
Abstract
Tau, a microtubule-associated protein predominantly localized in neuronal axons, plays a crucial role in promoting microtubule assembly, stabilizing their structure, and participating in axonal transport. Perturbations in tau’s structure and function are implicated in the pathogenesis of neurodegenerative diseases collectively known as tauopathies, the most common disorder of which is Alzheimer’s disease (AD). In tauopathies, it has been found that tau has a variety of post-translational modification (PTM) abnormalities and/or tau is cleaved into a variety of fragments by some specific proteolytic enzymes; however, the precise contributions of these abnormal modifications and fragments to disease onset and progression remain incompletely understood. Herein, we provide an overview about the involvement of distinctive abnormal tau PTMs and different tau fragments in the pathogenesis of AD and other tauopathies and discuss the involvement of proteolytic enzymes such as caspases, calpains, and asparagine endopeptidase in mediating tau cleavage while also addressing the intercellular transmission role played by tau. We anticipate that further exploration into PTMs and fragmented forms of tau will yield valuable insights for diagnostic approaches and therapeutic interventions targeting AD and other related disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD), a progressive neurodegenerative brain disease featured by cognitive impairment and memory loss, has become the most common reason for dementia among the elderly [1]. AD has two main features: the formation of β-amyloid plaques and hyperphosphorylated tau-caused neurofibrillary tangles (NFTs) [2, 3]. Tau is a microtubule-associated protein (MAP) present in neurons that improves microtubule assembly, stabilizes microtubules, and participates in axonal transport [4,5,6,7]. Studies found that tau, as a synaptic protein, had synaptic functions to promote dendrite elongation, spine formation, and synaptic plasticity [8, 9]. The abnormal misfolding of tau protein forms β-folded fibrils that aggregate in the central nervous system (CNS) neurons to form tau aggregates, leading to neurodegenerative diseases collectively termed as tauopathies [10]. Disorders included in this category are AD, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease, aging-associated tau astrocytosis, and primary age-related tauopathies [11, 12].
Under pathological conditions, Tau aggregation begins with tau detaching from microtubules, leading to microtubule disassembly. The detached tau protein aggregates to generate oligomers, which in turn form paired helical filaments (PHFs) with a double-helix bond structure, and finally form NFTs [13]. Tau affinity for tubulin is modulated by post-translational modifications (PTMs) of tau [13, 14], which include phosphorylation, glycosylation, O-linked N-acetylglucosamine (O-GlcNAc) glycosylation, acetylation, methylation, oxidation, nitrification, ubiquitination, and SUMOylation. Tau phosphorylation has been the most studied so far.
In addition to PTMs mentioned above, tau truncation is another important modification mediated by specific proteases that promote neurodegenerative variations via neurotoxic tau fragments that can aggregate and/or propagate across cells. To date, a number of proteolytic fragments of tau have already been identified in cerebrospinal fluid (CSF) and plasma from patients with neurodegenerative diseases, supporting their application as biomarkers of disease progression [15]. At present, there are corresponding therapeutic strategies to inhibit the toxicity of truncated tau, such as repressing protease activity, selectively weakening protease-substrate interaction, and preventing truncated tau from acting [16]. However, the protein breakdown process and pathological changes caused by the transformation of each tau isoform to distinct tau fragments remain to be further studied.
Therefore, this review aims to comprehensively understand PTMs of tau and has a special focus on protease-mediated tau truncation, including their formation process, distribution location, possible pathogenic mechanism, and pathological impacts. Furthermore, we also discuss tau fragments in intercellular secretion and transmission. It is desired to find the action points of inhibiting tau lesions and provide crucial targets for the diagnosis and treatment of tauopathies including AD.
Molecular Structure and Functions of Tau
Human tau is a MAP encoded by the MAPT gene, which is located on chromosome 17 and consists of 16 exons [17]. Tau is overall hydrophilic with its relatively low proportion of hydrophobic amino acids and thus has a high degree of thermal stability and solubility [18]. Tau can be subdivided into four functional domains: an N-terminal projection domain (amino acids 1–150), a proline-rich domain (PRD) encompassing residue 151–243, a microtubule-binding domain (MBD) encompassing residue 244–369, and a C-terminal region (amino acids 370–443), which are characterized by their diverse biochemical properties [19] (Fig. 1). Microtubule binding repeat sequences in MBD (termed R) and N-terminal exons (termed N) together determine the type and naming of six different major tau isoforms, and the expression of these isoforms is developmentally modulated [20]. In adults, all six main isoforms of tau are formed by alternative splicing around MBD and the N-terminal region in the CNS, while only 0N3R isoform is present in fetal brain [20]. The second and third MBD repeats tend to exhibit a well-ordered β-sheet structure [21]. Exon 10 encodes the R2 fragment, and thus, tau with exon 10 in MBD is collectively referred to as 4R tau, and vice versa as 3R tau. In the brains of adult humans, the proportion of 3R tau and 4R tau is equivalent, while there is only 4R tau in adult mice [17, 22]. Collectively, tau expresses in six main distinct isoforms in the human CNS via alternative splicing processes, and alternative splicing around exon 6 produces six additional isoforms [23]. For instance, a set of additional tau isoforms 6D and 6P generated by the alternative splicing of exon 6 may ameliorate the polymerization of full length tau (htau40) and thus are promising endogenous inhibitors of filamentous tau formation [24].

Organization of tau gene and isoforms of tau protein
Tau expression exists throughout the whole brain development, and six isoforms of tau eventually exist in human embryonic stem cell-derived neurons [25]; hence, severity of the increase in tau mRNA levels is synchronized with the degree of maturation of cells [26]. Specifically, tau expression levels are high in SYN + /TBR1 + mature neurons and low in intermediate precursor cells in deep white matter and radial glia in the subventricular zone, with minimal expression in the germinal matrix and subventricular zone [26]. Tau shares a similar expression pattern with induced pluripotent stem cell–derived cortical organisms, and both increase with neuronal maturation, being a promising target for the therapy of neurodevelopmental disorders [26, 27]. Under pathological conditions, tau may accumulate in the perinuclear region of neurons or be expressed in certain neuronal populations. For instance, all the six isoforms of tau exist in dystrophic olfactory epithelial neurites of AD patients [28].
Biophysical studies have shown that tau is a soluble unfolded protein with little secondary structure but highly flexible conformation under physiological conditions [29]. However, tau can perform its tertiary structure when tau is separated from microtubules: “paperclip” conformational folding via intramolecular interactions between charged regions, with the C-terminus folded on MBD and the N-terminus folded on the C-terminus [30,31,32]. Recent studies have revealed that phosphatase activation domain (PAD), the N-terminus region spanning amino acid 2–18, is tucked away beneath the native protein in a paperclip conformation. Aberrant modification of tau is likely to alter its conformation through abnormal exposure of PAD, thereby thwarting normal biological functions of tau [33] and exerting prominent effects on inhibiting tau aggregation and rapid axonal transport [34]. Proline-directed tau phosphorylation also tends to impair tau conformation, loosing or tightening its paperclip conformation [35]. The unfolding of the paperclip conformation may occur before tau oligomers and NFTs form, giving a hint that the unfolded paperclip is a precursor from NFTs [36].
As a multifunctional protein, tau can not only regulate microtubule dynamics and influence cytoskeletal components but also modulate signaling pathways through serving as a protein scaffold for signaling cascades. Numerous studies have found that tau has a novel and important role in multiple physiological functions.
Tau protein, as MAP, combines with microtubules to maintain neuronal health. The prevailing view is that the crucial physiological function of tau in neurons is to stabilize microtubules in axons. A new opinion suggested that tau do not stabilize axon microtubules but promote the assembly of labile domains while restricting its binding to microtubule stabilizers, resulting in long labile regions of axon microtubules [37]. The binding of tau to microtubules is interfered by PTMs, inducing the pathological aggregation of tau. Recently, one pseudo-repeat sequence R’ of MBD of tau were targeted. R’ outperforms the other four repeat sequences when combining to microtubules. Since R’ contains the most charged residues, charge-charge interactions could promote the combination of tau and microtubules [38]. Additionally, MAP has a pivotal role in the modulation of neuronal microtubule cytoskeleton and dynamics, among which tau is the most enriched. Tau bundles and cross-links actin filaments.
Tau is also located in the nucleus of neurons. It is mainly expressed in the nucleolus and pericentromeric heterochromatin (PCH), and nuclear chromatin proteins like tau may modulate chromosome stability and transcription. Tau could interact with nuclear lamina; hence, the nuclear transport in neurons would be influenced under pathogenic conditions [39]. In the nucleolus, the co-localization was found between tau and the related nucleolus proteins, for example, epigenetic factor BAZ2Aor upstream binding transcription factor [40], which suggests that tau may be involved in rDNA transcription and ribosome synthesis. Furthermore, it has been found that tau’s binding to the promotor region of rDNA locus could act as a potential role in regulating transcriptional processes [41]. By utilizing a tau gene knockout mouse model to alter tau expression, its involvement in transcriptional regulation could possibly confirmed. Nuclear tau may act as a protective role during stressful events, since PCH heat shock–induced DNA repair was damaged in tau KO mice [42].
The significance of tau in maintaining the normal structure and function of neurons has been further elucidated through extensive research, highlighting its multifaceted roles and indispensability.
Role of Post-translational Modification of Tau in Tauopathies
The affinity of tau to tubulin is regulated by various PTMs, encompassing phosphorylation, glycosylation, acetylation, methylation, oxidation, nitration, ubiquitination, and SUMOylation. Among these PTMs studied extensively thus far is tau phosphorylation. Acetylation and some other PTMs also impair the functions of tau and promote tau monomer aggregation [43]. The study of the pathological mechanisms of major PTMs is helpful for the targeted treatment of tauopathies.
Phosphorylation
Numerous amino acids of tau are potential phosphorylation sites. Tau phosphorylation is regulated by the activities of protein kinase and phosphatase. A large number of studies have shown that the activity of multiple protein kinases in the brain region of AD patients increases, while the activity of phosphatase decreases [44, 45]. A certain degree of phosphorylation of tau in its normal state does not interfere with the affinity with microtubules, but hyperphosphorylation can separate tau from microtubules in cell [46, 47] and destabilizes microtubules by making itself more likely to aggregate into insoluble inclusions [48]. Eventually, filamentous, insoluble tau aggregates and aggregated PHFs are formed, namely NFTs [49].
In recent years, many researches devoted to exploring which sites of tau phosphorylation can reduce its affinity for microtubules. Some of the known phosphorylation sites that alter microtubule stability are S258, S262, S324, and S356 [50,51,52]. A recent tau structure analysis done by Brotzakis et al. found that S262, S324, and S356 all had an effect on tau-microtubule complex stability, and among these sites, S262 had a relatively more significant effect [52]. Protein kinase R (PKR)-mediated phosphorylation of Thr181, Ser199/202, Thr231, Ser262, Ser396, Ser404, and Ser409 could remove tau from intracellular microtubules [53]. Tyrosine sites, which are less numerous than the other two amino acids, can also be phosphorylated but may also play a stronger role in the affinity between tau and microtubules. Phosphorylation of multiple N-terminal tyrosine residues, or specific phosphorylation at the residue of tyr-310 only, locally reduces the β-sheet tendency structural domain of tau in PHF6, thereby eliminating tau aggregation and inhibiting its properties of microtubule binding [54, 55]. Phosphorylation of Ser396-404, present in 50% of the total structure of early phosphorylated tau aggregates in brain sections from AD and Down syndrome patients [56], is also thought to be more important for tau events since being found to reduce the solubility of full-length tau and promote aggregation [57]. Site-specific phosphorylation of tau could lead to tau mislocalization and accumulation. Phosphorylated-T231-tau mislocalizes to the somatodendrites and accumulates there, which is a prerequisite for the formation of tau tangles in neurogenic fibers [58]. But the opposite is also reported. Biophysical studies have recently revealed that phosphorylated-Ser356-tau (pS356-tau) can affect the stability of β6 and β7 structures in protofibrils of PHFs and straight filament, blocking the interaction between tau and Aβ peptides. Thus, pS356-tau alters the structure of PHFs and disrupts the formation of NFTs [59].
Intracellularly, NFTs formed by hyperphosphorylated tau proteins can lead to neurodegeneration and cell death, by activating calpain and causing mitochondrial dysfunction [60]. However, there are arguments that tau protects against cell death. For instance, phosphorylated tau antagonizes apoptosis by stabilizing β-catenin [61, 62]. Hyperphosphorylation of tau could also negatively regulate the physical function of nuclear tau when protecting DNA from damage [63]. This may suggest that phosphorylation at distinct tau residues and different contexts may cause diverse effects on downstream signals, which may alter the course of cell death [64].
Acetylation
Acetylation belongs to one of the crucial PTMs of tau with various biological functions such as metabolism, histone regulation, and stress response. In transgenic AD mice models and AD patients, tau acetylation level is significantly increased [65]. Multiple research projects have focused on the potential pathogenesis and impact of tau hyperacetylation in AD.
Recently, tau acetylation at the lysine sites has been found significantly increased in brains with human tauopathies [66, 67] and influences numerous biological functions of tau, such as synaptic connection, mitochondrial function, tau aggregation, and diffusion of pathogenic tau [65, 67, 68]. Mitochondrial dysfunction is a clear early feature of AD [69]. Increased acetylation of tau at K274 and K281 has been identified in the brains of AD patients and has recently been found to have damaging effects on mitochondria [70]. However, BGP-15, a poly (ADP-ribose) polymerase (PARP) inhibitor and insulin sensitizer [71], is a potential therapeutic agent for AD as it mitigates these impairments in memory and learning via attenuating the aforementioned impairments caused by acetylation of tau at K274 and K281 [70, 72]. On the other hand, recent studies have shown that acetylation of tau sites K311 and K340 both reduced the rate of microtubule polymerization, so it may affect AD by promoting NFT formation [73]. Li et al. identified lysine residue acetylation patterns on tau fragments flanking the amyloidogenic motifs that contributed to fibril assembly and observed amyloid fibril structures composed of acetylated tau fragments. It suggests that specific lysine residue acetylation patterns cause pro-aggregation interactions of tau, which can assemble tau into different amyloid folds [74].
Tauopathies result in synaptic loss and are one of the earliest structure-related factors in cognitive dysfunction and AD progression. It is worth noting that the increase of phosphorylation and acetylation induces tau to mislocate the synapse, inhibit the release of synaptic vesicles, and affect the activity-dependent secretion of tau from neurons [75]. Therefore, the accumulation of acetylated tau at synapses has a negative effect on crucial neurons related to neuronal activity and synaptic function. Furthermore, most tau proteins are degraded through chaperone-mediated autophagy, while when tau undergoes acetylation, it is preferentially degraded through endosomal microautophagy and macroautophagy, in part because acetylated tau inhibits chaperone-mediated autophagy and leads to the extracellular release of tau [76].
cAMP-response element binding protein (CBP) mediates tau acetylation, while histone deacetylase 6 (HDAC6) and sirtuin 1 (SIRT1) mediate tau deacetylation [65]. According to reports, tau acetylation at the sites regulated by HDAC6 competes with tau phosphorylation, thus disrupting tau aggregation [50]. It has been found that HDAC6 inhibitor CKD-504 significantly alters the tau interactome in the brain of animal models with AD and brain organoids derived from AD patients [77]. Acetylated tau collects chaperone proteins including Hsp40, Hsp70, and Hsp110 to form a complex and then combines with new tau E3 ligases, including RNF14 and UBE2O [77]. Such tau interactome might degrade pathological tau via the proteasome pathway, thus improving cognitive capability and synaptic impairment of AD mice. Mounting evidence suggests that SIRT1 can protect neurons from neurodegenerative diseases including AD [78]. In the brains of patients with AD, the decrease of SIRT1 level elicits the hyperacetylation and accumulation of tau, thus aggravating the transmission of pathogenic tau [68, 79]. The latest research found that the activation of AMP-activated protein kinase (AMPK) reduces the tau acetylation level and attenuates memory damage, becoming a potential target for future AD treatment [80]. Furthermore, in tauP301S transgenic mice, a high level of SIRT1 significantly ameliorates the diffusion of tau pathology into anatomically associated brain regions, suggesting that SIRT1 not only regulates tau acetylation but also inhibits the propagation of tau pathology in vivo [81].
Methylation
In common proteins, lysine residues can be methylated three times at most, resulting in a mono-, di-, or trimethylated lysine, which lead to different biological outcomes. Each time methylation is conducted, a proton would be removed from the ε-amino group, therefore reducing the hydrogen bond potential of the Lys. Thus, methylation could also increase the hydrophobicity and bulk of the Lys side chain [82].
Tau proteins can be mono- or dimethylated in both healthy or AD brains. Methylated tau may be involved in the pathogenesis of tauopathies, which is related to aging, tau aggregation, and changes in microtubule dynamics. One study found that tau aggregated in AD brains was monomethylated at seven Lys sites in the PRD and MBD, where the relative abundance varied between 12 and 67% [83]. Later, they discovered that methylated tau was widely distributed in the AD brain and co-localized with neurogenic fibers in late AD [83]. Several types of methylation mimetics with tau mutation P301L show impairment of microtubule binding and enhancement of prion-like seeding aggregation in frontotemporal dementia [84]. It is interesting that the methylation status of tau changes qualitatively with the progression of aging and disease [84]. There is a possibility that aging alone determines tau methylation, which is similar to the “aging clock” described by DNA methylation [85]. However, the result of this conversion may be pathological. Lysine methylation is a physiological PTM of tau protein which shifts from predominantly dimethyl lysine to monomethyl lysine along with the aging and progress of disease, and pharmacologically enhancing tau methylation may provide a way of preventing pathological tau aggregation [86]. In addition, several studies show that residue-specific methylated tau proteins are more likely to assemble into insoluble structures. methylation of K317 reduces tau solubility, which facilitates efficient dimerization of tau proteins and thus promotes more advanced oligomerization [87,88,89]. Besides, K317 methylation may also increase microtubule dynamics and somewhat inhibit tau-dependent proliferation of Hela cells [87, 90,91,92].
Other PTMs
Other PTMs of tau include glycosylation, ubiquitination, and SUMOylation.
N-terminal glycosylation occurs in hyperphosphorylated tau proteins. The role of N-terminal glycosylation in AD is unclear, and some questions remain about its correlation with tau function [82]. Increased O-GlcNAcylization on serine and threonine residues on tau proteins may protect tau from being phosphorylated [93]; O-GlcNAcylization increases tau-microtubule interactions, increases tau degradation, and inhibits tau aggregation [94,95,96]. Therefore, O-GlcNAcylization of tau may have neuroprotective effects.
As a lysine-rich residue protein, tau is highly sensitive to ubiquitination. There are 17 residues of the 44 lysine residues from the 2N4R tau isoform that have been found to be ubiquitinated. And most of them are located in the MBD [97, 98]. Lysine 63 ubiquitinated tau oligomers play an important role in tau pathological aggregation, accumulation, and proliferation [99]. Insoluble tau from AD brains is predominantly ubiquitinated through K48-linked modifications [97], when soluble tau could also be ubiquitinated through K63 polyubiquitin coupling [100], which suggests that dissolvable and aggregated tau are degraded by different pathways [101]. There is no direct evidence to prove that tau ubiquitination has something to do with neurotoxicity. However, as AD progresses, proteasomal damage could lead to the accumulation of ubiquitinated proteins. It has been found that proteasome inhibition increases the accumulation and insolubility of tau proteins independent of tau phosphorylation and that proteasome inhibition may also indirectly lead to reduced relative tau phosphorylation in the rat brain through c-JUN terminal kinase (JNK) inhibition [102]. When E3 ligase attaches ubiquitin, deubiquitinases (dUbs) remove them. Cysteine protease Otub1 is the only dUbs reported to target tau, removing the K48 polyubiquitin chain from endogenous tau and preventing tau from degrading in transgenic mouse-derived primary neurons [103]. Overexpression of USP10, one of the important dUbs, directly leads to elevated levels of total and phosphorylated tau, inducing tau aggregation and delaying tau degradation [104].
SUMOylation may be a PTM that is highly correlated with tauopathies. SUMO1 modification, combining with tau truncation, may contribute to the pathogenesis of PSP [105]. Tau-SUMOylation could induce tau to be hyperphosphorylated at a series of AD-associated sites, and tau hyperphosphorylation can in turn promote its SUMOylation and inhibit tau degradation by reducing tau protein solubility and ubiquitination [106]. Phosphorylation of tau at residue S-214 promoted its SUMOylation at specific sites and improved its stability [107]. In an in vivo model of AD, few phospho-tau particles in neuritis plaques were ubiquitin-positive in cortical sections from Tg2576 mice, whereas all phospho-tau particles and punctate deposits were SUMO-1-positive [108].
Role of Tau Fragments in Neurodegeneration
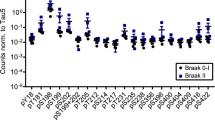
Truncation is one of the most important modifications in PTMs. Cleavage of tau by diverse proteolytic enzymes produces short, easily aggregated fragments that participate in the pathogenesis of neurodegenerative diseases such as AD, CBD, and PSP. Tau accumulation in neurons and glial cells is a crucial characteristic of tauopathies [6, 109]. Tau degradation is found to be the precipitating factor of tau accumulation, and different tau fragments are detected in CSF and brain extract [110, 111]. Tau in CSF is composed of a number of fragments, with the central region and N-terminal tau being the most affluent [112]. However, variants of CSF tau are of a complex variety, containing tau peptides from the N-terminal to the C-terminal, and the most reproducible phenomenon is a significantly increased level of C-terminal truncated tau in patients with AD [111, 113, 114]. Most CSF tau lack C-terminal and MBD parts, and the neuronal secretion of these tau fragments is induced by Aβ exposure, suggesting that CSF tau levels exhibit neuronal responses to Aβ pathology [115]. Numerous tau fragments cleaved by diverse proteolytic enzymes play different roles in tau pathology (Table 1).
Caspase
Caspases are a cysteine protease family that are primarily involved in cell death and inflammatory responses. On the one hand, caspase-8, -9, and -10 could initiate apoptosis by activating the executioner caspase-3, -6 and -7. On the other hand, caspase-1, -4, -5 and -11 are regarded as inflammatory caspases [140]. Especially, activated caspase-3 could lead to non-inflammatory cell death [141,142,143]. Caspase-3-cleaved tau could be possible markers of preclinical neurodegenerative disease [144]. The substrate for caspase-6 is lamin A, and its cleavage is crucial for apoptotic chromatin condensation [145]. Degenerative diseases such as AD are also associated with neuronal apoptosis and inflammation [146, 147], so it can be assumed that there is a relationship between caspases and the progression of AD. Activation of caspase induces the formation of NFTs via cleaving tau. Truncated tau cleaved by activated caspases recruits normal tau to form tangles, which in turn reduce caspase activity to inhibit acute neuronal death and make tangle-bearing neurons long-lived [148, 149]. Caspase-cleaved human tau fragments (tauΔD402, and tauΔD421) are proved in the pathogenesis of tau and induce cognitive impairment [16, 150].
TauΔD421
TauΔD421 is produced by caspase-3, caspase-6, and caspase-7, whereas tauDΔ402 is produced only by caspase-6. Tau is cleaved at aspartate 421 (D421) by these caspases to form tauΔD421. In individuals without cognitive impairment, active caspase-6 is present only in the regions where NFTs first appear: hippocampal CA1 and the entorhinal cortex [151]. Tau fragments cleaved by caspase-6 at amino acids 421 (tauΔD421), as well as active caspase-6, are present in pre- and mature NFTs composed mainly of tau filaments in the patients’ brains with mild cognitive impairment (MCI) or early stage of AD, while absent in the non-AD brains [150, 152]. Levels of tauΔD421-positive CA1 are negatively associated with mini-Mental State Examination scores, and the levels of serum distinguish patients with AD and MCI from those with other dements [116, 153].
Numerous studies have demonstrated that tauΔD421 participates in the pathological mechanisms of tau in many aspects, inducing axonal loss, mitochondrial dysfunction, and tau polymerization and aggregation in vitro [116,117,118,119]. TauΔD421-induced alterations in mitochondrial dynamics could abruptly affect normal synaptic communication. In AD mice, cells expressing tauΔD421 show fragmented mitochondria and impaired mitochondrial dynamics by inhibiting the optic atrophy protein (Opa1) [154]. In addition, tauΔD421 inhibits mitochondrial transport by increasing the co-localization of trafficking kinesin-binding protein 2 (TRAK2) with mitochondria and reducing ATP production, which in turn reduces the number of transporting mitochondria, leading to the deposition of mitochondria in the cytoplasm and a reduction of synaptic mitochondria, which leads to synaptic failure in AD [155].
Immunodepleting of tauΔD421 inhibits the aggregation of tau induced by high molecular weight protein fraction in AD brain, indicating that tauΔD421 affects the pathological spread of tau [156]. Furthermore, tauΔD421 may lead to tau oligomers, microgliosis, and neurodegeneration [148, 157]. However, if a tau mutant that cannot be cleaved by caspase in D421 is expressed, mice could develop memory deficits, synaptic plasticity defects, and prepathological tau changes [158]. The accumulation of tauΔD421 correlates with increasing age. Overexpression of tauΔD421 in middle-aged mice contributes to significant hippocampal long-term potentiation deficits and cognitive deficits, and the increasing age is positively associated with the neuronal degeneration affected by tauΔD421 accumulation [159]. Moreover, the apoptosis of rat cortical neurons is initiated, when treated with aggregated Aβ, and during the process, tau is cleaved at D421 with the onset of neurite death, indicating that tau cleavage is an important component of the deleterious cascade resulting in neuronal dysfunction and apoptosis [160]. Fasulo et al. found that tau151–421 cleaved by caspase-3 exerted significant apoptotic effects on rat hippocampal neurons [161]. But how the D421 could still remain in undead neurons and how it reverses the apoptotic effect and found in the AD brain are still unknown. TauΔD421 impairs neuronal firing, causing inefficient initiation of network bursts, consistent with reduced excitatory drive. Since reduced neuronal activity is coupled to proteasome dysfunction, it drives cleaved tau accumulation at the post synaptic density and subsequent synaptotoxicity [162].
As to possible treatment for this target, a study reveals that tauΔD421 expression significantly reduces dendritic spine density and synaptic vesicle number in hippocampal neurons. Also, neurons transfected with tauΔD421 showed a significant accumulation of synaptophysin protein in soma. All these synaptic changes could be prevented by cyclosporine A, a drug which could inhibit the function of mitochondrial permeability transition pore (mPTP). It means that mPTP may play a role in synaptic dysfunction derived from caspase-3-cleaved tauΔD421, and cyclosporine A may have a cure for AD and other neural dysfunctional diseases [163].
ΔTau314
Aspartate 314 (D314) of tau is another site for cleavage by caspase-2; this process could mislocalize tau to dendritic spines, which can facilitate the mislocalization of tau proteins, leading to reduced postsynaptic AMPA receptors and reduced excitatory neurotransmission [120], thereby affecting cognitive and synaptic function in animal and cellular models of tauopathies and reversibly damaging memory in animal models [121]. The soluble fragment Δtau314 brought about by this cleavage is significantly elevated in the brains of individuals with Huntington’s disease, Lewy body disease, and AD [120, 164], so Δtau314 could become a CSF biomarker to predict tauopathies. Δtau314 protein has now been found to have a distribution in the human striatum and prefrontal cortex [164] and has also been found to be present in the inferior temporal gyrus [165]. Using caspase-2 inhibitors could block the production of Δtau314, inhibiting the excessive dendritic spinal accumulation of tau and thus influencing excitatory neurotransmission in reverse in cultured primary hippocampal neurons of rats [166]. Another treatment for this target is the D314E mutation. Targeted insertion of D314E mutated htau into mice to express cleavage-resistant D314E mutant retards transgene-mediated accumulation of tau in postsynaptic densities [167].
Calpain
Calpains are a series of calcium-dependent proteases that are widely expressed in a variety of organisms and exert activity under neutral pH conditions [168]. Calpain activation is a crucial neurodegenerative factor leading to apoptosis and is closely associated with pathological processes of neurodegenerative diseases [169]. There is always a link between neurodegenerative diseases like AD, Huntington’s disease and PD, and an abnormal increase in calcium concentration, which leads to the abnormal activation of calpain [170]. Abnormal activation of calpain can nonspecifically cleave a variety of target proteins including tau proteolysis. Specifically, calpain-1 and -2 lead to the cleavage of 17 kDa tau, and calpain-2 mediates the formation of N224 tau [171, 172], both of which are common tau fragments with neurotoxic effects and mediation in the progression of degenerative diseases. Focus on possible treatment, calpain-1 activity may be downregulated by UB-ALT-EV, a new NMDA receptor antagonist, under which 5FXAD mice show a better cognitive performance, revealing that UB-ALT-EV could reduce cognitive alterations in the mice model of familial AD [173].
17 kDa tau (Tau45–230)
β-Amyloid oligomers could induce calpain activation and thus cleavage of tau. One of the more studied fragments is the 17 kDa tau fragment comprising 45–230, which arises due to the cleavage by calpain-1 at K44–K45 [174] and cleavage by calpain-1 and -2 at R230–T231 [172]. Tau45–230 occurs in the temporal cortex of AD patients and can also be detected in the brains of patients with other tauopathies such as Pick’s disease and Parkinson’s syndrome-17 [175]. Tau45–230 may exert toxicity by way of oligomerization. The oligomer can be incorporated by neurons and indirectly induce neurosynaptic degeneration. In vitro, tau45–230 can oligomerize to form heptamers and octamers [176]. Another study reported that tau45–230 oligomers can be internalized by cultured hippocampal neurons and induce neurosynaptic degeneration through a clathrin-mediated mechanism [177].
Tau45–230 can also function as a toxic agent by altering the composition of the normal neuronal cytoskeleton and affecting neurite growth and transport. In cultured hippocampal neurons, tau45–230 can exert toxicity by partially blocking axonal transport along the microtubules, markedly reducing the organelles transported along axons [178]. Tau45–230 can also trigger a transient increase in unstable tyrosine microtubules [123]. Significant synaptic deficits were detected in mice expressing tau45–230 at 6 months postnatally, accompanied by altered NMDA receptor expression [122]. Tau45–230 overexpression leads to apoptosis in Chinese hamster ovary cells CHO, hippocampal neurons, and mouse hippocampus [122, 179, 180]. Yet it has also been argued that the 17 kDa tau fragment may be actually a shorter tau (125–230) fragment [172]. The accumulation of both tau125–230 and tau45–230 fragments has recently been found in brain tissue from acute ischemic shock [181]. Thus, the 125–230 fragment may also be involved in pathological progression of neurodegenerative disease.
N224 tau
Tau in CSF is recently found to have a major calpain-2-induced cleavage at amino acid (aa) 224, and the N-terminal tau fragment terminating at aa224 (N224) is prominently upregulated in AD [182]. Levels of N224 tau from the soluble brain fraction of AD patients are significantly lower than that of controls, while in CSF, N224 tau is higher in AD patients than in controls [124]. High levels of N224 tau correlate with low Mini-Mental State Examination (MMSE) scores [126]. Cicognola et al. demonstrated that increased levels of N224 tau in CSF were connected with conversion from MCI to cognitive decline and AD [124]. Moreover, N224 tau levels are lower in PSP or corticobasal syndrome (CBS) than in AD [125]. N224 tau exists in tangles and neuronally derived extracellular vesicles (NDEV), with neuron-specific secretion in CSF and upregulation in AD, indicating its involvement in cognitive impairment and AD pathology [124]. In addition, a recent study has revealed that levels of N224 tau in AD patients were higher than that in MCI, subjective cognitive decline SCD, PD, PD dementia, multiple system atrophy, and PSP [126]. Therefore, N224 tau could contribute to distinguish AD from SCD and other dementias and may be an important tau biomarker to study species of tau fragments in CSF.
NH2-tau
NH2-tau is a 20–22 kDa fragment that predominantly involves in mitochondria from synaptosomes of the AD brain [134]. This fragment was discovered by Corsetti et al. while studying the toxic effects of the N-terminal fragment of tau. At that time, they found that some NH2-terminal fragments, such as 26–44 tau and 26–230 tau, induced N-methyl-D-aspartate receptor (NMDAR)-mediated cell death [183], and 26–230 tau was also found by others in cellular and animal models of AD, so they named this fragment NH2-26–230 tau (aka NH2-tau) [184]. The full length of NH2-tau comprises 26–230 aa, and the first 25 amino acids have been clearly sheared down by caspases, while amino acids after 231 are sheared by calpain-1 and -2 [185, 186]. NH2-tau is an essential component in the toxic cascade that leads to neurodegeneration or cell death [186]. Following apoptotic stimulation or neurodegenerative injury, aberrant activation of cysteinyl asparaginase may produce toxic NH2-tau fragments which could then propagate and also elaborate cellular dysfunction.
NH2-tau enrichment in hippocampal parenchyma revealed significant impairment of learning/memory capacity in treated healthy mice, which was associated with reduced synaptic linkages and neuroinflammatory responses [132, 133]. NH2-tau can accumulate in AD synapses and be detected in CSF [110], and this truncated tau protein in vitro can acutely cause presynaptic defects in K+-induced glutamate release from hippocampal synaptosomes as well as altered local calcium dynamics [133]. Downregulation of the CREB/c-fos pathway can be detected in the hippocampus following subchronic intracerebroventricular infusion of NH2-tau protein into wild-type mice [132]. A more widely reported function of NH2-tau is its damage to mitochondria. Extracts of Aβ oligomers may damage mitochondrial function by producing this kind of fragment in vitro in rat hippocampal neurons and mature human SY5Y cells [134]. NH2-tau can impair mitochondrial autophagy, directly through the inhibition of ADP/ATP exchange depending on ANT-1 [135, 137, 187] and indirectly through impairing mitochondrial autophagy [135]. One of the pathways is that NH2-tau causes abnormal recruiting of parkin and UCHL-1 to mitochondria, where deleterious autophagic clearance occurs [187].
NH2 26–44 tau acts as the minimum active moiety of NH2-tau. Experiments with the NH2-derived NH2 26–44 tau peptide revealed that both cytochrome C oxidase and adenine nucleotide translocators are targets of this fragment, but adenine nucleotide translocation is a characteristic target of the fragment, and this action significantly affects cellular access to ATP synthesized by mitochondria [136, 137]. In contrast, fragments 1–25 do not have such a significant toxic effect. Another example is that this truncated tau protein accumulates at synapses of AD individual and can be secreted into parenchyma. This could acutely trigger presynaptic defects in glutamate release evoked by K+ at synaptosomes in the hippocampus as well as alter local Ca2+ dynamics [133].
It has recently been shown that 12A12mAb can well neutralize NH2tau fragments [188], restoring synaptic linkage and cytoskeletal function and effectively improving learning and memory function in pathological AD mice [189]. NH2-tau proteins may be involved in pathological degeneration of the retina, but targeted injection of monoclonal antibodies can ameliorate the degenerative process [188] and improve visuo-spatial recognition memory [190]. Interestingly, 12A12mAb could downregulate the steady state expression levels of APP and beta-secretase 1 (BACE-1), thus limiting the Aβ production both in the hippocampus and retina, showing that 12A12mAb could also make an influence on APP involved pathological mechanisms [191].
AEP
Asparagine endopeptidase (AEP) is a lysosomal cysteine proteinase, also named δ-secretase, which is not expressed in neurons but in microglia [131, 192]. AEP specifically hydrolyses the carboxyl-terminal peptide bond of asparagine residues and is a crucial enzyme participating in the processing of antigens and autoantigen processing, since it is known to involve in the processing of antigens for MHC class II presentation in the lysosomes of antigen-presenting cells [193, 194]. A decrease in pH can activate inactive full-length pro-AEP [194]. AEP is related to neurodegeneration [192, 195]; it could mediate dementia by enhancing amyloid plaque and tau hyperphosphorylation, indicating that it played an important role in neurodegeneration [196]. It can specifically shear human synuclein (aSyn) at the N103 site and trigger its aggregation to produce neurotoxicity and mediate PD pathogenesis [197]. In the brain of patients with PD, Netrin-1 expression is downregulated, and UNC5C receptors are specifically sheared by activated AEP, leading to apoptosis of dopaminergic neurons [198]. In AD, AEP activity is upregulated, and the activation of AEP leads to hyperphosphorylation of tau protein, mediating the neurofibrillary pathology [192, 197, 198]. Tau168–368 and tau1–368 are important fragments processed by AEP.
Tau168–368
Tau is cleaved at N167 and N368 by AEP after endocytic uptake into microglia rather than neurons, forming tau168–368 fragment at similar concentrations in both AD and control groups, which does not accumulate in microglia [131]. AEP-mediated tau cleavage does not alter in AD compared with control groups. Therefore, AEP-cleaved tau168–368 is a component of the proteolytic cascade that induces tau degradation, and AEP-mediated cleavage of tau is a physiological response that occurs when secreted neuronal protein is degraded in microglia. Additionally, the amount of tau fragment cleaved at the site N368 in insoluble tau aggregates is very small (< 0.1%) compared with uncleaved tau, indicating that AEP-cleaved tau has a limited role in tau aggregation in AD [199]. Based on these results, AEP-mediated tau cleavage might not be a direct valid therapeutic target for AD. However, AEP-cleaved tau does participate in other important aspects of AD pathology.
Tau1–368
To date, effective fluid biomarkers for tau pathology in AD are still lacking. A new SIMOA® quantification assay has recently been established to assess the presence of tau1–368 in CSF. The results showed that tau1–368 is a tangle-rich fragment, and the tau1–368/total tau (t-tau) ratio is significantly decreased in AD patients, reflecting tangle pathology. This ratio in CSF is negatively associated with the retention of 18F-Genentech Tau Probe 1 (GTP1) [200]. This new tau biomarker might aid in the AD diagnosis and contribute to drug development targeting tau pathology. In addition to cleaving tau at N368, AEP can also cleave amyloid precursor protein (APP) to form APP 586–695, contributing to cognitive impairment and the pathogenesis of AD [127, 128]. Upon APP stimulation, tau1–368 significantly enhances beta-secretase1 (BACE1) expression and Aβ production, facilitating its nuclear translocation [127]. Notably, the activation of JAK2 or SGK1 kinases by Aβ phosphorylates STAT1 and triggers its binding to tau1–368 [201]. Thus, not only tau may be a downstream effector of Aβ, but also tau1–368 can exacerbate Aβ production. Notably, aberrant PTMs of truncated tau are equally important in endogenous tau pathology and neurodegeneration. Tau1–368 contains MBD and PRD, whose phosphorylation stimulates endogenous tau phosphorylation and aggregation. Interestingly, phosphorylated tau1–368 causes body weight loss and recognition memory impairment of C57BL/6 J mice, whereas non-phosphorylated tau1–368 does not [129, 130]. In addition, overexpression of phosphorylated tau1–368 elicits hippocampal neuronal loss and gliosis, but tau1–368 delivery and tau1–368-mediated neurotoxicity are phosphorylation independent [129, 192]. Spinal cord injury could stimulate AEP activation in mice. Activated AEP then cleaved APP and tau, resulting in tau1–368 formations, and consequentially accelerated Aβ deposit and Tau hyperphosphorylation, resulting in cognitive impairment [196].
Secretion and Transmission of Truncated Tau
The aggregation and diffusion of misfolded tau protein in the CNS is one of the hallmark features of tauopathies. In AD brains, tau aggregates accumulate first in the trans-entorhinal cortex and then spread to the anatomically connected hippocampus, leading to progressive cognitive deficits and neurodegeneration [202]. Numerous studies have revealed that tau can undergo prion-like pathological propagation between cells [203], that is, cellular secretion of tau protein, internalization of tau, and misfolding of normal tau in recipient cells, but currently, no epidemiological evidence shows that tau aggregates are infectious [204, 205]. Specifically, prion-like transmission of tau starts with the production of transmissible seed-competent tau monomers [206], which generate distinct biologically active and self-replicating assemblies called strains in distinct tauopathies. Secondly, pathological cells secrete and normal cells subsequently uptake seed-competent tau. Finally, templated tau misfolding occurs in recipient cells, resulting in the formation of new tau seeds. Under physiological and pathological conditions, tau seeds are actively secreted outside the cell through a variety of possible unconventional pathways, such as plasma membrane translocation, secretion based on membrane organelles, and ectosomal shedding [207,208,209]. The physiological role of extracellular tau needs to be further investigated, but preliminary studies revealed that tau in the extracellular space enhances electrical activity in primary neuronal cultures [210].
Hyperphosphorylation can promote tau detachment from microtubules and cell secretion of full-length tau [211]. Recently, however, truncated tau has been found to be transmitted between cells in a limited capacity, independent of phosphorylation. Although phosphorylation does not affect the cell-to-cell transmission of tau1–368, it mediates neurotoxicity when tau1–368 is over-expressed in the hippocampal CA1 region, resulting in anxiety, impaired recognition memory, and weight loss [129]. In addition, tau fibrils synthesized artificially with truncated tau were sufficient to deliver tau inclusions in mouse models. To be specific, synthetic preformed fibrils (pffs) assembled from truncated tau containing four microtubule-binding repeats induce the time-dependent propagation of NFT-like inclusions from the injection site to contiguous brain regions [212].
Tau degradation is normally completed via the ubiquitin–proteasome system and autophagy-lysosome system [213, 214]. However, phosphorylated tau in pathological states can be directly degraded through the autophagy-lysosome system [215]. As a further step, excess tau remains on the surface of the lysosomal membrane, which makes it possible for tau to oligomerize at or near the organelle surface [102, 216]. The accumulation of aggregated proteins could result in lysosomal dysfunction in multiple ways, including membrane rip and lysosomal cleavage, which can promote tau aggregation to form a vicious cycle. It is probable that lysosomal dysfunction results in the compensatory release of extracellular vesicles/exosomes in neurodegenerative diseases [217, 218]. As tau lacks conventional signal sequences, there are four possible unconventional protein secretion mechanisms leading to tau secretion [219]: (1) direct export across the plasma membrane; (2) secretion via ectosomes that are shed from the plasma membrane, or microvesicle shedding; (3) formation of intraluminal vesicles (ILVs) through late endosome membrane’s inward budding, free tau in cytosol can be sequestered in ILVs which could form endosomes of multivesicular vesicles (MVBs), and subsequently, tau can be packaged into exosomes and secreted extracellularly upon fusion of the MVB with the plasma membrane; and (4) direct release into the extracellular space in a vesicle-free manner via the organelle hitchhiking pathway.
To be inclusive, tau can enter extracellularly via ectosome, exosome, or without vesicles [220]. Tau-containing exosomes are derived from MVB and in AD patients, where exosomes have significantly higher levels of tau protein than normal exosomes [221]. Exosomal tau from neurons is hyperphosphorylated, and the release of tau-containing exosomes could be promoted through depolarization of neurons. These exosomes complete the transmission of tau between neurons through synaptic connection [222]. In human-induced multifunctional neurons derived from stem cell, the exosomes contain mutant tau (mTau) and contain the P301L and V337M mutations. In Podvin’s study, mTau is a dynamic regulator of exosome biosynthesis, leading to the acquisition, deletion, upregulation, or downregulation of protein cargo, resulting in pathological mTau exosomes that can cause p-tau neuropathology in the brain of mice intracellularly [223]. Microglia could also release tau-contained exosomes and cause the spread of abnormal tau. This kind of immune cell engulfs neurons containing pathological tau proteins, which are subsequently transmitted to exosomes involved in the pathological transmission of tau in the brain. This transmission can lead to tau propagation from the olfactory cortex to the dentate gyrus region. However, clearing microglia and inhibiting exosome synthesis with GW4869 is able to limit tau propagation in patient brains [224]. P2RX7 is an ATP-gated cation channel that triggers exosome secretion, and oral administration of a P2RX7 inhibitor to P301S tau transgenic mice inhibited exosome secretion; thus, MC1 + and Alz50 + tau proteins were significantly reduced in the hippocampus, and significant improvements in working and situational memory of mice were discovered [225]. Truncated tau species which lack MBD that is essential for seeding have been reported to undergo active secretion.
Both tau fragments or full-length tau could be secreted. The PRD of tau could contribute as a membrane-binding site when full-length tau was secreted [226]. However, extracellular species are predominantly composed of N-terminal fragments of tau, while there is no evidence that C-terminal tau fragments are involved [210]. The effect of expressing constructed N-terminal and full-length tau in transfected neuronal lines is examined, and secreted tau shows a cleavage pattern which is similar to tau in CSF from AD patients [227]. Htau was secreted by neuronal and non-neuronal cells when htau was upregulated. In HeLa cells, phosphorylation and cleavage of tau favored its secretion. A mutant form of tauΔD421, where caspase-3 prefer to cleave, was secreted more than wild-type tau, which could contribute to the pathological tau propagation in brain and its accumulation in the CSF [228].
The transcription factor EB (TFEB), a master of regulating lysosomal biogenesis, regulates the secretion of truncated mutant tau lacking MBD. Since lysosomal exocytosis mediated by TFEB could promote cellular clearance, it has been proved that reduced interstitial fluid tau without TFEB’s occurrence have something to do with enhanced cell-to-cell pathology and accelerated spreading; thus, exocytosis could act as a clearance mechanism, reducing intracellular tau on pathological conditions [229]. VAMP8, a late endosomal R-SNARE, increased tau secretion, which was cleaved at the C-terminal. Upon VAMP8 overexpression, an increase of caspase-3-cleaved tau in the cell lysate and medium was observed. However, this intracellular cleaving may not be necessary, since extracellular tau cleavage by caspase-3 could occur resulting from released active caspase-3, which reached the most when VAMP8 was upregulated [230]. This could explain why full-length tau could be secreted, but most of extracellular tau are C-terminal cleaved. Tau cleavage mediated by AEP is a physiological event occurring during secreted neuronal proteins are degraded in microglia [131]. N-224 tau fragment is specifically processed in neurons, discovered in NFT, secreted in CSF, and overexpressed in AD [231].
Few articles have discussed tau-containing ectosomes, but in principle, similar biogenesis mechanisms can be activated in distinctive regions of the cell to produce ectosomes and exosomes. For example, the endosomal sorting complex required for transport (ESCRT) mechanism participated in the biosynthesis of both exosomes and ectosomes, and the situation is similar for both exosomal and endosomal sorting mechanisms [232].
Conclusion
Tau protein is widely distributed within the neurons and has multiple functions, from modulating the cellular structure to regulating the expression of DNA or RNA, from connecting the transported organisms and the microtubules to controlling the transportation between nucleus and plasma. Tau is one of the most crucial molecules in tauopathies like AD, PSP, and CBD. Numerous studies proved tau structural abnormalities were often induced by post-translational modifications and abnormal protease cleavage, where they contribute to induce pathological lesions in the nervous system. The special role and pathological mechanism of tau abnormalities in these diseases are very complex and unclear till now. In fact, the precise understanding of whether tau is the causative factor underlying these diseases or a consequence of other pathogenic agents also remains elusive. Nevertheless, more in-depth investigation is still worthy of attention and expectation because that tau and its fragments can serve as fundamental basis for tauopathies diagnosis, novel biomarkers for assessing disease progression, and a potential target for therapeutic interventions.
Availability of Data and Materials
Not applicable.
References
2015 Alzheimer’s disease facts and figures (2015). Alzheimers Dement 11 (3):332–384. https://doi.org/10.1016/j.jalz.2015.02.003
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science (New York, NY) 297(5580):353–356. https://doi.org/10.1126/science.1072994
Eikelenboom P, Rozemuller AJ, Hoozemans JJ, Veerhuis R, van Gool WA (2000) Neuroinflammation and Alzheimer disease: clinical and therapeutic implications. Alzheimer Dis Assoc Disord 14(Suppl 1):S54-61. https://doi.org/10.1097/00002093-200000001-00009
Sergeant N, Delacourte A, Buée L (2005) Tau protein as a differential biomarker of tauopathies. Biochem Biophys Acta 1739(2–3):179–197. https://doi.org/10.1016/j.bbadis.2004.06.020
Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B et al (2005) Tau pathology in Alzheimer disease and other tauopathies. Biochem Biophys Acta 1739(2–3):198–210. https://doi.org/10.1016/j.bbadis.2004.09.008
Ballatore C, Lee VM, Trojanowski JQ (2007) Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci 8(9):663–672. https://doi.org/10.1038/nrn2194
Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW (1975) A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 72(5):1858–1862. https://doi.org/10.1073/pnas.72.5.1858
Zempel H, Dennissen FJA, Kumar Y, Luedtke J, Biernat J, Mandelkow EM, Mandelkow E (2017) Axodendritic sorting and pathological missorting of tau are isoform-specific and determined by axon initial segment architecture. J Biol Chem 292(29):12192–12207. https://doi.org/10.1074/jbc.M117.784702
Velazquez R, Ferreira E, Tran A, Turner EC, Belfiore R, Branca C, Oddo S (2018) Acute tau knockdown in the hippocampus of adult mice causes learning and memory deficits. Aging Cell 17(4):e12775. https://doi.org/10.1111/acel.12775
Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159. https://doi.org/10.1146/annurev.neuro.24.1.1121
Arendt T, Stieler JT, Holzer M (2016) Tau and tauopathies. Brain Res Bull 126(Pt 3):238–292. https://doi.org/10.1016/j.brainresbull.2016.08.018
Hernández F, Avila J (2007) Tauopathies. Cell Mol Life Sci 64(17):2219–2233. https://doi.org/10.1007/s00018-007-7220-x
Mandelkow EM, Biernat J, Drewes G, Steiner B, Lichtenberg-Kraag B, Wille H, Gustke N, Mandelkow E (1993) Microtubule-associated protein tau, paired helical filaments, and phosphorylation. Ann N Y Acad Sci 695:209–216. https://doi.org/10.1111/j.1749-6632.1993.tb23054.x
Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E (1993) Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron 11(1):153–163. https://doi.org/10.1016/0896-6273(93)90279-z
Shoji M (2019) Cerebrospinal fluid and plasma tau as a biomarker for brain tauopathy. Adv Exp Med Biol 1184:393–405. https://doi.org/10.1007/978-981-32-9358-8_29
Quinn JP, Corbett NJ, Kellett KAB, Hooper NM (2018) Tau proteolysis in the pathogenesis of tauopathies: neurotoxic fragments and novel biomarkers. J Alzheimer's Dis: JAD 63(1):13–33. https://doi.org/10.3233/jad-170959
Andreadis A (2006) Misregulation of tau alternative splicing in neurodegeneration and dementia. Prog Mol Subcell Biol 44:89–107. https://doi.org/10.1007/978-3-540-34449-0_5
Iqbal K, Grundke-Iqbal I (2008) Alzheimer neurofibrillary degeneration: significance, etiopathogenesis, therapeutics and prevention. J Cell Mol Med 12(1):38–55. https://doi.org/10.1111/j.1582-4934.2008.00225.x
Gasparini L, Crowther RA, Martin KR, Berg N, Coleman M, Goedert M, Spillantini MG (2011) Tau inclusions in retinal ganglion cells of human P301S tau transgenic mice: effects on axonal viability. Neurobiol Aging 32(3):419–433. https://doi.org/10.1016/j.neurobiolaging.2009.03.002
Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA (1989) Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 3(4):519–526. https://doi.org/10.1016/0896-6273(89)90210-9
Mukrasch MD, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, Mandelkow E, Zweckstetter M (2009) Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol 7(2):e34. https://doi.org/10.1371/journal.pbio.1000034
Guo T, Noble W, Hanger DP (2017) Roles of tau protein in health and disease. Acta Neuropathol 133(5):665–704. https://doi.org/10.1007/s00401-017-1707-9
Wei ML, Andreadis A (1998) Splicing of a regulated exon reveals additional complexity in the axonal microtubule-associated protein tau. J Neurochem 70(4):1346–1356. https://doi.org/10.1046/j.1471-4159.1998.70041346.x
Lapointe NE, Horowitz PM, Guillozet-Bongaarts AL, Silva A, Andreadis A, Binder LI (2009) Tau 6D and 6P isoforms inhibit polymerization of full-length tau in vitro. Biochemistry 48(51):12290–12297. https://doi.org/10.1021/bi901304u
Iovino M, Patani R, Watts C, Chandran S, Spillantini MG (2010) Human stem cell-derived neurons: a system to study human tau function and dysfunction. PLoS ONE 5(11):e13947. https://doi.org/10.1371/journal.pone.0013947
Fiock KL, Smalley ME, Crary JF, Pasca AM, Hefti MM (2020) Increased tau expression correlates with neuronal maturation in the developing human cerebral cortex. eNeuro 7 (3). https://doi.org/10.1523/eneuro.0058-20.2020
Jovanov-Milošević N, Petrović D, Sedmak G, Vukšić M, Hof PR, Simić G (2012) Human fetal tau protein isoform: possibilities for Alzheimer's disease treatment. Int J Biochem Cell Biol 44(8):1290–1294. https://doi.org/10.1016/j.biocel.2012.05.001
Lee JH, Goedert M, Hill WD, Lee VM, Trojanowski JQ (1993) Tau proteins are abnormally expressed in olfactory epithelium of Alzheimer patients and developmentally regulated in human fetal spinal cord. Exp Neurol 121(1):93–105. https://doi.org/10.1006/exnr.1993.1074
Jeganathan S, von Bergen M, Mandelkow EM, Mandelkow E (2008) The natively unfolded character of tau and its aggregation to Alzheimer-like paired helical filaments. Biochemistry 47(40):10526–10539. https://doi.org/10.1021/bi800783d
Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E (2006) Global hairpin folding of tau in solution. Biochemistry 45(7):2283–2293. https://doi.org/10.1021/bi0521543
Lathuiliere A, Valdes P, Papin S, Cacquevel M, Maclachlan C, Knott GW, Muhs A, Paganetti P et al (2017) Motifs in the tau protein that control binding to microtubules and aggregation determine pathological effects. Sci Rep 7(1):13556. https://doi.org/10.1038/s41598-017-13786-2
Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A (2002) Tau is essential to beta-amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A 99(9):6364–6369. https://doi.org/10.1073/pnas.092136199
Kanaan NM, Cox K, Alvarez VE, Stein TD, Poncil S, McKee AC (2016) Characterization of early pathological tau conformations and phosphorylation in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 75(1):19–34. https://doi.org/10.1093/jnen/nlv001
Hovakimyan A, Antonyan T, Shabestari SK, Svystun O, Chailyan G, Coburn MA, Carlen-Jones W, Petrushina I et al (2019) A MultiTEP platform-based epitope vaccine targeting the phosphatase activating domain (PAD) of tau: therapeutic efficacy in PS19 mice. Sci Rep 9(1):15455. https://doi.org/10.1038/s41598-019-51809-2
Leroy K, Boutajangout A, Authelet M, Woodgett JR, Anderton BH, Brion JP (2002) The active form of glycogen synthase kinase-3beta is associated with granulovacuolar degeneration in neurons in Alzheimer's disease. Acta Neuropathol 103(2):91–99. https://doi.org/10.1007/s004010100435
Rudenko LK, Wallrabe H, Periasamy A, Siller KH, Svindrych Z, Seward ME, Best MN, Bloom GS (2019) Intraneuronal tau misfolding induced by extracellular amyloid-beta oligomers. J Alzheimer's Dis 71(4):1125–1138. https://doi.org/10.3233/JAD-190226
Baas PW, Qiang L (2019) Tau: it’s not what you think. Trends Cell Biol 29(6):452–461. https://doi.org/10.1016/j.tcb.2019.02.007
El Mammeri N, Dregni AJ, Duan P, Wang HK, Hong M (2022) Microtubule-binding core of the tau protein. Sci Adv 8(29):eabo4459. https://doi.org/10.1126/sciadv.abo4459
Eftekharzadeh B, Daigle JG, Kapinos LE, Coyne A, Schiantarelli J, Carlomagno Y, Cook C, Miller SJ et al (2018) Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron 99(5):925-940 e927. https://doi.org/10.1016/j.neuron.2018.07.039
Maina MB, Bailey LJ, Wagih S, Biasetti L, Pollack SJ, Quinn JP, Thorpe JR, Doherty AJ et al (2018) The involvement of tau in nucleolar transcription and the stress response. Acta Neuropathol Commun 6(1):70. https://doi.org/10.1186/s40478-018-0565-6
Bou Samra E, Buhagiar-Labarchede G, Machon C, Guitton J, Onclercq-Delic R, Green MR, Alibert O, Gazin C et al (2017) A role for tau protein in maintaining ribosomal DNA stability and cytidine deaminase-deficient cell survival. Nat Commun 8(1):693. https://doi.org/10.1038/s41467-017-00633-1
Mansuroglu Z, Benhelli-Mokrani H, Marcato V, Sultan A, Violet M, Chauderlier A, Delattre L, Loyens A et al (2016) Loss of tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci Rep 6:33047. https://doi.org/10.1038/srep33047
Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, Lee VM (2011) The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun 2:252. https://doi.org/10.1038/ncomms1255
Iqbal K, Liu F, Gong CX, Alonso Adel C, Grundke-Iqbal I (2009) Mechanisms of tau-induced neurodegeneration. Acta Neuropathol 118(1):53–69. https://doi.org/10.1007/s00401-009-0486-3
Liu F, Iqbal K, Grundke-Iqbal I, Rossie S, Gong CX (2005) Dephosphorylation of tau by protein phosphatase 5: impairment in Alzheimer’s disease. J Biol Chem 280(3):1790–1796. https://doi.org/10.1074/jbc.M410775200
Johnson GV, Stoothoff WH (2004) Tau phosphorylation in neuronal cell function and dysfunction. J Cell Sci 117(Pt 24):5721–5729. https://doi.org/10.1242/jcs.01558
Kellogg EH, Hejab NMA, Poepsel S (2018) Near-atomic model of microtubule-tau interactions. 360(6394):1242-1246https://doi.org/10.1126/science.aat1780
Sferra A, Nicita F (2020) Microtubule dysfunction: a common feature of neurodegenerative diseases. 21 (19). https://doi.org/10.3390/ijms21197354
Goedert M, Klug A, Crowther RA (2006) Tau protein, the paired helical filament and Alzheimer’s disease. J Alzheimer’s Dis: JAD 9(3 Suppl):195–207. https://doi.org/10.3233/jad-2006-9s323
Carlomagno Y, Chung DC, Yue M, Castanedes-Casey M, Madden BJ, Dunmore J, Tong J, DeTure M et al (2017) An acetylation-phosphorylation switch that regulates tau aggregation propensity and function. J Biol Chem 292(37):15277–15286. https://doi.org/10.1074/jbc.M117.794602
Haj-Yahya M, Gopinath P, Rajasekhar K, Mirbaha H, Diamond MI, Lashuel HA (2020) Site-specific hyperphosphorylation inhibits, rather than promotes, tau fibrillization, seeding capacity, and its microtubule binding. 59 (10):4059–4067. https://doi.org/10.1002/anie.201913001
Brotzakis ZF, Lindstedt PR, Taylor RJ, Rinauro DJ, Gallagher NCT, Bernardes GJL (2021) A structural ensemble of a tau-microtubule complex reveals regulatory tau phosphorylation and acetylation mechanisms. 7(12):1986-1995https://doi.org/10.1021/acscentsci.1c00585
Reimer L, Betzer C, Kofoed RH, Volbracht C, Fog K, Kurhade C, Nilsson E, Överby AK et al (2021) PKR kinase directly regulates tau expression and Alzheimer’s disease-related tau phosphorylation. Brain Pathol (Zurich, Switzerland) 31(1):103–119. https://doi.org/10.1111/bpa.12883
Ait-Bouziad N, Chiki A, Limorenko G (2020) Phosphorylation of the overlooked tyrosine 310 regulates the structure, aggregation, and microtubule- and lipid-binding properties of Tau. 295 (23):7905–7922. https://doi.org/10.1074/jbc.RA119.012517
Lebouvier T, Scales TME, Williamson R, Noble W, Duyckaerts C, Hanger DP, Reynolds CH, Anderton BH et al (2009) The microtubule-associated protein tau is also phosphorylated on tyrosine. J Alzheimer’s Dis 18(1):1–9. https://doi.org/10.3233/JAD-2009-1116
Mondragon-Rodriguez S, Perry G, Luna-Munoz J, Acevedo-Aquino MC, Williams S (2014) Phosphorylation of tau protein at sites Ser(396–404) is one of the earliest events in Alzheimer’s disease and Down syndrome. Neuropathol Appl Neurobiol 40(2):121–135. https://doi.org/10.1111/nan.12084
Gu J, Congdon EE, Sigurdsson EM (2013) Two novel tau antibodies targeting the 396/404 region are primarily taken up by neurons and reduce tau protein pathology. J Biol Chem 288(46):33081–33095. https://doi.org/10.1074/jbc.M113.494922
Aragão Gomes L, Uytterhoeven V (2021) Maturation of neuronal AD-tau pathology involves site-specific phosphorylation of cytoplasmic and synaptic tau preceding conformational change and fibril formation. 141(2):173-192https://doi.org/10.1007/s00401-020-02251-6
Leonard C, Phillips C, McCarty J (2021) Insight into seeded tau fibril growth from molecular dynamics simulation of the Alzheimer’s disease protofibril core. Front Molec Biosci 8. https://doi.org/10.3389/fmolb.2021.624302
Bano D, Ankarcrona M (2018) Beyond the critical point: an overview of excitotoxicity, calcium overload and the downstream consequences. Neurosci Lett 663:79–85. https://doi.org/10.1016/j.neulet.2017.08.048
Liu XA, Liao K, Liu R, Wang HH, Zhang Y, Zhang Q, Wang Q, Li HL et al (2010) Tau dephosphorylation potentiates apoptosis by mechanisms involving a failed dephosphorylation/activation of Bcl-2. J Alzheimers Dis 19(3):953–962. https://doi.org/10.3233/JAD-2010-1294
Li HL, Wang HH, Liu SJ, Deng YQ, Zhang YJ, Tian Q, Wang XC, Chen XQ et al (2007) Phosphorylation of tau antagonizes apoptosis by stabilizing beta-catenin, a mechanism involved in Alzheimer’s neurodegeneration. Proc Natl Acad Sci U S A 104(9):3591–3596. https://doi.org/10.1073/pnas.0609303104
Ulrich G, Salvadè A, Boersema P, Calì T, Foglieni C, Sola M, Picotti P, Papin S, et al. (2018) Phosphorylation of nuclear tau is modulated by distinct cellular pathways. Sci Reports 8 (1). https://doi.org/10.1038/s41598-018-36374-4
Chi H, Chang HY, Sang TK (2018) Neuronal cell death mechanisms in major neurodegenerative diseases. Int J Molec Sci 19 (10). https://doi.org/10.3390/ijms19103082
Cook C, Carlomagno Y, Gendron TF, Dunmore J, Scheffel K, Stetler C, Davis M, Dickson D et al (2013) Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum Mol Genet 23(1):104–116. https://doi.org/10.1093/hmg/ddt402
Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y et al (2010) Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67(6):953–966. https://doi.org/10.1016/j.neuron.2010.08.044
Tracy TE, Sohn PD, Minami SS, Wang C, Min SW, Li Y, Zhou Y, Le D et al (2016) Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes tauopathy-related memory loss. Neuron 90(2):245–260. https://doi.org/10.1016/j.neuron.2016.03.005
Min S-W, Sohn PD, Li Y, Devidze N, Johnson JR, Krogan NJ, Masliah E, Mok S-A et al (2018) SIRT1 deacetylates tau and reduces pathogenic tau spread in a mouse model of tauopathy. J Neurosci 38(15):3680–3688. https://doi.org/10.1523/jneurosci.2369-17.2018
Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD (2009) Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 106(34):14670–14675. https://doi.org/10.1073/pnas.0903563106
Liu Q, Wang X, Hu Y, Zhao JN, Huang CH, Li T, Zhang BG, He Y et al (2023) Acetylated tau exacerbates learning and memory impairment by disturbing with mitochondrial homeostasis. Redox Biol 62:102697. https://doi.org/10.1016/j.redox.2023.102697
Racz I, Tory K, Gallyas F Jr, Berente Z, Osz E, Jaszlits L, Bernath S, Sumegi B et al (2002) BGP-15 - a novel poly(ADP-ribose) polymerase inhibitor - protects against nephrotoxicity of cisplatin without compromising its antitumor activity. Biochem Pharmacol 63(6):1099–1111. https://doi.org/10.1016/s0006-2952(01)00935-2
Bárdos G, Móricz K, Jaszlits L, Rabloczky G, Tory K, Rácz I, Bernáth S, Sümegi B et al (2003) BGP-15, a hydroximic acid derivative, protects against cisplatin- or taxol-induced peripheral neuropathy in rats. Toxicol Appl Pharmacol 190(1):9–16. https://doi.org/10.1016/s0041-008x(03)00155-8
Brotzakis ZF, Lindstedt PR, Taylor RJ, Rinauro DJ, Gallagher NCT, Bernardes GJL, Vendruscolo M (2021) A structural ensemble of a tau-microtubule complex reveals regulatory tau phosphorylation and acetylation mechanisms. ACS Cent Sci 7(12):1986–1995. https://doi.org/10.1021/acscentsci.1c00585
Li L, Nguyen B, Mullapudi V, Saelices L, Joachimiak LA (2023) Disease-associated patterns of acetylation stabilize tau fibril formation. bioRxiv: the preprint server for biology. https://doi.org/10.1101/2023.01.10.523459
Hanger DP, Goniotaki D, Noble W (2019) Synaptic localisation of tau. Adv Exp Med Biol 1184:105–112. https://doi.org/10.1007/978-981-32-9358-8_9
Alquezar C, Schoch KM, Geier EG, Ramos EM, Scrivo A, Li KH, Argouarch AR, Mlynarski EE et al (2021) TSC1 loss increases risk for tauopathy by inducing tau acetylation and preventing tau clearance via chaperone-mediated autophagy. Sci Adv 7(45):eabg3897. https://doi.org/10.1126/sciadv.abg3897
Choi H, Kim HJ, Yang J, Chae S, Lee W, Chung S, Kim J, Choi H et al (2020) Acetylation changes tau interactome to degrade tau in Alzheimer’s disease animal and organoid models. Aging Cell 19(1):e13081. https://doi.org/10.1111/acel.13081
Herskovits AZ, Guarente L (2014) SIRT1 in neurodevelopment and brain senescence. Neuron 81(3):471–483. https://doi.org/10.1016/j.neuron.2014.01.028
Chen C, Zhou M, Ge Y, Wang X (2020) SIRT1 and aging related signaling pathways. Mech Ageing Dev 187:111215. https://doi.org/10.1016/j.mad.2020.111215
Wang L, Shi FX, Li N, Cao Y, Lei Y, Wang JZ, Tian Q, Zhou XW (2020) AMPK ameliorates tau acetylation and memory impairment through Sirt1. Mol Neurobiol 57(12):5011–5025. https://doi.org/10.1007/s12035-020-02079-x
Min SW, Sohn PD, Li Y, Devidze N, Johnson JR, Krogan NJ, Masliah E, Mok SA et al (2018) SIRT1 deacetylates tau and reduces pathogenic tau spread in a mouse model of tauopathy. J Neurosci 38(15):3680–3688. https://doi.org/10.1523/jneurosci.2369-17.2018
Alquezar C, Arya S, Kao AW (2020) Tau post-translational modifications: dynamic transformers of tau function, degradation, and aggregation. Front Neurol 11:595532. https://doi.org/10.3389/fneur.2020.595532
Thomas SN, Funk KE, Wan Y, Liao Z, Davies P, Kuret J, Yang AJ (2012) Dual modification of Alzheimer’s disease PHF-tau protein by lysine methylation and ubiquitylation: a mass spectrometry approach. Acta Neuropathol 123(1):105–117. https://doi.org/10.1007/s00401-011-0893-0
Xia Y, Bell BM, Giasson BI (2023) Tau lysine pseudomethylation regulates microtubule binding and enhances prion-like tau aggregation. Int J Mol Sci 24 (9). https://doi.org/10.3390/ijms24098286
Bell CG, Lowe R, Adams PD, Baccarelli AA, Beck S, Bell JT, Christensen BC, Gladyshev VN et al (2019) DNA methylation aging clocks: challenges and recommendations. Genome Biol 20(1):249. https://doi.org/10.1186/s13059-019-1824-y
Huseby CJ, Hoffman CN, Cooper GL, Cocuron JC, Alonso AP, Thomas SN, Yang AJ, Kuret J (2019) Quantification of tau protein lysine methylation in aging and Alzheimer's disease. J Alzheimer's Dis 71(3):979–991. https://doi.org/10.3233/JAD-190604
Balmik AA, Chinnathambi S (2021) Methylation as a key regulator of tau aggregation and neuronal health in Alzheimer’s disease. Cell Commun Signal 19(1):51. https://doi.org/10.1186/s12964-021-00732-z
Alonso ADC, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K (2001) Hyperphosphorylation induces self-assembly of τ into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A 98(12):6923–6928. https://doi.org/10.1073/pnas.121119298
Friedhoff P, Von Bergen M, Mandelkow EM, Davies P, Mandelkow E (1998) A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc Natl Acad Sci USA 95(26):15712–15717. https://doi.org/10.1073/pnas.95.26.15712
Shams H, Matsunaga A, Ma Q, Mofrad MRK, Didonna A (2022) Methylation at a conserved lysine residue modulates tau assembly and cellular functions. Mol Cell Neurosci 120:103707. https://doi.org/10.1016/j.mcn.2022.103707
Flores-Rodríguez P, Harrington CR, Wischik CM, Ibarra-Bracamontes V, Zarco N, Navarrete A, Martínez-Maldonado A, Guadarrama-Ortíz P et al (2019) Phospho-tau protein expression in the cell cycle of SH-SY5Y neuroblastoma cells: a morphological study. J Alzheimer’s Dis 71(2):631–645. https://doi.org/10.3233/JAD-190155
Ossola B, Zhao C, Compston A, Pluchino S, Franklin RJM, Spillantini MG (2016) Neuronal expression of pathological tau accelerates oligodendrocyte progenitor cell differentiation. Glia 64(3):457–471. https://doi.org/10.1002/glia.22940
Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX (2004) O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc Natl Acad Sci U S A 101(29):10804–10809. https://doi.org/10.1073/pnas.0400348101
Arnold CS, Johnson GV, Cole RN, Dong DL, Lee M, Hart GW (1996) The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine. J Biol Chem 271(46):28741–28744. https://doi.org/10.1074/jbc.271.46.28741
Yu CH, Si T, Wu WH, Hu J, Du JT, Zhao YF, Li YM (2008) O-GlcNAcylation modulates the self-aggregation ability of the fourth microtubule-binding repeat of tau. Biochem Biophys Res Commun 375(1):59–62. https://doi.org/10.1016/j.bbrc.2008.07.101
Yuzwa SA, Cheung AH, Okon M, McIntosh LP, Vocadlo DJ (2014) O-GlcNAc modification of tau directly inhibits its aggregation without perturbing the conformational properties of tau monomers. J Mol Biol 426(8):1736–1752. https://doi.org/10.1016/j.jmb.2014.01.004
Cripps D, Thomas SN, Jeng Y, Yang F, Davies P, Yang AJ (2006) Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J Biol Chem 281(16):10825–10838. https://doi.org/10.1074/jbc.M512786200
Munari F, Barracchia CG (2020) Semisynthetic modification of tau protein with di-ubiquitin chains for aggregation studies. 21 (12). https://doi.org/10.3390/ijms21124400
Puangmalai N, Sengupta U, Bhatt N, Gaikwad S, Montalbano M, Bhuyan A, Garcia S, McAllen S et al (2022) Lysine 63-linked ubiquitination of tau oligomers contributes to the pathogenesis of Alzheimer's disease. J Biol Chem 298(4):101766. https://doi.org/10.1016/j.jbc.2022.101766
Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E et al (2004) CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet 13(7):703–714. https://doi.org/10.1093/hmg/ddh083
Li L, Jiang Y, Wang JZ, Liu R, Wang X (2021) Tau ubiquitination in Alzheimer’s disease. Front Neurol 12:786353. https://doi.org/10.3389/fneur.2021.786353
Liu YH, Wei W, Yin J, Liu GP, Wang Q, Cao FY, Wang JZ (2009) Proteasome inhibition increases tau accumulation independent of phosphorylation. Neurobiol Aging 30(12):1949–1961. https://doi.org/10.1016/j.neurobiolaging.2008.02.012
Wang P, Joberty G, Buist A, Vanoosthuyse A, Stancu IC, Vasconcelos B, Pierrot N, Faelth-Savitski M et al (2017) Tau interactome mapping based identification of Otub1 as tau deubiquitinase involved in accumulation of pathological tau forms in vitro and in vivo. Acta Neuropathol 133(5):731–749. https://doi.org/10.1007/s00401-016-1663-9
Wei Z, Zeng K, Hu J, Li X, Huang F, Zhang B, Wang JZ, Liu R et al (2022) USP10 deubiquitinates tau, mediating its aggregation. Cell Death Dis 13(8):726. https://doi.org/10.1038/s41419-022-05170-4
Takamura H, Nakayama Y, Ito H, Katayama T, Fraser PE, Matsuzaki S (2022) SUMO1 modification of tau in progressive supranuclear palsy. Mol Neurobiol 59(7):4419–4435. https://doi.org/10.1007/s12035-022-02734-5
Luo HB, Xia YY, Shu XJ, Liu ZC, Feng Y, Liu XH, Yu G, Yin G et al (2014) SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc Natl Acad Sci U S A 111(46):16586–16591. https://doi.org/10.1073/pnas.1417548111
Lee EJ, Hyun SH, Chun J, Ahn HR, Kang SS (2007) Facilitation of SUMO (small ubiquitin-like modifier) modification at Tau 340-Lys residue (a microtubule-associated protein) through phosphorylation at 214-Ser residue. Integr Biosci 11(1):39–50
Takahashi K, Ishida M, Komano H, Takahashi H (2008) SUMO-1 immunoreactivity co-localizes with phospho-tau in APP transgenic mice but not in mutant tau transgenic mice. Neurosci Lett 441(1):90–93. https://doi.org/10.1016/j.neulet.2008.06.012
Wolfe MS (2012) The role of tau in neurodegenerative diseases and its potential as a therapeutic target. Scientifica (Cairo) 2012:796024. https://doi.org/10.6064/2012/796024
Amadoro G, Corsetti V, Sancesario GM, Lubrano A, Melchiorri G, Bernardini S, Calissano P, Sancesario G (2014) Cerebrospinal fluid levels of a 20–22 kDa NH2 fragment of human tau provide a novel neuronal injury biomarker in Alzheimer’s disease and other dementias. J Alzheimers Dis 42(1):211–226. https://doi.org/10.3233/JAD-140267
Barthélemy NR, Fenaille F, Hirtz C, Sergeant N, Schraen-Maschke S, Vialaret J, Buée L, Gabelle A et al (2016) Tau protein quantification in human cerebrospinal fluid by targeted mass spectrometry at high sequence coverage provides insights into its primary structure heterogeneity. J Proteome Res 15(2):667–676. https://doi.org/10.1021/acs.jproteome.5b01001
Meredith JE, Sankaranarayanan S, Guss V, Lanzetti AJ, Berisha F, Neely RJ, Slemmon JR, Portelius E et al (2013) Characterization of novel CSF tau and ptau biomarkers for Alzheimer’s disease. PLoS ONE 8(10):e76523. https://doi.org/10.1371/journal.pone.0076523
Barthélemy NR, Gabelle A, Hirtz C, Fenaille F, Sergeant N, Schraen-Maschke S, Vialaret J, Buée L et al (2016) Differential mass spectrometry profiles of tau protein in the cerebrospinal fluid of patients with Alzheimer’s disease, progressive supranuclear palsy, and dementia with Lewy bodies. J Alzheimer’s Dis 51(4):1033–1043. https://doi.org/10.3233/jad-150962
Russell CL, Mitra V, Hansson K, Blennow K, Gobom J, Zetterberg H, Hiltunen M, Ward M et al (2017) Comprehensive quantitative profiling of tau and phosphorylated tau peptides in cerebrospinal fluid by mass spectrometry provides new biomarker candidates. J Alzheimer’s Dis 55(1):303–313. https://doi.org/10.3233/jad-160633
Sato C, Barthélemy NR, Mawuenyega KG, Patterson BW, Gordon BA, Jockel-Balsarotti J, Sullivan M, Crisp MJ et al (2018) Tau kinetics in neurons and the human central nervous system. Neuron 97(6):1284-1298.e1287. https://doi.org/10.1016/j.neuron.2018.02.015
Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, LaFerla FM, Rohn TT et al (2004) Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest 114(1):121–130. https://doi.org/10.1172/jci20640
Chung CW, Hong YM, Song S, Woo HN, Choi YH, Rohn T, Jung YK (2003) Atypical role of proximal caspase-8 in truncated tau-induced neurite regression and neuronal cell death. Neurobiol Dis 14(3):557–566. https://doi.org/10.1016/j.nbd.2003.08.017
Quintanilla RA, Dolan PJ, Jin YN, Johnson GV (2012) Truncated tau and Aβ cooperatively impair mitochondria in primary neurons. Neurobiol Aging 33(3):619.e625-635. https://doi.org/10.1016/j.neurobiolaging.2011.02.007
Quintanilla RA, Matthews-Roberson TA, Dolan PJ, Johnson GV (2009) Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. J Biol Chem 284(28):18754–18766. https://doi.org/10.1074/jbc.M808908200
Smith BR, Nelson KM, Kemper LJ, Leinonen-Wright K, Petersen A, Keene CD, Ashe KH (2019) A soluble tau fragment generated by caspase-2 is associated with dementia in Lewy body disease. Acta Neuropathol Commun 7(1):124. https://doi.org/10.1186/s40478-019-0765-8
Zhao X, Kotilinek LA, Smith B, Hlynialuk C, Zahs K, Ramsden M, Cleary J, Ashe KH (2016) Caspase-2 cleavage of tau reversibly impairs memory. Nat Med 22(11):1268–1276. https://doi.org/10.1038/nm.4199
Lang AE, Riherd Methner DN, Ferreira A (2014) Neuronal degeneration, synaptic defects, and behavioral abnormalities in tau(4)(5)(-)(2)(3)(0) transgenic mice. Neuroscience 275:322–339. https://doi.org/10.1016/j.neuroscience.2014.06.017
Afreen S, Ferreira A (2019) Altered cytoskeletal composition and delayed neurite elongation in tau(45–230)-expressing hippocampal neurons. Neuroscience 412:1–15. https://doi.org/10.1016/j.neuroscience.2019.05.046
Cicognola C, Brinkmalm G, Wahlgren J, Portelius E, Gobom J, Cullen NC, Hansson O, Parnetti L et al (2019) Novel tau fragments in cerebrospinal fluid: relation to tangle pathology and cognitive decline in Alzheimer’s disease. Acta Neuropathol 137(2):279–296. https://doi.org/10.1007/s00401-018-1948-2
Borroni B, Premi E, Agosti C, Alberici A, Cerini C, Archetti S, Lanari A, Paghera B et al (2011) CSF Alzheimer’s disease-like pattern in corticobasal syndrome: evidence for a distinct disorder. J Neurol Neurosurg Psychiatry 82(8):834–838. https://doi.org/10.1136/jnnp.2010.221853
Cicognola C, Hansson O, Scheltens P, Kvartsberg H, Zetterberg H, Teunissen CE, Blennow K (2021) Cerebrospinal fluid N-224 tau helps discriminate Alzheimer’s disease from subjective cognitive decline and other dementias. Alzheimer’s research & therapy 13(1):38. https://doi.org/10.1186/s13195-020-00756-6
Zhang Z, Li XG, Wang ZH, Song M, Yu SP, Kang SS, Liu X, Zhang Z et al (2021) δ-Secretase-cleaved tau stimulates Aβ production via upregulating STAT1-BACE1 signaling in Alzheimer’s disease. Mol Psychiatry 26(2):586–603. https://doi.org/10.1038/s41380-018-0286-z
Yao Y, Kang SS, Xia Y, Wang ZH, Liu X, Muller T, Sun YE, Ye K (2021) A delta-secretase-truncated APP fragment activates CEBPB, mediating Alzheimer’s disease pathologies. Brain 144(6):1833–1852. https://doi.org/10.1093/brain/awab062
Li L, Jiang Y, Wu G, Mahaman YAR, Ke D, Wang Q, Zhang B, Wang JZ et al (2022) Phosphorylation of truncated tau promotes abnormal native tau pathology and neurodegeneration. Mol Neurobiol 59(10):6183–6199. https://doi.org/10.1007/s12035-022-02972-7
Vogels T, Vargová G, Brezováková V, Quint WH, Hromádka T (2020) Viral delivery of non-mutated human truncated tau to neurons recapitulates key features of human tauopathy in wild-type mice. J Alzheimer’s Dis 77(2):551–568. https://doi.org/10.3233/jad-200047
Behrendt A, Bichmann M, Ercan-Herbst E, Haberkant P, Schondorf DC, Wolf M, Fahim SA, Murolo E et al (2019) Asparagine endopeptidase cleaves tau at N167 after uptake into microglia. Neurobiol Dis 130:104518. https://doi.org/10.1016/J.Nbd.2019.104518
Borreca A, Latina V, Corsetti V, Middei S, Piccinin S, Della Valle F, Bussani R, Ammassari-Teule M et al (2018) AD-related N-terminal truncated tau is sufficient to recapitulate in vivo the early perturbations of human neuropathology: implications for immunotherapy. Mol Neurobiol 55(10):8124–8153. https://doi.org/10.1007/s12035-018-0974-3
Florenzano F, Veronica C, Ciasca G, Ciotti MT, Pittaluga A, Olivero G, Feligioni M, Iannuzzi F et al (2017) Extracellular truncated tau causes early presynaptic dysfunction associated with Alzheimer's disease and other tauopathies. Oncotarget 8(39):64745–64778. https://doi.org/10.18632/oncotarget.17371
Amadoro G, Corsetti V, Stringaro A, Colone M, D'Aguanno S, Meli G, Ciotti M, Sancesario G et al (2010) A NH2 tau fragment targets neuronal mitochondria at AD synapses: possible implications for neurodegeneration. J Alzheimers Dis 21(2):445–470. https://doi.org/10.3233/JAD-2010-100120
Amadoro G, Corsetti V, Florenzano F, Atlante A, Ciotti MT, Mongiardi MP, Bussani R, Nicolin V et al (2014) AD-linked, toxic NH2 human tau affects the quality control of mitochondria in neurons. Neurobiol Dis 62:489–507. https://doi.org/10.1016/j.nbd.2013.10.018
Atlante A, Amadoro G, Bobba A, de Bari L, Corsetti V, Pappalardo G, Marra E, Calissano P et al (2008) A peptide containing residues 26–44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim Biophys Acta - Bioenerget 1777(10):1289–1300. https://doi.org/10.1016/j.bbabio.2008.07.004
Amadoro G, Corsetti V, Atlante A, Florenzano F, Capsoni S, Bussani R, Mercanti D, Calissano P (2012) Interaction between NH2-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol Aging 33(4):833.e831-833.e825. https://doi.org/10.1016/j.neurobiolaging.2011.08.001
Al-Hilaly YK, Hurt C, Rickard JE, Harrington CR, Storey JMD, Wischik CM, Serpell LC, Siemer AB (2022) Solid-state NMR of paired helical filaments formed by the core tau fragment tau(297–391). Front Neurosci 16:988074. https://doi.org/10.3389/fnins.2022.988074
Gu J, Xu W, Jin N, Li L, Zhou Y, Chu D, Gong CX, Iqbal K et al (2020) Truncation of tau selectively facilitates its pathological activities. J Biol Chem 295(40):13812–13828. https://doi.org/10.1074/jbc.RA120.012587
Van Opdenbosch N, Lamkanfi M (2019) Caspases in cell death, inflammation, and disease. Immunity 50(6):1352–1364. https://doi.org/10.1016/j.immuni.2019.05.020
McKenzie BA, Fernandes JP, Doan MAL, Schmitt LM, Branton WG, Power C (2020) Activation of the executioner caspases-3 and -7 promotes microglial pyroptosis in models of multiple sclerosis. J Neuroinflamm 17(1):253. https://doi.org/10.1186/s12974-020-01902-5
Boucher D, Blais V, Denault JB (2012) Caspase-7 uses an exosite to promote poly(ADP ribose) polymerase 1 proteolysis. Proc Natl Acad Sci U S A 109(15):5669–5674. https://doi.org/10.1073/pnas.1200934109
Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, Smulson M (1999) Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem 274(33):22932–22940. https://doi.org/10.1074/jbc.274.33.22932
Axelsen TM, Bager C, Bihlet A, Karsdal MA, Henriksen K, Tang MHE (2023) A genetic validation of the neurodegeneration biomarkers tau-A and tau-C - a Mendelian randomization study. J Prevent Alzheimer's Dis 10(3):536–542. https://doi.org/10.14283/jpad.2023.36
Ruchaud S, Korfali N, Villa P, Kottke TJ, Dingwall C, Kaufmann SH, Earnshaw WC (2002) Caspase-6 gene disruption reveals a requirement for lamin A cleavage in apoptotic chromatin condensation. EMBO J 21(8):1967–1977. https://doi.org/10.1093/emboj/21.8.1967
Calvo-Rodriguez M, Bacskai BJ (2021) Mitochondria and calcium in Alzheimer’s disease: from cell signaling to neuronal cell death. Trends Neurosci 44(2):136–151. https://doi.org/10.1016/j.tins.2020.10.004
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T et al (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14(4):388–405. https://doi.org/10.1016/s1474-4422(15)70016-5
De Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT (2010) Caspase activation precedes and leads to tangles. Nature 464(7292):1201–1204. https://doi.org/10.1038/nature08890
Avila J (2010) Alzheimer disease: caspases first. Nat Rev Neurol 6(11):587–588. https://doi.org/10.1038/nrneurol.2010.157
Noël A, Foveau B, LeBlanc AC (2021) Caspase-6-cleaved Tau fails to induce Tau hyperphosphorylation and aggregation, neurodegeneration, glial inflammation, and cognitive deficits. Cell Death Dis 12(3):227. https://doi.org/10.1038/s41419-021-03506-0
Ramcharitar J, Afonso VM, Albrecht S, Bennett DA, LeBlanc AC (2013) Caspase-6 activity predicts lower episodic memory ability in aged individuals. Neurobiol Aging 34(7):1815–1824. https://doi.org/10.1016/j.neurobiolaging.2013.01.007
Ramcharitar J, Albrecht S, Afonso VM, Kaushal V, Bennett DA, Leblanc AC (2013) Cerebrospinal fluid tau cleaved by caspase-6 reflects brain levels and cognition in aging and Alzheimer disease. J Neuropathol Exp Neurol 72(9):824–832. https://doi.org/10.1097/NEN.0b013e3182a0a39f
Inekci D, Henriksen K, Linemann T, Karsdal MA, Habib A, Bisgaard C, Eriksen FB, Vilholm OJ (2015) Serum fragments of tau for the differential diagnosis of Alzheimer’s disease. Curr Alzheimer Res 12(9):829–836. https://doi.org/10.2174/1567205012666150710111211
Perez MJ, Vergara-Pulgar K, Jara C, Cabezas-Opazo F, Quintanilla RA (2018) Caspase-cleaved tau impairs mitochondrial dynamics in Alzheimer’s disease. Mol Neurobiol 55(2):1004–1018. https://doi.org/10.1007/s12035-017-0385-x
Quintanilla RA, Tapia-Monsalves C, Vergara EH, Perez MJ, Aranguiz A (2020) Truncated tau induces mitochondrial transport failure through the impairment of TRAK2 protein and bioenergetics decline in neuronal cells. Front Cell Neurosci 14:175. https://doi.org/10.3389/Fncel.2020.00175
Nicholls SB, DeVos SL, Commins C, Nobuhara C, Bennett RE, Corjuc DL, Maury E, Eftekharzadeh B et al (2017) Characterization of TauC3 antibody and demonstration of its potential to block tau propagation. PLoS ONE 12(5):e0177914. https://doi.org/10.1371/journal.pone.0177914
Sandusky-Beltran LA, Kovalenko A, Ma C, Calahatian JIT, Placides DS, Watler MD, Hunt JB, Darling AL et al (2019) Spermidine/spermine-N1-acetyltransferase ablation impacts tauopathy-induced polyamine stress response. Alzheimer’s Res Ther 11(1):58. https://doi.org/10.1186/s13195-019-0507-y
Biundo F, d'Abramo C, Tambini MD, Zhang H, Del Prete D, Vitale F, Giliberto L, Arancio O et al (2017) Abolishing tau cleavage by caspases at Aspartate421 causes memory/synaptic plasticity deficits and pre-pathological tau alterations. Transl Psychiatry 7(8):e1198. https://doi.org/10.1038/tp.2017.165
Loon A, Zamudio F, Sanneh A, Brown B, Smeltzer S, Brownlow ML, Quadri Z, Peters M et al (2022) Accumulation of C-terminal cleaved tau is distinctly associated with cognitive deficits, synaptic plasticity impairment, and neurodegeneration in aged mice. Geroscience 44(1):173–194. https://doi.org/10.1007/s11357-021-00408-z
Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW (2005) Tau, tangles, and Alzheimer’s disease. Biochim Biophys Acta (BBA) - Molec Basis Dis 1739 (2):216–223. https://doi.org/10.1016/j.bbadis.2004.08.014
Fasulo L, Ugolini G, Cattaneo A (2005) Apoptotic effect of caspase-3 cleaved tau in hippocampal neurons and its potentiation by tau FTDP-mutation N279K. J Alzheimer's Dis 7(1):3–13. https://doi.org/10.3233/jad-2005-7102
Opland CK, Bryan MR, Harris B, McGillion-Moore J, Tian X, Chen Y, Itano MS, Diering GH et al (2023) Activity-dependent tau cleavage by caspase-3 promotes neuronal dysfunction and synaptotoxicity. iScience 26(6):106905. https://doi.org/10.1016/j.isci.2023.106905
Tapia-Monsalves C, Olesen MA, Villavicencio-Tejo F, Quintanilla RA (2023) Cyclosporine A (CsA) prevents synaptic impairment caused by truncated tau by caspase-3. Mol Cell Neurosci 125:103861. https://doi.org/10.1016/j.mcn.2023.103861
Liu P, Smith BR, Huang ES, Mahesh A, Vonsattel JPG, Petersen AJ, Gomez-Pastor R, Ashe KH (2019) A soluble truncated tau species related to cognitive dysfunction and caspase-2 is elevated in the brain of Huntington’s disease patients. Acta Neuropathol Commun 7(1):111. https://doi.org/10.1186/s40478-019-0764-9
Liu P, Smith BR, Montonye ML, Kemper LJ, Leinonen-Wright K, Nelson KM, Higgins L, Guerrero CR et al (2020) A soluble truncated tau species related to cognitive dysfunction is elevated in the brain of cognitively impaired human individuals. Sci Rep 10(1):3869. https://doi.org/10.1038/s41598-020-60777-x
Singh G, Liu P, Yao KR, Strasser JM, Hlynialuk C, Leinonen-Wright K, Teravskis PJ, Choquette JM, Ikramuddin J, Bresinsky M, Nelson KM, Liao D, Ashe KH, Walters MA, Pockes S (2022) Caspase-2 inhibitor blocks tau truncation and restores excitatory neurotransmission in neurons modeling FTDP-17 tauopathy. ACS Chem Neurosci. https://doi.org/10.1021/acschemneuro.2c00100
Steuer EL, Kemper LJ, Hlynialuk CJW, Leinonen-Wright K, Montonye ML, Lapcinski IP, Forster CL, Ashe KH et al (2022) Blocking site-specific cleavage of human tau delays progression of disease-related phenotypes in genetically matched tau-transgenic mice modeling frontotemporal dementia. J Neurosci 42(23):4737–4754. https://doi.org/10.1523/JNEUROSCI.0543-22.2022
Goll DE, Thompson VF, Li H, Wei W, Cong J (2003) The calpain system. Physiol Rev 83(3):731–801. https://doi.org/10.1152/physrev.00029.2002
Khan H, Garg N, Singh TG, Kaur A, Thapa K (2022) Calpain inhibitors as potential therapeutic modulators in neurodegenerative diseases. Neurochem Res 47(5):1125–1149. https://doi.org/10.1007/s11064-021-03521-9
Metwally E, Zhao G, Zhang YQ (2021) The calcium-dependent protease calpain in neuronal remodeling and neurodegeneration. Trends Neurosci 44(9):741–752. https://doi.org/10.1016/j.tins.2021.07.003
Park S-Y, Ferreira A (2005) The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J Neurosci 25(22):5365–5375. https://doi.org/10.1523/jneurosci.1125-05.2005
Garg S, Timm T, Mandelkow EM, Mandelkow E, Wang Y (2011) Cleavage of tau by calpain in Alzheimer’s disease: the quest for the toxic 17 kD fragment. Neurobiol Aging 32(1):1–14. https://doi.org/10.1016/j.neurobiolaging.2010.09.008
Companys-Alemany J, Turcu AL, Schneider M, Muller CE, Vazquez S, Grinan-Ferre C, Pallas M (2022) NMDA receptor antagonists reduce amyloid-beta deposition by modulating calpain-1 signaling and autophagy, rescuing cognitive impairment in 5XFAD mice. Cell Mol Life Sci 79(8):408. https://doi.org/10.1007/s00018-022-04438-4
Yang LS, Ksiezak-Reding H (1995) Calpain-induced proteolysis of normal human tau and tau associated with paired helical filaments. Eur J Biochem 233(1):9–17. https://doi.org/10.1111/j.1432-1033.1995.009_1.x
Ferreira A, Bigio EH (2011) Calpain-mediated tau cleavage: a mechanism leading to neurodegeneration shared by multiple tauopathies. Mol Med 17(7–8):676–685. https://doi.org/10.2119/molmed.2010.00220
Feinstein HE, Benbow SJ, LaPointe NE, Patel N, Ramachandran S, Do TD, Gaylord MR, Huskey NE et al (2016) Oligomerization of the microtubule-associated protein tau is mediated by its N-terminal sequences: implications for normal and pathological tau action. J Neurochem 137(6):939–954. https://doi.org/10.1111/jnc.13604
Afreen S, Ferreira A (2022) The formation of small aggregates contributes to the neurotoxic effects of tau(45–230). Neurochem Int 152:105252. https://doi.org/10.1016/j.neuint.2021.105252
Afreen S, Riherd Methner DN, Ferreira A (2017) Tau(45–230) association with the cytoskeleton and membrane-bound organelles: functional implications in neurodegeneration. Neuroscience 362:104–117. https://doi.org/10.1016/j.neuroscience.2017.08.026
Park SY, Ferreira A (2005) The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J Neurosci 25(22):5365–5375. https://doi.org/10.1523/JNEUROSCI.1125-05.2005
Reinecke JB, DeVos SL, McGrath JP, Shepard AM, Goncharoff DK, Tait DN, Fleming SR, Vincent MP et al (2011) Implicating calpain in tau-mediated toxicity in vivo. PLoS ONE 6(8):e23865. https://doi.org/10.1371/journal.pone.0023865
Chen YD, Huang PY, Chiang CS, Huang YS, Tang SC (2021) Generation and role of calpain-cleaved 17-kDa tau fragment in acute ischemic stroke. Mol Neurobiol 58(11):5814–5825. https://doi.org/10.1007/s12035-021-02519-2
Cicognola C, Satir TM, Brinkmalm G, Matečko-Burmann I, Agholme L, Bergström P, Becker B, Zetterberg H et al (2020) Tauopathy-associated tau fragment ending at amino acid 224 is generated by calpain-2 cleavage. J Alzheimer's Dis 74(4):1143–1156. https://doi.org/10.3233/jad-191130
Amadoro G, Ciotti MT, Costanzi M, Cestari V, Calissano P, Canu N (2006) NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc Natl Acad Sci U S A 103(8):2892–2897. https://doi.org/10.1073/pnas.0511065103
Corsetti V, Florenzano F, Atlante A, Bobba A, Ciotti MT, Natale F, Della Valle F, Borreca A et al (2015) NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: implications in Alzheimer’s disease. Hum Mol Genet 24(11):3058–3081. https://doi.org/10.1093/hmg/ddv059
Corsetti V, Amadoro G, Gentile A, Capsoni S, Ciotti MT, Cencioni MT, Atlante A, Canu N et al (2008) Identification of a caspase-derived N-terminal tau fragment in cellular and animal Alzheimer’s disease models. Mol Cell Neurosci 38(3):381–392. https://doi.org/10.1016/j.mcn.2008.03.011
Amadoro G, Serafino AL, Barbato C, Ciotti MT, Sacco A, Calissano P, Canu N (2004) Role of N-terminal tau domain integrity on the survival of cerebellar granule neurons. Cell Death Differ 11(2):217–230. https://doi.org/10.1038/sj.cdd.4401314
Corsetti V, Florenzano F, Atlante A, Bobba A, Ciotti MT, Natale F, Della Valle F, Borreca A et al (2014) NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: implications in Alzheimer’s disease. Hum Mol Genet 24(11):3058–3081. https://doi.org/10.1093/hmg/ddv059
Latina V, Giacovazzo G, Cordella F, Balzamino BO, Micera A, Varano M, Marchetti C, Malerba F et al. (2021) Systemic delivery of a specific antibody targeting the pathological N-terminal truncated tau peptide reduces retinal degeneration in a mouse model of Alzheimer’s disease. Acta Neuropathol Commun 9 (1). https://doi.org/10.1186/s40478-021-01138-1
Corsetti V, Borreca A, Latina V, Giacovazzo G, Pignataro A, Krashia P, Natale F, Cocco S, et al. (2020) Passive immunotherapy for N-truncated tau ameliorates the cognitive deficits in two mouse Alzheimer’s disease models. Brain Commun 2 (1). https://doi.org/10.1093/braincomms/fcaa039
Latina V, De Introna M, Caligiuri C, Loviglio A, Florio R, La Regina F, Pignataro A, Ammassari-Teule M, et al. (2023) Immunotherapy with cleavage-specific 12A12mAb reduces the tau cleavage in visual cortex and improves visuo-spatial recognition memory in Tg2576 AD mouse model. Pharmaceutics 15 (2). https://doi.org/10.3390/pharmaceutics15020509
Latina V, Atlante A, Malerba F, La Regina F, Balzamino BO, Micera A, Pignataro A, Stigliano E, et al. (2023) The cleavage-specific tau 12A12mAb exerts an anti-amyloidogenic action by modulating the endocytic and bioenergetic pathways in Alzheimer’s disease mouse model. Int J Mol Sci 24 (11). https://doi.org/10.3390/ijms24119683
Zhang Z, Song M, Liu X, Kang SS, Kwon IS, Duong DM, Seyfried NT, Hu WT, Liu Z, Wang JZ, Cheng L, Sun YE, Yu SP, Levey AI, Ye K (2014) Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat Med 20(11):1254–1262. https://doi.org/10.1038/nm.3700
Antoniou AN, Blackwood S-L, Mazzeo D, Watts C (2000) Control of antigen presentation by a single protease cleavage site. Immunity 12(4):391–398. https://doi.org/10.1016/S1074-7613(00)80191-0
Zhao L, Hua T, Crowley C, Ru H, Ni X, Shaw N, Jiao L, Ding W et al (2014) Structural analysis of asparaginyl endopeptidase reveals the activation mechanism and a reversible intermediate maturation stage. Cell Res 24(3):344–358. https://doi.org/10.1038/cr.2014.4
Wang ZH, Liu P, Liu X, Yu SP, Wang JZ, Ye K (2018) Delta-secretase (AEP) mediates tau-splicing imbalance and accelerates cognitive decline in tauopathies. 215 (12):3038–3056. https://doi.org/10.1084/jem.20180539
Wu Z, Zhu R, Yu Y, Wang J, Hu X, Xu W, Ren Y, Li C et al (2023) Spinal cord injury-activated C/EBPbeta-AEP axis mediates cognitive impairment through APP C586/Tau N368 fragments spreading. Prog Neurobiol 227:102467. https://doi.org/10.1016/j.pneurobio.2023.102467
Kang SS, Ahn EH, Zhang Z, Liu X, Manfredsson FP, Sandoval IM, Dhakal S, Iuvone PM, et al. (2018) alpha-Synuclein stimulation of monoamine oxidase-B and legumain protease mediates the pathology of Parkinson’s disease. EMBO J 37 (12). https://doi.org/10.15252/embj.201798878
Chen G, Ahn EH, Kang SS, Xia Y, Liu X, Zhang Z, Ye K (2022) UNC5C receptor proteolytic cleavage by active AEP promotes dopaminergic neuronal degeneration in Parkinson’s disease. Advanced Sci (Weinheim, Baden-Wurttemberg, Germany) 9 (7):e2103396. https://doi.org/10.1002/advs.202103396
Schlegel K, Awwad K, Heym RG, Holzinger D, Doell A, Barghorn S, Jahn TR, Klein C, et al. (2019) N368-Tau fragments generated by legumain are detected only in trace amount in the insoluble tau aggregates isolated from AD brain. Acta Neuropathol Commun 7 (1). https://doi.org/10.1186/s40478-019-0831-2
Blennow K, Chen C, Cicognola C, Wildsmith KR, Manser PT, Bohorquez SMS, Zhang Z, Xie B et al (2020) Cerebrospinal fluid tau fragment correlates with tau PET: a candidate biomarker for tangle pathology. Brain 143(2):650–660. https://doi.org/10.1093/brain/awz346
Zhang Y, Zhang Y, Aman Y, Ng CT, Chau W-H, Zhang Z, Yue M, Bohm C et al (2021) Amyloid-beta toxicity modulates tau phosphorylation through the PAX6 signalling pathway. Brain 144:2759–2770. https://doi.org/10.1093/brain/awab134
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82(4):239–259. https://doi.org/10.1007/bf00308809
Mudher A, Colin M, Dujardin S, Medina M, Dewachter I, AlaviNaini SM, Mandelkow EM, Mandelkow E et al (2017) What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol Commun 5(1):99. https://doi.org/10.1186/s40478-017-0488-7
Irwin DJ, Abrams JY, Schonberger LB, Leschek EW, Mills JL, Lee VM, Trojanowski JQ (2013) Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol 70(4):462–468. https://doi.org/10.1001/jamaneurol.2013.1933
Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI (2012) Trans-cellular propagation of tau aggregation by fibrillar species. J Biol Chem 287(23):19440–19451. https://doi.org/10.1074/jbc.M112.346072
Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC et al (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82(6):1271–1288. https://doi.org/10.1016/j.neuron.2014.04.047
Lonati E, Sala G, Tresoldi V, Coco S, Salerno D, Milani C, Losurdo M, Farina F et al (2018) Ischemic conditions affect rerouting of tau protein levels: evidences for alteration in tau processing and secretion in hippocampal neurons. J Molec Neurosci 66(4):604–616. https://doi.org/10.1007/s12031-018-1199-7
Kang S, Son SM, Baik SH, Yang J, Mook-Jung I (2019) Autophagy-mediated secretory pathway is responsible for both normal and pathological tau in neurons. J Alzheimer’s Dis 70(3):667–680. https://doi.org/10.3233/jad-190180
Pernègre C, Duquette A, Leclerc N (2019) Tau secretion: good and bad for neurons. Front Neurosci 13:649. https://doi.org/10.3389/fnins.2019.00649
Bright J, Hussain S, Dang V, Wright S, Cooper B, Byun T, Ramos C, Singh A et al (2015) Human secreted tau increases amyloid-beta production. Neurobiol Aging 36(2):693–709. https://doi.org/10.1016/j.neurobiolaging.2014.09.007
Katsinelos T, Zeitler M, Dimou E, Karakatsani A, Muller HM, Nachman E, Steringer JP, de Almodovar CR et al (2018) Unconventional secretion mediates the trans-cellular spreading of tau. Cell Rep 23(7):2039–2055. https://doi.org/10.1016/j.celrep.2018.04.056
Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM (2013) Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 33(3):1024–1037. https://doi.org/10.1523/jneurosci.2642-12.2013
Hamano T, Gendron TF, Causevic E, Yen SH, Lin WL, Isidoro C, DeTure M, Ko LW (2008) Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci 27(5):1119–1130. https://doi.org/10.1111/j.1460-9568.2008.06084.x
Rubinsztein DC (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443(7113):780–786. https://doi.org/10.1038/nature05291
Bondulich MK, Guo T, Meehan C, Manion J, Martin TR, Mitchell JC, Hortobagyi T, Yankova N et al (2016) Tauopathy induced by low level expression of a human brain-derived tau fragment in mice is rescued by phenylbutyrate. Brain 139:2290–2306. https://doi.org/10.1093/brain/aww137
Fraldi A, Klein AD, Medina DL, Settembre C (2016) Brain disorders due to lysosomal dysfunction. Annu Rev Neurosci 39(39):277–295. https://doi.org/10.1146/annurev-neuro-070815-014031
Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJA, Cooper JM (2011) Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis 42(3):360–367. https://doi.org/10.1016/j.nbd.2011.01.029
Eitan E, Suire C, Zhang S, Mattson MP (2016) Impact of lysosome status on extracellular vesicle content and release. Ageing Res Rev 32:65–74. https://doi.org/10.1016/j.arr.2016.05.001
Yan ML, Zheng TT (2021) Role of the endolysosomal pathway and exosome release in tau propagation. Neurochem Int 145:104988. https://doi.org/10.1016/J.Neuint.2021.104988
Medina M, Avila J (2014) The role of extracellular tau in the spreading of neurofibrillary pathology. Front Cell Neurosci 8:113. https://doi.org/10.3389/fncel.2014.00113
Fiandaca MS, Kapogiannis D, Mapstone M, Boxer A, Eitan E, Schwartz JB, Abner EL, Petersen RC et al (2015) Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: a case-control study. Alzheimers & Dementia 11(6):600–607. https://doi.org/10.1016/j.jalz.2014.06.008
Wang Y, Balaji V, Kaniyappan S, Kruger L, Irsen S, Tepper K, Chandupatla R, Maetzler W et al (2017) The release and trans-synaptic transmission of tau via exosomes. Mol Neurodegener 12:5. https://doi.org/10.1186/S13024-016-0143-Y
Podvin S, Jones A, Liu Q, Aulston B, Ransom L, Ames J, Shen G, Lietz CB et al (2020) Dysregulation of exosome cargo by mutant tau expressed in human-induced pluripotent stem cell (iPSC) neurons revealed by proteomics analyses. Mol Cell Proteom 19(6):1017–1034. https://doi.org/10.1074/mcp.RA120.002079
Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O et al (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18(11):1584–1593. https://doi.org/10.1038/nn.4132
Ruan Z, Delpech JC, Kalavai SV, Van Enoo AA, Hu JQ, Ikezu S, Ikezu T (2020) P2RX7 inhibitor suppresses exosome secretion and disease phenotype in P301S tau transgenic mice. Mol Neurodegener 15(1):47. https://doi.org/10.1186/S13024-020-00396-2
Perez M, Cuadros R, Hernandez F, Avila J (2016) Secretion of full-length tau or tau fragments in a cell culture model. Neurosci Lett 634:63–69. https://doi.org/10.1016/j.neulet.2016.09.026
Kim W, Lee S, Hall GF (2010) Secretion of human tau fragments resembling CSF-tau in Alzheimer’s disease is modulated by the presence of the exon 2 insert. FEBS Lett 584(14):3085–3088. https://doi.org/10.1016/j.febslet.2010.05.042
Plouffe V, Mohamed NV, Rivest-McGraw J, Bertrand J, Lauzon M, Leclerc N (2012) Hyperphosphorylation and cleavage at D421 enhance tau secretion. Plos One 7 (5):e36873 https://doi.org/10.1371/journal.pone.0036873
Xu Y, Du SQ, Marsh JA, Horie K, Sato C, Ballabio A, Karch CM, Holtzman DM et al (2021) TFEB regulates lysosomal exocytosis of tau and its loss of function exacerbates tau pathology and spreading. Mol Psychiatry 26(10):5925–5939. https://doi.org/10.1038/s41380-020-0738-0
Pilliod J, Gelinas-Faucher M, Leclerc N (2022) Unconventional secretion of tau by VAMP8 impacts its intra- and extracellular cleavage. Front Cell Dev Biol 10:912118. https://doi.org/10.3389/Fcell.2022.912118
Cicognola C, Brinkmalm G, Wahlgren J, Portelius E, Gobom J, Cullen NC, Hansson O, Parnetti L et al (2019) Novel tau fragments in cerebrospinal fluid: relation to tangle pathology and cognitive decline in Alzheimer’s disease. Acta Neuropathol 137(2):279–296. https://doi.org/10.1007/s00401-018-1948-2
Dixson AC, Dawson TR, Di Vizio D, Weaver AM (2023) Context-specific regulation of extracellular vesicle biogenesis and cargo selection. Nat Rev Mol Cell Bio. https://doi.org/10.1038/s41580-023-00576-0
Acknowledgements
Not applicable
Funding
This work was supported by grants from the National Natural Science Foundation of China [82071440, 31171028].
Author information
Authors and Affiliations
Contributions
H-LL had the idea of this review. JY and NS performed the literature search and wrote the original manuscript. JY and NS contributed equally to this work. JS, YY, and H-LL critically revised the work.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, J., Shen, N., Shen, J. et al. Complicated Role of Post-translational Modification and Protease-Cleaved Fragments of Tau in Alzheimer’s Disease and Other Tauopathies. Mol Neurobiol 61, 4712–4731 (2024). https://doi.org/10.1007/s12035-023-03867-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03867-x