
Abstract
Background
In Sub-Saharan Africa, Borassus aethiopum Mart. (African fan palm) is an important non-timber forest product-providing palm that faces multiple anthropogenic threats to its genetic diversity. However, this species is so far under-studied, which prevents its sustainable development as a resource. The present work is a first attempt at characterizing the genetic diversity and population structure of B. aethiopum across nine collection sites spanning the three climatic regions of Benin, West Africa, through the use of microsatellite markers.
Results
During a first phase we relied on the reported transferability of primers developed in other palm species. We find that, in disagreement with previously published results, only 22.5% of the markers tested enable amplification of B. aethiopum DNA and polymorphism detection is very low.
In a second phase, we generated a B. aethiopum-specific genomic dataset through high-throughput sequencing and used it for the de novo detection of microsatellite loci. Among the primer pairs targeting these, 11 detected polymorphisms and were further used for analyzing genetic diversity. Across the nine sites, expected heterozygosity (He) ranges from 0.263 to 0.451 with an overall average of 0.354, showing a low genetic diversity. Analysis of molecular variance (AMOVA) shows that within-site variation accounts for 53% of the genetic variation. Accordingly, the low number of migrants and positive values of the fixation index (F) in sites from both the Central (Sudano-Guinean) and the Southern (Guinean) climatic regions suggest limited gene flow between sites. The global correlation between genetic and geographic distances is weak; however, our clustering analyses indicate that B. aethiopum palms from Savè (Center) are genetically more similar to those from the North than to samples from other Central sites.
Conclusions
In the light of our results, we discuss the use of inter-species transfer vs. de novo development of microsatellite markers in genetic diversity analyses targeting under-studied species, and suggest future applications for our molecular resources. We propose that, while prominent short-range pollen and seed dispersal in Benin explain most of our results, gene flux between the Central and Northern regions, as a result of animal and/or human migrations, might underlie the Savè discrepancy.
Similar content being viewed by others


Background
Many plant species remain under-studied due to their low economic importance, complicated biology and/or the absence of available genome sequence information. Upon initiating a research project aimed at characterizing the genetic diversity of such a species, researchers may be confronted with the situation that some resources can be found in related taxa. In such cases, the first step is often to assess whether some of these resources, such as molecular markers, can be used to study the new species. Provided that the “source” species display enough genetic similarities to the “target” species and that marker transferability has been previously assessed, this first step may lead to quick progress in a cost-effective manner. Often, transferring markers between species is seen as a smarter investment than developing and testing new markers, especially if limited funding is available [1, 2].
Over the last three decades, molecular markers have been widely used to study genetic variation among and within populations of various plant species [3,4,5,6,7]. Among the different types of markers that are available, microsatellites or simple sequence repeats (SSRs) are often selected due to their high mutation frequency, which ranges from 10− 2 to 10− 6 nucleotides per locus per generation [2, 8] and generates multiple allelic forms, and their co-dominant nature. The combination of both characteristics makes them sensitive tools for the assessment of genetic diversity among species, determination of population structure, phylogenetic reconstruction, genetic mapping, evolutionary analyses, and molecular breeding [9,10,11,12]. From a practical perspective, the popularity of SSRs is also related to their low resource requirements (i.e. technical skills, laboratory equipments and consumables) that enable their easy implementation and the reproducibility of results in most research environments [2, 8]. However, the steps leading to the development of functional SSR markers, namely the initial identification of microsatellite loci, primer selection and assessment of amplification/polymorphism detection, require some prior knowledge of the genome of the target species and may prove to be expensive and time-consuming [11, 13]. In order to overcome this difficulty, approaches relying on the transfer of SSR markers between species or genera have therefore been implemented. They have been successful in many instances, as documented across Prunus species and among members of the Rosaceae family [14, 15]; between species of the Hevea genus and to other Euphorbiaceae [16]; among Lamiaceae [17]; among Legumes belonging to the Vicia genus [18] and from the Phaseolus genus to Vigna [19]. In other cases, the ever-increasing affordability of high-throughput sequencing technologies and the development of dedicated bioinformatics data mining tools have enabled the identification of microsatellite loci and the development of SSR markers, including in non-model plant species with limited or no background genetic information [20,21,22,23].
Borassus aethiopum Mart., also known as ron palm, toddy palm or African fan palm, is a dioecious species belonging to the Arecaceae family. It is widely distributed across West and Central Africa, where it is present as wild populations [24]. The species is classified as a non-timber forest products (NTFPs)-providing plant, since different parts of the plant are used for various purposes by local populations [24, 25]. In Benin (West Africa) for instance, 121 different uses distributed in seven categories (medicinal, handicrafts, food, construction, firewood, ceremonies and rituals) have been reported for the species [26]. Among these, the consumption of ripe fruits (fresh or roasted) and hypocotyls as food, the use of the weather- and pest-resistant stipe as construction wood and that of leaves and petioles in handicrafts, are the most widespread in local populations [26,27,28]. These different products are also sold in markets, mostly by women, to whom they provide additional income: it is indeed estimated that in Benin, sales of hypocotyls alone may represent 50% to nearly three times the minimum wage of 40,000 CFA Francs (ca. 61 euros) a month [27].
These multiple uses of products derived from B. aethiopum have put a strong anthropogenic pressure on the species, thus contributing to both fragmentations of its populations and their poor natural regeneration [27, 29,30,31,32]. Further fragmentation of the species’ habitat has been observed as a result of land clearing for agriculture or urban development [32,33,34]. As illustrated through similar examples in the literature [35, 36], such phenomena may lead to restricted gene flow and ultimately, to loss of genetic diversity among B. aethiopum populations. A sustainable management policy for B. aethiopum populations is therefore urgently needed and acquiring information on the genetic diversity of the species and population structure is a major step towards defining sustainable management actions. At the time of writing the present article, only a few chloroplast sequences are publicly available for B. aethiopum through NCBI (https://www.ncbi.nlm.nih.gov/search/all/?term=borassus%20aethiopum). By contrast, abundant molecular resources, including genome assemblies or drafts, are available for model palm species such as Elaeis guineensis Jacq [37]., Phoenix dactylifera L. [38,39,40] and Cocos nucifera L. [41, 42]. In each of these three palm species, large numbers of SSR markers have been identified and for a fraction of them, cross-species and cross-genera transferability tests among species belonging to the Palmaceae family have been performed [43,44,45,46,47,48,49]. In several instances [44,45,46,47, 49] these tests included samples from Borassus flabellifer, the Asian relative of B. aethiopum.
The primary objective of the present study is to generate the first set of genetic data on Borassus aethiopum, as a first step towards improving the management of this species through a better knowledge of its diversity. In order to achieve this, we first describe attempts to use SSR markers identified in these other palm species. Then, we describe the low-coverage sequencing of the B. aethiopum genome with the aim of developing the first set of specific SSR markers targeting this species. Finally, we used the novel SSR markers to assess the genetic diversity and population structure of B. aethiopum samples collected across the three different climatic regions of Benin, a country that was most readily accessible to us for sample collection, as an important first step towards more comprehensive studies spanning the West African sub-region.
Results
Assessment of palm SSR marker transferability to Borassus aethiopum and evaluation of their capacity for characterizing genetic diversity
Of the 80 microsatellite markers selected from the three model palm species Elaeis guineensis, Phoenix dactylifera and Cocos nucifera and tested for amplification on B. aethiopum DNA, 18 (22.5%) generate amplification products (Table 1). No amplification is observed using the 11 C. nucifera markers, whereas 7 (15.9%) and 11 (44%) of the P. dactylifera and E. guineensis markers, respectively, show a successful amplification. None of the amplification products generated with P. dactylifera primers display genetic polymorphism in our B. aethiopum test panel. Among E. guineensis-derived SSR markers however, two, namely ESSR566 and ESSR652, display polymorphism. However, it must be noted that depending on the DNA sample the ESSR566 primer pair generates a variable number of amplicons with distinct sizes, which may be an indication that more than one locus is targeted.
Overall, during this phase of the study we detect polymorphism in our B. aethiopum test panel with only 2 (11.1% of successfully amplified markers, 2.5% of total) of the palm SSR primer pairs assayed. Only one of these markers, namely ESSR652, enables unambiguous detection of microsatellite locus polymorphism in B. aethiopum, and might therefore be used for studying genetic diversity in this species.
De novo identification of microsatellite sequences in the B. aethiopum genome and assessment of potential SSR markers
In order to enable a more precise evaluation of genetic diversity in B. aethiopum, we developed specific B. aethiopum markers from de novo sequencing data. A total of 23,281,354 raw reads with an average length of 250 bp have been generated from one MiSeq run. Raw sequence reads have been trimmed resulting in 21,636,172 cleaned-up reads, yielding 493,636 high-quality reads after filtering (Q > 30) from which 216,475 contigs have been assembled.
From the contigs, the QDD software identifies a total of 1618 microsatellite loci (Additional file 1), of which 1327 (82.01%) are perfect (i.e. repeat size 4 bp or smaller and repeat number 10–20). Among the perfect microsatellite loci, 83.86% are composed of di-nucleotidic repeat units, 13.06% of tri-nucleotidic units, 2.39% of tetra-nucleotidic repeats and 0.67% of repeats with five nucleotides and over. From these, we selected SSR markers composed of di- (AG) or tri- nucleotide repeats, using the following criteria for specific amplification of easily scorable bands: primer lengths ranging from 18 to 22 bp, annealing temperatures 55–60 °C, and predicted amplicon sizes 90–200 bp.
The characteristics of the 57 selected primer pairs and the results of the test amplifications are presented in Table 2. Successful amplification of B. aethiopum DNA is obtained for 54 (94.7%) primer pairs and of these, 34 (60.0% of amplifying couples) show no polymorphism. The remaining 20 primer pairs enable the amplification of polymorphic products, however nine of them yield complex, ambiguous amplification profiles that prevent their use for reliable detection of genetic variation. As a result, 11 putative B. aethiopum SSR markers (representing 20.4% of primer pairs associated with successful amplification and 55.0% of those detecting polymorphic products in our study) are both polymorphic and unambiguously mono-locus in our amplification test panel and may therefore be used for further analyses.
Microsatellite-based characterization of genetic variation of B. aethiopum in Benin
The newly identified set of 11 B. aethiopum-specific SSR markers has been used for the characterization of genetic diversity in our full panel of 180 individual samples from nine locations distributed across Benin (Fig. 1).

Sampling locations of the Beninese B. aethiopum used in this study
Map generated from publicly available resources of the Institut Géographique National du Bénin (IGN; https://geobenin.bj/) and the “Major Rivers of the World” dataset from the World Bank Data Catalog (https://datacatalog.worldbank.org/dataset/major-rivers-world; Creative Commons Attribution 4.0 International license), using the ArcGIS software by ESRI (www.esri.com).
As shown in Table 3, among our sample set the number of alleles per microsatellite locus ranges from 2 for marker Mbo41 to 6 for markers Mbo34, Mbo35, and Mbo50, with an average value of 4.27, whereas expected heterozygosity (He) values range from 0.031 (marker Mbo56) to 0.571 (marker Mbo35). Using these markers, the analysis of genetic diversity (Table 4) shows that the number of polymorphic markers detected at the microsatellite loci investigated ranges from 8 (sites of Togbin and Malanville) to 10 (Savè, Agoua, Pendjari, Pingou and Trois Rivières), with a mean value of 9 ± 0.865. With the exception of Savè, Hounviatouin and Malanville, 1 to 3 private alleles of the targeted microsatellite loci are observed in most sampling locations. Regarding the genetic parameters, the number of effective alleles (Ne) ranges from 1.447 to 2.069 with an average number of 1.761. He values range from 0.263 (Hounviatouin) to 0.451 (Savè) with an average value of 0.354 whereas the observed heterozygosity (Ho) varied from 0.234 (Togbin) to 0.405 (Pingou) with an average value of 0.335. Negative values of Fixation index (F) are obtained for Pingou, Malanville and Trois Rivières whereas positive F values, indicating a deficit of heterozygosity, are observed in all other sites investigated.
Population structure of B. aethiopum in Benin
Nei’s genetic distance among locations (Table 5) ranges from 0.073, as observed between Togbin and Hounviatouin (Guineo-Congolian region), to 0.577 between Togbin (Guineo-Congolian region) and Trois Rivières (Sudanian region). Overall, genetic distances between B. aethiopum sampling locations are lowest within the same region, with the lowest genetic distances among the sites of Pendjari, Pingou, and Trois Rivières which are all located in the Northern part of the country. One interesting exception is the Central (Guineo-Sudanian) region of Benin, where we find that the most genetically distant location from Savè is the one from the Agoua forest reserve (0.339). Surprisingly, Savè displays its highest genetic identity value when compared to the other two collection sites located within protected areas, namely Pendjari (0.870) and Trois Rivières (0.882) which are both located in the Sudanian region. This is an unexpected finding considering the geographic distances involved.
A similar structure of genetic distances emerges from the analysis of pairwise location genetic differentiation (Fst) (Table 6), suggesting genetic differentiation according to geographic distances between collection sites, with the notable exception of the lower genetic differentiation between samples from Savè and those from either one of the forest reserves in the Northern region, namely Pendjari and Trois Rivières.
In order to assess the strength of the relationship between genetic and geographic distances, we plotted them as a linear regression and performed the Mantel permutation test. As shown in Fig. 2, the positive correlation between both variables is weak, but significant (R2 = 0.1139, P = 0.040).

Correlation between pairwise Fst vs. pairwise geographical distance
The results of the non-hierarchical AMOVA (Table 7) show that within-site variation underlies the major part (53%) of total variance, whereas among-sites and among-regions variations explain genetic variance to a similar extent (23 and 24%, respectively). Accordingly, the average Number of migrants between collection sites (Nm = 1.019) is low, indicating very limited gene flow.
Hierarchical analyses performed with K = 2 and K = 3, respectively, yield an identical proportion of genetic variation at the within-individual level (62% of total; Table 7). Analysis using K = 3 allows for a balanced representation of variation between the among-regions and among-sites scales (16% of total variance for each), whereas among-regions variation is not as well accounted for under K = 2 (7% of total variance, vs. 24% for among-sites variation).
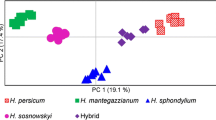
The Principal Coordinates Analysis (PCoA) of 180 B. aethiopum samples (Fig. 3a) shows that the first axis (accounting for 24% of total variation out of a sum of 33.90 for axes 1 and 2) roughly separates individual samples in two main groups, a result that is in agreement with the analysis of genetic distances. The sampling locations-based PCoA (Fig. 3b) confirms the genetic separation along the first axis (accounting for 44.08% of total variation over a total of 61.06% for the sum for axes 1 and 2) between sites from the Guineo-Congolian (Southern) region, plus the sites of Agoua and Biguina (Center) vs. sites from the Sudanian (Northern) region, plus the site of Savè (Center). Although the distinction is not as clearly marked, the second axis (accounting for 16.98% of total variation) further allows to distinguish two subgroups within the first group, corresponding to sites belonging to the Southern region and to those from the Central one, respectively.

Principal Coordinates Analysis (PCoA). a PCoA analysis of individual B. aethiopum samples. b Sampling locations-based PCoA
Likewise, the Bayesian analysis of our data indicates an optimal value of K = 2 for the clustering of the samples into two groups (Fig. 4a and b): one group that includes samples from Togbin and Hounviatouin in the Southern part of the country, as well as most samples from Biguina and Agoua at the Western (Togolese) border of the Center region; and one group composed of the majority of samples collected in Savè (Eastern part of the Center region) and from the Northern locations of Pendjari, Pingou, Malanville, and Trois Rivières. Since there is a possibility that the ΔK method used for estimating K leads to over- or under-estimated values [50], clustering with higher values of K have also been tested. As is apparent in Fig. 4b, for values of K = 4 and above standard deviations increase considerably, therefore we present results for both K = 2 and K = 3 (Fig. 4c; see also Additional Figure 4 for the summary of the complete analyses with K = 1 to K = 10). As previously observed with the location-based PCoA, under K = 3 further clustering emerges within the first group, involving samples from Togbin and Hounviatouin (South) and those from Biguina and Agoua (Center), respectively.

Bayesian cluster analysis. a Determination of the optimal value of K from Structure Harvester. b Evanno plot. c Bar plot representations of Bayesian STRUCTURE analysis of Beninese B. aethiopum samples with K = 2 (top) or with K = 3 (bottom) generated with CLUMPAK. Sampling sites are displayed along the horizontal axis
The Unweighted pair-group method with arithmetic mean (UPGMA) tree constructed from our data (Fig. 5) distinguishes two main groups matching the ones defined through the Bayesian analysis with K = 2, and which are supported by bootstrap values above 50. Within each of these groups, subgroups corresponding to those observed with K = 3 clustering and that globally match geo-climatic regions (Savè excepted) can further be defined. However, in this case most bootstrap values attached to these secondary branches are not significant.

UPGMA dendrogram based on genetic distances between Beninese B. aethiopum sampling locations. Bootstrap values supporting each branch are indicated on the nodes
Discussion
In flowering plant, the efficiency of cross-species transfer of SSR markers is highly variable among taxa, especially when important differences in genome complexity exist between the marker source and the target [51]. Nevertheless, this method has been used successfully for accelerating the analysis of genetic diversity in many plant species, including palms [11, 52,53,54]. In the present study, we find that the transferability rate of microsatellite markers developed in other palms genera to Borassus aethiopum, i.e. their ability to successfully amplify genomic DNA from the latter species, is very low. Indeed, among the 80 primer pairs designed on either Elaeis guineensis, Phoenix dactylifera or Cocos nucifera, we observe that only 18 (22.5%) produce amplicons from B. aethiopum. This percentage is very low when compared to both the inter-species and inter-genera transferability rates that have been found in similar studies targeting other palm species: from 17 to 93% in a panel of 32 palm species [49], 75% from E. oleifera to E. guineensis [54], 86% between the wooly jelly palm (Butia eriospatha Mart.) and related species Butia catarinensis [55] and up to 100% in the licuri palm (Syagrus coronate Mart) [56]. When considering other plant families, our transferability rate is also markedly lower than both the average rate of 50% found by Peakall et al. [57] within the Glycine genus and among Legumes genera, and the overall rate of 35.2% calculated by Rossetto [58] for within-family transferability among Gymnosperms and Angiosperms. The low transferability rate in our study might be explained in part by the fact that we used markers originating from genomic sequences. Indeed, as pointed out by Fan et al. [1], such markers have a lower transferability rate when compared to Expressed Sequence Tags (ESTs)-derived microsatellites due to the higher inter-species sequence variability within non-coding vs. coding sequences. Similarly, it is plausible that differences in genome size and complexity among palm species and genera account for our difficulty to identify palm SSR markers that successfully amplify in B. aethiopum. As a matter of fact, the size of the B. aethiopum genome, as determined by flow cytometry (1C = 7.73 Gb; Jaume Pellicer, unpublished data), is 3.2 to 11.5 times larger than those of the microsatellite source species used in the present study: P. dactylifera genome is estimated to be 671 Mb [39] whereas the E. guineensis genome is 1.8–1.9 Gb [37, 59] and C. nucifera genome is 2.42 Gb [42]. It is possible that these differences in genome sizes among related diploid plant species rely on differences in transposable element (TE) content, which in turn might have induced structural alterations throughout the genome through indels, copy number variations and recombinations [60, 61]. The illustration of such a mechanism working at the intra-genus level has been provided by cultivated rice species Oryza sativa L. and its wild relative Oryza australiensis [60]. Ultimately, TE-induced structural variations may have a negative effect on the cross-species amplification ability of some of the SSR primers. Indeed, in a recent study Xiao et al. [49] showed that over 70% of the conserved microsatellite loci between E. guineensis and P. dactylifera are located within genic regions of the genome with low TE content, and which are therefore less likely to be submitted to TE-dependent structural variations. More generally, gaining a better understanding of genome structures within the Borassus genus could also help reconcile our results with previous published reports of successful transfer of SSR markers developed from other palm sources to Borassus flabellifer (see references cited in Table 8, Methods section). Indeed, since the genome size of B. flabellifer (7.58 Gb; Jaume Pellicer, unpublished data) is only marginally smaller than that of B. aethiopum, significant differences in genome composition may be underlying the lack of SSR transferability between both species.
In any case, from the low number of successfully transferred microsatellite markers we could only identify one displaying polymorphism in our B. aethiopum test panel, making it impossible to rely on for analysis of genetic diversity. Still, the fact that so little microsatellite polymorphism (2 out of 18 amplifying primer pairs: 11.1%) could be detected in this subset of 20 palms sampled across different locations throughout Benin is somewhat surprising and its reasons remain to be elucidated. In addition to possibly being a symptom of habitat fragmentation, this low diversity might also result from the extremely long juvenile phase that has been attributed to this palm species. Indeed, floral maturity has been reported to occur 30 to 50 years after germination [68]. The manner of seed and pollen dispersal, which have so far not been studied extensively in B. aethiopum, might also play a role. Indeed, in pollen-mediated gene flow species, the distance the pollen travel is of importance in the occurrence of crossing between populations [69, 70].
Regarding the development of novel SSR markers, our results are similar to other studies based on the use of high-throughput sequencing techniques in species where very little information is available [22, 71]. We identified 57 microsatellite loci, from which we selected 11 markers displaying polymorphism that were used to assess the genetic structure of B. aethiopum sampled from different sites in Benin. We find low genetic diversity, with an average He value (0.354) that is substantially below those reported for B. flabellifer (0.417) [45] and for other non-timber forest products such as Khaya senegalensis (0.53) [72] and Phyllanthus sp. (0.607 and 0.582 for Phyllanthus emblica and Phyllanthus indofischeri respectively [73]. The positive F value that we observed in the majority (6 out of 9) of locations in the present study indicates an overall deficiency of heterozygotes across sites. This deviation from the Hardy-Weinberg equilibrium (HWE) might reflect low gene flow through pollen and seed dissemination, leading to crosses between related individuals, as supported by the low average number of migrants between sites. Accordingly, our data reveal limited genetic distances among collection sites, with values that are lower than those reported for others palm species. Indeed for B. flabellifer, genetic distances ranged from 0.716 to 0.957 [74] and among natural E. guineensis accessions an average of 0.769 was observed [75]. Both our Fst values and AMOVA analysis point to intra-site differentiation as being the main source of genetic variation.
As illustrated by the global agreement between our PCoA and Bayesian analyses, Beninese B. aethiopum samples cluster into two main groups that are mostly dependent on geo-climatic regions and geographic distances between collection sites, although the correlation between genetic and geographic distance is poorly significant. There might be further genetic separation between Southern B. aethiopum samples and those from the Central sites of Agoua and Biguina, resulting in the splitting of one group into two subgroups. However, with our current dataset it is not possible to achieve this level of discrimination in our analyses. Additional sampling campaigns from intermediate locations in the Central and Northern regions will be necessary in order to make progress on the subject.
Among the nine locations studied in Benin, samples from Savè appear to be the most diversified (He = 0.451) and constitute the exception to the general distribution according to geographical distances. This site located in the Sudano-Guinean transition zone of Benin is currently the most active for the production of B. aethiopum hypocotyls, and it acts as a supplier for the whole national territory ([76]; V.K. Salako, personal communication), suggesting that it might be the largest population of B. aethiopum in the country. Moreover, individuals sampled in Savè appear to be genetically distinct from those sampled in other locations of the Central region and closer to those originating from the Northern region, despite the considerable geographical distances involved in the latter case. A part of the explanation for the genetic distance observed between the Western (i.e. Biguina and Agoua) and the Eastern (Savè) collection sites within the Central region may reside in their physical separation by the Ouémé river, which further forms a natural corridor between Savè and the sites of Trois Rivières and Malanville in the North-East (see Fig. 1) [77]. We postulate that seed dispersal by humans and/or animals along this corridor might have played a major role in the observed pattern of genetic diversity and explain the singularity observed in Savè. As a matter of fact, members of the Bariba ethnic group, who live in the Eastern part of the country up to Malanville, share strong historical ties with the Shabè people from Savè, and exchanges between both groups are frequent [78]. The same corridor is also used annually for transhumance by the Fulani people [79], for whom B. aethiopum is an important plant: the role of their mobility in the dispersal of the plant, similar to what has been proposed for Caesalpina bonduc [80], is therefore plausible. Regarding the impact of animal migrations, Salako et al. [31, 32] detected the presence of B. aethiopum seeds in elephant dungs and hypothesized that elephants may have played important role in the seed dissemination for this species through fruit consumption and long-distance herd migrations. In support to this assumption, Savè is part of a continuous forest corridor connecting with the Northern region that was likely used by elephants in their migrations. Up until 1982, the seasonal occurrence of the animal has been reported in the Wari-Maro forest of Central Benin [81].
The specific microsatellite markers developed in this study from the partial genomic sequencing of B. aethiopum appear to be efficient to assess the genetic diversity and population structure of this species. Additionally, and provided that genome divergence is not too extensive to allow marker transferability, our SSR markers may also been used in a palm species that belongs to the same genus and that is reported to share parts of its distribution area, namely Borassus akeassii B.O.G., which has long been confused with B. aethiopum due to its similar morphology [82]. High-throughput sequencing techniques are an effective way of developing new microsatellite markers in plant species without significant molecular data. The increasing technical performances and financial affordability of these technologies make it feasible to overcome the difficulties arising in case studies such as ours, where marker transfer was proved to be limited or ineffective.
Conclusions
To our knowledge, the data presented in the present article constitute the first sizeable molecular resource available for Borassus aethiopum, which we have made available to the scientific community at large in order to facilitate the implementation of an increasing number of studies on this palm species. Using 11 newly identified SSR markers, we have also performed the first analysis of the genetic diversity of B. aethiopum in an African country, which we see as a first step towards the elaboration of an evidence-based strategy for sustainable resource management and preservation in Benin. Our results support the hypothesis that pollen and seed dispersal mainly occur within sites, leading to crosses among related individuals. The exception to this general rule in the region of Savè (Center) seems to indicate long-range transfer of genes as a result to animal and/or human movements towards and from forest reserves of the North. Further research into the characteristics of these migrations and their impact on gene flow among B. aethiopum populations is required in order to confirm this assumption. As a complement to the present work, the acquisition of agro-morphological data is currently under way, in a bid to elucidate the reproductive development and breeding system of the species. As a longer-term perspective, we also plan to extend our analysis of B. aethiopum diversity to the West African sub-region, and leverage the data acquired to improve knowledge of other species within the Borassus genus, and of palms diversity as a whole.
Methods
Plant material sampling and DNA extraction
Samples of Borassus aethiopum were collected from wild populations in nine distinct sites (three located in protected forest areas, six in farmlands) that were distant from each other by at least 50 km and which spanned the three main climatic regions encountered in Benin (Fig. 1). According to White [83], Benin covers three contrasted climatic regions which are the Sudanian region in the North, the Sudano-Guinean region in the Center and the Guineo-Congolian region in the South. Along a South-North gradient, the rainfall regime switches from bimodal to unimodal, the climate becomes globally drier [29] and the density of B. aethiopum distribution increases [31]. At each location, young leaves from 10 male and 10 female adult trees separated by at least 100 m were collected and stored in plastic bags containing silica gel until further processing. The complete list of samples and their characteristics is available in Additional file 2.
Genomic DNA was extracted from 250 mg of leaves ground to powder under liquid nitrogen using the Chemagic DNA Plant Kit (Perkin Elmer, Germany), according to the manufacturer’s instructions on a KingFisher Flex™ (Thermo Fisher Scientific, USA) automated DNA purification workstation. Final DNA concentration was assessed fluorometrically with the GENios Plus reader (TECAN) using bis-benzimide H 33258 (Sigma-Aldrich) as a fluorochrome.
Transferability of palms microsatellite markers: selection and amplification
A total of 80 SSR markers from previous studies were selected for assessment of their transferability to B. aethiopum: 44 developed for Phoenix dactylifera [67]; 25 developed for Elaeis guineensis [44, 62]; and 11 developed for Cocos nucifera [65]. The respective sequences and origins of these primer sets are displayed in Table 8.
Transferability of the 80 palm SSR markers was assessed on a representative subset of 20 B. aethiopum individuals sampled at the different locations, plus four positive controls from each.
source species for these markers (i.e. P. dactylifera, C. nucifera, and E. guineensis). Microsatellite amplification was performed with a modification of the M13-tailed Primers protocol [63] adapted to the use of fluorescent labelling [64]. The PCR reaction was performed on 20 ng of leaf DNA in volume of 20 μL with the following final concentrations or amounts: 1X PCR buffer, 200 μM dNTP, 2 mM MgCl2, 0.4 pmol M13-tailed forward primer, 4 pmol M13 primer, (5′-CACGACGTTGTAAAACGAC-3′) fluorescently labeled at the 5′ end with FAM, HEX or TAMR, 4 pmol reverse primer, and 0.5 U of KAPA Taq polymerase (Sigma-Aldrich). The following program was used: 3 min of initial denaturation at 95 °C, followed by 35 cycles of 30 s at 95 °C, 30 s at 50 °C and 72 °C for 1 min and a final extension at 72 °C for 5 min. The resulting amplification products were then diluted to 1/10th, mixed with 0.5 μL of an internal size standard (GeneScan 500 ROX, Thermo Fisher Scientific), and denatured for 5 min at 94 °C prior to separation through capillary electrophoresis on an Applied Biosystems 3500 Genetic Analyzer (Thermo Fisher Scientific). Amplification products visualization was performed using the GeneMapper software version 3.7 (Applied Biosystems).
De novo identification of microsatellite loci in the B. aethiopum genome, marker selection and diversity analysis
One B. aethiopum leaf sample (originating from the Togbin site) was randomly selected and used for genomic DNA purification according to the protocol of Mariac et al. [84]. The DNA was then used for the construction of an Illumina paired-end library, as described in Mariac et al. [85], before high-throughput sequencing on a MiSeq v3 platform (Illumina; average read size 250 bp). Demultiplexing of the raw data output was performed using the Maillol script (https://github.com/maillol/demultadapt), with a 0-mistmatch threshold. Adapters were eliminated using Cutadapt version 1.10 [86]. (http://code.google.com/p/cutadapt/) with the following parameters: overlap length = 7, minimum length = 35 and quality = 20. High-quality reads (Q > 30) were filtered using the following script: https://github.com/SouthGreenPlatform/arcad-hts/blob/master/scripts/arcad_hts_2_Filter_Fastq_On_Mean_Quality.pl and the resulting filtered reads were deposited into GenBank under BioProject ID PRJNA576413. Paired-end reads were then merged using FLASH version 1.2.11 (https://github.com/SouthGreenPlatform/arcad-hts/blob/master/scripts/arcad_hts_3_synchronized_paired_fastq.pl). Finally, microsatellite motif detection and specific primer design were carried out after elimination of redundant sequences using the QDD software version 3.1.2 [87] with default settings (detailed in Additional file 3).
Using selected primer pairs, test amplifications were performed with two randomly selected B. aethiopum DNA samples, then primers showing successful amplification were further tested for polymorphism detection among seven randomly selected DNA samples. The M13 Tailed Primers protocol described previously was used, with the following program: 3 min of initial denaturation at 95 °C, followed by 35 cycles of 30 s at 95 °C, 30 s at 55 °C and 72 °C for 1 min and a final extension at 72 °C for 5 min. Separation and vizualization of amplification products were performed as described previously. Finally, the primer pairs enabling successful and unambiguous amplification of polymorphic bands were used for the analysis of genetic diversity among the complete set of 180 B. aethiopum individuals under the same PCR conditions.
Data analysis
Amplification products were scored using the GeneMapper software version 3.7 (Applied Biosystems) and only unambiguous amplification products were considered for data analysis. Genetic diversity parameters were calculated for each locus and each sampling location using the GenAlEx software version 6.502 [88]. Expected heterozygosity (He) was calculated using the formula:
where pi is the frequency of each allele. The fixation index (F) was calculated as:
where Ho is observed heterozygosity and He is expected heterozygosity [89].
F-statistics analysis assessing genetic differentiation (Fst), genetic identity, number of migrants (Nm) [90] and non-hierarchical analysis of molecular variance (AMOVA) for estimating genetic differentiation within and among locations were performed with the same software. Allelic richness was calculated using the SPAGeDi software version 1.5 (http://ebe.ulb.ac.be/ebe/SPAGeDi.html [91];). Consecutively to K determination (see below), successive hierarchical AMOVA analyses were carried out with K = 2 and K = 3. The Mantel permutation test was used for assessing the correlation between genetic and geographic distances between sampling sites [92, 93]. Two Principal Coordinates Analyses (PCoA) enabling the visualization of genetic variation distribution across individuals and sampling sites, respectively, were performed using GenAlEx.
The STRUCTURE software version 2.3.4 [94] was used for the determination of the most probable number of clusters for population structure (K value). Using the admixture model, eight simulations were performed for each inferred K value, with a running length composed of 300,000 burn-in periods and 50,000 Markov chain Monte Carlo (MCMC) replicates. The output from this analysis was then used as input in the Structure HARVESTER online program version 0.6.94 (http://taylor0.biology.ucla.edu/structureHarvester/) to determine the optimal value of K using the ΔK method of Evanno et al. [95] and allowing for different estimates of K in accordance with Janes et al [50]. Based on the resulting values of K, a clustering analysis of the studied sampling sites was performed and graphical output was generated using CLUMPAK’s main pipeline (http://clumpak.tau.ac.il [96];). In order to further assess genetic clustering, a UPGMA tree based on Fst values using 1000 bootstrap replications was constructed using the POPTREE2 software [97].
Availability of data and materials
Data generated from genome sequencing (filtered reads) were deposited into GenBank under BioProject ID PRJNA576413. Capillary electrophoresis profiles are available upon reasonable request to the Corresponding Author. All other data generated or analyzed during this study are included in this published article (and its supplementary information files).
Abbreviations
- AMOVA:
-
Analysis of molecular variance
- F:
-
Fixation index
- Fst :
-
Inter-population genetic differentiation coefficient
- He:
-
Expected Heterozygosity
- Ho:
-
Observed Heterozygosity
- HWE:
-
Hardy-Weinberg equilibrium
- Na:
-
Average number of different alleles
- Ne:
-
Effective number of alleles
- Nm :
-
Number of migrants
- PCoA:
-
Principal coordinate analysis
- SSR:
-
Simple sequence repeat
- UPGMA:
-
Unweighted pair-group method with arithmetic mean
References
Fan L, Zhang M-Y, Liu Q-Z, Li L-T, Song Y, Wang L-F, et al. Transferability of newly developed pear SSR markers to other Rosaceae species. Plant Mol Biol Rep. 2013;31:1271–82.
Oliveira EJ, Pádua JG, Zucchi MI, Vencovsky R, Vieira MLC. Origin, evolution and genome distribution of microsatellites. Genet Mol Biol. 2006;29:294–307.
Huang XQ, Cöster H, Ganal MW, Röder MS. Advanced backcross QTL analysis for the identification of quantitative trait loci alleles from wild relatives of wheat (Triticum aestivum L.). Theor Appl Genet. 2003;106:1379–89.
Moon HSMS, Nicholson JSNS, RSLS L. Use of transferable Nicotiana tabacum L. microsatellite markers for investigating genetic diversity in the genus Nicotiana. Genome. 2008. https://doi.org/10.1139/G08-039.
Muriira NG, Muchugi A, Yu A, Xu J, Liu A. Genetic diversity analysis reveals genetic differentiation and strong population structure in Calotropis plants. Sci Rep. 2018;8:7832.
Mohamed A, García-Martínez S, Loumerem M, Carbonell P, Ruiz JJ, Boubaker M. Assessment of genetic diversity among local pea (Pisum sativum L.) accessions cultivated in the arid regions of Southern Tunisia using agro-morphological and SSR molecular markers. Genet Resour Crop Evol. 2019;66:1189–203.
Yuan Q-J, Zhang Z-Y, Hu J, Guo L-P, Shao A-J, Huang L-Q. Impacts of recent cultivation on genetic diversity pattern of a medicinal plant, Scutellaria baicalensis (Lamiaceae). BMC Genet. 2010;11:29.
Ellegren H. Microsatellite mutations in the germline:: implications for evolutionary inference. Trends Genet. 2000;16:551–8.
Kalia RK, Rai MK, Kalia S, Singh R, Dhawan AK. Microsatellite markers: an overview of the recent progress in plants. Euphytica. 2011;177:309–34.
Nadeem MA, Nawaz MA, Shahid MQ, Doğan Y, Comertpay G, Yıldız M, et al. DNA molecular markers in plant breeding: current status and recent advancements in genomic selection and genome editing. Biotechnol Biotechnol Equip. 2018;32:261–85.
Satya P, Paswan PK, Ghosh S, Majumdar S, Ali N. Confamiliar transferability of simple sequence repeat (SSR) markers from cotton (Gossypium hirsutum L.) and jute (Corchorus olitorius L.) to twenty two Malvaceous species. 3. Biotech. 2016;6:65.
Wang C, Jia G, Zhi H, Niu Z, Chai Y, Li W, et al. Genetic diversity and population structure of chinese foxtail millet [Setaria italica (L.) Beauv.] landraces. G3: genes, genomes. Genetics. 2012;2:769–77.
Squirrell J, Hollingsworth PM, Woodhead M, Russell J, Lowe AJ, Gibby M, et al. How much effort is required to isolate nuclear microsatellites from plants? Mol Ecol. 2003;12:1339–48.
Mnejja M, Garcia-Mas J, Audergon J-M, Arús P. Prunus microsatellite marker transferability across rosaceous crops. Tree Genet Genomes. 2010;6:689–700.
Wünsch A. Cross-transferable polymorphic SSR loci in Prunus species. Sci Hortic. 2009;120:348–52.
Yu F, Wang B-H, Feng S-P, Wang J-Y, Li W-G, Wu Y-T. Development, characterization, and cross-species/genera transferability of SSR markers for rubber tree (Hevea brasiliensis). Plant Cell Rep. 2011;30:335–44.
Karaca M, Ince AG, Aydin A, Ay ST. Cross-genera transferable e-microsatellite markers for 12 genera of the Lamiaceae family: cross-genera transferable e-microsatellite markers for Lamiaceae. J Sci Food Agric. 2013;93:1869–79.
Raveendar S, Lee G-A, Jeon Y-A, Lee Y, Lee J-R, Cho G-T, et al. Cross-amplification of Vicia sativa subsp. sativa microsatellites across 22 other Vicia species. Molecules. 2015;20:1543–50.
Pratap A, Gupta S, Tomar R, Malviya N, Maurya R, Pandey VR, et al. Cross-genera amplification of informative microsatellite markers from common bean and scarlet runner bean for assessment of genetic diversity in mungbean (Vigna radiata). Plant Breed. 2016;135:499–505.
Taheri S, Lee Abdullah T, Yusop M, Hanafi M, Sahebi M, Azizi P, et al. Mining and development of novel SSR markers using next generation sequencing (NGS) data in plants. Molecules. 2018;23:399.
Purwoko D, Cartealy IC, Tajuddin T, Dinarti D, Sudarsono S. SSR identification and marker development for sago palm based on NGS genome data. Breed Sci. 2019;69:1–10.
Grubisha LC, Nelson BA, Dowie NJ, Miller SL, Klooster MR. Characterization of microsatellite markers for pinedrops, Pterospora andromedea (Ericaceae), from Illumina MiSeq sequencing. Appl Plant Sci. 2014;2:1400072.
Sun Z-X, Ye L-J, Zhang F, Hu W, Fan D-M, Zhang Z-Y. Development of microsatellite markers for Sargentodoxa cuneata (Lardizabalaceae) using next-generation sequencing technology. Appl Plant Sci. 2016;4:1600003.
Gruca M, Yu W, Amoateng P, Nielsen MA, Poulsen TB, Balslev H. Ethnomedicinal survey and in vitro anti-plasmodial activity of the palm Borassus aethiopum Mart. J Ethnopharmacol. 2015;175:356–69.
Blach-Overgaard A, Svenning J-C, Dransfield J, Greve M, Balslev H. Determinants of palm species distributions across Africa: the relative roles of climate, non-climatic environmental factors, and spatial constraints. Ecography. 2010;33:380–91.
Salako KV, Moreira F, Gbedomon RC, Tovissodé F, Assogbadjo AE, Glèlè Kakaï RL. Traditional knowledge and cultural importance of Borassus aethiopum Mart in Benin: interacting effects of socio-demographic attributes and multi-scale abundance. J Ethnobiol Ethnomedicine. 2018;14:36.
Ouinsavi C, Gbémavo C, Sokpon N. Ecological structure and fruit production of African Fan palm (Borassus aethiopum) populations. Am J Plant Sci. 2011;02:733.
Gbesso F, Nassi KM, Gbesso GHF, Akoegninou A. Utilisation sociale de Borassus aethiopum Mart et de ses habitats dans les Communes de Savè et de Glazoué au Bénin. Int J Bio Chem Sci. 2017;11:1512.
Adomou AC, Agbani OP, Sinsin B. Plantes. In: Neuenschwander P, Sinsin B, Georg G, editors. Protection de la nature en Afrique de l’Ouest: une liste rouge pour le Bénin - Nature conservation in West Africa: red list for Benin. Ibadan: International Institute of Tropical Agriculture (IITA); 2011. p. 21–46.
Gbesso F, Yedomonhan H, Tente B, Akoegninou A. Distribution géographique des populations de rôniers (Borassus aethiopum Mart, Arecaceae) et caractérisation phytoécologique de leurs habitats dans la zone soudano-guinéenne du Bénin. J Appl Biosci. 2014;74:6099.
Salako VK, Assogbadjo AE, Adomou AC, Agbangla C, Glèlè Kakaï RL. Latitudinal distribution, co-occurring tree species and structural diversity of the threatened palm Borassus aethiopum (Arecaceae) in Benin, West Africa. Plant Ecol Evol. 2015;148:335–49.
Salako VK, Houehanou TH, Yessoufou K, Assogbadjo AE, Akoegninou A, Glèlè Kakaï RL. Patterns of elephant utilization of Borassus aethiopum Mart. And its stand structure in the Pendjari National Park, Benin, West Africa. Trop Ecol. 2017;58:425–37.
Montoya D. Habitat loss, dispersal, and the probability of extinction of tree species. Commun Integr Biol. 2008;1:146–7.
Ouédraogo A, Boussim J, Zongo J-D, Guinko S. Caracteristiques morphologiques des roniers (Borassus L.) du Burkina Faso. Etudes flor vég Burkina Faso. 2002;7:37–40.
Dixo M, Metzger JP, Morgante JS, Zamudio KR. Habitat fragmentation reduces genetic diversity and connectivity among toad populations in the Brazilian Atlantic coastal Forest. Biol Conserv. 2009;142:1560–9.
Jump AS, Peñuelas J. Genetic effects of chronic habitat fragmentation in a wind-pollinated tree. Proc Natl Acad Sci U S A. 2006;103:8096–100.
Singh R, Ong-Abdullah M, Low E-TL, Manaf MAA, Rosli R, Nookiah R, et al. Oil palm genome sequence reveals divergence of interfertile species in old and new worlds. Nature. 2013;500:335–9.
Al-Dous EK, George B, Al-Mahmoud ME, Al-Jaber MY, Wang H, Salameh YM, et al. De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nat Biotechnol. 2011;29:521–7.
Al-Mssallem IS, Hu S, Zhang X, Lin Q, Liu W, Tan J, et al. Genome sequence of the date palm Phoenix dactylifera L. Nat Commun. 2013;4. https://doi.org/10.1038/ncomms3274.
He Z, Zhang C, Liu W, Lin Q, Wei T, Aljohi HA, et al. DRDB: an online date palm genomic resource database. Front Plant Sci. 2017;8. https://doi.org/10.3389/fpls.2017.01889.
Huang Y-Y, Matzke AJM, Matzke M. Complete sequence and comparative analysis of the chloroplast genome of coconut palm (Cocos nucifera). PLoS One. 2013;8:e74736.
Xiao Y, Xu P, Fan H, Baudouin L, Xia W, Bocs S, et al. The genome draft of coconut (Cocos nucifera). Gigascience. 2017;6. https://doi.org/10.1093/gigascience/gix095.
Bazzo BR, de Carvalho LM, Carazzolle MF, Pereira GAG, Colombo CA. Development of novel EST-SSR markers in the macaúba palm (Acrocomia aculeata) using transcriptome sequencing and cross-species transferability in Arecaceae species. BMC Plant Biol. 2018;18:276.
Billotte N, Marseillac N, Risterucci A-M, Adon B, Brottier P, Baurens F-C, et al. Microsatellite-based high density linkage map in oil palm (Elaeis guineensis Jacq.). Theor Appl Genet. 2005;110:754–65.
Pipatchartlearnwong K, Swatdipong A, Vuttipongchaikij S, Apisitwanich S. Genetic evidence of multiple invasions and a small number of founders of Asian Palmyra palm (Borassus flabellifer) in Thailand. BMC Genet. 2017;18. https://doi.org/10.1186/s12863-017-0554-y.
Rivera R, Edwards KJ, Barker JHA, Arnold GM, Ayad G, Hodgkin T, et al. Isolation and characterization of polymorphic microsatellites in Cocos nucifera L. Genome. 1999;42:668–75.
Ukoskit K, Chanroj V, Bhusudsawang G, Pipatchartlearnwong K, Tangphatsornruang S, Tragoonrung S. Oil palm (Elaeis guineensis Jacq.) linkage map, and quantitative trait locus analysis for sex ratio and related traits. Mol Breed. 2014;33:415–24.
Wu Y, Yang Y, Qadri R, Iqbal A, Li J, Fan H, et al. Development of SSR Markers for Coconut (Cocos nucifera L.) by Selectively Amplified Microsatellite (SAM) and Its Applications. Trop Plant Biol. 2019;12:32–43.
Xiao Y, Xia W, Ma J, Mason AS, Fan H, Shi P, et al. Genome-wide identification and transferability of microsatellite markers between Palmae species. Front Plant Sci. 2016;7. https://doi.org/10.3389/fpls.2016.01578.
Janes JK, Miller JM, Dupuis JR, Malenfant RM, Gorrell JC, Cullingham CI, et al. The K = 2 conundrum. Mol Ecol. 2017;26:3594–602.
Barbará T, Palma-Silva C, Paggi GM, Bered F, Fay MF, Lexer C. Cross-species transfer of nuclear microsatellite markers: potential and limitations. Mol Ecol. 2007;16:3759–67.
Manju KP, Manimekalai R, Naganeeswaran SA, Arunachalam V, Karun A. Microsatellites mining in date palm (Phoenix dactylifera L.) and their cross transferability across Arecaceae family. Plant Omics. 2016;9:191–7.
Viana MVC, Miranda EA, de Francisco AK, Carvalho CAL, Waldschmidt AM. Transferability of microsatellite primers developed for stingless bees to four other species of the genus Melipona. Genet Mol Res. 2011;10:3942–7.
Zaki NM, Singh R, Rosli R, Ismail I. Elaeis oleifera genomic-SSR markers: exploitation in oil palm germplasm diversity and cross-amplification in Arecaceae. Int J Mol Sci. 2012;13:4069–88.
Nazareno AG, Zucchi MI, dos Reis MS. Microsatellite markers for Butia eriospatha (Arecaceae), a vulnerable palm species from the Atlantic rainforest of Brazil. Am J Bot. 2011;98:e198–200.
Simplicio RR, Pereira DG, Waldschmidt AM. Transferability of microsatellite markers in Syagrus coronata (Mart.) Becc. (Arecaceae), an iconic palm tree from the Brazilian semiarid region. Genet Mol Res. 2017;16. https://doi.org/10.4238/gmr16029699.
Peakall R, Gilmore S, Keys W, Morgante M, Rafalski A. Cross-species amplification of soybean (Glycine max) simple sequence repeats (SSRs) within the genus and other legume genera: implications for the transferability of SSRs in plants. Mol Biol Evol. 1998;15:1275–87.
Rossetto M. Sourcing of SSR markers from related plant species. In: Henry RJ, editor. Plant genotyping: the DNA fingerprinting of plants. Wallingford: CABI; 2001. p. 211–24. https://doi.org/10.1079/9780851995151.0211.
Rival A, Beulé T, Barre P, Hamon S, Duval Y, Noirot M. Comparative flow cytometric estimation of nuclear DNA content in oil palm (Elaeis guineensis Jacq) tissue cultures and seed-derived plants. Plant Cell Rep. 1997;16:884–7.
Piegu B, Guyot R, Picault N, Roulin A, Saniyal A, Kim H, et al. Doubling genome size without polyploidization: dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006;16:1262–9.
Hanin M, Paszkowski J. Plant genome modification by homologous recombination. Curr Opin Plant Biol. 2003;6:157–62.
Billotte N, Risterucci AM, Barcelos E, Noyer JL, Amblard P, Baurens FC. Development, characterisation, and across-taxa utility of oil palm (Elaeis guineensis Jacq.) microsatellite markers. Genome. 2001;44:413–25.
Rao MN, Ramesha BT, Ravikanth G, Ganeshaiah KN, Shaanker RU. Cross-species amplification of coconut microsatellite markers in rattans. Silvae Genet. 2007;56:282–6.
Anitha N, Jayaraj KL, Kumar EPA, George J, Rajesh MK. Assessment of cross-taxa utility of coconut microsatellite markers. Indian J Hortic. 2008;65:317–21.
Meerow AW, Wisser RJ, Brown SJ, Kuhn DN, Schnell RJ, Broschat TK. Analysis of genetic diversity and population structure within Florida coconut (Cocos nucifera L.) germplasm using microsatellite DNA, with special emphasis on the Fiji dwarf cultivar. Theor Appl Genet. 2003;106:715–26.
Perera L, Russell JR, Provan J, McNicol JW, Powell W. Evaluating genetic relationships between indigenous coconut (Cocos nucifera L.) accessions from Sri Lanka by means of AFLP profiling. Theor Appl Genet. 1998;96:545–50.
Aberlenc-Bertossi F, Castillo K, Tranchant-Dubreuil C, Chérif E, Ballardini M, Abdoulkader S, et al. In silico mining of microsatellites in coding sequences of the date palm (Arecaceae) genome, characterization, and transferability. Appl Plant Sci. 2014;2:1300058.
Willemin V, Cabannes Y, Chantry G. Le ronier et le palmier a sucre: production et mise en œuvre dans l’habitat. Paris: GRET; 1987.
Rasmussen IR, Brødsgaard B. Gene flow inferred from seed dispersal and pollinator behaviour compared to DNA analysis of restriction site variation in a patchy population of Lotus corniculatus L. Oecologia. 1992;89:277–83.
Szczecińska M, Sramko G, Wołosz K, Sawicki J. Genetic Diversity and Population Structure of the Rare and Endangered Plant Species Pulsatilla patens (L.) Mill in East Central Europe. PLoS ONE. 2016;11:e0151730.
Yamamoto M, Handa Y, Aihara H, Setoguchi H. Development and characterization of 43 microsatellite markers for the critically endangered primrose Primula reinii using MiSeq sequencing. Plant Divers. 2018;40:41–4.
Gaoue OG, Lemes MR, Ticktin T, Sinsin B, Eyog-Matig O. Non-timber Forest product harvest does not affect the genetic diversity of a tropical tree despite negative effects on population fitness. Biotropica. 2014;46:756–62.
Geethika E, Triveni HN, Srirama R, Siva R, Setty S, Ravikanth G. Development and characterization of microsatellite markers for Phyllanthus emblica Linn., important nontimber forest product species. J Genet. 2018;97:1001–6.
Vinayagam P, Dhandapani J, Raman P, Alagarsamy RK, Muthusamy P, Balaraman SS. Molecular characterization for genetic diversity of Palmyrah (Borassus flabellifer) accessions using inter simple sequence repeat (ISSR) markers. Asian Austral J Plant Sci Biotech. 2009;3:11–5.
Bakoumé C, Wickneswari R, Siju S, Rajanaidu N, Kushairi A, Billotte N. Genetic diversity of the world’s largest oil palm (Elaeis guineensis Jacq.) field genebank accessions using microsatellite markers. Genet Resour Crop Evol. 2014;62:349–60.
Michon L, Adéoti K, Koffi K, Ewedje E, Stauffer FW. Notes on Borassus aethiopum Mart., a multi- purpose palm in Togo and Benin. Palms. 2018;62:57–69.
Moniod F. Régime hydrologique de l’Ouémé (Dahomey). Cahiers ORSTOM Série Hydrologie. 1973;10:171–83.
Affouda EO. Le royaume Shabè et ses rapports socio-ethniques de voisinage jusqu’à la création de la colonie du Dahomey. In: Mama VJ, editor. Symposium culturel de Yaoui - Première édition 2018 - Peuplement du pays Shabè et occupation du site historique Etoo de Yaoui. Bénin: Yaoui; 2018. p. 20–8.
Houessou SO, Dossa LH, Diogo RVC, Houinato M, Buerkert A, Schlecht E. Change and continuity in traditional cattle farming systems of west African coast countries: a case study from Benin. Agric Syst. 2019;168:112–22.
Assogbadjo AE, Fandohan B, Kakaï RG, Kyndt T, Hardy OJ, Gheysen G, et al. Genetic evidence of the contribution of ethnic migrations to the propagation and persistence of the rare and declining scrambling shrub Caesalpinia bonduc L. Hum Ecol. 2012;40:117–28.
Roth HH, Douglas-Hamilton I. Distribution and status of elephants in West Africa. Mammalia. 1991;55:489–527.
Bayton RP, Ouédraogo A, Guinko S. The genus Borassus (Arecaceae) in West Africa, with a description of a new species from Burkina Faso. Bot J Linn Soc. 2006;150:419–27.
White F. The vegetation of Africa: a descriptive memoir to accompany the UNESCO/AETFAT/UNSO vegetation map of Africa. UNESCO. Paris: Natural resources research; 1983.
Mariac C, Luong V, Kapran I, Mamadou A, Sagnard F, Deu M, et al. Diversity of wild and cultivated pearl millet accessions (Pennisetum glaucum [L.] R. Br.) in Niger assessed by microsatellite markers. Theor Appl Genet. 2006;114:49–58.
Mariac C, Scarcelli N, Pouzadou J, Barnaud A, Billot C, Faye A, et al. Cost-effective enrichment hybridization capture of chloroplast genomes at deep multiplexing levels for population genetics and phylogeography studies. Mol Ecol Resour. 2014;14:1103–13.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 2011;17:10–2.
Meglécz E, Costedoat C, Dubut V, Gilles A, Malausa T, Pech N, et al. QDD: a user-friendly program to select microsatellite markers and design primers from large sequencing projects. Bioinformatics. 2010;26:403–4.
Peakall R, Smouse PE. GenAlEx 6: genetic analysis in excel. Population genetic software for teaching and research. Mol Ecol. 2006;6:288–95.
Hartl DL, Clark AG. Principles of population genetics. 3rd ed. Sunderland: Sinauer Associates Incorporated; 1997. https://www.cabdirect.org/cabdirect/abstract/19980108046.
Wright S. Evolution and the genetics of populations. Chicago: University of Chicago Press; 1969. https://press.uchicago.edu/ucp/books/book/chicago/E/bo5961634.html. Accessed 5 Nov 2020.
Hardy OJ, Vekemans X. SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes. 2002;2:618–20.
Slatkin M. Isolation by distance in equilibrium and non-equilibrium populations. Evolution. 1993;47:264–79.
Slatkin M. Gene flow and the geographic structure of natural populations. Science. 1987;236:787–92.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol. 2005;14:2611–20.
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I. CLUMPAK: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour. 2015;15:1179–91.
Takezaki N, Nei M, Tamura K. POPTREE2: software for constructing population trees from allele frequency data and computing other population statistics with windows Interface. Mol Biol Evol. 2010;27:747–52.
Acknowledgements
The Authors wish to thank Emira Cherif, Karina Castillo and Frédérique Aberlenc (IRD, UMR DIADE, Montpellier, France) for sharing their unpublished microsatellite markers data for transferability tests, and Dr. Jaume Pellicer (Comparative Plant & Fungal Biology, Jodrell Laboratory, Royal Botanical Gardens, Kew, UK), for allowing us to use the palm flow cytometry data.
Funding
The work described in this article was funded through travel grants to MJK and KA under the framework of the MooSciTIC project granted to EJ by Agropolis Fondation (ID 1501–011, “Investissements d’Avenir” Program - Labex Agro: ANR-10-LABX-0001-01). Additional funding was provided by the Sud Expert Plantes - Développement Durable (SEP2D) programme to KA (GenPhyB project, ID AAP3–64). MJK is the recipient of a PhD fellowship from the French Embassy in Benin. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
KVS, EJ, TB and KA conceived and designed the experiments and were responsible for funding acquisition. All authors were involved in defining the experimental strategy. MJK, SS, ML, CTC, KA, KVS performed the experiments. MJK, KA, SS, CM, LZ and TB processed and analyzed data. All authors contributed to writing and revising the manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
In accordance with the Nagoya Protocol on Access and Benefit Sharing (ABS), a field permit allowing access and non-commercial use for research purposes of the plant material used in the present study has been submitted to the competent national authority (Direction Générale des Eaux, Forêts et Chasse/Ministère du Cadre de Vie et du Développement Durable, Benin).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
List and characteristics of putative microsatellite loci identified in the genome of Borassus aethiopum through QDD analysis. Characteristics (basic motif, length) of the microsatellite loci and of the primer pairs (length, position, Tm, stability, amplicon size and sequence) designed for their targeted amplification.
Additional file 2.
List of sampled Borassus aethiopum individuals. M, F: male or female palm, respectively. All geographic coordinates are provided as North from the Equator (latitude) and East from the Greenwich meridian (longitude), respectively.
Additional file 3.
Default QDD software settings. Parameters used in the QDD detection of microsatellite loci.
Additional file 4.
Results of the Bayesian cluster analysis with variable values of K. Graphical summary generated from STRUCTURE results by CLUMPAK’s main pipeline with values of K ranging from 1 to 10.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kpatènon, M.J., Salako, K.V., Santoni, S. et al. Transferability, development of simple sequence repeat (SSR) markers and application to the analysis of genetic diversity and population structure of the African fan palm (Borassus aethiopum Mart.) in Benin. BMC Genet 21, 145 (2020). https://doi.org/10.1186/s12863-020-00955-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-020-00955-y