin, or Musashi) and FOXO1/FOXO3a decrease [136]. Another study has also shown that FOXO1 can increase the expression of a stem cell marker named OCT4, exerting an oncogenic impact in GBM cells (Fig. 5B) [137]. Moreover, the knockdown of FOXO1 was slightly able to increase response to the above-mentioned treatments, representing that for attaining higher levels of response, both FOXO1 and FOXO3a should be inhibited together. This gets more confusing when a recent study revealed that targeting FOXO1 by miR-5188 is necessary for the activation of PI3K/AKT/c-JUN signaling pathway in U87 and U251 glioma cell lines, supporting the tumor-suppressive side of FOXO1 [138]. Similarly, another study has shown that the anti-tumor features of herbal medicine named Xihuang Pill are exerted through dephosphorylation of Akt and mTOR, resulting in decreased phosphorylation of FOXO1 and its subsequent translocation to the nucleus to induce apoptosis [139]. The interplay between FOXOs and PI3K/AKT could be a possible factor in causing pro or anti-apoptotic effects; however, targeting PI3K/AKT alone is not enough to control FOXO1 expression since it can be phosphorylated independently by other upstream regulators, such as mTORC2 [140] or CDKs [141].
FOXO1 and metabolism in glioma
FOXO1 regulates processes related to energy homeostasis and glucose metabolism under physiological conditions in organs such as the pancreas, liver, skeletal muscle, and adipose tissue [142]. Recent studies also support its role in cancer metabolism as well [18]. In GBM cells, upon either FOXO1 or PI3K/mTOR inhibition, the expression of genes involved in glycolysis, such as LDHA, is reduced. However, surprisingly when both of them are inhibited, not only LDHA but ENO1 as glycolytic genes associated with poorer survival are increased, supporting the theory that for the efficacy of PI3/mTOR inhibitors against glycolysis, the intact activity of FOXO1 is necessary [143](Fig. 5B). Masui et al. [140] have shown that mTORC2, independent of PI3K/AKT activity, suppresses FOXO1/FOXO3 activity by promoting their acetylation (Fig. 5A). Activation of mTORC2 also leads to suppression of miR-34c, a miRNA that targets c-Myc. When c-Myc is upregulated, the Warburg effect (as a hallmark of cancer) is promoted and assists cell survival. Furthermore, another study has pointed out that treatment with Progesterone (a pleiotropic steroid hormone) in GBM cells can exert anti-tumor properties and suppress glycolysis and Warburg’s effect via inhibiting GLUT1, GAPDH, and cytoplasmic activity of FOXO1 [144].
FOXO1 and cell cycle regulation in glioma
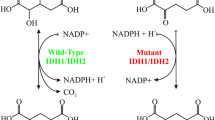
Regulation of the cell cycle has been proposed as an essential mechanism in which FOXO proteins exert their tumor-suppressive functions via repressing the activity of various proto-oncogenes, including cyclins (e.g., A, E, D) and cyclin-dependent kinases (CDKs including 2,4, and 6) [145]. The reciprocal interplay between cycle cell regulators and FOXOs in glioma has been reported in various studies. Restoring FOXO1 expression in gliomas can cause cell cycle arrest at the G2/M phase via phosphorylating CDK1 at s249, resulting in hindered cell proliferation and increased apoptosis [146]. Under metabolic stress conditions, cyclin F but not cyclin A, cyclin B, cyclin D, or cyclin E is expressed frequently in glioma cell lines. In more detail, the binding of FOXO1 but not FOXO3a, FOXO4, or FOXO6 to cyclin F promoter subsequently represses the expression of IDH1 as a crucial proto-oncogene in glioma [147]. In addition, it’s well established from a long time ago that cyclin-dependent kinases by phosphorylating FOXO1 on S249 cause its cytoplasmic localization and decreased activity [145]. Therefore, using CDK2 inhibitors was shown to increase the nuclear translocation of FOXO1 in U87 glioma cells more than in U251 cells [141]. Similarly, other cyclin-dependent kinase inhibitors (CDKi), such as p21Cip1, were shown to be activated by FOXO1/SMAD complex, following TGF-β signaling pathway activation in GBMs. However, PI3K/Akt signaling, as well as other forkhead transcription factors such as FOXG1, both acted as antagonists for FOXO1 by preventing TGF-β induced cytostasis(Fig. 4) [148].

Cell cycle regulation by FOXO1 in gliomas
FOXOs can induce cell cycle arrest at different stages by inhibiting cyclins and cyclin-dependent kinases. In addition, reciprocal phosphorylation of FOXO1 and CDKs is a crucial factor in regulating the cell cycle. Under metabolic stress, FOXO1 increases Cyclin F but not other Cyclins in gliomas, suppressing IDH1 expression, an essential tumor marker which its overexpression is implicated in glioma progression. Moreover, TFG-β/SMAD can form a complex with FOXO1 and induce the expression of CDK inhibitor p21Cip1. However, interference of PI3K/Akt signaling and other oncogenes, such as FOXG1, diminish this process.
Other upstream regulators and downstream targets of FOXO1 in glioma
Similar to FOXM1, various studies have identified upstream/downstream regulators of FOXO1 in gliomas. As seen in Table 3, most of these regulators are ncRNAs, and in their results, FOXO1 was proposed as a tumor suppressor, except in a study by Shi et al. [149] which showed the opposite result. It is noteworthy to mention that some ncRNAs form a positive feedback loop that constantly represses FOXO1 expression, leading to glioma progression [138, 150, 151] (Fig. 5A). According to two other studies which proposed FOXO1 as an oncogene, two hypotheses can be raised; a) FOXO1 can act as an oncogene in radioresistant or chemoresistant glioma cell lines that have not responded to conventional treatments [152], and b) in a context-dependent manner FOXO1 can act as an oncogene by increasing stem cell markers in glioma [137] (Fig. 5B).

Double-edged role of FOXO1 in glioma progression. A Tumor suppressive effects of FOXO1. B Oncogenic effects of FOXO1
A)Tumor suppressive effects of FOXO1
Transduction of growth-related signals and subsequent activation of PI3K/Akt signaling cascade prevents FOXO1 translocation to the nucleus via inducing its phosphorylation. While mTOR inhibits FOXO1 translocation to the nucleus upon its phosphorylation, mTORc2, and lncRNA-DANCR have been shown to exert similar effects on FOXO1 by promoting its acetylation and ubiquitination, respectively. Moreover, treatment with agents such as Progesterone suppresses EGFR-dependent activation of the PI3K/Akt signaling pathway. There are various upregulated miRNAs, including miR-21, miR-28-5p, miR-196a-5p, miR-374a, miR-486–5p, and miR-5188 in different glioma cell lines that target 3′ UTR of FOXO1 mRNA. In the nucleus, on the one hand, FOXO1 can regulate its downstream targets (e.g., PID1). On the other hand, transcription factors such as KLF4 control FOXO1 transcriptional activity by binding to its promoter.
B) Oncogenic effects of FOXO1
Few studies have mentioned the oncogenic capabilities of FOXO1 as a therapeutic target in glioma. Following combined treatment with PI3K/mTOR inhibitor and gamma ionizing radiation, the expression of FOXO1 and stem cell marker SOX2 is decreased in GBM stem cells with wild p53 phenotype [136]. Moreover, FOXO1 can bind to the promoter of two other proto-oncogenes, including OCT4 and RFC2, and increase their transcription [137, 152]. Shi et al. have discovered that miR-135a acts as a tumor suppressor in gliomas by hindering FOXO1 expression [149]. In addition, PI3K/mTOR or FOXO1 inhibitors could prevent glycolysis in gliomas. However, when both of them are concurrently inhibited, the expression of glycolysis-related genes, including LDHA and ENO1, is elevated [143].
FOXO1 and therapeutic opportunities in glioma
Given that many studies conducted up to now support the tumor suppressor role of FOXO1, restoring its expression may reverse glioma tumorigenesis in its early stages. Several studies have shown that restoration of FOXO1 could facilitate the efficacy of treatment with TMZ [129, 161, 164, 166], etoposide [154, 156], and radiotherapy [150, 162]. In addition, most of the pharmacological compounds which affect FOXO1 were shown to increase their expression in gliomas (Table 4). Some of these agents were shown to induce FOXO1 expression in a dose [144, 167] and/or time-dependent manner [144, 168].
FOXO3
Similar to FOXO1, FOXO3 (also known as FOXO3a) is generally considered a tumor suppressor in different cancers, and its sub-cellular localization was shown to be crucial for its activity. FOXO3 expression is regulated at different levels of gene expression, including post-transcriptional (mainly by miRNAs), post-translational modifications (such as phosphorylation, acetylation, methylation, ubiquitination and etc.), and protein–protein interaction [177]. As will be discussed below, the majority of studies have introduced FOXO3 as a tumor suppressor in gliomas, however; similar to FOXO1, there is a controversial role for FOXO3 in gliomas in terms of function and prognosis [178, 179]. While Qian et al. [178] have shown that in human GBM tissues, high nuclear FoxO3a expression is linked to a poor prognosis, a study with a smaller sample size by Shi et al. [179] demonstrated that in low-grade astrocytomas (grade II), the expression of FOXO3a is significantly higher than in anaplastic astrocytomas (grade III) and GBM (grade IV). However, the discrepancy in prognosis outcomes between the two studies could be attributed to factors such as sample size, patient selection, differences in FoxO3a expression levels, and the potential involvement of FoxO3a in resistance to radiotherapy and chemotherapy, which was not previously considered [178].
Protein kinases and FOXO3 in glioma
Similar to other forkhead box transcription factors, nuclear exportation of FOXO3 is dependent on its phosphorylation by protein kinase B (PKB, Akt) as a downstream member of PI3K/Akt signaling cascade [127, 180]; however, other protein kinases such as AMPK [181, 182], EGFR [183,184,185], and MAPK [186] can also regulate its activity in gliomas. Accumulating evidence supports the abovementioned proteins' role in regulating FOXO3a in gliomas, and various pharmacological compounds exert their inhibitory role by affecting these axes (Table 6 and Fig. 6).

A summary of interaction between protein kinases and FOXO3 in glioma
PKB(Akt)
Various upstream regulators of Akt such as CLK2 (oncogene) [187, 188], IGF1 (dual role) [189], SPHK1(oncogene) [190], CHAF1A (oncogene) [191], and importantly FOXM1B (oncogene) [192] was found to exert their function by affecting Akt/FOXO3 axis in gliomas. Moreover, our understanding of how PI3K/Akt inhibitors affect FOXOs is still insufficient in gliomas. While a previous study [179] has shown that using LY294002 as a PI3K/AKT inhibitor can activate FOXO3a in the nucleus, a recent study [137] has shown using NVP-BEZ235 (as another PI3K inhibitor) was not enough to induce its nuclear localization in GBM cells. Therefore, more studies are required to explore the mechanisms behind FOXO3a regulation by PI3K/Akt inhibitors.
EGFR
EGFR (a receptor tyrosine kinase) mutations are frequently seen in high-grade gliomas; therefore, its overexpression has prognostic importance in clinical diagnosis [193]. Although a significant correlation between EGFR and FOXO3a does not exist in GBM cell lines clinically [179], its inhibition can induce nuclear translocation of FoXO3a in GBM cells [185]. In fact, in GSCs with high expression of EGFR, FOXO3 is substantially upregulated, again supporting the hypothesis that FOXOs can induce stem cell proliferation. In contrast to cells with low EGFR expression, treatment with BMP4 (Bone morphogenic protein 4) alongside TMZ in GSCs with high EGFR triggers FOXO3a dephosphorylation and translocation to the nucleus to induce pro-apoptotic genes such as BCL2L11 [183]. These data show that the regulation of FOXO3 is complex and diverse factors are involved.
AMP-activated protein kinase (AMPK)
AMPK is a protein kinase sensitive to ADP and AMP changes in cells involved in energy homeostasis through switching anabolism to catabolism, and its activity has been well-studied in gliomas [194]. Given that AMPK can phosphorylate FOXO3a at Ser413, suppression of GGCT can be considered a promising strategy to promote the AMPK/FOXO3a/p21 axis and inhibit the proliferation of A172 GBM cells [182]. Moreover, activating the AMPK/FOXO3a axis by metformin was a desirable therapeutic strategy to prevent self-renewal and tumor formation of stem-like glioma-initiating cells [181].
MAPK
Activation of MAPK cascade, known as RAS/RAF/MEK/ERK signaling axis, has significant participation in gliomagenesis and tumor progression via inducing cell proliferation, metastasis, angiogenesis, and inhibition of apoptosis [195]. Sato, Sunayama, and colleagues have shown that concurrent inhibition of this pathway and PI3K/Akt/mTOR induces differentiation of undifferentiated glioma stem-like cells via activating FoxO3a transcriptional activity [196]. Their further investigation also highlighted the ROS-dependent mechanism of p38 MAPK/FOXO3 activation in GICs [186]. In addition, tumor necrosis factor related apoptosis inducing ligand (TRAIL) is a naturally occurring protein with tumor-suppressing features in various cancer cell lines. However, it suffers from efficient delivery to the brain tissue due to its chemical structure limitation for passing through the blood–brain barrier. Allen et al. could induce TRAIN expression in mice models with GBM significantly by deploying a novel TRAIL-inducing compound 10 (TIC10), and this mechanism was attributed to stimulant inhibition of Akt and ERK signaling pathways and subsequent transcriptional activity promotion of FOXO3a by TIC10 [197].
Cellular stress and FOXO3 in glioma
FOXO3a subcellular localization and its post-translational modification are highly dependent on a wide range of stress-related conditions, including starvation, oxidative stress, hypoxia, heat shock, and DNA damage. Energetic stress often affects FOXO3a phosphorylation through activators such as AMPK and Sirt-1 as well as suppressors like Akt and CREB binding protein and p300 (CBP/p300) signals. Furthermore, under oxidative or genotoxic stress, the MEK/ERK pathway (as a downstream member of MAPK signaling) regulates mitochondrial accumulation of degraded FOXO3a and cellular respiration [198].
ROS, hypoxia, and nutrition starvation
As mentioned earlier, hydrogen peroxide as a ROS can activate p38-MAPK and induce FOXO3a expression in GICs very efficiently, leading to cell differentiation and inhibited stem cell self-renewal capacity [186]. PTEN-induced kinase 1 (PINK1) negatively regulates GBM growth through activating FOXO3a and alleviating ROS and metabolic reprogramming while its loss promotes aerobic glycolysis (Warburg effect) via stabilizing HIF1α, a master modulator of hypoxia [199]. This evidence shows that FOXO3a is involved in the regulation of hypoxia. However, Hashimoto et al. have demonstrated that severe hypoxia, instead of affecting FoxO3a, increases the expression of Sp1. In addition, under hypoxic conditions, the knockdown of FOXO3a does not influence the activity of AMPK in both T98G and A172 GBM cells but suppresses Sp1 only in T98G cells [200]. Moreover, the authors have previously shown that nutrition starvation activates Akt in T98G GBM cells and slightly decreases FOXO3a expression, leading to radio-resistance. They also showed that DNA-PKcs act as an upstream regulator for FOXO3a and Akt under starvation conditions. Targeting DNA-PKcs by NU7026 can suppress their activation and slightly increase FOXO3a expression [201]. Intriguingly, Brucker et al. [202] have shown that FOXO3a expression is positively correlated with glioma WHO grade in peri-necrotic tumor lesions (where there is higher cellular stress) and under hypoxic conditions independent of HIF-1α, its upregulation causes cell death in GBM LNT-229 cells in a caspase-independent manner. More interestingly, when the FOXO3a gene was silenced, the intracellular level of ROS was significantly increased and facilitated cell death, followed by oxidative stress. Although silencing this gene saves glucose, but does not have an impact on cell proliferation. Moreover, abolishing FOXO3a lowers oxygen consumption to compensate for decreased glucose uptake of LNT-229 cells and reinforces the transcriptional activity of HIF-1α under hypoxia. More importantly, overactivation of Tp53 activity in cells with inhibited FOXO3 can improve cell survival in cellular stress conditions. These data showed that although FOXO3 is increased and results in cell death in perinecrotic tumors undergoing hypoxia, silencing its expression can also accelerate cell death via promoting excessive ROS production (Fig. 7). Taken together, several conclusions can be obtained from these studies regarding the different aspects of FOXO3 in gliomas: (a) The expression and function of FOXO3 differ in glioma depending on the spatial localization of tumor cells and tumors grade. (b) Cells tolerating hypoxia tend to promote FOXO3 activity. (c) Although FOXO3 can regulate hypoxia, there are more crucial modulators of hypoxia, such as HIF-1α, AMPK, Sp1, and Tp53. d) Role of FOXO3 in modulating oxidative stress is complicated and is highly dependent on the amount of available ROS produced by other regulators in tumor cells. (e) Under hypoxia, FOXO3 can regulate HIF1a, but the opposite is not true [202].

FOXO3 as a core component in regulating cellular stress: Various cellular stress conditions, including nutrition starvation, hypoxia, oxidative stress, and glucose metabolism, have a reciprocal relationship with FOXO3 in glioma. Nutrition starvation via activating Akt and deactivating FOXO3 causes radioresistance of glioma tumors. The relationship between ROS and FOXO3 is very complicated, and several molecules are involved. In GICs, the production of ROS induces p38-AMPK that, via degradation of Bmi1, activates FOXO3, resulting in differentiation and loss of self-renewal (red arrows). However, the role of ROS was shown to be double-edged in tumor progression in a way that their inhibition following PINK1-induced FOXO3 expression represses cell growth and prevents HIF-1α stabilization (blue lines). On the other hand, the study by He et al. showed that although TMZ, through producing ROS and inducing AIF1 expression, causes cell death followed by DNA double-strand breaks, in this condition, FOXO3 is activated and via upregulating BNIP3 and ATG5 prevents DNA from damage, therefore reverses this process (green lines). In addition, cells undergoing hypoxia in perinecrotic areas express FOXO3 more frequently, inhibiting the transcriptional activity of HIF-1α and p53. Since p53 can repress GLUT1 expression (a glucose transporter), inhibition of p53 by FOXO3 increases glucose consumption by tumor cells (dashed lines)
DNA damage
Repairing DNA at the G2-M checkpoint was shown to be stimulated by FOXO3a in mammalian cells [203]. In line with this, He et al. [204] have reported that, on the one hand, TMZ causes the production of mitochondrial superoxide (ROS) that subsequently, via increasing apoptosis-inducing factor (AIF), induces cell death. On the other hand, the excessively produced ROS elevates FOXO3a expression and gradually promotes BNIP3 and ATG5, two autophagy-related genes, and resulted in resistance to TMZ-induced DNA double-strand breaks (DSBs) caused by mitophagy.
Sirtuins
Sirtuins are deacetylase enzymes dependent on NAD+ for regulating cellular metabolism in response to stress. Dysfunction of sirtuins leads to various diseases, including cancer and neurodegeneration. Like FOXO transcription factors, these proteins have a dual oncogenic and tumor-suppressive function via regulating DNA repair, transcriptional modulation, and metabolism context-dependent depending on tissue type and cancer type [205, 206]. SIRT6 is a nuclear-residing protein that, due to its deacetylase activity, affects a variety of targets and substrates, including FOXO3, PARP1, MYC, and HIF-1α, involved in metabolism and chromatin/DNA repair [205]. MST1 is a downregulated protein kinase in GBM cells that inhibits cell viability, colony formation, and aerobic glycolysis but exerts apoptotic effects via directly increasing FOXO3a expression and its proposed downstream target SIRT6 [207, 208]. In contrast, SIRT1 was shown to inhibit acetylation of FOXO3a; however, treatment with betulinic acid (BA) as a natural pentacyclic triterpenoid could induce FOXO3a via repressing SIRT1, leading to mitochondrial dysfunction and cell death [209].
β-catenin and FOXO3 in glioma
Β-catenin is a protein with multiple functions that plays an important role in Wnt signal transduction pathway via regulating gene transcription and cell adhesion [210]. Upon β-catenin proteins translocation to the nucleus they form a complex with binding to transcription factors named lymphoid enhancer factor/T cell factor (LEF/TCF), activating the target genes of Wnt signaling pathway [210]. FOXO proteins (especially FOXO3a) were shown to compete with TCF for binding to β-catenin and suppress TCF transcriptional activity particularly under oxidative stress [211]. In line with the study conducted by Xu and colleagues [212], Sun et al. demonstrated that in U87 and U251 GBM cells resistant to TMZ, overexpression of FOXO3a positively regulates the amount of nuclear β-catenin via governing MMP9 expression [213]. In contrast, Lu et al. have shown that miR-370 as a downregulated tumor suppressor miRNA by targeting 3′ UTR of β-catenin mRNA, suppresses its expression in astrocytoma and GBM cells and subsequently promoting FOXO3a nuclear accumulation, suppressing cancer cell proliferation [214]. These data again support the oncogenic activity of FOXO proteins in therapy resistant cell lines.
Cell cycle regulation and FOXO3 in glioma.
Studies have reported that FOXO3 can control cell cycle via increasing transcriptional activity of two important pro-apoptotic genes including BIM [183, 190, 191, 209, 215,216,217]and p27 [184, 187, 188, 218,219,220] in gliomas, emphasizing the tumor suppressive impact of Akt/FOXO3a/BIM axis [183, 190, 191]. In addition, other transcription factors including SOX2 and FOXG1 can repress FOXO3a expression level in GBM stem cells leading to cell cycle re-entry and dedifferentiation [221].
FOXO3’ function in glioma stem cells
An important function of FOXO3 is its contribution to stem cell differentiation in both neural stem cells and glioma stem cells [222]. It has been proposed that nuclear accumulation of FOXO3a in GBM cancer stem-like cells could induce their differentiation. Due to the prominent role of PI3K/Akt/mTOR and MEK/ERK signaling pathways in the phosphorylation of FOXO3a, inhibiting these two signaling pathways can be an promising method for differentiation therapy against high-grade gliomas, especially GBM [137, 196, 221]. However, once GBM cells undergo chemotherapy and radiotherapy and maintain resistance to these treatments, FOXO3a overexpression exerts oncogenic function by increasing the expression of stem cell markers such as SOX2 [136]. A study suggests that following repeated radiation, continuous IGF1 stimulation ultimately induces FoxO3a activation, leading to slower proliferation and enhanced self-renewal. In contrast, after acute radiation in GBM stem cells, IGF1R/AKT/FOXO3a axis induce radioresistance [189]. It should be noted that the activity of FOXO proteins is highly dependent on the other upstream regulators. For example, BMP4 treatment only is effective in sensitizing those glioma stem cells with high EGFR expression to TMZ treatment, leading to the accumulation of FOXO3a in the nucleus [183]. As mentioned above, following radiotherapy and chemotherapy, FOXO3a induces the expression of stem cell markers. Therefore, the knockdown of FOXO3a in glioblastoma multiforme stem cells with intact p53 activity can significantly enhance the response to treatment with radiation therapy combined with PI3K/mTOR inhibition [136].
Regulation of FOXO3 by non-coding RNAs
Several studies have reported that 3′ UTR of FOXO3a is targeted by oncogenic microRNAs, including miR-10b [223], miR-27a [224], miR-93 [225], miR-155 [226], and miR-184 [218] that their expression is upregulated in glioma cell lines (Table 5). In addition, FOXO3a can also mediate the expression of non-coding RNAs. Temozolomide-associated lncRNA (lnc-TALC) is an overexpressed lncRNA in TMZ resistant cell lines that upregulates c-MET through competitively binding to its regulator miR-20b-3p. c-MET can promote cytoplasmic degradation of FOXO3a via activating Akt signaling pathway. In TMZ sensitive cell lines, there is much more nuclear levels of FOXO3a compared to resistant cells, which through binding to promoter of lnc-TALC inhibits its expression and results in MGMT silencing [227].
FOXO3a and therapeutic opportunities in glioma
Though the mechanism of many drugs on FOXO3 has been mentioned above, current conducted pharmacologic research with relying on its tumor suppressive properties has shown remarkable results. These treatments mainly include PI3K/mTOR inhibitors, metabolism related drugs (e.g., metformin and Fenofibrate), and natural derived compounds. However, as seen in Table 6, a limitation of these studies is that they are limited to in vitro studies rather than pre-clinical clinical or levels. Moreover, designing strategies against oncogenic activity of FOXO3 can be a step forward.
Conclusion
According to evidence collected up to now, FOXM1 acts as an absolute oncogene in gliomas, associated with poor survival, independent of the type of cell line, stage of the tumor, etc. The activity of protein kinases such as Akt, MELK, and growth factors (e.g., EGFs or FGFs) subsequently leads to phosphorylation of FOXM1 in gliomas, promoting transcriptional activity of a variety of targets, including STAT3, EZH2, β-catenin, MMP-2, Sox2, VEGF, PDGF-A, VEGF, UBE2C, Rad51, RFC5, BUB1B, Anxa1, SIRT1, ASPM, and ADAM17. Furthermore, several downregulated miRNAs, including miR-216b, miR-320, miR-370-3p, and miR-525-5p, have been verified to target 3’ UTR of FOXM1. More importantly, overexpression of FOXM1 has been strongly associated with increased proliferation, migration, angiogenesis, invasion, and resistance to radiation and TMZ in glioma through facilitating DNA repair response. Some studies have elucidated the anti-FOXM1 activity of MELK and proteasome inhibitors as well as natural products on glioma (Table 2). Therefore, it is suggested that more studies at clinical and pre-clinical levels should be conducted to assess the subsequences of FOXM1 pharmacological inhibition.
Like other FOXO subgroups, FOXO1 has a crucial role in regulating proliferation, metastasis, invasion, drug response/resistance, and apoptosis. Furthermore, while targeting the PI3K/Akt signaling pathway has a prominent role in restoring FOXO1 activity, other proteins, and transcription factors are involved in its regulation. More importantly, FOXO transcription factors exert their tumor-suppressive functions via forming a reciprocal interplay with cell cycle modulators such as CDKs and Cyclins. An overview of literature has demonstrated that FOXO1 has a controversial role in tumorgenesis of gliomas. The most oncogenic role of FOXO1 was mainly attributed to elevating the expression of stem cell markers such as OCT4 and SOX2. Altogether, focusing on the tumor suppressor role of FOXO1, most of the anticancer drugs that affect FOXO1 in glioma increase its expression, except EMAP-II and Progesterone, which their efficacy is dose or/and time-dependent. Also, the majority of these pharmacological compounds enhance FOXO1 expression through Akt, including Progesterone, Urolithin A, Xihuang Pill, and EMAP-II. Moreover, restoring FOXO1 expression could be utilized in the sensitization of tumor cells to etoposide, BCNU, or cisplatin. These findings shed light on a novel approach to conducting research and assessing FOXO1 role in the prognosis and treatment of glioma.
Finally, protein kinases such as EGFR, MAPK, IGF1R, and AKT were shown to phosphorylate FOXO3 directly or indirectly, repressing its transcriptional activity. This is while AMPK, via phosphorylating it at Ser413, causes its transactivation without affecting its subcellular localization. Moreover, MST1, via phosphorylating FOXO3, promotes its nuclear localization, leading to SIRT6 overexpression. In addition, FOXO3 was shown to act as a core component in the response of glioma cells to cellular stress, such as ROS production, hypoxia, glucose metabolism, and sirtuins. There are several upregulated ncRNAs in glioma, including miR-10b, miR-27a, miR-93, miR-155, miR-184, Circ-DONSON, and Inc-TALC that their oncogenic activity was shown to be exerted through repressing FOXO3. Therefore, suppressing their expression can be considered a step forward in restoring FOXO3 expression. Similar to FOXO1, most studies agree on the tumor-suppressive feature of FOXO3. However, under specific circumstances, both FOXO1 and FOXO3 were shown to be implicated in the occurrence of TMZ and radiation resistance.
Future perspectives
In this review, we have covered a variety of regulatory pathways, mechanisms, and effects of FOXM1 alteration on numerous subtypes of gliomas. Most ongoing studies of FOXM1 considered it as an oncogene in light of its function in regulating several cellular processes that have been reviewed throughout this review. Moreover, among the FOX transcription factors, the FOXO subfamily is another mostly investigated one in glioma, which seems to be a tumor suppressor. Despite the vast quantity of literature describing the different mechanisms linking FOXM1 to glioma, the fact that it is a transcription factor restricts its applicability as a target compound for the therapeutic approach of glioma. Even though an enormous quantity of in vitro studies has been conducted to clarify the role of FOXM1 in glioma, the application of FOXM1 inhibition by chemical inhibitors in clinical settings has been constrained due to a number of issues, including the need for precise concentrations, and a wide range of interacting pathways and FOXM1 regulators and unknown side effects. A recent clinical trial on 79 human glioma tissues unraveled that down-regulation of FOXM1 by siRNAs induced the apoptosis, cell cycle arrest, and EMT of glioma cells [41]. Clinical trials in phases 1 and II are required to analyze the safety, pharmacodynamics, and pharmacokinetics of FOXM1 inhibitors; therefore, additional investigation and extensive clinical trials need to be conducted in order to gain conclusive evidence and elaborate the clinical potency of FOXM1 in glioma. In addition, glioma cells with mutations of the IDH gene have a decreased expression of FOMX1 compared to wild-type phenotypes. Therefore, it is suggested that the function of FOXM1 in lower-grade gliomas with IDH mutations be studied in more detail [236]. The FOXO subfamily is notably regulated by epigenetic triggers and has a close association with the cell cycle. Therefore, these proteins are exciting candidates for developing new therapeutics related to epigenetics. Further studies should investigate the function of FOXO1 and FOXO3 before and after different treatments with chemotherapy and radiotherapy in more detail. In addition, the prognostic function of these proteins should be evaluated in studies with larger sample sizes and different glioma grades, as the number of studies that evaluated the function of FOXOs in gliomas is very few and mostly based on publicly available cohorts(TCGA). The FOXO-FOXM1 axis, in particular, should be further studied in translational and clinical research due to its effects on a variety of cellular activities, including carcinogenesis, progression, and treatment resistance. Given the significance of the FOXO and FOXM1 proteins, it may be possible to utilize these proteins as potential targeted therapies and prognostic markers for glioma if their regulation mechanisms and roles in cancer initiation, progression, and drug resistance are better understood. Moreover, a combination therapy targeting the FOXO subfamily and FOXM1 has a significant chance of creating beneficial synergistic effects, reducing adverse effects, and ultimately boosting clinical outcomes. Finally, since growing numbers of models are developing in the prediction of survival of GBM patients [237, 238], the construction of models using FOX proteins for prognosis evaluation of patients appears as a promising strategy in clinical settings, and future studies should consider this point.
Availability of data and materials
Data sharing does not apply to this article as no datasets were generated or analyzed during the current study.
References
Goodenberger ML, Jenkins RB. Genetics of adult glioma. Cancer Genet. 2012;205(12):613–21.
Komori T. The 2016 WHO classification of tumours of the central nervous system: the major points of revision. Neurol Med Chir. 2017;57(7):301–11.
Wen PY, Huse JT. 2016 World Health Organization classification of central nervous system tumors. Contin Lifelong Learn Neurol. 2017;23(6):1531–47.
Masui K, Cloughesy TF, Mischel P. Molecular pathology in adult high-grade gliomas: from molecular diagnostics to target therapies. Neuropathol Appl Neurobiol. 2012;38(3):271–91.
Tunthanathip T, Ratanalert S, Sae-Heng S, Oearsakul T, Sakarunchai I, Kaewborisutsakul A, et al. Prognostic factors and clinical nomogram predicting survival in high-grade glioma. J Cancer Res Ther. 2021;17(4):1052–8.
Gupta A, Dwivedi T. A simplified overview of World Health Organization classification update of central nervous system tumors 2016. J Neurosci Rural Pract. 2017;8(04):629–41.
Louis DN, Perry A, Reifenberger G, Von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–20.
Claus EB, Walsh KM, Wiencke JK, Molinaro AM, Wiemels JL, Schildkraut JM, et al. Survival and low-grade glioma: the emergence of genetic information. Neurosurg Focus. 2015;38(1):E6.
Sevastre A-S, Costachi A, Tataranu LG, Brandusa C, Artene SA, Stovicek O, et al. Glioblastoma pharmacotherapy: a multifaceted perspective of conventional and emerging treatments. Exp Ther Med. 2021;22(6):1–18.
Tabnak P, Masrouri S, Mafakheri A. Natural products in suppressing glioma progression: a focus on the role of microRNAs. Phyther Res. 2022;36(4):1576–99.
Papavassiliou KA, Papavassiliou AG. Transcription factors in glioblastoma—molecular pathogenesis and clinical implications. Biochim Biophys (BBA) Acta Rev Cancer. 2022;1877(1):188667.
Zhang W, Duan N, Song T, Li Z, Zhang C, Chen X. The emerging roles of forkhead box (FOX) proteins in osteosarcoma. J Cancer. 2017;8(9):1619.
Myatt SS, Lam EW-F. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 2007;7(11):847–59.
Benayoun BA, Caburet S, Veitia RA. Forkhead transcription factors: key players in health and disease. Trends Genet. 2011;27(6):224–32.
Jackson BC, Carpenter C, Nebert DW, Vasiliou V. Update of human and mouse forkhead box (FOX) gene families. Hum Genomics. 2010;4(5):1–8.
Furukawa-Hibi Y, Kobayashi Y, Chen C, Motoyama N. FOXO transcription factors in cell-cycle regulation and the response to oxidative stress. Antioxid Redox Signal. 2005;7(5–6):752–60.
Farhan M, Wang H, Gaur U, Little PJ, Xu J, Zheng W. FOXO signaling pathways as therapeutic targets in cancer. Int J Biol Sci. 2017;13(7):815.
Farhan M, Silva M, Xingan X, Huang Y, Zheng W. Role of FOXO transcription factors in cancer metabolism and angiogenesis. Cells. 2020;9(7):1586.
Cheong J-W, Eom JI, Maeng H-Y, Lee ST, Hahn JS, Ko YW, et al. Constitutive phosphorylation of FKHR transcription factor as a prognostic variable in acute myeloid leukemia. Leuk Res. 2003;27(12):1159–62.
Wu Y, Elshimali Y, Sarkissyan M, Mohamed H, Clayton S, Vadgama JV. Expression of FOXO1 is associated with GATA3 and annexin-1 and predicts disease-free survival in breast cancer. Am J Cancer Res. 2012;2(1):104.
Zhang B, Tomita Y, Ch’ng E, Qiu Y, He J, Jin Y-F, et al. Prognostic significance of phosphorylated FOXO1 expression in soft tissue sarcoma. Ann Surg Oncol. 2009;16:1925–37.
Trinh DL, Scott DW, Morin RD, Mendez-Lago M, An J, Jones SJM, et al. Analysis of FOXO1 mutations in diffuse large B-cell lymphoma. Blood, J Am Soc Hematol. 2013;121(18):3666–74.
Kim JH, Kim MK, Lee HE, Cho SJ, Cho YJ, Lee BL, et al. Constitutive phosphorylation of the FOXO1A transcription factor as a prognostic variable in gastric cancer. Mod Pathol. 2007;20(8):835–42.
Cheng S-X, Tu Y, Zhang S. FoxM1 promotes glioma cells progression by up-regulating Anxa1 expression. PLoS ONE. 2013;8(8):e72376.
Hamurcu Z, Ashour A, Kahraman N, Ozpolat B. FOXM1 regulates expression of eukaryotic elongation factor 2 kinase and promotes proliferation, invasion and tumorgenesis of human triple negative breast cancer cells. Oncotarget. 2016;7(13):16619.
Maachani UB, Shankavaram U, Kramp T, Tofilon PJ, Camphausen K, Tandle AT. FOXM1 and STAT3 interaction confers radioresistance in glioblastoma cells. Oncotarget. 2016;7(47):77365.
Li D, Zhang Z, Xia C, Niu C, Zhou W. Non-Coding RNAs in glioma microenvironment and angiogenesis. Front Mol Neurosci. 2021;261:763610.
Joshi K, Banasavadi-Siddegowda Y, Mo X, Kim S-H, Mao P, Kig C, et al. MELK-dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cells. 2013;31(6):1051–63.
Gong A, Huang S. FoxM1 and Wnt/β-catenin signaling in glioma stem CellsFoxM1 and Wnt/β-catenin in glioma and other cancers. Cancer Res. 2012;72(22):5658–62.
Liu C, Barger CJ, Karpf AR. FOXM1: a multifunctional oncoprotein and emerging therapeutic target in ovarian cancer. Cancers. 2021;13(12):3065.
Yang S, Pang L, Dai W, Wu S, Ren T, Duan Y, et al. Role of forkhead box O proteins in hepatocellular carcinoma biology and progression. Front Oncol. 2021;11:667730.
Liao G-B, Li X-Z, Zeng S, Liu C, Yang S-M, Yang L, et al. Regulation of the master regulator FOXM1 in cancer. Cell Commun Signal. 2018;16(1):57.
Liu M, Dai B, Kang S-H, Ban K, Huang F-J, Lang FF, et al. FoxM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer Res. 2006;66(7):3593–602.
Van den Boom J, Wolter M, Kuick R, Misek DE, Youkilis AS, Wechsler DS, et al. Characterization of gene expression profiles associated with glioma progression using oligonucleotide-based microarray analysis and real-time reverse transcription-polymerase chain reaction. Am J Pathol. 2003;163(3):1033–43.
Bai H, Harmancı AS, Erson-Omay EZ, Li J, Coşkun S, Simon M, et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat Genet. 2016;48(1):59–66.
Hodgson JG, Yeh R-F, Ray A, Wang NJ, Smirnov I, Yu M, et al. Comparative analyses of gene copy number and mRNA expression in glioblastoma multiforme tumors and xenografts. Neuro Oncol. 2009;11(5):477–87.
Stępniak K, Machnicka MA, Mieczkowski J, Macioszek A, Wojtaś B, Gielniewski B, et al. Mapping chromatin accessibility and active regulatory elements reveals pathological mechanisms in human gliomas. Nat Commun. 2021;12(1):3621.
Xie Q, Wu Q, Mack SC, Yang K, Kim L, Hubert CG, et al. CDC20 maintains tumor initiating cells. Oncotarget. 2015;6(15):13241.
Li X, Wu C, Chen N, Gu H, Yen A, Cao L, et al. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget. 2016;7(22):33440.
Borhani S, Gartel AL. FOXM1: a potential therapeutic target in human solid cancers. Expert Opin Ther Targets. 2020;24(3):205–17.
Zhang X, Lv Q-L, Huang Y-T, Zhang L-H, Zhou H-H. Akt/FoxM1 signaling pathway-mediated upregulation of MYBL2 promotes progression of human glioma. J Exp Clin Cancer Res. 2017;36(1):1–18.
Wang S, Zhang S, Li J, Xu X, Weng Y, Zheng M, et al. CXCL12-induced upregulation of FOXM1 expression promotes human glioblastoma cell invasion. Biochem Biophys Res Commun. 2014;447(1):1–6.
Pal S, Kozono D, Yang X, Fendler W, Fitts W, Ni J, et al. Dual HDAC and PI3K inhibition abrogates NFκB-and FOXM1-mediated DNA damage response to radiosensitize pediatric high-grade gliomasNFκB and FOXM1 inhibition radiosensitizes pediatric gliomas. Cancer Res. 2018;78(14):4007–21.
Ganguly R, Hong CS, Smith LGF, Kornblum HI, Nakano I. Maternal embryonic leucine zipper kinase: key kinase for stem cell phenotype in glioma and other cancersmelk, key kinase for stem cell phenotype in cancer stem cells. Mol Cancer Ther. 2014;13(6):1393–8.
Paskeh MDA, Mehrabi A, Gholami MH, Zabolian A, Ranjbar E, Saleki H, et al. EZH2 as a new therapeutic target in brain tumors: molecular landscape, therapeutic targeting and future prospects. Biomed Pharmacother. 2022;146:112532.
Kim S-H, Joshi K, Ezhilarasan R, Myers TR, Siu J, Gu C, et al. EZH2 protects glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner. Stem cell reports. 2015;4(2):226–38.
Thakur VS, Aguila B, Brett-Morris A, Creighton CJ, Welford SM. Spermidine/spermine N1-acetyltransferase 1 is a gene-specific transcriptional regulator that drives brain tumor aggressiveness. Oncogene. 2019;38(41):6794–800.
Reich NC, Liu L. Tracking STAT nuclear traffic. Nat Rev Immunol. 2006;6(8):602–12.
Ou A, Ott M, Fang D, Heimberger AB. The role and therapeutic targeting of JAK/STAT signaling in glioblastoma. Cancers. 2021;13(3):437.
Gong A, Wei P, Zhang S, Yao J, Yuan Y, Zhou A, et al. FoxM1 drives a feed-forward STAT3-activation signaling loop that promotes the self-renewal and tumorigenicity of glioblastoma stem-like cells. Cancer Res. 2015;75(11):2337–48.
Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS, et al. Preferential iron trafficking characterizes glioblastoma stem-like cells. Cancer Cell. 2015;28(4):441–55.
Han M, Xu R, Wang S, Yang N, Ni S, Zhang Q, et al. Six-transmembrane epithelial antigen of prostate 3 predicts poor prognosis and promotes glioblastoma growth and invasion. Neoplasia. 2018;20(6):543–54.
Xie P, Han Q, Liu D, Yao D, Lu X, Wang Z, et al. miR-525-5p modulates proliferation and epithelial–mesenchymal transition of glioma by targeting Stat-1. Onco Targets Ther. 2020;13:9957.
Tabnak P, Mafakheri A, Emsailpoor ZH, Kazemi T, Shekari N. Regulatory interplay between microRNAs and WNT pathway in glioma. Biomed Pharmacother. 2021;143:112187.
Zhang N, Wei P, Gong A, Chiu W-T, Lee H-T, Colman H, et al. FoxM1 promotes β-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell. 2011;20(4):427–42.
Zhang Y, Zhang N, Dai B, Liu M, Sawaya R, Xie K, et al. FoxM1B transcriptionally regulates vascular endothelial growth factor expression and promotes the angiogenesis and growth of glioma cells. Cancer Res. 2008;68(21):8733–42.
Zhang C, Han X, Xu X, Zhou Z, Chen X, Tang Y, et al. FoxM1 drives ADAM17/EGFR activation loop to promote mesenchymal transition in glioblastoma. Cell Death Dis. 2018;9(5):469.
Gouazé-Andersson V, Ghérardi M-J, Lemarié A, Gilhodes J, Lubrano V, Arnauduc F, et al. FGFR1/FOXM1 pathway: a key regulator of glioblastoma stem cells radioresistance and a prognosis biomarker. Oncotarget. 2018;9(60):31637.
Tabnak P, Ghasemi Y, Natami M, Khorram R, Ebrahimnezhad M. Role of m6A modification in dysregulation of Wnt/β-catenin pathway in cancer. Biomed Pharmacother. 2023;157:114023.
Qu J, Yan H, Hou Y, Cao W, Liu Y, Zhang E, et al. RNA demethylase ALKBH5 in cancer: from mechanisms to therapeutic potential. J Hematol Oncol. 2022;15(1):1–24.
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. m6A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31(4):591–606.
Takahashi H, Hase H, Yoshida T, Tashiro J, Hirade Y, Kitae K, et al. Discovery of two novel ALKBH5 selective inhibitors that exhibit uncompetitive or competitive type and suppress the growth activity of glioblastoma multiforme. Chem Biol Drug Des. 2022;100(1):1–12.
HajiEsmailPoor Z, Tabnak P, Ahmadzadeh B, Ebrahimi SS, Faal B, Mashatan N. Role of hedgehog signaling related non-coding RNAs in developmental and pathological conditions. Biomed Pharmacother. 2022;153:113507.
Xue J, Zhou A, Tan C, Wu Y, Lee H-T, Li W, et al. Forkhead box M1 is essential for nuclear localization of glioma-associated oncogene homolog 1 in glioblastoma multiforme cells by promoting importin-7 expression. J Biol Chem. 2015;290(30):18662–70.
Sasai K, Tabu K, Saito T, Matsuba Y, Saido TC, Tanaka S. Difference in the malignancy between RAS and GLI1-transformed astrocytes is associated with frequency of p27KIP1–positive cells in xenograft tissues. Pathol Pract. 2021;223:153465.
Du W, Feng Y, Wang X, Piao X, Cui Y, Chen L, et al. Curcumin suppresses malignant glioma cells growth and induces apoptosis by inhibition of SHH/GLI 1 signaling pathway in vitro and vivo. CNS Neurosci Ther. 2013;19(12):926–36.
Tabnak P, Masrouri S, Geraylow KR, Zarei M, Esmailpoor ZH. Targeting miRNAs with anesthetics in cancer: current understanding and future perspectives. Biomed Pharmacother. 2021;144:112309.
Wang S, Chen C, Li J, Xu X, Chen W, Li F. The CXCL12/CXCR4 axis confers temozolomide resistance to human glioblastoma cells via up-regulation of FOXM1. J Neurol Sci. 2020;414:116837.
Dai B, Gong A, Jing Z, Aldape KD, Kang S-H, Sawaya R, et al. Forkhead box M1 is regulated by heat shock factor 1 and promotes glioma cells survival under heat shock stress. J Biol Chem. 2013;288(3):1634–42.
Kowalski-Chauvel A, Gouaze-Andersson V, Baricault L, Martin E, Delmas C, Toulas C, et al. Alpha6-integrin regulates FGFR1 expression through the ZEB1/YAP1 transcription complex in glioblastoma stem cells resulting in enhanced proliferation and stemness. Cancers. 2019;11(3):406.
Quan J-J, Song J-N, Qu J-Q. PARP3 interacts with FoxM1 to confer glioblastoma cell radioresistance. Tumor Biol. 2015;36:8617–24.
Su X, Yang Y, Yang Q, Pang B, Sun S, Wang Y, et al. NOX4-derived ROS-induced overexpression of FOXM1 regulates aerobic glycolysis in glioblastoma. BMC Cancer. 2021;21(1):1181.
Zhang P, Chen X, Zhang L, Cao D, Chen Y, Guo Z, et al. POLE2 facilitates the malignant phenotypes of glioblastoma through promoting AURKA-mediated stabilization of FOXM1. Cell Death Dis. 2022;13(1):61.
Liu J, Guo S, Li Q, Yang L, Xia Z, Zhang L, et al. Phosphoglycerate dehydrogenase induces glioma cells proliferation and invasion by stabilizing forkhead box M1. J Neurooncol. 2013;111:245–55.
Li F, Jin D, Guan L, Zhang C-C, Wu T, Wang Y-J, et al. CEP55 promoted the migration, invasion and neuroshpere formation of the glioma cell line U251. Neurosci Lett. 2019;705:80–6.
Zhong X, Liu X, Li Y, Cheng M, Wang W, Tian K, et al. HMGA2 sustains self-renewal and invasiveness of glioma-initiating cells. Oncotarget. 2016;7(28):44365.
Dong Y, Xiong Y, Zhou D, Yao M, Wang X, Bi W, et al. TRIM56 reduces radiosensitization of human glioblastoma by regulating FOXM1-Mediated DNA repair. Mol Neurobiol. 2022;59(9):5312–25.
Xu K, Zhang K, Ma J, Yang Q, Yang G, Zong T, et al. CKAP4-mediated activation of FOXM1 via phosphorylation pathways regulates malignant behavior of glioblastoma cells. Transl Oncol. 2023;29:101628.
Chong YK, Sandanaraj E, Koh LWH, Thangaveloo M, Tan MSY, Koh GRH, et al. ST3GAL1-associated transcriptomic program in glioblastoma tumor growth, invasion, and prognosis. JNCI J Natl Cancer Inst. 2016;108(2):djv326.
Yang L, He K, Yan S, Yang Y, Gao X, Zhang M, et al. Metadherin/astrocyte elevated gene-1 positively regulates the stability and function of forkhead box M1 during tumorigenesis. Neuro Oncol. 2017;19(3):352–63.
Fu J, Peng J, Tu G. Knockdown MTDH inhibits glioma proliferation and migration and promotes apoptosis by downregulating MYBL2. Mediators Inflamm. 2022;2022:1–9.
Kao Y, Tsai W-C, Chen S-H, Hsu S-Y, Huang L-C, Chang C-J, et al. Shugoshin 2 is a biomarker for pathological grading and survival prediction in patients with gliomas. Sci Rep. 2021;11(1):18541.
Xie Z, Janczyk PŁ, Zhang Y, Liu A, Shi X, Singh S, et al. A cytoskeleton regulator AVIL drives tumorigenesis in glioblastoma. Nat Commun. 2020;11(1):3457.
Yokogami K, Kikuchi T, Watanabe T, Nakatake Y, Yamashita S, Mizuguchi A, et al. Methionine regulates self-renewal, pluripotency, and cell death of GIC through cholesterol—rRNA axis. BMC Cancer. 2022;22(1):1351.
Li Y-P, Liu Y, Xiao L-M, Chen L-K, Tao E-X, Zeng E-M, et al. Induction of cancer cell stemness in glioma through glycolysis and the long noncoding RNA HULC-activated FOXM1/AGR2/HIF-1α axis. Lab Investig. 2022;102(7):691–701.
Yang J, Tian S, Wang B, Wang J, Cao L, Wang Q, et al. CircPIK3C2A facilitates the progression of glioblastoma via targeting miR-877-5p/FOXM1 Axis. Front Oncol. 2021;11:801776.
Zhao P, Li T, Wang Y, Wang Y, Gu Q, Li Z. LncRNA MYCNOS promotes glioblastoma cell proliferation by regulating miR-216b/FOXM1 axis. Metab Brain Dis. 2021;36:1185–9.
Zhang T, Ma G, Zhang Y, Huo H, Zhao Y. miR-216b inhibits glioma cell migration and invasion through suppression of FoxM1. Oncol Rep. 2017;38(3):1751–9.
Xiong Q, Su H. MiR-325-3p functions as a suppressor miRNA and inhibits the proliferation and metastasis of glioma through targeting FOXM1. J Integr Neurosci. 2021;20(4):1019–28.
Li T, Ma J, Han X, Jia Y, Yuan H, Shui S, et al. MicroRNA-320 enhances radiosensitivity of glioma through down-regulation of sirtuin type 1 by directly targeting forkhead box protein M1. Transl Oncol. 2018;11(2):205–12.
Nadaradjane A, Briand J, Bougras-Cartron G, Disdero V, Vallette FM, Frenel J-S, et al. miR-370-3p is a therapeutic tool in anti-glioblastoma therapy but is not an intratumoral or cell-free circulating biomarker. Mol Ther Acids. 2018;13:642–50.
Li C, Guan X, Jing H, Xiao X, Jin H, Xiong J, et al. Circular RNA circBFAR promotes glioblastoma progression by regulating a miR-548b/FoxM1 axis. FASEB J. 2022;36(3):e22183.
Qi L, Wang W, Zhao G, Jiang H, Zhang Y, Zhao D, et al. Circular RNA circCCDC66 promotes glioma proliferation by acting as a ceRNA for miR-320a to regulate FOXM1 expression. Aging. 2021;13(13):17673.
Guo L, Ding Z, Huang N, Huang Z, Zhang N, Xia Z. Forkhead Box M1 positively regulates UBE2C and protects glioma cells from autophagic death. Cell Cycle. 2017;16(18):1705–18.
Zhang N, Wu X, Yang L, Xiao F, Zhang H, Zhou A, et al. FoxM1 inhibition sensitizes resistant glioblastoma cells to temozolomide by downregulating the expression of DNA-repair gene Rad51Targeting FoxM1-Rad51 axis enhances chemosensitivity. Clin cancer Res. 2012;18(21):5961–71.
Peng W, Han X, Zhang C, Ge L, Du F, Jin J, et al. FoxM1-mediated RFC5 expression promotes temozolomide resistance. Cell Biol Toxicol. 2017;33:527–37.
Ma Q, Liu Y, Shang L, Yu J, Qu Q. The FOXM1/BUB1B signaling pathway is essential for the tumorigenicity and radioresistance of glioblastoma. Oncol Rep. 2017;38(6):3367–75.
Lee Y, Kim KH, Kim DG, Cho HJ, Kim Y, Rheey J, et al. FoxM1 promotes stemness and radio-resistance of glioblastoma by regulating the master stem cell regulator Sox2. PLoS ONE. 2015;10(10):e0137703.
Dai B, Kang S-H, Gong W, Liu M, Aldape KD, Sawaya R, et al. Aberrant FoxM1B expression increases matrix metalloproteinase-2 transcription and enhances the invasion of glioma cells. Oncogene. 2007;26(42):6212–9.
Zhu GY, Shi BZ, Li Y. FoxM1 regulates Sirt1 expression in glioma cells. Eur Rev Med Pharmacol Sci. 2014;18(2):205–11.
Zeng W, Cheng Q, Wen Z, Wang J, Chen Y, Zhao J, et al. Aberrant ASPM expression mediated by transcriptional regulation of FoxM1 promotes the progression of gliomas. J Cell Mol Med. 2020;24(17):9613–26.
Guo L, Wu Z. FOXM1-mediated NUF2 expression confers temozolomide resistance to human glioma cells by regulating autophagy via the PI3K/AKT/mTOR signaling pathway. Neuropathology. 2022;42(5):430–46.
Chen S-H, Lin H-H, Li Y-F, Tsai W-C, Hueng D-Y. Clinical significance and systematic expression analysis of the thyroid receptor interacting protein 13 (TRIP13) as human gliomas biomarker. Cancers. 2021;13(10):2338.
Han X, Xue X, Zhou H, Zhang G. A molecular view of the radioresistance of gliomas. Oncotarget. 2017;8(59):100931.
Choudhary S, Burns SC, Mirsafian H, Li W, Vo DT, Qiao M, et al. Genomic analyses of early responses to radiation in glioblastoma reveal new alterations at transcription, splicing, and translation levels. Sci Rep. 2020;10(1):1–12.
Zona S, Bella L, Burton MJ, de Moraes GN, Lam EWF. FOXM1: an emerging master regulator of DNA damage response and genotoxic agent resistance. Biochim Biophys Acta (BBA) Gene Regul Mech. 2014;1839(11):1316–22.
Zhou Y-S, Wang W, Chen N, Wang L-C, Huang J-B. Research progress of anti-glioma chemotherapeutic drugs. Oncol Rep. 2022;47(5):1–12.
Bhat UG, Halasi M, Gartel AL. FoxM1 is a general target for proteasome inhibitors. PLoS ONE. 2009;4(8):e6593.
Tang J-H, Yang L, Chen J-X, Li Q-R, Zhu L-R, Xu Q-F, et al. Bortezomib inhibits growth and sensitizes glioma to temozolomide (TMZ) via down-regulating the FOXM1–Survivin axis. Cancer Commun. 2019;39(1):1–16.
Roth P, Mason WP, Richardson PG, Weller M. Proteasome inhibition for the treatment of glioblastoma. Expert Opin Investig Drugs. 2020;29(10):1133–41.
Takei J, Fukasawa N, Tanaka T, Yamamoto Y, Tamura R, Sasaki H, et al. Impact of neoadjuvant bevacizumab on neuroradiographic response and histological findings related to tumor stemness and the hypoxic tumor microenvironment in glioblastoma: paired comparison between newly diagnosed and recurrent glioblastomas. Front Oncol. 2022;12:898614.
Xu S, Tang L, Li X, Fan F, Liu Z. Immunotherapy for glioma: current management and future application. Cancer Lett. 2020;476:1–12.
Kikuchi R, Ueda R, Saito K, Shibao S, Nagashima H, Tamura R, et al. A pilot study of vaccine therapy with multiple glioma oncoantigen/glioma angiogenesis-associated antigen peptides for patients with recurrent/progressive high-grade glioma. J Clin Med. 2019;8(2):263.
Prinzing B, Schreiner P, Bell M, Fan Y, Krenciute G, Gottschalk S. MyD88/CD40 signaling retains CAR T cells in a less differentiated state. JCI insight. 2020. https://doi.org/10.1172/jci.insight.136093.
Zhang X, Wang J, Wang Y, Liu G, Li H, Yu J, et al. MELK inhibition effectively suppresses growth of glioblastoma and cancer stem-like cells by blocking AKT and FOXM1 pathways. Front Oncol. 2021;10:608082.
Tao W, Zhang A, Zhai K, Huang Z, Huang H, Zhou W, et al. SATB2 drives glioblastoma growth by recruiting CBP to promote FOXM1 expression in glioma stem cells. EMBO Mol Med. 2020;12(12):e12291.
Niu M, Cai W, Liu H, Chong Y, Hu W, Gao S, et al. Plumbagin inhibits growth of gliomas in vivo via suppression of FOXM1 expression. J Pharmacol Sci. 2015;128(3):131–6.
Liu X, Cai W, Niu M, Chong Y, Liu H, Hu W, et al. Plumbagin induces growth inhibition of human glioma cells by downregulating the expression and activity of FOXM1. J Neurooncol. 2015;121:469–77.
Li W, Liu J, Fu W, Zheng X, Ren L, Liu S, et al. 3-O-acetyl-11-keto-β-boswellic acid exerts anti-tumor effects in glioblastoma by arresting cell cycle at G2/M phase. J Exp Clin Cancer Res. 2018;37:1–15.
Wang W-X, Sun Z-H, Chen H-M, Xu B-N, Wang F-Y. Role and mechanism of Sophoridine on proliferation inhibition in human glioma U87MG cell line. Int J Clin Exp Med. 2015;8(1):464.
Li Y, Hu Y, Liu C, Wang Q, Han X, Han Y, et al. Human fibulin-3 protein variant expresses anti-cancer effects in the malignant glioma extracellular compartment in intracranial xenograft models. Oncotarget. 2017;8(63):106311.
Zhang M, Liu Y, Gao Y, Li S. Silibinin-induced glioma cell apoptosis by PI3K-mediated but Akt-independent downregulation of FoxM1 expression. Eur J Pharmacol. 2015;765:346–54.
Li C, Wang R, Guo W, Feng X, Guan N. Chalcone 9X contributed to repressing glioma cell growth and migration and inducing cell apoptosis by reducing FOXM1 expression in vitro and repressing tumor growth in vivo. Biomed Res Int. 2022;2022:1–13.
Shim J-K, Lim SH, Jeong JH, Choi RJ, Oh Y, Park J, et al. A lignan from alnus japonica inhibits glioblastoma tumorspheres by suppression of FOXM1. Sci Rep. 2022;12(1):13990.
Gandomkari MS, Ayat H, Ahadi AM. Recombinantly expressed MeICT, a new toxin from Mesobuthus eupeus scorpion, inhibits glioma cell proliferation and downregulates Annexin A2 and FOXM1 genes. Biotechnol Lett. 2022;44(5–6):703–12.
Coomans de Brachène A, Demoulin J-B. FOXO transcription factors in cancer development and therapy. Cell Mol life Sci. 2016;73:1159–72.
Hornsveld M, Dansen TB, Derksen PW, Burgering BMT. Re-evaluating the role of FOXOs in cancer. Semin cancer Biol. 2018;50:90–100.
Xing Y, Li A, Yang Y, Li X, Zhang L, Guo H. The regulation of FOXO1 and its role in disease progression. Life Sci. 2018;193:124–31.
Chen C, Han G, Li Y, Yue Z, Wang L, Liu J. FOXO1 associated with sensitivity to chemotherapy drugs and glial-mesenchymal transition in glioma. J Cell Biochem. 2019;120(1):882–93.
Chen C, Xu T, Zhou J, Yan Y, Li W, Yu H, et al. High cytoplasmic FOXO1 and pFOXO1 expression in astrocytomas are associated with worse surgical outcome. PLoS ONE. 2013;8(7):e69260.
Huang Z-Y, Hueng D-Y, Tsai W-C. The application of FOXO1A expression predicts aggressive behavior and poor prognosis in gliomas. Appl Immunohistochem Mol Morphol. 2020;28(1):74–82.
Wang C, Qiu J, Chen S, Li Y, Hu H, Cai Y, et al. Prognostic model and nomogram construction based on autophagy signatures in lower grade glioma. J Cell Physiol. 2021;236(1):235–48.
Liu S, Liu X, Zhang C, Shan W, Qiu X. T-cell exhaustion status under high and low levels of hypoxia-inducible factor 1α expression in glioma. Front Pharmacol. 2021;12:711772.
Choe G, Horvath S, Cloughesy TF, Crosby K, Seligson D, Palotie A, et al. Analysis of the phosphatidylinositol 3′-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 2003;63(11):2742–6.
Latonen L, Laiho M. Cellular UV damage responses—functions of tumor suppressor p53. Biochim Biophys Acta (BBA)-Rev Cancer. 2005;1755(2):71–89.
Firat E, Niedermann G. FoxO proteins or loss of functional p53 maintain stemness of glioblastoma stem cells and survival after ionizing radiation plus PI3K/mTOR inhibition. Oncotarget. 2016;7(34):54883.
Martinez E, Vazquez N, Lopez A, Fanniel V, Sanchez L, Marks R, et al. The PI3K pathway impacts stem gene expression in a set of glioblastoma cell lines. J Cancer Res Clin Oncol. 2020;146:593–604.
Yi R, Yang S, Lin X, Zhong L, Liao Y, Hu Z, et al. miR-5188 augments glioma growth, migration and invasion through an SP1-modulated FOXO1-PI3K/AKT-c-JUN-positive feedback circuit. J Cell Mol Med. 2020;24(20):11800–13.
Shao M, He Z, Yin Z, Ma P, Xiao Q, Song Y, et al. Xihuang pill induces apoptosis of human glioblastoma U-87 MG cells via targeting ROS-mediated Akt/mTOR/FOXO1 pathway. Evidence-Based Complement Altern Med. 2018;2018:1.
Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013;18(5):726–39.
Lau CJ, Koty Z, Nalbantoglu J. Differential response of glioma cells to FOXO1-directed therapy. Cancer Res. 2009;69(13):5433–40.
Kousteni S. FoxO1, the transcriptional chief of staff of energy metabolism. Bone. 2012;50(2):437–43.
Udawant S, Litif C, Lopez A, Gunn B, Schuenzel E, Keniry M. PI3K Pathway inhibition with NVP-BEZ235 hinders glycolytic metabolism in glioblastoma multiforme cells. Cells. 2021;10(11):3065.
Atif F, Yousuf S, Espinosa-Garcia C, Sergeeva E, Stein DG. Progesterone treatment attenuates glycolytic metabolism and induces senescence in glioblastoma. Sci Rep. 2019;9(1):988.
Ho KK, Myatt SS, Lam EWF. Many forks in the path: cycling with FoxO. Oncogene. 2008;27(16):2300–11.
Piao X, Li W, Li Z, Zhang N, Fang H, Zahid D, et al. Forced Fox O1: S249V expression suppressed glioma cell proliferation through G2/M cell cycle arrests and increased apoptosis. Neurol Res. 2019;41(2):189–98.
Das S, Deshmukh RS, Sharma S. Cyclin F-dependent degradation of RBPJ inhibits IDH1R132H-mediated tumorigenesis. Cancer Res. 2018;78:6386.
Seoane J, Le H-V, Shen L, Anderson SA, Massagué J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117(2):211–23.
Shi HZ, Wang DN, Xu F, Teng JH, Wang YL. miR-135a inhibits glioma cell proliferation and invasion by directly targeting FOXO1. Eur Rev Med Pharmacol Sci. 2018;22(13):4215–23.
Lopez-Bertoni H, Kotchetkov IS, Mihelson N, Lal B, Rui Y, Ames H, et al. A Sox2: miR-486-5p axis regulates survival of GBM cells by inhibiting tumor suppressor NetworksSox2: miR-486-5p axis controls survival of GBM cells. Cancer Res. 2020;80(8):1644–55.
Cao C, Zhang J, Zhang Z, Feng Y, Wang Z. Knockdown circular RNA circGFRA1 inhibits glioma cell proliferation and migration by upregulating microRNA-99a. NeuroReport. 2021;32(9):748–56.
Qiu X, Tan G, Wen H, Lian L, Xiao S. Forkhead box O1 targeting replication factor C subunit 2 expression promotes glioma temozolomide resistance and survival. Ann Transl Med. 2021;9(8):692.
Liu C-L, Zhao D, Li J-J, Liu S, An J-J, Wang D, et al. Inhibition of glioblastoma progression by urolithin A in vitro and in vivo by regulating Sirt1-FOXO1 axis via ERK/AKT signaling pathways. Neoplasma. 2021;69:80.
Han J, Yu X, Wang S, Wang Y, Liu Q, Xu H, et al. IGF2BP2 induces U251 glioblastoma cell chemoresistance by inhibiting FOXO1-mediated PID1 expression through stabilizing lncRNA DANCR. Front Cell Dev Biol. 2022;9:3283.
Zhu G, Wang Z, Mijiti M, Du G, Li Y, Dangmurenjiafu G. MiR-28-5p promotes human glioblastoma cell growth through inactivation of FOXO1. Int J Clin Exp Pathol. 2019;12(8):2972.
Ni W, Luo L, Zuo P, Li R, Xu X, Wen F, et al. Mir-374a inhibitor enhances etoposide-induced cytotoxicity against glioma cells through upregulation of foxo1. Oncol Res Featur Preclin Clin Cancer Ther. 2019;27(6):703–12.
Xie C, Xu M, Lu D, Zhang W, Wang L, Wang H, et al. Candidate genes and microRNAs for glioma pathogenesis and prognosis based on gene expression profiles. Mol Med Rep. 2018;18(3):2715–23.
Zhao X, Liu Y, Zheng J, Liu X, Chen J, Liu L, et al. GAS5 suppresses malignancy of human glioma stem cells via a miR-196a-5p/FOXO1 feedback loop. Biochim Biophys Acta (BBA)-Mol Cell Res. 2017;1864(10):1605–17.
Que T, Song Y, Liu Z, Zheng S, Long H, Li Z, et al. Decreased miRNA-637 is an unfavorable prognosis marker and promotes glioma cell growth, migration and invasion via direct targeting Akt1. Oncogene. 2015;34(38):4952–63.
Lei B-X, Liu Z-H, Li Z-J, Li C, Deng Y-F. miR-21 induces cell proliferation and suppresses the chemosensitivity in glioblastoma cells via downregulation of FOXO1. Int J Clin Exp Med. 2014;7(8):2060.
Tang H, Bian Y, Tu C, Wang Z, Yu Z, Liu Q, et al. The miR-183/96/182 cluster regulates oxidative apoptosis and sensitizes cells to chemotherapy in gliomas. Curr Cancer Drug Targets. 2013;13(2):221–31.
Huang C, Chen D, Zhu H, Lv S, Li Q, Li G. LITAF enhances radiosensitivity of human glioma cells via the FoxO1 pathway. Cell Mol Neurobiol. 2019;39:871–82.
Tang G, Liu D, Xiao G, Liu Q, Yuan J. Transcriptional repression of FOXO1 by KLF4 contributes to glioma progression. Oncotarget. 2016;7(49):81757.
Ranjit M, Hirano M, Aoki K, Okuno Y, Ohka F, Yamamichi A, et al. Aberrant active cis-regulatory elements associated with downregulation of RET finger protein overcome chemoresistance in glioblastoma. Cell Rep. 2019;26(9):2274–81.
Kesarwani P, Kant S, Zhao Y, Prabhu A, Buelow KL, Miller CR, et al. Quinolinate promotes macrophage-induced immune tolerance in glioblastoma through the NMDAR/PPARγ signaling axis. Nat Commun. 2023;14(1):1459.
Wang L, Wang J, Jin T, Zhou Y, Chen Q. FoxG1 facilitates proliferation and inhibits differentiation by downregulating FoxO/Smad signaling in glioblastoma. Biochem Biophys Res Commun. 2018;504(1):46–53.
Liu J, Liu L, Xue Y, Meng F, Li S, Wang P, et al. Anti-neoplastic activity of low-dose endothelial-monocyte activating polypeptide-II results from defective autophagy and G2/M arrest mediated by PI3K/Akt/FoxO1 axis in human glioblastoma stem cells. Biochem Pharmacol. 2014;89(4):477–89.
Cai R, Xue Y, Huang J, Wang J, Wang J, Zhao S, et al. NS1619 regulates the expression of caveolin-1 protein in a time-dependent manner via ROS/PI3K/PKB/FoxO1 signaling pathway in brain tumor microvascular endothelial cells. J Neurol Sci. 2016;369:109–18.
Cheng C, Jiao J, Qian Y, Guo X, Huang J, Dai M, et al. Curcumin induces G2/M arrest and triggers apoptosis via FoxO1 signaling in U87 human glioma cells. Mol Med Rep. 2016;13(5):3763–70.
Sun F, Han D, Cao B, Wang B, Dong N, Jiang D. Caffeine-induced nuclear translocation of FoxO1 triggers Bim-mediated apoptosis in human glioblastoma cells. Tumor Biol. 2016;37:3417–23.
Han D, Zhang J, Wei W, Tao T, Hu Q, Wang Y, et al. Fenofibrate induces G 0/G 1 phase arrest by modulating the PPARα/FoxO1/p27 kip pathway in human glioblastoma cells. Tumor Biol. 2015;36:3823–9.
Weng H-Y, Hsu M-J, Wang C-C, Chen B-C, Hong C-Y, Chen M-C, et al. Zerumbone suppresses IKKα, Akt, and FOXO1 activation, resulting in apoptosis of GBM 8401 cells. J Biomed Sci. 2012;19(1):1–11.
Chen X, Luo X, Cheng Y. Trifluoperazine prevents FOXO1 nuclear excretion and reverses doxorubicin-resistance in the SHG44/DOX drug-resistant glioma cell line. Int J Mol Med. 2018;42(6):3300–8.
Mota M, Sweha SR, Pun M, Natarajan SK, Ding Y, Chung C, et al. Targeting SWI/SNF ATPases in H3. 3K27M diffuse intrinsic pontine gliomas. Proc Natl Acad Sci. 2023;120(18):e2221175120.
Weng H-Y, Hsu M-J, Chen C-C, Chen B-C, Hong C-Y, Teng C-M, et al. Denbinobin induces human glioblastoma multiforme cell apoptosis through the IKKα–Akt–FKHR signaling cascade. Eur J Pharmacol. 2013;698(1–3):103–9.
Flores D, Lopez A, Udawant S, Gunn B, Keniry M. The FOXO1 inhibitor AS1842856 triggers apoptosis in glioblastoma multiforme and basal-like breast cancer cells. FEBS Open Bio. 2023;13:352.
Liu Y, Ao X, Ding W, Ponnusamy M, Wu W, Hao X, et al. Critical role of FOXO3a in carcinogenesis. Mol Cancer. 2018;17(1):1–12.
Qian Z, Ren L, Wu D, Yang X, Zhou Z, Nie Q, et al. Overexpression of FoxO3a is associated with glioblastoma progression and predicts poor patient prognosis. Int J cancer. 2017;140(12):2792–804.
Shi J, Zhang L, Shen A, Zhang J, Wang Y, Zhao Y, et al. Clinical and biological significance of forkhead class box O 3a expression in glioma: mediation of glioma malignancy by transcriptional regulation of p27 kip1. J Neurooncol. 2010;98:57–69.
Zhang J, Yang Z, Ou J, Xia X, Zhi F, Cui J. The F-box protein FBXL 18 promotes glioma progression by promoting K63-linked ubiquitination of Akt. FEBS Lett. 2017;591(1):145–54.
Sato A, Sunayama J, Okada M, Watanabe E, Seino S, Shibuya K, et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl Med. 2012;1(11):811–24.
Taniguchi K, Ii H, Kageyama S, Takagi H, Chano T, Kawauchi A, et al. Depletion of gamma-glutamylcyclotransferase inhibits cancer cell growth by activating the AMPK–FOXO3a–p21 axis. Biochem Biophys Res Commun. 2019;517(2):238–43.
Ciechomska IA, Gielniewski B, Wojtas B, Kaminska B, Mieczkowski J. EGFR/FOXO3a/BIM signaling pathway determines chemosensitivity of BMP4-differentiated glioma stem cells to temozolomide. Exp Mol Med. 2020;52(8):1326–40.
Gopinath S, Alapati K, Malla RR, Gondi CS, Mohanam S, Dinh DH, et al. Mechanism of p27 upregulation induced by downregulation of cathepsin B and uPAR in glioma. Mol Oncol. 2011;5(5):426–37.
Ramis G, Villalonga-Planells R, Serra-Sitjar M, Brell M, Fernández de Mattos S, Villalonga P. The tumor suppressor FOXO3a mediates the response to EGFR inhibition in glioblastoma cells. Cell Oncol. 2019;42:521–36.
Sato A, Okada M, Shibuya K, Watanabe E, Seino S, Narita Y, et al. Pivotal role for ROS activation of p38 MAPK in the control of differentiation and tumor-initiating capacity of glioma-initiating cells. Stem Cell Res. 2014;12(1):119–31.
Park SY, Mittal S, Dong J, Jeong K, Martinez-Ledesma E, Piao Y, et al. Depletion of CLK2 sensitizes glioma stem-like cells to PI3K/mTOR and FGFR inhibitors. Am J Cancer Res. 2020;10(11):3765.
Park SY, Piao Y, Thomas C, Fuller GN, De Groot JF. Cdc2-like kinase 2 is a key regulator of the cell cycle via FOXO3a/p27 in glioblastoma. Oncotarget. 2016;7(18):26793.
Osuka S, Sampetrean O, Shimizu T, Saga I, Onishi N, Sugihara E, et al. IGF1 receptor signaling regulates adaptive radioprotection in glioma stem cells. Stem Cells. 2013;31(4):627–40.
Guan H, Song L, Cai J, Huang Y, Wu J, Yuan J, et al. Sphingosine kinase 1 regulates the Akt/FOXO3a/Bim pathway and contributes to apoptosis resistance in glioma cells. PLoS ONE. 2011;6(5):e19946.
Peng H, Du B, Jiang H, Gao J. Over-expression of CHAF1A promotes cell proliferation and apoptosis resistance in glioblastoma cells via AKT/FOXO3a/Bim pathway. Biochem Biophys Res Commun. 2016;469(4):1111–6.
Dai B, Pieper RO, Li D, Wei P, Liu M, Woo SY, et al. FoxM1B regulates NEDD4-1 expression, leading to cellular transformation and full malignant phenotype in immortalized human astrocytesFoxM1B in astrocytoma transformation. Cancer Res. 2010;70(7):2951–61.
Taylor OG, Brzozowski JS, Skelding KA. Glioblastoma multiforme: an overview of emerging therapeutic targets. Front Oncol. 2019;9:963.
Strickland M, Stoll EA. Metabolic reprogramming in glioma. Front cell Dev Biol. 2017;5:43.
Pandey V, Bhaskara VK, Babu PP. Implications of mitogen-activated protein kinase signaling in glioma. J Neurosci Res. 2016;94(2):114–27.
Sunayama J, Sato A, Matsuda K-I, Tachibana K, Watanabe E, Seino S, et al. FoxO3a functions as a key integrator of cellular signals that control glioblastoma stem-like cell differentiation and tumorigenicity. Stem Cells. 2011;29(9):1327–37.
Allen JE, Krigsfeld G, Mayes PA, Patel L, Dicker DT, Patel AS, et al. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci Transl Med. 2013;5(171):171ra17-171ra17.
Fasano C, Disciglio V, Bertora S, Lepore Signorile M, Simone C. FOXO3a from the nucleus to the mitochondria: a round trip in cellular stress response. Cells. 2019;8(9):1110.
Agnihotri S, Golbourn B, Huang X, Remke M, Younger S, Cairns RA, et al. PINK1 is a negative regulator of growth and the warburg effect in GlioblastomaPINK1 inhibits glioblastoma growth. Cancer Res. 2016;76(16):4708–19.
Hashimoto T, Urushihara Y, Murata Y, Fujishima Y, Hosoi Y. AMPK increases expression of ATM through transcriptional factor Sp1 and induces radioresistance under severe hypoxia in glioblastoma cell lines. Biochem Biophys Res Commun. 2022;590:82–8.
Shiga S, Murata Y, Hashimoto T, Urushihara Y, Fujishima Y, Kudo K, et al. DNA-PKcs is activated under nutrient starvation and activates Akt, MST1, FoxO3a, and NDR1. Biochem Biophys Res Commun. 2020;521(3):668–73.
Brucker DP, Maurer GD, Harter PN, Rieger J, Steinbach JP. FOXO3a orchestrates glioma cell responses to starvation conditions and promotes hypoxia-induced cell death. Int J Oncol. 2016;49(6):2399–410.
Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ Jr, DiStefano PS, et al. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296(5567):530–4.
He C, Lu S, Wang X, Wang C, Wang L, Liang S, et al. FOXO3a protects glioma cells against temozolomide-induced DNA double strand breaks via promotion of BNIP3-mediated mitophagy. Acta Pharmacol Sin. 2021;42(8):1324–37.
Chalkiadaki A, Guarente L. The multifaceted functions of sirtuins in cancer. Nat Rev Cancer. 2015;15(10):608–24.
Carafa V, Altucci L, Nebbioso A. Dual tumor suppressor and tumor promoter action of sirtuins in determining malignant phenotype. Front Pharmacol. 2019;10:38.
Dong Z, Zhong X, Lei Q, Chen F, Cui H. Transcriptional activation of SIRT6 via FKHRL1/FOXO3a inhibits the warburg effect in glioblastoma cells. Cell Signal. 2019;60:100–13.
Qian X, Sun C, Zhu D. MST1 suppresses viability and promotes apoptosis of glioma cells via upregulating SIRT6 expression. J Integr Neurosci. 2019;18(2):117–26.
Huo L, Bai X, Wang Y, Wang M. Betulinic acid derivative B10 inhibits glioma cell proliferation through suppression of SIRT1, acetylation of FOXO3a and upregulation of Bim/PUMA. Biomed Pharmacother. 2017;92:347–55.
Wang B, Li X, Liu L, Wang M. β-Catenin: oncogenic role and therapeutic target in cervical cancer. Biol Res. 2020. https://doi.org/10.1186/s40659-020-00301-7.
Hoogeboom D, Essers MAG, Polderman PE, Voets E, Smits LMM, Boudewijn MT. Interaction of FOXO with β-catenin inhibits β-catenin/T cell factor activity. J Biol Chem. 2008;283(14):9224–30.
Xu K, Zhang Z, Pei H, Wang H, Li L, Xia Q. FoxO3a induces temozolomide resistance in glioblastoma cells via the regulation of β-catenin nuclear accumulation corrigendum in/10.3892/or. 2020.7842. Oncol Rep. 2017;37(4):2391–7.
Sun D, Yang S, Zhang X, Li S, Wang L, Chen J, et al. Forkhead box protein O3a promotes glioma cell resistance to temozolomide by regulating matrix metallopeptidase and β-catenin. Oncol Lett. 2021;21(4):1–9.
Lu M, Wang Y, Zhou S, Xu J, Li J, Tao R, et al. MicroRNA-370 suppresses the progression and proliferation of human astrocytoma and glioblastoma by negatively regulating β-catenin and causing activation of FOXO3a. Exp Ther Med. 2018;15(1):1093–8.
Aroui S, Dardevet L, Najlaoui F, Kammoun M, Laajimi A, Fetoui H, et al. PTEN-regulated AKT/FoxO3a/Bim signaling contributes to human cell glioblastoma apoptosis by platinum-maurocalcin conjugate. Int J Biochem Cell Biol. 2016;77:15–22.
Handrick R, Rübel A, Faltin H, Eibl H, Belka C, Jendrossek V. Increased cytotoxicity of ionizing radiation in combination with membrane-targeted apoptosis modulators involves downregulation of protein kinase B/Akt-mediated survival-signaling. Radiother Oncol. 2006;80(2):199–206.
Wilk A, Urbanska K, Grabacka M, Mullinax J, Marcinkiewicz C, Impastato D, et al. Fenofibrate-induced nuclear translocation of FoxO3A triggers Bim-mediated apoptosis in glioblastoma cells in vitro. Cell Cycle. 2012;11(14):2660–71.
Cui Q-K, Liu W-D, Zhu J-X, Wang Y-H, Wang Z-G. MicroRNA-184 promotes proliferation ability of glioma cells by regulating FOXO3. Asian Pac J Trop Med. 2014;7(10):776–9.
Sharma N, Colangelo NW, de Toledo SM, Azzam EI. Diffusible factors secreted by glioblastoma and medulloblastoma cells induce oxidative stress in bystander neural stem progenitors. ASN Neuro. 2016;8(4):1759091416662808.
Quan K, Zhang X, Fan K, Liu P, Yue Q, Li B, et al. Icariside II induces cell cycle arrest and apoptosis in human glioblastoma cells through suppressing Akt activation and potentiating FOXO3a activity. Am J Transl Res. 2017;9(5):2508.
Bulstrode H, Johnstone E, Marques-Torrejon MA, Ferguson KM, Bressan RB, Blin C, et al. Elevated FOXG1 and SOX2 in glioblastoma enforces neural stem cell identity through transcriptional control of cell cycle and epigenetic regulators. Genes Dev. 2017;31(8):757–73.
Audesse AJ, Karashchuk G, Gardell ZA, Lakis NS, Maybury-Lewis SY, Brown AK, et al. FOXO3 regulates a common genomic program in aging and glioblastoma stem cells. Aging and cancer. 2021;2(4):137–59.
Lin J, Teo S, Lam DH, Jeyaseelan K, Wang S. MicroRNA-10b pleiotropically regulates invasion, angiogenicity and apoptosis of tumor cells resembling mesenchymal subtype of glioblastoma multiforme. Cell Death Dis. 2012;3(10):e398–e398.
Ge Y-F, Sun J, Jin C-J, Cao B-Q, Jiang Z-F, Shao J-F. AntagomiR-27a targets FOXO3a in glioblastoma and suppresses U87 cell growth in vitro and in vivo. Asian Pacific J Cancer Prev. 2013;14(2):963–8.
Jiang L, Wang C, Lei F, Zhang L, Zhang X, Liu A, et al. miR-93 promotes cell proliferation in gliomas through activation of PI3K/Akt signaling pathway. Oncotarget. 2015;6(10):8286.
Ling N, Gu J, Lei Z, Li M, Zhao J, Zhang H-T, et al. microRNA-155 regulates cell proliferation and invasion by targeting FOXO3a in glioma. Oncol Rep. 2013;30(5):2111–8.
Wu P, Cai J, Chen Q, Han B, Meng X, Li Y, et al. Lnc-TALC promotes O6-methylguanine-DNA methyltransferase expression via regulating the c-Met pathway by competitively binding with miR-20b-3p. Nat Commun. 2019;10(1):2045.
De La Iglesia N, Konopka G, Puram SV, Chan JA, Bachoo RM, You MJ, et al. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008;22(4):449–62.
Zou Z, Dong Y, Liu J, Zhao Z, Li G, Liu D. Circ-DONSON promotes malignant progression of glioma through modulating FOXO3. Eur Rev Med Pharmacol Sci. 2020;24(2):749–57.
Qiu C, Ma J, Wang ML, Zhang Q, Li YB. MicroRNA-155 deficiency in CD8+ T cells inhibits its anti-glioma immunity by regulating FoxO3a. Eur Rev Med Pharmacol Sci. 2019;23(6):2486–96.
Du P, Meng L, Liao X, Liu Y, Mo X, Gong M, et al. The miR-27a-3p/FTO axis modifies hypoxia-induced malignant behaviors of glioma cells: miR-27a-3p/FTO affects glioma growth. Acta Biochim Biophys Sin. 2023;55(1):103.
Xu K, Pei H, Zhang Z, Dong S, Fu R-J, Wang W-M, et al. FoxO3a mediates glioma cell invasion by regulating MMP9 expression. Oncol Rep. 2016;36(5):3044–50.
Ohno M, Kitanaka C, Miyakita Y, Tanaka S, Sonoda Y, Mishima K, et al. Metformin with temozolomide for newly diagnosed glioblastoma: results of phase I study and a brief review of relevant studies. Cancers. 2022;14(17):4222.
Jung Y, Park H, Zhao H-Y, Jeon R, Ryu J-H, Kim W-Y. Systemic approaches identify a garlic-derived chemical, Z-ajoene, as a glioblastoma multiforme cancer stem cell-specific targeting agent. Mol Cells. 2014;37(7):547.
Morfouace M, Lalier L, Bahut M, Bonnamain V, Naveilhan P, Guette C, et al. Comparison of spheroids formed by rat glioma stem cells and neural stem cells reveals differences in glucose metabolism and promising therapeutic applications. J Biol Chem. 2012;287(40):33664–74.
Huang LE, Cohen AL, Colman H, Jensen RL, Fults DW, Couldwell WT. IGFBP2 expression predicts IDH-mutant glioma patient survival. Oncotarget. 2017;8(1):191.
Lv Y, Zhang S, Liu Z, Tian Y, Liang N, Zhang J. Prognostic value of preoperative neutrophil to lymphocyte ratio is superior to systemic immune inflammation index for survival in patients with glioblastoma. Clin Neurol Neurosurg. 2019;181:24–7.
Pasqualetti F, Giampietro C, Montemurro N, Giannini N, Gadducci G, Orlandi P, et al. Old and new systemic immune-inflammation indexes are associated with overall survival of glioblastoma patients treated with radio-chemotherapy. Genes. 2022;13(6):1054.
Funding
No fund was received for this study.
Author information
Authors and Affiliations
Contributions
All appropriate contributors are listed as authors, and all authors have agreed to the manuscript's content and its submission. PT wrote and edited the original draft (concept and design, manuscript preparation, manuscript editing, and manuscript review). MS and ME collected data and edited the manuscript (literature search, manuscript editing, and manuscript review). AHB assisted the authors in revising the manuscript (literature search, manuscript editing, and manuscript review).
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable (review study).
Consent for publication
Not applicable.
Competing interests
The authors have no competing interests to declare that they are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tabnak, P., Hasanzade Bashkandi, A., Ebrahimnezhad, M. et al. Forkhead box transcription factors (FOXOs and FOXM1) in glioma: from molecular mechanisms to therapeutics. Cancer Cell Int 23, 238 (2023). https://doi.org/10.1186/s12935-023-03090-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-023-03090-7