
Abstract
The Hypoxylaceae (Xylariales, Ascomycota) is a diverse family of mainly saprotrophic fungi, which commonly occur in angiosperm-dominated forests around the world. Despite their importance in forest and plant ecology as well as a prolific source of secondary metabolites and enzymes, genome sequences of related taxa are scarce and usually derived from environmental isolates. To address this lack of knowledge thirteen taxonomically well-defined representatives of the family and one member of the closely related Xylariaceae were genome sequenced using combinations of Illumina and Oxford nanopore technologies or PacBio sequencing. The workflow leads to high quality draft genome sequences with an average N50 of 3.0 Mbp. A backbone phylogenomic tree was calculated based on the amino acid sequences of 4912 core genes reflecting the current accepted taxonomic concept of the Hypoxylaceae. A Percentage of Conserved Proteins (POCP) analysis revealed that 70% of the proteins are conserved within the family, a value with potential application for the definition of family boundaries within the order Xylariales. Also, Hypomontagnella spongiphila is proposed as a new marine derived lineage of Hypom. monticulosa based on in-depth genomic comparison and morphological differences of the cultures. The results showed that both species share 95% of their genes corresponding to more than 700 strain-specific proteins. This difference is not reflected by standard taxonomic assessments (morphology of sexual and asexual morph, chemotaxonomy, phylogeny), preventing species delimitation based on traditional concepts. Genetic changes are likely to be the result of environmental adaptations and selective pressure, the driving force of speciation. These data provide an important starting point for the establishment of a stable phylogeny of the Xylariales; they enable studies on evolution, ecological behavior and biosynthesis of natural products; and they significantly advance the taxonomy of fungi.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fungal taxonomy is a constantly evolving area that has been greatly influenced by the advances in technology and research. Micro-morphological characters of many species were not accessible until the development of microscopes, which greatly enhanced the discovery of new species since the seventeenth century. With the application of physiological and biochemical characters since the nineteenth century including color reaction tests, nutrient utilization assays or thin layer chromatography for secondary metabolites, fungal classification became more reliable (Zhang et al. 2017). Subsequently, the establishment of gene sequencing as a means of barcoding, and phylogenetic reconstructions on various taxonomic levels substantially improved our concept of fungal species delimitation. The increasing availability of gene sequences derived from various genetic loci, such as ribosomal DNA (18s rDNA, internal transcribed spacer region 1 and 2 [ITS1/2], 5.8S rDNA, 28S rDNA) or protein-coding genes (beta-tubulin, translation elongation factor 1 alpha, DNA-directed RNA polymerase II subunit RPB2, etc.) transitioned phylogenetic inferences from single-locus to multi-loci datasets delivering more stable and reliable tree topologies (Dornburg et al. 2017; Zhang et al. 2017). Currently, the biological research of the twenty-first century is driven by the development of cost-efficient and very rapid methods to generate full genome sequences of organisms. In this respect, mycology is pushing into a new direction by implementing this new level of information to construct robust taxonomic frameworks, which are now referred to as phylogenomics (Nagy and Szöllősi 2017).
So far, phylogenomic studies have been implemented only in a limited number of taxonomic groups in the fungal kingdom, despite the fact that more than 1300 fungal genomes have been sequenced to date according to the 1000 Fungal Genomes Project website (https://1000.fungalgenomes.org/home/, Grigoriev et al. 2014). Given the enormous diversity of fungi (up to 5.1 million estimated species; Blackwell 2011), often only a few or single members of the families are covered, restricting the use of the public repository to address interspecific relationships on taxonomic levels below ordinal rank. Nevertheless, a few fungal groups were extensively genome sequenced due to their economic and scientific importance and hence offered the basis for phylogenomic analyses. Respective studies have been recently conducted on some members of the Hypocreales (Ascomycota) including a set of nine Trichoderma species (Druzhinina et al. 2018), and in particular, on the Aspergillaceae based on a dataset of 45 Aspergillus species and 33 Penicillium species (Steenwyk et al. 2019). The latter study comprises so far the largest phylogenomic reconstruction on the family level by using a 1669 gene matrix. Pizarro and coworkers for the first time generated a larger number of genome sequences only for the taxonomic purposes to resolve relationships of 51 lichenized fungi of the Parmeliaceae. Even though their overall genome quality for the majority of strains was quite low (average N50 of 20,000 bp), they obtained 2556 orthologous single-copy genes as the basis for tree construction, resulting in a highly supported stable tree topology with monophyletic subclades (Pizarro et al. 2018).
Except for the Hypocreales and Magnaporthales (Zhang et al. 2018b), no other significant taxonomic groups within the Sordariomycetes, one of the largest classes in the Ascomycota with 37 orders (Wijayawardene et al. 2018), were subject of comparative genomic studies. Among those, the Xylariales are of special importance as they harbor a great number of taxa distributed throughout 26 families including phytopathogens, endophytes, saprobionts and prolific secondary metabolite producers (Helaly et al. 2018). With more than 350 described species, the Hypoxylaceae contributes considerably to the Xylariales diversity (Helaly et al. 2018). This family currently contains 15 genera, where the sexual morph is characterized by carbonaceous ascostromata with KOH-extractable pigments usually associated with decaying hardwood (Wendt et al. 2018; Lambert et al. 2019). Members of the Hypoxylaceae are also frequently encountered in seed plants, lichens and algae and can even be found in insect nests and sponges (Pažoutová et al. 2013; U’Ren et al. 2016; Medina et al. 2016; Leman-Loubière et al. 2017; Sir et al. 2019). Due to their worldwide distribution with hotspots in tropical regions, these fungi play an important ecological role in particular in forested areas as wood-decomposers (Stadler 2011). In addition, they might have beneficial effects for their hosts during their endophytic life stage. Besides their biological relevance, family members have been found to produce a wide variety of natural products ranging from various types of pigments [e.g. azaphilones (Kuhnert et al. 2015b; Surup et al. 2018b), tetramic acids (Kuhnert et al. 2014b), binaphthalenes (Sudarman et al. 2016)] to compounds with unusual carbon skeletons [e.g. rickiols (Surup et al. 2018a), sporochartines (Leman-Loubière et al. 2017)] to highly bioactive molecules [e.g. nodulisporic acids (Bills et al. 2012), sporothriolide (Surup et al. 2014), rickenyls (Kuhnert et al. 2015a), cytochalasins (Yuyama et al. 2018; Wang et al. 2019) and concentricolide (Qin et al. 2006)]. Even though hundreds of genes of Hypoxylaceae species have been sequenced in the course of phylogenetic studies, taxonomically relevant genome sequences of these fungi have been scarce. In fact, only two genome sequences from unambiguously identified species have been published. This includes an isolate of Hypoxylon pulicicidum, which has been sequenced in the context of the investigation of the nodulisporic acid biosynthesis (Nicholson et al. 2018; Van de Bittner et al. 2018) and an Annulohypoxylon stygium strain (Wingfield et al. 2018). Further genomes are only available for environmental isolates mainly identified based on marker sequences and which therefore have limited value for phylogenomic analyses (Ng et al. 2012; Wu et al. 2017).
To close the knowledge gap and to offer a solid backbone for genomic investigations of the Hypoxylaceae, we attempted to create high-quality draft genomes (N50 > 1Mbp) of selected taxonomically well-characterized representatives of the major phylogenetic clades (cf. Wendt et al. 2018) within the family and one outgroup species from the related Xylariaceae. For the study, ex-epitype strains of Annulohypoxylon truncatum, Hypoxylon fragiforme, H. rickii, H. rubiginosum, Jackrogersella multiformis, Pyrenopolyporus hunteri, Xylaria hypoxylon and non-type strains of Daldinia concentrica, Entonaema liquescens, H. lienhwacheense, Hypomontagnella monticulosa and H. pulicicidum were chosen. In addition, we decided to sequence the genomes of a marine-derived Hypom. monticulosa (Leman-Loubière et al. 2017, as Hypoxylon monticulosum; see Lambert et al. 2019) and an endophytic isolate of Hypom. submonticulosa (Burgess et al. 2017, as Hypoxylon submonticulosum; see Lambert et al. 2019) for complementary follow-up studies. Various genome comparison methods were applied to estimate the similarity of the members and the conserved core genes were used to calculate a phylogenomic backbone tree of the Hypoxylaceae. A new species of Hypomontagnella is described based on the results and the authenticity of the solely available strain of E. liquescens is discussed on genome-based evidence. Other results of the current genome sequencing campaign, revealing interesting findings about the intragenomic polymorphisms of the ITS regions have recently been published elsewhere (Stadler et al. 2020).
Material and Methods
General
All scientific names of fungi follow the entries in MycoBank and Index Fungorum, hence no authorities and years of publications are given in the text.
Selection of fungal strains
For the genome sequencing, twelve morphologically well characterized ascospore-derived strains of the order Xylariales were chosen. The selection was comprised of eleven representatives of the Hypoxylaceae (Annulohypoxylon truncatum CBS 140778, Daldinia concentrica CBS 113277, Entonaema liquescens ATCC 46302, Hypoxylon fragiforme MUCL 51264, H. lienhwacheense MFLUCC 14-1231, H. pulicicidum ATCC 74245, H. rickii MUCL 53309, H. rubiginosum MUCL 52887, Jackrogersella multiformis CBS 119016, Pyrenopolyporus hunteri MUCL 49339) and one member of the Xylariaceae (Xylaria hypoxylon CBS 122620) for comparison. The majority of isolates was designated as ex-epitype strains of the respective species in previous studies (Stadler et al. 2014a; Kuhnert et al. 2014a, 2017; Wendt et al. 2018) except for D. concentrica, E. liquescens, H. lienhwacheense, Hypomontagnella monticulosa MUCL 54604 and H. pulicicidum, which do not represent type strains. Regarding upcoming studies, two endosymbiontic isolates of Hypomontagnella were included as well. This involves a marine sponge-derived strain designated as Hypom. monticulosa (originally referred to as Hypoxylon monticulosum, Leman-Loubière et al. 2017) and an endophytic isolate from Rubus idaeus identified as Hypom. submonticulosa (originally referred to as Hypoxylon submonticulosum, Burgess et al. 2017).
Genomic DNA preparation
All fungi were grown in 250 ml Erlenmeyer flasks containing 50 ml YMG media (10 g l−1 malt, 4 g l−1 glucose, 4 g l−1 yeast extract, pH 6.3) for 5 to 10 days (depending on growth speed) at 150 rpm and 25 °C in a shaking incubator. Afterwards, mycelia were harvested by vacuum filtration using a Büchner funnel with filter paper (MN 640 w, Macherey–Nagel, Düren, Germany). The biomass was then frozen with liquid nitrogen and ground to a fine powder in a mortar. The DNA extraction and purification were performed with the GenElute® Plant Genomic DNA Miniprep Kit (Sigma-Aldrich, St. Louis, MO, USA) according to manufacturer’s instructions.
Nanopore library preparation & MinION® sequencing
MinION sequencing library with genomic DNA from the different fungal strains was prepared using the Nanopore Rapid DNA Sequencing kit (SQK-RAD04, Oxford Nanopore Technologies, Oxford, UK) according to the manufacturer's instructions. Sequencing was performed on an Oxford Nanopore MinION Mk1b sequencer using a R9.5 flow cell, which was prepared according to the manufacturer's instructions.
Illumina library preparation & MiSeq sequencing
Whole-genome-shotgun PCR-free libraries were constructed from 5 µg of gDNA with the Nextera XT DNA Sample Preparation Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s protocol. The libraries were quality controlled by analysis on an Agilent 2000 Bioanalyzer with Agilent High Sensitivity DNA Kit (Agilent Technologies, Santa Clara, CA, USA) for fragment sizes of 500–1000 bp. Sequencing was performed on the MiSeq platform (Illumina; 2 × 300 bp paired-end sequencing, v3 chemistry). Adapters and low-quality reads were removed by an in-house software pipeline prior to polishing as recently described (Wibberg et al. 2016).
Base calling, reads processing, and assembly
MinKNOW (v1.13.1, Oxford Nanopore Technologies) was used to control the run using the 48 h sequencing run protocol; base calling was performed offline using albacore (v2.3.1, https://github.com/Albacore/albacore). The assembly was performed using canu v1.6 and v1.7 (Koren et al. 2017), resulting in a single, circular contig. This contig was then polished with Illumina short read data using Pilon (Walker et al. 2014), run for eight iterative cycles. BWA-MEM (Li 2013) was used for read mapping in the first four iterations and Bowtie2 v2.3.2 (Langmead and Salzberg 2012) in the second set of four iterations.
PacBio library preparation and sequencing
For Hypoxolon fragiforme a SMRTbell™ template library was prepared according to the instructions from Pacific Biosciences (Menlo Park, CA, USA), following the Procedure & Checklist – Greater Than 10 kb Template Preparation. Briefly, for preparation of 15 kb libraries genomic DNA was sheared using g-tubes™ from Covaris, Woburn, MA, USA according to the manufacturer's instructions. DNA was end-repaired and ligated overnight to hairpin adapters applying components from the DNA/Polymerase Binding Kit P6 from Pacific Biosciences. Reactions were carried out according to the manufacturer's instructions. BluePippin™ Size-Selection to greater than 7 kb was performed according to the manufacturer's instructions (Sage Science, Beverly, MA, USA). Conditions for annealing of sequencing primers and binding of polymerase to purified SMRTbell™ template were assessed with the Calculator in RS Remote (Pacific Biosciences). One SMRT cell was sequenced on the PacBio RSII (Pacific Biosciences) taking one 240-min movie. Two further SMRT cells were sequenced on the Sequel System (Pacific Biosciences) taking one 600-min movie for each SMRT cell.
For Hypoxolon rubiginosum a SMRTbell™ template library was prepared according to the instructions from Pacific Biosciences, following the Procedure & Checklist – Preparing Greater Than 30 kb Libraries Using SMRTBell® Express Template Preparation Kit. Briefly, for preparation of libraries genomic DNA was end-repaired and ligated overnight to hairpin adapters applying components from the SMRTBell® Express Template Preparation Kit 2.0 from Pacific Biosciences. Reactions were carried out according to the manufacturer´s instructions. BluePippin™ Size-Selection to greater than 15 kb was performed according to the manufacturer´s instructions (Sage Science). Conditions for annealing of sequencing primers and binding of polymerase to purified SMRTbell™ template were assessed with the SMRT® Link Software (PacificBiosciences). One SMRT cell was sequenced on the Sequel System taking one 600-min movie.
Genome assembly was performed within SMRTLink 6.0.0.47841 using the HGAP4 protocol and a target genome size of 40 Mbp.
Gene prediction and genome annotation
Gene prediction was performed by applying Augustus version 3.2 (Stanke et al. 2008) and GeneMark-ES 4.3.6. (Ter-Hovhannisyan et al. 2008) using default settings. For Augustus, species parameter sets were established based on GeneMark-ES fungal version predictions. Predicted genes were functionally annotated using a modified version of the genome annotation platform GenDB 2.0 (Meyer 2003) for eukaryotic genomes as previously described (Rupp et al. 2014). For automatic annotation within the platform, similarity searches against different databases including COG (Tatusov et al. 2003), KEGG (Kanehisa et al. 2004) and SWISS-PROT (Boeckmann et al. 2003) were performed. In addition to genes, putative tRNA genes were identified with tRNAscan-SE (Lowe and Eddy 1997). Completeness, contamination, and strain heterogeneity were estimated with BUSCO (v3.0.2 Simão et al. 2015), using the Pezizomycotina-specific single-copy marker genes database (odb9).
RNA-Isolation and transcriptome sequencing of Hypom. monticulosa
Hypomontagnella monticulosa MUCL 54604 was grown in two 250 ml flasks each containing 50 ml of a different medium (DPY: 20 g l−1 dextrin from potato starch, 10 g l−1 polypeptone, 5 g l−1 yeast extract, 5 g l−1 monopotassium phosphate, 0.5 g l−1 magnesium sulfate hexahydrate; PDB: 24 g l−1 potato dextrose broth) for 5 days at 25 °C and 150 rpm. Small quantities of mycelia (> 100 µl) were removed with a sterile inoculating loop and RNA was extracted from the samples using the Quick-RNA Fungal/Bacterial Miniprep Kit (Zymo Research, Irvine, CA, USA). Samples were treated with DNase I (Zymo Research) according to the manufacturer`s recommendations. In total, ~ 2 μg of RNA per sample was used for library preparation with the TruSeq mRNA Sample Preparation Kit (stranded) (Illumina). Sequencing of the prepared cDNA libraries was carried out on the Illumina HiSeq 1500 platform (2 × 75 bp) using the ‘Rapid Mode’. Data analysis and base calling were accomplished with in-house software (Wibberg et al. 2016). The sequencing raw data for all libraries have been stored on the EBI ArrayExpress server, accession E-MTAB-8948.
Improved gene prediction with BRAKER2 for Hypom. monticulosa
BRAKER2 (Hoff et al. 2019) allows fully automated training of the gene prediction tools GeneMark and AUGUSTUS from RNA-Seq by integrating the extrinsic evidence from RNA-Seq information into the prediction. Therefore, the RNAseq data was used for mapping to the Hypom. monticulosa reference genome. The sequenced reads were quality filtered (> Q30) by applying the FASTX tool kit. Data of each condition were subsequently mapped to the Hypom. monticulosa using tophat2 (Kim et al. 2013). Two mismatches were allowed. The resulting BAM-files were used for the BRAKER2 gene prediction with default settings.
Identification and analysis of carbohydrate-related proteins
Carbohydrate active enzymes (CAZymes) among the predicted proteins of the 13 fungi were analyzed with dbCAN “Data-Base for automated Carbohydrate-active enzyme Annotation” version 7 (Yin et al. 2012) and the HMMER 3.0 package (Mistry et al. 2013) under relaxed settings (E value: < 1e−5, coverage: 0.3). CAZy-family definitions were followed according to the CAZy database (https://www.cazy.org/) (Lombard et al. 2014).
Comparative genome analyses
The genomes of the sequenced and annotated fungal strains were used for comparative genome analyses. Comparative analyses between fungal genomes were accomplished using a modified version of the comparative genomics program EDGAR designed to handle eukaryotic genomes and their multi-exon genes (Blom et al. 2009, 2016) as described recently (Wibberg et al. 2015). During the analyses, identification of orthologous genes, classification of genes as core genes or singletons and visualization of Venn diagrams were performed. In addition, average nucleotide identity (ANI) and average amino acid identity analyses (AAI) were performed based on the GeneMark prediction similarly to previously described methods (Wibberg et al. 2015) to determine the relationship between the different species. Synteny analysis were performed by applying D-GENIES (Cabanettes and Klopp 2018).
Phylogenetic tree reconstruction
Based on the EDGAR analysis, 4912 core genes were identified. First, multiple alignments for all core protein sequences were created using MUSCLE (Edgar 2004). All amino acid alignments were subjected to automatic elimination of poorly aligned positions via the Castresana Lab Gblocks standalone application with default parameters; applying ‘with-hal’ gap treatment and minimum block length set to 5 (Talavera and Castresana 2007). For the Maximum-Likelihood (ML) tree inference, a super matrix approach was followed using IQTree v.1.70 (beta, Nguyen et al. 2015) with standard options except for the tree topology improvement strategy using nearest-neighbor interchange (NNI), which was extended to all possible interchanges instead of only looking for previously applied NNIs for further topology improvement (allnni). Protein evolutionary models and statistical support on the partitioned dataset (Chernomor et al. 2016) was assigned with Modelfinder (Kalyaanamoorthy et al. 2017) following Bayesian information criteria (BIC) with ultra-fast Bootstrapping option (UFB, Hoang et al. 2018). The latter was performed with 1000 UFB replicates in combination with 1000 replicates of SH-aLRT (Statistical significance in an approximate likelihood ratio test, Guindon et al. 2010).
For a coalescence-based phylogeny using the program ASTRAL III v.5.14.2 (Zhang et al. 2018a), single gene maximum-likelihood trees were inferred following the same procedure as stated before with IQTree except for the strategy of Bootstrap support calculation. Here, non-parametric bootstrapping was applied with fast tree search option to decrease the computational burden. Analysis of the resulting dataset with ASTRAL III followed standard parameters.
For an evaluation of the four-gene phylogeny methodology followed by Wendt et al. (2018), a whole-gene dataset compromised of α-actin, TEF1-α, RPB2 and TUB2 was subjected to molecular phylogenetic inference with prior curation via the gblocks Online Server and nucleotide substitution model selection with smart model selection (SMS, Lefort et al. 2017) following current literature methodology with the ATGC PhyML 3.0 Online Server (Wendt et al. 2018; Lambert et al. 2019; Guindon et al. 2010).
POCP analysis
POCP analysis was performed according to Qin et al. (2014) and as previously described (Adamek et al. 2018; Margos et al. 2018). Briefly, for each genome pair reciprocal BLASTP (Altschul 1997) was used to identify homologous proteins. Proteins were considered to be conserved if the BLAST matches had an E-value of < 1 × 10–5, > 40% sequence identity and > 50% of the query sequence in each of the reciprocal searches. The POCP value for a genome pair was then determined as [(C1 + C2)/(T1 + T2)] × 100, where C1 and C2 are the number of conserved proteins between the genome pair and T1 and T2 are the total number of proteins in each genome being compared (Qin et al. 2014).
Results and Discussion
Genomic data
To gain insights into the functional differences and the phylogenetic relationship between all fungal strains targeted in this work, their genomes were completely sequenced by application of 3rd generation sequencing technologies. The respective sequences are stored on the ENA (European Nucleotide Archive) portal of the EMBL-EBI (https://www.ebi.ac.uk/) under the bioproject numbers PRJEB36622 (A. truncatum), PRJEB36624 (D. concentrica), PRJEB36625 (E. liquescens), PRJEB36647 (Hypom. monticulosa), PRJEB37480 (Hypom. spongiphila) PRJEB36653 (Hypom. submonticulosa), PRJEB36654 (H. fragiforme), PRJEB36656 (H. lienhwacheense), PRJEB36657 (H. pulicicidum), PRJEB36658 (H. rickii), PRJEB36693 (J. multiformis), PRJEB36695 (H. rubiginosum), PRJEB36696 (P. hunteri) and PRJEB36697 (X. hypoxylon).
The genome sequences of the analyzed fungi were mainly established using a combination of Nanopore and short-read Illumina methods. The latter data were used to improve base accuracy and thus significantly reduce error rates in the final genomes. For comparative reasons two species (H. fragiforme and H. rubiginosum) were randomly chosen for PacBio sequencing. General genome features, e.g., size, contig number, GC content, and numbers of predicted genes, are summarized in Table 1.
The number of reads generated by Nanopore sequencing ranged from 83,869 to 937,388 with an average read count of 406,479. The mean read length averaged 9736 bp (5454–27,241 bp) with a maximum read length average of 470,823 bp (64,165–2,031,748 bp). Illumina polished assemblies resulted in a total contig number average of 55 (16–123) with a 61 fold average coverage (14.8–125.1 fold). Details on the genome sequencing statistics for the individual strains are listed in the supplementary information (Tab. S1). Established genomes range in size from 35 to 54 Mb and feature GC contents around 45%.
Sequencing on the Sequel System (PacBio) resulted in 727,768 (sub)reads with a mean length of 7823 bp for H. fragiforme. The genome assembly yielded 36 contigs summing up to a final genome size of 38.1 Mbp (3.6 Mbp N50 contig length, 127× Genome Coverage). Sequencing of H. rubiginosum resulted in 692,592 (sub)reads with a mean length of 14,154 bp. The genome assembly yielded 70 contigs summing up to a final genome size of 48.3 Mbp (1.1 Mbp N50 contig length, 202× Genome Coverage).
In general, the combinatorial approach (Illumina/Nanopore) performed equally to the newest generation of PacBio sequencing in terms of generated contigs, N50 contig lengths and average read lengths, which resulted in 14 high quality draft genome sequences. PacBio sequencing yielded higher read numbers and higher genome coverage, but maximum read length with Nanopore sequencing was longer. Based on the obtained data, we can state that the generation of high-quality fungal genome sequences can be achieved by different technologies leaving mycologists with options depending on their personal preferences.
The re-sequencing of H. pulicicidum (ATCC 74245) resulted in a substantially improved genome quality compared to the previous report (Nicholson et al. 2018). The genome size increased from 41.4 to 43.5 Mbp and contig numbers were reduced from 204 to 24. In addition, the N50 increased almost sevenfold from 580,679 to 3,855,590 bp. This demonstrates that long-read technologies enable access to additional genomic information (here more than 2 Mbp) offering a much more robust basis for genomic comparisons.
In the case of X. hypoxylon, the genome sequence of a different strain has been published recently (Büttner et al. 2019). The respective length of the Illumina assembly summed up to 42.8 Mbp, which is significantly shorter than what was obtained for the ex-epitype strain (54.3 Mbp) herein. Consequently, the number of predicted genes was lower as well (11,038 vs 12,704 here). These differences can be mainly attributed to the sequencing quality (N50 of 0.1 Mbp vs 3.8 Mbp, and 635 vs 88 contigs in our study, respectively). Similar to H. pulicicidum, third generation sequencing resulted in a substantial gain of genetic information (~ 15% of gene content), which makes subsequent analyses more reliable.
CAZyme analysis
The Hypoxylaceae are considered as endophytes that can switch to a saprobiontic lifestyle if necessary. However, there is no conclusive information on whether family members are able to become facultative parasites. To address this question at the genomic scale, a CAZyme analysis was conducted to investigate the number of enzymes possibly involved in carbohydrate interactions (Fig. 1). The number of related genes and modules ranged from 510 (H. lienhwacheense) to 780 (H. rubiginosum) with an average of 669. Glycoside hydrolases (GH) accounted for almost half of the identified enzymes (218–289), followed by the auxiliary activity proteins (AA, 96–187), glycosyl transferases (GT, 89–108) and carbohydrate esterases (CE, 76–136). Polysaccharide lyases (PL) comprised only a few genes (5–16) and carbohydrate-binding modules (CBM) accounted for approx. 7%of the hits (25–59). Depending on the strictness of the E-value settings in dbCAN (standard < 1e−15), a significant number of CBMs (8–21) and CEs (59–99) were not recognized, while the values for the other enzyme classes remained stable (see Supplementary information for more details). We want to highlight this result as many publications, which involve dbCAN analyses do not specify their parameters for CAZyme search. Therefore, comparisons with other datasets need to be treated carefully as they might lead to wrong conclusions.

Comparison of CAZymes classes within the Hypoxylaceae and Xylaria hypoxylon. Number of identified enzymes or related modules are displayed by horizontal bars (GT – glycosyl transferase, GH – glycoside hydrolase, CE – carbohydrate esterase, CBM – carbohydrate-binding module, PL – polysaccharide lyase, AA – auxiliary activity enzymes). AT – Annulohypoxylon truncatum, DC – Daldinia concentrica, EL – Entonaema liquescens., HF- Hypoxylon fragiforme, HL – Hypoxylon lienhwacheense, HM – Hypomontagnella monticulosa, HP – Hypoxylon pulicicidum, HSM – Hypomontagnella submonticulosa, HSP – Hypomontagnella spongiphila, HR – Hypoxylon rickii, HRUB – Hypoxylon rubiginosum, JM – Jackrogersella multiformis, PH – Pyrenopoylporus hunteri, XH – Xylaria hypoxylon
The relatively high numbers of CAZymes in this study are in the range of other known saprobiontic fungi, such as Aspergillus oryzae or Penicillium chrysogenum (Zhao et al. 2013) and also match those of previously analyzed X. hypoxylon and A. stygium strains (Wingfield et al. 2018; Büttner et al. 2019). In contrast, the P. fici genome (which is also used in this paper for subsequent analyses) was reported to contain much higher numbers of respective enzymes (460 GHs, 138 CEs, 121 GTs, 39 PLs; Wang et al. 2015). As no parameters for the dbCAN analysis were reported in the corresponding reference, the encoded proteins of P. fici were re-analyzed using the same settings as for the Hypoxylaceae proteins. The previous results could be mostly confirmed except for CE, where we found even higher numbers (243), demonstrating that the capabilities of utilizing carbohydrate sources within the Xylariales can strongly vary.
When analyzing the individual families of CAZymes (a detailed list of which can be found in the Supplementary Information), a slightly higher number of proteins are categorized as cutinases (CE5, 3–8 genes) in comparison with the average results obtained by Zhao et al. (2013) for saprobiontic fungi (Ø = 1; 0–7 genes). Cutinases are responsible for the digestion of cutin and are usually correlated with plant infection events enabling a fungus to invade its host (Lu et al. 2018). This expanded set of CE5 enzymes corresponds well with the proposed initial endophytic lifestyle, as the entry into the host plants is a crucial step to establish such an association. Unfortunately, little is yet known on how Hypoxylaceae recognize and enter their host. Only in the case of Hypoxylon fragiforme has it been demonstrated that ascospore eclosion and germination occurs at high frequency when triggered by monolignol glucosides released from the beech host (Chapela et al. 1993). Even though there is no supportive data available, it is likely that the site of infection is primarily located at leaf surfaces (which is also the main tissue to acquire endophytic isolates of Xylariales). The germinating spores (ascospores or conidia) can enter the plant either by using opened stoma or by lysing the protective cuticle layer to grow into or in-between the cells. In general, there is a lack of knowledge about the host range of the individual Hypoxylaceae species as the association of their stromata with certain tree species does not necessarily reflect the distribution of a given species. This became, in particular, evident when various family members have been isolated from different plant parts of Viscum album and Pinus sylvestris (Peršoh et al. 2010), which in contrast never harbor Hypoxylaceae stromata. It can therefore be assumed that these fungi have a rather broad host range. Thus, an extended number of cutinases might be required to sustain the capacity of entering various hosts.
Pectin degrading enzymes are important to break down the cell wall of plants and are especially abundant in pathogenic fungi. Members of the majority of corresponding CAZYme families (CE8, PL1, PL2, PL3, PL9, PL10, GH28, GH78, GH88) can be found within our genomes (7 out of 9), but the number of copies is lower than those identified for most phytopathogenic fungi (CE8: 2.7 vs 4.0; PL1: 1.9 vs 5.3; PL3: 0.8 vs 3.1; PL9: 0.1 vs 0.5; GH28: 3.1 vs 7.1; GH78: 3.1 vs 4.3; GH88: 0.6 vs 1.3) (for detailed numbers see Zhao et al. 2013). Hence, Hypoxylaceae and X. hypoxylon encode in general the capabilities to degrade pectin and related carbohydrates, but to a lesser extent than plant pathogens.
The CAZyme analysis supports the hypothesis of Hypoxylaceae being endophytes without a major pathogenic life stage that can easily switch to a saprobiotic lifestyle based on their carbohydrate degrading capacities. Even though it is unlikely that these fungi actively harm their host, the nature of the symbiosis still remains obscure, and probably also varies during the host life cycle. Commensalistic or mutualistic interactions are generally possible and depend on the effect of the fungus on its host. The respective genome sequences should enable more thorough future studies to resolve these cryptic fungus-host interactions.
Phylogenomic analysis
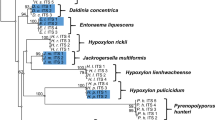
Previous phylogenetic studies have shown that the Hypoxylaceae are a well-conserved monophyletic family (Wendt et al. 2018; Daranagama et al. 2018). In particular, the inclusion of house-keeping genes and calculation of multi-locus trees has improved the topology of the corresponding reconstructions, which well-resolved the structure of the major clades. Nevertheless, the respective tree backbones lack support, preventing conclusive results on intergeneric evolutionary relationships. Therefore, we aimed to construct a backbone tree based on a core set of proteins derived from genomic data of the genome-sequenced strains and compare the clade relationships with those of previously published studies. As gene or protein trees do not necessarily reflect the evolutionary history of taxa, a coalescence-based ASTRAL species tree was calculated for comparison. Moreover, we wanted to estimate how well genomic trees perform in comparison to multilocus trees. For the calculations, Pestalotiopsis fici was chosen as the outgroup due to its taxonomic classification in the Xylariales (note that the taxonomic position of the species is still under debate, as recent studies suggest a placement into the new order Amphisphaeriales, a proposed sister taxon of the Xylariales, see Jaklitsch et al. 2016; Hongsanan et al. 2017; Liu et al. 2019; Hyde et al. 2020 for discussion on this topic) and the relatively high quality of the genome sequence (Wang et al. 2015). An EDGAR analysis revealed the presence of 4912 homologous genes located in all 15 genomes, the amino acid sequences of which were used to assess the relative phylogenetic position of the fungi. The concatenation of the 4912 GBLOCKS-cured amino acid alignments resulted in a data matrix with 2,532,758 sites, from which 49.0% were constant and 28.5% variable sites. Cured single loci alignments for species-tree inference ranged from 44 to 4781 amino acids (median 437.5 ± 358.4 aa). Alignment length, selected substitution models, distinct patterns, log-likelihood, constant and variable sites for each alignment are summarized in the supplementary material. The resulting gene and species tree showed identical topologies with four major clades (C1–C4, Fig. 2), which include all Hypoxylaceae species except H. rubiginosum. The backbone and all branches received maximum bootstrap support or posterior probability values in both phylogenomic reconstructions. As expected, X. hypoxylon forms a sister clade to the Hypoxylaceae, where H. rubiginosum is located at a separate branch at the base of the family clade.

Phylogenomic vs multigene phylogenetic reconstruction of the Hypoxylaceae. A, B: Comparison of a maximum-Likelihood phylogenetic tree inferred from a supermatrix approach (lL = − 21,719,985.1679) of 4912 curated concatenated protein alignments (A, IQTree) with a species tree (quartet score = 3,750,366, normalized = 66.61%) of coalescence-based phylogenetic inference (B, ASTRAL III). Bootstrap and SH-alRLT support values of 1000 replicates are given on nodes for the inferred consensus tree A. Multi-locus bootstrap values (MLBS) and posterior probability (pp) values for the species tree are given on nodes for tree B. Branch length for tree A is shown as nucleotide substitutions per site. C Four-gene phylogenetic inferences based on the full gene sequences of alpha-actin, beta-tubulin, RPB2 and TEF1-α with lLn = − 50,232.40147 prior curation of the alignment via gblocks and GTR + G + I nucleotide substitution model as determined by Smart Model Selection (SMS). Bootstrap support values of 1000 replicates above 50% are shown at corresponding nodes. Scale bar indicates nucleotide substitution rates. The different subclades (C1–C4) are highlighted in colors
Clade 1 (C1) is composed of H. fragiforme and H. rickii with both species displaying similar branch lengths. These taxa have been already demonstrated to be closely related based on morphological, chemotaxonomical and phylogenetic data (Kuhnert et al. 2014a, 2015c). They mainly differ in their stromata shape (hemispherical vs. effused-pulvinate) and distribution (temperate zones vs tropical zones).
The second clade (C2) contains H. pulicicidum, A. truncatum and J. multiformis. Before the erection of Jackrogersella, the latter two species were placed in the same genus based on their morphological similarities (carbonaceous layer surrounding the perithecia, ostioles higher than the stromatal surface, dehiscent perispore with conspicuous thickening). However, their strongly deviating secondary metabolite profiles in conjunction with completely reduced stromatal discs set them clearly apart (Kuhnert et al. 2017; Wendt et al. 2018). In contrast, the relationship with H. pulicicidum does not seem obvious in the first place, but the presence of papillate ostioles in overmature stromata and reduced discs in younger material morphologically resembles the main characteristics of Jackrogersella. In addition, the appearance of cohaerin-type stromatal pigments in H. pulicicidum (and closely related Hypoxylon species, see Sir et al. 2019) is completely restricted to these taxa and members of Jackrogersella. Based on the phylogenomic reconstruction and taxonomic significant similarities, it can be speculated that Annulohypoxylon is derived from Jackrogersella, which by itself shares a common ancestor with Hypoxylon. Hence, Annulohypoxylon in its current definition represents in an evolutionary context one of the derived genera in the Hypoxylaceae.
The genus Hypomontagnella forms the third clade (C3) in the phylogenomic tree. Two of the representatives are endosymbiontic isolates (Hypom. submonticulosa from plant and Hypom. spongiphila from a marine sponge) and only Hypom. monticulosa is stroma-derived. The marine isolate was originally identified as Hypom. monticulosa and is now treated as a distinct new species based on the genomic comparisons (see the taxonomic section for more details). The Hypomontagnella clade is located on a long branch supporting its status as a distinct genus. Due to the sister clade placement of C3 to C4 Hypomontagnella taxa are evolutionarily closer to Daldinia and Pyrenopolyporus than to other genera included in this study (note that the taxonomic position of H. lienhwacheense is unclear). This result contradicts a previous study where Hypomontagnella grouped with C2 in a multigene phylogenetic tree (based on the ITS, 28s rDNA, RPB2 and TUB2 loci) with statistical bootstrap support (Lambert et al. 2019). These differences might be caused by the restricted sample size in our analysis but could also indicate that phylogenomic data better resolve the taxonomic relationship of Hypomontagnella. Future studies with a larger taxon selection will prove whether the organization of this basic phylogenomic tree remains stable.
In clade 4 (C4), the representatives of Daldinia, Pyrenopolyporus and H. lienhwacheense clustered together. The peltate to hemispherical shape of Pyrenopolyporus stromata and its naphthalene content are reminiscent of Daldinia and underline their relationship. The position of H. lienhwacheense is inconclusive especially since the metabolite profile of this taxon is unique among all analyzed Hypoxylaceae species (unpublished data). In addition, its stromata are irregular in form with conspicuous elevations. The latter could be an evolutionary precursor of peltate ascomata, which further evolved into hemispherical shapes. In a recently published phylogenetic analysis with a much larger taxon selection, H. lienhwacheense also clustered with Daldinia species and Pyrenopolyporus species but lacked support and had no close relatives (Sir et al. 2019). Moreover, the genome of H. lienhwacheense is the smallest among all sequenced species herein with a size reduction of 12.5 Mbp compared to H. rubiginosum. Therefore, it is possible that H. lienhwacheense represents a derived lineage and has undergone heavy gene or chromosome loss in the course of adaptation. Due to its isolated position in the phylogenies, the genome characteristics and metabolite profile, its affinities with Hypoxylon are only supported by certain morphological features. The data would justify the erection of a new genus, but we refrain from this for the time being as we see the necessity to include the putative relatives H. lividipigmentum, H. lividicolor and H. brevisporum in a corresponding study. The lack of viable cultures for the latter two species currently prevent such an approach.
Hypoxylon rubiginosum appears as a basal branch within the Hypoxylaceae. Based on the branch length and position (sister group of the Xylariaceae), this fungus most likely represents an old lineage. Still, the morphological habitus of H. rubiginosum is typical for the family and has little in common with the genus Xylaria. To deduce the divergent evolution of the two families and understand the development of the distinct morphologies, many more representatives of the Xylariaceae (e.g. Nemania spp., Kretzschmaria spp., Rosellinia spp.), and also other related families (e.g. Graphostromataceae spp., Barrmaeliaceae spp., Lopadostomataceae spp.) should be sequenced.
To evaluate the strengths of our amino acid-based phylogenomic reconstruction compared to previous standard phylogenies, we calculated a multigene tree for the same species set based on the complete gene sequences of alpha-actin, beta-tubulin, RPB2 and TEF1-α (Fig. 2c). The concatenated dataset of the four genes had a final length of 8566 nucleotides of original 10,614 positions (80.7% of the original alignment). Unique site patterns were constituted by 3007 positions, while 2713 were parsimony informative and 4925 invariant sites. The substructure of the clades is resolved identically to the phylogenomic trees, but the clades are arranged in a different order and with lower support values for the backbone. Thus, we conclude that the multigene phylogenetic family reconstruction does only correctly reflect the evolutionary origin of closely related genera, while the relationship of major lineages (here equal to the clades) remains unresolved. Thus, previously published phylogenies of the Hypoxylaceae (Wendt et al. 2018; Lambert et al. 2019) can only provide a robust picture of infrageneric relationships. In addition, the position of certain family members such as H. trugodes and H. griseobrunneum remains unresolved in these calculations and will most likely only stabilize in phylogenomic reconstructions. Our results of the phylogenomic analysis are promising in this regard and the inclusion of more high-quality genome sequences will help to understand the evolution and speciation of Hypoxylaceae.
For the phylogenomic reconstructions, we have deliberately decided to use protein sequences rather than nucleotide sequences as these are directly correlated with structural and functional information, and thus better reflect evolutionary divergence (Chowdhury and Garai 2017). However, it is important to note that this approach has its limitation for very closely related organisms as synonymous changes of nucleotides do not alter the protein sequence and hence core proteins only provide restricted information (which is the case for the species pair Hypom. monticulosa and Hypom. spongiphila). In addition, protein level phylogenomic reconstructions are susceptible to sequencing errors, which can strongly influence the predictions of genes and therefore should only be considered if high-quality genome sequences are available. On the other hand, the slow evolution of protein sequences allows for a more robust inference of relationships between fungal families and higher ranks, where the alteration of the respective gene sequences is usually too strong for reliable alignments. Thus, we are convinced that protein-based phylogenetic reconstructions will become more important in the future to reevaluate current taxonomic concepts.
Comparative genomics
POCP analysis
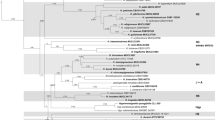
To better estimate the genomic differences between the fungi and to deduce the relative amount of individual genes, a percentage of conserved protein (POCP) analysis was conducted (Fig. 3). In advance, it is important to state that the results of POCP analyses depend on the reliability of the applied gene prediction models. The more accurate such models can call genes and their respective coding sequences, the higher the significance of POCP becomes. To test the quality of the gene prediction in our genomes, the transcriptome of Hypom. monticulosa under two different conditions was sequenced and reads were implemented into the Braker pipeline for improved gene prediction (Hoff et al. 2019). Braker called 12,744 genes, while GeneMark and Augustus predictions resulted in 12,477 and 11,204 genes, respectively. Sizes and intron mapping of predicted genes displayed high similarity between the Braker and GeneMark output. In contrast, roughly 30% of the genes predicted by Augustus deviated in these parameters. Thus, it can be concluded that GeneMark already delivers a good coverage of the gene content, which is comparable to RNA-Seq-based prediction pipelines. Consequently, protein sequences derived from GeneMark predictions were chosen as the starting point for subsequent analysis. Nevertheless, protein translations can still differ from the actual protein sequences, which however has usually little influences on the recognition of orthologous proteins in other organisms. In addition, the number of unpredicted genes cannot be evaluated by the applied methods and therefore some of the conserved proteins might be missed for the POCP analysis. As a result, the data presented in the following have to be treated as an approximation to the real values.

Pairwise Percentage of Conserved Proteins (POCP) analysis for members of the Hypoxylaceae and non-family members. Species located within dashed line belong to the Hypoxylaceae and those located within solid line belong to the subclass Xylariomycetidae. AN – Aspergillus nidulans, AT – Annulohypoxylon truncatum, DC – Daldinia concentrica, Dsp – Daldinia sp., HF – Hypoxylon fragiforme, HL – Hypoxylon lienhwacheense, HM – Hypomontagnella monticulosa, HP – Hypoxylon pulicicidum, HSM – Hypomontagnella submonticulosa, HSP – Hypomontagnella spongiphila, HR – Hypoxylon rickii, HRUB – Hypoxylon rubiginosum, JM – Jackrogersella multiformis, PC – Penicillium chrysogenum, PF – Pestalotiopsis fici, PH – Pyrenopoylporus hunteri, XH – Xylaria hypoxylon
To study how POCP numbers vary across families, P. fici (Sordariomycetes, Xylariomycetidae, Xylariales, Sporocadaceae), Aspergillus niger and Penicillium chrysogenum (both Eurotiomycetes, Eurotiomycetidae, Eurotiales, Aspergillaceae) were included in the dataset. The values ranged from 72.5 to 95.8% within the Hypoxylaceae and were in general higher for phylogenetically related species, albeit with some exceptions. For example, H. rickii shares 82.6% of its encoded proteins with P. hunteri, and Hypom. monticulosa shows a similar number compared to H. pulicicidum. Surprisingly, between the selected members of the genus Hypoxylon the POCP did not exceed values higher than 79.8%, but also did not fall below 72.5% confirming the already known heterogeneity of the genus (Wendt et al. 2018). As these numbers strongly overlap with those of species from different genera or are even sometimes smaller, there is no clear boundary between interspecific and intergeneric POCPs. In comparison, the values between other members of the Xylariales and Hypoxylaceae are located in the range of 62.5 to 66.8% for X. hypoxylon and 55.6 to 60.1% for P. fici. These values do not overlap with those calculated for species within the Hypoxylaceae in our taxon selection. The POCP values significantly drop when comparing unrelated species as demonstrated by A. niger and P. chrysogenum where the numbers were always below 50% (but not lower than 40.3%). The POCP analysis has been proposed as a measure to define genus boundaries in prokaryotes (Qin et al. 2014). The authors demonstrated that species of different genera share less than half of their proteins and consequently selected 50% as threshold for genus delimitations. As shown herein this value is not applicable for fungal taxonomy and overall proves that genes are much more conserved in fungi than in bacteria. However, POCP analysis might be helpful to delimit species or define family borders in fungi. Even very closely related species differed in at least 4.2% of their encoded conserved protein content. In order to determine how valuable this number is, various additional isolates of individual fungi should be genome-sequenced to compile a proper comparison and support the data by statistical means. In our example, the POCP analysis always yielded values between non-family members below 70%, while the values between Hypoxylaceae species were in all cases above 70%. Whether this threshold can be generally applied for fungi or whether it is even valid for the Xylariales cannot be answered yet as additional fungi of various families from this particular order need to be sequenced. Furthermore, sequences of a broad range of genera within other fungal families are scarce to conduct such a respective study on the class or even division level. Nevertheless, based on our data we propose that members of the Hypoxylaceae share on average at least 70% of their encoded proteins. To the best of our knowledge, this is the first study to apply POCP analysis to a larger taxon selection in fungi.
ANI analysis
Besides the POCP, we analyzed the Average Nucleotide Identity (ANI, Fig. 4) of the species which measures the nucleotide-level genomic similarity between two genomes (Arahal 2014). The ANI values between members of the Hypoxylaceae varied from 71.1 to 93.3%. The (by far) highest similarity was found between the genomes of Hypom. monticulosa and Hypom. spongiphila. All other genome comparisons showed maximum values of 80.4%. In contrast, the nucleotide difference between X. hypoxylon and Hypoxylaceae species remained relatively stable around 32% (ANI 68.5–67.8%). Based on our calculations, family members of the Hypoxylaceae share at least 70% of their nucleotide content. This threshold is identical with the one estimated for the POCP analysis. ANI analyses have been widely applied to check genomic variations between prokaryotic genomes and even revealed clear species boundaries (Jain et al. 2018). However, in fungi this method has only been used for very small taxon selections. In the case of Rhizoctonia solani, four isolates were studied which only showed approximately 80% sequence similarity (Wibberg et al. 2015). In a more recent study, ANI was used to compare four related dermatophytic fungal species (Arthrodermataceae). The generated numbers (76.4–90.0%) are comparable to those obtained in our study (Alshahni et al. 2018). Due to the lack of representative genome sequences for many fungal genera, the overall value of ANI analyses in fungal taxonomy, in particular for species and family delimitation, cannot be assessed by now. However, our study provides the first evidence that this method has an important application potential for mycologists in the future.

Pairwise Average Nucleotide Identity (ANI) analysis between genome-sequenced fungi in this study. Species located within dashed line belong to the Hypoxylaceae. AT – Annulohypoxylon truncatum, DC – Daldinia concentrica, Dsp – Daldinia sp., HF – Hypoxylon fragiforme, HL – Hypoxylon lienhwacheense, HM – Hypomontagnella monticulosa, HP – Hypoxylon pulicicidum, HSM – Hypomontagnella submonticulosa, HSP – Hypomontagnella spongiphila, HR – Hypoxylon rickii, HRUB – Hypoxylon rubiginosum, JM – Jackrogersella multiformis, PF – Pestalotiopsis fici, PH – Pyrenopolyporus hunteri, XH – Xylaria hypoxylon
POCP vs ANI
When comparing the ANI and POCP between species pairs in the Hypoxylaceae, it can be seen that the latter values are overall higher. This is an indicator that genomic variation within a family is more strongly influenced by the changes in nucleotide content of conserved genes (such as single nucleotide polymorphisms—SNP) than by changes in gene content (including gene loss, gene gain, gene duplication). Interestingly, this pattern changes completely when comparing family members of the Hypoxylaceae with non-family members, where the POCP numbers are much lower than the respective ANI numbers. In the case of the species pair X. hypoxylon/P. fici, the results become even more striking with an ANI of 70.8% compared to a POCP of 58.9%. Consequently, it can be assumed that, in these particular cases, gene gain and loss is much more impactful towards speciation (and in consequence the development of phenotypic characters). Whether this observation represents a general pattern in fungi cannot be deduced from our preliminary data as much more representative species from the different families of the Xylariales and also different orders need to be included. However, it points towards a weak spot in phylogenetic analysis for inferring taxonomic relationships as these calculations only reflect nucleotide differences in sequences of conserved genes. Future studies likely need to take variations on genome scale, such as genome size, gene content and genome rearrangement, into consideration to understand the evolution of fungi.
AAI analysis
The average amino acid identity (AAI, Fig. 5) measures the differences between orthologous proteins of different organisms and therefore well reflects their evolutionary distances. Within our selected Hypoxylaceae, the AAI ranged from 76.1 to 98.5% and X. hypoxylon showed values between 68.3 and 70.7% when compared to other species. For P. fici the numbers dropped slightly and were in the range of 65.3 to 67.8%. It appears that for this restricted taxon selection there is a clear difference between intergeneric and interfamilial values with an estimated threshold value of 75%. The AAI between Hypom. monticulosa and Hypom. spongiphila was by far the highest with 98.5% showing that the conserved proteins are nearly identical for species that have recently diverged. Whether this value overlaps with intraspecific AAI levels cannot be answered yet. This topic needs to be addressed in the future when more genome sequences of the same species become available. As expected, for all pairs the AAIs were higher than the ANIs as changes in the nucleotide sequence do not necessarily result in amino acid changes due to the genetic code (Castro-Chavez 2010). However, with increased taxonomic distance the AAI values approximated those of the ANI. This result can be explained by a stochastic effect, where the likelihood of silent mutations (i.e. those that do not change the encoded amino acid) decreases with increasing numbers of mutations. Our preliminary data suggest a potential application for AAI as means of taxonomic discrimination on family level. However, in fungi, in contrast to the POCP, AAI is more prone to errors in gene prediction and should only be considered for genomes with high quality intron mapping in an optimal way verified by RNA sequencing.

Pairwise Average Amino Acid Identity (AAI) analysis between genome-sequenced fungi in this study. Species located within dashed line belong to the Hypoxylaceae. AT – Annulohypoxylon truncatum, DC – Daldinia concentrica, Dsp – Daldinia sp., HF – Hypoxylon fragiforme, HL – Hypoxylon lienhwacheense, HM – Hypomontagnella monticulosa, HP – Hypoxylon pulicicidum, HSM – Hypomontagnella submonticulosa, HSP – Hypomontagnella spongiphila, HR – Hypoxylon rickii, HRUB – Hypoxylon rubiginosum, JM – Jackrogersella multiformis, PF – Pestalotiopsis fici, PH – Pyrenopolyporus hunteri, XH – Xylaria hypoxylon
Gene-based genome comparison
As the final step in our genomic comparison, we analyzed the number of shared (core) and individual genes between most closely related species of the Hypoxylaceae (corresponding to the phylogenomic clades) as depicted in Venn diagrams (Fig. 6). Similar to the POCP, the numbers strongly depend on the accuracy of gene prediction and hence are not exact measurements. Nevertheless, they give an insight into the general distribution of orthologous genes between related and unrelated species. It is also important to state that the identified core genes in this analysis are only partially identical with the identified conserved proteins of the POCP analysis (see Methods section for more details), therefore the values can significantly vary.

Venn diagrams displaying number of shared homologous genes between related species of the Hypoxylaceae. A: clade 1 (HF – H. fragiforme, HR – H. rickii, HRUB – H. rubiginosum), B: clade 2 (AT – A. truncatum, HP – H. pulicicidum, JM – J. multiformis), C: clade 3 (HM – Hypom. monticulosa, HSM – Hypom. submonticulosa, HSP – Hypom. spongiphila), D: clade 4 (DC – D. concentrica, EL – E. liquescens, HL – H. lienhwacheense, PH – P. hunteri)
Within clade 1, where H. rubiginosum was included for this analysis, the species had 7911 protein encoding genes in common. Despite the phylogenetic position of H. rubiginosum the individual pairs within the diagram contained approx. the same amount of shared genes ranging from 8405 (between H. fragiforme and H. rickii) to 8569 (between H. rickii and H. rubiginosum). When looking at the individual genes, H. fragiforme showed the least amount (1632), followed by H. rickii (2038) and H. rubiginosum (4187). It appears that the discrepancy between the unique genes of H. rubiginosum and the other species is not a consequence of a reduced amount of shared genes, but instead caused by heavy gene gain (or less likely gene loss in all other species). This is also reflected by the genome size of H. rubiginosum which is by far the largest (48 Mbp) among the sequenced Hypoxylaceae.
Species within clade 2 share 8656 core genes and contain between 1619 (J. multiformis) to 2386 (H. pulicicidum) singletons. The amount of common genes between the individual pairs in the clade varied only slightly in the range of 9107 to 9243 genes.
The three genome sequenced members of the Hypom. monticulosa species complex (clade 3) contain a core set of 10,253 genes. The gene overlap of Hypom. spongiphila with Hypom. submonticulosa (10,330) is smaller than with Hypom. monticulosa (11,916). The latter exhibits also the lowest number of unique genes (510) within the three species, while Hypom. spongiphila and Hypom. submonticulosa contain higher numbers (640 and 1271, respectively). This result clearly indicates that despite their close taxonomic (and evolutionary) relationship, these taxa have already diverged regarding the gene content.
The last gene-based comparison involves all four species of clade 4. Within this data subset, 6631 genes were shared among all taxa. A pairwise comparison reveals the presence of 7623 to 8563 common genes between H. lienhwacheense and other clade members. Similar numbers of conserved genes can be found for P. hunteri in relation to D. concentrica and E. liquescens (8008 and 7116, respectively). The latter two share the highest amount of genes (8791). Despite the relative small genome size of H. lienhwacheense (35.8 Mbp) and its low number of total genes (9924), this taxon still contains 1097 genes (11.1% of total gene content), which lack in the other three related organisms. In comparison, 1257, 1373 and 1490 singletons were identified for D. concentrica, E. liquescens and P. hunteri, respectively.
Based on these results, two main conclusions can be drawn. First of all, there is no obvious correlation between the number of shared genes and the genetic distance of two species indicating that presumably very closely related species can have less genes in common than distantly related species pairs. This is well reflected in Fig. 6a where H. rickii shares almost 200 fewer genes with its close relative H. fragiforme than with the more distantly related H. rubiginosum. Secondly, the relatively high number of singletons (genes exclusively present in a single genome within a given data matrix), in particular between genetically close species (e.g. Hypom. monticulosa vs. Hypom. spongiphila, H. fragiforme vs. H. rickii, D. concentrica vs. E. liquescens), suggests a high rate of gene turnover. Due to the small set of genome-sequenced species, it cannot be evaluated which are the major driving forces of this result, but we assume that gene loss and gene acquisition contribute equally to this outcome. In the latter event, lateral gene transfer has been shown to substantially contribute to this phenomenon especially in cases where fungal species (such as Trichoderma species or plant-pathogens in the Magnaporthales) can colonize a broad range of habitats and are in frequent contact with other fungi (Qiu et al. 2016; Druzhinina et al. 2018). As endophytes and saprotrophs the Hypoxylaceae are also in a constant competitive environment with other fungi offering an ideal opportunity to acquire new genes.
Authenticity of E. liquescens ATCC 46302
As a representative of a rather unusual genus inside the Hypoxylaceae, we decided to include Entonaema liquescens in our genome sequencing project. Members of Entonaema feature brightly colored stromata with usually orange KOH-extractable pigments and a liquid-filled cavity as a unique character and can therefore be distinguished from other hypoxyloid genera (Stadler et al. 2008). The type species of the genus is E. liquescens with a single existing culture which has been deposited at the ATCC (46302) more than 30 years ago. Different single loci of the strain were sequenced and included in various phylogenetic reconstructions of the family. In all of these studies, the fungus nested inside the genus Daldinia. As the massive stromata of Entonaema are reminiscent of those found in Daldinia and some species of the latter are known to possess gelatinous interiors (e.g. the Daldinia vernicosa group, and in particular D. gelatinoides; Stadler et al. 2014a, b), the taxonomic position of Entonaema appeared plausible (Triebel et al. 2005; Wendt et al. 2018). From a chemo-taxonomical point of view, E. liquescens and a few other members of the genus (e.g. E. cinnabarinum, E. globosum) show striking differences to the phylogenetically related Daldinia species due to the presence of mitorubrin-type azaphilones (Stadler et al. 2004). Within the Hypoxylaceae, this type of pigments can otherwise only be found in members of the genus Hypoxylon, thus offering a possibility to study conservation events and evolutionary aspects of the azaphilone biosynthesis on family level. In the context of a related study, we identified various sets of genes (i.e. biosynthetic gene clusters) putatively responsible for the assembly of mitorubrin-type compounds in H. fragiforme (Becker et al. in preparation) and found a high degree of gene conservation in other azaphilone-producing Hypoxylaceae (i.e. J. multiformis, H. pulicicidum, H. rickii, H. rubiginosum). In contrast, most of the species that are devoid of those pigments did not contain the respective genes with the exception of Hypom. monticulosa and Hypom. spongiphila (data not shown, but will be part of a subsequent study). When screening the genome of E. liquescens for the presence of homologous gene clusters, no hits could be found. As the genome sequence of this fungus provides one of the best resolutions (N50 of 3.5 Mbp, 31 contigs) within this study, it appears unlikely that the cluster has been missed due to sequencing gaps. This still leaves the possibility of convergent evolution of azaphilone biosynthetic genes in E. liquescens. However, as the genetic basis for azaphilone assembly seems to be conserved across various ascomycetes (see Monascus spp. and Aspergillus niger; Zabala et al. 2012; Chen et al. 2017), this is not a likely scenario. Consequently, the genomic investigation raised serious concerns about the authenticity of the E. liquescens strain ATCC 46302. To exclude the possibility that the strain has been contaminated on our side, the extracted ITS and TUB2 sequences of the genome were compared to those previously reported (AY616686, KX271248; Triebel et al. 2005; Wendt et al. 2018), which were in agreement with each other. In addition, the sequences do not fit with any known ITS-sequenced Daldinia species (Stadler et al. 2014b) (closest BLAST hits with 97.0% similarity is D. korfii, Sir et al. 2016) rendering identification to species rank impossible in case of a contaminant. In order to confirm whether the isolate it a true member of Entonaema or a contaminating Daldinia species, fresh specimens of the genus and in particular E. liquescens need to be recollected and multiple isolates have to be obtained for comparison. As this has not been achieved yet due to the rare occurrence of E. liquescens, ATCC 46302 is retained in the analysis herein, especially since it still constitutes a member of the Hypoxylaceae.
Taxonomy
Based on the genomic and ecological differences between Hypomontagnella monticulosa MUCL 54604 and tentatively identified Hypom. monticulosa CLL-205, a new species is proposed and described in the following for the latter fungus.
Hypomontagnella spongiphila Kuhnert, sp. nov.

Culture morphology of various members of the Hypomontagnella monticulosa species complex after 16 days of growth on different media (YMG, PDA, OA). HM – Hypom. monticulosa MUCL 54604, HS – Hypom. spongiphila MUCL 57903, HSM – Hypom. submonticulosa DAOMC 242471

Whole genome macro-synteny plot between Hypom. monticulosa and Hypom. spongiphila. Dotted lines represent contig borders
Holotype: French Polynesia, Tahiti, coastal area, cave of Ti-Pari, 20 m depth, 9°45.421′S, 139°08.275′W, isolated from a Sphaerocladina sponge, 17 Dec. 2015 (host material), leg. C. Debitus, UP-CLL-205 (ex-type culture MUCL 57903, GenBank Acc. No.: ITS—KY744359/ MK131719, LSU—MK131717, RPB2—MK135890, β-tubulin—MK135892).
Etymology: Refers to the sponge host from which it was isolated.
Known distribution/host preference: Only known from the holotype.
It differs from Hypomontagnella monticulosa by its marine habitat, sterile cultures and slow growth on PDA as well as radially furrowed cultures on OA.
Sexual and asexual morph: not observed.
Culture: After 16 d at room temperature. Colonies on YMG white, sterile, velvety to felty and flat; zonate only in the center, with entire margins, reaching > 60 mm diam., reverse uncolored; on PDA white, sterile, azonate, velvety to slightly cottony, with slightly undulate margins, reaching > 40 mm diam., reverse uncolored; on OA reaching > 50 mm diam., white, sterile, velvety with slightly cottony at the center, radially furrowed surface and filiform margins formed by aerial mycelium tufts, reverse uncolored. Mycelial cords are sporadically formed on all media. No sporulation observed.
Secondary metabolites: Cultures produce sporothriolide and derivatives, sporochartines A-E, trienylfuranol A in PDB.
Notes: Hypomontagnella spongiphila appears identical to Hypom. monticulosa based on phylogenetic and chemotaxonomic data as sequences only slightly differ and the production of metabolites is very similar (Lambert et al. 2019). Morphological characters for comparison are rare as the fungus remains sterile under different culture conditions and stromata formation cannot be induced artificially. It also appears unlikely that sexual structures are formed in a marine environment. Therefore, Hypom. spongiphila was grown on different media (YMG, PDA, OA) in comparison with its close relatives Hypom. monticulosa and Hypom. submonticulosa (Fig. 7). In general, growth speed of the strain on PDA was much slower than those observed for the other fungi. Both relatives formed sporulating regions after 14 days, while Hypom. spongiphila remained sterile even after 5 weeks of incubation. On YMG media morphology only slightly differed between Hypom. spongiphila and Hypom. monticulosa, whereas Hypom. submonticulosa differed substantially. The appearance of the strains on OA was strikingly different as Hypom. monticulosa only produced visual mycelia in the center, which started to grow subsurficially towards the edge of the plate. In contrast, Hypom. spongiphila formed radially furrowed surfaces with filiform margins, which appeared related to the morphology of Hypom. submonticulosa. The genomic comparison revealed obvious differences between the different Hypomontagnella isolates (see Fig. 6c) in terms of gene content. More than 600 genes were predicted to be restricted to Hypom. spongiphila in the Hypomontagnella subset, whereas the direct comparison of H. spongiphila and H. monticulosa shows more than 700 singletons corresponding to roughly 5.5% of the total gene content (note that this value slightly differs from the respective POCP dataset as both analyses are based on different models for homology search). The function of the majority of these singletons could not be identified as the respective protein sequences showed no known homologs in the BLAST databases. These genes might be involved in the adaptation to marine environments, in particular in osmotolerance and nutrient uptake. To study whether the genetic differences between Hypom. monticulosa and Hypom. spongiphila can be also seen in the organization of the chromosomes, a macrosynteny analysis between both genomes was carried out. In Fig. 8 a high genomic synteny can be observed between both organisms with occasional inversions of larger chromosomal areas. This suggests that the separation of the two species is a rather recent event, which is also supported by the 93.3% average nucleotide identity (Fig. 4) and 98.5% average amino acid identity.
The erection of a new fungal species evidenced by genomic information is a novel approach in fungal taxonomy. Such approaches are only feasible if genome data of related organism are available or are generated in the context of respective studies. We are aware that such sophisticated techniques are currently still not feasible for the majority of researchers. That is why for the time being other diagnostic features need to be included in the species description, which can be accessed by taxonomists. We see the ecological niche of Hypom. spongiphila and the morphological differences of the cultures as sufficient to fulfill such a criterion.
Conclusion and Outlook
Herein, we introduced the genomes of thirteen Hypoxylaceae species representing the major phylogenetic lineages of the family and the outgroup genome of Xylaria hypoxylon. The usage of third generation sequencing methods enabled the assembly of high-quality draft genome sequences with an average N50 of 3.0 Mbp, which served as the basis for phylogenomic reconstructions and thorough genomic comparisons. We created the first phylogenomic trees for the Hypoxylaceae, which for the first time in fungal taxonomy are based on a set of 4912 protein sequences per organism in place of nucleotide sequences. The tree topology is identical to previous multigene-based calculations, but with superior node support values rendering it the most stable phylogenetic reconstruction for this family. The application of POCP, ANI and AAI analyses on a larger set of related fungal species to investigate their relationships and to deduce taxonomic hierarchies is a novel approach in mycology. Species within the Hypoxylacae have around 70% of their protein content conserved and share an overall genome-wide nucleotide identity of at least 70%. The similarity of the conserved protein sequences shows slightly higher values with clear differences beyond family level. However, this analysis strongly depends on the accuracy of the gene prediction. These thresholds have application potential to define family level associations but need to be re-evaluated on a larger taxon selection and also different fungal families. The comparison of genome-sequences also enabled the differentiation of fungi on the species level, especially in cases where the morphological characters are scarce (e.g. in sterile environmental isolates) and the identity of marker (barcode) sequences is high. This led to the erection of Hypomontagnella spongiphila, a marine endosymbiotic isolate without sexual or asexual morph. Despite the high genomic synteny of the new species with Hypom. monticulosa, it possesses around 700 unique genes in comparison. This change of genome content is likely caused by selection and evolutionary pressure under different environmental conditions. The analysis of differences in gene content is in our opinion a comprehensible approach to set species apart as it reflects events of adaptation and thus evolution much better than changes in nucleotide sequences in genomic loci. Therefore, we suggest a threshold of at least 5% differences in total gene content to unambiguously recognize new fungal species when comparing closely related organisms.
The application of third generation sequencing methods to create high quality genome sequences for taxonomic purposes in mycology is a consequent step to keep taxonomy on the same level as other research areas. It might appear unrealistic that such technology will be available for a broad range of mycologists across most countries around the world and thus limiting the value of such approaches. However, the current development of sequencing technologies points towards the mainstream application of genome sequencing with the already available sequencers in the size of USB flash drives (see MinION sequencing from Oxford Nanopore Technologies). New generations of sequencers will allow fast sequencing of complete fungal genomes with low error rates in almost every lab environment and prices for sequencing kits (the main factor for increased sequencing costs) will strongly decrease driven by the increased demand. Prices of around $100 per genome sequence are close to being realistic (https://www.labiotech.eu/features/genome-sequencing-review-projects/), making genomic approaches affordable also for taxonomists.
The genome sequences generated in this work will enable a broad range of investigations including studies on fungal evolution, population dynamics, host-fungus interactions, biodegradation and also biosynthesis of secondary metabolites. We would like to point out, that in particular the latter is among our main interests. We are currently working on an in-depth evaluation of the biosynthetic capabilities of the Hypoxylaceae to get insights into the underlying enzyme machineries and the evolution of the respective pathways within the family. More than 750 biosynthesis related gene clusters (BGC) were found across the genome-sequenced species averaging 54 clusters per species. This value is comparable to other well-known secondary metabolite producers such as Fusarium, Aspergillus and Penicillium (Nielsen et al. 2017; Hoogendoorn et al. 2018; Theobald et al. 2018). The identified BGCs include various types of pathways, among others responsible for the formation of polyketides, terpenes, peptides, meroterpenoids and alkaloids. The effort of this investigation already resulted in the discovery and characterization of the cytochalasin and azaphilone gene cluster in H. fragiforme (Wang et al. 2019; Becker et al. in preparation). More publications are in preparation and will expand the knowledge about secondary metabolism in fungi.
References
Adamek M, Alanjary M, Sales-Ortells H et al (2018) Comparative genomics reveals phylogenetic distribution patterns of secondary metabolites in Amycolatopsis species. BMC Genomics 19:426. https://doi.org/10.1186/s12864-018-4809-4
Alshahni MM, Yamada T, Yo A et al (2018) Insight into the draft whole-genome sequence of the dermatophyte Arthroderma vanbreuseghemii. Sci Rep 8:15127. https://doi.org/10.1038/s41598-018-33505-9
Altschul S (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. https://doi.org/10.1093/nar/25.17.3389
Arahal DR (2014) Whole-genome analyses: average nucleotide identity. Method Microbiol 41:103–122. https://doi.org/10.1016/bs.mim.2014.07.002
Bills GF, González-Menéndez V, Martín J et al (2012) Hypoxylon pulicicidum sp. nov. (Ascomycota, Xylariales), a pantropical insecticide-producing endophyte. PLoS ONE 7:e46687. https://doi.org/10.1371/journal.pone.0046687
Blackwell M (2011) The fungi: 1, 2, 3 … 5.1 million species? Am J Bot 98:426–438. https://doi.org/10.3732/ajb.1000298
Blom J, Albaum SP, Doppmeier D et al (2009) EDGAR: A software framework for the comparative analysis of prokaryotic genomes. BMC Bioinform 10:154. https://doi.org/10.1186/1471-2105-10-154
Blom J, Kreis J, Spänig S et al (2016) EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res 44:W22–W28. https://doi.org/10.1093/nar/gkw255
Boeckmann B, Bairoch A, Apweiler R et al (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31:365–370. https://doi.org/10.1093/nar/gkg095
Burgess KMN, Ibrahim A, Sørensen D, Sumarah MW (2017) Trienylfuranol A and trienylfuranone A-B: metabolites isolated from an endophytic fungus, Hypoxylon submoniticulosum, in the raspberry Rubus idaeus. J Antibiot (Tokyo) 70:721–725. https://doi.org/10.1038/ja.2017.18
Büttner E, Liers C, Hofrichter M et al (2019) Draft genome sequence of Xylaria hypoxylon DSM 108379, a ubiquitous fungus on hardwood. Microbiol Resour Announc 8:9–11. https://doi.org/10.1128/MRA.00845-19
Cabanettes F, Klopp C (2018) D-GENIES: dot plot large genomes in an interactive, efficient and simple way. PeerJ 6:e4958. https://doi.org/10.7717/peerj.4958
Castro-Chavez F (2010) The rules of variation: Amino acid exchange according to the rotating circular genetic code. J Theor Biol 264:711–721. https://doi.org/10.1016/j.jtbi.2010.03.046
Chapela IH, Petrini O, Bielser G (1993) The physiology of ascospore eclosion in Hypoxylon fragiforme: mechanisms in the early recognition and establishment of an endophytic symbiosis. Mycol Res 97:157–162. https://doi.org/10.1016/S0953-7562(09)80237-2
Chen W, Chen R, Liu Q et al (2017) Orange, red, yellow: biosynthesis of azaphilone pigments in Monascus fungi. Chem Sci 8:4917–4925. https://doi.org/10.1039/C7SC00475C
Chernomor O, von Haeseler A, Minh BQ (2016) Terrace aware data structure for phylogenomic inference from supermatrices. Syst Biol 65:997–1008. https://doi.org/10.1093/sysbio/syw037
Chowdhury B, Garai G (2017) A review on multiple sequence alignment from the perspective of genetic algorithm. Genomics 109:419–431. https://doi.org/10.1016/j.ygeno.2017.06.007
Daranagama DA, Hyde KD, Sir EB et al (2018) Towards a natural classification and backbone tree for Graphostromataceae, Hypoxylaceae, Lopadostomataceae and Xylariaceae. Fungal Divers 88:1–165. https://doi.org/10.1007/s13225-017-0388-y
Dornburg A, Townsend JP, Wang Z (2017) Maximizing power in phylogenetics and phylogenomics: a perspective illuminated by fungal big data. Adv Genet 100:1–47. https://doi.org/10.1016/bs.adgen.2017.09.007
Druzhinina IS, Chenthamara K, Zhang J et al (2018) Massive lateral transfer of genes encoding plant cell wall-degrading enzymes to the mycoparasitic fungus Trichoderma from its plant-associated hosts. PLOS Genet 14:e1007322. https://doi.org/10.1371/journal.pgen.1007322
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Grigoriev IV, Nikitin R, Haridas S et al (2014) MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res 42:D699–D704. https://doi.org/10.1093/nar/gkt1183
Guindon S, Dufayard J-F, Lefort V et al (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. https://doi.org/10.1093/sysbio/syq010
Helaly SE, Thongbai B, Stadler M (2018) Diversity of biologically active secondary metabolites from endophytic and saprotrophic fungi of the ascomycete order Xylariales. Nat Prod Rep 35:992–1014. https://doi.org/10.1039/C8NP00010G
Hoang DT, Chernomor O, von Haeseler A et al (2018) UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol 35:518–522. https://doi.org/10.1093/molbev/msx281
Hoff KJ, Lomsadze A, Borodovsky M, Stanke M (2019) Whole-genome annotation with BRAKER. Methods Mol Biol 1962:65–95. https://doi.org/10.1007/978-1-4939-9173-0_5
Hongsanan S, Maharachchikumbura SSN, Hyde KD et al (2017) An updated phylogeny of Sordariomycetes based on phylogenetic and molecular clock evidence. Fungal Divers 84:25–41. https://doi.org/10.1007/s13225-017-0384-2
Hoogendoorn K, Barra L, Waalwijk C et al (2018) Evolution and diversity of biosynthetic gene clusters in Fusarium. Front Microbiol 9:1158. https://doi.org/10.3389/fmicb.2018.01158
Hyde KD, Norphanphoun C, Maharachchikumbura SSN et al (2020) Refined families of Sordariomycetes. Mycosphere 11:305–1059. https://doi.org/10.5943/mycosphere/11/1/7
Jain C, Rodriguez-R LM, Phillippy AM et al (2018) High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun 9:5114. https://doi.org/10.1038/s41467-018-07641-9
Jaklitsch WM, Gardiennet A, Voglmayr H (2016) Resolution of morphology-based taxonomic delusions: Acrocordiella, Basiseptospora, Blogiascospora, Clypeosphaeria, Hymenopleella, Lepteutypa, Pseudapiospora, Requienella, Seiridium and Strickeria. Persoonia 37:82–105. https://doi.org/10.3767/003158516X690475
Kalyaanamoorthy S, Minh BQ, Wong TKF et al (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589. https://doi.org/10.1038/nmeth.4285
Kanehisa M, Goto S, Kawashima S et al (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res 32:D277–D280. https://doi.org/10.1093/nar/gkh063
Kim D, Pertea G, Trapnell C et al (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. https://doi.org/10.1186/gb-2013-14-4-r36
Koren S, Walenz BP, Berlin K et al (2017) Canu: scalable and accurate long-read assembly via adaptive k -mer weighting and repeat separation. Genome Res 27:722–736. https://doi.org/10.1101/gr.215087.116
Kuhnert E, Fournier J, Peršoh D et al (2014a) New Hypoxylon species from Martinique and new evidence on the molecular phylogeny of Hypoxylon based on ITS rDNA and β-tubulin data. Fungal Divers 64:181–203. https://doi.org/10.1007/s13225-013-0264-3
Kuhnert E, Heitkämper S, Fournier J et al (2014b) Hypoxyvermelhotins A–C, new pigments from Hypoxylon lechatii sp. nov. Fungal Biol 118:242–252. https://doi.org/10.1016/j.funbio.2013.12.003
Kuhnert E, Surup F, Herrmann J et al (2015a) Rickenyls A–E, antioxidative terphenyls from the fungus Hypoxylon rickii (Xylariaceae, Ascomycota). Phytochemistry 118:68–73. https://doi.org/10.1016/j.phytochem.2015.08.004
Kuhnert E, Surup F, Sir EB et al (2015b) Lenormandins A–G, new azaphilones from Hypoxylon lenormandii and Hypoxylon jaklitschii sp. nov., recognised by chemotaxonomic data. Fungal Divers 71:165–184. https://doi.org/10.1007/s13225-014-0318-1
Kuhnert E, Surup F, Wiebach V et al (2015c) Botryane, noreudesmane and abietane terpenoids from the ascomycete Hypoxylon rickii. Phytochemistry 117:116–122. https://doi.org/10.1016/j.phytochem.2015.06.002
Kuhnert E, Sir EB, Lambert C et al (2017) Phylogenetic and chemotaxonomic resolution of the genus Annulohypoxylon (Xylariaceae) including four new species. Fungal Divers 85:1–43. https://doi.org/10.1007/s13225-016-0377-6
Lambert C, Wendt L, Hladki AI et al (2019) Hypomontagnella (Hypoxylaceae): a new genus segregated from Hypoxylon by a polyphasic taxonomic approach. Mycol Prog 18:187–201. https://doi.org/10.1007/s11557-018-1452-z
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. https://doi.org/10.1038/nmeth.1923
Lefort V, Longueville J-E, Gascuel O (2017) SMS: Smart Model Selection in PhyML. Mol Biol Evol 34:2422–2424. https://doi.org/10.1093/molbev/msx149
Leman-Loubière C, Le Goff G, Debitus C, Ouazzani J (2017) Sporochartines A-E, a new family of natural products from the marine fungus Hypoxylon monticulosum isolated from a Sphaerocladina sponge. Front Mar Sci 4:1–9. https://doi.org/10.3389/fmars.2017.00399
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv Preprint. arXiv:1303.3997 [q-bio.GN]
Liu F, Bonthond G, Groenewald JZ, Cai L, Crous PW (2019) Sporocadaceae, a family of coelomycetous fungi with appendage-bearing conidia. Stud Mycol 92:287–415. https://doi.org/10.1016/j.simyco.2018.11.001
Lombard V, Golaconda Ramulu H, Drula E et al (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495. https://doi.org/10.1093/nar/gkt1178
Lowe TM, Eddy SR (1997) tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:0955–0964. https://doi.org/10.1093/nar/25.5.0955
Lu L, Rong W, Massart S, Zhang Z (2018) Genome-wide identification and expression analysis of cutinase gene family in Rhizoctonia cerealis and functional study of an active cutinase RcCUT1 in the fungal–wheat interaction. Front Microbiol 9:1813. https://doi.org/10.3389/fmicb.2018.01813
Margos G, Gofton A, Wibberg D et al (2018) The genus Borrelia reloaded. PLoS ONE 13:e0208432. https://doi.org/10.1371/journal.pone.0208432
Medina R, Silva A, Andersen R et al (2016) Botryane sesquiterpenes and binaphthalene tetrols from endophytic fungi associated to the marine red algae Asparagopsis taxiformis. Planta Med 81:S1–S381. https://doi.org/10.1055/s-0036-1596629
Meyer F (2003) GenDB—an open source genome annotation system for prokaryote genomes. Nucleic Acids Res 31:2187–2195. https://doi.org/10.1093/nar/gkg312
Mistry J, Finn RD, Eddy SR et al (2013) Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res 41:e121–e121. https://doi.org/10.1093/nar/gkt263
Nagy LG, Szöllősi G (2017) Fungal phylogeny in the age of genomics: Insights into phylogenetic inference from genome-scale datasets. Adv Genet 100:49–72. https://doi.org/10.1016/bs.adgen.2017.09.008
Ng KP, Ngeow YF, Yew SM et al (2012) Draft genome aequence of Daldinia eschscholzii isolated from blood culture. Eukaryot Cell 11:703–704. https://doi.org/10.1128/EC.00074-12
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274. https://doi.org/10.1093/molbev/msu300
Nicholson MJ, Van de Bittner KC, Ram A et al (2018) Draft genome sequence of the filamentous fungus Hypoxylon pulicicidum ATCC 74245. Genome Announc 6:1–2. https://doi.org/10.1128/genomeA.01380-17
Nielsen JC, Grijseels S, Prigent S et al (2017) Global analysis of biosynthetic gene clusters reveals vast potential of secondary metabolite production in Penicillium species. Nat Microbiol 2:17044. https://doi.org/10.1038/nmicrobiol.2017.44
Pažoutová S, Follert S, Bitzer J et al (2013) A new endophytic insect-associated Daldinia species, recognised from a comparison of secondary metabolite profiles and molecular phylogeny. Fungal Divers 60:107–123. https://doi.org/10.1007/s13225-013-0238-5
Peršoh D, Melcher M, Flessa F, Rambold G (2010) First fungal community analyses of endophytic ascomycetes associated with Viscum album ssp. austriacum and its host Pinus sylvestris. Fungal Biol 114:585–596. https://doi.org/10.1016/j.funbio.2010.04.009
Pizarro D, Divakar PK, Grewe F et al (2018) Phylogenomic analysis of 2556 single-copy protein-coding genes resolves most evolutionary relationships for the major clades in the most diverse group of lichen-forming fungi. Fungal Divers 92:31–41. https://doi.org/10.1007/s13225-018-0407-7
Qin X-D, Dong Z-J, Liu J-K et al (2006) Concentricolide, an anti-HIV agent from the ascomycete Daldinia concentrica. Helv Chim Acta 89:127–133. https://doi.org/10.1002/hlca.200690004
Qin Q-L, Xie B-B, Zhang X-Y et al (2014) A proposed genus boundary for the prokaryotes based on genomic insights. J Bacteriol 196:2210–2215. https://doi.org/10.1128/JB.01688-14
Qiu H, Cai G, Luo J, Bhattacharya D, Zhang N (2016) Extensive horizontal gene transfers between plant pathogenic fungi. BMC Biol 14:41. https://doi.org/10.1186/s12915-016-0264-3
Rupp O, Becker J, Brinkrolf K et al (2014) Construction of a public CHO cell line transcript database using versatile bioinformatics analysis pipelines. PLoS ONE 9:e85568. https://doi.org/10.1371/journal.pone.0085568
Simão FA, Waterhouse RM, Ioannidis P et al (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. https://doi.org/10.1093/bioinformatics/btv351
Sir E, Lambert C, Wendt L et al (2016) A new species of Daldinia (Xylariaceae) from the Argentine subtropical montane forest. Mycosphere 7:1378–1388. https://doi.org/10.5943/mycosphere/7/9/11
Sir EB, Becker K, Lambert C et al (2019) Observations on Texas hypoxylons, including two new Hypoxylon species and widespread environmental isolates of the H. croceum complex identified by a polyphasic approach. Mycologia 111:832–856. https://doi.org/10.1080/00275514.2019.1637705
Stadler M (2011) Importance of secondary metabolites in the Xylariaceae as parameters for assessment of their taxonomy, phylogeny, and functional biodiversity. Curr Res Environ Appl Mycol 1:75–133. https://doi.org/10.5943/cream/1/2/1
Stadler M, Ju Y-M, Rogers JD (2004) Chemotaxonomy of Entonaema, Rhopalostroma and other Xylariaceae. Mycol Res 108:239–256. https://doi.org/10.1017/S0953756204009347
Stadler M, Fournier J, Læssøe T et al (2008) Recognition of hypoxyloid and xylarioid Entonaema species and allied Xylaria species from a comparison of holomorphic morphology, HPLC profiles, and ribosomal DNA sequences. Mycol Prog 7:53–73. https://doi.org/10.1007/s11557-008-0553-5
Stadler M, Hawksworth DL, Fournier J (2014a) The application of the name Xylaria hypoxylon, based on Clavaria hypoxylon of Linnaeus. IMA Fungus 5:57–66. https://doi.org/10.5598/imafungus.2014.05.01.07
Stadler M, Laessøe T, Fournier J et al (2014b) A polyphasic taxonomy of Daldinia. Stud Mycol 77:1–143. https://doi.org/10.3114/sim0016
Stadler M, Lambert C, Wibberg D, Kalinowski J, Cox RJ, Kolarik M, Kuhnert E (2020) Intragenomic polymorphisms in the ITS region of high quality genomes of the Hypoxylaceae (Xylariales, Ascomycota). Mycol Prog 19:235–245
Stanke M, Diekhans M, Baertsch R, Haussler D (2008) Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24:637–644. https://doi.org/10.1093/bioinformatics/btn013
Steenwyk JL, Shen X-X, Lind AL et al (2019) A robust phylogenomic time tree for biotechnologically and medically important fungi in the genera Aspergillus and Penicillium. MBio 10:1–25. https://doi.org/10.1128/mBio.00925-19
Sudarman E, Kuhnert E, Hyde KD et al (2016) Truncatones A-D, benzo[j]fluoranthenes from Annulohypoxylon species (Xylariaceae, Ascomycota). Tetrahedron 72:6450–6454. https://doi.org/10.1016/j.tet.2016.08.054
Surup F, Kuhnert E, Lehmann E et al (2014) Sporothriolide derivatives as chemotaxonomic markers for Hypoxylon monticulosum. Mycology 5:110–119. https://doi.org/10.1080/21501203.2014.929600
Surup F, Kuhnert E, Böhm A et al (2018a) The rickiols: 20-, 22-, and 24-membered macrolides from the ascomycete Hypoxylon rickii. Chemistry Eur J 24:2200–2213. https://doi.org/10.1002/chem.201704928
Surup F, Narmani A, Wendt L et al (2018b) Identification of fungal fossils and novel azaphilone pigments in ancient carbonised specimens of Hypoxylon fragiforme from forest soils of Châtillon-sur-Seine (Burgundy). Fungal Divers 92:345–356. https://doi.org/10.1007/s13225-018-0412-x
Talavera G, Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56:564–577. https://doi.org/10.1080/10635150701472164
Tatusov RL, Fedorova ND, Jackson JD et al (2003) The COG database: an updated version includes eukaryotes. BMC Bioinform 4:41. https://doi.org/10.1186/1471-2105-4-41
Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M (2008) Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res 18:1979–1990. https://doi.org/10.1101/gr.081612.108
Theobald S, Vesth TC, Rendsvig JK et al (2018) Uncovering secondary metabolite evolution and biosynthesis using gene cluster networks and genetic dereplication. Sci Rep 8:17957. https://doi.org/10.1038/s41598-018-36561-3
Triebel D, Peršoh D, Wollweber H, Stadler M (2005) Phylogenetic relationships among Daldinia, Entonaema, and Hypoxylon as inferred from ITS nrDNA analyses of Xylariales. Nov Hedwigia 80:25–43. https://doi.org/10.1127/0029-5035/2005/0080-0025
U’Ren JM, Miadlikowska J, Zimmerman NB et al (2016) Contributions of North American endophytes to the phylogeny, ecology, and taxonomy of Xylariaceae (Sordariomycetes, Ascomycota). Mol Phylogenet Evol 98:210–232. https://doi.org/10.1016/j.ympev.2016.02.010
Van de Bittner KC, Nicholson MJ, Bustamante LY et al (2018) Heterologous biosynthesis of nodulisporic acid F. J Am Chem Soc 140:582–585. https://doi.org/10.1021/jacs.7b10909
Walker BJ, Abeel T, Shea T et al (2014) Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9:e112963. https://doi.org/10.1371/journal.pone.0112963
Wang C, Becker K, Pfütze S et al (2019) Investigating the function of cryptic cytochalasan cytochrome P450 monooxygenases using combinatorial biosynthesis. Org Lett. https://doi.org/10.1021/acs.orglett.9b03372
Wang X, Zhang X, Liu L et al (2015) Genomic and transcriptomic analysis of the endophytic fungus Pestalotiopsis fici reveals its lifestyle and high potential for synthesis of natural products. BMC Genomics 16:28. https://doi.org/10.1186/s12864-014-1190-9
Wendt L, Sir EB, Kuhnert E et al (2018) Resurrection and emendation of the Hypoxylaceae, recognised from a multigene phylogeny of the Xylariales. Mycol Prog 17:115–154. https://doi.org/10.1007/s11557-017-1311-3
Wibberg D, Andersson L, Tzelepis G et al (2016) Genome analysis of the sugar beet pathogen Rhizoctonia solani AG2-2IIIB revealed high numbers in secreted proteins and cell wall degrading enzymes. BMC Genomics 17:245. https://doi.org/10.1186/s12864-016-2561-1
Wibberg D, Rupp O, Blom J et al (2015) Development of a Rhizoctonia solani AG1-IB specific gene model enables comparative genome analyses between phytopathogenic R. solani AG1-IA, AG1-IB, AG3 and AG8 isolates. PLoS ONE 10:e0144769. https://doi.org/10.1371/journal.pone.0144769
Wijayawardene NN, Hyde KD, Lumbsch HT et al (2018) Outline of Ascomycota: 2017. Fungal Divers 88:167–263. https://doi.org/10.1007/s13225-018-0394-8
Wingfield BD, Bills GF, Dong Y et al (2018) Draft genome sequence of Annulohypoxylon stygium, Aspergillus mulundensis, Berkeleyomyces basicola (syn. Thielaviopsis basicola), Ceratocystis smalleyi, two Cercospora beticola strains, Coleophoma cylindrospora, Fusarium fracticaudum, Phialophora cf. hyal. IMA Fungus 9:199–223. https://doi.org/10.5598/imafungus.2018.09.01.13
Wu W, Davis RW, Tran-Gyamfi MB et al (2017) Characterization of four endophytic fungi as potential consolidated bioprocessing hosts for conversion of lignocellulose into advanced biofuels. Appl Microbiol Biotechnol 101:2603–2618. https://doi.org/10.1007/s00253-017-8091-1
Yin Y, Mao X, Yang J et al (2012) dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 40:W445–W451. https://doi.org/10.1093/nar/gks479
Yuyama KT, Wendt L, Surup F, Kretz R, Chepkirui C, Wittstein K, Boonlarppradab C, Wongkanoun S, Luangsa-ard JJ, Stadler M, Abraham WR (2018) Cytochalasans act as inhibitors of biofilm formation of Staphylococcus aureus. Biomolecules 8:4
Zabala AO, Xu W, Chooi Y-H, Tang Y (2012) Characterization of a silent azaphilone gene cluster from Aspergillus niger ATCC 1015 reveals a hydroxylation-mediated pyran-ring formation. Chem Biol 19:1049–1059. https://doi.org/10.1016/j.chembiol.2012.07.004
Zhang C, Rabiee M, Sayyari E, Mirarab S (2018a) ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19:153. https://doi.org/10.1186/s12859-018-2129-y
Zhang N, Cai G, Price DC et al (2018b) Genome wide analysis of the transition to pathogenic lifestyles in Magnaporthales fungi. Sci Rep 8:5862. https://doi.org/10.1038/s41598-018-24301-6
Zhang N, Luo J, Bhattacharya D (2017) Advances in fungal phylogenomics and their impact on fungal systematics. Adv Genet 100:309–328. https://doi.org/10.1016/bs.adgen.2017.09.004
Zhao Z, Liu H, Wang C, Xu J-R (2013) Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics 14:274. https://doi.org/10.1186/1471-2164-14-274
Acknowledgements
Open Access funding provided by Projekt DEAL. This work was funded by the DFG (Deutsche Forschungsgemeinschaft) priority program “Taxon-Omics: New Approaches for Discovering and Naming Biodiversity” (SPP 1991). The bioinformatics support of the BMBF-funded project ‘Bielefeld-Gießen Center for Microbial Bioinformatics; BiGi (Grant Number 031A533)’ within the German Network for Bioinformatics Infrastructure (de.NBI) is gratefully acknowledged. The authors also would like to thank Dr. Mark Sumarah for providing the Hypom. submonticulosa strain and Dr. Jamal Ouazzani for providing the Hypom. spongiphila strain. In this regard we thank the French and French-Polynesian governments for their support in the sponge survey from French Polynesia (project Biopolyval). We are grateful to Dr. Esteban B. Sir for help in morphological descriptions. Dongsong Tian and Sen Yin are acknowledged for support in DNA and RNA isolation. Prof. Theresia Stradal is thanked for providing computational power for phylogenetic calculations.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article dedicated to Prof. Dr. Kevin David Hyde on the occasion of his 65th birthday.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wibberg, D., Stadler, M., Lambert, C. et al. High quality genome sequences of thirteen Hypoxylaceae (Ascomycota) strengthen the phylogenetic family backbone and enable the discovery of new taxa. Fungal Diversity 106, 7–28 (2021). https://doi.org/10.1007/s13225-020-00447-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13225-020-00447-5