
Abstract
Based on experimental observations about structure and dynamics of the reactive surface of the M1 phase some considerations are made about the nature and size of active sites. In analyzing the stoichiometry of the reactions following activation of propane it occurs that for dehydrogenation small sites are desirable being just sufficient to re-activate oxygen without kinetic hindrance. For deeper oxidation to acrylic acid (AA) the sites should be larger to accommodate all redox equivalents and oxygen species required for one transformation. A qualitative model for size and composition of the active site is made. It is likely that the active site consists on a VxOy species of strictly 2-D nature. This follows the structural suggestion for the active site on the a–b-plane of the M1 structure. The role of Te in moderating the active site is discussed. The suggestions are discussed in comparing requirements for oxidative dehydrogenation of propane (ODP) with those for AA synthesis.
Similar content being viewed by others
1 Introduction
The synthesis of functionalized small molecules from alkanes is an important area of research to prepare for the expected changeover from oil-based feedstock. Natural gas and in a more distant future alkanes from solar hydrogen processing may serve as basis for the chemical industry. Olefines are and will remain key building blocks for the industry. Thus the on-purpose generation of these molecules (ethene, propene and to a lesser extent butenes) is an important and long-studied topic. Its present technical realization is limited as the performance data of dehydrogenation and oxidative dehydrogenation processes are still [1] sub-optimal in comparison of petrochemical processing of crude oil. A smaller yet important share of reactions involves deeper oxidation of alkanes to oxygenates and also to acylonitrile [2]. One challenge in these reactions lies in finding environmentally benign integrated processes from alkanes to the oxygenates. Butane to maleic anhydride is the trendsetter, the synthesis of acrylic acid, of ethylene epoxide and adipic acid are further examples [3] for such processes.
The science required to tackle these challenges is largely confined to empirical [4] knowledge. Surface-science approaches work only with activated molecules such as methanol [5, 6] and theory [5, 7, 8] is confined to simplified yet highly valuable model systems [6, 9, 10]. It is still a great challenge to construct concepts of active sites [11, 12] for selective oxidation that are based upon experimental observations [13] of reacting surfaces.
For the case of propane oxidation a proposal has been made [14, 15] that can well serve as discussion basis. A highlight in the development process [16–22] of selective oxidation catalysis was the discovery of the orthorhombic M1 phase MoVNbTeOx. This truly complex mixed oxide is a versatile catalyst for several selective oxidation processes of C3 [17, 19, 23–26] and C2 [27, 28] feedstock. It works well as crystalline single phase. This fact renders the system suitable for trying understanding facts about active sites in selective oxidation. The structural and chemical complexity is not optimal for facile analysis, but the advantage of a single highly crystalline phase being the true catalyst for a complicated reaction is of invaluable advantage. The integrity of the system can be verified with various in situ techniques [29, 30] and the formation of activity is related to the termination of a stable solid and not correlated [31] to the formation of a 3-D disordered (“amorphous”) phase.
Although reports exist [23, 32] on attempts supported by high-throughput experimentation to improve the function of the M1 phase and to benefit from a suspected synergy [33–35] with the related hexagonal M2 [36, 37] phase, it is established [38] that a stable and long-term performance can also be achieved with the bare single [39] M1 phase. This gives hope that a detailed understanding of the mode of operation can be obtained based upon accurate oxide crystallography [40] forming the basis of a detailed understanding [41] of the nature of the active site.
It is not clear as yet [42–44] that the active sites of the M1 phase are exclusively located on the (001) basal faces (“a–b plane”) of the needle morphology [42] but there is strong and general belief that the (001) face contains the motif of the active site that may exist in the termination structure of the whole surface of the material. For this reason the exact crystallographic analysis [40, 45] of the basal plane structure not only in terms of its connectivity but also in terms of its cation ordering statistics [45–47] is of particular relevance. The high level of structural definition has inspired theoretical work [11, 32, 41, 48] aiming at a detailed quantum chemical explanation [11, 12, 41] of the mode of operation; such work is only possible with a reasonable knowledge about the local chemistry and with the atom coordinates available now in high precision from structure determination. Transmission electron microscopy, a technique since long of great value [2, 42, 47, 49, 50] in the analysis of the complex oxide catalysts, helped [51] also here to elucidate the surface termination [52] of M1.
It is the purpose of the present work to address selected issues in defining and understanding the nature of active sites for AA formation on M1. This will be done by interrogating active sites for AA formation chemistry contrasted with sites required to perform the ODP reaction as first step of the AA reaction sequence. The discussion rests upon the empirical concepts of site isolation and of concerted reactions. The catalytic reaction sequence should occur on an ensemble of atoms (“the active site”) that holds together with the educt molecule all other reactants and exchanges electrons between the reactants. For these operations neither the influx of active species during the unit conversion time (as long as the educt is adsorbed) nor the participation of electrons from outside of the active site are required. This is the consequence of the site isolation concept [53, 54] being so powerful in guiding the development of selective oxidation catalysts. It is expected that the active site behaves dynamical and changes its electronic and geometric structure during the catalytic reaction. The catalytic cycle is only finished when the site is regenerated into its initial active form being here fully oxidized and holding all oxygen atoms belonging to its initial structure. Such a rigorous definition excludes the operation of non-molecular oxidants (1/2 O2) and phenomena of spill-over or diffusion during the catalytic function. Such phenomena may well contribute to the formation of active sites, a process that is not considered here in depth.
2 Size and Functions of Active Sites
Although the M1 phase is composed of four different cations we have learned that the surface dynamics of this phase is such that the V + Te content is the relevant fraction in the surface to which correlates [13] the in situ measured AA abundance for a series of compositional variants of the single M1 phase. To specify this further we found by quantitative line profile analysis of the V 2p line that a good correlation exists during equilibration of a fresh catalyst to the abundance of V5+, which is combined with a constant amount of V4+ that we localize within the M1 structure. The resulting correlation is shown in Fig. 1. It is deduced from this result that the most relevant active sites contain predominantly V5+ as cations.

Selected result of an in situ XPS run of M1 at 625 K. The low temperature was chosen to avoid Te evaporation. Plotted is the fraction of V5+ from line fitting against the measured abundance of AA. Both quantities are normalized to 100. Between the first and all subsequent data points steam was added to the feed. The experimental run time was 24 h after which the performance and surface composition was constant. It is noted that this long time of equilibration may be due to the low temperature and the reduced total pressure of about 0.25 mbar. For experimental details see [56, 57]
It was shown [13] that the abundance of Mo scales inversely with the AA yield excluding that the active sites are MoxOy species. This does not exclude that Mo species hold the actives sites in place and this does also not deny the enormous importance of Mo as the most abundant structure-building cation of the bulk crystal. It is noted that Mo is the most abundant cation in the reactive surface occurring there about five times more often than V. The Mo species do not change their valence during equilibration and operation being in contrast to the observations on the V species. This reduces further the probability that Mo surface species are involved in the catalytic action.
The most abundant species is tellurate with Te ions being present in a ratio 1:2 with respect to Mo. It is noted that a qualitatively similar observation on function and oxidation states of surface species with hence a different interpretation was made [55] that was based on quite different analytical methods being, however, also sensitive to reactive sites.
The activation of propane to propene is a redox reaction involving two electrons per propane molecule. These and the two protons associated need to be reacted to water with the co-reactant oxygen. For oxidative dehydrogenation Eq. 1 describes the alkane oxidation.
We use a stoichiometrically correct notation for the transfer of species in Eqs. 1, 2 without making any statement about the atomistic nature of the atom transfer which is likely to occur via radicalic [8] activation. It is obvious that we need twice the reaction (2) in order to complete Eq. 1. This either requires two adjacent monomeric sites or one dimeric active species under the definition of an active site given above.
Eqs. 3–5 describe the regeneration of the active species. We see immediately that we need either 4 monomeric or two dimeric units of redox storage to deal with one molecule of di-oxygen.
We assume here for maximum efficiency that each redox centre exchanges only one redox equivalent (electron or hole) in order to allow for fast kinetics. This view has been criticised [56] using the example of methanol oxidation on the basis of theoretical models. We do not follow this view based on general chemical consideration on spin selection rules and based upon the argument that we still know too little about the elementary step kinetics of the complex reaction sequence involved here.
It is noted that a redox change on a metal centre requires some structural relaxation in order to accommodate for the modified connectivity. The facile change between edge- and corner sharing of two interconnected polyhedra (the “dimer”) is the prototype of such a reaction. It occurs further immediately that such restructuring is facile in a bulk with non-dense packing of polyhedra (such as in VPO with phosphate “hinges”) or better in a 2-D structure such as in oxide clusters, where the activation barrier for the motion of polyhedra is small.
The consecutive oxidation of propene requires a new quality of activated oxygen. In Eqs. 1, 2 the active oxygen is strongly nucleophilic [58, 59] with a propensity to accept hydrogen. When oxygen should be transferred onto an organic substrate this mode of strong oxygen-site interaction (M = O) is unsuitable but rather a weaker interaction of an oxygen atom being electrophilic to reach its stable electron configuration is desired for the reaction with a radicalic activated state. We further assume that the sites that can activate propene are no monomers but rather small clusters [60] represented here as dimers [61, 62]. Bridging oxygen atoms (V–O–V) may then act as electrophiles.
The first step for deeper oxidation is the oxidative adsorption of propene to the allylic alkoxide shown in Eq. 6. This is a one-electron oxidation leaving behind on the organic moiety one electron to enable the allylic stabilization. The active site has absorbed one redox equivalent but contains one V5+ centre, being responsible of holding the substrate in place.
The next combined step is a formal three-electron oxidation leading to the adsorbed unsaturated aldehyde still with allylic stabilization.
This oxidation step in Eq. 7 leads to electronically stabilized acrolein that is still held to the active site, as the electrons released from the organic substrate do not reduce the vanadium centre. These electrons are transferred to neighbouring sites. It is speculated that the large number of redox equivalents that need to be exchanged in this step presents a substantial hurdle to the overall reaction and hence may represent the kinetic bottleneck of the whole oxidation process.
According to the assumed concept of one redox equivalent per metal sites and according to the assumption of dimer sites being the prevalent species available as active site, the stoichiometrically correct oxidation reaction described in Eq. 8 is rather complex. It is obvious that if larger oligomers (a trimer for Eq. 8) being capable of accepting the redox equivalents in a single ensemble site would be available, then the overall reaction may be simplified and the kinetically complex co-operation of a minimum of three dimer sites to achieve one turnover of the substrate could be avoided. At this point it becomes clear, how the details of the catalyst synthesis controlling the connectivity of the active sites may have a strong influence on the kinetics of the selective oxidation of propane.
Likewise, the concept of a functional heterodimer [63, 64] would help to simplify the reaction; the redox equivalents may be stored onto the redox-active “support”. Silica or alumina are thus no optimal supports for partial oxidation catalysts but molybdenum oxide (in the form of the M1 phase) or other reducible mixed oxides with a stable geometric structure [65–72] are suitable choices for reactions involving both C–H activation and oxygen transfer. The in situ analytical results described above and the concept of site isolation preclude, however, the view that a reactive dimer “antenna site” may be connected to the bulk of the M1 phase acting as collective electron storage unit. Such an idea [73] put forward for other mixed metal species contradicts the site isolation concept as it would not allow to control the timing of substrate oxidation and oxygen reduction and thus violate the concept of minimization of reactive species for maximising selectivity.
The exchange of electrophilic oxygen for redox equivalents is a facile process [56, 74–76] when the site can easily change its oxidation state without having to break bonds to many neighbouring atoms. Handing over an oxygen atom to the adsorbate requires a change in local coordination such as from a corner sharing to an edge sharing form. An adsorbate-type structure of the active site allowing dynamical response will be useful whereas a closely packed 3-D crystalline state will hinder such a process with high activation barriers. Such “adsorbate-like” active phases [77, 78] can be identified with probe molecule reactions [79–86] and have been rationalized for covering some bulk catalysts [13, 42, 87–91] as consequence of surface energy minimization.
The regeneration of the active reduced dimers can occur according to Eq. 5. This may happen in parallel to the continuation of the oxidation. It can be expected that the intermediate aldehyde is easily oxidized [92, 93] to the more stable carboxylic acid. In a facile one-electron oxidation this can be achieved by a mobile oxidation equivalent as shown in Eq. 9. The resulting redox equivalent can be stored in the active site that would loose its ability to hold the substrate.
The species in brackets does nor occur as product but is represented to illustrate how the auto-reduction of the binding site of the organic substrate gives rise to the liberation of the product.
The change of the redox state of the fixing metal site is characteristic of the concept of an adaptive site. In a sequence of reaction steps the bond between substrate and active site is adapted by the electronic structure modification of the substrate: in the present case the allylic stabilization is only lost in the last step enabling the redox equivalent from the substrate to auto-reduce its own binding site. Electron-accepting sites must be easily available to avoid pre-mature reduction of the adaptive binding site. This means that the number of surface locations where several active sites (if assumed that dimers are the units of active sites and not oligomers thereof) are at the same time available in the correct oxidation state will be quite low, giving a hint to the low levels of productivity [94, 95] observed for such catalysts.
The formal description did not make any distinction in the type of active oxygen that must fulfil two fundamentally different functions: one type acts as deprotonating agent and is referred to as “nucleophilic” whereas another type inserts into C–H bonds and reacts thus “electrophilic”. In the equations above both types are denoted with “OH−”. But the stoichiometry of these redox equivalents indicates their different function when ending either as water (ex nucleophilic) or as oxygen in the organic molecule (ex electrophilic). The formulation of redox equivalents as “OH−” pays tribute to the necessary management of hydrogen species [59] that is frequently neglected in other formulations of selective oxidation reactions. It further is inspired by the critical role that water plays in all selective oxidation reactions.
In summary, these simple considerations show that an active site for AA synthesis should consist of the following ingredients. It may be a dual site performing first as dimer the activation of propane and then rapidly continue with the oxidation of propene to AA. This part would require a trimeric unit for the most difficult step of allylic oxidation plus storage sites for three additional redox equivalents. We know from the original concept [15, 96] and from the analysis of the dynamics of reactive surfaces by in situ spectroscopy [13] that vanadium is essential in the active site. From the correlation of Fig. 1 we assume that the binding site producing AA is a vanadium species. As the complete conversion of propane to AA requires the exchange of eight redox equivalents, the active site ensemble should be able to store these eight equivalents. The suggested pentameric unit [15] within the a–b plane of the M1 structure may store five redox equivalents; as the pentameric units also coordinate two tellurate anions, these may complete the electron storage capacity if we assume that the surface tellurate are hexa-valent.
In the inorganic chemistry literature we find several examples of vanadium–tellurates (VI) whereby two building principles can be distinguished. Tellurate can heterogenize isopolyvanadate such as decavanadate [97], or more relevant in the present context, can form chain structures [98, 99] with VxOy dimers. The fact that such structures exist as quite stable materials (accessible through hydrothermal synthesis) qualifies the structural motifs as low-energy alternatives for the termination of the complex M1 structure. This would provide the driving force for a surface-restricted restructuring of the M1 surface. Such an energy-lowering surface termination would provide active sites following both the derived concept of a multi-centre active site and following the empirically derived suggestions in the literature.
In such a picture it is also clear why the intermediate acrolein, that is a desirable product in its own right, has little chance [100] to be found on a catalyst with the pentameric active site as it is the M1 phase. The difficult step is the generation of the less-stable acrolein whereas its subsequent oxidation to a much more stable product AA is a facile step.
3 Termination of the M1 Phase
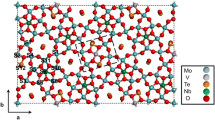
It is more than a co-incidence that the crystal structure of M1 provides for such a pentameric unit composed of sites with some deviation from perfect octahedral symmetry in the metal–oxygen coordination [39, 46] creating inner strain and so chemical energy to support the breaking between the backbone units of the structure. In adaptive active sites the inner strain between the polyhedra would support the necessary reversible change in connectivity accounting for different oxidation states during catalytic action. This pentameric site is located within the motif of M1 as the bridge between two adjacent heptagonal channels. Figure 2 illustrates that in agreement with experimental derivations [42, 49, 53, 71] the assumed active site does not only occur on the basal plane (001) but also on the stepped large prism faces of the oxide needles if we assume a low-energy process for creating a surface in the M1 structure.

Structure of the M1 phase following the determination in the literature [15]. The different colours of the polyhedra denote crystallographically in-equivalent oppositions and not cation occupation sites. Model a shows 4 unit cells with the Te sites omitted for clarity, model b reveals a low-energy termination of the rigid structure (a) including the Te sites. The boxes indicate the active site ensembles. Note that the active site ensembles (light green–red) are located between crystallographic unit cells. The gold-blue oxo-clusters represent the rigid backbone of the structure
Figure 2a reminds us on the basal plane projection of the orthorhombic structure of M1. Relevant for the present discussion are the empty spaces in the structure ordering to heptagonal and hexagonal channels. They are filled to a variable extent with TeOx units. The pentagonal sites are located between the channel systems and intersect the boundaries of the crystallographic unit cells.
Considering the complex bonding situation and the high covalency of the metal–oxygen interaction indicated by the non-dense filling of the unit cell it is likely to assume that a surface termination may be constructed through joining weak parts of the structure. A rational way of doing so is to join the centroids of the channels by lines representing the locations of weakest interaction. It is the specificity of this structural motif (and hence maybe its unique catalytic function) that these lines never intersect the backbone of the structure formed by the stacks of pentagonal bi-pyramids and their immediate octahedral surrounding (oxo-clusters visible in Fig. 2a). Removing the material outside of this joined lines leads to the structure shown in Fig. 2b. One can see that the resulting surface contains infinite rows of polyhedra with the same local chemistry as the active sites located at the basal plane. There is, however, insufficient site isolation from the translational surface structure. The required nano-domains can form, however if through site occupation statistics (“wrong” cations at the light green or red sites) sufficient chemical in-equivalence occurs within the chain structure at the surface so that oxygen mobility would allow for a reconstruction into structurally isolated nano-domains. This occupation statistics may also apply to the sites where the connecting lines of the channel centroids intersect and thus weaken through strained polyhedra the respective break positions. It occurs that the bulk crystalline phase provides the rigid base. The variation of the site occupation [16, 45, 46] as an ingredient or reactivity leading to metastability controls the formation and abundance of isolated nanodomains with the correct chemical composition [13, 26, 49, 53] yielding the active catalyst.
The cation composition of the pentameric site is of a mixture of V and Mo according to the original literature [15] with highly specific requirements for assumed catalytic function; not all VxMoY combinations would be active based upon assumptions of rigid oxidation states and neglecting the concept of adaptive sites. The model of essential cation occupation statistics achieving site isolation would be equivalent in its consequences to the statements in the literature based, however, on quite different arguments. In the present notion cation statistics is essential to create strain helping to terminate the bulk crystal in a maximum of situations as depicted in Fig. 2b and simultaneously in allowing for site isolation. The experimentally verified surface dynamical response [13] calls for a surface termination different from a simple cut through the perfectly translational structure; the cation distribution at the working surface is far outside the bulk composition but only in the surface layer and only under the chemical potential of the working system and e.g. not under vacuum [13, 42]. The undeniable fact [15] that a pure rigid V5+ active phase would be over-oxidizing but that a redox change between V5+ and V4+ is essential for propane activation leads together with the above arguments to the picture that the reactive surface contains active sites of VxOy pentamers separated from each other by patches of MoxOy.
These patches seem to be detrimental for selective operation as the negative correlation of AA yield with surface Mo content [13] shows. Here comes into play now one essential role of TexOy that may shield the MoxOy part better from reacting with the organic substrates than the VxOy part. This specificity is empirically expressed in the positive correlation of AA yield with the V–Te surface abundance in the working state [13] of the catalyst. The solid-state chemistry of Mo–Te cluster ions [101–103] supports the formation of poly-oxo species in a similar way as we see stable bonding between tellurate [104, 105] and the Mo polyhedra within the M1 structure. This structure-directing function of tellurate does not exclude multiple other catalytic functions in selective oxidation discussed [106–108] in the literature. The shielding function of Te in the M1 phase is essential for maintaining sufficient selectivity at the relatively harsh reaction conditions required for propane activation; combinations of nanostructured Mo–V oxides are good catalysts [109–112] for deeper oxidation of activated propane but operate at much milder conditions. The synergistic effect of mixed Mo–V compounds assumed in explaining [15] the mixed metal site in M1 is thus not wanted in order to avoid over-oxidation and to obtain in the one-pot synthesis from propane to some useful oxygenates.
4 Structure Function Relation of the Active Site
We have seen now that the active site needs to be of a substantial size to effectively catalyze the course of a complex selective oxidation of propane to AA. Likewise, the harsh conditions required for the first activation step requires the moderating action of tellurate upon the mixed metal oxide for controlling over-oxidation in the subsequent steps. This conceptual contradiction either calls for a separation of the AA synthesis in a ODP stage followed by a (conventional) selective oxidation stage operating at lower temperature or for an analysis of the origin of the initial harsh reaction conditions. This issue leads to the analysis of ODP catalysis.
The M1 system plus its optimized reaction conditions are the best compromise found so far to deal with the conceptual contradiction. This is possible as the enormous structural stability of the M1 crystal that we ascribe to its unique architecture of rigid backbones (the pentagonal-centered clusters) and flexible linkers (the active site pentamers) allows operating the system under reducing and hydrothermally corrosive conditions. In addition, the creation of an active surface layer of strictly 2-D extension is achieved in the terminating layer. The creation and restructuring of the active phase or the lack of reactivity of the oxygen species in the active site may create the necessity of the harsh reaction conditions.
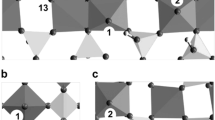
A model of a site described so far is given in Fig. 3. The motif shown in Fig. 3a is taken from an existing compound [99] translating the bond orders discussed there into single- and double bonded oxo-ligands. Fig. 3a is the simplest representation of an oxidized form of the active site. The docking position for the substrate would be the central vanadyl group. The oxidation equivalents are the two bridging oxygen atoms. The four peripheral vanadyl groups coordinate to meta-tellurate VI groups that can easily accept two redox equivalents each. The central vanadium site is strongly coordinated to the support and hence will not change its oxidation state as discussed with Eq. (5). Figure 3b would be the reduced form. four redox equivalents are stored with the tellurate groups and four additional equivalents can be stored with the four peripheral vanadyl groups. Figure 3c is an alternative to Fig. 3a where additional tellurates are assumed to achieve the site isolation from the supporting M1 phase. The peripheral double bonds may partly be connected to surrounding Mo-sites being the most abundant neighboring sites to this form of active site (most abundant transition metal cation at the surface).

Models for active sites terminating part of the surface of the M1 phase. The central dashed line symbolizes a bond to the supporting oxide matrix
Comparing such a model of a complex active site to a simple di-vanadyl dimer or even to a di-oxygen active site can provide insight into the effects arising from the local surrounding of the active oxygen on its reactivity. This comparison can only be done for the ODP reaction as the simpler forms of active sites may not perform the eight-electron deep oxidation to AA.
5 Active Sites for ODP
In Fig. 4 we compare the performance of several materials in the ODP reaction. These are: the M1 phase, two species of grafted vanadium oxide on silica as SBA 15 [113] and on the same mesoporous system pre-covered with amorphous titanium oxide. These two species contain monomeric and low oligomeric sites of vanadia in pentavalent states [114, 115]. Whereas on SBA a non-reducible support would represent electronically isolated species, the amorphous titania ad layer represents a prototype of reducible support to which the vanadia does form chemical bonds and does exchange [116] electrons. In addition, carbon nanotubes without any transition metal in the active site are also included in the comparison. The latter catalyst is of interest as it allows studying oxygen species in the form of nucleophilic and electrophilic species [117, 118] without having to analyze the large background of lattice oxygen [119] being the most abundant surface species on oxide catalysts.

Conversion–selectivity plot for ODP over various catalysts. The performance was varied by changing the GHSV from 120,000 to 25,000 h−1. The feed composition was stoichiometric on the basis of 17.2% C3H8 with N2 as balance gas
The comparison of different catalyst systems in a performance plot as shown in Fig. 4 must be taken with some reservation. The test conditions are not identical and the relation of the catalyst chemistry to the chosen chemical potential of reactants was not optimized; such comparisons would require projections of the performance on the parameter space defining the chemical potential. Nevertheless, some qualitative statements may be deduced from the figure. The general trend for the non-M1 catalysts is within expectation revealing a loss in selectivity with increasing conversion.
The reaction products competing with propene formations are exclusively CO and CO2. The much larger loss in propene selectivity with increasing conversion for M1 is traced back to the occurrence of the kinetically more demanding consecutive reaction of initially formed propene to AA. Nevertheless, when comparing the performance at fixed conversion (10%) we see that M1 is the best catalyst. The active sites of M1 are thus very productive for propane activation. The practical use of M1 may be limited by the unwanted consecutive reaction to AA, a feature, that was not attempted to minimize by modifying the reaction conditions. In any case, the intrinsic activity of the active oxygen species sems not to be limiting as long as they are in a chemical environment prevailing at moderate temperatures and conversions.
We see that reducible metal centers are not essential for performance in ODP when comparing vanadia on silica and pure carbon. It is noted that under no experimental conditions applied it was possible to observe AA formation from propane with CNT catalysts. This is in agreement with the experiment showing that diketonic nucleophilic oxygen [120] forms the active site on CNT that would not allow reactions of the type as indicated in Eq. 8 with more than two redox equivalents being exchanged in one reaction step. The rapid loss in selectivity at higher conversion is ascribed to a reduced efficiency of the moderating boron oxide in preventing unwanted oxygen functional groups to form upon hydrolysis of the C–O–B bonds.
The grafted vanadia species also did not form any AA in our reaction systems being in line with dimer sites [121] as predominant active species. Larger oligomers [122], mimicking the active site in M1 seem not to be frequently present on these systems. This is also true for the titania–vanadia mixed system where EPR clearly showed [116] redox exchange between vanadium and titanium sites. Here unwanted combustion of activated propane at bare titania sites leads to a rapid decay in selectivity.
Dimers can store only two redox equivalents according to the above-mentioned idea leading to the formation of peroxo species during regenerative oxygen activation. A careful EXAFS analysis [122] of grafted monolayer systems revealed that the expected dimers are clearly compatible with the observations. In addition, the dimers seem to occur in an ordered form with close proximity as to form dimers of dimers. Such a structure would facilitate the oxygen regeneration, as a bridge location of the activating oxygen could access four electrons for its activation.
6 Transition Between ODP and Selective Oxidation
The analysis shows that a dimer of nucleophilic oxygen species is necessary in ODP but is insufficient to explain the reactivity of actives sites for the AA synthesis. We interpret this as sign of the operation of a complex sequence of elementary steps where some of them are slow rather than of a single difficult reaction followed by a series of only fast consecutive processes. This is in line with the specificity [123] of oxidation catalysts for different products [124–126] excluding that different catalyst performing all the same rate-determining step would bring about different product distributions (see also Fig. 4). The statement [7, 8] about the rate-determining nature [55] of alkane activation is not inconsistent with this notion if we consider the first intermediate as a rare species limiting the equilibration of all consecutive steps, much in the way as a conventional Langmuir–Hinshelwood reaction may be limited by a pre-equilibrium.
A complicating fact precluding the facile modeling of such a kinetic situation is the dynamic response of the catalyst. For the case of the M1 system we have shown that the chemical composition depends on the composition of the gas phase. This can be inferred from Fig. 1 as well as from the data [13] in the literature. It may consequently be expected that the function of M1 as OPD catalyst and as AA catalyst may be changed with chemical potential. Without the direct spectroscopic evidence it may be argued that such flexibility would arise from different surface coverage imposed by different residence times. In the absence of precise microkinetic data the contributions of coverage effects and of changes in surface chemistry are hard to evaluate. From Fig. 5 where an experiment on the controlling the function of M1 was carried out, it occurs that there is not a smooth transition between regimes of ODP and of AA formation. Rather a steep change at about 10% conversion of propane takes place followed by a Langmuir-type dependence of AA productivity on conversion.

Flexibility of M1 as catalyst for ODP or AA synthesis. The control variable is the space velocity ranging from 60,000 to 6,000 for max AA yield. Feed composition was C3H8/O2/H2O/N2 = 3/6/40/51 vol%, reaction temperature was 673 K. The vertical bars denote the errors for different experiments, the horizontal bars indicate the variability for different feed conditions
The conditions for the experiment in Fig. 5 were chosen such that the conversion was limited by complete oxygen conversion. This left the combustion pathway to CO2 low. It is remarkable that under oxidation catalysis conditions a significant fraction of CO arises in the output stream. This indicates that the catalyst is either a very poor system for CO oxidation and/or that the reactor filling is inhomogeneous with respect to its oxidation state as the complete oxygen consumption may have occurred already within the active bed. This would give rise to reduced catalyst forms after removal of hypothetical lattice oxygen from the M1 material. Then non-oxidative reactions of steam reforming and reverse water gas shift reactions may take place, producing CO from non-combustion reaction pathways. We show such reaction conditions in Fig. 5, as the situation of complete oxygen conversion is frequently found in operating selective oxidation catalysts at high performance conditions.
We may then think of three different types of sites on M1 that is operated under conditions as shown with Fig. 5. One site would perform ODP at high oxygen potentials. Another site would convert the initially formed olefin directly into AA and into small amounts of deep oxidation products. A third site occurring in the reactor at locations with low oxygen chemical potential would perform reforming and shift chemistry as reduced species and produce CO (but no hydrogen from the reforming). The complex interplay of such sites with some parallel reactions likely to occur is summarized schematically in Fig. 6. A similar dynamical situation was assumed for ODP catalysts only [127] when subjected to varying chemical potentials of oxygen.

Schematic representation of reactions occurring upon activation of propane to a radicalic state in the presence of oxygen and of water
It is clear that all reactions of propane require initial activation to the C3 radical. This also includes combustion rendering the customary triangle of kinetic analysis [128, 129] (alkane goes to olefin and to COx) an oversimplification that also cannot account for the fact that CO should occur in parallel to CO2 even in the presence of unreacted oxygen. This radical may desorb at high temperatures into the gas phase and perform several reactions not discussed here. It should be noted that at temperatures above 673 K the occurrence of gas phase chemistry cannot be ruled out when larger hydrocarbons than methane are involved. The desired set of reactions leads directly or via re-adsorption to the formation of an allylic [58, 130] species. In this process undesired addition of water to primary or secondary alcohols can occur leading to highly reactive intermediates. From the allylic species oxidation to the target product can occur if sufficient oxygen is present. This oxygen competes with reaction channels [131] converting the alcohol species. All these reactions are superimposed with a reaction network occurring under non-oxidative conditions producing CO and eventually oxygenates with less than three carbon atoms. It emerges that several reaction channels compete for the conversion of initially activated propane. In the presence of oxygen and water it will thus not be feasible to reach simultaneously very high conversions and selectivity explaining qualitatively the trends seen with Fig. 4.
A meaningful attempt to model such a reaction network requires at least some kinetic constants and adsorption energies and surface coverage data to be measured in order to discriminate sets of fitting parameters. As these are unfortunately still unavailable for M1 or similar systems, much more work on well-defined materials is needed before the qualitative arguing indicated above can be transformed into a reliable model.
The present analysis is in line with observations on M1 supported on alumina. As consequence of thermal treatment some constituents of M1 reacted with alumina leading to a spatial disintegration of the active surface and its tailored support. The effects were drastic on the reactivity [132] of the system verifying that the spatial integrity of the M1 bulk phase and the active surface phase is essential and that the functions of active phase and optimized support cannot be separated. This again is in line with the observations discussed with Fig. 4 where no AA formation was obtained from ODP catalysts although the reaction channel may be open when propene is re-activated for deep oxidation. The reaction profile of Fig. 5 is thus a very special property of the phase-stable M1 system that survives a wide range of chemical potentials and is still an active catalyst without being over-oxidized or over-reduced.
7 Conclusions
The paper tried to rationalize several experimental observations about surface properties and reactivity of the M1 phase with a structural model of an active site. This model is topologically identical to the active site model on the a-b plane of the M1 phase but is of different chemical composition. The difference would be hard to reconcile from structural or crystallographic considerations.
The proposed active site is thought to survive its internal structural dynamics during red-ox cycling, if it is strictly 2-D. Thus the support of the site and the shielding of the strongly interacting M1 matrix phase from reactive molecules through peripheral tellurate are important additional ingredients into a description of the surprising and unique catalytic performance of the M1 phase. To this end a stable bulk phase is critical withstanding continuous segregation of lattice oxygen and/or cations under the harsh conditions of propane oxidation.
The present functional model is in line with many experimental observations and not in contradiction to experimental facts. The model is based upon experiments but remains speculative, as the desirable quantitative description of the reaction of propane is still an enormous challenge. The theoretical efforts addressing prediction of observable kinetic values from structural properties of active sites may be stimulated by the present considerations. Additional experiments aiming at observing adsorption properties and reaction intermediates are underway. These experiments may bracket the parameter space for modeling efforts and in such a way help to support or falsify the concepts presented here.
Finally, it is noted that the present ideas have many aspects in common with the traditional concept put forward by Grasselli and others. The present derivation, is, however, based on completely different arguments and observations. The demonstrated fact that many in situ observations could be brought in line with the predictions from earlier structural considerations is indication that we are approaching a coherent model. This makes it now worthwhile to invoke serious theoretical efforts to explain function and limiting potential of the highly advanced selective oxidation catalyst M1.
References
Cavani F, Ballarini N, Cericola A (2007) Catal Today 127:113
Grasselli RK, Buttrey DJ, Burrington JD, Andersson A, Holmberg J, Ueda W, Kubo J, Lugmair CG, Volpe AF Jr (2006) Top Catal 38:7
Fabrizio C, Joaquim Henrique T (2009) ChemSusChem 2:508
Grasselli RK (2002) Top Catal 21:79
Ganduglia-Pirovano MV, Popa C, Sauer J, Abbott H, Uhl A, Baron M, Stacchiola D, Bondarchuk O, Shaikhutdinov S, Freund H-J (2010) J Am Chem Soc 132:2345
Goebke D, Romanyshyn Y, Guimond S, Sturm J-M, Kuhlenbeck H, Doebler J, Reinhardt U, Ganduglia-Pirovano M-V, Sauer J, Freund H-J (2009) Angew Chem Int Ed 48:3695
Rozanska X, Sauer J (2009) J Phys Chem A 113:11586
Rozanska X, Fortrie R, Sauer J (2007) J Phys Chem C 111:6041
Baron M, Abbott H, Bondarchuk O, Stacchiola D, Uhl A, Shaikhutdinov S, Freund H-J, Popa C, Ganduglia-Pirovano MV, Sauer J (2009) Angew Chem Int Ed 48:8006
Romanyshyn Y, Guimond S, Kuhlenbeck H, Kaya S, Blum R, Niehus H, Shaikhutdinov S, Simic-Milosevic V, Nilius N, Freund HJ, Ganduglia-Pirovano M, Fortrie R, Döbler J, Sauer J (2008) Top Catal 50:106
Goddard WA III, Chenoweth K, Pudar S, Duin ACT, Cheng M-J (2008) Top Catal 50:2
Pudar S, Oxgaard J, Chenoweth K, van Duin ACT, Goddard WA (2007) J Phys Chem C 111:16405
Celaya Sanfiz A, Hansen TW, Teschner D, Schnörch P, Girgsdies F, Trunschke A, Schlögl R, Looi MH, Hamid SBA (2010) J Phys Chem C 114:1912
Guliants VV, Bhandari R, Al-Saeedi JN, Vasudevan VK, Soman RS, Guerrero-Perez O, Banares MA (2004) Appl Catal A 274:123
Grasselli RK, Buttrey DJ, DeSanto P, Burrington JD, Lugmair CG, Volpe AF Jr, Weingand T (2004) Catal Today 91–92:251
DeSanto P, Buttrey D, Grasselli R, Pyrz W, Lugmair C, Volpe A, Vogt T, Toby B (2006) Top Catal 38:31
Kubo J, Watanabe N, Ueda W (2008) Chem Eng Sci 63:1648
Moro-oka Y, Ueda W, Lee KH (2003) J Mol Catal A 199:139
Murayama H, Vitry D, Ueda W, Fuchs G, Anne M, Dubois JL (2007) Appl Catal A 318:137
Ueda W, Li W, Chen N, Kida M, Oshihara K (2000) Res Chem Intermed 26:137
Ueda W (2004) Shokubai 46:2
Vitry D, Dubois JL, Ueda W (2004) J Mol Catal A 220:67
Grasselli RK, Lugmair CG, Volpe AF Jr (2008) Top Catal 50:66
Celaya Sanfiz A, Hansen TW, Girgsdies F, Timpe O, Rödel E, Ressler T, Trunschke A, Schlögl R (2008) Top Catal 50:19
Ueda W, Vitry D, Katou T (2004) Catal Today 96:235
Wagner JB, Othman ND, Su DS, Abd Hamid SB, Schlögl R (2006) J Microsc 223:216
Ueda W, Kato T, Watanabe N, Kuranishi T, Kodato K, Sadakane M (2007) Stud Surf Sci Catal 172:91
Botella P, Dejoz A, Abello MC, Váquez MI, Arrúa L, Lopez Nieto JM (2009) Catal Today 142:272
ter Veen HRJ, Kim T, Wachs IE, Brongersma HH (2009) Catal Today 140:197
Jehng JM, Wachs IE, Ueda W (2004) Abstracts of papers, 228th ACS national meeting, Philadelphia, 22–26 Aug 2004, COLL
Grasselli RK, Lugmair CG, Volpe AF Jr, Andersson A, Burrington JD (2008) Catal Lett 126:231
Grasselli RK, Burrington JD (2008) In: Ertl G, Knoezinger H, Weitkamp J (eds) Handbook of heterogeneous catalysis, vol 7, 2nd edn. VCH-Wiley, Weinheim, p 3479
Lin MM (2003) Appl Catal A 250:287
Vieira Soares AP, Dimitrov LD, Andrede Oliveira MC-R, Hilaire L, Portela MF, Grasselli RK (2003) Appl Catal A 253:191
Baca M, Aouine M, Dubois JL, Millet JMM (2005) J Catal 233:234
Holmberg J, Hansen S, Grasselli RK, Andersson A (2006) Top Catal 38:17
Millet JMM, Roussel H, Pigamo A, Dubois JL, Jumas JC (2002) Appl Catal A 232:77
Deniau B, Bergeret G, Jouguet B, Dubois JL, Millet JMM (2008) Top Catal 50:33
DeSanto P, Buttrey DJ, Grasselli RK, Lugmair CG, Volpe AF, Toby BH, Vogt T (2003) Top Catal 23:23
DeSanto P Jr, Buttrey DJ, Grasselli RK, Lugmair CG, Volpe AF Jr, Toby BH, Vogt T (2004) Zeitschrift fuer Kristallographie 219:152
Govindasamy A, Muthukumar K, Yu J, Xu Y, Guliants VV (2010) J Phys Chem C 114:4544
Celaya Sanfiz A, Hansen TW, Sakthivel A, Schlögl R, Knoester A, Brongersma HH, Looi MH, Hamid SBA (2008) J Catal 258:35
Shiju NR, Liang X, Weimer AW, Liang C, Dai S, Guliants VV (2008) J Am Chem Soc 130:5850
Guliants VV, Brongersma HH, Knoester A, Gaffney AM, Han S (2006) Top Catal 38:41
Pyrz WD, Blom DA, Shiju NR, Guliants VV, Vogt T, Buttrey DJ (2009) Catal Today 142:320
Pyrz WD, Blom DA, Shiju NR, Guliants VV, Vogt T, Buttrey DJ (2008) J Phys Chem C 112:10043
Pyrz WD, Blom DA, Vogt T, Buttrey DJ (2008) Angew Chem Int Ed 47:2788
Grasselli RK, Tenhover MA (2008) In: In: Ertl G, Knoezinger H, Weitkamp J (eds) Handbook of heterogeneous catalysis, vol 7, 2nd edn. p 3489
Wagner JB, Timpe O, Hamid FA, Trunschke A, Su DS, Widi RK, Abd Hamid SB, Schlögl R (2006) Top Catal 38:51
Al-Saeedi JN, Vasudevan VK, Guliants VV (2003) Catal Commun 4:537
Kiely CJ, Hutchings GJ (2007) Appl Catal A 325:194
Zhang W, Trunschke A, Schlogl R, Su DS (2010) Angew Chem Int Ed 49:6084
Wagner JB, Timpe O, Hamid FA, Trunschke A, Su DS, Widi RK, Abd Hamid SB, Schlögl R (2006) Chem Commun 38:51
Grasselli RK, Andersson A, Buttrey DJ, Burrington JD, Lugmair CG, Volpe AF (2004) Abstracts of papers, 228th ACS national meeting, Philadelphia, 22–26 Aug 2004, COLL
Wachs IE, Jehng JM, Ueda W (2005) J Phys Chem B 109(6):2275–2284
Knop-Gericke A, Kleimenov E, Havecker M, Blume R, Teschner D, Zafeiratos S, Schlogl R, Bukhtiyarov VI, Kaichev VV, Prosvirin IP, Nizovskii AI, Bluhm H, Barinov A, Dudin P, Kiskinova M (2009) Adv Catal 52:213
Hävecker M, Cavalleri M, Herbert R, Follath R, Knop-Gericke A, Hess C, Hermann K, Schlögl R (2009) Phys Status Solidi B 246:1459
Haber J, Turek W (2000) J Catal 190:320
Vedrine JC, Millet JMM, Volta J-C (1996) Catal Today 32:115
Macht J, Iglesia E. (2008) Phys Chem Chem Phys 10:5331
Todorova TK, Doebler J, Sierka M, Sauer J (2009) J Phys Chem C 113:8336
Agaskar PA, DeCaul L, Grasselli RK (1994) Catal Lett 23:339
Yang S, Iglesia E, Bell AT (2006) J Phys Chem B 110:2732
Dai H, Bell AT, Iglesia E (2004) J Catal 221:491
Afanasiev P (2005) J Phys Chem B 109:18293
Botella P, Garcia-Gonzalez E, Lopez Nieto JM, Gonzalez-Calbet JM (2005) Solid State Sci 7:507
Haddad N, Bordes-Richard E, Barama A (2009) Catal Today 142:215
Kardash T, Plyasova L, Bondareva V, Shmakov A (2008) J Struct Chem 49:701
Ovsitser O, Uchida Y, Mestl G, Weinberg G, Blume A, Jäger J, Dieterle M, Hibst H, Schlögl R (2002) J Mol Catal A 185:291
Ueda W, Vitry D, Kato T, Watanabe N (2003) Abstracts of papers, 226th ACS national meeting, New York, 7–11 Sep 2003, COLL
Wagner JB, Su DS, Schunk SA, Hibst H, Petzoldt J, Schlögl R (2004) J Catal 224:28
Zenkovets GA, Kryukova GN, Gavrilov VY, Tsybulya SV, Anufrienko VA, Larina TA, Khabibulin DF, Lapina OB, Rödel E, Trunschke A, Ressler T, Schlögl R (2007) Mater Chem Phys 103:295
Mestl G (2006) Top Catal 38:69
Lei Y, Mehmood F, Lee S, Greeley J, Lee B, Seifert S, Winans RE, Elam JW, Meyer RJ, Redfern PC, Teschner D, Schlögl R, Pellin MJ, Curtiss LA, Vajda S (2010) Science 328:224
Bao X, Muhler M, Schedel-Niedrig T, Schlögl R (1996) Phys Rev B 54:2249
Schubert H, Tegtmeyer U, Herein D, Bao X, Muhler M, Schlögl R (1995) Catal Lett 33:305
Hävecker M, Knop-Gericke A, Bluhm H, Kleimenov E, Mayer W, Fait M, Schlögl R (2004) Appl Surf Sci 230:272
Brückner A (2006) Top Catal 38:133
Cavani F, Trifiro F (1997) Appl Catal A 157:195
Guliants Vadim V, Bhandari R, Hughett Andrew R, Bhatt S, Schuler Benjamin D, Brongersma Hidde H, Knoester A, Gaffney Anne M, Han S (2006) J Phys Chem B 110:6129
Guliants VV, Holmes SA (2001) J Mol Catal A 175:227
Haber J, Lalik E (1997) Catal Today 33:119
Kudelski A (2009) Surf Sci 603:1328
Luan Z, Fournier JA (2005) Microporous Mesoporous Mater 79:235
Venkov TV, Hess C, Jentoft FC (2007) Langmuir 23:1768
Wachs IE (1996) Catal Today 27:437
Brongersma H, ter Veen R, Knoester A, Trunschke A, Schlögl R. (2007) Abstracts of papers, 234th ACS national meeting, Boston, 19–23 Aug 2007, PETR
Hutchings GJ, Lopez-Sanchez JA, Bartley JK, Webster JM, Burrows A, Kiely CJ, Carley AF, Rhodes C, Havecker M, Knop-Gericke A, Mayer RW, Schlogl R, Volta JC, Poliakoff M (2002) J Catal 208:197
Ilkenhans T, Herzog B, Braun T, Schlogl R (1995) J Catal 153:275
Mestl G, Ilkenhans T, Spielbauer D, Dieterle M, Timpe O, Krohnert J, Jentoft F, Knozinger H, Schlogl R (2001) Appl Catal A 210:13
Schlogl R, Knop-Gericke A, Havecker M, Wild U, Frickel D, Ressler T, Jentoft RE, Wienold J, Mestl G, Blume A, Timpe O, Uchida I (2001) Top Catal 15:219
Andrushkevich TV (1993) Heterogeneous catalytic oxidation of acrolein to acrylic acid: mechanism and catalysts, vol 35. Taylor & Francis, New York, p 213
Kampe P, Giebeler L, Samuelis D, Kunert J, Drochner A, Haass F, Adams AH, Ott J, Endres S, Schimanke G, Buhrmester T, Martin M, Fuess H, Vogel H (2007) Phys Chem Chem Phys 9:3577
Berndt H, Martin A, Bruckner A, Schreier E, Muller D, Kosslick H, Wolf GU, Lucke B (2000) J Catal 191:384
Botella P, Lopez Nieto JM, Solsona B, Mifsud A, Marquez F (2002) J Catal 209:445
Grasselli RK (2005) Catal Today 99:23
Konaka S, Ozawa Y, Yagasaki A (2008) Inorg Chem Commun 11:1267
Yun G, Hwang Y, Yun H, Do J, Jacobson AJ (2010) Inorg Chem 49:229
Kim H, Cho Y, Yun H, Do J (2007) Zeitschrift Fur Anorganische und Allgemeine Chemie 633:473
Hibst H, Rosowski F, Cox G (2006) Catal Today 117:234
Himeno S, Sano K, Niiya H, Yamazaki Y, Tadaharu U, Hori T (1998) Inorg Chim Acta 281:214
Sun YH, Liu JF, Wang EB (1986) Inorg Chim Acta 117:23
Siebert H (1959) Zeitschrift Fur Anorganische und Allgemeine Chemie 301:161
Beato P, Blume A, Girgsdies F, Jentoft RE, Schlögl R, Timpe O, Trunschke A, Weinberg G, Basher Q, Hamid FA, Hamid SBA, Omar E, Mohd Salim L (2006) Appl Catal A 307:137
Baca M, Millet J-MM (2005) Appl Catal A 279:67
Haeggblad R, Wagner JB, Deniau B, Millet J-MM, Holmberg J, Grasselli RK, Hansen S, Andersson A (2008) Top Catal 50:52
Grasselli RK, Burrington JD, Buttrey DJ, De Santo P Jr, Lugmair CG, Volpe AF Jr, Weingand T (2003) Top Catal 23:5
Grasselli RK, Brazdil JF, Burrington JD (1986) Appl Catal 25:335
Sadakane M, Kodato K, Kuranishi T, Nodasaka Y, Sugawara K, Sakaguchi N, Nagai T, Matsui Y, Ueda W (2008) Angew Chem Int Ed 47:2493
Ueda W, Kobayashi Y, Mangaya Y (2008) Mo–V composite metal oxide catalysts for preparation of unsaturated acids from unsaturated aldehydes by vapor phase catalytic oxidation and their manufacture. Nippon Kayaku Co., Ltd., Tokyo, Application: JP JP
Ueda W, Sadakane M, Ogihara H (2008) Catal Today 132:2
Wang F, Ueda W (2008) Appl Catal A 346:155
Gruene P, Wolfram T, Pelzer K, Schlogl R, Trunschke A (2010) Catal Today 157:137
Hess C (2007) J Catal 248:120
Hess C, Schlögl R (2006) Chem Phys Lett 432:139
Dinse A, Ozarowski A, Hess C, Schomäcker R, Dinse K-P (2008) J Phys Chem C 112:17664
Frank B, Rinaldi A, Blume R, Schlogl R, Su DS (2010) Chem Mater 22:4462
Frank B, Zhang J, Blume R, Schlögl R, Su DS (2009) Angew Chem Int Ed 48:6913
Zhang J, Liu X, Blume R, Zhang AH, Schlogl R, Su DS (2008) Science 322:73
Zhang J, Wang X, Su Q, Zhi L, Thomas A, Feng X, Su DS, Schlögl R, Müllen K (2009) J Am Chem Soc 131:11296
Cavalleri M, Hermann K, Knop-Gericke A, Hävecker M, Herbert R, Hess C, Oestereich A, Döbler J, Schlögl R (2009) J Catal 262:215
Walter A, Herbert R, Hess C, Ressler T (2010) Chem Central J 4:3
Yang S, Iglesia E, Bell AT (2005) J Phys Chem B 109:8987
Lopez Nieto JM (2006) Top Catal 41:3
Argyle MD, Chen K, Bell AT, Iglesia E (2002) J Catal 208:139
Centi G, Trifiro F (1988) Catal Today 3:151
Ovsitser O, Cherian M, Brückner A, Kondratenko EV (2009) J Catal 265:8
Fushimi R, Shekhtman SO, Gaffney A, Han S, Yablonsky GS, Gleaves JT (2005) Ind Eng Chem Res 44:6310
Chen K, Iglesia E, Bell AT (2001) J Phys Chem B 105:646
Chen K, Iglesia E, Bell AT (2000) J Catal 192:197
Zhao C, Wachs IE (2006) Abstracts of papers, 232nd ACS national meeting, San Francisco, 10–14 Sep 2006, COLL
Lopez-Medina R, Golinska H, Ziolek M, Guerrero-Perez MO, Banares MA (2010) Catal Today 158:139
Acknowledgments
The author is greatly indebted to A Trunscke for multiple discussions and for providing with her group the results reported here. M. Hävecker is acknowledged for the efforts in performing in situ ambient pressure XPS experiments. F Girgsdies provided the termination model of the M1 structure. R. Schomäcker is gratefully acknowledged for collaboration and discussions about the ODP part. The work discussed here was supported by the Deutsche Forschungsgemeinschaft DFG through SFB 546.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper is dedicated to Bob Grasselli on the occasion of his 80th birthday. With his imaginative work on selective oxidation he paved the way not only towards substantial economic developments but also to fruitful conceptual insights into one of the more enigmatic areas of heterogeneous catalysis. The community will have to work for quite some time to bring the physical chemistry of selective oxidation to the level of insight provided by the “pillars of selective oxidation” erected by the laureate.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Schlögl, R. Active Sites for Propane Oxidation: Some Generic Considerations. Top Catal 54, 627–638 (2011). https://doi.org/10.1007/s11244-011-9683-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-011-9683-0