
Abstract
Simarouba, a neotropical genus in the family Simaroubaceae, currently lacks comprehensive genomic data in existing databases. This study aims to fill this gap by providing genomic resources for three Simarouba species, S. amara, S. versicolor, and S. glauca. It also aims to perform comparative molecular evolutionary analyses in relation to other species within the order Sapindales. The analysis of these three Simarouba species revealed the presence of the typical quadripartite structure expected in plastomes. However, some pseudogenization events were identified in the psbC, infA, rpl22, and ycf1 genes. In particular, the CDS of the psbC gene in S. amara was reduced from 1422 bp to 584 bp due to a premature stop codon. Nucleotide diversity data pointed to gene and intergenic regions as promising candidates for species and family discrimination within the group, specifically matK, ycf1, ndhF, rpl32, petA-psbJ, and trnS-trnG. Selection signal analyses showed strong evidence for positive selection on the rpl23 gene. Phylogenetic analyses indicated that S. versicolor and S. glauca have a closer phylogenetic relationship than S. amara. We provide chloroplast genomes of three Simaruba species and use them to elucidate plastome evolution, highlight the presence of pseudogenization, and identify potential DNA barcode regions.
Similar content being viewed by others

Introduction
In comparison to the nuclear genome, chloroplast genomes have shown distinctive characteristics that enhance their utility. These features include a significantly reduced size and complexity, as well as a high copy number1. Chloroplast genomes are less susceptible to recombination events, an attribute that enhances their ability to preserve and perpetuate valuable evolutionary imprints2,3. This genomic stability, as well as the specific structural features, make plastomes invaluable tools for understanding the evolutionary history and the phylogenetic relationships of plant species.
The increased accessibility of chloroplast genome sequences has led to a deeper understanding of the evolutionary intricacies underlying these genomes. It has also facilitated the development of sophisticated analytical tools aimed at species identification through comparative and evolutionary studies. Illustrative examples of analyses related to plastid genomes include studies of nucleotide diversity and assessments of nucleotide and amino acid substitution rates measured by the dN/dS ratio4,5,6, as well as the identification of genomic rearrangements associated with inverted repeats (IR), which have proven valuable for phylogenomic investigations across different taxonomic levels, often serving to substantiate or improve the resolution of phylogenetic relationships7,8.
The family Simaroubaceae, order Sapindales9, comprises seven subfamilies, 22 genera, and approximately 189 species with formally recognized names 10. These taxa are predominantly distributed within tropical regions, with the Neotropics being the focal point of the Simaroubaceae species diversity11,12. However, some species are known to extend their distribution range into subtropical or temperate climates 9,13. They are distinguished by the presence of a bitter taste in their bark and branches, which is attributed to the presence of quassinoid compounds. These compounds are synthesized in secretory cells that are distributed throughout their vegetative structures 11,14,15. Species belonging to the Simaroubaceae family have attracted considerable interest due to their manifold multiple traditional medicinal applications. These include improving conditions such as malaria, cancer, helminthiasis, viral infections, gastritis, ulcers, diarrhea, and diabetes. The intrinsic insecticidal and fungicidal properties of these botanical taxa have also been exploited 16,17.
The genus Simarouba Aubl. stands as an established monophyletic clade within the family Simaroubaceae18,19,20. This genus incorporates six neotropical species, including three continental species: Simarouba amara Aubl., Simarouba versicolor A. St.-Hil., and Simarouba glauca DC.21. S. amara boasts a wide distribution across Central and South America, with geographic boundaries extending into Bolivia and Brazil, where the southeastern region represents its distribution limit 21,22,23. Simarouba versicolor is largely restricted to South America, particularly in Brazil and Bolivia, while S. glauca occurs in Central America and Florida (USA) 21,24.
Notably, there is recurrent challenge of discerning between the three continental species, recognized by Cronquist (1994)21 and Franceschinelli et al. (1999)22. According to the authors, this is because these species have very similar morphological characteristics and are parapatric, as evidenced in regions such as Costa Rica, where S. amara and S. glauca occur, and Brazil, where S. amara and S. versicolor may overlap in their geographic distributions. Phylogenetic assessments of the genus have analyzed morphological data from flowers and leaves, and one hypothesis postulates that S. versicolor may have evolved from S. glauca or vice versa, or they may share a more recent common ancestor compared to S. amara21,25.
The three species of Simarouba, S. amara, S. versicolor, and S. glauca, are dioecious plant species, primarily undergoing pollination facilitated by small insects, such as small nocturnal moths26,27,28. The basic chromosome number of the Simaroubaceae family is postulated to be x = 12. The possibility of dysploidy events may lead to the establishment of fundamental numbers, x = 14 and x = 15, within most lineages29. However, the basic chromosome number for the family remains undefined. Cytogenetic records for the genus Simarouba are scarce, with data available for a single species, S. glauca, presenting n = 1530. Along with the limited cytogenetic studies, it is pertinent to emphasize the scarcity of genomic resources available for the family Simaroubaceae. A search NCBI, conducted on February 01, 2024, revealed the presence of complete chloroplast genomes for only five species within the family. Unfortunately, none of these datasets included a representative of the genus Simarouba. This situation highlights the urgent need to generate genomic data for the Simaroubaceae family, with the primary aim of elucidating its molecular and evolutionary characteristics. This will allow for a better understanding of the biology of this taxonomic group.
In this study, a dataset with a total of 17 species belonging to the families Simaroubaceae, Rutaceae, and Meliaceae was used to perform comparative genome analyses among their plastomes. Two new complete plastid sequences were added for representatives of the family Simaroubaceae, specifically S. amara and S. versicolor, and the availability of the assembly and annotation of the genome of S. glauca. The main objectives of this research were the characterization of plastomes within the family, the comparative exploration of plastid genome structures in Sapindales species, the investigation of genomic diversity, and the identification of selection signals within the coding sequences of plastids. It also contributes to a better understanding of the phylogenetic relationships of this botanical group. The results of this study strengthen our knowledge of plastid characteristics and molecular evolution within the order Sapindales. In addition, they provide new insights into the evolutionary relationships within the Simaroubaceae family and improve our understanding of the phylogenetic landscape within the order Sapindales.
Results
Characterization of the chloroplast genomes
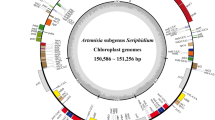
The chloroplast genome sizes of S. versicolor, S. amara, and S. glauca are 159,693 bp, 159,906 bp, and 160,294 bp, respectively, and exhibited the typical quadripartite circular structure: LSC (87,419 bp, 87,692 bp, and 88,077 bp) and an SSC (17,516 bp, 17,470 bp, and 17,525 bp) separated by two IR regions (27,379 bp, 27,372 bp, and 27,346 bp) (Fig. 1).

Circular genome maps of the plastomes of S. versicolor, S. amara, and S. glauca, showing genome sizes, LSC, IR and SSC regions. Gene classes are grouped by colors as indicated in the legend at the top and left of the image.
S. amara presented 131 genes (86 CDS, 37 tRNA and 8 rRNA), while S. versicolor and S. glauca presented 132 genes (87 CDS, 37 tRNA and 8 rRNA), similar to other species of the family Simaroubaceae. The genes were categorized according to their functions (Table 1). The discreet difference in the number of genes between the species was due to the pseudogenization of the psbC gene in S. amara.
In all three species, a total of 19 duplicated genes are located within the Inverted Repeats (IRs). Among them, eight are CDS, seven are tRNA genes, and four are rRNA genes. Additionally, 18 genes contain introns, and two genes, ycf3 and clpP, possess two introns each. The rps12 gene was trans-spliced, exhibiting three exons and one intron (Figure S4. Supplementary material).
We considerate pseudogenes one of the copies of Ψrpl22 (boundary LSC/IRa), Ψycf1 (boundary IRb/SSC), ΨpsbC (only in S. amara) and ΨinfA. Despite the presence of start and end codons in the pseudogenized copies of the Ψrpl22 and Ψycf1 genes, both exhibited notable nucleotide and amino acid substitution rates, as well as insertion/deletion events (Figure S5). These variations contributed to size discrepancies observed across different species. In addition, the Ψrpl22 (LSC/IRa) is absent in Ailanthus altissima (Mill.) Swingle, another Simaroubaceae species from China and cultivated in other countries mainly in temperate zones (Fig. 3).
The ΨpsbC gene in S. amara underwent a reduction in size, decreasing from 1422 bp to 586 bp. This reduction in size resulted from the insertion of a premature stop codon, which occurred due to the deletion of a single base at position 582 within the gene (see Supplementary Figures S1, S2 and S3 for details). Alternative start codons, GTG and ACG, were found in the rps19 and ndhD genes, respectively.
Benchmarking
The cpDNAs of the 17 species were systematically compared in terms of sizes and gene counts (Table S1). The cpDNA size ranged from 161,172 bp (Clausena excavata) to 157,434 bp (Ruta graveolens). The LSC region exhibited a range from 88,382 bp (A. altissima) to 85,387 bp (R. graveolens), while the IRs spanned from 2868 bp (R. graveolens) to 27,923 bp (B. javanica), and the SSC region ranged from 17,470 bp (S. amara) to 18,709 bp (Melia azedarach). The total number of genes varied between 129 (Cedrela odorata) and 134 (C. excavata), with the number of tRNA genes ranging from 36 (C. odorata) to 39 (C. excavata).
Long and short repeat regions
The predominant classes of long repeat sequences were identified as palindromic and forward, constituting 52.4% and 39.3%, respectively. S. amara and Ailanthus excelsa exhibited an absence of reverse and complementary repeats (Fig. 2a). In 11 out of the 17 surveyed species, the majority of long repeats were localized within the IRs (Fig. 2b). The total number of microsatellite repeats ranged from 256 in Leitneria floridana to 185 in R. graveolens. The mononucleotide repeat was the most frequent (Fig. 2c) and the penta and hexanucleotide repeats were absent in some species. The LSC region harbored the largest number of microsatellite repeats, followed by the SSC and IR regions (Fig. 2d). The most frequent motif types were those rich in A/T, such as the repetition of the mononucleotide type A/T and the di-, tri- and tetranucleotide motifs, AT/AT, AAT/ATT and AAAT/ATTT (Supplementary Table S2).

(a) Types of long repeats (forward, reverse, complementary, palindromic). (b) Distribution of long repeats in the SC and IR regions. (c) Types and total number of microsatellites. (d) Distribution of microsatellites in the SC and IR regions. The purple, green, and yellow highlights are the species of the families Simaroubaceae, Rutaceae, and Meliaceae, respectively.
Boundary regions and genomic rearrangements
The chloroplast genome of the 17 species exhibited slight differences in the SC/IR boundary regions (Fig. 3). The rpl22 gene is located at the LSC/IRb boundary in most species, with a size between 489 and 396 bp, and extends into the IR between 6 and 322 bp.

Boundaries between regions of the chloroplast genomes of the 17 species. LSC-IRb: boundary between large single copy (LSC in gray) and inverted repeat B (IRb in yellow). IRb-SSC: boundary between inverted repeat B and small single copy (SSC in green). SSC-IRa: boundary between SSC and inverted repeat A (in yellow). IRa-LSC: boundary between inverted repeat A and LSC. The purple, green, and yellow highlights are the species of the families Simaroubaceae, Rutaceae, and Meliaceae, respectively. ψ-pseudogenes (rpl22 and ycf1).
At the border of IRs/LCS the Ψrpl22 pseudogene exhibited size range of 177–429 bp, qualifying it as a pseudogene (ψ) in this study. In A. altíssima, the Ψrpl22 gene was absent, while in Brucea javanica and C. excavata it was fully localized within the IRs, measuring 486 bp and 177 bp, respectively (Fig. 3). At the IRb/SSC boundary, the Ψycf1 gene showed size variations ranging 1083 bp to 1488 bp, while at the SSC/IRa boundary it showed a size of 5697–5343 bp.
Progressive alignment using the MAUVE revealed the presence of three collinear blocks (LCBs) (Fig. 4). The LCBs harboring tRNA, rRNA and CDS genes showed the absence of genomic rearrangements in the plastomes, as a consequence, the position, direction, and order of the genes were preserved. We also highlight the formation of a collinear block (purple block) shared only by the species of the genus Simarouba, located in the ycf1 gene (SSC/IRa).

Progressive alignment performed using the MAUVE tool v.20150226 in the 17 chloroplast genomes representing the Sapindales family, including S. amara, S. versicolor, and S. glauca. Collinear blocks (LCB) in each species are represented as blocks of green, purple, and pink colors connected by lines. The rRNA, tRNA, and CDS annotations are displayed in red, green, and white, respectively, below the collinear blocks.
Nucleotide diversity (π), selection signal, and codon usage bias
The genomic regions exhibiting the highest nucleotide diversity (π) across the complete genomes were identified mainly within the rps16, ndhF, matK, rpl32, and ycf1 genes, displaying values exceeding twice the median (Fig. 5). Hotspots of nucleotide diversity in the intergenic regions were identified upon examination of the species of the Simaroubaceae family and the Simarouba genus (Fig. 5b, c). It is noteworthy the identification of a nucleotide diversity hotspot in the intergenic region petA-psbJ, persists across both analyses. In the order Sapindales, family Simaroubaceae, and genus Simarouba, the regions of the ndhF and ycf1 genes showed a nucleotide diversity hotspot (Fig. 5a–c). The regions exhibiting low values of nucleotide diversity (π = 0.01) correspond to genes associated with ribosomal RNA.

Nucleotide diversity (π) considering (a) the complete genomes of the seventeen species; (b) the complete genomes of the nine species of the family Simaroubaceae; (c) the complete genomes of the three species of the genus Simarouba. The blue highlighted π peaks indicate intergenic regions. The red horizontal line corresponds to twice the median of the π values.
The analysis of selection signals, as indicated by the dN/dS ratio (ω), revealed that the ribosomal protein large gene, rpl23, exhibited a dN/dS ratio of 2.33, suggesting positive selection (ω > 1) acting on this gene (Fig. 6). In contrast, other CDS demonstrated signatures of negative selection (ω < 1). This observation indicates that natural selection has preserved the amino acid sequence of proteins encoded by these genes. It is noteworthy, however, that genes responsible for encoding other ribosomal proteins and proteins constituting photosystems exhibited minimal changes in their nucleotide compositions.

Selection signal analysis in 77 CDS, dN/dS ratio in the seventeen species analyzed. The CDS were grouped into functional categories and ordered in descending order of the dN/dS values. The dashed horizontal line indicates dN/dS ratio = 1.
The prevalent amino acids in S. amara, S. versicolor and S. glauca were leucine, isoleucine and serine. Codon usage bias, measured by the Relative Synonymous Codon Usage (RSCU), was observed across a majority of amino acids. In S. amara the codons with the highest RSCU values were UUA-Leu, AGA-Arg, in S. versicolor AGA-Arg, GUU-Val, and in S. glauca, AGA-Arg and UUG-Leu (Figure S6). It should be noted that approximately 30 codons exhibited RSCU values greater than one (RSCU > 1), and most of them terminated with base U or A.
Phylogenetic analyses
Phylogenetics relationships exhibited the consistent formation of three distinct groups corresponding to the families Simaroubaceae, Rutaceae, Meliaceae, and the outgroup, Sapindaceae (Fig. 7). Regarding the Simarouba genus, the phylogenetic relationships revealed that S. versicolor is phylogenetically closer to S. glauca than S. amara (Fig. 7).

Reconstruction of the phylogenetic tree among 19 species within the order Sapindales, using 78 CDSs inferred through the maximum likelihood method, according to the TVM + F + I + G4 substitution model and estimated by Bayesian analysis criteria (BIC). Nodes represent the bootstrap values. The colored shades represent the analyzed families, and the vertical bars to the right correspond to the botanical tribes or subfamilies. Symbols according to the legend denote missing and pseudogenized genes.
Discussion
The chloroplast genomes of S. amara, S. versicolor, and S. glauca exhibited a circular, quadripartite structure, characterized by two copies of the repeated inverted region (IR) separated by single-copy regions (LSC and SSC). The size, gene order, and content of these plastomes were similar to those observed in other species within the family Simaroubaceae31,32,33 and across the order Sapindales13,34,35. Although electron microscopy-based analyses have revealed cpDNAs in linear, multi-branched structures in certain angiosperm species3, the predominant configuration of angiosperm cpDNAs is typically circular, and the majority of these circular molecules range in size from 135 to 160 kb 36,37.
A slight variation in the amount of CDS, tRNA and CG content was observed within the group (Table S1). The variation in the amount of CDS and tRNA observed in our analyses was due to at least one of the following factors: i) expansion and retraction of IR regions resulting in duplication and pseudogenization; ii) pseudogenization events of the psbC gene. The hypothesis of plastidial gene transfer to the nucleus has been used to answer some questions about pseudogenization or absence of genes in the plastidial genome, such as the rpl22 gene in the genera Passiflora, Castanea, Prunus, Theobroma 38,39, the accD gene in Primulaceae 40. However, few characterized functional genes have been transferred in angiosperms 39, and experiments are needed to search for functional copies of these genes in the nuclear genome (for review, see Millen et al.,200141 & Ueda et al.,200742). Daniell et al. (2016)43 add that once the gene is transferred to the nucleus, it must acquire sequences to regulate its transcription, as well as the signaling of the final peptide for its correct targeting, that is, from the cytoplasm to the chloroplast.
In S. amara, the pseudogenization of the psbC gene was observed, a gene which constitutes the photosystem II complex and is responsible for encoding the CP43 protein. The CP43 plays a role in energy transfer from the outer antenna complex to the reaction center, contributing to a light-harvesting, PSII stabilization, and as a tertiary electron donor and acceptor44,45,46. Intergenic regions proximal to the psbC gene have been identified with elevated rates of nucleotide diversity as has identified by Jo et al., 201947, Maurya et al., 202348, and Xu et al., 202349. Additionally, the gene has shown a positive selection signal, indicating nucleotide and amino acid sequence changes in selected angiosperm species 50,51. It's worth noting that the CP47 protein encoded by the psbB gene performs functions similar to those of the CP43 protein52,53,54.
The use of alternative start codons, specifically GUG in the rps19 gene and ACG in the ndhD gene, was identified in both S. amara and S. versicolor. Our data corroborate studies in species of the same family31,33,55 and in other angiosperm families such as Passifloraceae, Arecaceae and Apocynaceae56,57,58. In both prokaryotes and eukaryotes, some genes are known to be initiated with non-AUG codons derived from a single base substitution59, such as the GUG-Val, UUG-Leu, and AUU-Ile codons in Escherichia coli, and the ACG-Thr codon in adeno-associated codons and Sendai viruses60,61.
The trans-splicing pattern observed in the rps12 (ribosomal protein) gene, and the loss of its intron found in the species analyzed have been documented in other taxonomic groups, encompassing both in angiosperms and gymnosperms62,63. This bipartite gene is encoded at two locations in the plastidial genome, giving rise to two mRNA precursors that subsequently undergo trans-splicing events to form the complete functional transcript, molecular phenomenon known as exon shuffling (for more information see Long et al., 200364). The occurrence of trans-splicing in this rps12 gene was first documented in Nicotiana tabacum65,66. Across all 17 species examined, palindromic and direct repeats were the predominant types of long repeats. Comparable findings have been reported in species belonging to other taxonomic groups, such as Malpighiales 67,68 and Fabales 6,69. The microsatellite data revealed the presence of numerous SSR repeat sites in the analyzed plastid genomes. These data can be used as a valuable resource for subsequent analyses of polymorphisms in plastid microsatellite regions, facilitating investigations into evolutionary dynamics, genetic diversity, and population structure70,71,72. Furthermore, they can contribute to delineate the conservation status of the species within their geographic range73,74.
Analysis of the boundary regions revealed that the rpl22 and ycf1 genes are situated at the single copy/IR boundary in the majority of species. Variations in its size were observed in both pseudogenized and non-pseudogenized copies of these genes (Fig. 3). Similar findings have been documented in other species within the order Sapindales, including representatives of the families Sapindaceae75,76 and Fabaceae77. In these taxa, pseudogenization events involving one of the copies of the rpl22 and ycf1 genes have been reported. The authors attributed these occurrences to cases of partial gene duplication and events involving contraction and retraction of the IRs. These molecular dynamics likely contributed to the reduction in the size of the gene copies, exemplified, in our study by ycf1, which ranged from 1092 to 1600 bp, and the other copy of the same gene, measuring approximately 5300 bp. In the boundary regions of plastid genomes, episodes of IR contraction and expansion have been frequently reported in angiosperms78,79,80. These dynamic events play a role in the observed variation in both the size of chloroplast genomes and those of genes located at or near genome boundaries as reported by Jansen & Ruhlman (2012)39 and Dobrogojski, et al. (2020)36.
Genomic rearrangement data support the notion that both the order and orientation of genes within angiosperm chloroplast genomes have been relatively conserved across evolutionary history81,82,83, and high collinearity has been observed in Sapindales. While instances of genomic rearrangement events have been documented84,85,86, some cases have been attributed to the loss or reduction of inverted repeats (IRs)87,88,89. Palmer (1983)90 and Mower & Vickrey (2018)3 suggest that the presence of IRs promotes the stabilization of plastome structure, perhaps imposing structural constraints and mitigating major genomic rearrangements.
Our findings indicate the presence of nucleotide diversity hotspots within the rpl32, ycf1 and matK genes across and intergenic regions petA-psbJ, a majority of interspecific group comparisons (Fig. 5). The rpl32 gene encodes a protein component of the large ribosomal subunit91. The ycf1 gene encodes a protein integral to the Translocon on the Inner Chloroplast membrane (TIC) complex located in the inner membrane of the chloroplast92,93. Lastly, the matK gene encodes a putative maturase responsible for catalyzing the removal of introns from premature RNAs94. Nucleotide diversity hotspots within the rpl32, ycf1, and matK genes, and have been extensively documented in various taxonomic groups95,96,97. Recent research recommends their utility in species identification (DNA barcoding)98,99,100 and in the study of phylogenetic relationships101,102. The intergenic regions petA—psbJ have been discerned to exhibit elevated nucleotide substitution rates in species belonging to the order Sapindales103. Species of the genus Simarouba exhibit a parapatric distribution, and challenges in species identification have been documented, particularly in the frequent confusion between S. glauca and S. versicolor with S. amara by Franceschinelli et al. (1999)22. By analyzing the nucleotide diversity of the genus (Fig. 5c), we identified 13 nucleotide diversity hotspots. These regions hold promise for molecular differentiation among species within the genus Simarouba (DNA barcode), in the management and conservation of genomic resources, and in the analysis of phylogenetic relationships.
The rpl23 gene is under positive selective pressure in the species examined in this study and is located in the IR region, belonging to a gene family responsible for encoding proteins that contribute to the structural composition of the large ribosomal subunit. Hypervariable regions and signals of positive selection have been found in the rpl23 gene across the Celastraceae, Styracaceae, and Fabaceae families104,105,106. In addition, instances of rpl23 gene loss and pseudogenization have been documented in species belonging to the Podostemaceae107, Lauraceae108, Araceae109, and Hypericaceae86 families. Therefore, our results, combined with other evidence for positive selection on the rpl23 gene, call for further evolutionary studies of this gene within the order Sapindales.
The preferential use of amino acids identified in our investigation, particularly Leu, Ile, and Ser, showed patterns similar to other angiosperms4,110,111. In prokaryotes, branched-chain amino acids, including Leu and Ile, are involved in protein synthesis and maintenance of metabolic processes112,113. In addition, Ile is often found in the formation of beta sheets (β-sheets), while Leu contributes to the formation of leucine α-helices, loops, and zippers114. In the three species studied, S. amara, S. versicolor and S. glauca, AGA-Arg and UUA-Leu were the most frequently used codons (Figure S6). Our findings are consistent with previous investigations of codon usage bias in Rutaceae115 and other families of the order Sapindales116,117. According to Prosdocimi & Ortega (2007)118, the amino acids arginine and leucine have codons responsible for maintaining protein stability against DNA mutations. Moreover, factors such as translation optimization, genes with high expression rates, nucleotide composition of the genome, and the less stringent matching in the first base of tRNA (wobble base pairing) can influence the preferential use of specific codons119,120,121,122,123.
Phylogenetic analysis reveals that S. versicolor exhibits a closer evolutionary relationship to S. glauca more than to S. amara. The distinct geographic distributions and vegetation preferences of these three continental species warrant emphasis. S. versicolor, native to South America, and S. glauca, found in Central America, are frequently found in dry forests and savannas. In contrast, S. amara, distributed throughout Central and South America, is usually found in riparian and ombrophilous forests21,22,23. Both, S. versicolor and S. glauca occupy similar drier vegetation types than S. amara, raising the possibility that the tropical forests of Panama and South America, particularly the Amazon region, may act as an ecological barrier, influencing the geographical separation of these two species. This observation supports the hypothesis that S. glauca and S. versicolor may share a common evolutionary ancestor, or alternatively, that S. versicolor may have evolved from S. glauca and vice versa21. The families Simaroubaceae, Meliaceae, and Rutaceae exhibit a polytomy, characterized by unresolved phylogenetic relationships, according to APG (2006)9. This finding is consistent with recent chloroplast genome studies within the botanical group conducted by Liu et al. (2021)124, Majure et al. (2021)19 and Yang et al. (2022)76. Therefore, the phylogenetic insights presented in this study not only contribute to the discourse on the evolutionary relationships of S. amara, S. versicolor, and S. glauca within the family Simaroubaceae, but also enrich the discussion on the evolutionary relationships within the order Sapindales.
In this study, the chloroplast genomes of S. amara, S. versicolor, and S. glauca were successfully sequenced. The gene order (collinearity) and structure closely resembled those observed in chloroplast genomes of other species within the family Simaroubaceae and the order Sapindales, with the absence of genomic rearrangements. Pseudogenization events of a gene important for the photosynthetic pathway were detected. In addition, the investigation revealed highly divergent genomic regions, future candidates for the development of DNA barcode genetic markers, particularly within the genus Simarouba. We found that the majority of the CDSs are under negative pressure selection and observed the preferences in codon and amino acid usage and amino acids in the chloroplast of S. amara, S. versicolor and S. glauca. Phylogenetic analyses based on molecular plastome data elucidated evolutionary relationships, positioning S. versicolor closer to S. glauca than to S. amara. Furthermore, Simaroubaceae exhibited a close evolutionary relationship with Rutaceae. This work not only contributed novel genomic resources to the Simaroubaceae family, but also advanced comparative genomics and the understanding of evolutionary dynamics within the plastomes of Sapindales representatives. The findings presented herein also provide valuable insights into the discussions of phylogenetic relationships within the genus Simarouba and the order Sapindales.
Methods
Sequencing, assembly, and characterization of the chloroplast genome
Fresh leaves of S. amara were collected in the Serra of Pirineus, city of Pirenópolis, and of S. versicolor in the city of Goiânia, both in the state of Goiás, Brazil (geographical coordinates: − 48.84820, − 15.80294 and − 50.15408289, − 15.92941545, respectively). Total DNA was isolated using the CTAB protocol 2% 125. Library preparation was performed using the Illumina DNA Prep Kit and sequencing was performed on the Illumina MiSeq platform in paired-end using the V3 600 cycles and V2 300 cycles kits for sequencing S. amara and S. versicolor, respectively. Raw reads were subjected to a sequencing quality control processing step using the Trimommatic software126. Raw reads of S. glauca and Ailanthus excelsa were obtained from databases. For S. amara reads the parameters used were SLIDINGWINDOW: 4:20, CROP:289, HEADCROP:15, ILLUMINACLIP: NexteraPE-PE.fa:2:15:10, LEADING:10, TRAILING:10, MINLEN:100. For S. versicolor, S. glauca, and A. excelsa, the following parameters were applied: SLIDINGWINDOW: 4:15, CROP:150, HEADCROP:15, ILLUMINACLIP: NexteraPE-PE.fa:2:15:10, LEADING:10, TRAILING:10, MINLEN:100.
De novo assemblies of the chloroplast genomes were performed using the software NOVOPlasty v. 4.2.1127, using the rbcL gene of S. amara as seed (access number EU043036.1) for the three Simarouba species and the rbcL gene of A. altissima (access number EU043036.1) for A. excelsa. The genes were predicted and annotated using the GeSeq tool128 on the ChloroBox-MPI-MPI online version platform. The annotations were manually checked for correct annotation of the single copy (LSC and SSC) and inverted repeat (IR) regions, as well as the start and end codons of the CDS. This step was performed using the tools Geneious Prime v. 2021.1.1129 and Ugene v.48.1130. After the manual inspection and curation of the annotations, circular maps of the chloroplast genomes were drawn using the program OGDRAW v.1.3.1131.
Benchmarking
For comparative analyses, we used cpDNAs from the species assembled in this work, S. amara, S. versicolor, S. glauca and A. excelsa, and 13 other species from three families of the order Sapindales, for a total of 17 species (Table S1). Note that the plastomes of all species were annotated with the same parameters for comparison purposes.
Repetitive regions
Long repeats were detected on the plastomes using the tool Online Reputer132. The parameters were: Hamming distance = 3 and minimum repeat size 20 bp. The single sequence repeat (SSR) was identified using the program MISA Web Version133, and the parameters were: ≥ 8 mononucleotide, ≥ 5 dinucleotide, ≥ 4 trinucleotide, ≥ 3 tetra, penta and hexanucleotide. We consider the maximum allowable size of 100 bp between two microsatellites, to be registered as a composite microsatellite. The distribution of microsatellite patterns was compared and analyzed graphically.
Boundary regions and genomic rearrangements
After annotation and curation of the plastomes of all species included in this study, information on the gene composition of the boundaries of IRs and SCs was obtained using Geneious Prime v.2023.1.2. To determine the presence of genomic rearrangements, we used the tool Mauve v.2.4.0134, whose analyses were performed from a multiple alignment of the 17 genomes.
Nucleotide diversity, selection signal and codon bias usage
To quantify the degree of polymorphism among populations or species, proposed the metric of nucleotide diversity (π)135. This measure corresponds to the estimate of the number of nucleotide substitutions obtained by comparing sequences from different individuals and species. Nucleotide diversity (π) was calculated to identify the sites of mutational hotspots. For this purpose, the analyses were performed in three groups, the first including all 17 species of the three families, the second including only the 9 species of the family Simaroubaceae, and finally we analyzed the nucleotide diversity within the genus Simarouba. The complete plastomes were aligned using the MAFFT v.7136, and the nucleotide diversity was calculated using the tool DnaSP137 with the following parameters: sliding window 600 bp e step size 200 bp. The generated data were plotted using the R environment138.
The presence of a selection signal in species of the order Sapindales with nonsynonymous (dN) and synonymous (dS) substitutions was estimated by the value of omega (ω) = dN/dS, which was obtained using 77 CDS. We aligned the CDS using MAFFT v.7136 and then estimated ω using the Codeml tool included in the PAML 4.9 program139. We first estimated the value of ω under two neutral models, using species trees (runmode = 0; model = 0; NSsites = 0) and no tree information (nullmode = 0; NSsites = 0), and then contrasted groups of models (neutral and positive selection model), M2a contrast (positive selection model; model = 0; NSsites = 2) x M1a (neutral selection model; model = 0; NSsites = 1) and M8 (positive selection under beta distribution; model = 0; NSsites = 8) x M7 (neutral model under beta distribution; model = 0; NSsites = 7). The p-value and FDR (false discovery rate) were calculated for each contrast and for each gene (Supplementary Table S2). Values of ω > 1 indicate positive selection, ω = 1 indicates neutral selection and ω < 1 indicates negative selection (Jeffares et al., 2015). To identify the codon bias usage, the RSCU index (Relative Synonymous Codon Usage)140 was obtained using the MEGA X v.10.2.2 tool141, and the results were plotted in heat map using the R environment138 (pheatmap package v.1.0.12).
Phylogenomics analyses
Phylogenomic analyses were performed using 78 CDSs present in the 17 species. The outgroup consists of two species of the family Sapindaceae (Pometia tomentosa and Dodonea viscosa). The CDSs were aligned using the MAFFT v.7 tool and then concatenated into a single matrix using the catfasta2phyml software142. The Gblocks v.0.91b tool143 was used to identify conserved blocks, eliminate poorly aligned positions and divergent regions of non-homologous sites, and eliminate saturated substitutions. The phylogenetic tree was constructed using the IQ-TREE multicore version 1.6.12 tool144 and the best evolutionary model was TVM + F + I + G4 inferred using the ModelFinder tool, according to the Bayesian information criterion (BIC). The maximum likelihood method was used to access the evolutionary relationships, with 1000 replications.
Data availability
The Simarouba amara, Simarouba versicolor and Simarouba glauca chloroplast genome are being processed at NCBI, with the following submission code: S. amara (2799504), S. versicolor (2798048) and S. glauca (2798158).
References
Herrmann, R. G. & Possingham, J. V. Plastid DNA-the plastome. In: Results and Problems in Cell Differentiation: Chloroplasts. 10, (Springer-Verlag, Berlin, 1980).
Ravi, V., Khurana, J. P., Tyagi, A. K. & Khurana, P. An update on chloroplast genomes. Plant Syst. Evol. 271, 101–122. https://doi.org/10.1007/s00606-007-0608-0 (2008).
Mower J. P & Vickrey T. L. Structural diversity among plastid genomes of land plants. In: Plastid genome evolution. In Advances in Botanical Research. Ed. Chaw Shu-Miaw, Jansen Robert) 106p (Elsevier, Cambridge, MA, USA: Academic Press, 2018).
Sobreiro, M. B. et al. Chloroplast genome assembly of Handroanthus impetiginosus: Comparative analysis and molecular evolution in Bignoniaceae. Planta 252, 1–16 (2020).
Carvalho, L. R. et al. The complete chloroplast genome sequence of Eugenia klotzschiana O. Berg unveils the evolutionary dynamics in plastomes of Myrteae DC. Tribe (Myrtaceae). Gene 876, 147488 (2023).
Li, C., Liu, Y., Lin, F., Zheng, Y. & Huang, P. Characterization of the complete chloroplast genome sequences of six Dalbergia species and its comparative analysis in the subfamily of Papilionoideae (Fabaceae). PeerJ 10, e13570 (2022).
Xi, Z. et al. Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. Proc. Natl. Acad. Sci. U.S.A. 109, 17519–17524 (2012).
Kuo, W. H. et al. Plastome phylogenomics of Allaeanthus, Broussonetia and Malaisia (Dorstenieae, Moraceae) and the origin of B. × kazinoki. J. Plant Resour. 135, 203–220 (2022).
Iv, A. P. G. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20 (2016).
WFO. World Flora Online. https://wfoplantlist.org/ (2023).
Clayton, J. W. Simaroubaceae. In The Families and Genera of Vascular Plants. Springer. 11, (Springer, Hamburg-Germany, 2011).
Thomas, W. W. The American genera of Simaroubaceae and their distribution. Acta Bot. Bras. 4, 11–18 (1990).
Wang, T. et al. Complete chloroplast genome sequence of Fortunella venosa (Champ. ex benth.) (Rutaceae): Comparative analysis, phylogenetic relationships, and robust support for its status as an independent species. Forests 12, 996 (2021).
Polonsky, J. Quassinoid bitter principles II. In Progress in the Chemistry of Organic Natural Products 222–259 (1985).
Alves, I. A. B. S., Miranda, H. M., Soares, L. A. L. & Randau, K. P. Simaroubaceae family: Botany, chemical composition and biological activities. Revista Brasileira de Farmacognosia 24, 481–501 (2014).
Gazoni, V. F. et al. Assessment of toxicity and differential antimicrobial activity of methanol extract of rhizome of Simaba ferruginea A. St.-Hil. and its isolate canthin-6-one. J. Ethnopharmacol. 223, 122–134 (2018).
Gomes, M. C. A. R., Paula, V. F., Moreira, A. A., Castellani, M. A. & Macedo, G. E. L. Toxicity of plant extracts from Bahia, Brazil, to Atta sexdens sexdens (Hymenoptera: Formicidae) workers. Sociobiology 63, 770–776 (2016).
Devecchi, M. F., Thomas, W. W., Plunkett, G. M. & Pirani, J. R. Testing the monophyly of Simaba (Simaroubaceae): Evidence from five molecular regions and morphology. Mol. Phylogenet. Evol. 120, 63–82 (2018).
Majure, L. C., Clase, T., Blankenship, A. & Noa-Monzón, A. A new species of Picrasma, P. nanophylla (Simaroubaceae), from the Dominican Republic. Brittonia 73, 334–342 (2021).
Clayton, J. W., Soltis, P. S. & Soltis, D. E. Recent long-distance dispersal overshadows ancient biogeographical patterns in a pantropical angiosperm family (Simaroubaceae, Sapindales). Syst. Biol. 58, 395–410 (2009).
Cronquist, A. Studies in the Simaroubaceae-II. The genus Simarouba. Bull. Torrey Bot. Club 71, 226–234 (1944).
Franceschinelli, E. V., Yamamoto, K. & Shepherd, G. J. Distinctions among three Simarouba species. Syst. Bot. 23, 479 (1999).
Devecchi, M. F., Pirani, J. R. & Thomas, W. W. Simaroubaceae in Flora do Brasil. Jardim Botânico do Rio de Janeiro (2020).
Pirani, J. R., Majure, L. C. & Devecchi, M. F. An updated account of Simaroubaceae with emphasis on American taxa. Revista Brasileira de Botanica https://doi.org/10.1007/s40415-021-00731-x (2021).
Franceschinelli, E. V. & Yamamoto, K. Taxonomic use of leaf anatomical characters in the genus Simarouba Aublet (Simaroubaceae). Flora 188, 117–124 (1993).
Hardesty, B. D., Dick, C. W., Kremer, A., Hubbell, S. & Bermingham, E. Spatial genetic structure of Simarouba amara Aubl. (Simaroubaceae), a dioecious, animal-dispersed Neotropical tree, on Barro Colorado Island, Panama. Heredity (Edinb.) 95, 290–297 (2005).
Waghmode, V. A., Kute, N. S. & Ban, Y. G. Reproductive biology of oilseed tree Simarouba glauca DC. Acta Hortic. 1241, 459–463 (2019).
Ferreira, I. N. M. et al. Two dioecious Simarouba species with a specialized pollination system and low reproductive efficacy in Central Brazil. Rodriguésia https://doi.org/10.1590/2175-7860202273030 (2022).
Romero-da-Cruz, M. V., Guimarães, R., Devecchi, M. F., Pirani, J. R. & Forni-Martins, E. R. Chromosome numbers in Homalolepis Turcz. and their significance in Simaroubaceae evolution. Revista Brasileira de Botanica https://doi.org/10.1007/s40415-021-00729-5 (2021).
Baratakke, R. C. & Patil, C. G. Cytological investigations in poly-gamo-dioecious tree Simarouba glauca DC. The Nucleus 53, 33–36 (2010).
Saina, J. K., Li, Z. Z., Gichira, A. W. & Liao, Y. Y. The complete chloroplast genome sequence of tree of heaven (Ailanthus altissima (mill.) (sapindales: Simaroubaceae), an important pantropical tree. Int. J. Mol. Sci. 19, 929 (2018).
Ng, W. L., Lee, S. Y. & Yeap, S. K. Characterization of the complete chloroplast genome of an important Southeast Asian medicinal plant, Eurycoma longifolia (Simaroubaceae). Mitochondrial DNA B Resour. 4, 128–129 (2019).
Qin, L. et al. The complete chloroplast genome sequence of Picrasma quassioides (D. Don) Benn. 1844 (Simaroubaceae). Mitochondrial DNA B Resour. 7, 1114–1116 (2022).
Mader, M. et al. Complete chloroplast genome sequences of four Meliaceae species and comparative analyses. Int. J. Mol. Sci. 19, 701 (2018).
Zhang, J., Li, Y. & Wang, Y. The complete chloroplast genome sequence of Aglaia odorata. Mitochondrial DNA B Resour. 5, 472–473 (2020).
Dobrogojski, J., Adamiec, M. & Luciński, R. The chloroplast genome: a review. Acta Physiol. Plant 42, 1–13 (2020).
Bock, R. & Knoop, V. Genomics of Chloroplasts and Mitochondria Vol. 35 (Springer, 2012).
Pacheco, T. G. et al. The complete plastome of Passiflora cirrhiflora A. Juss.: Structural features, RNA editing sites, hotspots of nucleotide diversity and molecular markers within the subgenus Deidamioides. Revista Brasileira de Botanica 43, 839–853 (2020).
Jansen, R. K. & Ruhlman, T. A. Plastid Genomes of Seed Plants.103–126 (2012). https://doi.org/10.1007/978-94-007-2920-9_5.
Liu, T. J. et al. Complete plastid genome sequence of Primula sinensis (Primulaceae): Structure comparison, sequence variation and evidence for accD transfer to nucleus. PeerJ 2016, e2101 (2016).
Millen, R. S. et al. Many parallel losses of InfA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 13, 645 (2001).
Ueda, M. et al. Loss of the rpl32 gene from the chloroplast genome and subsequent acquisition of a preexisting transit peptide within the nuclear gene in Populus. Gene 402, 51–56 (2007).
Daniell, H., Lin, C. S., Yu, M. & Chang, W. J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 1–29 (2016).
Iwata, S. & Barber, J. Structure of photosystem II and molecular architecture of the oxygen-evolving centre. Current Opin. Struct. Biol. 14, 447–453. https://doi.org/10.1016/j.sbi.2004.07.002 (2004).
Chisholm, D. & Williams, J. G. K. Nucleotide sequence of psbC, the gene encoding the CP-43 chlorophyll a-binding protein of Photosystem II, in the cyanobacterium synechocystis 6803. Plant Mol. Biol. 10, 293–301 (1988).
Hankamer, B., Barber, J. & Boekema, E. J. Structure and membrane organization of photosystem II in green plants. Annu. Rev. Plant Biol. 48, 641–671 (1997).
Jo, S., Kim, Y. K., Cheon, S. H., Fan, Q. & Kim, K. J. Characterization of 20 complete plastomes from the tribe Laureae (Lauraceae) and distribution of small inversions. PLoS ONE 14, e0224622 (2019).
Maurya, S. et al. Plastome characterization of Musa indandamanensis, an endemic banana in Andaman and Nicobar Islands, India. Nucleus 66, 117–126 (2023).
Xu, W. et al. Comparative plastome analyses and evolutionary relationships of all species and cultivars within the medicinal plant genus Atractylodes. Ind. Crops Prod. 201, 116974 (2023).
Sawicki, J., Krawczyk, K., Ślipiko, M., Szandar, K. & Szczecińska, M. Comparative analysis of Apopellia endiviifolia plastomes reveals a strikingly high level of differentiation between its terrestrial and water form. Diversity (Basel) 13, 674 (2021).
Liu, M. L. et al. Evolutionary analysis of plastid genomes of seven Lonicera L. species: Implications for sequence divergence and phylogenetic relationships. Int. J. Mol. Sci. 19, 4039 (2018).
Komenda, J. et al. Accumulation of the D2 protein is a key regulatory step for assembly of the photosystem II reaction center complex in synechocystis PCC 6803. J. Biol. Chem. 279, 48620–48629 (2004).
Fu, H. Y. et al. The availability of neither D2 nor CP43 limits the biogenesis of photosystem II in tobacco. Plant Physiol. 185, 1111–1130 (2021).
Bricker, T. M. & Frankel, L. K. The structure and function of CP47 and CP43 in photosystem II. Photosynth. Res. 72, 131–146 (2002).
Logacheva, M. D. & Shipunov, A. B. Phylogenomic analysis of Picramnia, Alvaradoa, and Leitneria supports the independent Picramniales. J. Syst. Evol. 55, 171–176 (2017).
Cauz-Santos, L. A. et al. The chloroplast genome of Passiflora edulis (Passifloraceae) assembled from long sequence reads: Structural organization and phylogenomic studies in malpighiales. Front. Plant Sci. 8, 334 (2017).
Da Silva, R. S. et al. The plastome sequence of Bactris gasipaes and evolutionary analysis in tribe Cocoseae (Arecaceae). PLoS ONE 16, e0256373 (2021).
Yu, X. et al. Transcriptome and comparative chloroplast genome analysis of vincetoxicum versicolor: Insights into molecular evolution and phylogenetic implication. Front. Genet. 12, 1–13 (2021).
Belinky, F., Rogozin, I. B. & Koonin, E. V. Selection on start codons in prokaryotes and potential compensatory nucleotide substitutions. Sci. Rep. 7, 12422 (2017).
Watanabe, K. & Suzuki, T. Genetic code and its variants. eLS https://doi.org/10.1038/npg.els.0000810 (2001).
Looman, A. C. & van Knippenberg, P. H. Effects of GUG and AUG initiation codons on the expression of lacZ in Escherichia coli. FEBS Lett. 197, 315–320 (1986).
Ping, J. et al. The molecular evolution pattern of rps12 gene in gymnosperms. Kexue Tongbao/Chin. Sci. Bull. 66, 3182–3193 (2021).
Yan, W., Shi, W., Tian, Q., Li, Z. & Gao, H. The complete chloroplast genome sequence of Melilotoides ruthenica: structural comparative and phylogenetic analysis in Leguminosae. In IOP Conference Series: Earth and Environmental Science vol. 697 (IOP Publishing Ltd, 2021).
Long, M., Betrán, E., Thornton, K. & Wang, W. The origin of new genes: Glimpses from the young and old. Nat. Rev. Genet. 4, 865–875. https://doi.org/10.1038/nrg1204 (2003).
Zaita, N., Torazawa, K., Shinozaki, K. & Sugiura, M. Trans splicing in vivo: Joining of transcripts from the ‘divided’ gene for ribosomal protein S12 in the chloroplasts of tobacco. FEBS Lett. 210, 153–156 (1987).
Hildebrand, M., Hallick, R. B., Passavant, C. W. & Bourque, D. P. Trans-splicing in chloroplasts: The rps 12 loci of Nicotiana tabacum. Proc. Natl. Acad. Sci. U.S.A. 85, 372–376 (1988).
Nunes, R. et al. Complete chloroplast genome sequence of Caryocar brasiliense camb. (caryocaraceae) and comparative analysis brings new insights into the plastome evolution of Malpighiales. Genet Mol. Biol. 43, 1–7 (2020).
Trad, R. J., Cabral, F. N., Bittrich, V., Silva, S. R. D. & Amaral, M. D. C. E. D. Calophyllaceae plastomes, their structure and insights in relationships within the clusioids. Sci. Rep. 11(1), 20712 (2021).
de Souza, U. J. B., Nunes, R., Targueta, C. P., Diniz-Filho, J. A. F. & de Telles, M. P. C. The complete chloroplast genome of Stryphnodendron adstringens (Leguminosae-Caesalpinioideae): Comparative analysis with related Mimosoid species. Sci. Rep. 9, 1–12 (2019).
Kim, K. S. & Sappington, T. W. Microsatellite data analysis for population genetics. Methods Mol. Biol. 1006, 271–295 (2013).
Singh, K. P., Kumari, P., Raipuria, R. K. & Rai, P. K. Development of genome-specific SSR markers for the identification of introgressed segments of Sinapis alba in the Brassica juncea background. 3 Biotech 12, 332 (2022).
Borlay, A. J., Mweu, C. M., Nyanjom, S. G., Omolo, K. M. & Omire, A. Molecular characterization of doum palm (Hyphaene compressa) from selected regions of Kenya using chloroplast simple sequence repeats (cpSSR) markers. Pak. J. Bot. 55, 1013 (2023).
Hardesty, B. D., Hubbell, S. P. & Bermingham, E. Genetic evidence of frequent long-distance recruitment in a vertebrate-dispersed tree. Ecol. Lett. 9, 516–525 (2006).
Hardesty, B. D. et al. Geographic influence on genetic structure in the widespread Neotropical tree Simarouba amara (Simaroubaceae). Trop. Plant Biol. 3, 28–39 (2010).
Areces-Berazain, F., Wang, Y., Hinsinger, D. D. & Strijk, J. S. Plastome comparative genomics in maples resolves the infrageneric backbone relationships. PeerJ 8, e9483 (2020).
Yang, J., Hu, G. & Hu, G. Comparative genomics and phylogenetic relationships of two endemic and endangered species (Handeliodendron bodinieri and Eurycorymbus cavaleriei) of two monotypic genera within Sapindales. BMC Genomics 23, 27 (2022).
Zhou, S. M. et al. Phylogenomics and plastome evolution of Indigofera (Fabaceae). Front. Plant Sci. 14, 1186598 (2023).
Sun, K. et al. Comparative analysis and phylogenetic implications of plastomes of five genera in subfamily amyridoideae (Rutaceae). Forests 12, 1–14 (2021).
Ahmad, W., Asaf, S., Al-Rawahi, A., Al-Harrasi, A. & Khan, A. L. Comparative plastome genomics, taxonomic delimitation and evolutionary divergences of Tetraena hamiensis var. qatarensis and Tetraena simplex (Zygophyllaceae). Sci. Rep. 13, 7436 (2023).
Guisinger, M. M., Kuehl, J. V., Boore, J. L. & Jansen, R. K. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: Rearrangements, repeats, and codon usage. Mol. Biol. Evol. 28, 583–600 (2011).
Zhao, Y. J., Liu, J., Yin, G. S. & Gong, X. Characteristics of plastid genomes in the genus Ceratostigma inhabiting arid habitats in China and their phylogenomic implications. BMC Plant Biol. 23, 303 (2023).
Quiroga, M. P., Zattara, E. E., Souza, G., Pedrosa-Harand, A. & Premoli, A. C. Plastome sequencing of South American Podocarpus species reveals low rearrangement rates despite ancient Gondwanan disjunctions. Mol. Biol. Rep. 50, 309–318 (2023).
Mauad, A. V. S. R. et al. Plastid phylogenomics of Pleurothallidinae (Orchidaceae): Conservative plastomes, new variable markers, and comparative analyses of plastid, nuclear, and mitochondrial data. PLoS ONE 16, e0256126 (2021).
Kim, Y. K., Cheon, S. H., Hong, J. R. & Kim, K. J. Evolutionary patterns of the chloroplast genome in Vanilloid Orchids (Vanilloideae, Orchidaceae). Int. J. Mol. Sci. 24, 3808 (2023).
Raman, G., Nam, G. H. & Park, S. J. Extensive reorganization of the chloroplast genome of Corydalis platycarpa: A comparative analysis of their organization and evolution with other Corydalis plastomes. Front. Plant Sci. 13, 132 (2022).
Claude, S. J., Park, S. & Park, S. J. Gene loss, genome rearrangement, and accelerated substitution rates in Plastid genome of Hypericum ascyron (Hypericaceae). BMC Plant Biol. https://doi.org/10.1186/s12870-022-03515-x (2022).
Yu, J. et al. Plastome variations reveal the distinct evolutionary scenarios of plastomes in the subfamily Cereoideae (Cactaceae). BMC Plant Biol. 23, 132 (2023).
Wang, Z. X., Wang, D. J. & Yi, T. S. Does IR-loss promote plastome structural variation and sequence evolution?. Front. Plant Sci. https://doi.org/10.3389/fpls.2022.888049 (2022).
Jin, D. M. et al. The loss of the inverted repeat in the Putranjivoid clade of Malpighiales. Front. Plant Sci. https://doi.org/10.3389/fpls.2020.00942 (2020).
Palmer, J. D. Chloroplast DNA exists in two orientations. Nature 301, 92–93 (1983).
Vera, A., Matsubayashi, T. & Sugiura, M. Active transcription from a promoter positioned within the coding region of a divergently oriented gene: The tobacco chloroplast Rp132 gene. Mol. General Genet. MGG 233, 151–156 (1992).
Boudreau, E. et al. A large open reading frame (Orf1995) in the chloroplast DNA of Chlamydomonas reinhardtii encodes an essential protein. Mol. General Genet. MGG 253, 649–653. https://doi.org/10.1007/s004380050368 (1997).
Drescher, A., Stephanie, R., Calsa, T., Carrer, H. & Bock, R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 22, 97–104 (2000).
Neuhaus, H. & Link, G. The chloroplast tRNALys (UUU) gene from mustard (Sinapis alba) contains a class II intron potentially coding for a maturase-related polypeptide. Curr. Genet. https://doi.org/10.1007/BF00355398 (1987).
Xie, H. et al. Comparative analysis of the complete chloroplast genomes of six threatened subgenus Gynopodium (Magnolia) species. BMC Genomics 23, 716 (2022).
Castro, A. A. et al. Chloroplast genome characterization of Uncaria guianensis and Uncaria tomentosa and evolutive dynamics of the Cinchonoideae subfamily. Sci. Rep. https://doi.org/10.1038/s41598-023-34334-1 (2023).
Moghaddam, M., Wojciechowski, M. F. & Kazempour-Osaloo, S. Characterization and comparative analysis of the complete plastid genomes of four Astragalus species. PLoS ONE 18, e0286083 (2023).
Dong, W. et al. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 5, 8348 (2015).
Li, H. et al. The specific DNA barcodes based on chloroplast genes for species identification of Orchidaceae plants. Sci. Rep. 11, 1424 (2021).
CBOL Pant Working Group. A DNA barcode for land plants. PNAS 106, 12794–12797 (2009).
Neubig, K. M. et al. Phylogenetic utility of ycf1 in orchids: A plastid gene more variable than matK. Plant Syst. Evolut. 277, 75–84 (2009).
Amar, M. H. ycf1-ndhF genes, the most promising plastid genomic barcode, sheds light on phylogeny at low taxonomic levels in Prunus persica. J. Genetic Eng. Biotechnol. 18, 42 (2020).
Corvalán, L. C. J. et al. Chloroplast genome assembly of Serjania erecta Raldk: comparative analysis reveals gene number variation and selection in protein-coding plastid genes of Sapindaceae. Front. Plant Sci. 14, 1258794 (2023).
Wang, Y. C., Zhou, H. Y. & Liu, X. Q. Chloroplast genomic variation in Euonymus maackii Rupr. and its differentiation time in Euonymus. Forests 13, 265 (2022).
Cai, X. L. et al. Plastome structure and phylogenetic relationships of Styracaceae (Ericales). BMC Ecol. Evol. 21, 103 (2021).
Yu, X., Tan, W., Gao, H., Miao, L. & Tian, X. Development of a specific mini-barcode from plastome and its application for qualitative and quantitative identification of processed herbal products using DNA metabarcoding technique: A case study on Senna. Front. Pharmacol. 11, 585687 (2020).
Bedoya, A. M. et al. Plastid genomes of five species of riverweeds (podostemaceae): Structural organization and comparative analysis in malpighiales. Front. Plant. Sci. 10, 1–14 (2019).
Yang, Z., Ferguson, D. K. & Yang, Y. New insights into the plastome evolution of Lauraceae using herbariomics. BMC Plant Biol. 23, 387 (2023).
Yun, S. & Kim, H. The complete plastome sequence of Monstera deliciosa (Araceae), an ornamental foliage plant. Mitochondrial. DNA B Resour. 8, 1301–1305 (2023).
Qin, H. H. et al. The plastid genome of twenty-two species from Ferula, Talassia, and Soranthus: Comparative analysis, phylogenetic implications, and adaptive evolution. BMC Plant. Biol. 23, 9 (2023).
Pham, M. H. et al. The complete chloroplast genome of an Ophiorrhiza baviensis drake species reveals its molecular structure, comparative, and phylogenetic relationships. Genes (Basel) 14, 227 (2023).
Kaiser, J. C. & Heinrichs, D. E. Branching out: Alterations in bacterial physiology and virulence due to branched-chain amino acid deprivation. MBio 9(5), 10–1128 (2018).
Amorim Franco, T. M. & Blanchard, J. S. Bacterial branched-chain amino acid biosynthesis: Structures, mechanisms, and drugability. Biochemistry 56, 5849–5865. https://doi.org/10.1021/acs.biochem.7b00849 (2017).
Brosnan, J. T. & Brosnan, M. E. Branched-chain amino acids: Metabolism, physiological function, and application. Renal Fail. 1, 2 (2006).
Xu, C. et al. Analysis of synonymous codon usage patterns in seven different citrus species. Evolut. Bioinform. 2013, 215–228 (2013).
Chi, X., Zhang, F., Dong, Q. & Chen, S. Insights into comparative genomics, codon usage bias, and phylogenetic relationship of species from biebersteiniaceae and nitrariaceae based on complete chloroplast genomes. Plants 9, 1–15 (2020).
Wang, L., He, N., Li, Y., Fang, Y. & Zhang, F. Complete chloroplast genome sequence of Chinese lacquer tree (Toxicodendron vernicifluum, Anacardiaceae) and its phylogenetic significance. Biomed. Res. Int. 2020, 9014873 (2020).
Prosdocimi, F. & Ortega, J. M. The codon usage of Leucine, Serine and Arginine reveals evolutionary stability of proteomes and protein-coding genes. In Brazilian Symposium on Bioinfomatics (2007).
Salim, H. M. W. & Cavalcanti, A. R. O. Factors influencing codon usage bias in genomes. J. Braz. Chem. Soc. 19, 257–262 (2008).
Shah, P. & Gilchrist, M. A. Explaining complex codon usage patterns with selection for translational efficiency, mutation bias, and Genetic drift. Proc. Natl. Acad. Sci. U.S.A. 108, 10231–10236 (2011).
Rapino, F. et al. Wobble tRNA modification and hydrophilic amino acid patterns dictate protein fate. Nat. Commun. 12, 2170 (2021).
Alberts, B. et al. Molecular biology of the cell. Ed. Artmed. 5ed, 1396 (2010).
Whittle, C. A., Kulkarni, A., Chung, N. & Extavour, C. G. Adaptation of codon and amino acid use for translational functions in highly expressed cricket genes. BMC Genom. 22, 1 (2021).
Liu, C. et al. Plastome phylogenomics of the East Asian endemic genus Dobinea. Plant Divers. 43, 35–42 (2021).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15 (1987).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucl. Acids Res. 45, e18 (2017).
Tillich, M. et al. GeSeq—Versatile and accurate annotation of organelle genomes. Nucl. Acids Res. 45, W6–W11 (2017).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Okonechnikov, K. et al. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 28, 1166–1167 (2012).
Greiner, S., Lehwark, P. & Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucl. Acids Res. 47, W59–W64 (2019).
Kurtz, S. et al. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucl. Acids Res. 29, 4633–4642 (2001).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 33, 2583–2585 (2017).
Darling, A. C. E., Mau, B., Blattner, F. R. & Perna, N. T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements Aaron. Genome Res. 14, 1394–1403 (2004).
Nei, M. & Li, W. H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. U.S.A. 76, 5269–5273 (1979).
Katoh, K., Rozewicki, J. & Yamada, K. D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166 (2018).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302 (2017).
R Core Team. R: A language and enviroment for statistical computing. Preprint at (2016).
Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Sharp, P. M., Tuohy, T. M. F. & Mosurski, K. R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucl. Acids Res. 14, 5125–5143 (1986).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Nylander, J. A. A. catfasta2phyml. Preprint at https://github.com/nylander/catfasta2phyml (2010).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Acknowledgements
This work was developed in the context of Instituto Nacional de Ciência Tecnologia em Ecologia, Evolução e Conservação da Biodiversidade (INCT – EECBio), supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq – process 465610/20145) and Fundação de Amparo à Pesquisa do Estado de Goiás (FAPEG – process 201810267000023). We are also thankful for the support from PPGS Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES)/FAPEG (#08/2014) and CNPq (MCTIC/CNPq #28/2018, 435477/2018-8). This work was also developed in the context of PPBio Araguaia (CNPq – Processo: 441114/2023-7) and Araguaia Vivo 2030 (TWRA/FAPEG Processo: 202210267000536).
Author information
Authors and Affiliations
Contributions
M.A.A.S., R.N. and M.P.C.T. conceived and designed research, M.P.C.T. provided financial resources. C.M.S.N. sample collect. C.M.S.N., and E.V.F. taxonomic identification. M.A.A.S., R.S.B.F. and C.P.T. performed the experiments., M.A.A.S., L.C.J.C. and M.B.S. did computational analysis. M.A.A.S., L.C.J.C. and R.N. analyzed data. M.A.A.S. wrote the paper. All authors reviewed the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Legality in the access of biological samples
The collection and management of biological samples were carried out following the legal guidelines of SisGen—National System for the Management of Genetic Heritage and associated traditional knowledge and are available under the access code AFBD2DB.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Almeida-Silva, M.A., Braga-Ferreira, R.S., Targueta, C.P. et al. Chloroplast genomes of Simarouba Aubl., molecular evolution and comparative analyses within Sapindales. Sci Rep 14, 21358 (2024). https://doi.org/10.1038/s41598-024-71956-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-71956-5
- Springer Nature Limited