
Abstract
RNA demethylase ALKBH5 takes part in the modulation of N6-methyladenosine (m6A) modification and controls various cell processes. ALKBH5-mediated m6A demethylation regulates gene expression by affecting multiple events in RNA metabolism, e.g., pre-mRNA processing, mRNA decay and translation. Mounting evidence shows that ALKBH5 plays critical roles in a variety of human malignancies, mostly via post-transcriptional regulation of oncogenes or tumor suppressors in an m6A-dependent manner. Meanwhile, increasing non-coding RNAs are recognized as functional targets of ALKBH5 in cancers. Here we reviewed up-to-date findings about the pathological roles of ALKBH5 in cancer, the molecular mechanisms by which it exerts its functions, as well as the underlying mechanism of its dysregulation. We also discussed the therapeutic implications of targeting ALKBH5 in cancer and potential ALKBH5-targeting strategies.
Similar content being viewed by others
Introduction
More than 170 post-transcriptional modifications in different types of RNA have been reported [1, 2], with N6-methyladenosine (m6A) being a well-studied RNA modification thus far. m6A was firstly described about five decades ago (1970s) and was identified as the commonest internal modification of polyadenylated mRNAs in most eukaryotic species, including mammals [3, 4]. In 2010s, the development of various next-generation sequencing (NGS)-based m6A sequencing techniques provided further insights into such epigenetic mark, revealing m6A presence in virtually all RNA types, such as mRNAs, ribosomal RNAs (rRNAs), microRNAs (miRNAs), long non-coding RNAs (lncRNAs), circular RNAs (circRNAs) and small nuclear RNAs (snRNAs) [5,6,7,8,9,10,11,12]. m6A undergoes a dynamic regulation by RNA methyltransferases (“writers”) and demethylases (“erasers”). The deposition of m6A modification into RNAs is catalyzed by the m6A methyltransferase complex (MTC) comprising a METTL3-METTL14 heterodimer core alongside other ligands, including WTAP, VIRMA, RBM15A/B, ZC3H13 and HAKAI [13,14,15,16,17,18,19]. m6A removal from an RNA could be performed by m6A demethylases, including fat mass and obesity-associated protein (FTO) and alpha-ketoglutarate-dependent dioxygenase alkB homolog 5 (ALKBH5) (Fig. 1a) [20, 21]. The reversible m6A modification contributes to multiple pathways controlling mRNA metabolism and have critical functions in gene regulation. Indeed, m6A regulates mRNA splicing [12, 22,23,24,25], promotes mRNA nuclear export [26], alters mRNA stability [27,28,29,30,31,32], increases translation efficiency [30,31,32,33,34,35,36] and facilitates non-canonical translation initiation [37]. Most of these effects are controlled by m6A “readers”, which selectively interact with m6A and exert a regulatory function on the m6A-marked mRNA (Table 1). Additionally, m6A modifications of non-coding RNAs (ncRNAs) are critical for their expression and functions, with some of them using the same machinery employed by mRNAs for writing, erasing and reading [7, 11, 12, 22, 38,39,40,41]. Given its wide-ranging functions in RNA metabolism and gene regulation, m6A is unsurprisingly required for numerous biological processes in normal physiology and disease conditions.

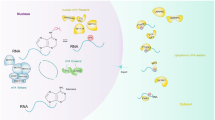
Dynamic regulation of N6-methyladenosine (m6A) and structure of RNA demethylase ALKBH5. a The m6A modification is added to the RNA by the methyltransferase complex (writer) composed of the METTL3-METTL14 heterodimer core subunit and additional cofactors such as WTAP. The RNA m6A modification could be removed by RNA demethylases (erasers), FTO and ALKBH5 in a reversible manner. b Domain structures of ALKBH5. c Cartoon representation of the ALKBH566–292 structure (PDB ID: 4NJ4) in complex with Mn(II) [67]. Mn(II) is a substitute for Fe(II). d Schematic form of the ALKBH566–292 structure [67]. Arrows represent β-strands and circles represent α-helices. The DSBH is highlighted in yellow, the extra loop (β9-10 loop) in purple and the NRLs in red
ALKBH5 is a member of the well-conserved AlkB family of non-heme Fe(II)/α-KG-dependent dioxygenases, which mediate the repair of N-alkylated nucleobases by oxidative demethylation [42, 43]. There are nine human AlkB members, including ALKBH1–8 and FTO [44], which have the same 2-oxoglutarate (2OG, also known as α-KG) oxygenase double-stranded β-helix (DSBH) and associated 2OG and iron binding sites, but differ in substrates and functions [42, 45]. This difference exists even between two m6A demethylases: while FTO has been demonstrated to demethylate internal m6A on mRNAs and U6 RNAs, N6, 2-O-dimethyladenosine (m6Am) on mRNAs and snRNAs, N1-methyladenosine (m1A) on tRNAs, 3-methylthymine (m3T) on single-stranded DNAs (ssDNAs) and 3-methyluracil (m3U) on single-stranded RNAs (ssRNAs) [20, 46,47,48], ALKBH5 was found to be only responsible for catalyzing the removal of m6A on ssRNAs [21]. ALKBH5 appears to be localized to nuclear speckles that contribute to the assembly of mRNA-processing factors, which supports the notion that nuclear nascent RNAs are major substrates of ALKBH5 [21]. ALKBH5 preferentially interacts with the distal 5′ region of coding sequences [49], with specific preference for the consensus sequence Pu[G>A]m6AC[A/C/U] (Pu is any purine base) [21]. In normal condition, ALKBH5 shows high expression in the testis and lung, followed by spleen, kidney and liver, with low cardiac and cerebral amounts [21]. Consistently, ALKBH5-deficient mice show impaired spermatogenesis because of abnormal levels of major genes controlling spermatogenic maturation [21].
In addition to spermatogenesis [21, 50], significant biomolecular involvement of ALKBH5 has been demonstrated in osteogenic differentiation [51, 52], heart regeneration [53], embryonic stem cell cardiac commitment [54], brain development [55], post-ischemic angiogenesis [56], ROS-induced DNA damage response [57] and immune response [58,59,60,61,62]. Abnormal expression of ALKBH5 is tightly associated with human pathologies. ALKBH5 was found to be involved in the ossification of the ligamentum flavum [63], Hirschsprung's disease [64] and cerebral ischemia–reperfusion injury [65]. In addition, the role of ALKBH5 in disease has been most extensively studied in the context of cancer. This review summarized recently reported progress in understanding ALKBH5’s structure and up-to-date findings concerning ALKBH5’s roles in cancer, with a main focus on the regulatory effects of ALKBH5 on diverse targets in cancer and how ALKBH5 is dysregulated in cancer. We also highlighted advances in ALKBH5-targeting approaches and the related clinical potential in tumor therapy.
ALKBH5 structure
The full-length human ALKBH5 has 394 amino acids and contains an active site motif HXDXnH (X = any amino acid) for Fe(II) binding, RXnR for 2OG binding and substrate recognition, and an extra loop resulting in preferential interaction with single-stranded over double-stranded nucleic acids (Fig. 1b) [66,67,68,69]. In detail, the catalytic core of ALKBH5 exhibits a well-conserved DSBH, in which β6, β8, β11 and β13 form the major β-sheet, whereas β7, β9, β10 and β12 form the minor one (Fig. 1c, d) [67]. The DSBH functions as a scaffold for three Fe(II)-ligating amino acids, including His204, Asp206 and His266 (Fig. 1c), which constitute the well-conserved HXDXnH motif coordinating metal ions [67]. The 2OG binding site is found in a cavity surrounded by the two DSBH’s β-sheets, with the more open end of this cavity allowing the interaction between substrate and active site [67]. The m6A base likely packs against His204 in a pocket comprising Arg130 and Tyr139 [68]. An unanticipated disulfide bond linking Cys230 and Cys267 is found in ALKBH5, which impedes the access of dsDNAs and dsRNAs to its active site [69]. Meanwhile, an extra loop (β9-10 loop) in ALKBH5’s steric clash (amino acids 229–243), causes steric hindrance of dsDNAs (Fig. 1c, d) [67, 68]. Moreover, the nucleotide recognition lid (NRL) outside the DSBH fold, which is further divided into two β hairpin-like loops named NRL1 (β2–3) and NRL2 (β4–5), plays an important role in substrate recognition and catalysis (Fig. 1c, d) [67, 69]. Furthermore, the N-terminus of ALKBH5 has multiple alanine residues and a coiled-coil structure, which may determine its localization [66]. Its C-terminus is likely disordered and comprises Arg–Ser-rich regions, which might control RNA interactions [67]. The knowledge of ALKBH5 structure provides clues for rationally designing specific AKBH5 inhibitors and activators.
Roles of ALKBH5 in cancer
Recent findings suggested ALKBH5 is commonly dysregulated in multiple malignancies, and plays important roles as an m6A demethylase. Altered ALKBH5 expression could both promote and suppress carcinogenesis, based on cancer type (Fig. 2; Table 2).

Roles of ALKBH5 in cancers. GM Glioma, GBM Glioblastoma, OVC Ovarian cancer, ENC Endometrial cancer, CVC Cervical cancer, BRC Breast cancer, AML Acute myeloid leukemia, UM Uveal melanoma, PAM Pituitary adenomas, OSCC Oral squamous cell carcinoma, ESCC Esophageal squamous cell carcinoma, GC Gastric cancer, HBV-HCC Hepatitis B virus (HBV) related hepatocellular carcinoma, ICC Intrahepatic cholangiocarcinoma, LAM Lung adenocarcinoma, NSCLC Non-small cell lung cancer, OST Osteosarcoma, RCC Renal cell carcinoma, CC Colon cancer, PC Pancreatic cancer, HCC Hepatocellular carcinoma, BC Bladder cancer. ALKBH5 plays an oncogenic role in cancers in red, and plays a tumor-suppressive role in cancers in green; ALKBH5 has controversial roles reported in cancers with both colors
ALKBH5 as an oncogene
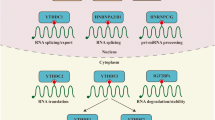
ALKBH5 was shown to induce the formation and development of various malignancies by removing m6A modifications on essential RNAs. ALKBH5 overexpression and its contributions to carcinogenesis are presented in Table 2 and Fig. 3, which include the core target RNAs and/or pathways regulated by ALKBH5.

Molecular mechanisms underpinning ALKBH5 regulation on target RNAs in cancers. HuR Human antigen R, YTHDF1/2/3 YTH domain-containing family protein 1/2/3, IGF2BP1/2/3 Insulin-like growth factor 2 mRNA binding protein 1/2/3
Breast cancer
Breast cancer represents the major female malignancy, affecting one in 20 women worldwide, with an even higher incidence in high-income countries [70]. ALKBH5 amounts are increased in immortalized and oncogenically-transformed human mammary epithelial cells in comparison with their primary cell precursors [71]. Besides, ALKBH5 is upregulated in breast cancer samples compared to noncancerous tissue [72]. Knockdown of ALKBH5 inhibits breast cancer cell viability, colony formation and migration, confirming the oncogenic properties of ALKBH5 in this malignancy [72]. In breast cancer, there is a small subset of cells, called breast cancer stem cells (BCSCs), which could both self-renew and differentiate into cancer cells [73]. BCSCs are resistant to chemotherapy and survive the treatment, leading to tumor recurrence and metastasis [73]. Zhang et al. found that NANOG, a core pluripotency factor, is regulated by ALKBH5 in breast cancer cells [74]. Indeed, ALKBH5 overexpression decreases m6A methylation of NANOG mRNA at 3′ untranslated region (3′UTR), increasing NANOG mRNA stability and levels, and induces BCSC enrichment in hypoxic breast cancer cells [74]. Later, these authors found another pluripotency factor, KLF4, is also positively regulated by ALKBH5-mediated m6A methylation in breast cancer, contributing to the overall regulation of the BCSC phenotype by ALKBH5 [75].
Glioma and glioblastoma
Glioma is a central nervous system tumor that seriously endangers the physical and mental health of patients [76]. ALKBH5 is upregulated in glioma and of significance in regulating the metabolism and development of glioma [77]. Specifically, ALKBH5 demethylates the transcript of glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme of the pentose phosphate pathway (PPP), and enhances its mRNA stability, thus promoting oxidative PPP flux and stimulating the aggravation of glioma [77]. In addition, AKT2 mRNA was identified as another critical target of ALKBH5 that contributes to the tumor-promoting effects of ALKBH5 in glioma cells [78].
Glioblastoma (GBM) represents the commonest primary brain cancer with high heterogeneity and poor prognosis, making up 54% of all gliomas [79, 80]. GBM stem-cell like cells (GSCs) constitute an important factor in maintaining tumor growth and inducing tumor recurrence [81]. ALKBH5 is overexpressed in GSCs and correlated with reduced patient survival in GBM [82]. It demethylates the nascent transcript of FOXM1 and promotes the interaction of FOXM1 pre-mRNA with the nuclear RNA binding protein HuR, resulting in increased FOXM1 expression, which is critical for GSC growth and self-renewal [82]. ALKBH5 also plays an important role in radioresistance and invasiveness of GSCs [83]. ALKBH5 enhances radioresistance by modulating genes involved in homologous recombination (HR) and contributes to the aggressiveness of GBM by upregulating YAP1 expression [83]. Temozolomide, an alkylating agent, is a first-line chemotherapeutic orally administered following surgical excision in GBM [83]. However, temozolomide resistance is frequently found in the middle and late stages of chemotherapy [84]. ALKBH5 could demethylate SOX2 mRNA, enhancing SOX2 expression and promoting temozolomide resistance in GBM [85].
Lung cancer
Lung cancer ranks first among cancers in terms of morbidity and mortality globally, with non-small-cell lung cancer (NSCLC) representing the main histological subtype [86]. ALKBH5 undergoes ectopic upregulation in NSCLC, and is tightly associated with reduced patient survival [87]. ALKBH5 promotes proliferation and reduces apoptosis in NSCLC cells by repressing TIMP3 mRNA stability and translation [87]. UBE2C, an oncogene that selectively represses autophagy and promotes cell proliferation in NSCLC, is epitranscriptionally stabilized with reduced m6A amounts within its mRNA because of ALKBH5 upregulation in NSCLC [88]. Meanwhile, m6A demethylation caused by ALKBH5 stabilizes oncogenic drivers, including SOX2, SMAD7 and MYC, via the m6A reader protein YTHDF2 in KRAS mutant NSCLC [89].
In addition to NSCLC, ALKBH5 is elevated in lung adenocarcinoma cells submitted to intermittent hypoxia [90]. ALKBH5 knockout in lung adenocarcinoma cells increases m6A abundance in FOXM1 transcript and decreases its translation, leading to reduced cell proliferation and invasion [90]. ALKBH5-dependent m6A demethylation of the lncRNA RMRP also has an oncogenic effect in lung adenocarcinoma [91]. RMRP deficiency in lung adenocarcinoma cell lines suppresses cell proliferation, migration and invasion, and promotes cell apoptosis, mimicking the effects of ALKBH5 inhibition [91].
Ovarian carcinoma
As a malignant neoplasm affecting women with a high death rate [92], the development ovarian cancer was reported to be tightly associated with m6A modification. A study examined the expression profiles of AlkB family members in ovarian serous carcinoma, and revealed that cases highly expressing ALKBH5 have reduced overall survival (OS) and progression-free survival (PFS) [93]. ALKBH5 expression is increased in epithelial ovarian cancer in comparison with noncancerous ovarian tissue [94]. ALKBH5 inhibits autophagy and promotes malignancy in ovarian cancer cells by stabilizing BCL2 mRNA and promoting BCL2 binding to BECN1 [94]. Besides, ALKBH5-mediated mRNA demethylation increases NANOG expression and enhances aggressiveness of ovarian cancer cells [95].
Acute myeloid leukemia
Acute myeloid leukemia (AML) represents a fatal hematologic cancer featuring uncontrolled expansion of poorly differentiated myeloid cells, with hematopoietic stem and progenitor cells (HSPCs) retaining their self-renewal ability and impaired myeloid differentiation [96]. Kwok and collaborators demonstrated ALKBH5 is commonly deleted in AML, particularly in individuals with TP53 mutations, based on the cancer genome atlas (TCGA) data [97], implying ALKBH5’s tumor-suppressive function in AML. However, recent studies showed that ALKBH5 is actually upregulated in human AML, with its elevated amounts associated with reduced patient survival [98, 99]. These studies all demonstrated that ALKBH5 is selectively required for self-renewal of leukemia stem cells (LSCs) but not for normal hematopoiesis [98, 99]. Mechanistically, ALKBH5 enhances malignancy in AML by post-transcriptionally regulating TACC3 and AXL [98, 99].
Gastric cancer and colon cancer
Gastric cancer (GC) represents a very common malignancy of the digestive tract, ranking fifth and fourth in terms of incidence and mortality among all cancers, respectively [100]. ALKBH5 is overexpressed in GC and downregulates m6A levels of the lncRNA NEAT1, leading to the upregulation of NEAT1 expression [101]. Increased NEAT1 then positively affects the expression of EZH2, a subunit of the polycomb repressive complex, by acting as a scaffold and thus promoting GC invasion and metastasis [101]. Colorectal cancer is the third incident and second deadliest malignancy worldwide, representing about one in 10 cancer cases and deaths [100]. ALKBH5 knockdown suppresses the proliferative and migratory abilities of colorectal cancer cells partially through ALKBH5-related NEAT1 downregulation [102], which is consistent with the finding in GC cells [101].
Other cancers
Besides the above cancers, ALKBH5 is also correlated with poor prognosis and/or plays an oncogenic role in other malignancies, including endometrial cancer [103, 104], renal cell carcinoma [105], hepatitis B virus (HBV) related hepatocellular carcinoma (HCC) [106], osteosarcoma [107], uveal melanoma [108], pituitary adenomas [109], esophageal squamous cell carcinoma [110], cervical cancer [111] and oral squamous cell carcinoma [112]. During the investigation of underlying mechanisms, a set of functional targets of ALKBH5 were identified in these cancers (Table 2; Fig. 3).
ALKBH5 as a tumor suppressor
While ALKBH5 has overt oncogenic roles in most cancers, it is a tumor suppressor in others (Fig. 2; Table 2). Reduced ALKBH5 amounts has been described in the following malignancies.
Bladder cancer
Bladder cancer (BC) is among the most prevalent cancers globally, with approximately 573,000 new cases reported in 2020 [100]. Chen et al. demonstrated that ALKBH5 expression in BC shows positive correlations with grade and M1 stage, indicating ALKBH5 might be an oncogene in BC [113]. However, other studies demonstrated that ALKBH5 is actually a tumor suppressor in BC according to cell and animal assays. Yu et al. found that reduced ALKBH5 amounts in BC are associated with decreased patient survival [114]. Functional assays showed ALKBH5 silencing increases the proliferative, migratory and invasive abilities of BC cells and reduces their cisplatin chemosensitivity via a CK2α-related glycolytic pathway [114]. Jin and colleagues examined the role of m6A modification in BC, and revealed ALKBH5, but not METTL3, inhibits cell adhesion of BC by repressing ITGA6 expression [115]. Specifically, ALKBH5 decreases m6A methylations in the 3′UTR of the ITGA6 transcript, suppressing ITGA6 protein synthesis mediated by YTHDF1 and YTHDF3 [115].
Pancreatic cancer
Pancreatic cancer (PC) represents one of the rare cancers whose mortality and incidence rates are almost identical, ranking seventh among deadliest cancers globally [116]. Studies revealed enhanced survival in PC cases highly expressing ALKBH5 [117, 118]. ALKBH5 exerts tumor-suppressive effects via the demethylation of the lncRNA KCNK15-AS1, controlling KCNK15-AS1-dependent cell migration and invasion in PC [119]. In addition, ALKBH5 post-transcriptionally enhances PER1 expression in an m6A-YTHDF2-dependent fashion, activating ATM-CHK2-P53/CDC25C signaling and suppressing PC cell proliferation [120]. Moreover, ALKBH5 acts as a tumor suppressor in pancreatic ductal adenocarcinoma (PDAC) and sensitizes PDAC cells to gemcitabine by reducing WIF1 mRNA methylation and modulating the Wnt pathway, which leads to the downregulation of C-MYC, Cyclin D1, MMP-2 and MMP-9 [121].
NSCLC and osteosarcoma
Although most reports showed that ALKBH5 is an oncogene in NSCLC [87,88,89], as stated above, another work reported its tumor suppressive role in NSCLC. Jin et al. showed ALKBH5 inhibits tumor cell proliferation and metastasis by decreasing YTHDFs-dependent YAP1 expression in NSCLC [122]. Meanwhile, ALKBH5 decreases YAP1 activity by HuR-dependently controlling miR-107/LATS2 expression [122]. In osteosarcoma, Yuan and co-workers discovered downregulated levels of ALKBH5 in osteosarcoma cell/tissue specimens in comparison with noncancerous osteoblast cell/tissue samples [123], which was contrary to the observation of a previous study [107]. According to Yuan’s study, ALKBH5 overexpression markedly reduces the proliferative, migratory and invasive features of osteosarcoma cells, while triggering apoptosis [123]. Similar to the finding in NSCLC [122], ALKBH5 suppresses osteosarcoma progression via m6A-related direct/indirect YAP1 regulation [123]. Specifically, YAP1 expression is directly suppressed through mRNA m6A methylation, alongside indirect suppression by ALKBH5-regulated miR-181b-1 [123].
Other cancers
ALKBH5 downregulation was demonstrated in clinical colon cancer tissue samples, which was tightly associated with distant metastasis and disease stage [124]. Further functional assays confirmed ALKBH5 overexpression inhibits colon cancer cell invasion in vitro and metastasis in experimental animals [124]. However, the underlying mechanism remains to be elucidated. In addition, ALKBH5 also functions as a tumor suppressor in clear cell renal carcinoma (ccRCC) and HCC. In ccRCC, reduced ALKBH5 gene expression is associated with shorter OS, indicating that ALKBH5 could be used as a prognostic biomarker [125]. In HCC, ALKBH5 suppresses the proliferative and invasive properties of cancer cells by m6A-mediated inhibition of LYPD1 [126].
ALKBH5 and cancer immunity
Recent works have demonstrated that m6A modification affects immune response and cancer immunity [127, 128]. As an m6A modulator, ALKBH5 have biological significance for both innate and adaptive immune responses. First, ALKBH5 impacts virus infection and antiviral innate immune responses by demethylating the transcripts of genes encoding essential molecules of the innate immune system, including TRAF3, TRAF6, MAVSI, FNB1 and OGDH [58, 59]. Second, ALKBH5-mediated NR4A1 mRNA demethylation is required for group 3 innate lymphoid cell (ILC3) homeostasis and gut immunity [60]. Third, ALKBH5-dependent upregulation of HMGB1 mediates STING-IRF3 innate immune response in radiation-induced liver diseases [61]. Fourth, ALKBH5 contributes to the regulation of CD4 + T cell homeostasis and function during the induced neuroinflammation by increasing interferon-γ (IFN-γ) and C-X-C motif chemokine ligand 2 (CXCL2) mRNA stability in an m6A-dependent manner, leading to enhanced responses of CD4 + T cells and increased recruitment of neutrophils into the central nervous system [62].
Besides, studies have shown that ALKBH5 is involved in the induction of cancer immune evasion. Over the course of cancer development, tumor cells evolve and exhibit multiple mechanisms to inhibit antitumor immune responses and evade tumor immunosurveillance [129]. The expression of inhibitory checkpoint molecules on tumor cells is a major mechanism underlying tumor immune evasion [129]. For instance, programmed cell death 1 ligand 1 (PD-L1) on cancer cells suppress antitumor effector T cells and enables immune evasion through interaction with programmed cell death receptor 1 (PD-1) on T cells [130]. Intrahepatic cholangiocarcinoma (ICC) is a highly aggressive and lethal hepatobiliary malignancy [131]. In ICC, tumor-intrinsic ALKBH5 removes m6A modification in PDL1 mRNA’s 3′UTR and reduces its degradation in a YTHDF2-dependent manner, thus inhibiting cytotoxicity of T cells and mediating immune escape of ICC cells [132].
Apart from tumor-intrinsic ALKBH5–PD-L1 regulating axis, accumulating evidence confirmed the critical role of ALKBH5 in remodeling immune microenvironment in cancer. The immunosuppressive tumor microenvironment (TME) represents another major cause of immune evasion and low immune therapy response in cancer [129]. Regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) are the dominant subsets of immune cells with immunosuppressive activity in the TME [129]. During Anti–PD-1 treatment for melanoma and colorectal carcinoma, tumor cell expression of ALKBH5 regulates extracellular lactate content and then affects Tregs and MDSCs accumulation in the TME [133]. Specifically, MCT4/SLC16A3, an ALKBH5 target gene, is responsible for lactate secretion and recruitment of immunosuppressive Tregs and MDSCs [133]. Moreover, tumor-associated macrophages (TAMs), another critical component of the TME, can be recruited and educated by tumor-expressed ALKBH5 under hypoxic conditions [134]. Macrophages are broadly categorized as M1 or M2 types, and TAMs have been shown to exhibit immunosuppressive and tumor-promoting M2-like phenotypes [129, 135]. In glioblastoma multiforme, TAMs are one of the most abundant TME cell types, contributing up to 30% to 50% of brain tumor mass [134, 136]. Through m6A demethylation and stabilization of lncRNA NEAT1, hypoxia-induced ALKBH5 facilitates paraspeckle assembly and sequesters transcription repressor SFPQ from the CXCL8/IL8 promoter, whose expression and secretion recruit TAMs [134]. In this scenario, tumor-intrinsic ALKBH5 drives TAM infiltration and induces M2 polarization, leading to immunosuppression and tumor progression in glioblastoma multiforme [134].
Regulation of ALKBH5 expression and function in cancer
Increasing research has focused on exploring the mechanisms responsible for the dysregulation of ALKBH5 in cancer. Recent findings suggested that hypoxia, as well as some epigenetic modulators, transcription factors and non-coding RNAs are main contributors to ALKBH5 dysregulation in cancer (Table 2; Fig. 4).

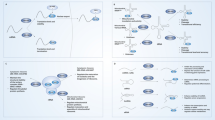
Regulation on the expression and/or function of ALKBH5 in cancers. a Epigenetic modulators of ALKBH5. Histone modifications involved in ALKBH5 transcriptional activation and their modulators (left); LKB1 loss upregulates ALKBH5 by inducing 5mC DNA hypermethylation of the CTCF motif on ALKBH5’s promoter, preventing CTCF binding and enhancing active histone modifications (middle); Histone modifications involved in ALKBH5 transcriptional inactivation and their modulators (right). HBx HBV X protein, MLL Myeloid/lymphoid or mixed-lineage leukemia protein, WDR5 WD-40 Repeat Protein 5, EP300 E1A-associated protein p300, LKB1 Serine/threonine-protein kinase STK11, CTCF CCCTC-binding factor, KDM4C Lysine-specific demethylase 4C, JMJD2B/1C Jumonji domain-containing protein 2B/1C. b Transcription factors of ALKBH5. Transcription activators and repressors bind to promoter or enhancer of ALKBH5 DNA to control ALKBH5 transcription; Hypoxia-inducible factors (HIFs) activate ALKBH5 transcription in response to local hypoxia in cancer. MYB Proto-oncogene c-Myb, RUNX2 Runt-related transcription factor 2, PBX3 Pre-B-cell leukemia transcription factor 3, p53 Cellular tumor antigen p53, HIF-1/2α Hypoxia-inducible factor 1/2-alpha. c Non-coding RNA partners of ALKBH5. Long non-coding RNAs (lncRNAs) function as scaffolds to enhance ALKBH5 binding to their antisense mRNAs; microRNA (miRNA) miR-193a-3 interacts with 3′UTR of ALKBH5 mRNA and promotes its degradation, while ALKBH5 demethylates and suppresses miR-193a-3p maturation in turn; Circular RNA (circRNA) cIARS interacts with ALKBH5 and represses its regulatory effect on BCL2 mRNA. d Other regulators of ALKBH5. ALKBH5 is positively regulated by TLR4 in ovarian cancer cells after co-culture with M2 macrophages; DDX3 interacts with ALKBH5 and enhances demethylation activity of ALKBH5. TLR4 Toll-like receptor 4, DDX3 DEAD box protein 3, X-chromosomal
Hypoxia
Hypoxia, a feature of most tumors, results from fast cell division and abnormal angiogenesis [137]. Hypoxia-inducible factors (HIFs) control transcriptional responses to local hypoxia in cancer and induce tumor progression by affecting cell metabolism, promoting angiogenesis, controlling stem cell state and other cellular processes [137]. HIFs consist of α (HIF-α) and β (HIF-β) subunits and interact with canonical DNA sequences termed hypoxia-response elements (HREs) in the promoters or enhancers of target genes, upregulating 150 genes or more [137]. An early study found that ALKBH5 is a direct target of HIF-1α, indicating that ALKBH5 may be involved in the regulation of cellular responses to hypoxia [138]. Later, it was demonstrated in human breast cancer cells that ALKBH5 is significantly upregulated under hypoxic conditions, while knockdown of HIF-1α and/or HIF-2α abrogates this effect [74, 75]. Similarly, hypoxia and HIFs elevate ALKBH5 expression in lung adenocarcinoma cells [90], renal cell carcinoma cells [105], endometrial cancer stem cells [104] and pituitary adenoma cells [109], leading to invasive tumor phenotype, enhanced tumor initiation capacity, sustained stem-like state and poor clinical outcomes.
Epigenetic modulators
Epigenetic modifications, including DNA methylation, histone modifications and nucleosome positioning, affect gene expression with no alteration of the underlying DNA sequence [139]. Dysregulation of these processes is frequently observed in human cancers, some of which are responsible for ALKBH5 dysregulation in cancers.
Histone modifications and associated enzymes are involved in chromatin compaction, nucleosome dynamics and transcription activation or suppression [140]. In leukemia-initiating cells, the promoter regions of ALKBH5 have markedly elevated amounts of active histone markers, including H3K9ac, H3K4me3, H3K4me2 and H3K79me2, and reduced levels of the repressive histone marker H3K27me3 and H3K9me3, leading to a higher degree of chromatin openness [99]. Consistently, a set of H3K4 histone methyltransferases (MLL1 and MLL2) and H3K9 histone demethylases (JMJD2B and JMJD1C) were identified as ALKBH5 promoter-binding proteins in AML cells [99]. Further investigation confirmed knockdown of MLL1 and MLL3, but not MLL2 and MLL4, markedly reduces ALKBH5 amounts in leukemia cells [99]. Moreover, histone demethylase KDM4C controls ALKBH5 expression in leukemia by reducing H3K9me3 levels and enhancing chromatin accessibility of the ALKBH5 locus, which promotes the binding of the transcriptional factor MYB and the C-terminal domain (CTD) of the RNA polymerase II to the ALKBH5 promoter [99]. These results showed ALKBH5 overexpression is controlled by chromatin state in the pathogenesis of AML.
Epigenetic dynamics of ALKBH5 has also been researched in HBV-HCC, uveal melanoma (UM) and NSCLC. The HBV X protein (HBx) was found to induce an elevated degree of epigenetic H3K4me3 modification of chromatin by upregulating WD-40 Repeat Protein 5 (WDR5), contributing to HBV-induced hepatocellular carcinogenesis [141]. It was demonstrated that highly expressed ALKBH5 in HBV-HCC occurs via HBx- and WDR5-dependent H3K4me3 modification of the ALKBH5 promoter following HBV infection [106]. Elevated ALKBH5 amounts then enhance HBx mRNA expression, forming a positive-feedback loop that contributes to HBV-associated liver carcinogenesis [106]. In UM, EP300-induced H3K27 acetylation activation upregulates ALKBH5 expression [108]. ALKBH5 level is starkly decreased in UM cells after treatment with C646, a histone acetyltransferase suppressor specific to EP300 [108]. Many NSCLC cases contain concurrent KRAS mutation, alongside loss of LKB1, which negatively regulates the total 5mC DNA methylation [142,143,144]. LKB1 deficiency upregulates ALKBH5 by inducing DNA hypermethylation of the transcriptional repressor CTCF binding region on ALKBH5 gene’s promoter, preventing CTCF binding while enhancing active histone modifications such as H3K4me3, H3K9ac and H3K27ac [89].
Transcription factors
Transcription factors bind DNA in a sequence-specific manner to control chromatin and transcription, forming a complicated system that impact gene expression [145]. During the investigation of ALKBH5 promoter-binding proteins in AML cells, a set of transcription factors was identified, including RUNX2, MYB and PBX3 [99]. Further investigation confirmed MYB silencing markedly decreased ALKBH5 mRNA and protein amounts in leukemia cells [99]. The well-known transcription activator P53 is mutated or suppressed in about half of cancers [120]. A TCGA dataset assessment demonstrated elevated and reduced ALKBH5 amounts in the P53 wild-type and mutant groups of PC cells, respectively [120]. P53 interacts with the ALKBH5 promoter, transcriptionally activating ALKBH5 and indirectly reducing m6A amounts in PC cells [120].
Non-coding RNAs
While ALKBH5 acts as an important factor controlling the expression and functions of a number of ncRNAs, some ncRNAs also influence its expression and function. The crosstalk between ALKBH5 and ncRNAs appears to form a feedback loop with extensive impacts in cancers.
LncRNAs, i.e., noncoding transcripts of more than 200 nucleotides, might play the roles of enhancers, scaffolds or decoys by physically binding other RNAs or proteins [146]. In GBM cells, the lncRNA FOXM1-AS (antisense to FOXM1) enhances ALKBH5 binding to FOXM1 nascent mRNA [82]. FOXM1-AS suppression mimics ALKBH5 depletion in modulating FOXM1 methylation and biosynthesis [82]. Similarly, the lncRNA GAS5-AS (antisense to GAS5) enhances GAS5 stability by binding to ALKBH5 and controlling m6A modifications of GAS5 in cervical cancer [147]. In GBM cells with temozolomide-resistance, the lncRNA SOX2OT functions as a scaffold that recruits ALKBH5, positively regulating its function and expression [85].
MiRNAs are 18–25 nucleotide-long ncRNAs that inhibit gene expression by directly interacting with complementary target mRNAs, which are then degraded or translationally inhibited [148]. ALKBH5 mRNA was recognized as a miR-193a-3 target in both glioma and esophageal squamous cell carcinoma (ESCC), with its 3′UTR being a miR-193a-3p binding site [78, 149]. miR-193a-3p transfection results in substantial ALKBH5 downregulation, while miR-193a-3p inhibition upregulates ALKBH5 [78, 149]. Meanwhile, in ESCC, ALKBH5 suppresses the maturation of primary miR-193a-3p by decreasing its m6A modification in turn, suggesting a feedback regulation between miR-193a-3p and ALKBH5 [149].
CircRNAs represent single-stranded, covalently closed RNAs [150]. They interact with and sequester target proteins to adequate subcellular locations, and modulate some protein–protein and protein-RNA interactions [150]. CircRNA cIARS (hsa_circ_0008367) controls sorafenib-induced autophagy and ferroptosis in HCC cells through interaction with ALKBH5 [151], which inhibits autophagy by demethylating BCL2 mRNA and promoting BCL2-BECN1 binding [94]. cIARS interacts with ALKBH5 directly and represses its regulatory effect on BCL2-BECN1 autophagy regulatory complex, leading to increased autophagic flux and ferritinophagy [151].
Other regulators of ALKBH5
DDX3, a DEAD-box RNA helicase, interacts with ALKBH5 in an RNA-independent manner and modulates the demethylation activities of ALKBH5 [152]. The ATP and DSBH domains of DDX3 and ALKBH5, respectively, were required for this interaction [152]. Later, another study confirmed that ALKBH5 is directly regulated by DDX3 in oral squamous cell carcinoma cells [112]. However, the study showed that ATP domain of DDX3 was not essential for DDX3 mediated up-regulation of ALKBH5 [112]. In human ovarian cancer, ALKBH5 and TLR4 amounts are elevated in cancer cells after co-culture with alternatively activated (M2) macrophages [95]. Specifically, TLR4 positively regulates ALKBH5 through NF-κB signaling [95].
Dual roles of ALKBH5 in cancer
Data from human malignancies, as noted above, suggest a contradictory role for ALKBH5: that of an oncogene in some cancers and that of a tumor suppressor in other cancer types. It should not be a surprising event, considering the fact that ALKBH5 post-transcriptionally regulates a great number of target genes that have distinct functions, either positively or negatively, controlling cell cycle, survival, apoptosis, DNA repair, metabolism, autophagy, and other cellular processes determining the fate of tumor cells. It can be expected that some of those effects might stimulate cell growth, while others might induce negative regulation of cell growth. Thus, the role of ALKBH5 in cancer is mainly dependent on its functional targets in a specific cancer type or cellular context. Theoretically, ALKBH5 may regulate a similar set of target genes in different cells. However, cancer is a complex and heterogeneous disease. The independent studies of different cancer types or studies using different models suggested that the regulatory network of ALKBH5 in cancer varies from case to case. The presence or absence of various protein/RNA partners may account for the variable behavior of ALKBH5 in different cellular contexts.
m6A reader proteins are key factors responsible for determining and altering the effect of ALKBH5 on a certain target. For example, YAP1, a transcription co-activator whose activation is important for cancer cell proliferation, survival, migration and invasion [153, 154], is one of the critical targets that mediate ALKBH5’s function in multiple cancers. It was demonstrated that YTHDF1 and YTHDF2 competitively bind m6A-modified YAP1 mRNA to positively and negatively regulate YAP1 expression, respectively [122]. Specifically, YTHDF2 facilitates YAP1 mRNA degradation through the AGO2 system, whereas YTHDF1 promotes YAP1 mRNA translation by interacting with eIF3a [122]. In NSCLC, the expression of YTHDF1 is higher in tumor tissues than in normal tissues, while the expression of YTHDF2 is lower in tumor tissues than in normal tissues, indicating that YTHDF1 is more likely to bind to methylated YAP1 mRNA to promote its translation. As a result, ALKBH5-mediated YAP1 demethylation negatively regulates YAP1 translation and inhibits the growth of tumor cells in NSCLC [122]. In contrast, ALKBH5 positively regulates YAP1 mRNA expression in GBM cells, and thus exerts its oncogenic role in GBM [83]. Although the study did not investigate the reader protein of methylated YAP1 mRNA in GBM, one possibility can be advanced that YTHDF2 is responsible for affecting YAP1 mRNA stability in GBM cells because numerous studies have shown that YTHDF2 is upregulated in GBM [155,156,157]. Therefore, potential tissue- or cell-specific differences in m6A reader proteins may alter ALKBH5’s regulation of downstream targets and explain the conflicting functions of ALKBH5 in cancers.
The interaction of ALKBH5 with auxiliary ncRNAs is another important aspect of ALKBH5’s regulation on its targets. For instance, lncRNA FOXM1-AS functions as a scaffold to increase the ability of ALKBH5 to bind to its antisense mRNA FOXM1, providing both tumorigenic and proliferative advantages to cancer cells [82]. In addition, the finding that the interaction of circRNA cIARS with ALKBH5 alters the autophagy inhibitory behavior of ALKBH5 may allow us to understand the complexity of this epigenetic regulation [94, 151]. These findings show that cell-specific differences in ncRNAs may alter ALKBH5’s binding affinity for different targets and change the final phenotypic outcome.
Following these possible reasons and examples, it remains difficult to tell clearly what natural cellular contexts allow ALKBH5 to act as an oncogene or tumor-suppressor. However, it is increasingly clear that the effect of ALKBH5 on the cancer cell is determined by the cellular context in which it is present and the functional targets it regulates. Particularly, the presence or absence of other regulatory partners are likely to be critical confounding factors that change the role of ALKBH5 in cancer.
Therapeutic implications of targeting ALKBH5 in cancer
The findings that ALKBH5 is dysregulated and plays critical roles in multiple cancers support the hypothesis that targeting ALKBH5 can act as an approach to cure different types of cancer. Since ALKBH5 has opposite, context-dependent functions in cancer, approaches to both induce and suppress ALKBH5 could represent viable treatment options for malignancies. For example, ALKBH5 is overexpressed and plays an oncogenic role in many tumors, such as breast cancer [72, 74, 75], Glioma [77, 78, 82] and AML [98, 99]. Thus, pharmacological inhibition of ALKBH5 may exert antitumor effects in these cancer types. In contrast, restoration or augmentation of ALKBH5 expression is a potential therapeutic strategy for the treatment of those cancers in which ALKBH5 plays a tumor-suppressive role, such as bladder cancer [114, 115] and pancreatic cancer [119,120,121].
Additionally, dysregulation of ALKBH5 was found to mediate drug- or radio-resistance in GBM [85, 158], oral squamous cell carcinoma [112], bladder cancer [114], pancreatic cancer [121] and BRCA-mutated epithelial ovarian cancers [159] (Table 3). Such data suggest that ALKBH5 might serve as a predictive marker for personalized treatment of malignancies, providing insights into overcoming therapeutic resistance in cancer by combining drugs targeting ALKBH5 with chemotherapy, targeted therapy, and/or radiation therapy. Notably, the existence of cancer stem cells with self-renewal ability is a major reason for drug or radio resistance and tumor recurrence [160]. Considering ALKBH5’s functions in cancer stem cell maintenance in breast cancer [74, 75], GBM [82], AML [98, 99] and endometrial cancer [104], ALKBH5 could be a potential target for suppressing cancer stem cells and promoting complete remission in cancer treatment.
What’s more, immunotherapy, e.g., using antibodies that mediate immune checkpoint blockade, has emerged in recent years as an innovative treatment option for tumors [161]. However, the response rates to immunotherapy for the majority of tumors remain low, due to primary, adaptive, or acquired resistance [161]. ALKBH5 overexpression induces immunosuppressive TME and promotes immune evasion in multiple cancers, including ICC [132], melanoma [133], colorectal carcinoma [133] and glioblastoma multiforme [134], suggesting that ALKBH5 could affect tumor response to immunotherapy. Indeed, ALKBH5 mutation or expression are associated with response to anti–PD-1 immunotherapy in ICC and melanoma (Table 3) [132, 133]. ALKBH5 deletion or inhibition enhances the efficacy of anti–PD-1 treatment in colorectal cancer and melanoma [133], suggesting ALKBH5 could represent a promising therapeutic target to enhance immunotherapy outcome in cancer patients.
Potential strategies for targeting ALKBH5
To the best of our knowledge, the reported functions of ALKBH5 in cancer rely on m6A demethylation activity in all cases. Potential strategies including small-molecule modulators, proteolysis targeting chimera (PROTAC), programmable m6A-editing systems, compounds targeting the regulatory machinery of ALKBH5, as well as gene therapy, could be applied to manipulate ALKBH5-mediated m6A demethylation in cancer (Table 4; Fig. 5).

Potential approaches for ALKBH5 targeting in cancers. a Small-molecule inhibitors of ALKBH5. b Proteolysis targeting chimera (PROTAC) for ALKBH5 protein degradation. c CRISPR-based site-specific m6A editing systems. M3 methyltransferase-like protein 3 (METTL3), M14 methyltransferase-like protein 14 (METTL14). d Compounds targeting the upstream regulators of ALKBH5. e ALKBH5 gene therapy. ALKBH5 is delivered within a viral vector followed by direct injection to the tumor
Small-molecule modulators
To identify specific ALKBH5 suppressors, a high-throughput in silico screening of 144,000 preselected molecules was performed, with further analysis by the enzyme inhibition assay [162]. The authors ultimately identified two ALKBH5 inhibitors, including 2-[(1-hydroxy-2-oxo-2-phenylethyl)sulfanyl]acetic acid (Fig. 5a, Inhibitor A; IC50 = 0.84 μM) and 4-[(furan-2-yl)methyl]amino-1,2-diazinane-3,6-dione (Fig. 5a, Inhibitor B; IC50 = 1.79 μM), which were tested for activity in leukemia and GBM cell lines [162]. In leukemia HL-60, CCRF-CEM and K562 cells, theses inhibitors blunted cell proliferation in the low micromolar range (IC50 = 1.38–16.5 μM) [162]. However, another leukemia cell line, Jurkat, and glioblastoma A-172 cells had low to no susceptibility, suggesting ALKBH5 suppression by small-molecule inhibitors as a feasible cancer-cell-type-selective anti-proliferative approach [162]. A novel sodium channel blocker, named MV1035 (Fig. 5a, Inhibitor C), potently inhibits ALKBH5 activity by an alternative off-target interaction with ALKBH5, with the binding site of MV1035 in ALKBH5 partially overlapping with that of 2OG [163]. It was demonstrated that the inhibitory effects of MV1035 on glioma cell migration and invasion rely on ALKBH5 inhibition but not sodium channel blocking [163]. Besides, a specific inhibitor of ALKBH5, named ALK-04, could potentiate the efficacy of anti–PD-1 immunotherapy in melanoma treatment, providing a rational for future combination drug therapy [133].
In addition to naturally-derived small-molecule inhibitors of ALKBH5, developing synthetic small-molecule inhibitors represent another approach to inhibit ALKBH5. ALKBH5 requires an interaction with 2OG and ferrous iron to function as an m6A demethylase [68]. Therefore, it would be reasonable to design suppressors by taking into consideration the reported binding pocket of this protein. Particularly, a 2OG-tethering approach of concurrently occupying both the 2OG- and substrate-binding sites is of interest. Linking 2OG derivatives with substrate analogs was successfully utilized to develop selective histone demethylase suppressors [164, 165] and AlkB enzyme inhibitors [166], which can be used to design specific ALKBH5 inhibitors in the future.
Given the tumor suppressive role of ALKBH5 in a small number of cancers, activators of ALKBH5 are needed in such cases. However, pharmacologic activators specifically targeting ALKBH5 have not been reported to date, raising the need for developing ALKBH5 activators. The ability to bind metal cofactor (Fe2+) and the secondary substrate (2OG) precedes binding of the m6A-containing oligonucleotide to ALKBH5 and is crucial for demethylation activity of ALKBH5 [167]. Therefore, the binding sites of Fe2+ and 2OG could be used as target areas for identifying potential compounds as activators of ALKBH5 by high-throughput screens. Flavin mononucleotide, a metabolite produced by riboflavin kinase, was found to mediate substantial photochemical demethylation of m6A residues of RNAs in live cells, indicating that regulation of m6A modification by external small organic molecules is possible [168]. It also provided insights into the development of powerful small molecules as RNA demethylases and specific functional probes targeting m6A modification for biomolecular and treatment purposes [168].
Proteolysis targeting chimera
Besides small molecule-based occupancy-driven pharmacology as an approach to inhibit ALKBH5 functions, PROTAC represents a new technological method for degrading proteins of interest (Fig. 4b). A PROTAC is a heterobifunctional molecule that consists of a protein comprising a ligand for interacting with a target protein and another interacting with an E3 ubiquitin ligase, with a linker utilized for connection (Fig. 5b, right panel) [169, 170]. Indeed, a PROTAC recruits the E3 ligase to the protein of interest and induces its ubiquitination and degradation [169, 170]. The main advantages of PROTACs include their high selectivity, the potential to target non-druggable proteins, and the capacity of overcoming resistance to small-molecule suppressors via targeting of mutant proteins [170]. Using PROTACs for degrading proteins critical for tumorigenesis represents a potential therapeutic approach in cancer. PROTACs have be successfully used in solid (targeting AR, ER, FAK and P38) and hematological (targeting BRD4, BTK, BCR-ABL and CDK-6) tumors [170]. Importantly, the first PROTACs against AR and ER have been applied clinically [170]. Whether such approach would degrade ALKBH5 in cancer deserves further attention.
CRISPR-based site-specific m6A editing
A potential concern is that targeting ALKBH5 utilizing the abovementioned techniques or reagents would affect the global demethylation of mRNAs, including both oncogenes and tumor suppressor genes, or genes with other functions, which may lead to potential deleterious effects [171]. According to previous studies (Table 2; Fig. 3), the tumor-promoting or suppressing role of ALKBH5 is largely mediated by a few critical targets, suggesting that modulating the m6A on specific RNAs could alleviate tumor progression with reduced side effects. Therefore, developing a site-specific m6A modulation system may provide a more potent tool for correcting dysregulated m6A modifications in cancers.
The development of Cas13, a novel RNA-targeting CRISPR-associated nuclease, enables the targeting of dynamics of endogenous RNAs [172, 173]. Li et al. have recently developed a CRISPR–Cas13b-based tool, named dm6ACRISPR, to specifically demethylate target mRNAs [171]. The dm6ACRISPR fusion protein was constructed by linking catalytically inactive Cas13 enzyme (dCas13b) to ALKBH5 (Fig. 5c, left panel) [171]. dm6ACRISPR effectively demethylated target transcripts without significant off-target effects, and was successfully applied to target oncogenic mRNAs, such as EGFR and MYC mRNAs, to decrease HeLa cell proliferation [171]. In addition, another team designed programmable m6A modification utilizing the CRISPR-based METTL3 methyltransferase domain and METTL3:METTL14 methyltransferase complex, named dCas13-M3nls and dCas13-M3M14nes respectively, which can install m6A on targeted mRNAs (Fig. 5c, right panel) [174]. Together, these technologies could work together to add or delete functional essential m6A sites on specific RNAs. Considering that CRISPR-based gene therapy has achieved great success, with many ongoing phase I/II clinical studies [175], CRISPR-based m6A targeting may provide a powerful strategy for m6A-based precision medicine.
Targeting ALKBH5 regulators
The finding of upstream regulators of ALKBH5 in cancers (Table 2; Fig. 4) provides an alternative approach to inhibit ALKBH5 by targeting its regulatory machinery. These ALKBH5 regulators, including multiple epigenetic modulators, transcription factors and non-coding RNAs, are also dysregulated in specific cancers and exert similar effects on cellular phenotypes as ALKBH5. Inhibitors of these regulators, like DNA methylation inhibitor 5-azacytidine (5-aza) [89] and histone acetyltransferase EP300 inhibitor C646 [108], as well as miRNA miR-193a-3p mimic [78, 149] are able to indirectly or directly suppress ALKBH5 expression in cancer cells (Fig. 5d). Numerous epigenetic, HIF-targeted and miRNA-directed therapies have been examined in clinical trials [176,177,178], showing potential of targeting these kinds of molecules for cancer treatment, which might exert anti-tumor effects partly through altering ALKBH5-mediated m6A demethylation.
In the setting that ALKBH5 functions as a tumor suppressor, inhibiters targeting negative regulators of ALKBH5 demonstrate great pharmacological potential as cancer remedy. This approach has proven to be beneficial in the case of activating tumor suppressor p53 for cancer treatment. Several compounds have been developed to specifically target MDM2, a negative regulator of p53, leading to p53 stabilization and activation [179]. For ALKBH5, epigenetic modulators, e.g., protein arginine methyltransferase PRMT7, that function to repress ALKBH5 transcription have been investigated with mechanistic insights gained. PRMT7 binds to the BRG1-related hSWI/SNF chromatin remodeling complex and carries out the methylation of histones H2A Arg-3 (H2AR3) and H4 Arg-3 (H4R3), leading to negative regulation of ALKBH5 expression [180]. Thus, inhibitors targeting PRMT7 may effectively reverse the inhibition of ALKBH5 transcription. In addition, SUMO E2 conjugation enzyme UBC9 and E3 ligase PIAS4 mediates ALKBH5 SUMOylation at lysine residues K86 and K321, which markedly inhibits demethylase activity of ALKBH5 by blocking its binding to m6A RNA species [57], indicating the design of small-molecule inhibitors targeting the modulators of ALKBH5 post-translational modifications might be another intriguing strategy to reactivate ALKBH5. Further understanding of regulatory mechanisms on the expression, activation and stabilization of ALKBH5 may provide more ideal targets to up-regulate its level and enhance its tumor-suppressive activities in some cancers.
Gene therapy
Gene therapy is the treatment of a genetic disease by repairing or reconstructing defective genetic material [181]. For cancers caused by a deficiency in ALKBH5, such as bladder cancer [114, 115] and pancreatic cancer [119,120,121], the most direct way to restore the expression and function of ALKBH5 is to introduce wild-type ALKBH5 into cancer cells. This can be achieved by in vivo delivery of ALKBH5 gene into the target tumor cells by vectors based on retroviruses, adenoviruses or adeno-associated viruses (AAVs) [181]. Due to their gene delivery efficacy, lack of pathogenicity, and strong safety profile, AAV vectors may be an excellent platform for delivering therapeutic genes [182]. Recombinant AAV vectors are generated by replacing the endogenous rep and cap genes with an expression cassette consisting of a promoter driving a transgene of interest and a poly(A) tail (Fig. 5e) [183]. The viral vectors can be administered intratumorally and are able to infect cancer cells to achieve ectopic expression of the therapeutic transgene [183]. AAV vector-mediated gene delivery was recently approved for the treatment of inherited blindness and spinal muscular atrophy [183, 184]. Additionally, AAV vectors have been increasingly employed in a variety of preclinical tumor models to deliver a wide array of therapeutic genes and tumor suppressor genes, such as TP53 [182]. The application of gene therapy to restore ALKBH5 expression in cancers as well as its clinical efficacy and biosafety await further validation.
Conclusion and perspectives
As one of the m6A regulators, ALKBH5 has a critical function in keeping the balance between methylation and demethylation of RNAs. It is well established that ALKBH5-mediated post-transcriptional regulation of gene expression is crucial for both normal physiological and pathophysiological events. Studies that implicate ALKBH5 in the pathogeneses of multiple malignancies have emerged at a rapid pace in recent years, providing novel insights into ALKBH5’s functions. ALKBH5 could modulate the epitranscriptome in cancer, causing alterations in cell proliferation, survival, invasion and metastasis, drug sensitivity, cancer stem cell state and cancer immunity. However, there remain some issues to be addressed. First, it is noticeable that in some cancers, including NSCLC [87,88,89, 122], colon cancer [101, 102, 124], renal cell carcinoma [105, 125] and osteosarcoma [107, 122, 123], ALKBH5 plays oncogenic and/or tumor-suppressive roles, which might result from tumor heterogeneity and/or various models. Thus, a comprehensive investigation is required for exploring the exact context in which ALKBH5 promotes or inhibits carcinogenesis, especially for those cancers in which the role of ALKBH5 is highly contentious. Second, it is reasonable that m6A methyltransferases and demethylases exhibit opposite effects in a given pathology. However, exceptions have been reported. For instance, both m6A writers and erasers, including METTL3, METTL14, FTO and ALKBH5, are aberrantly upregulated, with major oncogenic functions in AML [98, 99, 185,186,187]. How enzymes with opposite functions play similar roles in a given cancer deserves further investigation. Third, recent studies showed that many m6A regulators, such as METTL3 [35, 36, 188] and VIRMA [189, 190], function in cancers independently of catalytic activity. However, m6A-independent functions of ALKBH5 and their potential roles in cancer remain unknown, representing another research direction for future study.
Given the functional importance of ALKBH5 in multiple malignancies, targeting dysregulated ALKBH5 is an attractive approach for treating cancer. ALKBH5 has been shown to exhibit strict substrate specificity by only demethylating m6A on ssRNAs [21], and its modulation on m6A modification is reversible, making it an ideal therapeutic target. To date, studies have identified a few inhibitors of ALKBH5. However, issues with potency, selectivity and cytotoxicity need to be addressed. In addition, CRISPR-based m6A targeting tools have been developed to achieve site-specific m6A modulation of target oncogenes or tumor suppressors. Besides, PROTACs, gene therapy, as well as inhibitors against regulators of ALKBH5 could also be considered as efficacious therapeutic candidates. Remarkably, several companies have successfully developed small-molecule suppressors of METTL3 and FTO, with high efficacy and selectivity [191,192,193,194,195], and some of them have yielded encouraging preliminary findings in preclinical investigations. Due to the recent finding of ALKBH5 as an m6A eraser and its roles in cancer, targeting of ALKBH5 for clinical application is much less examined than METTL3 and FTO, and is still in its infancy. The discovered differences in 2OG binding pockets between ALKBH5 and FTO could help develop selective compounds targeting these two RNA demethylase. Indeed, crystallographic and biochemical analyses applying multiple 2OG analogs demonstrated ALKBH5’s active site cavity is starkly smaller compared with FTO’s, with ALKBH5 preferentially binding small-molecule inhibitors [67, 69]. Collectively, continued efforts are required for designing and optimizing approaches for ALKBH5 targeting in cancer therapy.
In conclusion, we summarized the recently described critical functions and therapeutic potential of ALKBH5 in cancer. The therapeutic targeting of ALKBH5 is just in its early stage. With increased knowledge of ALKBH5 structure, mechanisms behind ALKBH5-mediated carcinogenesis and drug response, alongside comprehensive preclinical analysis, ALKBH5-targeting therapeutics may be applied clinically in the future.
Availability of data and materials
Not applicable.
Abbreviations
- m6A:
-
N6-Methyladenosine
- NGS:
-
Next-generation sequencing
- rRNA:
-
Ribosomal RNA
- miRNA:
-
MicroRNA
- lncRNA:
-
Long non-coding RNAs
- circRNA:
-
Circular RNA
- snRNA:
-
Small nuclear RNA
- MTC:
-
Methyltransferase complex
- FTO:
-
Fat mass and obesity-associated protein
- ALKBH5:
-
Alpha-ketoglutarate-dependent dioxygenase alkB homolog 5
- ncRNA:
-
Non-coding RNA
- 2OG:
-
2-Oxoglutarate
- DSBH:
-
Double-stranded β-helix
- m6Am:
-
N6,2-O-dimethyladenosine
- m1A:
-
N1-methyladenosine
- m3T:
-
3-Methylthymine
- ssDNA:
-
Single-stranded DNAs
- m3U:
-
3-Methyluracil
- ssRNA:
-
Single-stranded RNA
- NRL:
-
Nucleotide recognition lid
- BCSC:
-
Breast cancer stem cell
- 3′UTR:
-
3′ Untranslated region
- G6PD:
-
Glucose-6-phosphate dehydrogenase
- PPP:
-
Pentose phosphate pathway
- GBM:
-
Glioblastoma
- GSC:
-
Glioblastoma stem-cell like cell
- HR:
-
Homologous recombination
- NSCLC:
-
Non-small cell lung cancer
- OS:
-
Overall survival
- PFS:
-
Progression-free survival
- AML:
-
Acute myeloid leukemia
- HSPC:
-
Hematopoietic stem and progenitor cell
- TCGA:
-
The cancer genome atlas
- LSC:
-
Leukemia stem cells
- GC:
-
Gastric cancer
- HBV:
-
Hepatitis B virus
- HCC:
-
Hepatocellular carcinoma
- BC:
-
Bladder cancer
- PC:
-
Pancreatic cancer
- PDAC:
-
Pancreatic ductal adenocarcinoma
- ccRCC:
-
Clear cell renal carcinoma
- ILC3:
-
Group 3 innate lymphoid cell
- IFN-γ:
-
Interferon-γ
- CXCL2:
-
C-X-C motif chemokine ligand 2
- PD-L1:
-
Programmed cell death 1 ligand 1
- PD-1:
-
Programmed cell death receptor 1
- ICC:
-
Intrahepatic cholangiocarcinoma
- TME:
-
Tumor microenvironment
- Treg:
-
Regulatory T cell
- MDSC:
-
Myeloid-derived suppressor cell
- TAM:
-
Tumor-associated macrophage
- HIF:
-
Hypoxia-inducible factor
- HRE:
-
Hypoxia-response element
- H3K9ac:
-
Acetylation of lysine 9 on histone 3
- H3K4me3:
-
Tri-methylation of lysine 4 on histone H3
- H3K4me2:
-
Dimethylation of lysine 4 on histone H3
- H3K79me2:
-
Dimethylation of lysine 79 on histone H3
- H3K27me3:
-
Tri-methylation of lysine 27 on histone H3
- H3K9me3:
-
Tri-methylation of lysine 9 on histone H3
- CTD:
-
C-terminal domain
- UM:
-
Uveal melanoma
- HBx:
-
HBV X protein
- WDR5:
-
WD-40 Repeat Protein 5
- H3K27ac:
-
Acetylation of lysine 27 on histone 3
- ESCC:
-
Esophageal squamous cell carcinoma
- PROTAC:
-
Proteolysis targeting chimera
- 5-aza:
-
5-Azacytidine
- H2AR3:
-
Histone H2A Arg-3
- H4R3:
-
Histone H4 Arg-3
- AAV:
-
Adeno-associated virus
References
Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46:D303–7.
Xuan JJ, Sun WJ, Lin PH, Zhou KR, Liu S, Zheng LL, et al. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018;46:D327–34.
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71:3971–5.
Perry R, Kelley D. Existence of methylated messenger RNA in mouse L cells. Cell. 1974;1:37–42.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m(6)A RNA methylomes revealed by m(6)A-seq. Nature. 2012;485:201–6.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–46.
Huang H, Weng H, Chen J. m6A modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell. 2020;37:270–88.
Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12:767–72.
Liu N, Parisien M, Dai Q, Zheng G, He C, Pan T. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA. 2013;19:1848–56.
Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, et al. The U6 snRNA m(6)A Methyltransferase METTL16 Regulates SAM synthetase intron retention. Cell. 2017;169:824-835.e814.
Zhou C, Molinie B, Daneshvar K, Pondick JV, Wang J, Van Wittenberghe N, et al. Genome-wide maps of m6A circRNAs identify widespread and cell-type-specific methylation patterns that are distinct from mRNAs. Cell Rep. 2017;20:2262–76.
Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. 2015;162:1299–308.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–5.
Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–89.
Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014;8:284–96.
Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537:369–73.
Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, et al. VIRMA mediates preferential m(6)A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018;4:10.
Wen J, Lv R, Ma H, Shen H, He C, Wang J, et al. Zc3h13 regulates nuclear RNA m(6)A methylation and mouse embryonic stem cell self-renewal. Mol Cell. 2018;69:1028-1038.e1026.
Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev. 2018;32:415–29.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N6-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature. 2015;518:560–4.
Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45:6051–63.
Zhou KI, Shi H, Lyu R, Wylder AC, Matuszek Z, Pan JN, et al. Regulation of co-transcriptional pre-mRNA splicing by m(6)A through the low-complexity protein hnRNPG. Mol Cell. 2019;76:70-81.e79.
Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61:507–19.
Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife. 2017;6:31311.
Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, et al. YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4–NOT deadenylase complex. Nat Commun. 2016;7:12626.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–20.
Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16:191–8.
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017;27:315–28.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–95.
Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an N6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27:1115–27.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–99.
Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, et al. Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res. 2017;27:444–7.
Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m6A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62:335–45.
Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, et al. mRNA circularization by METTL3–eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561:556–60.
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5′ UTR m6A promotes cap-independent translation. Cell. 2015;163:999–1010.
Yang D, Qiao J, Wang G, Lan Y, Li G, Guo X, et al. N6-methyladenosine modification of lincRNA 1281 is critically required for mESC differentiation potential. Nucleic Acids Res. 2018;46:3906–20.
Alarcon CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519:482–5.
Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L, et al. N6-methyladenosine modification controls circular RNA immunity. Mol Cell. 2019;76:96-109.e109.
Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, et al. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 2017;27:626–41.
Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature. 2002;419:174–8.
Tsujikawa K, Koike K, Kitae K, Shinkawa A, Arima H, Suzuki T, et al. Expression and sub-cellular localization of human ABH family molecules. J Cell Mol Med. 2007;11:1105–16.
Yu B, Edstrom WC, Benach J, Hamuro Y, Weber PC, Gibney BR, et al. Crystal structures of catalytic complexes of the oxidative DNA/RNA repair enzyme AlkB. Nature. 2006;439:879–84.
Falnes PO, Johansen RF, Seeberg E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature. 2002;419:178–82.
Gerken T, Girard CA, Tung YC, Webby CJ, Saudek V, Hewitson KS, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318:1469–72.
Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC, et al. Differential m6A, m6Am, and m1A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol Cell. 2018;71:973-985.e975.
Jia G, Yang CG, Yang S, Jian X, Yi C, Zhou Z, et al. Oxidative demethylation of 3-methylthymine and 3-methyluracil in single-stranded DNA and RNA by mouse and human FTO. FEBS Lett. 2008;582:3313–9.
Baltz AG, Munschauer M, Schwanhausser B, Vasile A, Murakawa Y, Schueler M, et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. 2012;46:674–90.
Tang C, Klukovich R, Peng H, Wang Z, Yu T, Zhang Y, et al. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3′-UTR mRNAs in male germ cells. Proc Natl Acad Sci USA. 2018;115:E325–33.
Feng L, Fan Y, Zhou J, Li S, Zhang X. The RNA demethylase ALKBH5 promotes osteoblast differentiation by modulating Runx2 mRNA stability. FEBS Lett. 2021;595:2007–14.
Yu J, Shen L, Liu Y, Ming H, Zhu X, Chu M, et al. The m6A methyltransferase METTL3 cooperates with demethylase ALKBH5 to regulate osteogenic differentiation through NF-κB signaling. Mol Cell Biochem. 2020;463:203–10.
Han Z, Wang X, Xu Z, Cao Y, Gong R, Yu Y, et al. ALKBH5 regulates cardiomyocyte proliferation and heart regeneration by demethylating the mRNA of YTHDF1. Theranostics. 2021;11:3000–16.
Han Z, Xu Z, Yu Y, Cao Y, Bao Z, Gao X, et al. ALKBH5-mediated m(6)A mRNA methylation governs human embryonic stem cell cardiac commitment. Mol Ther Nucleic Acids. 2021;26:22–33.
Du T, Li G, Yang J, Ma K. RNA demethylase Alkbh5 is widely expressed in neurons and decreased during brain development. Brain Res Bull. 2020;163:150–9.
Zhao Y, Hu J, Sun X, Yang K, Yang L, Kong L, et al. Loss of m6A demethylase ALKBH5 promotes post-ischemic angiogenesis via post-transcriptional stabilization of WNT5A. Clin Transl Med. 2021;11:e402.
Yu F, Wei J, Cui X, Yu C, Ni W, Bungert J, et al. Post-translational modification of RNA m6A demethylase ALKBH5 regulates ROS-induced DNA damage response. Nucleic Acids Res. 2021;49:5779–97.
Rubio RM, Depledge DP, Bianco C, Thompson L, Mohr I. RNA m6 A modification enzymes shape innate responses to DNA by regulating interferon β. Genes Dev. 2018;32:1472–84.
Zheng Q, Hou J, Zhou Y, Li Z, Cao X. The RNA helicase DDX46 inhibits innate immunity by entrapping m6A-demethylated antiviral transcripts in the nucleus. Nat Immunol. 2017;18:1094–103.
Liu B, Liu N, Zhu X, Yang L, Ye B, Li H, et al. Circular RNA circZbtb20 maintains ILC3 homeostasis and function via Alkbh5-dependent m6A demethylation of Nr4a1 mRNA. Cell Mol Immunol. 2021;18:1412–24.
Chen G, Zhao Q, Yuan B, Wang B, Zhang Y, Li Z, et al. ALKBH5-modified HMGB1-STING activation contributes to radiation induced liver disease via innate immune response. Int J Radiat Oncol Biol Phys. 2021;111:491–501.
Zhou J, Zhang X, Hu J, Qu R, Yu Z, Xu H, et al. m(6)A demethylase ALKBH5 controls CD4(+) T cell pathogenicity and promotes autoimmunity. Sci Adv. 2021;7:eabg0470.
Wang HF, Kuang MJ, Han SJ, Wang AB, Qiu J, Wang F, et al. BMP2 modified by the m(6)A demethylation enzyme ALKBH5 in the ossification of the ligamentum flavum through the AKT signaling pathway. Calcif Tissue Int. 2020;106:486–93.
Wang B, Fang X, Sun X, Du C, Zhou L, Lv X, et al. m6A demethylase ALKBH5 suppresses proliferation and migration of enteric neural crest cells by regulating TAGLN in Hirschsprung’s disease. Life Sci. 2021;278:119577.
Xu K, Mo Y, Li D, Yu Q, Wang L, Lin F, et al. N6-methyladenosine demethylases Alkbh5/Fto regulate cerebral ischemia-reperfusion injury. Ther Adv Chronic Dis. 2020;11:1–15.
Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet. 2014;15:293–306.
Aik W, Scotti JS, Choi H, Gong L, Demetriades M, Schofield CJ, et al. Structure of human RNA N6-methyladenine demethylase ALKBH5 provides insights into its mechanisms of nucleic acid recognition and demethylation. Nucleic Acids Res. 2014;42:4741–54.
Xu C, Liu K, Tempel W, Demetriades M, Aik W, Schofield CJ, et al. Structures of human ALKBH5 demethylase reveal a unique binding mode for specific single-stranded N6-methyladenosine RNA demethylation. J Biol Chem. 2014;289:17299–311.
Feng C, Liu Y, Wang G, Deng Z, Zhang Q, Wu W, et al. Crystal structures of the human RNA demethylase Alkbh5 reveal basis for substrate recognition. J Biol Chem. 2014;289:11571–83.
Britt KL, Cuzick J, Phillips KA. Key steps for effective breast cancer prevention. Nat Rev Cancer. 2020;20:417–36.
Fry NJ, Law BA, Ilkayeva OR, Carraway KR, Holley CL, Mansfield KD. N(6)-methyladenosine contributes to cellular phenotype in a genetically-defined model of breast cancer progression. Oncotarget. 2018;9:31231–43.
Wu L, Wu D, Ning J, Liu W, Zhang D. Changes of N6-methyladenosine modulators promote breast cancer progression. BMC Cancer. 2019;19:326.
De Angelis ML, Francescangeli F, Zeuner A. Breast cancer stem cells as drivers of tumor chemoresistance, dormancy and relapse: new challenges and therapeutic opportunities. Cancers. 2019;11:1569.
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA. 2016;113:E2047-2056.
Zhang C, Zhi WI, Lu H, Samanta D, Chen I, Gabrielson E, et al. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 2016;7:64527–42.
Goodenberger ML, Jenkins RB. Genetics of adult glioma. Cancer Genet. 2012;205:613–21.
Liu Z, Chen Y, Wang L, Ji S. ALKBH5 promotes the proliferation of glioma cells via enhancing the mRNA stability of G6PD. Neurochem Res. 2021;46:3003–11.
Cui Y, Wang Q, Lin J, Zhang L, Zhang C, Chen H, et al. miRNA-193a-3p regulates the AKT2 pathway to inhibit the growth and promote the apoptosis of glioma cells by targeting ALKBH5. Front Oncol. 2021;11:600451.
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–401.
Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 2013;15:ii1–56.
Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–17.
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591-606.e596.
Kowalski-Chauvel A, Lacore MG, Arnauduc F, Delmas C, Toulas C, Cohen-Jonathan-Moyal E, et al. The m6A RNA demethylase ALKBH5 promotes radioresistance and invasion capability of glioma stem cells. Cancers. 2021;13:40.
Lee SY. Temozolomide resistance in glioblastoma multiforme. Genes dis. 2016;3:198–210.
Liu B, Zhou J, Wang C, Chi Y, Wei Q, Fu Z, et al. LncRNA SOX2OT promotes temozolomide resistance by elevating SOX2 expression via ALKBH5-mediated epigenetic regulation in glioblastoma. Cell Death Dis. 2020;11:384.
Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. 2017;17:637–58.
Zhu Z, Qian Q, Zhao X, Ma L, Chen P. N6-methyladenosine ALKBH5 promotes non-small cell lung cancer progress by regulating TIMP3 stability. Gene. 2020;731:144348.
Guo J, Wu Y, Du J, Yang L, Chen W, Gong K, et al. Deregulation of UBE2C-mediated autophagy repression aggravates NSCLC progression. Oncogenesis. 2018;7:49.
Zhang D, Ning J, Okon I, Zheng X, Satyanarayana G, Song P, et al. Suppression of m6A mRNA modification by DNA hypermethylated ALKBH5 aggravates the oncological behavior of KRAS mutation/LKB1 loss lung cancer. Cell Death Dis. 2021;12:518.
Chao Y, Shang J, Ji W. ALKBH5-m6A-FOXM1 signaling axis promotes proliferation and invasion of lung adenocarcinoma cells under intermittent hypoxia. Biochem Biophys Res Commun. 2020;521:499–506.
Yu H, Zhang Z. ALKBH5-mediated m6A demethylation of lncRNA RMRP plays an oncogenic role in lung adenocarcinoma. Mamm Genome. 2021;32:195–203.
Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin. 2018;68:284–96.
Cai Y, Wu G, Peng B, Li J, Zeng S, Yan Y, et al. Expression and molecular profiles of the AlkB family in ovarian serous carcinoma. Aging. 2021;13:9679–92.
Zhu H, Gan X, Jiang X, Diao S, Wu H, Hu J. ALKBH5 inhibited autophagy of epithelial ovarian cancer through miR-7 and BCL-2. J Exp Clin Cancer Res. 2019;38:163.
Jiang Y, Wan Y, Gong M, Zhou S, Qiu J, Cheng W. RNA demethylase ALKBH5 promotes ovarian carcinogenesis in a simulated tumour microenvironment through stimulating NF-κB pathway. J Cell Mol Med. 2020;24:6137–48.
Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373:1136–52.
Kwok CT, Marshall AD, Rasko JE, Wong JJ. Genetic alterations of m(6)A regulators predict poorer survival in acute myeloid leukemia. J Hematol Oncol. 2017;10:39.
Shen C, Sheng Y, Zhu AC, Robinson S, Jiang X, Dong L, et al. RNA demethylase ALKBH5 selectively promotes tumorigenesis and cancer stem cell self-renewal in acute myeloid leukemia. Cell Stem Cell. 2020;27:64-80.e69.
Wang J, Li Y, Wang P, Han G, Zhang T, Chang J, et al. Leukemogenic chromatin alterations promote AML leukemia stem cells via a KDM4C-ALKBH5-AXL signaling axis. Cell Stem Cell. 2020;27:81-97.e88.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Zhang J, Guo S, Piao HY, Wang Y, Wu Y, Meng XY, et al. ALKBH5 promotes invasion and metastasis of gastric cancer by decreasing methylation of the lncRNA NEAT1. J Physiol Biochem. 2019;75:379–89.
Guo T, Liu DF, Peng SH, Xu AM. ALKBH5 promotes colon cancer progression by decreasing methylation of the lncRNA NEAT1. Am J Transl Res. 2020;12:4542–9.
Pu X, Gu Z, Gu Z. ALKBH5 regulates IGF1R expression to promote the proliferation and tumorigenicity of endometrial cancer. J Cancer. 2020;11:5612–22.
Chen G, Liu B, Yin S, Li S, Ye Guo, Wang M, et al. Hypoxia induces an endometrial cancer stem-like cell phenotype via HIF-dependent demethylation of SOX2 mRNA. Oncogenesis. 2020;9:81.
Zhang X, Wang F, Wang Z, Yang X, Yu H, Si S, et al. ALKBH5 promotes the proliferation of renal cell carcinoma by regulating AURKB expression in an m(6)A-dependent manner. Ann Transl Med. 2020;8:646–646.
Qu S, Jin L, Huang H, Lin J, Gao W, Zeng Z. A positive-feedback loop between HBx and ALKBH5 promotes hepatocellular carcinogenesis. BMC Cancer. 2021;21:686.
Chen S, Zhou L, Wang Y. ALKBH5-mediated m6A demethylation of lncRNA PVT1 plays an oncogenic role in osteosarcoma. Cancer Cell Int. 2020;20:34.
Hao L, Yin J, Yang H, Li C, Zhu L, Liu L, et al. ALKBH5-mediated m(6)A demethylation of FOXM1 mRNA promotes progression of uveal melanoma. Aging. 2021;13:4045–62.
Qian Y, Zhang C, Wang W, Lu D, Li J, Li L, et al. Hypoxia promotes proliferation of pituitary adenomas by HIF-1α/ALKBH5 signaling in vitro. Int J Clin Exp Pathol. 2020;13:1030–4.
Nagaki Y, Motoyama S, Yamaguchi T, Hoshizaki M, Sato Y, Sato T, et al. m6A demethylase ALKBH5 promotes proliferation of esophageal squamous cell carcinoma associated with poor prognosis. Genes Cells. 2020;25:547–61.
Wang X, Li Z, Kong B, Song C, Cong J, Hou J, et al. Reduced m(6)A mRNA methylation is correlated with the progression of human cervical cancer. Oncotarget. 2017;8:98918–30.
Shriwas O, Priyadarshini M, Samal SK, Rath R, Panda S, Das Majumdar SK, et al. DDX3 modulates cisplatin resistance in OSCC through ALKBH5-mediated m6A-demethylation of FOXM1 and NANOG. Apoptosis. 2020;25:233–46.
Chen M, Nie ZY, Wen XH, Gao YH, Cao H, Zhang SF. m6A RNA methylation regulators can contribute to malignant progression and impact the prognosis of bladder cancer. Biosci Rep. 2019;39:66.
Yu H, Yang X, Tang J, Si S, Zhou Z, Lu J, et al. ALKBH5 inhibited cell proliferation and sensitized bladder cancer cells to cisplatin by m6A-CK2alpha-mediated glycolysis. Mol Ther Nucleic Acids. 2021;23:27–41.
Jin H, Ying X, Que B, Wang X, Chao Y, Zhang H, et al. N6-methyladenosine modification of ITGA6 mRNA promotes the development and progression of bladder cancer. EBioMedicine. 2019;47:195–207.
Rawla P, Sunkara T, Gaduputi V. Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J Oncol. 2019;10:10.
Cho SH, Ha M, Cho YH, Ryu JH, Yang K, Lee KH, et al. ALKBH5 gene is a novel biomarker that predicts the prognosis of pancreatic cancer: a retrospective multicohort study. Ann Hepatobiliary Pancreat Surg. 2018;22:305–9.
Geng Y, Guan R, Hong W, Huang B, Liu P, Guo X, et al. Identification of m6A-related genes and m6A RNA methylation regulators in pancreatic cancer and their association with survival. Ann Transl Med. 2020;8:387–387.
He Y, Hu H, Wang Y, Yuan H, Lu Z, Wu P, et al. ALKBH5 inhibits pancreatic cancer motility by decreasing long non-coding RNA KCNK15-AS1 methylation. Cell Physiol Biochem. 2018;48:838–46.
Guo X, Li K, Jiang W, Hu Y, Xiao W, Huang Y, et al. RNA demethylase ALKBH5 prevents pancreatic cancer progression by posttranscriptional activation of PER1 in an m6A-YTHDF2-dependent manner. Mol Cancer. 2020;19:91.
Tang B, Yang Y, Kang M, Wang Y, Wang Y, Bi Y, et al. m6A demethylase ALKBH5 inhibits pancreatic cancer tumorigenesis by decreasing WIF-1 RNA methylation and mediating Wnt signaling. Mol Cancer. 2020;19:3.
Jin D, Guo J, Wu Y, Yang L, Wang X, Du J, et al. m6A demethylase ALKBH5 inhibits tumor growth and metastasis by reducing YTHDFs-mediated YAP expression and inhibiting miR-107/LATS2–mediated YAP activity in NSCLC. Mol Cancer. 2020;19:40.
Yuan Y, Yan G, He M, Lei H, Li L, Wang Y, et al. ALKBH5 suppresses tumor progression via an m6A-dependent epigenetic silencing of pre-miR-181b-1/YAP signaling axis in osteosarcoma. Cell Death Dis. 2021;12:60.
Yang P, Wang Q, Liu A, Zhu J, Feng J. ALKBH5 holds prognostic values and inhibits the metastasis of colon cancer. Pathol Oncol Res. 2020;26:1615–23.
Strick A, von Hagen F, Gundert L, Klumper N, Tolkach Y, Schmidt D, et al. The N6-methyladenosine (m6A) erasers alkylation repair homologue 5 (ALKBH5) and fat mass and obesity-associated protein (FTO) are prognostic biomarkers in patients with clear cell renal carcinoma. BJU Int. 2020;125:617–24.
Chen Y, Zhao Y, Chen J, Peng C, Zhang Y, Tong R, et al. ALKBH5 suppresses malignancy of hepatocellular carcinoma via m6A-guided epigenetic inhibition of LYPD1. Mol Cancer. 2020;19:123.
Wang YN, Yu CY, Jin HZ. RNA N(6)-methyladenosine modifications and the immune response. J Immunol Res. 2020;2020:6327614.
Zhang M, Song J, Yuan W, Zhang W, Sun Z. Roles of RNA methylation on tumor immunity and clinical implications. Front Immunol. 2021;12:600.
Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell. 2021;184:5309–37.
Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48:434–52.
Sirica AE, Gores GJ, Groopman JD, Selaru FM, Strazzabosco M, Wei Wang X, et al. Intrahepatic cholangiocarcinoma: continuing challenges and translational advances. Hepatology. 2019;69:1803–15.
Qiu X, Yang S, Wang S, Wu J, Zheng B, Wang K, et al. M(6)A demethylase ALKBH5 regulates PD-L1 expression and tumor immunoenvironment in intrahepatic cholangiocarcinoma. Cancer Res. 2021;81:4778–93.
Li N, Kang Y, Wang L, Huff S, Tang R, Hui H, et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci USA. 2020;117:20159–70.
Dong F, Qin X, Wang B, Li Q, Hu J, Cheng X, et al. ALKBH5 facilitates hypoxia-induced paraspeckle assembly and IL8 secretion to generate an immunosuppressive tumor microenvironment. Cancer Res. 2021;81:5876.
Xue J, Schmidt Susanne V, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–88.
Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. 2017;31:326–41.
Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465–72.
Thalhammer A, Bencokova Z, Poole R, Loenarz C, Adam J, O’Flaherty L, et al. Human AlkB homologue 5 is a nuclear 2-oxoglutarate dependent oxygenase and a direct target of hypoxia-inducible factor 1alpha (HIF-1alpha). PLoS ONE. 2011;6:e16210.
Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–68.
Zhao Z, Shilatifard A. Epigenetic modifications of histones in cancer. Genome Biol. 2019;20:245.
Gao W, Jia Z, Tian Y, Yang P, Sun H, Wang C, et al. HBx protein contributes to liver carcinogenesis by H3K4me3 modification through stabilizing WD repeat domain 5 protein. Hepatology. 2020;71:1678–95.
Kottakis F, Nicolay BN, Roumane A, Karnik R, Gu H, Nagle JM, et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature. 2016;539:390–5.
Matsumoto S, Iwakawa R, Takahashi K, Kohno T, Nakanishi Y, Matsuno Y, et al. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene. 2007;26:5911–8.
Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5:860.
Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The human transcription factors. Cell. 2018;172:650–65.
Sanchez Calle A, Kawamura Y, Yamamoto Y, Takeshita F, Ochiya T. Emerging roles of long non-coding RNA in cancer. Cancer Sci. 2018;109:2093–100.
Wang X, Zhang J, Wang Y. Long noncoding RNA GAS5-AS1 suppresses growth and metastasis of cervical cancer by increasing GAS5 stability. Am J Transl Res. 2019;11:4909–21.
Treiber T, Treiber N, Meister G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol. 2019;20:5–20.
Xue J, Xiao P, Yu X, Zhang X. A positive feedback loop between AlkB homolog 5 and miR-193a-3p promotes growth and metastasis in esophageal squamous cell carcinoma. Hum Cell. 2021;34:502–14.
Huang A, Zheng H, Wu Z, Chen M, Huang Y. Circular RNA-protein interactions: functions, mechanisms, and identification. Theranostics. 2020;10:3503–17.
Liu Z, Wang Q, Wang X, Xu Z, Wei X, Li J. Circular RNA cIARS regulates ferroptosis in HCC cells through interacting with RNA binding protein ALKBH5. Cell Death Discov. 2020;6:72.
Shah A, Rashid F, Awan HM, Hu S, Wang X, Chen L, et al. The DEAD-box RNA helicase DDX3 interacts with m(6)A RNA demethylase ALKBH5. Stem Cells Int. 2017;2017:8596135.
Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163:811–28.
Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. 2016;29:783–803.
Dixit D, Prager BC, Gimple RC, Poh HX, Wang Y, Wu Q, et al. The RNA m6A reader YTHDF2 maintains oncogene expression and is a targetable dependency in glioblastoma stem cells. Cancer Discov. 2021;11:480.
Fang R, Chen X, Zhang S, Shi H, Ye Y, Shi H, et al. EGFR/SRC/ERK-stabilized YTHDF2 promotes cholesterol dysregulation and invasive growth of glioblastoma. Nat Commun. 2021;12:177.
Chai RC, Chang YZ, Chang X, Pang B, An SY, Zhang KN, et al. YTHDF2 facilitates UBXN1 mRNA decay by recognizing METTL3-mediated m(6)A modification to activate NF-kappaB and promote the malignant progression of glioma. J Hematol Oncol. 2021;14:109.
Kowalski-Chauvel A, Lacore MG, Arnauduc F, Delmas C, Toulas C, Cohen-Jonathan-Moyal E, et al. The m6A RNA demethylase ALKBH5 promotes radioresistance and invasion capability of glioma stem cells. Cancers. 2020;13:66.
Fukumoto T, Zhu H, Nacarelli T, Karakashev S, Fatkhutdinov N, Wu S, et al. N6-methylation of adenosine of FZD10 mRNA contributes to PARP inhibitor resistance. Cancer Res. 2019;79:2812–20.
Phi LTH, Sari IN, Yang YG, Lee SH, Jun N, Kim KS, et al. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018;2018:5416923.
Tan S, Li D, Zhu X. Cancer immunotherapy: pros, cons and beyond. Biomed Pharmacother. 2020;124:109821.
Selberg S, Seli N, Kankuri E, Karelson M. Rational design of novel anticancer small-molecule RNA m6A demethylase ALKBH5 inhibitors. ACS Omega. 2021;6:13310–20.
Malacrida A, Rivara M, Di Domizio A, Cislaghi G, Miloso M, Zuliani V, et al. 3D proteome-wide scale screening and activity evaluation of a new ALKBH5 inhibitor in U87 glioblastoma cell line. Bioorg Med Chem. 2020;28:115300.
Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488:404–8.
Woon EC, Tumber A, Kawamura A, Hillringhaus L, Ge W, Rose NR, et al. Linking of 2-oxoglutarate and substrate binding sites enables potent and highly selective inhibition of JmjC histone demethylases. Angew Chem. 2012;124:1663–6.
Woon EC, Demetriades M, Bagg EA, Aik W, Krylova SM, Ma JH, et al. Dynamic combinatorial mass spectrometry leads to inhibitors of a 2-oxoglutarate-dependent nucleic acid demethylase. J Med Chem. 2012;55:2173–84.
Purslow JA, Nguyen TT, Khatiwada B, Singh A, Venditti V. N6-methyladenosine binding induces a metal-centered rearrangement that activates the human RNA demethylase Alkbh5. Sci Adv. 2021;7:eabi8215.
Xie LJ, Yang XT, Wang RL, Cheng HP, Li ZY, Liu L, et al. Identification of flavin mononucleotide as a cell-active artificial N(6)-methyladenosine RNA demethylase. Angew Chem Int Ed Engl. 2019;58:5028–32.
Pettersson M, Crews CM. PROteolysis TArgeting Chimeras (PROTACs)—past, present and future. Drug Discov Today Technol. 2019;31:15–27.
Khan S, He Y, Zhang X, Yuan Y, Pu S, Kong Q, et al. PROteolysis TArgeting Chimeras (PROTACs) as emerging anticancer therapeutics. Oncogene. 2020;39:4909–24.
Li J, Chen Z, Chen F, Xie G, Ling Y, Peng Y, et al. Targeted mRNA demethylation using an engineered dCas13b-ALKBH5 fusion protein. Nucleic Acids Res. 2020;48:5684–94.
Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DBT, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353:aaf5573.
Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, et al. RNA targeting with CRISPR–Cas13. Nature. 2017;550:280–4.
Wilson C, Chen PJ, Miao Z, Liu DR. Programmable m6A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat Biotechnol. 2020;38:1431–40.
Sharma G, Sharma AR, Bhattacharya M, Lee SS, Chakraborty C. CRISPR-Cas9: a preclinical and clinical perspective for the treatment of human diseases. Mol Ther. 2021;29:571–86.
Fallah J, Rini BI. HIF inhibitors: status of current clinical development. Curr Oncol Rep. 2019;21:6.
Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203–22.
Bennett RL, Licht JD. Targeting epigenetics in cancer. Annu Rev Pharmacol Toxicol. 2018;58:187–207.
Hong B, Pieter J. van den Heuvel A, V. Prabhu V, Zhang S, S. El-Deiry W. Targeting tumor suppressor p53 for cancer therapy: strategies, challenges and opportunities. Curr Drug Targets. 2014;15:80–89.
Karkhanis V, Wang L, Tae S, Hu YJ, Imbalzano AN, Sif S. Protein arginine methyltransferase 7 regulates cellular response to DNA damage by methylating promoter histones H2A and H4 of the polymerase delta catalytic subunit gene, POLD1. J Biol Chem. 2012;287:29801–14.
Bulcha JT, Wang Y, Ma H, Tai PWL, Gao G. Viral vector platforms within the gene therapy landscape. Sig Transduct Target Ther. 2021;6:53.
Santiago-Ortiz JL, Schaffer DV. Adeno-associated virus (AAV) vectors in cancer gene therapy. J Control Release. 2016;240:287–301.
Li C, Samulski RJ. Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet. 2020;21:255–72.
Dunbar Cynthia E, High Katherine A, Joung JK, Kohn Donald B, Ozawa K, Sadelain M. Gene therapy comes of age. Science. 2018;359:eaan4672.
Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature. 2017;552:126–31.
Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell. 2018;22:191-205.e199.
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N6-methyladenosine RNA demethylase. Cancer Cell. 2017;31:127–41.
Hua W, Zhao Y, Jin X, Yu D, He J, Xie D, et al. METTL3 promotes ovarian carcinoma growth and invasion through the regulation of AXL translation and epithelial to mesenchymal transition. Gynecol Oncol. 2018;151:356–65.
Qian JY, Gao J, Sun X, Cao MD, Shi L, Xia TS, et al. KIAA1429 acts as an oncogenic factor in breast cancer by regulating CDK1 in an N6-methyladenosine-independent manner. Oncogene. 2019;38:6123–41.
Miao R, Dai CC, Mei L, Xu J, Sun SW, Xing YL, et al. KIAA1429 regulates cell proliferation by targeting c-Jun messenger RNA directly in gastric cancer. J Cell Physiol. 2020;235:7420–32.
Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593:597–601.
Bedi RK, Huang D, Eberle SA, Wiedmer L, Sledz P, Caflisch A. Small-molecule inhibitors of METTL3, the major human epitranscriptomic writer. ChemMedChem. 2020;15:744–8.
Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43:373–84.
Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. 2018;172:90-105.e123.
Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell. 2019;35:677-691.e610.
Acknowledgements
Not applicable.
Funding
This study was funded by the National Natural Science Foundation of China (81872322) and the Zhejiang Key Research and Development Project (2020C03014).
Author information
Authors and Affiliations
Contributions
J.Q. and H.Y. collected related literature and wrote the manuscript. Y.H., W.C., and Y.L. helped in drafting figures. Z.C., J.H., and E.Z. provided guidance and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qu, J., Yan, H., Hou, Y. et al. RNA demethylase ALKBH5 in cancer: from mechanisms to therapeutic potential. J Hematol Oncol 15, 8 (2022). https://doi.org/10.1186/s13045-022-01224-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-022-01224-4