
Abstract
Purpose of Review
Isocitrate dehydrogenase (IDH) mutant gliomas are a distinct type of primary brain tumors with unique characteristics, behavior, and disease outcomes. This article provides a review of standard of care treatment options and innovative, therapeutic approaches that are currently under investigation for these tumors.
Recent Findings
Extensive pre-clinical data and a variety of clinical studies support targeting IDH mutations in glioma using different mechanisms, which include direct inhibition and immunotherapies that target metabolic and epigenomic vulnerabilities caused by these mutations.
Summary
IDH mutations have been recognized as an oncogenic driver in gliomas for more than a decade and as a positive prognostic factor influencing the research for new therapeutic methods including IDH inhibitors, DNA repair inhibitors, and immunotherapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gliomas are the most common malignant primary brain tumors in adults [1, 2]. Classification of these tumors has evolved in recent years with the identification of molecular features including isocitrate dehydrogenase (IDH) mutations in gliomas in 2008 [3]. In humans, the IDH family includes three isoforms: IDH1, IDH2, and IDH3. All three forms are essential for several metabolic processes, such as the Krebs cycle. Recognized as an oncogenic event [4,5,6], IDH mutations are highly prevalent in gliomas and confer significant improved survival when compared to the IDH wild-type (IDH-WT) glioma [7,8,9].
The World Health Organization (WHO) Classification of Central Nervous System (CNS) Tumors (2021) classifies adult-type diffuse glioma based on the presence of IDH mutations and other key molecular alterations in astrocytoma IDH mutant, oligodendroglioma IDH mutant and 1p/19q-co-deleted, and glioblastoma IDH-WT [10].
Standard of care therapy for IDH mutant gliomas starts with maximal safe resection when feasible. Surgery has both diagnostic and therapeutic objectives and, in the majority of the cases, is followed by a combination of radiation and chemotherapy. The radiation dose depends on the grade of glioma, and the chemotherapy relies on a variety of factors including, but not limited to, histopathological categories, age at diagnosis, comorbidities, and physician preference [11,12,13].
Although significant advances have been made in the molecular characterization of gliomas, clinicians face challenges in managing this disease. Standard of care options offer limited survival benefits and may result in long-term toxicities including cognitive decline. This affects quality of life and remains a significant burden for patients and their families [14]. This article provides an overview of IDH mutant gliomas, standard of care treatment, and novel therapeutic strategies, including IDH inhibitors, agents targeting DNA repair mechanisms and epigenetic vulnerabilities, and immunotherapy.
Molecular Aspects and Classification
Historically, glioma classification relied on histology and immunohistochemistry to describe the tumors’ microscopic appearance and classify a spectrum of tumors with overlapping features. However, in 2016 the World Health Organization (WHO) introduced molecular markers in its classification for the first time, allowing these tumors to be defined by their molecular features resulting in a more accurate classification and prognosis [15,16,17,18]. These changes were expanded in the 2021 WHO Classification of CNS Tumors [10].
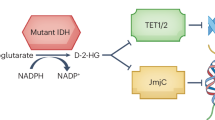
One of the key molecular alterations incorporated into the WHO Classifications of CNS Tumors in 2016 are mutations in IDH enzymes, which were originally discovered in 2006 in colorectal cancer and reported in gliomas in 2008 [3, 19, 20]. The IDH enzyme family consists of three isoforms that catalyze the oxidative decarboxylation of isocitrate to produce α-ketoglutarate. IDH mutations, occurring early in glioma oncogenesis, lead to the accumulation of the oncometabolite D-2-hydroxyglutarate (2-HG), which is linked to metabolic and epigenetic dysregulation and includes inhibition of normal cellular differentiation and hypermethylation that leads to disease [21,22,23] (Fig. 1).

The IDH enzymes family and their role in cell metabolism as well as the effect of IDH mutations on the Krebs cycle and the accumulation of D-2-HG

The 2021 WHO classification of adult-type diffuse gliomas
The 2021 WHO classification includes two adult-type diffuse IDH mutant gliomas, oligodendrogliomas (grades 2 or 3) and astrocytomas (grades 2, 3, or 4). The system also classifies tumors that do not carry IDH mutations (IDH-wildtype) in the presence of other molecular features, such as Epidermal growth factor receptor (EGFR) amplification, TERT promoter mutation, or the gain of chromosome seven with loss of chromosome 10 (+7/-10) to grade 4 glioblastomas regardless of their histologic appearance [10]. The 2021 WHO classification also defines oligodendrogliomas by the loss of chromosomes 1p and 19q (1p19q co-deletion) compared to astrocytomas that do not carry that feature (Fig. 2). Classifying IDH mutant tumors into grades 2 and 3 in oligodendrogliomas and 2, 3, and 4 in astrocytomas continues to rely on histologic appearance, except when astrocytic tumors carry homozygous deletion of CDKN2A/B making them grade 4 tumors independent of histologic characteristics. [24].
Clinical Manifestation and Prognosis
Patients with gliomas harboring IDH mutations typically present at a younger age compared to IDH wild type gliomas, with a peak incidence between ages of 35 and 44, according to the Central Brain Tumor Registry of the United States (CBTRUS) data base report. IDH mutant gliomas have a higher incidence rate in males with an approximate male to female ratio of 1.3 [25, 26]. Low-grade IDH mutant gliomas have a variety of presenting symptoms. Reports show that up to 80% of the patients have seizures as an initial symptom [13, 27]. Less frequently, patients present with focal neurological deficits, cognitive and behavioral changes, and signs of increased intracranial pressure. These tumors are often found incidentally with brain imaging while patients are undergoing evaluation for unrelated symptoms or during trauma assessments [28, 29]. Conversely, high-grade gliomas harboring IDH mutations may present more aggressively with a faster clinical deterioration [30].
IDH mutant gliomas appear as expansive lesions on magnetic resonance images (MRI) with hyperintensity on T2-weighted images and T2-fluid-attenuated inversion recovery sequences (FLAIR). Grade 2 IDH mutant gliomas do not commonly enhance upon the administration of IV gadolinium, unlike grade 3 and 4 IDH mutant tumors that often enhance or have an enhancing component within a larger non-enhancing tumor. IDH mutations are prognostic factors for longer survival independent of the histologic phenotype, and they improve progression free survival (PFS) [5, 6, 9, 31]. Moreover, IDH mutations are associated with better outcomes in high-grade gliomas as well. The median overall survival for patients with IDH mutant astrocytoma, CNS WHO grade 3 (previously known as anaplastic astrocytoma), is 65 months versus 20 months for IDH wild type [32, 33].
Standard of Care Therapies
In discussing therapy modalities for IDH mutant gliomas, this review differentiates between low-grade gliomas carrying these mutations as grade 2 oligodendrogliomas and astrocytomas versus IDH mutant high-grade gliomas, which include grade 3 oligodendrogliomas and grades 3 and 4 astrocytomas. This acknowledges the different behavior, and subsequently, the different management modalities between these low- and high-grade gliomas.
Surgery
Surgery remains the most important first step for glioma diagnosis and management. Historically, surgery was performed on patients presenting with large or symptomatic lesions, or when the initial imaging revealed features of high-grade pathology, such as the presence of edema, enhancement, or necrosis [34, 35]. The timing of surgery in small and non-enhancing tumors had been debated, especially in tumors that are asymptomatic and found incidentally; however, recently the field has moved towards performing early surgical resection [36].
A Norwegian study published in 2017 retrospectively analyzed the cases of 153 patients in two centers with different surgical treatment strategies. The first center favored early surgical resection of low-grade tumors, and the other favored observation following a diagnostic biopsy. The researchers found that the overall survival (OS) of patients treated at the center that favored early surgical resection was 14.4 years (95% CI 10.4–18.5) compared to 5.8 years (95% CI 4.5–7.2) in the center that favored observation (p=0.001) [37]. Multiple other retrospective and observational studies confirmed improved survival with early surgical resection, even for the small and asymptomatic tumor [38, 39]. In addition, early surgical intervention allows for a definitive pathologic and molecular diagnosis that informs treatment selection and determines prognosis.
Clinical experts agree on the benefit of surgical resection compared to a needle biopsy for an accurate diagnosis given the heterogeneity of gliomas [40, 41]. From the therapeutic perspective, the extent of surgical resection has a significant effect on the clinical outcomes of both high- and low-grade gliomas. In non-enhancing IDH mutant gliomas, extensive resection that targets all the non-enhancing disease area remains the standard first-line strategy. For enhancing tumors, the goal is to resect all of the enhancing component. Many of the studies supporting surgical resection were conducted prior to the molecular characterization of these tumors [42, 43]. However, modern era studies confirm these resection strategies impact clinical outcomes. One recent study retrospectively evaluated 228 tumors histologically classified as low-grade gliomas. The researchers molecularly re-evaluated these tumors to update their classification based on the 2016 WHO classification and found that the residual post-operative volume negatively affected OS with a hazard ratio of 1.01 (95% CI: 1.002–1.02; P = 0.016) per cm3 increase in volume [44]. Certain studies advocate for supramaximal resection of gliomas beyond the non-enhancing disease component and include a prospective study with 449 patients by Rossi et al. [45, 46]. However, the extension of the resection needed outside the border of visible tumor to achieve better outcomes is still unclear and long-term survival data is still needed [47]. The benefit of extensive surgical resection also applies to high-grade gliomas harboring IDH mutations. Multiple studies have shown that extensive resections of both enhancing and non-enhancing tumor components are associated with significant survival benefit [48, 49].
Radiation Therapy
Radiation therapy (RT) continues to play a significant role in managing IDH mutant gliomas; however, there are long-term side effects on cognition. To address this, researchers are evaluating the timing of RT during treatment course. More specifically, they are studying the safety effects of delaying RT in patients with “low-risk” features defined as younger than 40 years old, gross total resection of their tumor, positive for 1p-19q co-deletion, and good performance status and neurological function [50,51,52].
The timing of post-operative radiation in low grade IDH mutant gliomas has been controversial, but there is growing evidence for delaying radiation in certain cases. The EORTC 22845, a study by the European Organization for Research and Treatment of Cancer, was conducted in the pre-IDH era before gliomas were classified by IDH mutations. The study compared immediate radiation versus salvage radiation at progression in histologically-defined grade 2 gliomas. Results published in 2005 reported no difference between the two treatments in OS [53]. A study by the Radiation Therapy Oncology Group (RTOG 9802) followed low-risk grade 2 glioma patients who were defined as younger than 40 years old, had a gross total resection of their tumor, good performance status and neurological function, and received delayed radiation at progression. In addition, serial MRI observation did not adversely affect OS in these patients [52]. Based on this evidence, low-risk patients with IDH mutant low-grade gliomas can be observed with serial MRI scans, and radiation can be deferred till progression.
However, for the treatment of high-risk grade 2 glioma patients and patients with grade 3 and 4 gliomas harboring IDH mutations, immediate radiation post-operatively is recommended [26, 54].
The modality of radiation is also being assessed for impact on long-term side effects by comparing proton and photon therapy. A prospective single-arm study observed 20 patients with grade 2 gliomas, who were treated with proton therapy, and followed them over a median of 5.1 years. Results revealed no decline in the patients’ quality of life or cognitive deficits [55]. An on-going phase II clinical trial by the NRG Oncology Group (previously RTOG) is comparing proton versus photon radiation therapy in IDH mutant grade 2 or 3 gliomas.
Chemotherapy
Studies in the late 1990s and early 2000s began reporting a response in low-grade gliomas using chemotherapy alkylating agents, such as temozolomide and regimens of procarbazine, lomustine, and vincristine (PCV) [56,57,58,59,60]. The RTOG 9802 protocol (discussed above) included another phase II study for high-risk patients with grade 2 gliomas [57]. The study defined these high-risk patients as age 40 or older and those who had a sub-total resection or biopsies of their tumors. The study reported tumor regression in a meaningful proportion of patients using PCV. Based on these results, a phase III trial took place randomizing patients with high-risk grade 2 glioma to radiation alone versus radiation followed by six cycles of PCV. Patients treated with radiation alone had a median OS of 7.8 years versus 13.3 years in the radiation plus PCV arm (HR, 0.59; p = .003) [52]. Because these initial studies took place prior to the molecular era, the researchers in RTOG 9802 later performed a subset analysis and found that the improvement in overall median survival was statistically significant in patients with tumors harboring IDH1 mutations. Bell et al. assessed the same tumors from RTOG 9802 that had sufficient tissue with genomic sequencing to identify IDH mutation and 1p/19q co-deletion. The survival improvement seen in the original analysis was reported in patients with tumors harboring these mutations and not in the IDH wild-type [61].
Similar observations were seen with RTOG 9402 and EORTC 26951 studies that looked at patients with IDH mutant high-grade gliomas and tumors formerly known as anaplastic oligodendrogliomas (grade 3). In these randomized studies, patients received radiation alone versus radiation and PCV. Both trials reported improvement in OS in the PCV arm, which was limited to the tumors harboring 1p/19q co-deletion [62, 63].
Moreover, the addition of chemotherapy improves outcomes in high-grade gliomas with IDH mutations and intact 1p/19q confirming the predictive value of IDH mutations independently from the 1p/19q co-deletion status. The CATNON trial looked at grade 3 gliomas with IDH mutations and intact 1p/19q. Seven hundred fifty-one patients were randomly assigned (1:1:1:1) to radiotherapy alone, radiotherapy with concurrent oral temozolomide, radiotherapy with adjuvant oral temozolomide (12 cycles), or radiotherapy with both concurrent and adjuvant temozolomide. Adjuvant temozolomide chemotherapy, but not concurrent temozolomide chemotherapy, was associated with a survival benefit in patients with 1p/19q non-co-deleted anaplastic glioma. The clinical benefit was dependent on IDH1 and IDH2 mutational status [64, 65].
The ongoing CODEL phase III trial evaluates newly-diagnosed patients with IDH mutant, 1p/19q co-deleted gliomas grades 2 and 3, and compares the use of radiation followed by PCV versus radiation with concurrent temozolomide followed by adjuvant temozolomide [66]. Although the efficacy between temozolomide and PCV is still unclear, guidelines, including those recently published by an expert panel of the American Society of Clinical Oncology (ASCO) and the Society for Neuro-Oncology (SNO), allow both treatments to be used [54, 67]. Temozolomide’s toxicity profile is more manageable, and physicians may consider patient’s age and co-morbidities among other factors when deciding on chemotherapy options [64].
Despite the above-mentioned advances in managing gliomas with IDH mutations, there is still no known curative therapy, and patients continue to suffer premature death and cognitive decline.
Advances in Therapy Modalities
Without a curative therapy for IDH mutant gliomas regardless of the grade, research is ongoing for novel therapies. Several approaches are being investigated, including targeted therapies, immunotherapies, and other approaches.
IDH Inhibitors
In 2013, researchers published pre-clinical data on the prototype mIDH1 inhibitor AGI-5198 reporting that it inhibited both biochemical and cellular production of D-2-HG. In vivo studies revealed that AG1-5198 impaired growth of IDH mutant glioma cells and induction of glial differentiation [68, 69]. However, AGI-5198 had poor pharmaceutical properties that prevented its further use in clinical trials.
Subsequently, ivosidenib (mIDH1 inhibitor) and enasidenib (mIDH2 inhibitor) were developed and tested in patients with hematologic malignancies with IDH mutations, such as acute myeloid leukemia (AML). The Food and Drug Administration (FDA) approved these treatments for that indication [70]. More recently, olutasidenib has also obtained regulatory approval in recurrent AML.
The role of IDH inhibitors in glioma treatment, however, is still under investigation. Two phase I studies evaluating ivosidenib (mIDH1 inhibitor) and vorasidenib (mIDH1/2 inhibitor), in 66 and 93 patients respectively, reported a benign safety profile. Results showed patients treated with ivosidenib had prolonged stable disease and reduced growth of the non-enhancing tumors, while vorasidenib showed an overall response rate of 18% in non-enhancing gliomas [71, 72]. Another recent phase 1b/2 study tested olutasidenib, a selective mIDH1 inhibitor, on 26 patients with recurrent, mIDH1 gliomas (mainly enhancing tumors) and reported tolerability as well as preliminary clinical activity in a heavily pre-treated group of patients [73]. Other mIDH inhibitors are currently under clinical investigation, such as BAY1436032, DS-1001, LY3410738, and more [74, 75].
Despite the mIDH inhibitors’ therapeutic promise, reports suggest potential limitations. Some studies found that while different IDH inhibitors reduced the accumulation of D-2-HG, they failed to reverse global DNA or histone hypermethylation responsible for oncogenesis [76, 77]. Other studies suggest that there is potential for resistance to DNA-damage-inducing therapies, such as radiation and chemotherapy [78, 79]. Additional clinical trials looking at the efficacy of IDH inhibitors in glioma are ongoing. These include a phase III trial studying vorasidenib versus a placebo in patients with residual or recurrent IDH mutant gliomas grades 2 or 3 (NCT04164901) and a study of DS-1001 in patients with chemotherapy- and radiotherapy-naïve IDH1-mutated WHO grade 2 gliomas (NCT04458272). These clinical studies may help elucidate the precise role of these therapies in cancer treatment and the optimal timing for therapy.
DNA Methyltransferase Inhibitors (DNMTi)
As discussed earlier, IDH mutations lead to a hypermethylation phenotype that results in epigenetic alterations in glioma cells and is possibly linked to gliomagenesis [77, 80]. Correcting this epigenetic dysregulation was studied as a potential therapeutic strategy for IDH mutant gliomas. In 2013, two papers reported that 5-azacytidine and decitabine (both DNMTi agents) induced differentiation of glioma cells and reduced tumor growth in vivo and in vitro [81, 82]. In 2017, Yamashita et al reported that when 5-azacytidine was combined with temozolomide it increased survival in glioma models and further decreased tumor volumes [83]. Clinical trials evaluating DNMTi monotherapy with azacytidine (NCT03666559) and other DNMTi are investigating the role of these drugs for mIDH gliomas (NCT03922555).
PARP Inhibitors and DNA Repair Enzymes
IDH mutations and subsequent accumulation of D-2-HG impair the integrity of homologous recombination-mediated double strand DNA break repair. This impairment forces IDH mutant glioma cells to use alternative repair mechanisms, such as those mediated by poly-ADP ribose polymerase (PARP) [78, 84].
PARP-mediated DNA repair is one of the mechanisms that remain intact in IDH mutant cells. These cells use this repair mechanism to maintain genomic integrity and survival during exposure to genotoxic therapy, such as radiation and chemotherapy [78, 85]. PARP inhibitors, such as olaparib, can induce a lethality to these cells by depriving them of this essential repair mechanism [86, 87].
A recent pre-clinical study reported that using PARP inhibitors in addition to radiation-induced DNA damage was highly effective in killing cells both in vivo and in vitro [88]. However, preliminary results from two clinical studies with olaparib in gliomas (NCT03561870 and NCT03212274) showed limited clinical benefit [89, 90]. Multiple clinical trials using PARP inhibitors as a single agent or in addition to temozolomide are ongoing (NCT03212742, NCT05297864, NCT03749187).
Immunotherapies
IDH mutations are potential targets for immunotherapies as a tumor-specific neoantigen [91]. Moreover, D-2-HG induced DNA hypermethylation in gliomas results in silencing of programmed cell death-1 (PD-1) and its ligand (PDL-1) compared to IDH wild-type gliomas, which implies a stronger T-cell activation [92].
Multiple studies have reported on the suppression of the genes responsible for immune cell attraction in IDH mutant gliomas and the contributing role of D-2-HG as an inhibitor of anti-tumor immunity in the glioma microenvironment [93, 94]. In attempt to bypass this issue, IDH inhibitors have been added to checkpoint inhibitors and vaccine therapies to reduce the accumulation of D-2-HG and to subsequently to reverse the effect D-2-HG has on the immunity resulting in tumor volume reduction and prolonged OS [93, 94]. A recent preclinical study combined an IDH inhibitor with radiation and temozolomide in addition to a checkpoint inhibitor and reported improved survival in a mouse model [95]. Currently, multiple studies are looking at checkpoint inhibitors as monotherapy or in combination with other agents including IDH inhibitors, PARP inhibitors, or alkylating agents (NCT05188508, NCT04056910, NCT05484622).
Another immune-mediated strategy being investigated is using peptide vaccines that target the IDH neoantigen, which has shown positive results in mice [91, 96]. NOA-16 is a first-in-human, multicenter, phase I clinical trial using an IDH1 R132H peptide vaccine for IDH mutant high-grade astrocytomas. Recent results from this trial validate safety with no deaths, as well as vaccine-induced immunity in 93.3% of the patients enrolled and a three-year OS rate of 84% [97]. Two other peptide vaccines against mIDH1 are being studied: PEPIDH1M in patients with recurrent grade 2 gliomas (RESIST trial NCT02193347) and IDH1 R132H dendritic cell vaccine in patients with IDH mutant gliomas (NCT02771301).
Conclusion
The discovery of the IDH mutations represents a hallmark in the field of neuro-oncology, leading to significant progress in terms of glioma classification and prognosis and potential novel therapeutic approaches for these tumors. Guidelines for managing IDH mutant gliomas rely on studies conducted prior to the molecular era. These recommend surgical resection, radiation, and chemotherapy with variabilities in the timing of treatment and choice of chemotherapy between the different glioma grades and based on additional molecular alterations. While those guidelines still apply to current practice, the field is moving towards a more tailored approach in therapy strategies. Clinical researchers continue to seek new management methods that reflect the complexity and heterogeneity of these tumors while utilizing the advances made in understanding their behavior. Promising strategies continue to be developed, including targeting IDH mutations with mIDH inhibitors, focusing on DNA damage mechanisms with PARP inhibitors, and immunotherapies with checkpoint inhibitors, and peptide vaccines. Ongoing trials will help clarify the role of each of these therapies in the management of mIDH gliomas with a better understanding of their survival benefit as well as long-term toxicities. An examination of these strategies on a larger scale is needed to explore their benefit to the patients’ clinical course and overall survival in powered studies, and subsequently to influence a change in the existing guidelines.
References
Lapointe S, Perry A, Butowski NA. Primary brain tumours in adults. Lancet. 2018;392(10145):432–46.
Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005;109(1):93–108.
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–12.
Kayabolen A, Yilmaz E, Bagci-Onder T. IDH mutations in glioma: double-edged sword in clinical applications? Biomedicines. 2021;9(7):799.
Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. 2009;174(4):1149–53.
Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep. 2013;13(5):345.
Zong H, Verhaak RGW, Canoll P. The cellular origin for malignant glioma and prospects for clinical advancements. Expert Rev Mol Diagn. 2012;12(4):383–94.
Claus EB, Walsh KM, Wiencke JK, Molinaro AM, Wiemels JL, Schildkraut JM, et al. Survival and low-grade glioma: the emergence of genetic information. Neurosurg Focus. 2015;38(1):E6.
Leu S, von Felten S, Frank S, Vassella E, Vajtai I, Taylor E, et al. IDH/MGMT-driven molecular classification of low-grade glioma is a strong predictor for long-term survival. Neuro Oncol. 2013;15(4):469–79.
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23(8):1231–51.
MJvd B, Smits M, Kros JM, Chang SM. Diffuse infiltrating oligodendroglioma and astrocytoma. J Clin Oncol. 2017;35(21):2394–401.
Bush NAO, Chang S. Treatment strategies for low-grade glioma in adults. J Oncol Pract. 2016;12(12):1235–41.
Schiff D. Low-grade gliomas. CONTIN: Lifelong Learn Neurol. 2015;21(2):345–54.
Liu R, Page M, Solheim K, Fox S, Chang SM. Quality of life in adults with brain tumors: current knowledge and future directions. Neuro-oncology. 2009;11(3):330–9.
Louis DN. WHO classification of tumours of the central nervous system. WHO Regional Office Europe; 2007.
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109.
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathologica. 2016;131(6):803–20.
Perry A, Wesseling P. Histologic classification of gliomas. Handb Clin Neurol. 2016;134:71–95.
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–73.
Han S, Liu Y, Cai SJ, Qian M, Ding J, Larion M, et al. IDH mutation in glioma: molecular mechanisms and potential therapeutic targets. Br J Cancer. 2020;122(11):1580–9.
Dang L, Su SM. Isocitrate dehydrogenase mutation and (r)-2-hydroxyglutarate: from basic discovery to therapeutics development. Annu Rev Biochem. 2017;86:305–31.
Parker SJ, Metallo CM. Metabolic consequences of oncogenic IDH mutations. Pharmacol Ther. 2015;152:54–62.
Yen KE, Bittinger MA, Su SM, Fantin VR. Cancer-associated IDH mutations: biomarker and therapeutic opportunities. Oncogene. 2010;29(49):6409–17.
Wen PY, Packer RJ. The 2021 WHO classification of tumors of the central nervous system: clinical implications. Neuro-Oncology. 2021;23(8):1215–7.
Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro Oncol. 2015;17(suppl_4):iv1–iv62.
Miller JJ, Gonzalez Castro LN, McBrayer S, Weller M, Cloughesy T, Portnow J, et al. Isocitrate dehydrogenase (IDH) mutant gliomas: a Society for Neuro-Oncology (SNO) consensus review on diagnosis, management, and future directions. Neuro-Oncology. 2022;25(1):4–25.
Gonzalez Castro LN, Milligan TA. Seizures in patients with cancer. Cancer. 2020;126(7):1379–89.
Whittle IR. The dilemma of low grade glioma. J Neurol Neurosurg Psychiatry. 2004;75 Suppl 2(Suppl 2):ii31–6.
Grier JT, Batchelor T. low-grade gliomas in adults. The Oncologist. 2006;11(6):681–93.
Behin A, Hoang-Xuan K, Carpentier AF, Delattre J-Y. Primary brain tumours in adults. The Lancet. 2003;361(9354):323–31.
Sabha N, Knobbe CB, Maganti M, Al Omar S, Bernstein M, Cairns R, et al. Analysis of IDH mutation, 1p/19q deletion, and PTEN loss delineates prognosis in clinical low-grade diffuse gliomas. Neuro Oncol. 2014;16(7):914–23.
Juratli TA, Kirsch M, Geiger K, Klink B, Leipnitz E, Pinzer T, et al. The prognostic value of IDH mutations and MGMT promoter status in secondary high-grade gliomas. J Neurooncol. 2012;110(3):325–33.
Kizilbash SH, Giannini C, Voss JS, Decker PA, Jenkins RB, Laack NN, et al. Impact of adjuvant temozolomide and IDH mutation status among patients with anaplastic astrocytoma. American Society of Clinical Oncology; 2013.
Juratli TA, Cahill DP, McCutcheon IE. Determining optimal treatment strategy for diffuse glioma: the emerging role of IDH mutations. Expert Rev Anticancer Ther. 2015;15(6):603–6.
Wen PY, Kesari S. Malignant Gliomas in Adults. N Engl J Med. 2008;359(5):492–507.
Whittle IR. What is the place of conservative management for adult supratentorial low-grade glioma? Adv Tech Stand Neurosurg. 2010;35:65–79.
Jakola AS, Skjulsvik AJ, Myrmel KS, Sjåvik K, Unsgård G, Torp SH, et al. Surgical resection versus watchful waiting in low-grade gliomas. Ann Oncol. 2017;28(8):1942–8.
Aghi MK, Nahed BV, Sloan AE, Ryken TC, Kalkanis SN, Olson JJ. The role of surgery in the management of patients with diffuse low grade glioma: a systematic review and evidence-based clinical practice guideline. J Neurooncol. 2015;125(3):503–30.
Jakola AS, Myrmel KS, Kloster R, Torp SH, Lindal S, Unsgård G, et al. Comparison of a strategy favoring early surgical resection vs a strategy favoring watchful waiting in low-grade gliomas. Jama. 2012;308(18):1881–8.
Muragaki Y, Chernov M, Maruyama T, Ochiai T, Taira T, Kubo O, et al. Low-grade glioma on stereotactic biopsy: how often is the diagnosis accurate? Minim Invasive Neurosurg. 2008;51(5):275–9.
Jackson RJ, Fuller GN, Abi-Said D, Lang FF, Gokaslan ZL, Shi WM, et al. Limitations of stereotactic biopsy in the initial management of gliomas. Neuro-oncology. 2001;3(3):193–200.
McGirt MJ, Chaichana KL, Gathinji M, Attenello FJ, Than K, Olivi A, et al. Independent association of extent of resection with survival in patients with malignant brain astrocytoma. J Neurosurg. 2009;110(1):156–62.
Smith JS, Chang EF, Lamborn KR, Chang SM, Prados MD, Cha S, et al. Role of extent of resection in the long-term outcome of low-grade hemispheric gliomas. J Clin Oncol. 2008;26(8):1338–45.
Wijnenga MMJ, French PJ, Dubbink HJ, Dinjens WNM, Atmodimedjo PN, Kros JM, et al. The impact of surgery in molecularly defined low-grade glioma: an integrated clinical, radiological, and molecular analysis. Neuro Oncol. 2018;20(1):103–12.
de Leeuw CN, Vogelbaum MA. Supratotal resection in glioma: a systematic review. Neuro-Oncology. 2018;21(2):179–88.
Rossi M, Ambrogi F, Gay L, Gallucci M, Conti Nibali M, Leonetti A, et al. Is supratotal resection achievable in low-grade gliomas? Feasibility, putative factors, safety, and functional outcome. J Neurosurg. 2020;132(6):1692–705.
Karschnia P, Vogelbaum MA, van den Bent M, Cahill DP, Bello L, Narita Y, et al. Evidence-based recommendations on categories for extent of resection in diffuse glioma. Eur J Cancer. 2021;149:23–33.
Beiko J, Suki D, Hess KR, Fox BD, Cheung V, Cabral M, et al. IDH1 mutant malignant astrocytomas are more amenable to surgical resection and have a survival benefit associated with maximal surgical resection. Neuro-oncology. 2014;16(1):81–91.
Kawaguchi T, Sonoda Y, Shibahara I, Saito R, Kanamori M, Kumabe T, et al. Impact of gross total resection in patients with WHO grade III glioma harboring the IDH 1/2 mutation without the 1p/19q co-deletion. J Neurooncol. 2016;129(3):505–14.
Douw L, Klein M, Fagel SS, van den Heuvel J, Taphoorn MJ, Aaronson NK, et al. Cognitive and radiological effects of radiotherapy in patients with low-grade glioma: long-term follow-up. Lancet Neurol. 2009;8(9):810–8.
Lanese A, Franceschi E, Brandes AA. The risk assessment in low-grade gliomas: an analysis of the european organization for research and treatment of cancer (eortc) and the radiation therapy oncology group (RTOG) criteria. Oncol Ther. 2018;6(2):105–8.
Shaw EG, Berkey B, Coons SW, Brachman D, Buckner JC, Stelzer KJ, et al. Initial report of radiation therapy oncology group (rtog) 9802: prospective studies in adult low-grade glioma (lgg). J Clin Oncol. 2006;24(18_suppl):1500.
van den Bent MJ, Afra D, de Witte O, Hassel MB, Schraub S, Hoang-Xuan K, et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. The Lancet. 2005;366(9490):985–90.
Mohile NA, Messersmith H, Gatson NT, Hottinger AF, Lassman A, Morton J, et al. Therapy for diffuse astrocytic and oligodendroglial tumors in adults: asco-sno guideline. J Clin Oncol. 2021;40(4):403–26.
Shih HA, Sherman JC, Nachtigall LB, Colvin MK, Fullerton BC, Daartz J, et al. Proton therapy for low-grade gliomas: Results from a prospective trial. Cancer. 2015;121(10):1712–9.
Mason WP, Krol GS, DeAngelis LM. Low-grade oligodendroglioma responds to chemotherapy. Neurology. 1996;46(1):203–7.
Buckner JC, Gesme D Jr, O’Fallon JR, Hammack JE, Stafford S, Brown PD, et al. Phase II trial of procarbazine, lomustine, and vincristine as initial therapy for patients with low-grade oligodendroglioma or oligoastrocytoma: efficacy and associations with chromosomal abnormalities. J clin oncol. 2003;21(2):251–5.
Quinn JA, Reardon DA, Friedman AH, Rich JN, Sampson JH, Provenzale JM, et al. Phase II trial of temozolomide in patients with progressive low-grade glioma. J clin oncol. 2003;21(4):646–51.
Brada M, Viviers L, Abson C, Hines F, Britton J, Ashley S, et al. Phase II study of primary temozolomide chemotherapy in patients with WHO grade II gliomas. Annals of oncology. 2003;14(12):1715–21.
Cairncross JG, Macdonald DR. Successful chemotherapy for recurrent malignant oligodendroglioma. Ann Neurol. 1988;23(4):360–4.
Bell EH, Zhang P, Shaw EG, Buckner JC, Barger GR, Coons SW, et al. ACTR-37. Predictive significance of idh1/2 mutation and 1p/19q co-deletion status in a post-hoc analysis of nrg oncology/rtog 9802: a phase iii trial of rt vs rt + pcv in high risk low-grade gliomas. Neuro-Oncol. 2017;19(suppl_6):vi8–vi.
Cairncross G, Wang M, Shaw E, Jenkins R, Brachman D, Buckner J, et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J clinical oncol. 2013;31(3):337.
van den Bent MJ, Brandes AA, Taphoorn MJ, Kros JM, Kouwenhoven MC, Delattre J-Y, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J clin oncol. 2013;31(3):344–50.
van den Bent MJ, Baumert B, Erridge SC, Vogelbaum MA, Nowak AK, Sanson M, et al. Interim results from the CATNON trial (EORTC study 26053-22054) of treatment with concurrent and adjuvant temozolomide for 1p/19q non-co-deleted anaplastic glioma: a phase 3, randomised, open-label intergroup study. The Lancet. 2017;390(10103):1645–53.
van den Bent MJ, Tesileanu CMS, Wick W, Sanson M, Brandes AA, Clement PM, et al. Adjuvant and concurrent temozolomide for 1p/19q non-co-deleted anaplastic glioma (CATNON; EORTC study 26053-22054): second interim analysis of a randomised, open-label, phase 3 study. Lancet Oncol. 2021;22(6):813–23.
Jaeckle KA, Ballman KV, van den Bent M, Giannini C, Galanis E, Brown PD, et al. CODEL: phase III study of RT, RT + TMZ, or TMZ for newly diagnosed 1p/19q codeleted oligodendroglioma. Analysis from the initial study design. Neuro Oncol. 2021;23(3):457–67.
Nabors LB, Portnow J, Ammirati M, Baehring J, Brem H, Butowski N, et al. NCCN guidelines insights: central nervous system cancers, version 1.2017. J Natl Compr Canc Netw. 2017;15(11):1331–45.
Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant idh1 delays growth and promotes differentiation of glioma cells. Science. 2013;340(6132):626–30.
Popovici-Muller J, Lemieux RM, Artin E, Saunders JO, Salituro FG, Travins J, et al. Discovery of AG-120 (Ivosidenib): a first-in-class mutant IHD1 inhibitor for the treatment of IHD1 mutant cancers. ACS Med Chem Lett. 2018;9(4):300–5.
Okoye-Okafor UC, Bartholdy B, Cartier J, Gao EN, Pietrak B, Rendina AR, et al. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat Chem Biol. 2015;11(11):878–86.
Mellinghoff IK, Penas-Prado M, Peters KB, Burris HA 3rd, Maher EA, Janku F, et al. vorasidenib, a dual inhibitor of mutant IHD1/2, in recurrent or progressive glioma; results of a first-in-human phase i trial. Clin Cancer Res. 2021;27(16):4491–9.
Mellinghoff IK, Ellingson BM, Touat M, Maher E, De La Fuente MI, Holdhoff M, et al. Ivosidenib in isocitrate dehydrogenase 1-mutated advanced glioma. J Clin Oncol. 2020;38(29):3398–406.
de la Fuente MI, Colman H, Rosenthal M, Van Tine BA, Levacic D, Walbert T, et al. Olutasidenib (FT-2102) in patients with relapsed or refractory IDH1-mutant glioma: a multicenter, open-label, phase 1b/2 trial. Neuro Oncol. 2022;
Wick A, Bähr O, Schuler M, Rohrberg K, Chawla SP, Janku F, et al. Phase I assessment of safety and therapeutic activity of bay1436032 in patients with idh1-mutant solid tumors. Clin Cancer Res. 2021;27(10):2723–33.
Natsume A, Arakawa Y, Narita Y, Sugiyama K, Hata N, Muragaki Y, et al. The first-in-human phase I study of a brain penetrant mutant IDH1 inhibitor DS-1001 in patients with recurrent or progressive IDH1-mutant gliomas. Neuro Oncol. 2022;
Johannessen TA, Mukherjee J, Viswanath P, Ohba S, Ronen SM, Bjerkvig R, et al. Rapid conversion of mutant idh1 from driver to passenger in a model of human gliomagenesis. Mol Cancer Res. 2016;14(10):976–83.
Turcan S, Makarov V, Taranda J, Wang Y, Fabius AWM, Wu W, et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat Genet. 2018;50(1):62–72.
Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med. 2017;9(375)
Molenaar RJ, Botman D, Smits MA, Hira VV, van Lith SA, Stap J, et al. Radioprotection of IDH1-mutated cancer cells by the idh1-mutant inhibitor agi-5198idh1r132h inhibition radioprotects idh1r132h cells. Cancer research. 2015;75(22):4790–802.
Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529(7584):110–4.
Turcan S, Fabius AW, Borodovsky A, Pedraza A, Brennan C, Huse J, et al. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget. 2013;4(10):1729–36.
Borodovsky A, Salmasi V, Turcan S, Fabius AW, Baia GS, Eberhart CG, et al. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget. 2013;4(10):1737–47.
Yamashita AS, da Costa RM, Borodovsky A, Festuccia WT, Chan T, Riggins GJ. Demethylation and epigenetic modification with 5-azacytidine reduces IDH1 mutant glioma growth in combination with temozolomide. Neuro-oncology. 2019;21(2):189–200.
Chen F, Bian K, Tang Q, Fedeles BI, Singh V, Humulock ZT, et al. Oncometabolites d-and l-2-hydroxyglutarate inhibit the AlkB family DNA repair enzymes under physiological conditions. Chem Res Toxicol. 2017;30(4):1102–10.
Pang Y, Lu Y, Caisova V, Liu Y, Bullova P, Huynh T-T, et al. Targeting NAD+/PARP DNA repair pathway as a novel therapeutic approach to sdhb-mutated cluster i pheochromocytoma and paragangliomacombination therapy for pheochromocytoma and paraganglioma. Clinical Cancer Research. 2018;24(14):3423–32.
Lu Y, Liu Y, Pang Y, Pacak K, Yang C. Double-barreled gun: Combination of PARP inhibitor with conventional chemotherapy. Pharmacol Ther. 2018;188:168–75.
Lu Y, Kwintkiewicz J, Liu Y, Tech K, Frady LN, Su YT, et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in parp1-mediated dna repair. Cancer Res. 2017;77(7):1709–18.
Wang Y, Wild AT, Turcan S, Wu WH, Sigel C, Klimstra DS, et al. Targeting therapeutic vulnerabilities with PARP inhibition and radiation in IDH-mutant gliomas and cholangiocarcinomas. Sci Adv. 2020;6(17):eaaz3221.
Ducray F, Sanson M, Chinot OL, Fontanilles M, Rivoirard R, Thomas-Maisonneuve L, et al. Olaparib in recurrent IDH-mutant high-grade glioma (OLAGLI). J Clin Oncol. 2021;39(15_suppl):2007.
Fanucci K, Pilat MJP, Shah R, Boerner SA, Li J, Durecki DE, et al. Multicenter phase 2 trial of the PARP inhibitor (PARPi) olaparib in recurrent IDH1 and IDH2-mutant contrast-enhancing glioma. J Clin Oncol. 2022;40(16_suppl):2035–5.
Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512(7514):324–7.
Wang Z, Zhang C, Liu X, Wang Z, Sun L, Li G, et al. Molecular and clinical characterization of PD-L1 expression at transcriptional level via 976 samples of brain glioma. Oncoimmunology. 2016;5(11):e1196310.
Kohanbash G, Carrera DA, Shrivastav S, Ahn BJ, Jahan N, Mazor T, et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest. 2017;127(4):1425–37.
Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M, et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med. 2018;24(8):1192–203.
Kadiyala P, Carney SV, Gauss JC, Garcia-Fabiani MB, Haase S, Alghamri MS, et al. Inhibition of 2-hydroxyglutarate elicits metabolic reprogramming and mutant IDH1 glioma immunity in mice. J Clin Invest. 2021;131(4):e139542.
Pellegatta S, Valletta L, Corbetta C, Patanè M, Zucca I, Riccardi Sirtori F, et al. Effective immuno-targeting of the IDH1 mutation R132H in a murine model of intracranial glioma. Acta Neuropathol Commun. 2015;3(1):1–12.
Platten M, Schilling D, Bunse L, Wick A, Bunse T, Riehl D, et al. ATIM-33. NOA-16: a first-in-man multicenter phase I clinical trial of the German Neurooncology working group evaluating a mutation-specific peptide vaccine targeting IDH1R132H in patients with newly diagnosed malignant ASTROCYTOMAS. Neuro-oncology. 2018;20(suppl_6):vi8–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
RAN has no disclosures. MID reports participation as an advisory board member for Agios Pharmaceuticals and Forma Therapeutics.
Human and Animal Rights Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alshiekh Nasany, R., de la Fuente, M.I. Therapies for IDH-Mutant Gliomas. Curr Neurol Neurosci Rep 23, 225–233 (2023). https://doi.org/10.1007/s11910-023-01265-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11910-023-01265-3