
Abstract
Fibronectin (FN) is a ubiquitous extracellular matrix (ECM) glycoprotein that plays vital roles during tissue repair. The plasma form of FN circulates in the blood, and upon tissue injury, is incorporated into fibrin clots to exert effects on platelet function and to mediate hemostasis. Cellular FN is then synthesized and assembled by cells as they migrate into the clot to reconstitute damaged tissue. The assembly of FN into a complex three-dimensional matrix during physiological repair plays a key role not only as a structural scaffold, but also as a regulator of cell function during this stage of tissue repair. FN fibrillogenesis is a complex, stepwise process that is strictly regulated by a multitude of factors. During fibrosis, there is excessive deposition of ECM, of which FN is one of the major components. Aberrant FN-matrix assembly is a major contributing factor to the switch from normal tissue repair to misregulated fibrosis. Understanding the mechanisms involved in FN assembly and how these interplay with cellular, fibrotic and immune responses may reveal targets for the future development of therapies to regulate aberrant tissue-repair processes.
Similar content being viewed by others


Introduction
Fibronectin (FN) is a ubiquitous and essential component of the extracellular matrix (ECM). It functions both as a regulator of cellular processes and an important scaffolding protein to maintain and direct tissue organization and ECM composition.
During tissue repair, the body acts in a series of tightly regulated steps to rapidly reconstitute damaged tissue: the formation of a fibrin clot acts as the platform for granulation tissue assembly, with subsequent contraction and remodeling of the ECM to reform normal tissue [1, 2]. Different forms of FN play differential and temporally discrete roles during tissue repair. Plasma FN and cellular FN possess distinct structures and rates of assembly into three-dimensional matrices.
In this paper, we discuss the differences in the structure of plasma and cellular FN, and their roles during the different stages of tissue repair. We summarize current theories of how FN is assembled into a three-dimensional matrix, and how this process is regulated. Understanding this complex matrix-assembly process may highlight potential targets for therapeutic advancement in the treatment of aberrant tissue-repair conditions.
Structure of fibronectin isoforms
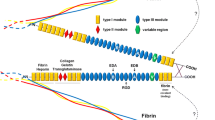
FN is a multi-domain glycoprotein composed of an array of multiple repeated modular structures: twelve FN type I repeats (FNI), two FN type II repeats (FNII), fifteen constitutively expressed and two alternatively spliced (in this paper, referred to as EIIIA and EIIIB) FN type III (FNIII) repeats, and a non-homologous variable (V) or type III connecting segment (IIICS) region. The multimodular structure and intermodular regions allow flexibility of the FN molecule, which is involved in regulating its function [3–8]. These modules are organized into functional domains, including the N-terminal 70-kDa domain (FNI1-9), the 120-kDa central binding domain (CBD; FNIII1-12) and the heparin-binding domain HepII (FNIII12-14). The specific domains of FN can interact with multiple binding partners, including other ECM components and cell-surface receptors [9]. FN is secreted as a dimer maintained by two disulfide S-S bonds at its C-terminus [7, 10–12] (Figure 1).

Fibronectin (FN) and FN fragments. FN is composed of a series of FNI repeats (dark-gray boxes), FNII repeats (circles), conserved FNIII repeats (light-gray boxes) and alternatively spliced FNIII repeats (EDA).
Plasma fibronectin
Plasma FN is synthesized by hepatocytes and secreted into the blood plasma, where it circulates at 300-400 μg/ml [13] in a soluble, compact, inactive form. In soluble plasma FN, only one subunit possesses a V domain, and the EIIIB and EIIIA modules are absent [14–16]. Only very low levels (1.3-1.4 μg/ml) of FN possessing the EIIIA and/or EIIIB modules (cellular FN) have been reported to circulate in the blood plasma [17], but blood plasma levels have been shown to increase after major trauma resulting in vascular tissue damage, after inflammation, and in diseases such as atherosclerosis, ischaemic heart disease and stroke [18–21].
Cellular fibronectin
Cellular FN is synthesized by many cell types, including fibroblasts, endothelial cells, chondrocytes, synovial cells and myocytes [22]. Cellular FN is a mixture of FN isoforms. The alternative splicing of EIIIB and EIIIA and more complex splicing of the V or IIICS domain during transcription of cellular FN allows different isoforms of FN to be expressed in a tissue-dependent, temporally regulated, and cell-type-specific manner [15, 23–26]. In humans, 20 potential FN isoforms can be generated [27]. Increased expression of the EIIIA+ and EIIIB+ isoforms of FN are associated with areas of physiological or pathological tissue remodeling, including wound healing and tissue repair. The observed isoforms of FN, and their association with physiological or pathological conditions, are outlined in Table 1. These isoforms modulate the properties of the ECM, and affect cellular processes.
The orientation and rotational interdomain flexibilities of FNIII modules are known to be affected by neighboring domains, so the inclusion or exclusion of alternatively spliced domains may change the global conformation of FN, affecting the presentation of FNIII loop structures and binding sequences to modulate FN-cell signaling and FN-FN interactions during matrix assembly [9, 28–33]. The structural composition of the different FN isoforms are important, as they play distinct roles in early and late wound-healing events [16].
Fibronectin in early wound-healing responses
Plasma FN is a major component of the fibrin clot. Multiple mechanisms allow FN to be incorporated into the fibrin matrix. FN can interact via non-covalent interactions with fibrin via its FNI1-5 and FNI10-12 domains [34]. FN is also covalently crosslinked to fibrin by activation of the blood coagulation cascade involving activated Factor XIIIa (plasma transglutaminase or coagulation factor XIII) [2, 14, 16, 35]. This crosslinks FN via glutamine residues within its N-terminus [36] to the fibrin α chains via ε-(γ-glutamyl) lysine cross links [37]. Furthermore, plasma FN can also be bound and then assembled into a high-molecular-weight multimeric matrix on the platelet surface. Platelet activation by thrombin induces increased cell-surface expression of the major platelet integrin αIIbβ3, which binds and assembles FN via a fibrin-independent mechanism [38–44]. Platelets also express a lower number of α5β1 and αvβ3 receptors on their surface, which mediate platelet adhesion [45]. Aggregated platelets can also assemble FN via a fibrin-dependent pathway [44]. The polymerization of fibrin into a three-dimensional network has been shown to be essential for FN assembly, as the γ chain of unprocessed fibrinogen signals via αIIbβ3 to inhibit this process [38].
Platelet-producing megakaryocytes endocytose and pinocytose FN from the plasma, which becomes packaged into the α granules of platelets [46]. This has been shown to involve the αIIbβ3 integrin; mice with a mutation in the gene encoding the fibrinogen γ chain (which prevents fibrinogen interactions with αIIbβ3), von Willebrand factor null mice, and fibrinogen null mice show increased FN levels in their α granules [47–49]. Upon platelet activation, FN is released from α granules in a process called degranulation. The FN released from platelets can also assemble on the surface of platelets [42, 46, 50]. FN incorporation into the fibrin matrix is important for various platelet functions, including adhesion, migration and aggregation (Figure 2) [16, 36, 38, 45, 46, 48, 49, 51–98]. However, in vitro fibrin assembly was shown to be unaffected by a complete absence of plasma FN [51]. Furthermore, plasma FN conditional knockout mice, or mutant mice with reduced plasma FN levels of 50% or 70-80%, were shown to have normal clotting and bleeding times. Minimal effects on wound healing in these mice were seen in vivo[17, 51, 52]. Cellular FN isoforms possessing the alternatively spliced EIIIA and EIIIB domains, derived from platelets, are thought to compensate for the loss of plasma FN in these conditions [51]. Although it has been shown that cellular FN isoforms are not efficiently incorporated into the fibrin clots in vitro, it is thought that they may be sufficient to allow normal wound-healing events in vivo.

Functions of plasma and cellular fibronectin (FN) during wound healing. The different forms of FN play distinct roles during the different stages of wound healing.
The fibrin-FN provisional matrix allows FN to adopt extended conformations within the fibrin-FN matrix, which leads to the exposure of cryptic cell binding domains to facilitate cellular processes (Figure 2) [36]. For example, fibroblast activation by various growth factors such as platelet-derived growth factor requires specific sequences within the heparin domain and IIICS region [99].
Fibronectin in the late wound-healing responses
Endothelial cells and fibroblasts repopulate the wound and deposit cellular FN, an important and abundant component of granulation tissue [1, 2, 66, 100]. FN organizes into fibrillar structures within the stroma of granulation tissue, and forms a dense network around fibroblasts, which polarize along the FN fibrils, parallel to the epidermis [66, 95]. FN assembly into a three-dimensional fibrillar network on the cell surface is vital for establishing and maintaining tissue architecture and for regulating cellular processes including adhesion [57–60], spreading [101], proliferation [58, 101–103], migration [99, 104–106] and apoptosis [107, 108] (Figure 2). The three-dimensional FN structural matrix plays an important role in regulating both ECM composition [61, 67] and the deposition of other ECM molecules, including collagen types I and III [61, 68–74], fibrinogen [75], fibrillins 1 and 2 [76–78], fibulin [79], laminin [61, 73, 80] and tenascin (TN)-C [81, 82]. Reticulin has also been shown to colocalize with FN fibrils within the granulation tissue [66]. The FN matrix can also sequester growth factors and associated proteins, including bone morphogenetic protein-1 [83], vascular endothelial growth factor (VEGF) [84] and latent transforming growth factor (TGF)-β binding proteins (LTBP) 1, 3 and 4 [67, 85–87] to regulate cell signaling events. While the FN matrix is continuously assembled, remodeled and turned over by cells, a more mature and stable ECM network assembles on this FN-matrix scaffold [61, 85, 88] (Figure 2).
FN in the wound site is also vital for regulating the neovascularisation of granulation tissue during the resolution of tissue injury. Exposure of different ECM protein conformations in the vascular basement during wounding acts as an important cue to regulate angiogenesis [55, 96]. FN expression, especially of isoforms possessing the alternatively spliced EIIIA+ and EIIIB+ modules are highly upregulated around neovessels and capillary sprouts within the highly vascularized granulation tissue [95, 97, 98]. An FN fibrillar network has been shown to be required for proliferation and migration of human umbilical-vein endothelial cells in a three-dimensional environment [109], and functions to promote endothelial-cell survival [24, 110] (Figure 2).
Myofibroblast differentiation is dependent on the presence of both TGF-β and EIIIA+FN and on the dramatic changes in the mechanical properties of the wound microenvironment [89, 111, 112]. Myofibroblasts form specialized actin-associated fibronexus adhesion complexes, which function in mechano-transduction to allow transmission of intracellular actin-generated contractile forces and the sampling of extracellular tension [111–114]. These specialized adhesion complexes may also function in myofibroblast-dependent contracture of the wound, which acts to 'shorten' and remodel the collagen-rich matrix, resulting in closure of the wound and recapitulation of normal tissue architecture and function.
The data highlighted here indicate distinct and discrete roles for the two different forms of FN during tissue injury and repair. Plasma FN and cellular FN are differentially expressed both temporally and spatially during wound healing: plasma FN circulates in the blood and functions during early wound-healing responses, whereas cellular FN is expressed and assembled locally and functions during later wound-healing responses. However, despite this non-overlapping expression and localization of the FN isoforms during wound healing, exogenous plasma FN can be assembled into pre-existing or newly assembling cellular FN matrices even if the plasma FN is isolated from a different species [115–117]. Although plasma FN shows slower initial kinetics of assembly than cellular FN [12], these data imply that plasma and cellular FN could potentially perform the same functions. Supporting this hypothesis, conditional plasma FN knockout mice were found to have normal wound healing and hemostasis [51], suggesting possible compensation by cellular isoforms of FN. However, in other physiological and pathological processes, these isoforms have been shown to be distinct and unique in their functions; plasma FN was found to be essential for protecting neuronal and non-neuronal cells from apoptosis after transient focal cerebral tissue ischaemia [51] and after traumatic brain injury [118], as cellular FN is not expressed in these damaged brain tissues. Furthermore, EIIIA-FN null mice were shown to have impaired abnormal skin wound-healing responses with reduced cell compaction and edematous-like areas within the granulation tissue and delayed re-epithelialization [90]. These results would suggest that EIIIA+FN plays an important role in the resolution of late wound-healing processes. Tan et al. reported that EIIIA-FN null mice on a different background strain showed no effect on wound healing, but did show reduced atherosclerosis, suggesting that cellular isoforms of FN contribute to pathological conditions [119]. These studies suggest independent roles for the different isoforms of FN, which cannot always be compensated for in their absence.
Fibronectin-matrix assembly
There is still much that we do not know about how FN is assembled or how the rate of deposition is controlled. Understanding the mechanisms that regulate FN-matrix assembly will allow us to control this process when it becomes misregulated.
Plasma FN in solution alone will not polymerize [120] and will not form a three-dimensional matrix in the absence of cells [116]. Both plasma and cellular FN are expressed and secreted in a soluble, compact form, which is maintained by intramolecular electrostatic interactions between the FNI1-5, FNIII1-2, FNIII2-3 and FNIII12-14 domains [7, 10–12] (Figure 3A). In low-salt conditions, this compact quaternary structure can be seen by electron microscopy [6, 8], and Förster resonance energy transfer studies have shown that the arms of soluble FN overlap each other [121]. FN mutants lacking FNIII12-14 or having this region replaced by the alternatively spliced FN type III domains A1-A3 from tenascin-C (TN-C), had lower sedimentation coefficient values, reflecting adoption of a more open conformation and highlighting the importance of FNIII12-14 in maintaining these intramolecular interactions [7].

Stages of fibronectin (FN)-matrix assembly: initiation, unfolding and fibrillar assembly. (A) FN initiation involves interactions with cell-surface receptors: (i) FNI1-5 within the 70-kDa domain binds to cell-surface receptors possibly including integrins, (ii) FNIII9-10 binds to integrin α5β1, (iii) integrin activation by outside-in or inside-out signaling induces integrins to adopt a high-affinity state and allow FN binding and (iv) FNIII12-14 binds to heparan sulfate proteoglycans (HSPGs). (B) FN unfolding: (i) FN binding to cell-surface receptors induces cyctoskeletal reorganization of the actin cytoskeleton and myosin II-dependent contractility that results in (ii) receptor clustering and translation. This causes the tethered FN molecules to become unfolded. (C) Unfolding of FN results in the exposure of FN binding sites that allow FN-FN intermolecular interactions to occur. The domains important for each step are circled and denoted.
FN-matrix assembly is a stepwise, cell-mediated process [122, 123]. The process seems to be rapid, as initial FN deposits appear at the cell surface within 10 minutes of plating, and in the conditioned medium within 30 minutes [122]. FN dimerisation is required for FN assembly, as mutation of C-terminal cysteines results in the loss of fibrillogenesis [124]. This process is briefly discussed below, but is discussed in more depth elsewhere [125].
Initiation of fibronectin-matrix assembly
Initiation of assembly involves FN binding to cell-surface receptors (Figure 3A(i, ii). Integrins that bind FN are summarized in Table 2, although some (for example, αvβ1) do not support FN fibrillogenesis. Mice engineered to be deficient in FN, α5, β1 or αv, or to express FN-RGE (FNRGE/RGE; the RGD integrin-binding sequence of FNIII10 is mutated to RGE) all exhibit embryonic lethality, highlighting the importance of FN fibrillogenesis and the FN fibrillar matrix during development [126–128].
FN is thought to bind first to the cell surface via FNI1-5 within the 70-kDa N-terminal domain of FN (hereafter referred to as '70-kDa'; Figure 3Ai) [58, 124, 129–135]. Initiation of FN-matrix assembly occurs at focal contacts, which are initial sites of ECM contact on the cell periphery, which are rich in paxillin, vinculin, phosphotyrosines, and β1 and β3 integrins [136, 137]. The receptor(s) for 70-kDa have not yet been clearly elucidated. Interactions with αvβ3 integrins have been shown to interact with the novel Gly-Asn-Gly-Arg-Gly (GNGRG) motif in FNI1-5[126, 138]. However, recent work has shown that mutation of the two GNGRG sequences within 70-kDa has no effect on the binding of 70-kDa or FN to adherent cells or on the ability of 70-kDa to compete for FN binding [139]. However, this study did show that interactions of cells with 70-kDa did require activated αvβ3 integrins [139], and it has also been shown that in vivo, the presence of pre-existing three-dimensional FN matrices also stimulates αvβ3 activation to induce FN-matrix assembly [116]. Another possible integrin-binding site is the Ile-Gly-Asp (IGD) sequence in FNI9[140]. It has been suggested that initiation of FN-matrix assembly may be a process that is not dependent on a single integrin or region within FN, but may involve many different molecules [125].
Other integrins may also be involved in the initiating the assembly of specific FN isoforms. In an interesting study, which demonstrated the essential role of FNIIIA+FN in lymphatic-valve development,α9β1 integrin was shown to be required for the assembly of EIIIA+FN by lymphatic endothelial cells [80]; however, this assembly mechanism may be isoform-specific, as a separate study found that overall FN-matrix assembly by EIIIA-FN null fibroblasts was unaffected [119].
It is well established that an essential step in the progression of FN-matrix assembly involves α5β1 integrin binding of the RGD loop on FNIII10 and the neighboring PHSRN sequence in FNIII9 in the CBD of FN [116, 141–146] (Figure 3Aii). α5β1 integrin binding to CBD is a high-affinity interaction (Kd = 0.2 μmol/l by solid-phase binding) [28]. FN mutants with loss of the RGD site or a mutation within the RGD site were found to be unable to assemble a complex fibrillar network, forming only linear fibrillar arrays at the periphery of cells [12, 139]. Antibodies against the RGD domain or small RGD peptides can also inhibit FN-matrix assembly [126, 133, 134, 147, 148]. Furthermore, FNRGE/RGE-expressing mouse embryos had normal FN distribution, and isolated embryonic fibroblasts could assemble short, thick, FN fibrils, confirming that FNIII10 interactions with α5β1 are not essential for assembly initiation [126].
The interaction of integrins with FN provides 'outside-in' signals [149], and induces integrin clustering in focal adhesions [60]. Ligation of integrins can also induce intracellular signaling and the activation of integrins to a higher-affinity binding state by 'inside-out' signaling. Phosphorylation of integrin cytoplasmic domains and subsequent integrin activation by signaling molecules such as talin requires myosin II-dependent cell-generated tension, which is activated by force-induced conformational changes [60, 146, 150, 151]. 'Outside-in' signals can be translated into 'inside-out' signals via a feedback loop [141, 149, 151] to regulate adhesion-complex formation, integrin affinity and FN-matrix assembly [116, 146, 151] (Figure 3Aiii). 'Inside-out' signaling and integrin activation and clustering may precede FN binding during embryogenesis: integrin α5β1 activation and clustering was shown to be initiated by Eph/Ephrin signaling with subsequent FN-matrix assembly [152].
Cell-surface heparan sulfate proteoglycans (HSPGs) have also been shown to play an important role in FN-matrix assembly (Figure 3A (iv)) [153, 154]. The HepII domain of FN (FNIII12-14) can interact with heparin [155], and the FNIII13 module is the primary binding site [156–158], suggesting that this FN domain is involved in the binding of HSPGs. Syndecan-2, which is the major syndecan of fibroblasts, has been shown to be an important HSPG for FN fibrillogenesis during zebrafish development [159], and supports FN-matrix assembly [160].
It should be noted that models of fibril initiation are often elucidated from two-dimensional cellular experiments, in which cells also form distinct adhesive structures. In three-dimensional cultures, cells that assemble FN do not form distinct focal or fibrillar adhesions, but instead form long, slender ECM attachments that contain both classic focal-adhesion and fibrillar-adhesion components: α5, paxillin, vinculin, focal-adhesion kinase, phosphotyrosine, and activated β1 [62]. These FN-assembly complexes probably involve a multitude of cell-surface receptors, HSPGs and signaling molecules that act in concert to facilitate FN-assembly initiation [125, 161].
Fibronectin unfolding, elongation and fibril formation
Upon binding to integrins and other cell-surface receptors, FN then has to be unfolded from its compact structure into an extended structure (Figure 3B) [3, 4, 121].
An intact actin cytoskeleton is essential to generate cell tension and a cytoskeletal force, which translates into a temporal change in traction force that induces conformational changes of the cell-surface-bound FN and allows integrin crosslinking, clustering and subsequent translocation [4, 60, 121, 136, 162–164] (Figure 3Bi). FN unfolding is dependent on β1-FN translocation from focal contacts to central tensin-rich fibrillar-adhesion complexes [136] (Figure 3Bii), and FN fibrils are shown to form from the continuous extension of FN molecules and their intermolecular associations [136, 148, 162]. FN unfolding exposes binding sites buried in the soluble structure to promote the interaction of FN with other FN molecules and ECM components [165]. Cell contractility may require a submembranous pool of myosin II, controlled by myosin light chain kinase via RhoA-Rho kinase II-dependent [165–167] or RhoA-independent pathways [97, 166, 168]. Cells in three-dimensional microenvironments may favor different isoforms of myosin II for generating intracellular forces compared with cells cultured in two-dimensional environments [169]. The transduction of intracellular cytoskeleton-generated forces and extracellular mechanical stresses are communicated via cell-ECM contacts, allowing the cell to respond to changes in its microenvironment [161, 170, 171].
During embryogenesis, an alternative mechanism for production of intracellular tension involves the activation of the non-canonical Wnt/PCP signaling pathway, Rho GTPase Rac, p21 activated kinase (Pak) and cadherins, which generate cytoskeletal tension via cell-cell adhesion. This tension is transmitted via β1 to cell-surface-bound FN to mediate fibril formation [172]. In support of this process, inhibition of β1 function inhibits convergence extension tissue morphogenesis and cadherin-mediated cell adhesion [173]. The presence of multiple pathways to mediate cell contractility demonstrates the importance of this generated force in FN-matrix assembly.
FNI1-5[58, 124, 174, 175], FNIII1-2[10, 134, 165, 174, 176], FNIII4-5[177] and FNIII12-14[132] are important domains for FN fibrillogenesis, and play a role in mediating and regulating intermolecular FN-FN interactions [12, 124] (Figure 3C). The EIIIA or EIIIB domains can also promote FN-matrix assembly, possibly by exposing integrin-binding sites in neighboring domains: FNIII10 (EIIIB) and IIICS region (EIIIA) [94, 178].
FN module unfolding and exposure of cryptic FN-FN binding sites further promote FN fibrillogenesis [121]. FN linker domains and the FNIII modules possess inherent elasticity [179–182]. Domains shown to possess cryptic binding sites include FNIII1[183–185], FNIII1-2[10], FNIII10[184], FNIII7 and FNIII15[186] (Figure 3C). A peptide sequence derived from FNIII1 (FNIII1-C) can induce superfibronectin formation; that is, high-molecular-weight, crosslinked aggregates of FN, which resemble cell-assembled FN fibrils [185, 187], indicating how effective these cryptic binding sequences are in promoting FN-FN interactions.
FN molecules are organized into thin fibrils of 5 nm in diameter, formed from the overlapping and staggering of extended FN dimers and the crosslinking of FN into stable multimers [69]. FN fibrils then become laterally associated into thicker fibrils of 6-22 nm in diameter [69]. Further FN-fibril interactions allow the formation of high-molecular-weight, complex, branched, fibrillar FN matrices, which are detergent-insoluble [122, 123, 129]. FN cross-linking and multimerization may occur via a partially cryptic endogenous protein disulfide isomerase activity present in FNI12, as reported by Lagenbach et al. in RNase refolding experiments [69, 188]. However, there is some question as to whether covalent disulfide crosslinking is a true phenomenon; there are suggestions that FN associates via non-covalent protein-protein interactions [189, 190]. The high-molecular-weight multimers seen in non-reduced SDS-PAGE gels are suggested to be a mixture of other ECM proteins such as fibrillin, which 'block' FN dimers from migrating through the gel [190]. This is not unexpected as FN is known to interact with many different ECM components, and these protein-protein interactions may well attenuate its migration through SDS-PAGE gels.
Once assembled, FN fibrils are continuously polymerized and remodeled within the fibrillar matrix on the cell surface [61]. FN remodeling is a dynamic process, in which fibrils continuously detach, contract, bend, stretch, extend, retract and anneal to neighboring fibrils [85, 87, 162, 164, 191, 192]. In vivo, the fibrillar ECM structure is deformed by local cell migratory and protrusive activities [76, 85, 192, 193], and large-scale tissue motion, particularly during embryogenesis [76, 85]. This strongly extensible behavior is due to mechanical unfolding and refolding of FNIII modules [194].
Pre-existing three-dimensional matrices act as scaffolds for further FN deposition; new fibrils colocalize with pre-existing matrix [116, 193, 195]. Cells cultured in native ECM scaffolds deposit more soluble FN onto pre-existing FN fibers, requiring lower concentrations of FN for initiation [116, 193]. New FN molecules deposited by cells onto a mature matrix are unfolded more than cells cultured on 'soft' polyacrylamide substrates [195]. ECM crosslinking has also been shown to increase the rate of de novo FN stretching by cells but to reduce overall deposition of soluble FN molecules [193, 195], suggesting that ECM maturation is a mechanism that regulates the rate of FN deposition.
Fibronectin and aberrant wound-healing conditions
FN-matrix assembly has to be tightly regulated. Cellular FN homeostasis is maintained by continual FN assembly and loss from the pericellular matrix [61]. Persistently high levels of FN promote cell proliferation, survival signals, and strong cell-ECM adhesions. Conversely, loss of FN-matrix assembly is observed in some transformed cells [168, 196], and may be important during cell-migration events such as metastasis. In addition, loss of FN deposition [61, 72] would accentuate the loss in the assembly of other ECM components within the wound bed, as seen in chronic non-healing wounds, even though FN expression persists and is actually increased in the surrounding dermis [197, 198].
Fibronectin in fibrotic conditions
FN plays an important role in the development of fibrotic disease [27]. Fibrosis is characterized by an excessive deposition of connective tissue that leads to the impairment of organ structure or function, and is considered a chronic inflammatory tissue-repair response, similar in structure and composition to granulation tissue [199, 200]. In fibrosis, there is a key interplay between the immune response, fibroblastic-cell response and ECM, which results in the formation of fibrotic lesions.
Although collagen is the most predominant ECM component of fibrotic tissue, excessive deposition of FN also occurs, and precedes the collagen deposition (Table 1) [201–208]. In glomerular and interstitial fibrosis, there is markedly increased expression of total FN levels, with increased levels of EIIIA+, EIIIB+ and oncofetal (IIICS+) isoforms detected in distinct areas of the kidney and in areas of fibrosis [26, 202–204]. Fibrogenesis is driven by fibroblasts and myofibroblasts, which show increased migration, proliferation, ECM synthesis and assembly within affected tissues [114, 199, 209]. The presence of both alternatively spliced EIIIA+FN and pro-inflammatory cytokines such as TGF-β1 have been shown to be required in the differentiation or transdifferentiation of cells to myofibroblasts [210]. For example, experiments with EIIIA null mice suggest that EIIIA+ isoform of FN induces α-smooth-muscle actin myofibroblast differentiation in the presence of TGF-β1. In the absence of EIIIA+ FN, there is continuous interstitial fibrosis after bleomycin treatment, but no switch to a chronic fibrotic response mediated by myofibroblasts. Furthermore, these experiments also showed that EIIIA+FN is required for latent TGF-β1 activation, and plays a response in fibroblast response to TGF-β1 [201, 211]. TGF-β1 and the TGF-β family have been shown to activate the Smad family of transcription factors (including Smad3), which are involved in the expression of profibrotic genes [212]. Furthermore, mouse models of atherosclerosis have shown the essential role of FN in initima-media thickening in vivo[68]. Using a 49-residue sequence from the FUD domain of the F1 adhesin protein produced by Streptococcus pyogenes (pUR4), which has been shown to inhibit FN-matrix assembly by binding to the N-terminal 70-kDa domain of FN [213], this study demonstrated that inhibiting FN-matrix assembly in vivo significantly reduced intimal, medial and adventitial thickening, collagen deposition, cell proliferation, and inflammatory-cell infiltration after induction of atherosclerosis [68]. The results confirm the essential role FN plays in mediating ECM deposition and inflammatory response, which are corollaries of fibrosis. This study also showed promise for possible in vivo applications for inhibiting FN assembly.
Recently clinical studies have shown that use of a 585 nm flashlamp-pumped pulsed-dye laser resulted in the regression or arrest of keloid development by reducing the expression of TGF-β1in keloid tissues and increasing the expression of matrix metalloproteinase (MMP)-13 (also called collagenase-3) [214]. As many fibrotic conditions are largely untreatable, it is imperative that the mechanisms involved in the development and maintenance of these diseases are understood so that effective therapies can be developed.
Regulation of fibronectin-matrix assembly
FN expression and assembly is stimulated in a cell-specific manner by a multitude of molecules (Table 3). Stimulation of FN expression, secretion and assembly by these agents emphasizes the complexity and requirement for tight regulation of this process. As mentioned earlier, TGF-β1 and connective tissue growth factor are key cytokines involved in upregulating FN expression during fibrogenesis [212, 215, 216]. FN deposition also requires RhoA-mediated cell contractility [4, 121, 165–167] (Table 3). FN can also be degraded by multiple proteases including MMP-9 [217]. FN fragments and modules can also inhibit FN-matrix assembly by competing for FN-assembly sites [187], which could act as a feedback system to regulate FN levels on the cell surface. The increased levels of proteases such as neutrophil elastase in chronic wound exudate, which can act to degrade FN [218–222], could further contribute to this condition. Aged clots in chronic wounds contain highly crosslinked fibrin, which is stripped of other functional proteins by the strongly proteolytic environment [223]. These examples illustrate the importance of FN and its assembly in regulating and resolving wound-healing processes to maintain tissue architecture.
FN fibrils are continuously remodeled and turned over, which is mediated via a β1-dependent, caveolin-1-dependent and low-density lipoprotein receptor-related protein (LRP)-independent endocytic mechanism [85, 224]. FN becomes targeted to the lysosomes and degraded intracellularly [61, 88]. As FN is assembled into high-molecular-weight multimers by an endogenous disulfide isomerase activity [188], some reverse proteolytic activity must occur to allow FN endocytosis [61, 88]. Furthermore, β1 integrin clustering can induce polarized expression of membrane type 1 (MT1)-MMP to invasive structures to cause localized ECM degradation [225]. Indeed, endocytosis of fibrillar FN from pre-assembled matrices was shown to be much slower than endocytosis of soluble FN [224]. Interestingly, mature FN matrices are as highly dynamic as immature FN matrices, but ECM maturation has been reported to assemble less dynamic ECM networks over time, which loses this dynamic remodeling [85].
Other ECM components can also influence FN-matrix assembly. Low levels of vitronectin (VN) have been shown to enhance FN-matrix assembly by increasing the expression of matrix-assembly sites on the cell surface [136, 226]. However, high concentrations of VN are inhibitory for FN-matrix assembly [226–228]. The HepII domain of VN has been shown to interact with αvβ3 and αvβ5 integrins, preventing actin microfilament reorganization and causing loss of FN-matrix assembly sites [228]. Loss of collagen type VI also impairs complex FN-matrix assembly; FN fibrils become oriented parallel to the long axis of the cell [229]. As discussed earlier, the individual FN domains including 70-kDa can also inhibit FN-matrix assembly by interfering with fibril formation by the full-length molecules.
Work carried out in our laboratory has also shown that smaller domains of TN-C, but not the full-length protein, can inhibit FN-matrix assembly [230, 231]. As TN-C is only coexpressed with FN in areas of physiological and pathological tissue remodeling and the presence of encrypted inhibitory activity within the individual TN-C domains suggests TN-C may also play an important role in regulating FN-matrix assembly. This highlights how ECM composition and the breakdown of ECM components can also act as a further level of control, which could be exploited to control pathological wound-healing events.
Future perspectives
Current therapies used to treat fibrotic conditions are well summarized and discussed elsewhere [232–234]. It is now appreciated that fibrosis can be considered an aberrant wound-healing response as the understanding of the mechanism of its development is better understood [234]. For example, anti-TGF-β has been successfully shown to reduce skin and pulmonary fibrosis in mice with sclerodermatous graft-versus-host disease, a mouse model of the systemic fibrotic condition scleroderma [235]. However, anti-TGF-β therapy has not been as successful in human systemic sclerosis (SSc): in a placebo-controlled phase I/II trial, systemic and repeated dosing of CAT-192, an antibody developed against active TGF-β1, showed no efficacy in a cohort of 45 patients with early-stage diffuse cutaneous SSc [236]. That study also had higher mortality rates than shown in trials of other drugs to treat diffuse cutaneous SSc, although whether this is due to other factors was not clear [236]. The lack of success might also have been due to the fact that other TGF-β isoforms have profibrotic effects [212], highlighting the complex interplay between the immune system, the ECM and cell signaling during wound healing and aberrant wounding responses. The interplay between cells and the ECM in the regulation of homeostasis and response to physiological and pathological events is complex, and it will be vital to understand these in order to develop therapies that can modify these processes.
Further research into the mechanisms that regulate FN-matrix assembly will help us understand how we can regulate it to prevent aberrant deposition that contributes to pathological conditions. In particular, the effect of other ECM proteins on the FN-assembly process may comprise a regulatory mechanism that could be further explored and exploited therapeutically. For example, ECM proteins such as TN-C are re-expressed only in tissues undergoing active remodeling, such as in fibrotic lesions. However, the role of these molecules in fibrotic tissues and their effects on FN expression, deposition or assembly are still unclear. Elucidation of the complex interplay between resident ECM constituents is likely to reveal how physiological, synergistic control of matrix remodeling is mediated.
Conclusions
The plasma and cellular forms of FN play temporally and spatially distinct and vital roles during the progression of wound healing. Plasma FN circulates in the blood plasma in an inactive form, and is stored in the α-granules of platelets until activated by the wound response and stimulation of the coagulation cascade. Plasma FN is then deposited and crosslinked to the provisional FN-rich matrix, and functions to stimulate platelet adhesion and aggregation, and fibroblast spreading and invasion into the clot. Cellular FN is then synthesized by the migrated cells within the clot, and assembled into a complex, fibrillar matrix on the cell surface, which directs the deposition of other ECM proteins and the migration, adhesion and differentiation of fibroblasts. Many mechanisms are involved to regulate FN-matrix assembly, and there is now also growing evidence that in addition to regulation via molecules, the ECM composition and structure itself are also important.
As ECM assembly is such a complex process, understanding the mechanisms involved is vital if we are to manipulate this process. The fact that other ECM components can affect the deposition and assembly levels of FN suggests further levels of control that could be exploited in cases of dysfunctional wound-healing events. It may be that modifying the microenvironment by altering the expression of other ECM components may be sufficient to induce the resolution of such aberrant tissue-repair processes, which can lead to conditions such as fibrosis.
Abbreviations
- ECM:
-
extracellular matrix
- EIIIA:
-
alternatively spliced fibronectin type III repeat A
- EIIIB:
-
alternatively spliced fibronectin type III repeat B
- FN:
-
fibronectin
- FNI:
-
fibronectin type I repeat
- FNII:
-
fibronectin type II repeat
- FNIII:
-
fibronectin type III repeat
- HSPG:
-
heparan sulfate proteoglycan
- MMP:
-
matrix metalloproteinase
- PDGF:
-
platelet-derived growth factor
- SSc:
-
systemic sclerosis
- TGF:
-
transforming growth factor
- VEGF:
-
vascular endothelial growth factor.
References
Lorenz HP, Longaker MT: Wounds: biology, pathology and management. Surgery, Second Edition Basic Science and Clinical Evidence. 2003, Springer New York, 77-88. Second
Midwood KS, Williams LV, Schwarzbauer JE: Tissue repair and the dynamics of the extracellular matrix. Int J Biochem Cell Biol. 2004, 36: 1031-1037. 10.1016/j.biocel.2003.12.003.
Baneyx G, Baugh L, Vogel V: Coexisting conformations of fibronectin in cell culture imaged using fluorescence resonance energy transfer. Proc Natl Acad Sci USA. 2001, 98: 14464-14468. 10.1073/pnas.251422998.
Baneyx G, Baugh L, Vogel V: Fibronectin extension and unfolding within cell matrix fibrils controlled by cytoskeletal tension. Proc Natl Acad Sci USA. 2002, 99: 5139-5143. 10.1073/pnas.072650799.
Erickson HP, Carrell N, McDonagh J: Fibronectin molecule visualized in electron microscopy: a long, thin, flexible strand. J Cell Biol. 1981, 91: 673-678. 10.1083/jcb.91.3.673.
Erickson HP, Carrell NA: Fibronectin in extended and compact conformations. Electron microscopy and sedimentation analysis. J Biol Chem. 1983, 258: 14539-14544.
Johnson KJ, Sage H, Briscoe G, Erickson HP: The compact conformation of fibronectin is determined by intramolecular ionic interactions. J Biol Chem. 1999, 274: 15473-15479. 10.1074/jbc.274.22.15473.
Rocco M, Infusini E, Daga MG, Gogioso L, Cuniberti C: Models of fibronectin. EMBO J. 1987, 6: 2343-2349.
Pickford AR, Campbell ID: NMR studies of modular protein structures and their interactions. Chem Rev. 2004, 104: 3557-3566. 10.1021/cr0304018.
Karuri NW, Lin Z, Rye HS, Schwarzbauer JE: Probing the conformation of the fibronectin III1-2 domain by fluorescence resonance energy transfer. J Biol Chem. 2009, 284: 3445-3452.
Rocco M, Carson M, Hantgan R, McDonagh J, Hermans J: Dependence of the shape of the plasma fibronectin molecule on solvent composition. Ionic strength and glycerol content. J Biol Chem. 1983, 258: 14545-14549.
Sechler JL, Takada Y, Schwarzbauer JE: Altered rate of fibronectin matrix assembly by deletion of the first type III repeats. J Cell Biol. 1996, 134: 573-583. 10.1083/jcb.134.2.573.
Zardi L, Cecconi C, Barbieri O, Carnemolla B, Picca M, Santi L: Concentration of fibronectin in plasma of tumor-bearing mice and synthesis by Ehrlich ascites tumor cells. Cancer Res. 1979, 39: 3774-3779.
Magnusson MK, Mosher DF: Fibronectin: structure, assembly, and cardiovascular implications. Arterioscler Thromb Vasc Biol. 1998, 18: 1363-1370. 10.1161/01.ATV.18.9.1363.
Tressel T, McCarthy JB, Calaycay J, Lee TD, Legesse K, Shively JE, Pande H: Human plasma fibronectin. Demonstration of structural differences between the A- and B-chains in the III CS region. Biochem J. 1991, 274 (Pt 3): 731-738.
Wilson CL, Schwarzbauer JE: The alternatively spliced V region contributes to the differential incorporation of plasma and cellular fibronectins into fibrin clots. J Cell Biol. 1992, 119: 923-933. 10.1083/jcb.119.4.923.
Chauhan AK, Kisucka J, Cozzi MR, Walsh MT, Moretti FA, Battiston M, Mazzucato M, De Marco L, Baralle FE, Wagner DD, Muro AF: Prothrombotic effects of fibronectin isoforms containing the EDA domain. Arterioscler Thromb Vasc Biol. 2008, 28: 296-301.
Peters JH, Loredo GA, Chen G, Maunder R, Hahn TJ, Willits NH, Hynes RO: Plasma levels of fibronectin bearing the alternatively spliced EIIIB segment are increased after major trauma. J Lab Clin Med. 2003, 141: 401-410. 10.1016/S0022-2143(03)00042-8.
Castellanos M, Leira R, Serena J, Blanco M, Pedraza S, Castillo J, Davalos A: Plasma cellular-fibronectin concentration predicts hemorrhagic transformation after thrombolytic therapy in acute ischemic stroke. Stroke. 2004, 35: 1671-1676. 10.1161/01.STR.0000131656.47979.39.
Song KS, Kim HK, Shim W, Jee SH: Plasma fibronectin levels in ischemic heart disease. Atherosclerosis. 2001, 154: 449-453. 10.1016/S0021-9150(00)00490-1.
Claudepierre P, Allanore Y, Belec L, Larget-Piet B, Zardi L, Chevalier X: Increased Ed-B fibronectin plasma levels in spondyloarthropathies: comparison with rheumatoid arthritis patients and a healthy population. Rheumatology (Oxford). 1999, 38: 1099-1103. 10.1093/rheumatology/38.11.1099.
Mao Y, Schwarzbauer JE: Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 2005, 24: 389-399. 10.1016/j.matbio.2005.06.008.
Hershberger RP, Culp LA: Cell-type-specific expression of alternatively spliced human fibronectin IIICS mRNAs. Mol Cell Biol. 1990, 10: 662-671.
Khan ZA, Chan BM, Uniyal S, Barbin YP, Farhangkhoee H, Chen S, Chakrabarti S: EDB fibronectin and angiogenesis-a novel mechanistic pathway. Angiogenesis. 2005, 8: 183-196. 10.1007/s10456-005-9017-6.
Rybak JN, Roesli C, Kaspar M, Villa A, Neri D: The extra-domain A of fibronectin is a vascular marker of solid tumors and metastases. Cancer Res. 2007, 67: 10948-10957. 10.1158/0008-5472.CAN-07-1436.
Van Vliet A, Baelde HJ, Vleming LJ, de Heer E, Bruijn JA: Distribution of fibronectin isoforms in human renal disease. J Pathol. 2001, 193: 256-262. 10.1002/1096-9896(2000)9999:9999<::AID-PATH783>3.0.CO;2-P.
ffrench-Constant C: Alternative splicing of fibronectin-many different proteins but few different functions. Exp Cell Res. 1995, 221: 261-271. 10.1006/excr.1995.1374.
Altroff H, Schlinkert R, van der Walle CF, Bernini A, Campbell ID, Werner JM, Mardon HJ: Interdomain tilt angle determines integrin-dependent function of the ninth and tenth FIII domains of human fibronectin. J Biol Chem. 2004, 279: 55995-56003. 10.1074/jbc.M406976200.
Bencharit S, Cui CB, Siddiqui A, Howard-Williams EL, Sondek J, Zuobi-Hasona K, Aukhil I: Structural insights into fibronectin type III domain-mediated signaling. J Mol Biol. 2007, 367: 303-309. 10.1016/j.jmb.2006.10.017.
Leahy DJ, Aukhil I, Erickson HP: 2.0 A crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell. 1996, 84: 155-164. 10.1016/S0092-8674(00)81002-8.
Pickford AR, Smith SP, Staunton D, Boyd J, Campbell ID: The hairpin structure of the (6)F1(1)F2(2)F2 fragment from human fibronectin enhances gelatin binding. EMBO J. 2001, 20: 1519-1529. 10.1093/emboj/20.7.1519.
Spitzfaden C, Grant RP, Mardon HJ, Campbell ID: Module-module interactions in the cell binding region of fibronectin: stability, flexibility and specificity. J Mol Biol. 1997, 265: 565-579. 10.1006/jmbi.1996.0736.
Ventura E, Sassi F, Parodi A, Balza E, Borsi L, Castellani P, Carnemolla B, Zardi L: Alternative splicing of the angiogenesis associated extra-domain B of fibronectin regulates the accessibility of the B-C loop of the type III repeat 8. PLoS One. 5: e9145.
Pankov R, Yamada KM: Fibronectin at a glance. J Cell Sci. 2002, 115: 3861-3863. 10.1242/jcs.00059.
Armstrong PB, Armstrong MT: Intercellular invasion and the organizational stability of tissues: a role for fibronectin. Biochim Biophys Acta. 2000, 1470: O9-20.
Corbett SA, Lee L, Wilson CL, Schwarzbauer JE: Covalent cross-linking of fibronectin to fibrin is required for maximal cell adhesion to a fibronectin-fibrin matrix. J Biol Chem. 1997, 272: 24999-25005. 10.1074/jbc.272.40.24999.
Matsuka YV, Migliorini MM, Ingham KC: Cross-linking of fibronectin to C-terminal fragments of the fibrinogen alpha-chain by factor XIIIa. J Protein Chem. 1997, 16: 739-745. 10.1023/A:1026307731751.
Cho J, Degen JL, Coller BS, Mosher DF: Fibrin but not adsorbed fibrinogen supports fibronectin assembly by spread platelets. Effects of the interaction of alphaIIb beta3 with the C terminus of the fibrinogen gamma-chain. J Biol Chem. 2005, 280: 35490-35498. 10.1074/jbc.M506289200.
Plow EF, Birdwell C, Ginsberg MH: Identification and quantitation of platelet-associated fibronectin antigen. J Clin Invest. 1979, 63: 540-543. 10.1172/JCI109334.
Olorundare OE, Peyruchaud O, Albrecht RM, Mosher DF: Assembly of a fibronectin matrix by adherent platelets stimulated by lysophosphatidic acid and other agonists. Blood. 2001, 98: 117-124. 10.1182/blood.V98.1.117.
Niewiarowska J, Cierniewski CS, Tuszynski GP: Association of fibronectin with the platelet cytoskeleton. J Biol Chem. 1984, 259: 6181-6187.
Thurlow PJ, Kenneally DA, Connellan JM: The role of fibronectin in platelet aggregation. Br J Haematol. 1990, 75: 549-556. 10.1111/j.1365-2141.1990.tb07797.x.
Gardner JM, Hynes RO: Interaction of fibronectin with its receptor on platelets. Cell. 1985, 42: 439-448. 10.1016/0092-8674(85)90101-1.
Plow EF, Marguerie GA, Ginsberg MH: Fibronectin binding to thrombin-stimulated platelets: evidence for fibrin(ogen) independent and dependent pathways. Blood. 1985, 66: 26-32.
Watson SP: Platelet activation by extracellular matrix proteins in haemostasis and thrombosis. Curr Pharm Des. 2009, 15: 1358-1372. 10.2174/138161209787846702.
Schick PK, Wojensk CM, Bennett V, Denisova L: Fibronectin isoforms in megakaryocytes. Stem Cells. 1996, 14 (Suppl 1): 212-219.
Ni H, Denis CV, Subbarao S, Degen JL, Sato TN, Hynes RO, Wagner DD: Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. J Clin Invest. 2000, 106: 385-392. 10.1172/JCI9896.
Ni H, Papalia JM, Degen JL, Wagner DD: Control of thrombus embolization and fibronectin internalization by integrin alpha IIb beta 3 engagement of the fibrinogen gamma chain. Blood. 2003, 102: 3609-3614. 10.1182/blood-2003-03-0850.
Ni H, Yuen PS, Papalia JM, Trevithick JE, Sakai T, Fassler R, Hynes RO, Wagner DD: Plasma fibronectin promotes thrombus growth and stability in injured arterioles. Proc Natl Acad Sci USA. 2003, 100: 2415-2419. 10.1073/pnas.2628067100.
Wencel-Drake JD, Painter RG, Zimmerman TS, Ginsberg MH: Ultrastructural localization of human platelet thrombospondin, fibrinogen, fibronectin, and von Willebrand factor in frozen thin section. Blood. 1985, 65: 929-938.
Sakai T, Johnson KJ, Murozono M, Sakai K, Magnuson MA, Wieloch T, Cronberg T, Isshiki A, Erickson HP, Fassler R: Plasma fibronectin supports neuronal survival and reduces brain injury following transient focal cerebral ischemia but is not essential for skin-wound healing and hemostasis. Nat Med. 2001, 7: 324-330. 10.1038/85471.
Matuskova J, Chauhan AK, Cambien B, Astrof S, Dole VS, Piffath CL, Hynes RO, Wagner DD: Decreased plasma fibronectin leads to delayed thrombus growth in injured arterioles. Arterioscler Thromb Vasc Biol. 2006, 26: 1391-1396. 10.1161/01.ATV.0000216282.58291.c6.
Kamykowski GW, Mosher DF, Lorand L, Ferry JD: Modification of shear modulus and creep compliance of fibrin clots by fibronectin. Biophys Chem. 1981, 13: 25-28. 10.1016/0301-4622(81)80021-X.
Cho J, Mosher DF: Enhancement of thrombogenesis by plasma fibronectin cross-linked to fibrin and assembled in platelet thrombi. Blood. 2006, 107: 3555-3563. 10.1182/blood-2005-10-4168.
Tonnesen MG, Feng X, Clark RA: Angiogenesis in wound healing. J Investig Dermatol Symp Proc. 2000, 5: 40-46. 10.1046/j.1087-0024.2000.00014.x.
Knox P, Crooks S, Rimmer CS: Role of fibronectin in the migration of fibroblasts into plasma clots. J Cell Biol. 1986, 102: 2318-2323. 10.1083/jcb.102.6.2318.
Manabe R, Ohe N, Maeda T, Fukuda T, Sekiguchi K: Modulation of cell-adhesive activity of fibronectin by the alternatively spliced EDA segment. J Cell Biol. 1997, 139: 295-307. 10.1083/jcb.139.1.295.
Sottile J, Mosher DF: N-terminal type I modules required for fibronectin binding to fibroblasts and to fibronectin's III1 module. Biochem J. 1997, 323 (Pt 1): 51-60.
Wang R, Clark RA, Mosher DF, Ren XD: Fibronectin's central cell-binding domain supports focal adhesion formation and Rho signal transduction. J Biol Chem. 2005, 280: 28803-28810. 10.1074/jbc.M501421200.
Friedland JC, Lee MH, Boettiger D: Mechanically activated integrin switch controls alpha5beta1 function. Science. 2009, 323: 642-644. 10.1126/science.1168441.
Sottile J, Hocking DC: Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol Biol Cell. 2002, 13: 3546-3559. 10.1091/mbc.E02-01-0048.
Cukierman E, Pankov R, Stevens DR, Yamada KM: Taking cell-matrix adhesions to the third dimension. Science. 2001, 294: 1708-1712. 10.1126/science.1064829.
Johansson S, Svineng G, Wennerberg K, Armulik A, Lohikangas L: Fibronectin-integrin interactions. Front Biosci. 1997, 2: d126-146.
Schnapp LM, Hatch N, Ramos DM, Klimanskaya IV, Sheppard D, Pytela R: The human integrin alpha 8 beta 1 functions as a receptor for tenascin, fibronectin, and vitronectin. J Biol Chem. 1995, 270: 23196-23202. 10.1074/jbc.270.39.23196.
Liao YF, Gotwals PJ, Koteliansky VE, Sheppard D, Van De Water L: The EIIIA segment of fibronectin is a ligand for integrins alpha 9beta 1 and alpha 4beta 1 providing a novel mechanism for regulating cell adhesion by alternative splicing. J Biol Chem. 2002, 277: 14467-14474. 10.1074/jbc.M201100200.
Repesh LA, Fitzgerald TJ, Furcht LT: Fibronectin involvement in granulation tissue and wound healing in rabbits. J Histochem Cytochem. 1982, 30: 351-358. 10.1177/30.4.6174568.
Dallas SL, Sivakumar P, Jones CJ, Chen Q, Peters DM, Mosher DF, Humphries MJ, Kielty CM: Fibronectin regulates latent transforming growth factor-beta (TGF beta) by controlling matrix assembly of latent TGF beta-binding protein-1. J Biol Chem. 2005, 280: 18871-18880. 10.1074/jbc.M410762200.
Chiang HY, Korshunov VA, Serour A, Shi F, Sottile J: Fibronectin is an important regulator of flow-induced vascular remodeling. Arterioscler Thromb Vasc Biol. 2009, 29: 1074-1079. 10.1161/ATVBAHA.108.181081. Epub 2009 Apr 1030
Dzamba BJ, Peters DM: Arrangement of cellular fibronectin in noncollagenous fibrils in human fibroblast cultures. J Cell Sci. 1991, 100 (Pt 3): 605-612.
Dzamba BJ, Wu H, Jaenisch R, Peters DM: Fibronectin binding site in type I collagen regulates fibronectin fibril formation. J Cell Biol. 1993, 121: 1165-1172. 10.1083/jcb.121.5.1165.
Li S, Van Den Diepstraten C, D'Souza SJ, Chan BM, Pickering JG: Vascular smooth muscle cells orchestrate the assembly of type I collagen via alpha2beta1 integrin, RhoA, and fibronectin polymerization. Am J Pathol. 2003, 163: 1045-1056. 10.1016/S0002-9440(10)63464-5.
McDonald JA, Kelley DG, Broekelmann TJ: Role of fibronectin in collagen deposition: Fab' to the gelatin-binding domain of fibronectin inhibits both fibronectin and collagen organization in fibroblast extracellular matrix. J Cell Biol. 1982, 92: 485-492. 10.1083/jcb.92.2.485.
Sottile J, Shi F, Rublyevska I, Chiang HY, Lust J, Chandler J: Fibronectin-dependent collagen I deposition modulates the cell response to fibronectin. Am J Physiol Cell Physiol. 2007, 293: C1934-1946. 10.1152/ajpcell.00130.2007.
Velling T, Risteli J, Wennerberg K, Mosher DF, Johansson S: Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins alpha 11beta 1 and alpha 2beta 1. J Biol Chem. 2002, 277: 37377-37381. 10.1074/jbc.M206286200.
Pereira M, Rybarczyk BJ, Odrljin TM, Hocking DC, Sottile J, Simpson-Haidaris PJ: The incorporation of fibrinogen into extracellular matrix is dependent on active assembly of a fibronectin matrix. J Cell Sci. 2002, 115: 609-617.
Czirok A, Zamir EA, Filla MB, Little CD, Rongish BJ: Extracellular matrix macroassembly dynamics in early vertebrate embryos. Curr Top Dev Biol. 2006, 73: 237-258.
Kinsey R, Williamson MR, Chaudhry S, Mellody KT, McGovern A, Takahashi S, Shuttleworth CA, Kielty CM: Fibrillin-1 microfibril deposition is dependent on fibronectin assembly. J Cell Sci. 2008, 121: 2696-2704. 10.1242/jcs.029819.
Sabatier L, Chen D, Fagotto-Kaufmann C, Hubmacher D, McKee MD, Annis DS, Mosher DF, Reinhardt DP: Fibrillin assembly requires fibronectin. Mol Biol Cell. 2009, 20: 846-858.
Godyna S, Mann DM, Argraves WS: A quantitative analysis of the incorporation of fibulin-1 into extracellular matrix indicates that fibronectin assembly is required. Matrix Biol. 1995, 14: 467-477. 10.1016/0945-053X(95)90004-7.
Bazigou E, Xie S, Chen C, Weston A, Miura N, Sorokin L, Adams R, Muro AF, Sheppard D, Makinen T: Integrin-alpha9 is required for fibronectin matrix assembly during lymphatic valve morphogenesis. Dev Cell. 2009, 17: 175-186. 10.1016/j.devcel.2009.06.017.
Chung CY, Zardi L, Erickson HP: Binding of tenascin-C to soluble fibronectin and matrix fibrils. J Biol Chem. 1995, 270: 29012-29017. 10.1074/jbc.270.48.29012.
Ramos DM, Chen B, Regezi J, Zardi L, Pytela R: Tenascin-C matrix assembly in oral squamous cell carcinoma. Int J Cancer. 1998, 75: 680-687. 10.1002/(SICI)1097-0215(19980302)75:5<680::AID-IJC4>3.0.CO;2-V.
Huang G, Zhang Y, Kim B, Ge G, Annis DS, Mosher DF, Greenspan DS: Fibronectin binds and enhances the activity of bone morphogenetic protein 1. J Biol Chem. 2009, 284: 25879-25888. 10.1074/jbc.M109.024125.
Wijelath ES, Rahman S, Namekata M, Murray J, Nishimura T, Mostafavi-Pour Z, Patel Y, Suda Y, Humphries MJ, Sobel M: Heparin-II domain of fibronectin is a vascular endothelial growth factor-binding domain: enhancement of VEGF biological activity by a singular growth factor/matrix protein synergism. Circ Res. 2006, 99: 853-860. 10.1161/01.RES.0000246849.17887.66.
Dallas SL, Chen Q, Sivakumar P: Dynamics of assembly and reorganization of extracellular matrix proteins. Curr Top Dev Biol. 2006, 75: 1-24.
Koli K, Hyytiainen M, Ryynanen MJ, Keski-Oja J: Sequential deposition of latent TGF-beta binding proteins (LTBPs) during formation of the extracellular matrix in human lung fibroblasts. Exp Cell Res. 2005, 310: 370-382. 10.1016/j.yexcr.2005.08.008.
Sivakumar P, Czirok A, Rongish BJ, Divakara VP, Wang YP, Dallas SL: New insights into extracellular matrix assembly and reorganization from dynamic imaging of extracellular matrix proteins in living osteoblasts. J Cell Sci. 2006, 119: 1350-1360. 10.1242/jcs.02830.
Sottile J, Chandler J: Fibronectin matrix turnover occurs through a caveolin-1-dependent process. Mol Biol Cell. 2005, 16: 757-768.
Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, Gabbiani G: The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol. 1998, 142: 873-881. 10.1083/jcb.142.3.873.
Muro AF, Chauhan AK, Gajovic S, Iaconcig A, Porro F, Stanta G, Baralle FE: Regulated splicing of the fibronectin EDA exon is essential for proper skin wound healing and normal lifespan. J Cell Biol. 2003, 162: 149-160. 10.1083/jcb.200212079.
Hashimoto-Uoshima M, Yan YZ, Schneider G, Aukhil I: The alternatively spliced domains EIIIB and EIIIA of human fibronectin affect cell adhesion and spreading. J Cell Sci. 1997, 110 (Pt 18): 2271-2280.
Chen W, Culp LA: Adhesion mediated by fibronectin's alternatively spliced EDb (EIIIB) and its neighboring type III repeats. Exp Cell Res. 1996, 223: 9-19. 10.1006/excr.1996.0053.
Fukuda T, Yoshida N, Kataoka Y, Manabe R, Mizuno-Horikawa Y, Sato M, Kuriyama K, Yasui N, Sekiguchi K: Mice lacking the EDB segment of fibronectin develop normally but exhibit reduced cell growth and fibronectin matrix assembly in vitro. Cancer Res. 2002, 62: 5603-5610.
Guan JL, Trevithick JE, Hynes RO: Retroviral expression of alternatively spliced forms of rat fibronectin. J Cell Biol. 1990, 110: 833-847. 10.1083/jcb.110.3.833.
Singh P, Reimer CL, Peters JH, Stepp MA, Hynes RO, Van De Water L: The spatial and temporal expression patterns of integrin alpha9beta1 and one of its ligands, the EIIIA segment of fibronectin, in cutaneous wound healing. J Invest Dermatol. 2004, 123: 1176-1181. 10.1111/j.0022-202X.2004.23485.x.
Kalluri R: Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. 2003, 3: 422-433. 10.1038/nrc1094.
Fernandez-Sauze S, Grall D, Cseh B, Van Obberghen-Schilling E: Regulation of fibronectin matrix assembly and capillary morphogenesis in endothelial cells by Rho family GTPases. Exp Cell Res. 2009, 315: 2092-2104. 10.1016/j.yexcr.2009.03.017.
Jarnagin WR, Rockey DC, Koteliansky VE, Wang SS, Bissell DM: Expression of variant fibronectins in wound healing: cellular source and biological activity of the EIIIA segment in rat hepatic fibrogenesis. J Cell Biol. 1994, 127: 2037-2048. 10.1083/jcb.127.6.2037.
Clark RA, An JQ, Greiling D, Khan A, Schwarzbauer JE: Fibroblast migration on fibronectin requires three distinct functional domains. J Invest Dermatol. 2003, 121: 695-705. 10.1046/j.1523-1747.2003.12484.x.
Wakui S, Furusato M, Nikaido T, Yokota K, Sekiguchi J, Ohmori K, Kano Y, Ushigome S: Ultrastructural localization of fibronectin and laminin in human granulation tissue in relation to capillary development. Cell Struct Funct. 1990, 15: 201-210. 10.1247/csf.15.201.
Gui L, Wojciechowski K, Gildner CD, Nedelkovska H, Hocking DC: Identification of the heparin-binding determinants within fibronectin repeat III1: role in cell spreading and growth. J Biol Chem. 2006, 281: 34816-34825. 10.1074/jbc.M608611200.
Jiang ST, Chiang HC, Cheng MH, Yang TP, Chuang WJ, Tang MJ: Role of fibronectin deposition in cystogenesis of Madin-Darby canine kidney cells. Kidney Int. 1999, 56: 92-103.
Sechler JL, Schwarzbauer JE: Control of cell cycle progression by fibronectin matrix architecture. J Biol Chem. 1998, 273: 25533-25536. 10.1074/jbc.273.40.25533.
Couchman JR, Rees DA, Green MR, Smith CG: Fibronectin has a dual role in locomotion and anchorage of primary chick fibroblasts and can promote entry into the division cycle. J Cell Biol. 1982, 93: 402-410. 10.1083/jcb.93.2.402.
Hocking DC, Chang CH: Fibronectin matrix polymerization regulates small airway epithelial cell migration. Am J Physiol Lung Cell Mol Physiol. 2003, 285: L169-179.
Rozario T, Dzamba B, Weber GF, Davidson LA, DeSimone DW: The physical state of fibronectin matrix differentially regulates morphogenetic movements in vivo. Dev Biol. 2009, 327: 386-398. 10.1016/j.ydbio.2008.12.025.
Natal C, Oses-Prieto JA, Pelacho B, Iraburu MJ, Lopez-Zabalza MJ: Regulation of apoptosis by peptides of fibronectin in human monocytes. Apoptosis. 2006, 11: 209-219. 10.1007/s10495-006-3761-y.
Tafolla E, Wang S, Wong B, Leong J, Kapila YL: JNK1 and JNK2 oppositely regulate p53 in signaling linked to apoptosis triggered by an altered fibronectin matrix: JNK links FAK and p53. J Biol Chem. 2005, 280: 19992-19999. 10.1074/jbc.M500331200.
Zhou X, Rowe RG, Hiraoka N, George JP, Wirtz D, Mosher DF, Virtanen I, Chernousov MA, Weiss SJ: Fibronectin fibrillogenesis regulates three-dimensional neovessel formation. Genes Dev. 2008, 22: 1231-1243. 10.1101/gad.1643308.
Kaczmarek J, Castellani P, Nicolo G, Spina B, Allemanni G, Zardi L: Distribution of oncofetal fibronectin isoforms in normal, hyperplastic and neoplastic human breast tissues. Int J Cancer. 1994, 59: 11-16. 10.1002/ijc.2910590104.
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA: Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002, 3: 349-363. 10.1038/nrm809.
Gabbiani G: The Evolution of the Myofibroblast Concept: a Key Cell for Wound Healing and fibrotic diseases. I processi di repiarazione e fibrosi. Giornale di Gerontologia. 2004, 52: 280-282.
Hinz B: Masters and servants of the force: the role of matrix adhesions in myofibroblast force perception and transmission. Eur J Cell Biol. 2006, 85: 175-181. 10.1016/j.ejcb.2005.09.004.
Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G: The myofibroblast: one function, multiple origins. Am J Pathol. 2007, 170: 1807-1816. 10.2353/ajpath.2007.070112.
Hayman EG, Ruoslahti E: Distribution of fetal bovine serum fibronectin and endogenous rat cell fibronectin in extracellular matrix. J Cell Biol. 1979, 83: 255-259. 10.1083/jcb.83.1.255.
Mao Y, Schwarzbauer JE: Stimulatory effects of a three-dimensional microenvironment on cell-mediated fibronectin fibrillogenesis. J Cell Sci. 2005, 118: 4427-4436. 10.1242/jcs.02566.
Peters DM, Portz LM, Fullenwider J, Mosher DF: Co-assembly of plasma and cellular fibronectins into fibrils in human fibroblast cultures. J Cell Biol. 1990, 111: 249-256. 10.1083/jcb.111.1.249.
Tate CC, Garcia AJ, LaPlaca MC: Plasma fibronectin is neuroprotective following traumatic brain injury. Exp Neurol. 2007, 207: 13-22. 10.1016/j.expneurol.2007.05.008. Epub 2007 May 2018
Tan MH, Sun Z, Opitz SL, Schmidt TE, Peters JH, George EL: Deletion of the alternatively spliced fibronectin EIIIA domain in mice reduces atherosclerosis. Blood. 2004, 104: 11-18. 10.1182/blood-2003-09-3363. Epub 2004 Feb 2019
Mosher DF, Johnson RB: Specificity of fibronectin-fibrin cross-linking. Ann N Y Acad Sci. 1983, 408: 583-594. 10.1111/j.1749-6632.1983.tb23275.x.
Smith ML, Gourdon D, Little WC, Kubow KE, Eguiluz RA, Luna-Morris S, Vogel V: Force-induced unfolding of fibronectin in the extracellular matrix of living cells. PLoS Biol. 2007, 5: e268-10.1371/journal.pbio.0050268.
Choi MG, Hynes RO: Biosynthesis and processing of fibronectin in NIL.8 hamster cells. J Biol Chem. 1979, 254: 12050-12055.
McKeown-Longo PJ, Mosher DF: Binding of plasma fibronectin to cell layers of human skin fibroblasts. J Cell Biol. 1983, 97: 466-472. 10.1083/jcb.97.2.466.
Schwarzbauer JE: Identification of the fibronectin sequences required for assembly of a fibrillar matrix. J Cell Biol. 1991, 113: 1463-1473. 10.1083/jcb.113.6.1463.
Singh P, Carraher C, Schwarzbauer JE: Assembly of fibronectin extracellular matrix. Annu Rev Cell Dev Biol. 26: 397-419.
Takahashi S, Leiss M, Moser M, Ohashi T, Kitao T, Heckmann D, Pfeifer A, Kessler H, Takagi J, Erickson HP, Fassler R: The RGD motif in fibronectin is essential for development but dispensable for fibril assembly. J Cell Biol. 2007, 178: 167-178. 10.1083/jcb.200703021.
Yang JT, Rayburn H, Hynes RO: Embryonic mesodermal defects in alpha 5 integrin-deficient mice. Development. 1993, 119: 1093-1105.
Yang JT, Bader BL, Kreidberg JA, Ullman-Cullere M, Trevithick JE, Hynes RO: Overlapping and independent functions of fibronectin receptor integrins in early mesodermal development. Dev Biol. 1999, 215: 264-277. 10.1006/dbio.1999.9451.
McKeown-Longo PJ, Mosher DF: Interaction of the 70,000-mol-wt amino-terminal fragment of fibronectin with the matrix-assembly receptor of fibroblasts. J Cell Biol. 1985, 100: 364-374. 10.1083/jcb.100.2.364.
Tomasini-Johansson BR, Annis DS, Mosher DF: The N-terminal 70-kDa fragment of fibronectin binds to cell surface fibronectin assembly sites in the absence of intact fibronectin. Matrix Biol. 2006, 25: 282-293. 10.1016/j.matbio.2006.02.002.
Allio AE, McKeown-Longo PJ: Extracellular matrix assembly of cell-derived and plasma-derived fibronectins by substrate-attached fibroblasts. J Cell Physiol. 1988, 135: 459-466. 10.1002/jcp.1041350313.
Bultmann H, Santas AJ, Peters DM: Fibronectin fibrillogenesis involves the heparin II binding domain of fibronectin. J Biol Chem. 1998, 273: 2601-2609. 10.1074/jbc.273.5.2601.
McDonald JA, Quade BJ, Broekelmann TJ, LaChance R, Forsman K, Hasegawa E, Akiyama S: Fibronectin's cell-adhesive domain and an amino-terminal matrix assembly domain participate in its assembly into fibroblast pericellular matrix. J Biol Chem. 1987, 262: 2957-2967.
Morla A, Ruoslahti E: A fibronectin self-assembly site involved in fibronectin matrix assembly: reconstruction in a synthetic peptide. J Cell Biol. 1992, 118: 421-429. 10.1083/jcb.118.2.421.
Zhang Q, Mosher DF: Cross-linking of the NH2-terminal region of fibronectin to molecules of large apparent molecular mass. Characterization of fibronectin assembly sites induced by the treatment of fibroblasts with lysophosphatidic acid. J Biol Chem. 1996, 271: 33284-33292. 10.1074/jbc.271.52.33284.
Pankov R, Cukierman E, Katz BZ, Matsumoto K, Lin DC, Lin S, Hahn C, Yamada KM: Integrin dynamics and matrix assembly: tensin-dependent translocation of alpha(5)beta(1) integrins promotes early fibronectin fibrillogenesis. J Cell Biol. 2000, 148: 1075-1090. 10.1083/jcb.148.5.1075.
Zamir E, Katz BZ, Aota S, Yamada KM, Geiger B, Kam Z: Molecular diversity of cell-matrix adhesions. J Cell Sci. 1999, 112 (Pt 11): 1655-1669.
Curnis F, Longhi R, Crippa L, Cattaneo A, Dondossola E, Bachi A, Corti A: Spontaneous formation of L-isoaspartate and gain of function in fibronectin. J Biol Chem. 2006, 281: 36466-36476. 10.1074/jbc.M604812200.
Xu J, Maurer LM, Hoffmann BR, Annis DS, Mosher DF: iso-DGR sequences do not mediate binding of fibronectin N-terminal modules to adherent fibronectin-null fibroblasts. J Biol Chem. 285: 8563-8571.
Millard CJ, Ellis IR, Pickford AR, Schor AM, Schor SL, Campbell ID: The role of the fibronectin IGD motif in stimulating fibroblast migration. J Biol Chem. 2007, 282: 35530-35535. 10.1074/jbc.M707532200.
Green JA, Berrier AL, Pankov R, Yamada KM: beta1 integrin cytoplasmic domain residues selectively modulate fibronectin matrix assembly and cell spreading through talin and Akt-1. J Biol Chem. 2009, 284: 8148-8159. 10.1074/jbc.M805934200.
Huveneers S, Truong H, Fassler R, Sonnenberg A, Danen EH: Binding of soluble fibronectin to integrin alpha5 beta1-link to focal adhesion redistribution and contractile shape. J Cell Sci. 2008, 121: 2452-2462. 10.1242/jcs.033001.
Takagi J, Strokovich K, Springer TA, Walz T: Structure of integrin alpha5beta1 in complex with fibronectin. EMBO J. 2003, 22: 4607-4615. 10.1093/emboj/cdg445.
Wennerberg K, Lohikangas L, Gullberg D, Pfaff M, Johansson S, Fassler R: Beta 1 integrin-dependent and -independent polymerization of fibronectin. J Cell Biol. 1996, 132: 227-238. 10.1083/jcb.132.1.227.
Wu C, Bauer JS, Juliano RL, McDonald JA: The alpha 5 beta 1 integrin fibronectin receptor, but not the alpha 5 cytoplasmic domain, functions in an early and essential step in fibronectin matrix assembly. J Biol Chem. 1993, 268: 21883-21888.
Wu C, Keivens VM, O'Toole TE, McDonald JA, Ginsberg MH: Integrin activation and cytoskeletal interaction are essential for the assembly of a fibronectin matrix. Cell. 1995, 83: 715-724. 10.1016/0092-8674(95)90184-1.
Darribere T, Guida K, Larjava H, Johnson KE, Yamada KM, Thiery JP, Boucaut JC: In vivo analyses of integrin beta 1 subunit function in fibronectin matrix assembly. J Cell Biol. 1990, 110: 1813-1823. 10.1083/jcb.110.5.1813.
Winklbauer R, Stoltz C: Fibronectin fibril growth in the extracellular matrix of the Xenopus embryo. J Cell Sci. 1995, 108 (Pt 4): 1575-1586.
Disatnik MH, Rando TA: Integrin-mediated muscle cell spreading. The role of protein kinase c in outside-in and inside-out signaling and evidence of integrin cross-talk. J Biol Chem. 1999, 274: 32486-32492. 10.1074/jbc.274.45.32486.
Lee SE, Kamm RD, Mofrad MR: Force-induced activation of talin and its possible role in focal adhesion mechanotransduction. J Biomech. 2007, 40: 2096-2106. 10.1016/j.jbiomech.2007.04.006.
Somanath PR, Kandel ES, Hay N, Byzova TV: Akt1 signaling regulates integrin activation, matrix recognition, and fibronectin assembly. J Biol Chem. 2007, 282: 22964-22976. 10.1074/jbc.M700241200.
Julich D, Mould AP, Koper E, Holley SA: Control of extracellular matrix assembly along tissue boundaries via Integrin and Eph/Ephrin signaling. Development. 2009, 136: 2913-2921. 10.1242/dev.038935.
Woods A, Longley RL, Tumova S, Couchman JR: Syndecan-4 binding to the high affinity heparin-binding domain of fibronectin drives focal adhesion formation in fibroblasts. Arch Biochem Biophys. 2000, 374: 66-72. 10.1006/abbi.1999.1607.
Chung CY, Erickson HP: Glycosaminoglycans modulate fibronectin matrix assembly and are essential for matrix incorporation of tenascin-C. J Cell Sci. 1997, 110 (Pt 12): 1413-1419.
Ingham KC, Brew SA, Atha DH: Interaction of heparin with fibronectin and isolated fibronectin domains. Biochem J. 1990, 272: 605-611.
Bloom L, Ingham KC, Hynes RO: Fibronectin regulates assembly of actin filaments and focal contacts in cultured cells via the heparin-binding site in repeat III13. Mol Biol Cell. 1999, 10: 1521-1536.
Busby TF, Argraves WS, Brew SA, Pechik I, Gilliland GL, Ingham KC: Heparin binding by fibronectin module III-13 involves six discontinuous basic residues brought together to form a cationic cradle. J Biol Chem. 1995, 270: 18558-18562. 10.1074/jbc.270.31.18558.
Ingham KC, Brew SA, Migliorini MM, Busby TF: Binding of heparin by type III domains and peptides from the carboxy terminal hep-2 region of fibronectin. Biochemistry. 1993, 32: 12548-12553. 10.1021/bi00097a035.
Arrington CB, Yost HJ: Extra-embryonic syndecan 2 regulates organ primordia migration and fibrillogenesis throughout the zebrafish embryo. Development. 2009, 136: 3143-3152. 10.1242/dev.031492.
Klass CM, Couchman JR, Woods A: Control of extracellular matrix assembly by syndecan-2 proteoglycan. J Cell Sci. 2000, 113 (Pt 3): 493-506.
Geiger B, Bershadsky A, Pankov R, Yamada KM: Transmembrane crosstalk between the extracellular matrix-cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2001, 2: 793-805. 10.1038/35099066.
Ohashi T, Kiehart DP, Erickson HP: Dual labeling of the fibronectin matrix and actin cytoskeleton with green fluorescent protein variants. J Cell Sci. 2002, 115: 1221-1229.
Lemmon CA, Chen CS, Romer LH: Cell traction forces direct fibronectin matrix assembly. Biophys J. 2009, 96: 729-738. 10.1016/j.bpj.2008.10.009.
Davidson LA, Dzamba BD, Keller R, Desimone DW: Live imaging of cell protrusive activity, and extracellular matrix assembly and remodeling during morphogenesis in the frog, Xenopus laevis. Dev Dyn. 2008, 237: 2684-2692. 10.1002/dvdy.21600.
Zhong C, Chrzanowska-Wodnicka M, Brown J, Shaub A, Belkin AM, Burridge K: Rho-mediated contractility exposes a cryptic site in fibronectin and induces fibronectin matrix assembly. J Cell Biol. 1998, 141: 539-551. 10.1083/jcb.141.2.539.
Yoneda A, Ushakov D, Multhaupt HA, Couchman JR: Fibronectin matrix assembly requires distinct contributions from Rho kinases I and -II. Mol Biol Cell. 2007, 18: 66-75.
Zhang Q, Magnusson MK, Mosher DF: Lysophosphatidic acid and microtubule-destabilizing agents stimulate fibronectin matrix assembly through Rho-dependent actin stress fiber formation and cell contraction. Mol Biol Cell. 1997, 8: 1415-1425.
Brenner KA, Corbett SA, Schwarzbauer JE: Regulation of fibronectin matrix assembly by activated Ras in transformed cells. Oncogene. 2000, 19: 3156-3163. 10.1038/sj.onc.1203626.
Meshel AS, Wei Q, Adelstein RS, Sheetz MP: Basic mechanism of three-dimensional collagen fibre transport by fibroblasts. Nat Cell Biol. 2005, 7: 157-164. 10.1038/ncb1216.
Geiger B, Spatz JP, Bershadsky AD: Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. 2009, 10: 21-33. 10.1038/nrm2593.
Lutz R, Sakai T, Chiquet M: Pericellular fibronectin is required for RhoA-dependent responses to cyclic strain in fibroblasts. J Cell Sci. 123: 1511-1521.
Dzamba BJ, Jakab KR, Marsden M, Schwartz MA, DeSimone DW: Cadherin adhesion, tissue tension, and noncanonical Wnt signaling regulate fibronectin matrix organization. Dev Cell. 2009, 16: 421-432. 10.1016/j.devcel.2009.01.008.
Marsden M, DeSimone DW: Integrin-ECM interactions regulate cadherin-dependent cell adhesion and are required for convergent extension in Xenopus. Curr Biol. 2003, 13: 1182-1191. 10.1016/S0960-9822(03)00433-0.
Aguirre KM, McCormick RJ, Schwarzbauer JE: Fibronectin self-association is mediated by complementary sites within the amino-terminal one-third of the molecule. J Biol Chem. 1994, 269: 27863-27868.
Limper AH, Quade BJ, LaChance RM, Birkenmeier TM, Rangwala TS, McDonald JA: Cell surface molecules that bind fibronectin's matrix assembly domain. J Biol Chem. 1991, 266: 9697-9702.
Sechler JL, Rao H, Cumiskey AM, Vega-Colon I, Smith MS, Murata T, Schwarzbauer JE: A novel fibronectin binding site required for fibronectin fibril growth during matrix assembly. J Cell Biol. 2001, 154: 1081-1088. 10.1083/jcb.200102034.
Maqueda A, Moyano JV, Hernandez Del Cerro M, Peters DM, Garcia-Pardo A: The heparin III-binding domain of fibronectin (III4-5 repeats) binds to fibronectin and inhibits fibronectin matrix assembly. Matrix Biol. 2007, 26: 642-651. 10.1016/j.matbio.2007.06.001.
Abe Y, Bui-Thanh NA, Ballantyne CM, Burns AR: Extra domain A and type III connecting segment of fibronectin in assembly and cleavage. Biochem Biophys Res Commun. 2005, 338: 1640-1647. 10.1016/j.bbrc.2005.10.134.
Chen Y, Wu Y, Cai J: Atomic force microscopic investigation on the potential early intermediate stages of fibrillogenesis of fibronectin within fibrils. Biochem Biophys Res Commun. 2007, 361: 391-397. 10.1016/j.bbrc.2007.07.014.
Craig D, Krammer A, Schulten K, Vogel V: Comparison of the early stages of forced unfolding for fibronectin type III modules. Proc Natl Acad Sci USA. 2001, 98: 5590-5595. 10.1073/pnas.101582198.
Gao M, Craig D, Lequin O, Campbell ID, Vogel V, Schulten K: Structure and functional significance of mechanically unfolded fibronectin type III1 intermediates. Proc Natl Acad Sci USA. 2003, 100: 14784-14789. 10.1073/pnas.2334390100.
Oberhauser AF, Badilla-Fernandez C, Carrion-Vazquez M, Fernandez JM: The mechanical hierarchies of fibronectin observed with single-molecule AFM. J Mol Biol. 2002, 319: 433-447. 10.1016/S0022-2836(02)00306-6.
Hocking DC, Sottile J, McKeown-Longo PJ: Fibronectin's III-1 module contains a conformation-dependent binding site for the amino-terminal region of fibronectin. J Biol Chem. 1994, 269: 19183-19187.
Hocking DC, Smith RK, McKeown-Longo PJ: A novel role for the integrin-binding III-10 module in fibronectin matrix assembly. J Cell Biol. 1996, 133: 431-444. 10.1083/jcb.133.2.431.
Morla A, Zhang Z, Ruoslahti E: Superfibronectin is a functionally distinct form of fibronectin. Nature. 1994, 367: 193-196. 10.1038/367193a0.
Ingham KC, Brew SA, Huff S, Litvinovich SV: Cryptic self-association sites in type III modules of fibronectin. J Biol Chem. 1997, 272: 1718-1724. 10.1074/jbc.272.3.1718.
Bourdoulous S, Orend G, MacKenna DA, Pasqualini R, Ruoslahti E: Fibronectin matrix regulates activation of RHO and CDC42 GTPases and cell cycle progression. J Cell Biol. 1998, 143: 267-276. 10.1083/jcb.143.1.267.
Langenbach KJ, Sottile J: Identification of protein-disulfide isomerase activity in fibronectin. J Biol Chem. 1999, 274: 7032-7038. 10.1074/jbc.274.11.7032.
Chen H, Mosher DF: Formation of sodium dodecyl sulfate-stable fibronectin multimers. Failure to detect products of thiol-disulfide exchange in cyanogen bromide or limited acid digests of stabilized matrix fibronectin. J Biol Chem. 1996, 271: 9084-9089. 10.1074/jbc.271.15.9084.
Ohashi T, Erickson HP: Revisiting the mystery of fibronectin multimers: the fibronectin matrix is composed of fibronectin dimers cross-linked by non-covalent bonds. Matrix Biol. 2009, 28: 170-175. 10.1016/j.matbio.2009.03.002. Epub 2009 Mar 2012
De Jong KL, MacLeod HC, Norton PR, Petersen NO: Fibronectin organization under and near cells. Eur Biophys J. 2006, 35: 695-708. 10.1007/s00249-006-0081-7.
Ohashi T, Kiehart DP, Erickson HP: Dynamics and elasticity of the fibronectin matrix in living cell culture visualized by fibronectin-green fluorescent protein. Proc Natl Acad Sci USA. 1999, 96: 2153-2158. 10.1073/pnas.96.5.2153.
Kubow KE, Klotzsch E, Smith ML, Gourdon D, Little WC, Vogel V: Crosslinking of cell-derived 3D scaffolds up-regulates the stretching and unfolding of new extracellular matrix assembled by reseeded cells. Integr Biol (Camb). 2009, 1: 635-648. 10.1039/b914996a.
Klotzsch E, Smith ML, Kubow KE, Muntwyler S, Little WC, Beyeler F, Gourdon D, Nelson BJ, Vogel V: Fibronectin forms the most extensible biological fibers displaying switchable force-exposed cryptic binding sites. Proc Natl Acad Sci USA. 2009, 106: 18267-18272. 10.1073/pnas.0907518106.
Antia M, Baneyx G, Kubow KE, Vogel V: Fibronectin in aging extracellular matrix fibrils is progressively unfolded by cells and elicits an enhanced rigidity response. Faraday Discuss. 2008, 139: 229-249. discussion 309-225, 419-220
Sakai T, Larsen M, Yamada KM: Fibronectin requirement in branching morphogenesis. Nature. 2003, 423: 876-881. 10.1038/nature01712.
Loots MA, Lamme EN, Zeegelaar J, Mekkes JR, Bos JD, Middelkoop E: Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds. J Invest Dermatol. 1998, 111: 850-857. 10.1046/j.1523-1747.1998.00381.x.
Ongenae KC, Phillips TJ, Park HY: Level of fibronectin mRNA is markedly increased in human chronic wounds. Dermatol Surg. 2000, 26: 447-451. 10.1046/j.1524-4725.2000.99281.x.
Diegelmann RF: Cellular and biochemical aspects of normal and abnormal wound healing: an overview. J Urol. 1997, 157: 298-302. 10.1016/S0022-5347(01)65364-3.
van der Straaten HM, Canninga-van Dijk MR, Verdonck LF, Castigliego D, Borst HP, Aten J, Fijnheer R: Extra-domain-A fibronectin: a new marker of fibrosis in cutaneous graft-versus-host disease. J Invest Dermatol. 2004, 123: 1057-1062. 10.1111/j.0022-202X.2004.23474.x.
Muro AF, Moretti FA, Moore BB, Yan M, Atrasz RG, Wilke CA, Flaherty KR, Martinez FJ, Tsui JL, Sheppard D, et al: An essential role for fibronectin extra type III domain A in pulmonary fibrosis. Am J Respir Crit Care Med. 2008, 177: 638-645. 10.1164/rccm.200708-1291OC.
Barnes VL, Musa J, Mitchell RJ, Barnes JL: Expression of embryonic fibronectin isoform EIIIA parallels alpha-smooth muscle actin in maturing and diseased kidney. J Histochem Cytochem. 1999, 47: 787-798. 10.1177/002215549904700608.
Oomura A, Nakamura T, Arakawa M, Ooshima A, Isemura M: Alterations in the extracellular matrix components in human glomerular diseases. Virchows Arch A Pathol Anat Histopathol. 1989, 415: 151-159. 10.1007/BF00784353.
Buyukbabani N, Droz D: Distribution of the extracellular matrix components in human glomerular lesions. J Pathol. 1994, 172: 199-207. 10.1002/path.1711720207.
Babu M, Diegelmann R, Oliver N: Fibronectin is overproduced by keloid fibroblasts during abnormal wound healing. Mol Cell Biol. 1989, 9: 1642-1650.
Chin GS, Liu W, Steinbrech D, Hsu M, Levinson H, Longaker MT: Cellular signaling by tyrosine phosphorylation in keloid and normal human dermal fibroblasts. Plast Reconstr Surg. 2000, 106: 1532-1540. 10.1097/00006534-200012000-00014.
McCarty SM, Syed F, Bayat A: Influence of the human leukocyte antigen complex on the development of cutaneous fibrosis: an immunogenetic perspective. Acta Derm Venereol. 90: 563-574.
Kischer CW, Hendrix MJ: Fibronectin (FN) in hypertrophic scars and keloids. Cell Tissue Res. 1983, 231: 29-37.
Strutz F, Muller GA: Renal fibrosis and the origin of the renal fibroblast. Nephrol Dial Transplant. 2006, 21: 3368-3370. 10.1093/ndt/gfl199.
White ES, Lazar MH, Thannickal VJ: Pathogenetic mechanisms in usual interstitial pneumonia/idiopathic pulmonary fibrosis. J Pathol. 2003, 201: 343-354. 10.1002/path.1446.
White ES, Baralle FE, Muro AF: New insights into form and function of fibronectin splice variants. J Pathol. 2008, 216: 1-14. 10.1002/path.2388.
Leask A, Abraham DJ: TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18: 816-827. 10.1096/fj.03-1273rev.
Tomasini-Johansson BR, Kaufman NR, Ensenberger MG, Ozeri V, Hanski E, Mosher DF: A 49-residue peptide from adhesin F1 of Streptococcus pyogenes inhibits fibronectin matrix assembly. J Biol Chem. 2001, 276: 23430-23439. 10.1074/jbc.M103467200. Epub 22001 Apr 23425
Kuo YR, Wu WS, Jeng SF, Wang FS, Huang HC, Lin CZ, Yang KD: Suppressed TGF-beta1 expression is correlated with up-regulation of matrix metalloproteinase-13 in keloid regression after flashlamp pulsed-dye laser treatment. Lasers Surg Med. 2005, 36: 38-42. 10.1002/lsm.20104.
Gabrielli A, Avvedimento EV, Krieg T: Scleroderma. N Engl J Med. 2009, 360: 1989-2003. 10.1056/NEJMra0806188.
Ihn H: Scleroderma, fibroblasts, signaling, and excessive extracellular matrix. Curr Rheumatol Rep. 2005, 7: 156-162. 10.1007/s11926-005-0069-9.
Kusubata M, Hirota A, Ebihara T, Kuwaba K, Matsubara Y, Sasaki T, Kusakabe M, Tsukada T, Irie S, Koyama Y: Spatiotemporal changes of fibronectin, tenascin-C, fibulin-1, and fibulin-2 in the skin during the development of chronic contact dermatitis. J Invest Dermatol. 1999, 113: 906-912. 10.1046/j.1523-1747.1999.00802.x.
Grinnell F, Zhu M: Fibronectin degradation in chronic wounds depends on the relative levels of elastase, alpha1-proteinase inhibitor, and alpha2-macroglobulin. J Invest Dermatol. 1996, 106: 335-341. 10.1111/1523-1747.ep12342990.
Latijnhouwers MA, Bergers M, Veenhuis RT, Beekman B, Ankersmit-Ter Horst MF, Schalkwijk J: Tenascin-C degradation in chronic wounds is dependent on serine proteinase activity. Arch Dermatol Res. 1998, 290: 490-496. 10.1007/s004030050341.
Palolahti M, Lauharanta J, Stephens RW, Kuusela P, Vaheri A: Proteolytic activity in leg ulcer exudate. Exp Dermatol. 1993, 2: 29-37. 10.1111/j.1600-0625.1993.tb00196.x.
Rao CN, Ladin DA, Liu YY, Chilukuri K, Hou ZZ, Woodley DT: Alpha 1-antitrypsin is degraded and non-functional in chronic wounds but intact and functional in acute wounds: the inhibitor protects fibronectin from degradation by chronic wound fluid enzymes. J Invest Dermatol. 1995, 105: 572-578. 10.1111/1523-1747.ep12323503.
Trengove NJ, Stacey MC, MacAuley S, Bennett N, Gibson J, Burslem F, Murphy G, Schultz G: Analysis of the acute and chronic wound environments: the role of proteases and their inhibitors. Wound Repair Regen. 1999, 7: 442-452. 10.1046/j.1524-475X.1999.00442.x.
Clark RAF: The Molecular and Cellular Biology of Wound Repair. 1996, New York, N. Y.: Plenum Pres, 2
Shi F, Sottile J: Caveolin-1-dependent beta1 integrin endocytosis is a critical regulator of fibronectin turnover. J Cell Sci. 2008, 121: 2360-2371. 10.1242/jcs.014977.
Bravo-Cordero JJ, Marrero-Diaz R, Megias D, Genis L, Garcia-Grande A, Garcia MA, Arroyo AG, Montoya MC: MT1-MMP proinvasive activity is regulated by a novel Rab8-dependent exocytic pathway. EMBO J. 2007, 26: 1499-1510. 10.1038/sj.emboj.7601606.
Zheng M, Ambesi A, Yu L, McKeown-Longo PJ: Quantification of fibronectin matrix assembly sites using a novel ELISA assay. Matrix Biol. 2007, 26: 330-333. 10.1016/j.matbio.2006.12.004.
Rivera-Torres A: Fibronectin matrix assembly: localization of β1 integrin and effects of laminin on formation of 70-kda fragment linear arrays or fibronectin fibrils. Integrated Biological Sciences Summer Research Program. 2001, University of Wisconsin
Hocking DC, Sottile J, Reho T, Fassler R, McKeown-Longo PJ: Inhibition of fibronectin matrix assembly by the heparin-binding domain of vitronectin. J Biol Chem. 1999, 274: 27257-27264. 10.1074/jbc.274.38.27257.
Sabatelli P, Bonaldo P, Lattanzi G, Braghetta P, Bergamin N, Capanni C, Mattioli E, Columbaro M, Ognibene A, Pepe G, et al: Collagen VI deficiency affects the organization of fibronectin in the extracellular matrix of cultured fibroblasts. Matrix Biol. 2001, 20: 475-486. 10.1016/S0945-053X(01)00160-3.
To WS, Midwood KS: Cryptic domains of tenascin-C differentially control fibronectin fibrillogenesis. Matrix Biol. 2010
To WS, Midwood KS: Identification of novel and distinct binding sites within tenascin-C for soluble and fibrillar fibronectin. J Biol Chem.
Hospenthal MA: Diagnosis and management of idiopathic pulmonary fibrosis: Implications for respiratory care. Respir Care. 2006, 51: 382-391.
Horan GS, Wood S, Ona V, Li DJ, Lukashev ME, Weinreb PH, Simon KJ, Hahm K, Allaire NE, Rinaldi NJ, et al: Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med. 2008, 177: 56-65.
Maher TM, Wells AU: Optimal treatment for idiopathic pulmonary fibrosis. Thorax. 2008, 63: 1120-1121. 10.1136/thx.2008.102566. author reply 1121
McCormick LL, Zhang Y, Tootell E, Gilliam AC: Anti-TGF-beta treatment prevents skin and lung fibrosis in murine sclerodermatous graft-versus-host disease: a model for human scleroderma. J Immunol. 1999, 163: 5693-5699.
Denton CP, Merkel PA, Furst DE, Khanna D, Emery P, Hsu VM, Silliman N, Streisand J, Powell J, Akesson A, et al: Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 2007, 56: 323-333. 10.1002/art.22289.
Fujimoto T, Fujigaki Y, Sun DF, Togawa A, Yonemura K, Hishida A: Important role for fibronectin-EIIIA during renal tubular repair and cellular recovery in uranyl acetate-induced acute renal failure of rats. Virchows Arch. 2003, 443: 194-205. 10.1007/s00428-003-0846-7.
Maniscalco WM, Watkins RH, Chess PR, Sinkin RA, Horowitz S, Toia L: Cell-specific expression of fibronectin and EIIIA and EIIIB splice variants after oxygen injury. Am J Physiol. 1998, 274: L599-609.
Havrlikova K, Mellott M, Kaufman AH, Loredo GA, Peters JH, Colvin RB, Foster CS: Expression of fibronectin isoforms bearing the alternatively spliced EIIIA, EIIIB, and V segments in corneal alkali burn and keratectomy wound models in the rat. Cornea. 2004, 23: 812-818. 10.1097/01.ico.0000133988.34274.24.
Tominaga K, Higuchi K, Watanabe T, Fujiwara Y, Kim S, Arakawa T, Iwao H, Kuroki T: Expression of gene for EIIIA- and EIIIB- fibronectin, fetal types of fibronectin, during gastric ulcer healing in rats. Dig Dis Sci. 2001, 46: 311-317. 10.1023/A:1005541305255.
Vitale AT, Pedroza-Seres M, Arrunategui-Correa V, Lee SJ, DiMeo S, Foster CS, Colvin RB: Differential expression of alternatively spliced fibronectin in normal and wounded rat corneal stroma versus epithelium. Invest Ophthalmol Vis Sci. 1994, 35: 3664-3672.
Matsui S, Takahashi T, Oyanagi Y, Takahashi S, Boku S, Takahashi K, Furukawa K, Arai F, Asakura H: Expression, localization and alternative splicing pattern of fibronectin messenger RNA in fibrotic human liver and hepatocellular carcinoma. J Hepatol. 1997, 27: 843-853. 10.1016/S0168-8278(97)80322-4.
Carnemolla B, Balza E, Siri A, Zardi L, Nicotra MR, Bigotti A, Natali PG: A tumor-associated fibronectin isoform generated by alternative splicing of messenger RNA precursors. J Cell Biol. 1989, 108: 1139-1148. 10.1083/jcb.108.3.1139.
Birchler MT, Milisavlijevic D, Pfaltz M, Neri D, Odermatt B, Schmid S, Stoeckli SJ: Expression of the extra domain B of fibronectin, a marker of angiogenesis, in head and neck tumors. Laryngoscope. 2003, 113: 1231-1237. 10.1097/00005537-200307000-00023.
Kosmehl H, Berndt A, Strassburger S, Borsi L, Rousselle P, Mandel U, Hyckel P, Zardi L, Katenkamp D: Distribution of laminin and fibronectin isoforms in oral mucosa and oral squamous cell carcinoma. Br J Cancer. 1999, 81: 1071-1079. 10.1038/sj.bjc.6690809.
Mhawech P, Dulguerov P, Assaly M, Ares C, Allal AS: EB-D fibronectin expression in squamous cell carcinoma of the head and neck. Oral Oncol. 2005, 41: 82-88. 10.1016/j.oraloncology.2004.07.003.
Astrof S, Crowley D, George EL, Fukuda T, Sekiguchi K, Hanahan D, Hynes RO: Direct test of potential roles of EIIIA and EIIIB alternatively spliced segments of fibronectin in physiological and tumor angiogenesis. Mol Cell Biol. 2004, 24: 8662-8670. 10.1128/MCB.24.19.8662-8670.2004.
D'Ovidio MC, Mastracchio A, Marzullo A, Ciabatta M, Pini B, Uccini S, Zardi L, Ruco LP: Intratumoral microvessel density and expression of ED-A/ED-B sequences of fibronectin in breast carcinoma. Eur J Cancer. 1998, 34: 1081-1085. 10.1016/S0959-8049(98)00041-0.
Shiozawa K, Hino K, Shiozawa S: Alternatively spliced EDA-containing fibronectin in synovial fluid as a predictor of rheumatoid joint destruction. Rheumatology (Oxford). 2001, 40: 739-742. 10.1093/rheumatology/40.7.739.
Takasaki I, Chobanian AV, Mamuya WS, Brecher P: Hypertension induces alternatively spliced forms of fibronectin in rat aorta. Hypertension. 1992, 20: 20-25.
Wierzbicka-Patynowski I, Schwarzbauer JE: The ins and outs of fibronectin matrix assembly. J Cell Sci. 2003, 116: 3269-3276. 10.1242/jcs.00670.
Kauf AC, Hough SM, Bowditch RD: Recognition of fibronectin by the platelet integrin alpha IIb beta 3 involves an extended interface with multiple electrostatic interactions. Biochemistry. 2001, 40: 9159-9166. 10.1021/bi010503x.
Hocevar BA, Brown TL, Howe PH: TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999, 18: 1345-1356. 10.1093/emboj/18.5.1345.
Qi W, Chen X, Holian J, Mreich E, Twigg S, Gilbert RE, Pollock CA: Transforming growth factor-beta1 differentially mediates fibronectin and inflammatory cytokine expression in kidney tubular cells. Am J Physiol Renal Physiol. 2006, 291: F1070-1077. 10.1152/ajprenal.00013.2006.
Roberts CJ, Birkenmeier TM, McQuillan JJ, Akiyama SK, Yamada SS, Chen WT, Yamada KM, McDonald JA: Transforming growth factor beta stimulates the expression of fibronectin and of both subunits of the human fibronectin receptor by cultured human lung fibroblasts. J Biol Chem. 1988, 263: 4586-4592.
Zhang J, Lo C: Regulation of fibronectin expression by PDGF-BB and IGF-I in cultured rat thoracic aortic adventitial fibroblasts. Cell Biol Int. 1995, 19: 517-525. 10.1006/cbir.1995.1096.
Liu Y, Centracchio JN, Lin L, Sun AM, Dworkin LD: Constitutive expression of HGF modulates renal epithelial cell phenotype and induces c-met and fibronectin expression. Exp Cell Res. 1998, 242: 174-185. 10.1006/excr.1998.4107.
Lam S, Verhagen NA, Strutz F, van der Pijl JW, Daha MR, van Kooten C: Glucose-induced fibronectin and collagen type III expression in renal fibroblasts can occur independent of TGF-beta1. Kidney Int. 2003, 63: 878-888. 10.1046/j.1523-1755.2003.00824.x.
Oh JH, Ha H, Yu MR, Lee HB: Sequential effects of high glucose on mesangial cell transforming growth factor-beta 1 and fibronectin synthesis. Kidney Int. 1998, 54: 1872-1878. 10.1046/j.1523-1755.1998.00193.x.
Oliver N, Babu M, Diegelmann R: Fibronectin gene transcription is enhanced in abnormal wound healing. J Invest Dermatol. 1992, 99: 579-586. 10.1111/1523-1747.ep12667776.
Zhang Q, Peyruchaud O, French KJ, Magnusson MK, Mosher DF: Sphingosine 1-phosphate stimulates fibronectin matrix assembly through a Rho-dependent signal pathway. Blood. 1999, 93: 2984-2990.
Quinn JA, Graeber CT, Frackelton AR, Kim M, Schwarzbauer JE, Filardo EJ: Coordinate regulation of estrogen-mediated fibronectin matrix assembly and epidermal growth factor receptor transactivation by the G protein-coupled receptor, GPR30. Mol Endocrinol. 2009, 23: 1052-1064. 10.1210/me.2008-0262.
Vial D, Monaghan-Benson E, McKeown-Longo PJ: Coordinate regulation of fibronectin matrix assembly by the plasminogen activator system and vitronectin in human osteosarcoma cells. Cancer Cell Int. 2006, 6: 8-10.1186/1475-2867-6-8.
Vial D, McKeown-Longo PJ: PAI1 stimulates assembly of the fibronectin matrix in osteosarcoma cells through crosstalk between the alphavbeta5 and alpha5beta1 integrins. J Cell Sci. 2008, 121: 1661-1670. 10.1242/jcs.020149.
Monaghan-Benson E, Mastick CC, McKeown-Longo PJ: A dual role for caveolin-1 in the regulation of fibronectin matrix assembly by uPAR. J Cell Sci. 2008, 121: 3693-3703. 10.1242/jcs.028423.
Weston BS, Wahab NA, Mason RM: CTGF mediates TGF-beta-induced fibronectin matrix deposition by upregulating active alpha5beta1 integrin in human mesangial cells. J Am Soc Nephrol. 2003, 14: 601-610. 10.1097/01.ASN.0000051600.53134.B9.
Acknowledgements
Our research was funded by Arthritis Research UK (ARC).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
WST and KSM drafted the manuscript. Both authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
To, W.S., Midwood, K.S. Plasma and cellular fibronectin: distinct and independent functions during tissue repair. Fibrogenesis Tissue Repair 4, 21 (2011). https://doi.org/10.1186/1755-1536-4-21
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-1536-4-21