
Abstract
Gliomas are the most lethal primary brain tumors in adults. These highly invasive tumors have poor 5-year survival for patients. Gliomas are principally characterized by rapid diffusion as well as high levels of cellular heterogeneity. However, to date, the exact pathogenic mechanisms, contributing to gliomas remain ambiguous. MicroRNAs (miRNAs), as small noncoding RNAs of about 20 nucleotides in length, are known as chief modulators of different biological processes at both transcriptional and posttranscriptional levels. More recently, it has been revealed that these noncoding RNA molecules have essential roles in tumorigenesis and progression of multiple cancers, including gliomas. Interestingly, miRNAs are able to modulate diverse cancer-related processes such as cell proliferation and apoptosis, invasion and migration, differentiation and stemness, angiogenesis, and drug resistance; thus, impaired miRNAs may result in deterioration of gliomas. Additionally, miRNAs can be secreted into cerebrospinal fluid (CSF), as well as the bloodstream, and transported between normal and tumor cells freely or by exosomes, converting them into potential diagnostic and/or prognostic biomarkers for gliomas. They would also be great therapeutic agents, especially if they could cross the blood–brain barrier (BBB). Accordingly, in the current review, the contribution of miRNAs to glioma pathogenesis is first discussed, then their glioma-related diagnostic/prognostic and therapeutic potential is highlighted briefly.
Similar content being viewed by others


Background
Noncoding RNAs (ncRNAs) are a group of RNA molecules that either have a limited ability to encode a protein or lack it. Currently, ncRNAs are classified into two major groups of long noncoding RNAs (> 200 nts) and small noncoding RNAs (18–200 nts), according to their length. The family of small noncoding RNAs consists of small nucleolar RNAs (snoRNAs), Piwi-interacting RNAs (piRNAs), small nuclear RNAs (snRNAs), tRNA-derived small RNA (tsRNA), and microRNAs (miRNAs). NcRNAs were previously thought to be genomic dark matter, but studies have shown that these structures make up about 60% of the human genome [1]. To date, numerous evaluations have also revealed that ncRNAs involve in regulating the physiological and pathological processes of many human diseases, including multiple cancers [2].
miRNAs, as noncoding, small endogenous RNA molecules with an average length of 20–22 nucleotides, are widely distributed and highly conserved, with cell- and tissue-specific expression patterns. These RNA molecules principally contribute to the regulation of transcription, translation, or epigenetic modification of various genes [3,4,5]. The discovery of the first miRNA, lin-4, in Caenorhabditis elegans by the research teams of Ambros and Ruvkun was a milestone in molecular biology. Since then, several investigations have focused on this topic, providing evidence for the involvement of different miRNAs in biological processes, such as cell proliferation, cellular differentiation, apoptosis, and inflammation [2, 6]. miRNAs can posttranscriptionally regulate gene expression by binding to specific sites, acting as miRNA response elements (MREs), in their target transcripts, which result in transcript eradication or translation suppression. More interestingly, the expression levels of miRNAs may be deregulated in multiple cancers, converting them into oncogenes or tumor suppressors.
miRNAs have been reported to be associated with the formation and development of gliomas [7]. Gliomas are known to be the most lethal and common malignant brain tumors, having poor prognosis. Unfortunately, the 5-year survival for patients diagnosed with glioblastoma is only 10%. Through affecting the onset and progression of gliomas, miRNAs regulate the expression of particular genes involved in cancer-related processes. Considering the expression profile of miRNAs in glioblastoma, it has been proposed that miRNAs can impact genes, regulating cell proliferation, invasion, angiogenesis, apoptosis, and even chemoresistance. Furthermore, several miRNAs have been shown to exhibit tumor-suppressive effects in glioblastoma pathophysiology. Surprisingly, miRNAs have also been considered to be suitable tools for identifying the origin of gliomas, as well as improving the prognosis of patients and monitoring their response to treatment. Moreover, these ncRNAs have also been suggested to function as appropriate targets for glioma therapy [8, 9].
In this context, the current review aims to discuss the substantial relationship between particular miRNAs and the pathogenesis and progression of gliomas, with a special focus on their roles in the diagnosis and treatment of glial tumors. Although a significant number of reviews have highlighted the role of miRNAs in the pathogenesis of gliomas, while others have discussed the therapeutic/diagnostic significance of miRNAs, no recently published review has focused on both the pathogenesis and diagnostic/therapeutic aspects. To the best of the authors’ knowledge, this review thus provides an update that focuses on the role(s) of miRNAs in gliomas, from pathogenesis to diagnosis and treatment. To achieve the objective, the most recent publications in the field are reviewed and fully covered (Table 1).
Gliomas
Gliomas are referred to as primary central nervous system (CNS) (esp. brain) tumors, resulting in substantial degrees of morbidity and mortality owing to their origin and locally invasive growth. These brain tumors represent approximately 30% of all primary CNS tumors, and 80% of all malignant ones. Based on their histological and immunobiological characteristics, gliomas can be categorized into astrocytomas, brain stem gliomas, ependymomas, mixed gliomas (so-called oligoastrocytomas), oligodendrogliomas, and glioblastomas [10]. In 2016, the updated version of the World Health Organization (WHO) grading system classified brain tumors principally based on the absence or presence of anaplastic features into four different grades from I to IV, including nuclear atypia, mitotic activity, microvascular proliferation, and/or necrosis. However, gliomas are classified as either low-grade (grades I or II) or high-grade (grades III and IV) tumors, depending on their growth potential and invasiveness [11].
Astrocytomas are slow-growing tumors that originate from astrocytes. These tumors can be either circumscribed, pilocytic astrocytomas, or infiltrative, diffuse astrocytomas. Histologically, pilocytic astrocytomas exhibit low to moderate cellularity and reduced mitotic activity, whereas diffuse tumors are characterized by a moderate increase in cellularity, mild to moderate nuclear atypia, and low degrees of mitotic activity. Oligodendrogliomas are generally low-grade, slow-growing tumors that develop from oligodendrocytes. Nevertheless, grade III oligodendrogliomas, which are also called anaplastic oligodendrogliomas, are considered to be malignant, fast-growing tumors [12]. Low-grade oligodendrogliomas have a relatively low proliferative index and well-delineated borders, while high-grade anaplastic oligodendrogliomas exhibit increased cell proliferation, nuclear atypia, and an altered rate of mitotic activity. On the other hand, tumors originating from ependymal cells can be classified into three distinct subgroups based on the WHO classification; grade I, including sub-ependymomas and myxopapillary; grade II, as slow-growing benign tumors; grade III, as invasive malignant cancer cells [13].
The most common gliomas in adults are the most infiltrative ones, including diffuse astrocytomas (WHO grade II), anaplastic astrocytomas (WHO grade III), glioblastomas (WHO grade IV), oligodendrogliomas (WHO grade II), and the controversial group of mixed oligoastrocytomas. Other tumors, such as pilocytic astrocytomas, pleomorphic xanthoastrocytomas, and ependymomas, are less common and have more favorable prognosis. In children, gliomas most commonly present in the form of pilocytic astrocytomas and diffuse midline gliomas in multiple grades, including diffuse intrinsic pontine gliomas [11, 13]. Nevertheless, respective studies of CNS tumors, such as ependymomas, have reported that patients cannot be reliably classified by employing only histological classifications, meaning that tumors with similar histological grades may result in substantially different survival outcomes. The fifth edition of the WHO classification of CNS tumors in 2021 employed a histo-molecular-based approach with confirmed prognostic and therapeutic advantages [14]. Indeed, the newly edited guideline in 2021 enhanced the previous classification system by employing novel molecular diagnostics, such as DNA methylome profiling [14].
Glioblastoma multiforme (GBM), the most common and mortal subtype of glioma, even after routine therapeutic methods, including resection surgery, followed by radiochemotherapy, has an average survival of approximately 6–12 months from detection [15, 16]. GBM is commonly categorized into two different clinical subtypes: primary and secondary. Approximately 95% of GBMs are primary, typically occurring de novo within a period of 3–6 months in the elderly. Meanwhile, secondary GBMs usually develop in younger patients with positive history of low-grade astrocytoma. Although the primary and secondary subtypes differ at the molecular level, their outcomes are almost identical because the same pathways are usually affected, thus the response to treatment also remains the same [15].
In the case of glioma therapy, which is guided by the WHO classification system, surgery is generally required, by either complete resection or a biopsy [15]. However, the treatment of patients with gliomas consists of a combination of resection, radiotherapy, and chemotherapy [17]. The goal of tumor resection is to remove all tumor tissue while minimizing the neurological risk to the patient. In this context, surgical resection has been reported to be more beneficial for glioblastoma than for WHO grade II and III gliomas [17]. The resection of recurrent glioblastoma cannot be beneficial alone and should be performed as part of an overall therapeutic approach. Since most recurrences of glioma arise in the immediate vicinity of the tumor, local radiotherapy of the tumor region or its bed can be employed following surgical resection [16, 18, 19]. In most cases, chemotherapy is also needed to maximize the efficiency of oncological treatment. The standard chemotherapeutic agent for gliomas is temozolomide (TMZ), while a combination of procarbazine, lomustine, and vincristine can be used for primary chemotherapy, as well as the treatment of recurrent gliomas [20, 21]. Although neurooncological studies are currently working on immunotherapeutic strategies and several trials have focused on these therapeutic approaches, no significantly positive outcomes have been obtained yet [17].
In the case of glioma detection, the final diagnosis is obtained by direct tissue assessment of glial tumors through surgical biopsies, which has inevitable risks. Thus, noninvasive strategies for the identification of brain tumors have attracted great attention to limit biopsy-related risks [22]. Although the BBB presents some challenges to accessing genetic material, some molecules have been reported to be secreted from tumor cells into the peripheral blood and the CSF of patients with glioblastoma [23]. miRNAs are perfect molecules in this context, with the ability to be secreted from tumor cells within extracellular vesicles [8, 24]. These ncRNAs are also reported to have particular regulatory roles in the tumor microenvironment [8, 22]. Therefore, miRNAs could be considered as potential diagnostic means, as well as promising therapeutic targets for the management of gliomas. The following sections provide much information about miRNAs and their functions in physiological as well as cancerous conditions.
miRNAs
miRNAs are endogenous small single-stranded ncRNAs that are about 17–25 nucleotides long, involved in gene modulation, as well as epigenetic modifications. Given their prominent roles in many biological events, the potential of miRNAs as novel diagnostic, prognostic, and therapeutic targets has been constantly studied [25,26,27]. More than 50% of the genes responsible for the biogenesis of miRNAs are located at fragile chromosomal sites or cancer-associated genomic regions, suggesting key roles for miRNAs in the development of human malignancies [28]. Moreover, miRNA dysregulation is implicated in the progression of cancers by disrupting the expression of tumor suppressors and/or oncogenes [28, 29].
Molecular analyses have demonstrated that almost 50% of all human miRNAs are intragenic, originated from introns, while only a few derive from exons of protein-coding transcripts. The rest of miRNAs are generated from intergenic noncoding precursor-miRNA transcripts that are strictly regulated by their promoters [28, 30]. Note that miRNAs can regulate gene expression levels not only through translational repression but also by degradation of target mRNAs [31]. The biogenesis of miRNAs is a process that takes place in both the nucleus and the cytoplasm [32]. The initial step of transcription of miRNA genes in the nucleus is mostly done by RNA polymerase II to primary transcripts (pri-miRNAs) that are polyadenylated and have a cap [33, 34]. The pri-miRNAs generated may have several hairpin-like structures. These structures are then processed into characteristic stem-loop precursor miRNAs that are approximately 70 nucleotides in length and then modified by Drosha ribonuclease III in the microprocessor complex and its binding partner DGCR8 to the pre-miRNAs. Pre-miRNAs produced by the Ran-GTP/exportin 5 complex leave the nucleus for the cytoplasm [35]. In the cytoplasm, the structure of the Pre-miRNAs stem loop is further cleaved by the other RNase III enzyme, Dicer, and its cofactors, TAR RNA-binding protein (TRBP), resulting in duplex miRNAs of approximately 22 base pairs (bp). One strand of miRNA is rapidly degraded, while the other strand (the guide strand) is loaded into the RNA-induced silencing complex [36]. The RISC is a multiprotein assemblage of the Argonaute [37] family of proteins that along with mature miRNA, is essential for miRNA-mediated gene silencing. The generated miRNA–RISC complex binds to the target 3′-untranslated region (UTR) mRNA by identifying specific binding sites. Then, depending on the degree of complementarity of the linkages between miRNA and the target mRNA, effects such as translation inhibition or cleavage of mRNA will occur [38,39,40] (Fig. 1).

Processing of miRNAs, from biogenesis to therapeutic strategies. miRNAs are transcribed from particular genes inside the nucleus through the action of RNA polymerase II. After the formation of pri-miRNA, this is developed to pre-miRNA by the Drosha–DGCR complex. Then, the pre-miRNA leaves the nucleus for the cytoplasm via exportin 5. Following particular cytoplasmic processing and the action of the Dicer–TRBP complex, pre-miRNA is converted into duplex miRNA, which then undergoes an unwinding to produce mature miRNA. Mature miRNA, using the Ago-2 protein, forms a complex with RISC to cleave the mRNA of interest, or suppresses the translation process. In the case of therapeutic strategies attributed to miRNAs, there are four distinct strategies: A AntagomiRs that bind to and inhibit the action of oncomiRs by blocking miRNA-to-mRNA attachment, through a process called antisense action. AntagomiRs are also responsible for further degradation of miRNAs; B miRNA mimics, which help anticarcinogenic miRNAs to induce tumor-suppressive activities by reversing the epigenetic silencing; C miRNA masks that prevent miRNAs from acting on mRNAs by masking the 3′-UTR sequence on the mRNA strand; D miRNA sponges, whose behavior prevents miRNAs from acting on mRNAs by occupying the binding sites of a particular miRNA or even a set of miRNAs with similar seed sequences by a complementary RNA sequence. Ago2 Argonaute RISC catalytic component 2; DGCR DiGeorge syndrome critical region; GTP guanosine triphosphate; RISC RNA-induced silencing complex; RNA poly RNA polymerase; TRBP TAR RNA binding protein
Specifically, miRNA-mediated gene expression control is crucial for the cellular response to environmental stresses, thereby being implicated in human diseases such as cancers. Hence, the expression of miRNAs may be altered as disease progresses. Furthermore, among all small ncRNAs, miRNAs are the most well-studied RNA molecules as promising biomarkers for diseases such as cancers, aging, and neurodegenerative disorders [41].
miRNAs in cancer
Several studies have shown that miRNAs play fundamental roles not only in the normal functions of cells but also in the pathogenesis of malignant diseases. Indeed, miRNAs can behave as tumor suppressor or oncogene molecules via regulation of other protein-coding genes [28].
So far, multiple large-scale studies have demonstrated that miRNA profiling of cancers may be beneficial for staging, prognosis, and monitoring the therapeutic response. Microarray investigations on human cancers, cell lines, and nonmalignant cells have revealed 217 miRNAs involved in the tumorigenesis of multiple human malignancies [42]. Mounting evidence also indicates an important role for miRNAs in the development of cancers including breast, prostate, lung, pancreatic, and colon cancers, retinoblastoma, and glioblastoma [43]. The mechanisms of miRNA dysregulation in these malignancies include chromosomal abnormalities, amplification or deletion of miRNAs, transcriptional regulator alterations, epigenetic modifications, and alterations in the miRNA biogenesis machinery [28, 43]. Deletion or downregulation of two miRNA genes, miR-15a/16-1, which frequently occurs in patients with chronic lymphocytic leukemia (CLL), was the first evidence of miRNA involvement in human cancers [44].
Additional studies on lung cancer have reported that underexpression of miR-143 and miR-145 occurs as a result of deletion of the 5q33 region [45]. In contrast, the gene cluster miR-17–92 is amplified in lung cancers and B-cell lymphomas, as well as T-cell acute lymphoblastic leukemia [46, 47]. Overexpression of these miRNAs has been suggested to result from miRNA cluster gene translocation.
Since miRNAs are strictly regulated by different transcription factors, altered expression of miRNAs in cancer may be caused by dysregulation of some transcription factors, such as c-Myc and p53 [48]. The proto-oncogene c-Myc plays a fundamental regulatory role in growth control, cell proliferation, differentiation, and apoptosis, and alterations in its expression are associated with many tumors. A growing body of evidence has shown that there is a close interaction between c-Myc and miRNAs, with c-Myc being one of the main regulators of miRNA expression; transcripts of a number of tumor suppressor miRNAs such as miR-15a/16-1, miR-26, miR-29, miR-30, miR-34a, and the let-7 family are repressed by c-Myc [49, 50]. Another example of the regulatory role of c-Myc is reported in aggressive B-cell lymphoma (BCL) and acute myeloid leukemia (AML), where c-Myc is implicated in the repression of miR-26a. Conversely, in GBM, c-Myc upregulates miR-26a expression, promoting tumor cell proliferation [51].
The p53 tumor suppressor continues to be known as the most frequently mutated gene in human cancers. Extensive literature has revealed a close relationship between p53 and miRNAs, with p53 regulating miRNAs at the transcriptional level and promoting the processing/maturation of a group of miRNAs (or certain miRNAs) [52]. Members of the miR-34 family were the first reported and the most prevalent p53-induced miRNAs, in this context [53]. Both 1p36.22 and 11q23.1 chromosome genes, which encode members of the miR-34 family, harbor several p53-responsive elements to which p53 can attach to activate transcription [53]. In human cancers, epigenetic alterations include DNA methylation, histone or chromatin posttranscriptional modifications (PTM), miRNAs, and nucleosome remodeling (or regulation) [54]. Interestingly, miR-127 levels were dramatically increased in cancer cell lines after treatment with DNA methylation, and histone deacetylase inhibitors [55].
Gene mutations and signaling pathways contributing to glioma
In-depth understanding of the molecular characteristics of cancer, genetic and epigenetic abnormalities, and disturbances in signal transduction pathways can provide new insights into the identification and application of molecular biomarkers, as well as the design and development of potential chemotherapeutic approaches [50].
The generally accepted theory is that cancers are principally the result of progressive genetic and epigenetic alterations involved in cell proliferation and homeostasis [56]. Similarly, the pathogenesis of gliomas is also characterized by the sequential accumulation of genetic changes and abnormal regulation of growth factor signaling pathways that eventually lead to malignant transformation [57, 58]. Low-grade gliomas are commonly presented by two genetic abnormalities: (I) mutations in the TP53 tumor suppressor gene (associated with astrocytomas), and concurrent deletion of chromosomes 1p and 19q, seen in oligodendrogliomas [59]. On the other hand, high-grade gliomas exhibit altered expression of any of the p16INK4a, cyclin-dependent kinase 4 (CDK4), or retinoblastoma 1 (RB1) genes, resulting in the loss of normal RB1 function [60, 61].
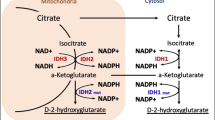
Mutations in isocitrate dehydrogenase (IDH), vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), platelet-derived growth factor (PDGF), and hepatocyte growth factor (HGF) have been recognized to be involved in aberrant proliferation of glioma cells [62]. In general, genes encoding IDH enzymes are commonly mutated in various types of human cancer, including gliomas. Indeed, according to the WHO classification, diffuse astrocytic and oligodendroglial tumors are classified based on IDH mutations. Additionally, IDH mutations are frequently seen in secondary GBM [14, 62].
There are three different isoforms of IDH, which mediate multiple primary cellular metabolic functions; IDH1 is mainly found in the cytoplasm and peroxisomes, while IDH2 and IDH3 are located within the inner mitochondrial membrane [63]. Acquisition of mutant IDH1/2 would result in incomplete reprogramming of cellular metabolism [63]. IDH-mutant enzymes exhibit low affinity for isocitrate, thus preventing the formation of a-ketoglutarate (a-KG). However, these mutations also cause an increased binding affinity for nicotinamide adenine dinucleotide phosphate (NADPH), leading to a considerable increase in cytoplasmic levels of 2-hydroxyglutarate (2-HG), which is an oncometabolite, responsible for tumorigenesis. Indeed, 2-HG itself unloads carbohydrates from the citric acid cycle, making other non-Krebs-cycle sources of metabolites such as glutamine, glutamate, and branched-chain amino acids (BCAA) serve as alternative sources to support cellular metabolism [64, 65]. These findings have substantial implications for cancer therapy, as IDH inhibitors successfully use 2-HG suppression to hinder IDH-mutant glioma cells, which are more sensitive to glutaminase inhibitors [64].
The oncometabolite 2-HG not only changes the tumor metabolism but also involves in epigenetic alterations. In primary gliomas, high levels of 2-HG are associated with increased histone methylation and induction of differentiation [66]. Hence, by altering the status of histone and DNA methylation, IDH mutations alter gene expression patterns, leading to the inhibition of progenitor cell differentiation and the promotion of tumorigenesis along with subsequent oncogenic mutations [66, 67]. In addition, primary GBMs usually exhibit amplification of mouse double minute 2 (MDM2), which is a negative regulator of the tumor suppressor p53, phosphate and tensin homolog (PTEN) mutations, and homozygous deletion involving the CDKN2A tumor suppressor gene, whereas secondary GBMs are more likely to exhibit p53 mutations, IDH mutations, MET amplification, and overexpression of PDGFRA [68].
Signal transduction is a multistep process by which a cell responds to an extracellular stimulus using intracellular signaling molecules, initiated via binding of an extracellular messenger to a cell surface receptor. The signal transduction pathways highly regulate cell proliferation, differentiation, the cell cycle, and cell death, thus playing a central role in the search for molecular targets that could be exploited for therapeutic purposes [69]. Therefore, signal transduction regulates the cellular process via transmembrane receptor kinase, which is activated by growth factors, cytokines, and hormones. These cellular receptors include epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), insulin-like growth factor 1 receptor (IGF-1R), and fibroblast growth factor receptor (FGFR), which all are receptor tyrosine kinases (RTKs) [70]. RTKs are a family of cell surface receptors that can regulate cell proliferation, differentiation, and metabolic pathways as well as the cell cycle [69, 70].
Three main signaling pathways involve in gliomas pathogenesis, including RB1 (retinoblastoma 1 gene), p53 pathway, and signaling mediated by RTK/Ras/phosphoinositide 3-kinase (PI3K) and downstream molecules [71]. Molecular mutations, turning protooncogenes into oncogenes can overactivate these signaling pathways, whereas inactivation of tumor suppressors would remove these crucial negative regulators [72].
As mentioned above, aberrations in the components of the RB pathway such as CDK4/6, RB, CCND, INK4A, and E2Fs have been implicated in gliomagenesis or transformation/progression from low- to high-grade astrocytoma [73]. The tumor suppressor Rb plays a critical role in regulating the cell cycle through its interaction with the transcription factors of the E2F family and various chromatin modifiers and remodelers with a contributing role in cell cycle progression [74]. Once Rb binds to E2F proteins, it negatively regulates the G1-to-S phase transition by suppressing E2F target genes such as CCNA1 and CCNE1. Conversely, in response to growth stimuli (or mitogen stimulation), the cyclin-dependent kinases (CDK4/6 and CDK2) phosphorylate Rb, releasing E2F, which allows the use of transcription factors and facilitates G1-to-S phase progression [74, 75]. However, as revealed by the Cancer Genome Atlas (TCGA) project, about 80% of primary GBMs exhibit abnormalities in this pathway, including RB1, CDKN2A gene deletion or mutation, and/or CDK4 gene amplification [76].
The most frequently altered gene is TP53, associated with human cancers. A deregulated p53/ARF/Mdm2 pathway has been proved to be central to GBM cell proliferation, invasion and migration, apoptosis evasion, and stemness [77]. In unstressed cells, p53 activity is low and negatively regulated through constant ubiquitination and proteasomal degradation by Mdm2/MdmX [78]. In response to stress, the interaction between p53 and Mdm2 is lost, leading to p53 accumulation, which further induces cell cycle arrest and/or apoptosis [78, 79]. According to the TCGA report, p53/ARF/Mdm2 is deregulated in nearly 84% of GBMs, which results in diminished tumor suppressor activity [76]. The most common mutations in the p53 gene are considered to be missense mutations, leading to the overexpression of oncogenic variants of p53 protein and homozygous deletions of CDKN2A/ARF, which cause degradation of p53 and/or amplification of Mdm2 and Mdm4, further resulting in loss of p53’s various tumor-suppressive activities [80].
Collectively, GBMs principally exhibit amplification and/or mutations of RTKs, either the EGFRs or the PDGFRAs. The amplification of the EGFR gene that encodes an altered EGFR protein occurs mostly in primary GBMs, whereas secondary GBMs exhibit dysregulation of PDGFR signaling. As the result of both mutations, the activity of multiple signaling pathways downstream to these RTKs, such as PI3K/Akt/rapamycin-sensitive mTOR complex (mTOR) pathways, is increased [81]. The PI3K/Akt/mTOR pathway is critical to many aspects of normal cellular functions as well as pathologic conditions such as oncogenesis and cancer progression [82]. In GBMs, overactivation of this pathway is found to be correlated with poor patient survival and tumor aggressiveness as it overstimulates processes responsible for cell proliferation, survival, and migration in malignant glial cells [83].
Epigenetic modifications are also present in different types of cancer, including gliomas. Mutations in epigenetic regulator genes are now known to drive certain types of glioma phenotype [84]. These changes include IDH1 or IDH2 mutations in low-grade gliomas, recurrent somatic heterozygous mutations in the gene encoding the histone variant H3 in high-grade childhood gliomas that are connected with DNA methylation profile [56, 85].
miRNAs and gliomas
During the last few decades, much effort has been invested in identifying miRNAs that have abnormal expression patterns in gliomas and selecting the most promising ones to be evaluated for use in therapeutic strategies. Several studies have identified various miRNAs with potential functions in gliomas [86]. One of the earliest and most extensively studied cancer-promoting “onco-miRNAs” is miRNA-21. Interestingly, it was the only one to be found to be increased in all types of solid cancers, as well as the first to be found to be deregulated in human glioblastoma [37]. Its overexpression is linked to tumor growth by impairing apoptosis in malignant glial cells [87]. To date, multiple evaluations have focused on the miRNA signature in glioma tissue samples versus noncancerous brain tissues, to identify the role of miRNAs in the glioma pathogenesis in an effort to illustrate the underlying mechanisms [86].
The expression profiles of miRNAs in tumor margin and non-tumor brain tissue have been found to be significantly different, as 256 miRNAs were significantly upregulated in GBM tissues compared with normal brains. These miRNAs include miR-21, members of the miR-17/92 cluster (miR-17, miR-18a, miR-19a, miR-19b-1, miR-20a, and miR-92a-1), miR-93, miR-221, and miR-222, with the ability to modulate several hallmarks of cancer, including cell proliferation, angiogenesis, invasion, metastasis, and drug resistance, while 95 miRNAs including miR-7, miR-34a, miR-128, miR-137, miR-181, etc. were downregulated in GBM, where their over expression might inhibit GBM development [88]. In the context of the glioma–miRNA relationship, the current review generally focuses on the contribution of miRNAs to the pathogenesis and progression of gliomas, as highlighted in the following sections (Fig. 2).

Schematic view of the relationship between miRNAs and the glioma progression
miRNAs involved in cell proliferation and apoptosis
The continuous cell proliferation ability in cancer represents an imbalance between cellular proliferation and apoptosis, which increases as the grade of differentiation decreases [89]. As mentioned above, one of the first miRNAs found to be deregulated in glioma was miR-21, which is extremely upregulated in glioma and its expression levels are strongly associated with tumor grade and prognosis [90]. A variety of processes are modulated by miR-21, and studies have determined that the regulation of this miRNA promotes glial cell proliferation [90,91,92]. In greater detail, miR-21 exerts its antiapoptotic effects by destabilizing TNF-α receptors (TNFRs) on the cell surface by deregulation of the TIMP3–TNFR interaction (the extrinsic pathway of apoptosis), and/or decreasing caspase-3, caspase-9, and cytosolic apoptotic peptidase activating factor 1 (APAF1) (intrinsic-mitochondrial pathway), or even induction of lower caspase-3/7 activity as key components of all cellular apoptotic pathways [93].
miR-21 targets several tumor suppressor genes known to be critical regulators of apoptosis or cell proliferation, including programmed cell death 4 (PDCD4) [92], acidic nuclear phosphoprotein 32 family member A (ANP32A) [91], PTEN [90], and Sprouty 2 (SPRY2) [94]. Additionally, miR-21 can also direct several components of p53, TGF-β pathways, namely TAp63 (tumor suppressor homolog of p53), p53 activating cofactors and homologs, heterogeneous nuclear ribonucleoprotein K (HNPRK), key TGF-β factors such as TGF-β receptors, and proapoptotic death-domain-associated protein Daxx, leading to promotion of glioma cell proliferation and inhibition of apoptosis [95].
miR-221 and miR-222, which are defined as gene clusters (miR-221/222), are notably upmodulated in many human disorders, including cancers. In glioma, miR-221/222 act as oncomiRs and target the cell cycle inhibitors p27 and/or p57, thus regulating cell cycle progression from the G1 to S phase [96]. In addition, miR-221/222 can promote cell proliferation and inhibit cell death by targeting of p53-upregulated modulator of apoptosis (PUMA) which is a proapoptotic protein. Under physiological conditions, PUMA binds to Bcl-2-like proteins such as Bcl-2, B-cell lymphoma-extra-large (Bcl-xL), and myeloid cell leukemia sequence 1 (Mcl-1), thereby freeing Bax and/or Bak, which are then able to trigger apoptosis through mitochondrial dysfunction and caspase activation [97].
On the other hand, miR-34a is a tumor-suppressive miRNA with low expression levels in various human cancers, and also is the first miRNA demonstrated to be directly regulated by the tumor suppressor p53 [98]. In glioma, miR-34a suppresses cell cycle progression and proliferation of glioma cells through direct inhibition of the expression of MET receptor tyrosine kinases (c-MET RTK), Notch-1, Notch-2, cyclin-dependent kinase 6 (CDK6), cyclin 1 (CCND1), and silent information regulator 1 (SIRT1) [99, 100].
The other miRNA that mediates cell proliferation and apoptosis in GBM is miR-210 [101]. miR-210 is a particular target of hypoxia-inducible factor-1 (HIF-1), which is overexpressed as an adaptive response to hypoxic conditions within a tumor [102]. Bcl-2 19 kD interacting protein (BNIP3), one of the enhancers of cell apoptosis, was identified as a direct functional target of miR-210 [103]. When BNIP3 gene expression is induced, it localizes to mitochondria, resulting in a loss of membrane integrity, oxidative stress, mitochondrial damage, and eventually cell death. Through direct inhibition of BNIP3 expression, miR-210 reduces cell death [103, 104]. Furthermore, regulator of differentiation 1 (ROD1) is a miR-210 target, being involved in GBM progression. MiR-210 inhibits the proliferation of tumor cells and induces the apoptotic flux by negative regulation of ROD1 [105].
MiR-124-3p is also recognized as a mediator of the cell cycle and survival/apoptosis in glioma cells. Flow cytometric analyses of cancer tissues and relevant cell lines have shown overexpression of miR-124-3p. Surprisingly, this miRNA has been verified to be a negative regulator of Neuropilin-1 (NRP-1), which is a multifunctional receptor involved in glioma cell proliferation, invasion, and migration through binding to various extracellular receptors [106, 107].
Xia et al. demonstrated a positive relationship between overexpression of miR-125b and glioma cell proliferation, as well as suppressed apoptosis [108]. This brain-enriched miRNA was reported to be distributed among neurons and astrocytes [109]. Prior to this exploration, miR-125b overexpression had been detected in oligodendroglial tumors and brain specimens of fetuses with Down syndrome [110, 111]. In vitro evaluations have indicated that miR-125b depletion would repress the proliferation of human neuroblastoma cells [112]. In the context of the roles of miR-125b in provoking glioma cell proliferation and inhibiting all-trans-retinoic acid (ATRA)-induced cell apoptosis, underexpression of miR-125b sensitizes cells to ATRA-induced apoptosis. Furthermore, negative crosstalk was found between miR-125b and the apoptosis-related protein Bcl-2 modifying factor (BMF), in which miR-125b can link to 3′-untranslated region (UTR) of BMF [108].
ICAT, which is defined as the inhibitor of β-catenin and T cell factor (TCF), is a perfect negative regulator of Wnt signaling, functioning by blocking the TCF-to-β-catenin attachment [113]. Although ICAT has been found to be deregulated in a group of human malignancies, its carcinogenic functions remain undetermined [59]. In GBM, ICAT downmodulation has been reported to inhibit cell proliferation, migration, and invasion, while stimulate the apoptotic flux [114]. More interestingly, the expression of ICAT can be modulated by different miRNAs in various different types of cancer, including hepatocellular carcinoma (HCC), breast cancer, and GBM [115, 116]. On the other hand, miR-296-3p is also downmodulated in GBM tissues compared to normal brain [117].
The overexpression of miR-296-3p has been shown to be linked to lower survival rates according to the Cancer Genome Atlas GBM dataset [118]. Surprisingly, the expression of ICAT has been revealed to be inversely associated with miR-296-3p expression in GBM tissues. Consistently, ICAT is underexpressed in grade II gliomas, while miR-296-3p is not, reflecting the complexity of ICAT modulation in GBM, which suggests that ICAT might be controlled by other mechanisms in grade II GBM [119].
Other possible signaling pathways targeted by miRNAs are the FOXG1 (Forkhead Box G1) and Smad6 pathways; FOXG1 is known to be an essential transcriptional factor in telencephalon development and was shown to be upregulated in multiple cancer cells, including gliomas [120]. One of the first studies connecting the dots between miRNAs and the FOXG1 pathway was carried out by Shibata et al. [121]. They suggested that the differentiation of neurons in the medial pallium of mice was regulated by miR-9 through FOXG1 signaling; more exactly, Zhen et al. suggested that miR-9-3p could be downmodulated in glioma cells, while the protein expression levels of FOXG1 were increased. They also identified FOXG1 as a direct target of miR-9-3p [122]. Furthermore, they observed suppressed proliferation and increased apoptosis of glioma cells transfected with miR-9-3p [122].
Likewise, miR-186 is significantly inhibited in tumor cells compared with adjacent nontumor tissues. Transfection of miR-186 into human glioma cells showed increased apoptosis through direct suppression of Smad6, which was negatively regulated by miR-186 levels [123]. Similarly, miR-142-3p also has antiproliferative functions and mitigates apoptosis of U87 and U251 glioma cells by blocking the transcription and translation of the high-mobility group box 1 (HMBG1) gene. By targeting HMBG1, miR-142-3p can inhibit the Wnt/β-catenin signaling pathway, and simultaneously initiates the caspase-3 pathway [124].
miRNAs contributing to invasion and migration
Cancer progression begins with cell invasion, the migration of tumor cells from the primary origin to distant organs. Several miRNAs have been found to be valuable for the detection of the presence of metastases [125]. Recent studies using glioma cells have suggested that miR-21 is a key regulator of the expression of multiple genes and can thus induce several cellular programs in glioma cells, including invasion and microvascular proliferation of GBM [126]. miR-21 mediates these processes through inhibition of matrix remodeling genes, a reversion-inducing cysteine-rich protein with Kazal motifs [127], and tissue inhibitor of metalloproteinases 3 (TIMP3), which normally regulates the levels of matrix metalloproteinases (MMPs). MMPs are proteolytic enzymes that promote tumor cell invasion by disrupting the extracellular matrix [128]. It was found that miR-21 could negatively regulate the mRNA and protein levels of RECK and TIMP3 in glioblastoma cells [129]. Overexpression of miR-21 is also related to the upmodulated Sox2 (SRY-Box transcription factor 2), and Sox2 downmodulation can inhibit miR-21-enhanced glioma cell migration and invasion [130]. miR-21 expression levels are also associated with the grade of the glioma, being predominantly present at lower concentrations in grade II and III gliomas. GBM, in contrast, exhibits higher levels of miR-21, which is assumed to be correlated with its higher microvascular texture and ECM reorganization [24, 131]. In support of these findings, Gabriely et al. reported that vascular morphogenesis- and angiogenesis-related genes were significantly downregulated in response to miR-21 suppression in A172 glioma cells [24].
Matrix metalloproteinases (MMPs) are enzymes that catalyze the degradation of ECM and cell adhesion molecules (CADMs) that are essential in the cell-to-ECM attachment during the process of cell adhesion [132]. Interestingly, MMPs and CADMs have been identified as emerging targets for miRNAs; for instance, the aforementioned miR-21 could target MMP inhibitors such as RECK, enhancing the ECM degradation, and increasing the motility and cell invasion [24]. Also, miR-25 is upregulated in glioblastomas, where its expression levels are closely linked to the stage of the disease. miR-25 knockdown markedly decreases tumor cell migration and invasiveness through enhancing the expression of cell adhesion molecule 2 (CADM2). CADM2 is significantly underexpressed in glioma cells, a finding that confirms CADM2 as a promising target for miR-25 [133].
Another potential oncogenic miRNA is miR-720, which directly upmodulates the transcription of invasion- and migration-related genes based on a study performed by Liu et al. This was the first study to demonstrate the inhibition of threonyl-tRNA synthetase like-2 (TARSL2) as a direct target of miR-720 [134]. Moreover, t-RNA synthases have previously been evaluated in immune disorders for their therapeutic roles [135]. Moreover, plasma lysyl-tRNA synthases have been shown to be associated with colorectal cancer [136, 137], enhancing cell adhesion for migration and metastasis [138]. In contrast, it seems that in GBM, threonyl-tRNA synthetase acts against miR-720 effects, promoting anti-invasive properties [134].
Conversely, tumor-suppressive miR-146b-5p has been proved to be negatively associated with its targeted gene, MMP16. The expression levels of miR-146b-5p in glioma cells are significantly low, while MMP16 show a surge in the same cells. Furthermore, the overexpression of miR-146b-5p was shown to promote degradation of MMP16-related mRNA. In this regard, the transfected U87 glioma cells presented shorter protrusions in response to miR-146b-5p mimics; also, direct blocking of MMP-16 using exogenous siRNA restored the miRNA activity against migration and invasion of glioma cells [139].
miR-379-5p has also been reported to be downmodulated in gliomas; the overexpression of this miRNA leads to the suppression of tumor cell viability, migration, invasion, and epithelial-to-mesenchymal transition (EMT). Notably, miR-379-5p exerts it effects by detoxifying microsomal glutathione transferase 1 (MGST1) [140]. High levels of glutathione transferase undesirably correlate with chemoresistance and poor survival, as these enzymes may bind to chemotherapy-induced cytotoxic agents, blocking their tumoricidal effects [141, 142]. Consistently, lower levels of MGST1 induced by miR-379-5p are linked to the improved survival, according to a study performed by Yang et al. [140].
Over recent years, HMGB1 has been discussed as a key mediator of proliferation and inhibition of apoptosis in tumor cells via the Wnt/β-catenin pathway. The suppression of this mediator by miR-665 is closely associated with the blockade of tumor cell migration and invasion in glioma patients. In line with this finding, lower levels of miR-665 in glioma cells correlate with developed grades and lower performance scales [143].
Among many other signaling cascades involved in cell proliferation, EMT, etc., the PI3K/Akt pathway is known to enable oncogenic signaling in multiple cancer cells, due to its effects on the aforementioned processes [144,145,146,147]. In this context, miR-3175 and miR-134 have been shown to affect cell proliferation, as well as EMT, in gliomas; the former miRNA activates the signaling of interest and leads to invasion, whereas the latter blocks the pathway, resulting in tumor cell apoptosis, which is a desirable result in cancerous conditions [148, 149].
Moreover, miR-296 has been revealed to be increased in primary cancerous endothelial cells, and is associated with cell invasion and multidrug resistance of glioma cells [150]. Finally, miR-320a has been reported to impede glioma cell invasion and migration by targeting aquaporin 4 (AQP4), which was recently defined as a strong regulator of cell invasion and migration in glioma subjects [151].
miRNAs affecting angiogenesis
Angiogenesis, in brief, is the formation of a new vascular network from the preexisting vasculature. Angiogenesis is an intricate process involving the proliferation, migration, and differentiation of vascular endothelial cells (ECs) as a result of the complex involvement of proangiogenic and antiangiogenic factors [152]. As in other solid tumors, the growth of cells in glioma greatly relies on an adequate supply of oxygen and vital nutrients, which is principally provided through new blood vessels. Glioma angiogenesis is triggered by a variety of angiogenic factors and genes. More interestingly, miRNAs have been found to act as modulators of angiogenesis by activating and/or inhibiting the relevant molecular pathways. These types of miRNAs are named “angiomiRs,” playing significant roles in controlling the vascular network properties of gliomas [153].
An angiomiR characterized by being markedly downregulated in GBM-associated endothelial cells (ECs) is miR-125b, which targets myc-associated zinc finger protein [154]. Indeed, MAZ is a transcription factor that can regulate the transcriptional activation of vascular endothelial growth factor (VEGF) and VEGF receptors. Therefore, the overstimulation of miR-125b inhibits tumor angiogenesis [155, 156]. A hypoxic microenvironment typically surrounds glioma cells and results in the promotion of tumor angiogenesis through the regulation of particular miRNAs. In this setting, miR-210-3P is a hypoxia-regulated miRNA that is defined as a positive modulator of the activity of VEGF, carbonic anhydrase 9 (CA9), and HIF [90, 102].
Functional in vitro analysis has shown that miR-218 expression is considerably decreased in GBM compared to normal tissues; miR-218 indirectly targets HIF-2α through RTK signaling pathways. Thus, miR-218 downregulation could further influence GBM tumor neovascularization [157]. On the other hand, in vivo data suggest that miR-93 supports angiogenesis and EC activities via the induction of new blood vessel formation and the enhancement of EC survival through suppression of integrin-β8 [158].
Some other miRNAs, such as miR-124-3p, can control GBM cell proliferation, migration, and angiogenesis through activating the PI3K/Akt/NF-κB signaling pathway; it has been reported that the overexpression of miR-124-3p restricts glioma angiogenesis [152].
miRNAs on the journey to drug resistance
Despite valuable advances in chemotherapeutic strategies, achieving successful therapeutic outcomes in glioblastoma patients has remained a major clinical hurdle, mostly owing to multiple resistance mechanisms [159]. It has been concluded on the basis of several studies that specific miRNAs involve in the chemoresistance of tumor cells through targeting drug-resistance-related genes or alterations in gene expression patterns in relation to distinct cellular processes, such as apoptosis, cell proliferation, and cell cycle modifications [160]. Hence, if such miRNAs are impaired and/or deregulated, they might act as deregulated gene modulators, leading to the drug resistance [161]. Depending on their specific target gene or mRNA, these miRNAs can possess cell specific activities, meaning that a single miRNA may have both tumor-suppressive and oncogenic potential [162,163,164].
Temozolomide (TMZ) has been widely accepted as the standard chemotherapeutic agent for GBM and astrocytoma. Since the introduction of TMZ as the first-line chemotherapeutic drug of choice for gliomas, high levels of drug resistance have occurred in at least half of all treated cases [165]. One of the accepted explanations is linked to miRNAs, as the expression profiles of several miRNAs have been shown to be upregulated in TMZ-resistant cells [160]. For instance, miR-548m is a downstream target of circular RNA-GLIS3 sponging activity, which upregulates MED31 mRNA expression, causing tumor cell invasion in TMZ-resistant cells [166].
The overexpression of miR-21 has been demonstrated to be linked to attenuated TMZ-induced apoptosis in human glioblastoma U87MG cells [167]. In particular, miR-21 mediates this by impairing the balance between proapoptotic Bax and antiapoptotic Bcl-2 proteins, through exerting a decrease in the Bax-to-Bcl-2 ratio, and also by decreasing the activity of caspase-3 [167, 168]. Similarly, miR-125b-2 confers GBM stem cells with resistance to TMZ; in TMZ-treated GBM stem cells, inhibition of miR-125b-2 activity using peptide nucleic acid (PNA) decreases the Bax-to-Bcl-2 ratio compared to treatment with TMZ alone, which further impedes apoptosis and confers drug resistance [169].
In human glioma cell lines with different p53 expression patterns, miR-221/222 contribute to a remarkable therapeutic resistance against TMZ by induction of the expression of Bax, cytochrome c, and caspase-3 [170]. On the other hand, miR-128 and miR-149 are also mediators of drug resistance; they both contribute to TMZ chemosensitivity by Rap-1B-mediated cytoskeletal remodeling [171]. In a similar pattern, miR-181a/b/c/d are also reported to sensitize tumor cells and minimize TMZ resistance through the same mechanism [172]. On the other hand, Chen et al. reported that miR-29a can also sensitize the response of glioma cells to TMZ through regulating the p53/MDM2 feedback loop [173]. Indeed, p53-induced expression of miR-29a can cause aberrant expression of MDM2 targeted by the corresponding miRNA, thereby affecting the activity of the p53-miR-29a-MDM2 loop [173].
Microarray analyses of miRNAs in GBM cell lines have shown that miR-10a, miR-195, and miR-455-3p involve in the acquisition of TMZ resistance. Although the suppression of miR-445-3p and miR-10a moderately influences the effectiveness of TMZ, miR-195 inhibition could effectively reverse the TMZ resistance [174].
Oncogenic miR-155-3p is reported to be overexpressed in gliomas, with the ability to induce cell growth in A172 and U87 cell lines, while its inhibition promotes sensitivity to TMZ by direct upregulation of Six1 at the translational level, thus inducing cell arrest at the same point at which TMZ acts on the G1/S phase [175]. Zhou et al. demonstrated that miR-141-3p increased cell growth and drug resistance through suppressing p53 and its downstream proteins such as cyclin-dependent kinase 2 (CDK2) and cyclin E1/B1, thus inhibiting cell cycle arrest at the G1 to S phases [176].
In contrast, miR-195 can decrease and reverse U251 glioma cells’ resistance by targeting oncogenic CCNE1 [177]. CCNE1 (cyclin E1) is considered to be a potential subunit for CDK2, promoting cell progress from the G1 to S phase [178]. In this regard, miR-524-5p and miR-324-5p also inhibit U251 and U87 cell growth, and increase tumor cells’ chemosensitivity by downregulating the methyltransferase enhancer of zeste homolog 2 (EZH2) oncogene, which was previously identified to play a crucial role in TMZ resistance [179, 180]. Additionally, miR-524-5p has been confirmed to be an independent prognostic factor for improved survival [180].
In the case of TMZ resistance, miRNAs have also been indicated to be sponged during the process of resistance. In this setting, the upregulation of long- noncoding RNA MSC-AS1 in TMZ-resistant glioma cells has been linked to the PI3K/Akt oncogenic pathway, and its knockdown could profoundly suppress cell proliferation and drug resistance through sponging miR-373-3p. Cytoplasmic polyadenylation element-binding 4 (CPEB4), which encodes a cell survival protein, is a possible target gene for this miRNA [181]. In other words, MSC-AS1-induced inhibition of miR-373-3p causes CPEB4 upmodulation, and therefore promotes cell growth and TMZ resistance through activation of the mentioned signaling pathway [182].
miRNAs in self-renewal and differentiation of glioma stem cells (or cell stemness)
Recent studies support that miRNAs are critical regulators of stem-like features. miRNA profiles of glioma stem cells (GSCs) have been evaluated, revealing unique miRNA signatures in CD133+ GSC population compared to CD133− non-stem-cell populations [183]. As described above, particular miRNAs could regulate major signaling pathways in the evolution of gliomas, including EGFR/RAS/NF1/PTEN/PI3K signaling and those related to p53, i.e. MDM2/MDM4/p14ARF and p16INK4a/CD4/RB1 pathways. Furthermore, miRNAs are key molecular players in relation to biological characteristics of GSCs such as self-renewal and differentiation [184].
According to the roles of miRNAs in the differentiation and self-renewal of GSCs, they can be categorized into two subgroups: pluripotent miRNAs and prodifferentiation miRNAs. Pluripotent miRNAs are capable of increasing the self-renewal capacity and proliferation of stem cells but impede cell differentiation [185]. miR-18 and miR-137 are clear examples in this regard. Conversely, prodifferentiation miRNAs, including miR-9/9, miR-17, miR-34a, miR-124, miR-128, miR-137, miR-141, miR-145, miR-202, miR-326, miR-302–367 cluster, miR-451, and miR-504, can stimulate or stabilize differentiation of GSCs to more mature phenotypes [186]. As an example, in CD133+ A172 GBM cells transfected with miR-451 in combination with imatinib mesylate treatment, GSC growth and neurosphere formation were markedly inhibited [187]. miR-137 is another prodifferentiation miRNA with low expression levels in GSCs, caused by promoter hypermethylation. miR-137 exerts its effects by targeting RTVP-1, which itself suppresses the expression of CXCR4 [188]. C-X-C chemokine receptor type 4 (CXCR4) involves in several GSC characteristics, including generation and self-renewal. Moreover, miR-504 expression levels are significantly downregulated in GSCs compared to NSCs. However, transducing GSCs with miR-504 inhibits their self-renewal by targeting Grb10, which is considered to be an oncogene in GSCs and GBM [189].
Glioblastoma chemoresistance and recurrence are also considered to be the results of the activation and self-renewal of cancer stem cells [190, 191]. Researchers have shown that Nanog can negatively associate with AP-2α protein levels in human and mouse glioma cells and tissues [192]. As an aside, Nanog is a protein contributing to mutual crosstalk between GSCs and miRNAs, and is also linked to tumor aggressiveness, especially at high levels [193]. Nanog was identified to be the target of oncogenic miR-26a and was associated with the chemosensitivity of U251 cells. This oncogenic miRNA inhibits AP-2α expression, resulting in upmodulation of Nanog, and thus allowing for GSC renewal and drug resistance [192].
In contrast, miR-30a has anti-GSC activities, as it downregulates ecto-5′-nucleotidase (NT5E) levels, which further inhibits the Akt signaling pathway and ultimately decreases GSC clone formation, as well as the proliferation in vitro and in vivo [194]. Investigation of rat-derived GSCs (C6 cells) has demonstrated that miR-30c overexpression can result in C6 cell sphere formation and neural differentiation to astrocytes through activation of the JAK–STAT signaling pathway [195]. miR-33a acts as another oncomiR, which is upregulated in GSCs and directly increases phosphodiesterase 8A (PDE8A) and ultraviolet (UV) radiation resistance-associated gene/protein (UVRAG) mRNAs’ expression; the encoded proteins have been reported to be responsible for regulating protein kinase A (PKA) and Notch endocytosis signaling pathways, respectively [196].
miR-300, which is extremely prevalent in glioma cells and tissues, and exists in even higher amounts in GSCs [197], increases cell proliferation and self-renewal-related activities in patient-derived GSCs; however, it inhibits stem cell differentiation, maintaining the GSCs’ undifferentiated status. Interestingly, it seems that all these effects are mediated by direct suppression of leucine zipper putative tumor suppressor 2 (LZTS2) [197].
miRNAs as promising diagnostic/prognostic biomarkers for gliomas
Early diagnosis of glioblastoma can remarkably reduce mortality and improve patient outcomes and quality of life. However, identifying early-stage glioblastoma is still very complicated. Molecular prognostic and diagnostic biomarkers have been introduced recently by the World Health Organization (WHO) into the classification of CNS tumors. Recognition of these biomarkers in tissues and/or body fluids is crucial to characterizing subclasses of tumors that cannot be precisely classified and/or cannot be analyzed due to scant tissue samples [198].
Several studies have shown a correlation between the expression patterns of specific miRNAs and the development/growth of glioblastomas. Moreover, circulating miRNAs could serve as potential cancer biomarkers through exosome-mediated intercellular communication. Exosomes containing miRNAs are released either from viable cells or apoptotic bodies, and can circulate through serum and/or cerebrospinal fluid (CSF) [199]. Therefore, measurement of these circulating miRNAs may provide reliable, accessible diagnostic tools for multiple malignancies, including gliomas. To date, tremendous effort has been made to establish a miRNA panel for glioma models with capability for diagnostic and prognostic purposes [199, 200]. A recent study in this regard reported that the expression levels of miR-21, miR-222, and miR-124-3p are increased in serum exosomes derived from patients with aggressive high-grade glioblastoma, suggesting that these miRNAs may serve as biomarkers for predicting tumor progression at early stages [201].
Another study demonstrated that coexpression of miR-15b and miR-21 could be a valuable diagnostic tool in gliomas. Additionally, it has been reported that miR-16 could be exploited to discriminate glioblastoma from other types of glioma [202].
A specific analysis of Cancer Genome Atlas data revealed an inverse connection between miR-196b/miR-10b levels and overall survival of glioblastoma patients [203]. Another prognostic miRNA in this field is miR-328, whose low expression levels correlated with poor patient survival [204]. Similarly, the expression levels of miR-549a and miR-502-5p have prognostic significance in patients with tumors of glial origin [205]. Some miRNAs could also be used for monitoring tumor progression. In this context, studies evaluating miR-205 in glioma patients have indicated that following surgery, miR-205 content is increased, whereas its serum concentrations decrease after the recurrence period [206].
The implications of using miRNAs as diagnostic and predictive biomarkers for glioblastoma have been reviewed comprehensively [207]; nevertheless, few assessments have evaluated their prognostic potential in lower-grade gliomas [208]. Meanwhile, individual prognostic miRNAs have been investigated in multiple studies; for instance, a meta-analysis suggested that the upmodulation of miR-15b, miR-21, miR-148a, miR-196, miR-210, and miR-221 and downregulation of miR-106a and 124 are consistent with poor survival in glioma patients [209].
However, there are controversial results regarding prognostic miRNA signatures in literature, which could be due to small sample size, shorter follow-up times, and the use of different assays and normalization techniques. This is more obvious in lower-grade gliomas, as these entities are less common and are often lost to follow-up owing to longer survival times. A recent analysis evaluating a 30-miRNA prognostic model suggested that glioblastomas (n = 35) can be classified into two subgroups with early death (< 450 days) versus long-term survival (> 450 days) [210]. A more comprehensive analysis using a larger patient population (n = 563) from the TCGA cohort identified three miRNAs (incl. miR-222, miR-302d, and miR-646) that independently acted as survival biomarkers [211]. Using a TCGA cohort, Hayes et al. also offered a risk assessment score according to nine miRNAs that were significantly associated with survival [212]. Surprisingly, miR-222 was found to be a common contributor in all three studies; however, it was not confirmed to be a disease-free survival biomarker in another study that used the same dataset [213]. These inconsistencies raise the need for not only larger patient cohorts but also adjustment for many confounders such as the grade and histology of gliomas [214].
Hayes et al. identified eight miRNAs (incl. miR-124a, miR-202, miR-7, miR-222, miR-363, miR-630, miR-663, and miR-204) with the ability to predict overall survival in those treated with bevacizumab [207]. Interestingly, miR-7, an antiangiogenic miRNA discussed above, has been reported to correlate with poor response to bevacizumab, suggesting that tumors not enriched with vascular networks will not exhibit an ideal response to this VEGF-targeting chemotherapeutic agent. Additional assessments have also investigated individual miRNAs and miRNA profiles for predicting responsiveness to TMZ, in addition to radiation therapy [215] or alone [216]; however, it remains to be concluded whether their predictive potentials are generalizable to clinical settings or not.
Therapeutic potential of miRNAs
miRNAs simultaneously regulate multiple genes and proteins across different signaling pathways. This regulatory characteristic makes miRNAs promising therapeutic targets for multiple diseases; therapeutics act as either miRNA mimics or miRNA inhibitors. miRNA inhibitors are single-stranded oligonucleotide antagonists [217, 218], while anticarcinogenic miRNAs could be induced through double-stranded miRNAs, formed synthetically according to the biological miRNA twin sequence. Accordingly, different aspects of miRNAs’ therapeutic potential in gliomas are discussed in this section (Fig. 1).
The first miRNA-based therapeutic approach is to inhibit the action of miRNAs; miRNA antagonists (antagomiRs) usually bind to oncomiRs and inhibit their action by blocking miRNA-to-mRNA attachment, a process which is called antisense action. Indeed, antagomiRs are specific and irreversible miRNA antagonists, responsible for further degradation of miRNAs [219]. Conversely, miRNA masks bind to mRNAs and mask the target 3′-UTR sequence, preventing the miRNA from acting on mRNA. Therefore, miRNA masks do not degrade miRNAs and allow their off-target actions on other genes to be left intact [220]. Another mechanism for inhibiting a miRNA is obtained through sponging of miRNAs [221, 222]. A sequence of RNA complementary to the binding site of a particular miRNA, or even a set of miRNAs with similar seed sequences, occupies this binding site and prevents all biological miRNAs from acting on mRNAs. Due to the fact that these sponges are not chemically modified for specific miRNAs, they may have low affinity and require higher concentrations to act as a proper inhibitor. Additionally, there is a requirement for strong promoters and the necessity for multiple vector integration [222, 223].
When using the second mechanism, which is defined as induction of the activities of miRNAs, anticarcinogenic miRNAs could induce tumor-suppressive activities by miRNA mimics, through reversing epigenetic silencing. Small-molecule modulators are considered to be excellent cancer therapeutics, based on their high stability [224]. miRNA silencing in carcinogenic signaling pathways is reversed by hypomethylating agents such as 5-azacytidine [225]. Interestingly, these agents can reexpress multiple mRNAs, miRNAs, as well as the other noncoding RNAs. Synthetic RNAs acting as native miRNA twins include one RNA strand (the guide strand) that is identical to the target miRNA and act by “mimicking” the functions of the mature miRNAs. The passenger strand may be either completely or partially compatible with the guide strand [226, 227]. Any type of incompatibility or side-compatibility with other miRNAs could result in side effects; therefore, the design process should be performed carefully and monitored before introduction into viable tumor cells [228].
In the case of the third therapeutic strategy, i.e., monitoring the delivery systems, a precise evaluation of the delivery of therapeutic miRNAs is essential, since many potential side effects may occur during in vivo systemic administration. For instance, chemically modified miRNAs are crucial for conserving their viability and stability while passing through the bloodstream. These alterations actually protect miRNAs from nuclease-induced degradation, as well as their renal clearance or reticuloendothelial removal [229,230,231]. Furthermore, researchers have also suggested the use of viral vectors for this purpose, in which regard careful consideration of immune system responses is definitely required. Accordingly, most approaches prefer nonviral vectors for successful delivery of miRNA therapeutics [218, 232]. Nanocarriers can also protect oligonucleotides, such as miRNA mimics or antioligomers, from sequestration by nucleases. Nanoparticles made of Gold (Au) or other inorganic materials such as silicon/iron oxide are considered to be safe carriers for oligonucleotides. Au, as an inert element, has been shown to cross the blood–brain barrier (BBB), which could make it particularly suitable for glioma patients [233, 234]. Other nanoparticles, such as polyethylene glycol [235]–polyethylenimine (PEI) liposomes, have been reported to exhibit longer viability and lower immunogenicity, and thus could be employed to treat various cancers. However, minimal changes in their cationic head or hydrophobic chain may result in dramatic immune responses or hinder their transfection [234, 236].
Despite all the developments in miRNA-based therapeutic strategies, designing appropriate delivery systems for patients with glioblastoma remains challenging due to the presence of the BBB. However, cell-penetrating peptides and immunoliposomes have been shown to be promising in terms of crossing the BBB [237]. Finally, after introducing miRNAs into tumor cells, there are many concerns regarding the targeting of specific actions or side effects, and many other biological barriers such as high interstitial tumor pressure or alterations in ECM may affect the final outcome [238].
Conclusions
The modulatory significance of miRNAs in glioma pathogenesis and progression is no longer unknown. The role of these ncRNAs in different glioma-related cellular and molecular processes, such as cell proliferation and apoptosis, migration and invasion, angiogenesis, drug resistance, and cell stemness and differentiation, as discussed comprehensively in this review, has been revealed. Based on this suggested correlation, any type of disturbance in the miRNA modulatory plexus could influence the pathogenesis of glioma. Since various human miRNAs have been identified to date, it is worth precisely profiling miRNAs that are highly expressed in glioma cell and tissue specimens. Moreover, establishing molecular targets, as well as relevant cellular pathways, is required to define the miRNA–glioma relationship. On the other hand, miRNAs have also provided novel capacities in the clinical setting; they can potentially act as glioma biomarkers for both diagnostic and prognostic purposes. Additionally, miRNA-based therapeutic approaches are very promising for the management of gliomas. The introduction of newly identified systems for the delivery of miRNAs and their transfer across the BBB heralds a bright future for glioma and GBM therapy. Notwithstanding, further investigations, especially in large cohorts at different laboratories and research centers, are still needed to further clarify the diagnostic and therapeutic potential of miRNAs in relation to gliomas and glioma patients.
Availability of data and materials
Not applicable.
Abbreviations
- CADM2:
-
Cell adhesion molecule 2
- CDK2:
-
Cyclin-dependent kinase 2
- ECM:
-
Extracellular matrix
- EGF:
-
Epidermal growth factor
- EMT:
-
Epithelial-to-mesenchymal transition
- GBM:
-
Glioblastoma multiforme
- HGF:
-
Hepatocyte growth factor
- IDH:
-
Isocitrate dehydrogenase
- MGST1:
-
Microsomal glutathione transferase 1
- miRNAs:
-
MicroRNAs
- MMPs:
-
Matrix metalloproteinases
- NcRNAs:
-
Noncoding RNAs
- PDGF:
-
Platelet-derived growth factor
- PiRNAs:
-
Piwi-interacting RNAs
- RISC:
-
RNA-induced silencing complex
- TMZ:
-
Temozolomide
- TNFRs:
-
TNF-α receptors
- TRBP:
-
TAR RNA-binding protein
- VEGF:
-
Vascular endothelial growth factor
References
Kapranov P, St Laurent G, Raz T, Ozsolak F, Reynolds CP, Sorensen PH, et al. The majority of total nuclear-encoded non-ribosomal RNA in a human cell is “dark matter” un-annotated RNA. BMC Biol. 2010;8:149.
Chan JJ, Tay Y. Noncoding RNA: RNA regulatory networks in cancer. Int J Mol Sci. 2018;19(5):1–8.
Bian Z, Ji W, Xu B, Huo Z, Huang H, Huang J, et al. Noncoding RNAs involved in the STAT3 pathway in glioma. Cancer Cell Int. 2021;21(1):445.
Letafati A, Najafi S, Mottahedi M, Karimzadeh M, Shahini A, Garousi S, et al. MicroRNA let-7 and viral infections: focus on mechanisms of action. Cell Mol Biol Lett. 2022;27(1):1–47.
Sun W, Li Y, Ma D, Liu Y, Xu Q, Cheng D, et al. ALKBH5 promotes lung fibroblast activation and silica-induced pulmonary fibrosis through miR-320a-3p and FOXM1. Cell Mol Biol Lett. 2022;27(1):1–17.
Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–54.
Bartel DP. Metazoan microRNAs. Cell. 2018;173(1):20–51.
Shi R, Wang P-Y, Li X-Y, Chen J-X, Li Y, Zhang X-Z, et al. Exosomal levels of miRNA-21 from cerebrospinal fluids associated with poor prognosis and tumor recurrence of glioma patients. Oncotarget. 2015;6(29):26971.
Riddick G, Fine HA. Integration and analysis of genome-scale data from gliomas. Nat Rev Neurol. 2011;7(8):439–50.
Ho VK, Reijneveld JC, Enting RH, Bienfait HP, Robe P, Baumert BG, et al. Changing incidence and improved survival of gliomas. Eur J Cancer. 2014;50(13):2309–18.
Louis DN, Perry A, Reifenberger G, Von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–20.
Parpura V, Heneka MT, Montana V, Oliet SH, Schousboe A, Haydon PG, et al. Glial cells in (patho) physiology. J Neurochem. 2012;121(1):4–27.
Komori T. The 2016 WHO classification of tumours of the central nervous system: the major points of revision. Neurol Med Chir (Tokyo). 2017;57(7):301–11.
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23(8):1231–51.
Tanase CP, Enciu AM, Mihai S, Neagu AI, Calenic B, Cruceru ML. Anti-cancer therapies in high grade gliomas. Curr Proteomics. 2013;10(3):246–60.
Salami R, Salami M, Mafi A, Vakili O, Asemi Z. Circular RNAs and glioblastoma multiforme: focus on molecular mechanisms. Cell Commun Signal. 2022;20(1):1–17.
Goldbrunner R, Ruge M, Kocher M, Lucas CW, Galldiks N, Grau S. The treatment of gliomas in adulthood. Dtsch Arztebl Int. 2018;115(20–21):356.
Baumert BG, Hegi ME, van den Bent MJ, von Deimling A, Gorlia T, Hoang-Xuan K, et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033–26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol. 2016;17(11):1521–32.
Niyazi M, Brada M, Chalmers AJ, Combs SE, Erridge SC, Fiorentino A, et al. ESTRO-ACROP guideline “target delineation of glioblastomas.” Radiother Oncol. 2016;118(1):35–42.
Weller M, Van Den Bent M, Tonn JC, Stupp R, Preusser M, Cohen-Jonathan-Moyal E, et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017;18(6):e315–29.
van den Bent MJ, Brandes AA, Taphoorn MJ, Kros JM, Kouwenhoven MC, Delattre J-Y, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol. 2013;31(3):344–50.
Ames H, Halushka MK, Rodriguez FJ. miRNA regulation in gliomas: usual suspects in glial tumorigenesis and evolving clinical applications. J Neuropathol Exp Neurol. 2017;76(4):246–54.
Butler TM, Spellman PT, Gray J. Circulating-tumor DNA as an early detection and diagnostic tool. Curr Opin Genet Dev. 2017;42:14–21.
Gabriely G, Wurdinger T, Kesari S, Esau CC, Burchard J, Linsley PS, et al. MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol Cell Biol. 2008;28(17):5369–80.
Mafi A, Aghadavod E, Mirhosseini N, Mobini M, Asemi Z. The effects of expression of different microRNAs on insulin secretion and diabetic nephropathy progression. J Cell Physiol. 2018;234(1):42–50.
Homayoonfal M, Asemi Z, Yousefi B. Targeting microRNAs with thymoquinone: a new approach for cancer therapy. Cell Mol Biol Lett. 2021;26(1):1–22.
Bahmyari S, Jamali Z, Khatami SH, Vakili O, Roozitalab M, Savardashtaki A, et al. MicroRNAs in female infertility: an overview. Cell Biochem Funct. 2021;39(8):955–69.
Peng Y, Croce CM. The role of microRNAs in human cancer. Signal Transduct Target Ther. 2016;1:15004.
Ohtsuka M, Ling H, Doki Y, Mori M, Calin GA. MicroRNA processing and human cancer. J Clin Med. 2015;4(8):1651–67.
Wu K, He J, Pu W, Peng Y. The role of exportin-5 in microRNA biogenesis and cancer. Genomics Proteomics Bioinf. 2018;16(2):120–6.
Yao Q, Chen Y, Zhou X. The roles of microRNAs in epigenetic regulation. Curr Opin Chem Biol. 2019;51:11–7.
Oliveto S, Mancino M, Manfrini N, Biffo S. Role of microRNAs in translation regulation and cancer. World J Biol Chem. 2017;8(1):45–56.
Chen X, Li X, Guo J, Zhang P, Zeng W. The roles of microRNAs in regulation of mammalian spermatogenesis. J Anim Sci Biotechnol. 2017;8:35.
Olejniczak M, Kotowska-Zimmer A, Krzyzosiak W. Stress-induced changes in miRNA biogenesis and functioning. Cell Mol Life Sci. 2018;75(2):177–91.
Kato M, Castro NE, Natarajan R. MicroRNAs: potential mediators and biomarkers of diabetic complications. Free Radic Biol Med. 2013;64:85–94.
Rokad D, Ghaisas S, Harischandra DS, Jin H, Anantharam V, Kanthasamy A, et al. Role of neurotoxicants and traumatic brain injury in α-synuclein protein misfolding and aggregation. Brain Res Bull. 2017;133:60–70.
Bautista-Sánchez D, Arriaga-Canon C, Pedroza-Torres A, De La Rosa-Velázquez IA, González-Barrios R, Contreras-Espinosa L, et al. The promising role of miR-21 as a cancer biomarker and its importance in RNA-based therapeutics. Mol Therapy. 2020;20:409–20.
Natarajan R, Putta S, Kato M. MicroRNAs and diabetic complications. J Cardiovasc Transl Res. 2012;5(4):413–22.
McClelland AD, Kantharidis P. MicroRNA in the development of diabetic complications. Clin Sci (Lond). 2014;126(2):95–110.
Kato M, Natarajan R. MicroRNAs in diabetic nephropathy: functions, biomarkers, and therapeutic targets. Ann N Y Acad Sci. 2015;1353:72–88.
Condrat CE, Thompson DC, Barbu MG, Bugnar OL, Boboc A, Cretoiu D, et al. MiRNAs as biomarkers in disease: latest findings regarding their role in diagnosis and prognosis. Cells. 2020;9(2):1–14.
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–8.
Zhou Q, Liu J, Quan J, Liu W, Tan H, Li W. MicroRNAs as potential biomarkers for the diagnosis of glioma: a systematic review and meta-analysis. Cancer Sci. 2018;109(9):2651–9.
Aqeilan RI, Calin GA, Croce CM. miR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ. 2010;17(2):215–20.
Dimitrova N, Gocheva V, Bhutkar A, Resnick R, Jong RM, Miller KM, et al. Stromal expression of miR-143/145 promotes neoangiogenesis in lung cancer development. Cancer Discov. 2016;6(2):188–201.
Zhang X, Li Y, Qi P, Ma Z. Biology of miR-17-92 cluster and its progress in lung cancer. Int J Med Sci. 2018;15(13):1443.
Mu P, Han Y-C, Betel D, Yao E, Squatrito M, Ogrodowski P, et al. Genetic dissection of the miR-17∼ 92 cluster of microRNAs in myc-induced b-cell lymphomas. Genes Dev. 2009;23(24):2806–11.
Li N, Jiang S, Shi J, Fu R, Wu H, Lu M. Construction of a potential microRNA, transcription factor and mRNA regulatory network in hepatocellular carcinoma. Transl Cancer Res. 2020;9(9):5528.
Mitsis T, Efthimiadou A, Bacopoulou F, Vlachakis D, Chrousos GP, Eliopoulos E. Transcription factors and evolution: an integral part of gene expression. World Acad Sci J. 2020;2(1):3–8.
Pu M, Chen J, Tao Z, Miao L, Qi X, Wang Y, et al. Regulatory network of miRNA on its target: coordination between transcriptional and post-transcriptional regulation of gene expression. Cell Mol Life Sci. 2019;76(3):441–51.
Ashrafizadeh M, Zarabi A, Hushmandi K, Moghadam ER, Hashemi F, Daneshi S, et al. C-myc signaling pathway in treatment and prevention of brain tumors. Curr Cancer Drug Targets. 2021;21(1):2–20.
Slattery ML, Mullany LE, Wolff RK, Sakoda LC, Samowitz WS, Herrick JS. The p53-signaling pathway and colorectal cancer: interactions between downstream p53 target genes and miRNAs. Genomics. 2019;111(4):762–71.
Jauhari A, Singh T, Singh P, Parmar D, Yadav S. Regulation of miR-34 family in neuronal development. Mol Neurobiol. 2018;55(2):936–45.
Autin P, Blanquart C, Fradin D. Epigenetic drugs for cancer and microRNAs: a focus on histone deacetylase inhibitors. Cancers. 2019;11(10):1530.
Farooqi AA, Fuentes-Mattei E, Fayyaz S, Raj P, Goblirsch M, Poltronieri P, et al editors. Interplay between epigenetic abnormalities and deregulated expression of microRNAs in cancer. Seminars in cancer biology. Amsterdam: Elsevier; 2019.
Deng D, Liu Z, Du Y. Epigenetic alterations as cancer diagnostic, prognostic, and predictive biomarkers. Adv Genet. 2010;71:125–76.
Shi J, Dong B, Cao J, Mao Y, Guan W, Peng Y, et al. Long non-coding RNA in glioma: signaling pathways. Oncotarget. 2017;8(16):27582.
Han W, Shi J, Cao J, Dong B, Guan W. Current advances of long non-coding RNAs mediated by Wnt signaling in glioma. Pathol Res Pract. 2020;216(8): 153008.
Imai M, Nakamura T, Akiyama T, Horii A. Infrequent somatic mutations of the ICAT gene in various human cancers with frequent 1p-LOH and/or abnormal nuclear accumulation of beta-catenin. Oncol Rep. 2004;12(5):1099–103.
Siegal T. Clinical relevance of prognostic and predictive molecular markers in gliomas. Adv Tech Stand Neurosurg. 2016;43:91–108.
Holdhoff M. Role of molecular pathology in the treatment of anaplastic gliomas and glioblastomas. J Natl Compr Canc Netw. 2018;16(5s):642–5.
Jones DTW, Bandopadhayay P, Jabado N. The power of human cancer genetics as revealed by low-grade gliomas. Annu Rev Genet. 2019;53:483–503.
Keum Y-S, Choi BY. Isocitrate dehydrogenase mutations: new opportunities for translational research. BMB Rep. 2015;48(5):266.
Ju H-Q, Lin J-F, Tian T, Xie D, Xu R-H. NADPH homeostasis in cancer: functions, mechanisms and therapeutic implications. Signal Transduct Target Ther. 2020;5(1):1–12.
Alexander BM, Mehta MP. Role of isocitrate dehydrogenase in glioma. Expert Rev Neurother. 2011;11(10):1399–409.
Wang T-X, Liang J-Y, Zhang C, Xiong Y, Guan K-L, Yuan H-X. The oncometabolite 2-hydroxyglutarate produced by mutant IDH1 sensitizes cells to ferroptosis. Cell Death Dis. 2019;10(10):1–12.
Zhang L, He L, Lugano R, Roodakker K, Bergqvist M, Smits A, et al. IDH mutation status is associated with distinct vascular gene expression signatures in lower-grade gliomas. Neuro Oncol. 2018;20(11):1505–16.
Han X, Xue X, Zhou H, Zhang G. A molecular view of the radioresistance of gliomas. Oncotarget. 2017;8(59):100931–41.
Croce CM, Zhang K, Wei Y. Announcing signal transduction and targeted therapy. Signal Transd Target Therapy. 2016;1(1):15006.
Levitzki A, Klein S. Signal transduction therapy of cancer. Mol Aspects Med. 2010;31(4):287–329.
Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010;60(3):166–93.
Dang Y, Wei X, Xue L, Wen F, Gu J, Zheng H. Long non-coding RNA in glioma: target miRNA and signaling pathways. Clin Lab. 2018;64(6):887–94.
Noorani I. Genetically engineered mouse models of gliomas: technological developments for translational discoveries. Cancers. 2019;11(9):1335.
Guzman F, Fazeli Y, Khuu M, Salcido K, Singh S, Benavente CA. Retinoblastoma tumor suppressor protein roles in epigenetic regulation. Cancers. 2020;12(10):2807.
Chen L, Liu S, Tao Y. Regulating tumor suppressor genes: post-translational modifications. Signal Transduct Target Ther. 2020;5(1):1–25.
Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8.
Olafson LR, Gunawardena M, Nixdorf S, McDonald KL, Rapkins RW. The role of tp53 gain-of-function mutation in multifocal glioblastoma. J Neurooncol. 2020;147(1):37–47.
Jiang L, Zawacka-Pankau J. The p53/mdm2/mdmx-targeted therapies—a clinical synopsis. Cell Death Dis. 2020;11(4):1–4.
Spiegelberg D, Mortensen AC, Lundsten S, Brown CJ, Lane DP, Nestor M. The MDM2/MDMX-p53 antagonist PM2 radiosensitizes wild-type p53 tumors. Can Res. 2018;78(17):5084–93.
Fontana R, Ranieri M, La Mantia G, Vivo M. Dual role of the alternative reading frame ARF protein in cancer. Biomolecules. 2019;9(3):87.
Rahme GJ, Luikart BW, Cheng C, Israel MA. A recombinant lentiviral PDGF-driven mouse model of proneural glioblastoma. Neuro Oncol. 2018;20(3):332–42.
Shorning BY, Dass MS, Smalley MJ, Pearson HB. The PI3K-AKT-mTOR pathway and prostate cancer: At the crossroads of AR, MAPK, and WNT signaling. Int J Mol Sci. 2020;21(12):4507.
Kalhori MR, Arefian E, Fallah Atanaki F, Kavousi K, Soleimani M. miR-548x and miR-4698 controlled cell proliferation by affecting the PI3K/AKT signaling pathway in glioblastoma cell lines. Sci Rep. 2020;10(1):1558.
Was H, Krol SK, Rotili D, Mai A, Wojtas B, Kaminska B, et al. Histone deacetylase inhibitors exert anti-tumor effects on human adherent and stem-like glioma cells. Clin Epigenet. 2019;11(1):1–13.
Romani M, Pistillo MP, Banelli B. Epigenetic targeting of glioblastoma. Front Oncol. 2018;8:448.
He J, Jiang Y, Liu L, Zuo Z, Zeng C. Circulating microRNAs as promising diagnostic biomarkers for patients with glioma: a meta-analysis. Front Neurol. 2021;11:8.
Qu K, Lin T, Pang Q, Liu T, Wang Z, Tai M, et al. Extracellular miRNA-21 as a novel biomarker in glioma: evidence from meta-analysis, clinical validation and experimental investigations. Oncotarget. 2016;7(23):33994.
Møller HG, Rasmussen AP, Andersen HH, Johnsen KB, Henriksen M, Duroux M. A systematic review of microRNA in glioblastoma multiforme: micro-modulators in the mesenchymal mode of migration and invasion. Mol Neurobiol. 2013;47(1):131–44.
Garcia-Bermudez J, Baudrier L, La K, Zhu XG, Fidelin J, Sviderskiy VO, et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat Cell Biol. 2018;20(7):775–81.
Zhou X, Ren Y, Moore L, Mei M, You Y, Xu P, et al. Downregulation of miR-21 inhibits EGFR pathway and suppresses the growth of human glioblastoma cells independent of PTEN status. Lab Invest. 2010;90(2):144–55.
Schramedei K, Mörbt N, Pfeifer G, Läuter J, Rosolowski M, Tomm JM, et al. MicroRNA-21 targets tumor suppressor genes ANP32A and SMARCA4. Oncogene. 2011;30(26):2975–85.
Gaur AB, Holbeck SL, Colburn NH, Israel MA. Downregulation of PDCD4 by miR-21 facilitates glioblastoma proliferation in vivo. Neuro Oncol. 2011;13(6):580–90.
Zhou X, Zhang J, Jia Q, Ren Y, Wang Y, Shi L, et al. Reduction of miR-21 induces glioma cell apoptosis via activating caspase 9 and 3. Oncol Rep. 2010;24(1):195–201.
Kwak HJ, Kim YJ, Chun KR, Woo YM, Park SJ, Jeong JA, et al. Downregulation of Spry2 by miR-21 triggers malignancy in human gliomas. Oncogene. 2011;30(21):2433–42.
Papagiannakopoulos T, Shapiro A, Kosik KS. MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res. 2008;68(19):8164–72.
Zhang J, Han L, Ge Y, Zhou X, Zhang A, Zhang C, et al. MiR-221/222 promote malignant progression of glioma through activation of the AKT pathway. Int J Oncol. 2010;36(4):913–20.
Zhang C-Z, Zhang J-X, Zhang A-L, Shi Z-D, Han L, Jia Z-F, et al. miR-221 and miR-222 target PUMA to induce cell survival in glioblastoma. Mol Cancer. 2010;9(1):1–9.
Xu H, Zhang Y, Qi L, Ding L, Jiang H, Yu H. NFIX circular RNA promotes glioma progression by regulating miR-34a-5p via notch signaling pathway. Front Mol Neurosci. 2018;11:225.
Xue F, Shen R, Chen X. Analysis of gene profiles in glioma cells identifies potential genes, miRNAs, and target sites of migratory cells. Tumori. 2015;101(5):542–8.
Li Q, Wang C, Cai L, Lu J, Zhu Z, Wang C, et al. miR-34a derived from mesenchymal stem cells stimulates senescence in glioma cells by inducing DNA damage. Mol Med Rep. 2019;19(3):1849–57.
Liu S, Jiang T, Zhong Y, Yu Y. miR-210 inhibits cell migration and invasion by targeting the brain-derived neurotrophic factor in glioblastoma. J Cell Biochem. 2019;120(7):11375–82.
Liu H, Chen C, Zeng J, Zhao Z, Hu Q. MicroRNA-210-3p is transcriptionally upregulated by hypoxia induction and thus promoting EMT and chemoresistance in glioma cells. PLoS ONE. 2021;16(7): e0253522.
Toraih EA, El-Wazir A, Abdallah HY, Tantawy MA, Fawzy MS. Deregulated microRNA signature following glioblastoma irradiation. Cancer Control. 2019;26(1):1073274819847226.
Shang C, Hong Y, Guo Y, Liu Y-H, Xue Y-X. MiR-210 up-regulation inhibits proliferation and induces apoptosis in glioma cells by targeting SIN3A. Med Sci Monit. 2014;20:2571.
Qin Q, Furong W, Baosheng L. Multiple functions of hypoxia-regulated miR-210 in cancer. J Exp Clin Cancer Res. 2014;33(1):1–10.
Zhang G, Chen L, Sun K, Khan AA, Yan J, Liu H, et al. Neuropilin-1 (NRP-1)/GIPC1 pathway mediates glioma progression. Tumour Biol. 2016;37(10):13777–88.
Zhang G, Chen L, Khan AA, Li B, Gu B, Lin F, et al. MiRNA-124-3p/neuropilin-1 (NRP-1) axis plays an important role in mediating glioblastoma growth and angiogenesis. Int J Cancer. 2018;143(3):635–44.
Xia HF, He TZ, Liu CM, Cui Y, Song PP, Jin XH, et al. MiR-125b expression affects the proliferation and apoptosis of human glioma cells by targeting Bmf. Cell Physiol Biochem. 2009;23(4–6):347–58.
Smirnova L, Gräfe A, Seiler A, Schumacher S, Nitsch R, Wulczyn FG. Regulation of miRNA expression during neural cell specification. Eur J Neurosci. 2005;21(6):1469–77.
Swellam M, Bakr NM, El Magdoub HM, Hamza MS, Ezz El Arab LR. Emerging role of miRNAs as liquid biopsy markers for prediction of glioblastoma multiforme prognosis. J Mol Neurosci. 2021;71(4):836–44.
Kuhn DE, Nuovo GJ, Terry AV, Martin MM, Malana GE, Sansom SE, et al. Chromosome 21-derived microRNAs provide an etiological basis for aberrant protein expression in human Down syndrome brains. J Biol Chem. 2010;285(2):1529–43.
Lee YS, Kim HK, Chung S, Kim K-S, Dutta A. Depletion of human micro-RNA miR-125b reveals that it is critical for the proliferation of differentiated cells but not for the down-regulation of putative targets during differentiation. J Biol Chem. 2005;280(17):16635–41.
Jiang Y, Ren W, Wang W, Xia J, Gou L, Liu M, et al. Inhibitor of β-catenin and TCF (ICAT) promotes cervical cancer growth and metastasis by disrupting e-cadherin/β-catenin complex. Oncol Rep. 2017;38(5):2597–606.
Zhang K, Zhu S, Liu Y, Dong X, Shi Z, Zhang A, et al. ICAT inhibits glioblastoma cell proliferation by suppressing Wnt/β-catenin activity. Cancer Lett. 2015;357(1):404–11.
Zhang Y, Li T, Guo P, Kang J, Wei Q, Jia X, et al. MiR-424-5p reversed epithelial-mesenchymal transition of anchorage-independent HCC cells by directly targeting ICAT and suppressed HCC progression. Sci Rep. 2014;4:6248.
Lin WH, Li J, Zhang B, Liu LS, Zou Y, Tan JF, et al. MicroRNA-1301 induces cell proliferation by downregulating ICAT expression in breast cancer. Biomed Pharmacother. 2016;83:177–85.
Zhou J, Du G, Fu H. MiR-296-3p promotes the proliferation of glioblastoma cells by targeting ICAT. Mol Med Rep. 2020;21(5):2151–61.
Lee H, Hwang SJ, Kim HR, Shin CH, Choi KH, Joung JG, et al. Neurofibromatosis 2 (NF2) controls the invasiveness of glioblastoma through YAP-dependent expression of CYR61/CCN1 and miR-296-3p. Biochim Biophys Acta. 2016;1859(4):599–611.
Shin J, Shin Y, Oh SM, Yang H, Yu WJ, Lee JP, et al. MiR-29b controls fetal mouse neurogenesis by regulating ICAT-mediated Wnt/β-catenin signaling. Cell Death Dis. 2014;5(10): e1473.
Chen J, Wu X, Xing Z, Ma C, Xiong W, Zhu X, et al. FOXG1 expression is elevated in glioma and inhibits glioma cell apoptosis. J Cancer. 2018;9(5):778–83.
Shibata M, Kurokawa D, Nakao H, Ohmura T, Aizawa S. MicroRNA-9 modulates Cajal-Retzius cell differentiation by suppressing FOXG1 expression in mouse medial pallium. J Neurosci. 2008;28(41):10415–21.
Zhen J, Zhang H, Dong H, Tong X. MiR-9-3p inhibits glioma cell proliferation and apoptosis by directly targeting FOXG1. Oncol Lett. 2020;20(2):2007–15.
Xu YF, Liu J, Wang J, Guo YC, Shen YZ. MiR-186 promotes the apoptosis of glioma U87 cells by down-regulating the expression of Smad6. Eur Rev Med Pharmacol Sci. 2020;24(14):7681–9.
Li C, Feng S, Chen L. MicroRNA-142-3p inhibits proliferation and induces apoptosis by targeting the high-mobility group box 1 via the Wnt/β-catenin signaling pathway in glioma. Int J Clin Exp Pathol. 2018;11(9):4493–502.
Gao C, Shen J, Meng Z-X, He X-F. Sevoflurane inhibits glioma cells proliferation and metastasis through miRNA-124-3p/ROCK1 axis. Pathol Oncol Res. 2020;26(2):947–54.
Singh A, Singh AK, Giri R, Kumar D, Sharma R, Valis M, et al. The role of microRNA-21 in the onset and progression of cancer. Future Med Chem. 2021;13(21):1885–906.
Aufiero S, Reckman YJ, Tijsen AJ, Pinto YM, Creemers EE. Circrnaprofiler: an R-based computational framework for the downstream analysis of circular RNAs. BMC Bioinform. 2020;21(1):1–9.
Mummidi S, Das NA, Carpenter AJ, Yoshida T, Yariswamy M, Mostany R, et al. RECK suppresses interleukin-17/TRAF3IP2-mediated MMP-13 activation and human aortic smooth muscle cell migration and proliferation. J Cell Physiol. 2019;234(12):22242–59.
Shen K-H, Hung J-H, Liao Y-C, Tsai S-T, Wu M-J, Chen P-S. Sinomenine inhibits migration and invasion of human lung cancer cell through downregulating expression of miR-21 and MMPs. Int J Mol Sci. 2020;21(9):3080.
Guan N, Wang R, Feng X, Li C, Guo W. Long non-coding RNA NBAT1 inhibits the progression of glioma through the miR-21/SOX7 axis. Oncol Lett. 2020;20(3):3024–34.
Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1:97–117.
Löffek S, Schilling O, Franzke CW. Series “matrix metalloproteinases in lung health and disease”: biological role of matrix metalloproteinases: a critical balance. Eur Respir J. 2011;38(1):191–208.
Peng G, Liu Y, Yang C, Shen C. MicroRNA-25 promotes cell proliferation, migration and invasion in glioma by directly targeting cell adhesion molecule 2. Exp Ther Med. 2022;23(1):16.
Liu Y, Jiang K, Zhi T, Xu X. MiR-720 is a key regulator of glioma migration and invasion by controlling TARSL2 expression. Hum Cell. 2021;34(5):1504–16.
Kwon NH, Fox PL, Kim S. Aminoacyl-tRNA synthetases as therapeutic targets. Nat Rev Drug Discov. 2019;18(8):629–50.
Suh JH, Park MC, Goughnour PC, Min BS, Kim SB, Lee WY, et al. Plasma lysyl-tRNA synthetase 1 (KARS1) as a novel diagnostic and monitoring biomarker for colorectal cancer. J Clin Med. 2020;9(2):533.
Nam SH, Kim D, Lee D, Lee H-M, Song D-G, Jung JW, et al. Lysyl-tRNA synthetase–expressing colon spheroids induce M2 macrophage polarization to promote metastasis. J Clin Investig. 2018;128(11):5034–55.
Nam SH, Kang M, Ryu J, Kim H-J, Kim D, Kim DG, et al. Suppression of lysyl-tRNA synthetase, KRS, causes incomplete epithelial-mesenchymal transition and ineffective cell-extracellular matrix adhesion for migration. Int J Oncol. 2016;48(4):1553–60.
Li Y, Wang Y, Yu L, Sun C, Cheng D, Yu S, et al. MiR-146b-5p inhibits glioma migration and invasion by targeting MMP16. Cancer Lett. 2013;339(2):260–9.
Yang B, Xia S, Ye X, Jing W, Wu B. MiR-379-5p targets microsomal glutathione transferase 1 (MGST1) to regulate human glioma in cell proliferation, migration and invasion and epithelial-mesenchymal transition (EMT). Biochem Biophys Res Commun. 2021;568:8–14.
De Luca A, Federici L, De Canio M, Stella L, Caccuri AM. New insights into the mechanism of JNK1 inhibition by glutathione transferase P1–1. Biochemistry. 2012;51(37):7304–12.
Cheng SY, Chen NF, Wen ZH, Yao ZK, Tsui KH, Kuo HM, et al. Glutathione S-transferase M3 is associated with glycolysis in intrinsic temozolomide-resistant glioblastoma multiforme cells. Int J Mol Sci. 2021;22(13):807.
Shen H, Xu L, You C, Tang H, Wu H, Zhang Y, et al. MiR-665 is downregulated in glioma and inhibits tumor cell proliferation, migration and invasion by targeting high mobility group box 1. Oncol Lett. 2021;21(2):156.
Shi X, Wang J, Lei Y, Cong C, Tan D, Zhou X. Research progress on the PI3K/AKT signaling pathway in gynecological cancer (review). Mol Med Rep. 2019;19(6):4529–35.
Mirzadeh Azad F, Naeli P, Malakootian M, Baradaran A, Tavallaei M, Ghanei M, et al. Two lung development-related microRNAs, miR-134 and miR-187, are differentially expressed in lung tumors. Gene. 2016;577(2):221–6.
Li X, Wu C, Chen N, Gu H, Yen A, Cao L, et al. PI3K/AKT/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget. 2016;7(22):33440–50.
Li B, Xu WW, Lam AKY, Wang Y, Hu HF, Guan XY, et al. Significance of PI3K/AKT signaling pathway in metastasis of esophageal squamous cell carcinoma and its potential as a target for anti-metastasis therapy. Oncotarget. 2017;8(24):38755–66.
Xu B, Mei J, Ji W, Huo Z, Bian Z, Jiao J, et al. MicroRNAs involved in the EGFR pathway in glioblastoma. Biomed Pharmacother. 2021;134: 111115.
Zhong X, Tang J, Li H, Shi X, Wu Y, Xia D, et al. MiR-3175 promotes epithelial-mesenchymal transition by targeting Smad7 in human conjunctiva and pterygium. FEBS Lett. 2020;594(7):1207–17.
Bai Y, Liao H, Liu T, Zeng X, Xiao F, Luo L, et al. MiR-296-3p regulates cell growth and multi-drug resistance of human glioblastoma by targeting ether-à-go-go (eag1). Eur J Cancer. 2013;49(3):710–24.
Xiong W, Ran J, Jiang R, Guo P, Shi X, Li H, et al. MiRNA-320a inhibits glioma cell invasion and migration by directly targeting aquaporin 4. Oncol Rep. 2018;39(4):1939–47.
Zhang G, Chen L, Khan AA, Li B, Gu B, Lin F, et al. MiRNA-124-3p/neuropilin-1 (NRP-1) axis plays an important role in mediating glioblastoma growth and angiogenesis. Int J Cancer. 2018;143(3):635–44.
Buruiană A, Florian ȘI, Florian AI, Timiș T-L, Mihu CM, Miclăuș M, et al. The roles of miRNA in glioblastoma tumor cell communication: diplomatic and aggressive negotiations. Int J Mol Sci. 2020;21(6):1950.
Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science. 2017;357(6357):1255–61.
Yuan M, Da Silva ACA, Arnold A, Okeke L, Ames H, Correa-Cerro LS, et al. MicroRNA (miR) 125b regulates cell growth and invasion in pediatric low grade glioma. Sci Rep. 2018;8(1):1–14.
Smits M, Wurdinger T, van het Hof B, Drexhage JA, Geerts D, Wesseling P, et al. Myc-associated zinc finger protein (MAZ) is regulated by miR-125b and mediates VEGF-induced angiogenesis in glioblastoma. FASEB J. 2012;26(6):2639–47.
Mathew LK, Skuli N, Mucaj V, Lee SS, Zinn PO, Sathyan P, et al. MiR-218 opposes a critical RTK-HIF pathway in mesenchymal glioblastoma. Proc Natl Acad Sci. 2014;111(1):291–6.
Malekpour Afshar R, Mollaei HR, Shokrizadeh M, Iranpour M. Evaluation expression of microRNA-93 and integrin β8 in different types of glioma tumors. Asian Pac J Cancer Prev. 2017;18(3):603–8.
Yi G-Z, Huang G, Guo M, Zhang X, Wang H, Deng S, et al. Acquired temozolomide resistance in MGMT-deficient glioblastoma cells is associated with regulation of DNA repair by DHC2. Brain. 2019;142(8):2352–66.
Jiapaer S, Furuta T, Tanaka S, Kitabayashi T, Nakada M. Potential strategies overcoming the temozolomide resistance for glioblastoma. Neurol Med Chir. 2018;58(10):405.
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20.
Garofalo M, Croce CM. MicroRNAs as therapeutic targets in chemoresistance. Drug Resist Updat. 2013;16(3–5):47–59.
Jayaraj R, Nayagam SG, Kar A, Sathyakumar S, Mohammed H, Smiti M, et al. Clinical theragnostic relationship between drug-resistance specific miRNA expressions, chemotherapeutic resistance, and sensitivity in breast cancer: a systematic review and meta-analysis. Cells. 2019;8:10.
Si W, Shen J, Zheng H, Fan W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin Epigenet. 2019;11(1):25.
Lee SY. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016;3(3):198–210.
Li G, Lan Q. Exosome-mediated transfer of CIRC-GLIS3 enhances temozolomide resistance in glioma cells through the miR-548m/MED31 axis. Cancer Biother Radiopharm. 2021.
Shi L, Chen J, Yang J, Pan T, Zhang S, Wang Z. MiR-21 protected human glioblastoma U87MG cells from chemotherapeutic drug temozolomide induced apoptosis by decreasing Bax/Bcl-2 ratio and caspase-3 activity. Brain Res. 2010;1352:255–64.
Movahedpour A, Khatami SH, Khorsand M, Salehi M, Savardashtaki A, Mirmajidi SH, et al. Exosomal noncoding RNAs: key players in glioblastoma drug resistance. Mol Cell Biochem. 2021;476(11):4081–92.
Shi L, Zhang S, Feng K, Wu F, Wan Y, Wang Z, et al. MicroRNA-125b-2 confers human glioblastoma stem cells resistance to temozolomide through the mitochondrial pathway of apoptosis. Int J Oncol. 2012;40(1):119–29.
Chen L, Zhang J, Han L, Zhang A, Zhang C, Zheng Y, et al. Downregulation of miR-221/222 sensitizes glioma cells to temozolomide by regulating apoptosis independently of p53 status. Oncol Rep. 2012;27(3):854–60.
She X, Yu Z, Cui Y, Lei Q, Wang Z, Xu G, et al. MiR-128 and miR-149 enhance the chemosensitivity of temozolomide by Rap1B-mediated cytoskeletal remodeling in glioblastoma. Oncol Rep. 2014;32(3):957–64.
She X, Yu Z, Cui Y, Lei Q, Wang Z, Xu G, et al. MiR-181 subunits enhance the chemosensitivity of temozolomide by Rap1B-mediated cytoskeleton remodeling in glioblastoma cells. Med Oncol. 2014;31(4):892.
Chen Q, Wang W, Chen S, Chen X, Lin Y. MiR-29a sensitizes the response of glioma cells to temozolomide by modulating the p53/MDM2 feedback loop. Cell Mol Biol Lett. 2021;26(1):1–15.
Ujifuku K, Mitsutake N, Takakura S, Matsuse M, Saenko V, Suzuki K, et al. miR-195, miR-455-3p and miR-10a(*) are implicated in acquired temozolomide resistance in glioblastoma multiforme cells. Cancer Lett. 2010;296(2):241–8.
Chen G, Chen Z, Zhao H. MicroRNA-155-3p promotes glioma progression and temozolomide resistance by targeting Six1. J Cell Mol Med. 2020;24(9):5363–74.
Zhou X, Wu W, Zeng A, Nie E, Jin X, Yu T, et al. MicroRNA-141-3p promotes glioma cell growth and temozolomide resistance by directly targeting p53. Oncotarget. 2017;8(41):71080–94.
Wang H, Ren S, Xu Y, Miao W, Huang X, Qu Z, et al. MicroRNA-195 reverses the resistance to temozolomide through targeting cyclin E1 in glioma cells. Anticancer Drugs. 2019;30(1):81–8.
Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005;24(17):2776–86.
Fan TY, Wang H, Xiang P, Liu YW, Li HZ, Lei BX, et al. Inhibition of EZH2 reverses chemotherapeutic drug TMZ chemosensitivity in glioblastoma. Int J Clin Exp Pathol. 2014;7(10):6662–70.
Zhi T, Yu T, Pan M, Nie E, Wu W, Wang X, et al. EZH2 alteration driven by microRNA-524-5p and microRNA-324-5p promotes cell proliferation and temozolomide resistance in glioma. Oncotarget. 2017;8(56):96239–48.
Kan MC, Oruganty-Das A, Cooper-Morgan A, Jin G, Swanger SA, Bassell GJ, et al. CPEB4 is a cell survival protein retained in the nucleus upon ischemia or endoplasmic reticulum calcium depletion. Mol Cell Biol. 2010;30(24):5658–71.
Li C, Feng S, Chen L. MSC-AS1 knockdown inhibits cell growth and temozolomide resistance by regulating miR-373-3p/CPEB4 axis in glioma through PI3K/AKT pathway. Mol Cell Biochem. 2021;476(2):699–713.
Morgenroth A, Vogg AT, Ermert K, Zlatopolskiy B, Mottaghy FM. Hedgehog signaling sensitizes glioma stem cells to endogenous nano-irradiation. Oncotarget. 2014;5(14):5483–93.
Jin J, Grigore F, Chen CC, Li M. Self-renewal signaling pathways and differentiation therapies of glioblastoma stem cells (review). Int J Oncol. 2021;59(1):1–11.
Diana A, Gaido G, Murtas D. MicroRNA signature in human normal and tumoral neural stem cells. Int J Mol Sci. 2019;20(17):4123.
Sana J, Busek P, Fadrus P, Besse A, Radova L, Vecera M, et al. Identification of microRNAs differentially expressed in glioblastoma stem-like cells and their association with patient survival. Sci Rep. 2018;8(1):2836.
Balachandran AA, Larcher LM, Chen S, Veedu RN. Therapeutically significant microRNAs in primary and metastatic brain malignancies. Cancers (Basel). 2020;12(9):1–9.
Bier A, Giladi N, Kronfeld N, Lee HK, Cazacu S, Finniss S, et al. MicroRNA-137 is downregulated in glioblastoma and inhibits the stemness of glioma stem cells by targeting RTVP-1. Oncotarget. 2013;4(5):665–76.
Bier A, Hong X, Cazacu S, Goldstein H, Rand D, Xiang C, et al. MiR-504 modulates the stemness and mesenchymal transition of glioma stem cells and their interaction with microglia via delivery by extracellular vesicles. Cell Death Dis. 2020;11(10):899.
Bradshaw A, Wickremesekera A, Brasch HD, Chibnall AM, Davis PF, Tan ST, et al. Cancer stem cells in glioblastoma multiforme. Front Surg. 2016;3:48.
Auffinger B, Spencer D, Pytel P, Ahmed AU, Lesniak MS. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev Neurother. 2015;15(7):741–52.
Huang W, Zhong Z, Luo C, Xiao Y, Li L, Zhang X, et al. The miR-26a/AP-2α/Nanog signaling axis mediates stem cell self-renewal and temozolomide resistance in glioma. Theranostics. 2019;9(19):5497–516.
Guo Y, Liu S, Wang P, Zhao S, Wang F, Bing L, et al. Expression profile of embryonic stem cell-associated genes OCT4, SOX2 and Nanog in human gliomas. Histopathology. 2011;59(4):763–75.
Peng L, Ming Y, Zhang L, Zhou J, Xiang W, Zeng S, et al. MicroRNA-30a suppresses self-renewal and tumorigenicity of glioma stem cells by blocking the NT5E-dependent Akt signaling pathway. FASEB J. 2020;34(4):5128–43.
Chao CC, Kan D, Lu KS, Chien CL. The role of microRNA-30c in the self-renewal and differentiation of C6 glioma cells. Stem Cell Res. 2015;14(2):211–23.
Wang H, Sun T, Hu J, Zhang R, Rao Y, Wang S, et al. MiR-33a promotes glioma-initiating cell self-renewal via PKA and NOTCH pathways. J Clin Invest. 2014;124(10):4489–502.
Zhang D, Yang G, Chen X, Li C, Wang L, Liu Y, et al. MiR-300 promotes self-renewal and inhibits the differentiation of glioma stem-like cells. J Mol Neurosci. 2014;53:637–44.
Kopkova A, Sana J, Fadrus P, Slaby O. Cerebrospinal fluid microRNAs as diagnostic biomarkers in brain tumors. Clin Chem Lab Med. 2018;56(6):869–79.
Piwecka M, Rolle K, Belter A, Barciszewska AM, Żywicki M, Michalak M, et al. Comprehensive analysis of microRNA expression profile in malignant glioma tissues. Mol Oncol. 2015;9(7):1324–40.
Xiao Y, Zhang L, Song Z, Guo C, Zhu J, Li Z, et al. Potential diagnostic and prognostic value of plasma circulating microRNA-182 in human glioma. Med Sci Monit. 2016;22:855–62.
Santangelo A, Imbrucè P, Gardenghi B, Belli L, Agushi R, Tamanini A, et al. A microRNA signature from serum exosomes of patients with glioma as complementary diagnostic biomarker. J Neurooncol. 2018;136(1):51–62.
Ivo D’Urso P, Fernando D’Urso O, Damiano Gianfreda C, Mezzolla V, Storelli C, Marsigliante S. MiR-15b and miR-21 as circulating biomarkers for diagnosis of glioma. Curr Genomics. 2015;16(5):304–11.
Akers JC, Hua W, Li H, Ramakrishnan V, Yang Z, Quan K, et al. A cerebrospinal fluid microRNA signature as biomarker for glioblastoma. Oncotarget. 2017;8(40):68769–79.
Yuan J, Zheng Z, Zheng Y, Lu X, Xu L, Lin L. MicroRNA-328 is a favorable prognostic marker in human glioma via suppressing invasive and proliferative phenotypes of malignant cells. Int J Neurosci. 2016;126(2):145–53.
Drusco A, Fadda P, Nigita G, Fassan M, Bottoni A, Gardiman MP, et al. Circulating microRNAs predict survival of patients with tumors of glial origin. EBioMedicine. 2018;30:105–12.
Yue X, Lan F, Hu M, Pan Q, Wang Q, Wang J. Downregulation of serum microRNA-205 as a potential diagnostic and prognostic biomarker for human glioma. J Neurosurg. 2016;124(1):122–8.
Hayes J, Thygesen H, Gregory W, Westhead DR, French PJ, Van Den Bent MJ, et al. A validated microRNA profile with predictive potential in glioblastoma patients treated with bevacizumab. Mol Oncol. 2016;10(8):1296–304.
Son JC, Jeong HO, Park D, No SG, Lee EK, Lee J, et al. MiR-10a and miR-204 as a potential prognostic indicator in low-grade gliomas. Cancer Inform. 2017;16:1176935117702878.
Zhang Y, Chen J, Xue Q, Wang J, Zhao L, Han K, et al. Prognostic significance of microRNAs in glioma: a systematic review and meta-analysis. Biomed Res Int. 2019;2019:4015969.
Niyazi M, Zehentmayr F, Niemöller OM, Eigenbrod S, Kretzschmar H, Schulze-Osthoff K, et al. MiRNA expression patterns predict survival in glioblastoma. Radiat Oncol. 2011;6:153.
Yuan Y, Zhang H, Liu X, Lu Z, Li G, Lu M, et al. MicroRNA signatures predict prognosis of patients with glioblastoma multiforme through the Cancer Genome Atlas. Oncotarget. 2017;8(35):58386–93.
Hayes J, Thygesen H, Tumilson C, Droop A, Boissinot M, Hughes TA, et al. Prediction of clinical outcome in glioblastoma using a biologically relevant nine-microRNA signature. Mol Oncol. 2015;9(3):704–14.
Chen W, Yu Q, Chen B, Lu X, Li Q. The prognostic value of a seven-microRNA classifier as a novel biomarker for the prediction and detection of recurrence in glioma patients. Oncotarget. 2016;7(33):53392.
Beyer S, Fleming J, Meng W, Singh R, Haque SJ, Chakravarti A. The role of miRNAs in angiogenesis, invasion and metabolism and their therapeutic implications in gliomas. Cancers (Basel). 2017;9(7):87.
Slaby O, Lakomy R, Fadrus P, Hrstka R, Kren L, Lzicarova E, et al. MicroRNA-181 family predicts response to concomitant chemoradiotherapy with temozolomide in glioblastoma patients. Neoplasma. 2010;57(3):264–9.
Haemmig S, Baumgartner U, Glück A, Zbinden S, Tschan MP, Kappeler A, et al. miR-125b controls apoptosis and temozolomide resistance by targeting TNFAIP3 and NKIRAS2 in glioblastomas. Cell Death Dis. 2014;5(6): e1279.
Shah MY, Ferrajoli A, Sood AK, Lopez-Berestein G, Calin GA. MicroRNA therapeutics in cancer—an emerging concept. EBioMedicine. 2016;12:34–42.
Thorsen SB, Obad S, Jensen NF, Stenvang J, Kauppinen S. The therapeutic potential of microRNAs in cancer. Cancer J. 2012;18(3):275–84.
Preethi KA, Lakshmanan G, Sekar D. AntagomiR technology in the treatment of different types of cancer. Epigenomics. 2021;13(7):481–4.
Raue R, Frank A-C, Syed SN, Brüne B. Therapeutic targeting of microRNAs in the tumor microenvironment. Int J Mol Sci. 2021;22(4):2210.
Liang AL, Zhang TT, Zhou N, Wu CY, Lin MH, Liu YJ. MiRNA-10b sponge: an anti-breast cancer study in vitro. Oncol Rep. 2016;35(4):1950–8.
Tay FC, Lim JK, Zhu H, Hin LC, Wang S. Using artificial microRNA sponges to achieve microRNA loss-of-function in cancer cells. Adv Drug Deliv Rev. 2015;81:117–27.
Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. 2004;10(3):544–50.
Ramalingam P, Palanichamy JK, Singh A, Das P, Bhagat M, Kassab MA, et al. Biogenesis of intronic miRNAs located in clusters by independent transcription and alternative splicing. RNA. 2014;20(1):76–87.
Datta J, Kutay H, Nasser MW, Nuovo GJ, Wang B, Majumder S, et al. Methylation mediated silencing of microRNA-1 gene and its role in hepatocellular carcinogenesis. Can Res. 2008;68(13):5049–58.
To KKW, Fong W, Tong CWS, Wu M, Yan W, Cho WCS. Advances in the discovery of microRNA-based anticancer therapeutics: latest tools and developments. Expert Opin Drug Discov. 2020;15(1):63–83.
Auffinger B, Thaci B, Ahmed A, Ulasov I, Lesniak MS. MicroRNA targeting as a therapeutic strategy against glioma. Curr Mol Med. 2013;13(4):535–42.
Inoue J, Inazawa J. Cancer-associated miRNAs and their therapeutic potential. J Hum Genet. 2021;66(9):937–45.
Wullner U, Neef I, Tur MK, Barth S. Targeted delivery of short interfering RNAs-strategies for in vivo delivery. Recent Pat Anti-Cancer Drug Discov. 2009;4(1):1–8.
Ventura A, Jacks T. MiRNAs and cancer: a little RNA goes a long way. Cell. 2009;136(4):586.
Zhang Y, Wang Z, Gemeinhart RA. Progress in microRNA delivery. J Control Release. 2013;172(3):962–74.
Yang N. An overview of viral and nonviral delivery systems for microRNA. Int J Pharm Investig. 2015;5(4):179.
Sela H, Cohen H, Elia P, Zach R, Karpas Z, Zeiri Y. Spontaneous penetration of gold nanoparticles through the blood brain barrier (BBB). J Nanobiotechnol. 2015;13(1):1–9.
Lee SWL, Paoletti C, Campisi M, Osaki T, Adriani G, Kamm RD, et al. MicroRNA delivery through nanoparticles. J Control Release. 2019;313:80–95.
Pegtel DM, Gould SJ. Exosomes. Annu Rev Biochem. 2019;88:487–514.
Chen Y, Zhu X, Zhang X, Liu B, Huang L. Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol Ther. 2010;18(9):1650–6.
Xin H, Jiang Y, Lv W, Xu J. Liposome-based drug delivery for brain tumor theranostics. Nanotechnol-Based Target Drug Deliv Syst Brain Tumors. 2018;1:245–66.
Wang S, Yin Y, Liu S. Roles of microRNAs during glioma tumorigenesis and progression. Histol Histopathol. 2018;34(3):213–22.
Zhang D, Yang G, Chen X, Li C, Wang L, Liu Y, et al. MiR-300 promotes self-renewal and inhibits the differentiation of glioma stem-like cells. J Mol Neurosci. 2014;53(4):637–44.
Acknowledgements
Not applicable.
Funding
No specific source of funding is associated with this work.
Author information
Authors and Affiliations
Contributions
The authors confirm contributions to this paper as follows; AM: conceptualization, investigation, writing, and draft preparation; AR: investigation and writing; ZBA: investigation and writing; RS: investigation and writing; MS: investigation and writing; EA; supervision and validation; OV: supervision and review and editing and validation. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mafi, A., Rahmati, A., Babaei Aghdam, Z. et al. Recent insights into the microRNA-dependent modulation of gliomas from pathogenesis to diagnosis and treatment. Cell Mol Biol Lett 27, 65 (2022). https://doi.org/10.1186/s11658-022-00354-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11658-022-00354-4