
Abstract
In this study, an answer has been sought to several questions: Can substituents’ impact on the electronic properties of molecules in the excited singlet states be summed up by a set of substituent constants? Are the well known, ground state Hammett σ constants practical for this purpose? To answer these questions, the potentials and charges on atoms of the functional groups in two classes of compounds, p-substituted benzoic acids and p-substituted nitrosobenzenes, were regressed against σp. It appeared that all correlations found in the ground state of the molecules also hold in the excited state, with lower correlation coefficients in the latter. An attempt has also been made to find a molecular characteristic which could be useful in a comparison of the acidic properties of benzoic acids (in gas phase) in the first excited and ground states.
Similar content being viewed by others

Introduction
Countless experimental and theoretical results have proven that electronic excitation of molecules is accompanied by redistribution of their electron density. Therefore, one may expect that electronic excitation will be accompanied by reactions, the rate constants of which will depend on the substituents in a different manner than recognized for reactions in the ground state. Indeed, such reactions are known which proceed to the same products in the ground and excited states but with different rates or equilibrium constants. This effect is manifested persuasively by the modification of acidity of some groups of compounds, to name for example phenols [1–4], carboxylic acids [3, 5, 6], and amines [3]. The acid–base properties of these and other groups of compounds are thoroughly presented in [3]; some earlier investigations into the problem of substituent effects on excited state reactivity are also described in [2]. As it is generally recognized that the Hammett constants reflect the electronic properties of substituents [7–9] in the ground state of molecules, it seems justifiable that similar correlations may also be searched for in the excited states. Indeed, the rates of some reactions in the excited state for a series of molecules differing in substituents could be correlated with the Hammett constants of the substituents, which were derived for reactions proceeding in the ground state. Of the rich array of photochemical phenomena one can mention the reaction of photoisomerization of 4,4′-disubstituted stilbenes [10] as well as their photochemical characterization [11] which have been studied from the point of view of the impact of substituents on excited state processes. It was shown that for the compounds fluorescence quantum yield Φ and lifetime τf were mainly determined by the cis–trans photoisomerization rate and about a half of the log(1/τf) and log Φ data points (in different solvents) show a linear dependence on (σX − σY), where X and Y are substituents at 4 and 4′ positions, and σX, σY—the Hammett constants for substituents in the para position to a functional group in benzene ring, generally known as σp. The derivatives which include a strong donor–acceptor X–Y pair followed another trend of reactivity. The Hammett relationship has also been found for hemithioindigo Z/E photoisomerization [12]. There are other reports concerning the experimentally investigated substituent dependence of the reaction kinetics in the first excited state; for example, proton transfer in 3-hydroxyflavones [13] and dissociation of phosphate esters [14]. It appeared, however, that the interpretation of some experimental results required the introduction of new sets of substituent constants, different from those devised for reaction proceeding in the ground state. For example, the new substituent constants for the first excited states were proposed based on the substituent effect on the acidity of phenols and benzoic acids [2, 4]. For the dissociation of phosphate esters the new set of special substituent constants, σexc, proposed by Baldry [2] was tested to account for the substituent-dependent rates of the heterocyclic cleavage of the carbon–oxygen bond. However, it was found that, in this case, the ground state σ gave better correlations that σexc [14].
As far as the theoretical results are concerned, statistically valid correlations were observed between the calculated energy barriers for intramolecular proton transfer processes in both the S0 and S1 states of substituted 2-(2-hydroxyphenyl)-5-phenyl-1,3,4-oxadiazoles [15].
Unfortunately, there are relatively few reports on the systematic investigation of the impact of substituents on the reaction rates in the excited states. Therefore, it remains an open question whether the Hammett relation can be applied to other reactions in the excited states, e.g., photochemical reactions.
Apart from the excited state reaction rates, the impact of substituents on the spectroscopic properties in solution was also investigated. While the frequency maxima for the S0–S1 transition of the base and acid forms of a dissociating compound showed very erratic variations with σ, a good correlation was obtained between σ and the difference of their shifts. The modified constants have also provided some improvement [3]. In this vein, more recently, for the interpretation of the experimental UV spectra for substituted stilbenes, another new set of substituent constants was applied; the constants were determined as the difference in maxima of absorption energy between a substituted molecule (M–Y) and unsubstituted one (M) [16]. It appeared that the set of constants thus obtained did not correlate with the Hammett constants. Therefore, it was recommended to apply the new excited-state substituent constants in a quantitative structure–activity relationship (QSPR) study of organic compounds in the excited state.
These results were completely at variance with other results of theoretical study of spectroscopic data, where linear relationships between the substituent constants and the calculated absorption, fluorescence, and phosphorescence wavelengths for 2-(2-hydroxyphenyl)-5-phenyl-1,3,4-oxadiazoles were found [15].
Taking into consideration that the reactivity/spectroscopic data are not in full agreement, we decided to study the impact of substituents on the electronic properties of two sets of compounds: p-substituted benzoic acids and p-substituted nitrosobenzenes. The first group was selected because the empirical Hammett equation had originally been used to correlate the influence of substituents in the aromatic ring upon the acidity of benzoic acids in the ground electronic state. The second group was selected because nitrosobenzene is an attractive electrophile in catalytic C–N and/or C–O bond forming reaction based on the polarization of the N–O bond [17] and thus its reactivity should be directly modified by the changes of electron density.
It has previously been shown that potentials on atoms of the COOH group in a series of acids correlate very well with σp, therefore, may serve as an acidity measure in acids ([18, 19]. We were interested in finding out whether similar correlations of the constants with atomic potential at the atoms of a functional group would hold in the first excited state as in the ground state.
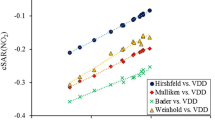
On the other hand, it has been shown that the charge of the substituent active region, qSAR(X) (which is a sum of charges of all atoms of the substituent and of the Cipso it is attached to) [20] is also well correlated with sigma constants, either in monosubstituted benzenes [20] or in p-substituted benzoic acids [21]. It has been shown that also charges transferred between the active regions of a functional group and of substituents correlate with substituent constants. Namely, we found such correlations of calculated qSAR(X)–qSAR(NO) in p- and m-substituted nitrosobenzenes and the values of σp or σm [22], where qSAR(NO) means the charge of the active region of the NO functional group. Therefore, we aimed to investigate whether the amount of the transfer in the excited singlet state of the two series of compounds, p-substituted benzoic acids and p-substituted nitrosobenzenes, also correlates with the Hammett constants.
Theoretical methods
Calculations concerning molecules in the ground state were carried out using density functional theory (DFT) and the Becke three-parameter exchange functional combined with the dynamic (non-local) Lee–Yang–Parr correlation functional (LYP), as implemented in the Gaussian 03 package [23]. The initial three-dimensional structures of the compounds were built using Spartan Pro software [24], followed by preliminary semi-empirical geometry optimization at the Austin model version 1 (AM1) level. Subsequently, geometries were optimized and energies calculated using the Dunning’s valence double zeta cc-pVDZ basis set, followed by the calculation of vibrational frequencies at the same level. Free energies of benzoic acids and their anions at 298.15 K were provided by the thermodynamic analysis included in frequency calculations; their difference ΔG was taken as the free energy of dissociation. The excited state geometries were optimized using CIS/cc-pVDZ and time-dependent B3LYP, with the G09 package [25].
The atomic charges were calculated by applying Natural Population Analysis (NPA) [26]. This is a Class II method of determination of partial atomic charges, according to the Cramer classification [27] (which involves direct partitioning of the molecular wave function into atomic contributions). The charge of a molecular fragment was calculated as the sum of the individual charges of the atoms of which it was composed.
The Atoms in Molecule properties were calculated with the AIM 2000 program [28].
The set of substituents in both series of compounds was comprised of CN, CF3, Cl, H, CH3, NH2, NC, N(CH3)2, SiH3, CH2Cl, CCH, COCl, NHCH3, COF, OH, CHF2, and NO for the series of benzoic acids, or COOH for the series of nitrosobenzenes. The Hammet constants σp were taken from the compilation of Taft et al. [9].
Results
p-Substituted benzoic acids
First, a comparison of the correlations involving Hammett constants was made for a group of p-substituted benzoic acids. It appeared that for 15 out of 17 acids investigated here, the first optimized excited states were of ππ* type, whereas for two, p-NO– and p-COCl-benzoic acid, the first excited state was of the nπ* type. Therefore, for the latter two acids, the results were taken for the S2 excited state. This way, for all acids, the first ππ* states were considered. The atomic potential and charges were calculated for the optimized as well as for the vertical excited states and compared with those in the ground state. The results of correlations of the potentials on the H atom of the COOH group with the Hammett constants are shown in Fig. 1a, where the potentials calculated for the optimized ππ* states are compared to those in the ground state, and 1b, which is similar to 1a, but for the vertical ππ* states. For the rest of the atoms of the COOH group, the corresponding correlation coefficients R are shown in Table 1.

Plots of potential at the H atom of the –COOH group in the series of p-substituted benzoic acids against the Hammett constants of the substituents in the first ππ* excited state (plus), optimized (a) and vertical (b), as compared to those in the ground state (triangle)
Figure 1a and b shows that the VH potentials in the ππ* excited state were generally lower than those in the ground state (except for NO substituent) and that in the ground state, the less negative potentials correspond to stronger acids, for which the substituent constants were large and positive (For example, for p-cyanobenzoic acid, the pK a in water equals 3.55, whereas for p-dimethylaminobenzoic acid it equals 5.03 [29]; the Hammett constants for –CN and –N(CH3)2 substituents are 0.66 and −0.83, respectively). In case the same tendency holds for the comparison of acidities in the ground and excited states, one could expect the molecules in the ππ* state be weaker acids than in the S0 state, assuming that the sequence of acid strength is the same in the water as in the gas phase.
It turned out, however, that the calculated values of the dissociation free energy (here in the gas phase) do not univocally confirm this conclusion—for eight acids (with the CN, CF3, Cl, H, SiH3, NO, CH2Cl, and CHF2 substituents) the calculated ΔG values for dissociation reaction are lower in the excited state, and for seven (with the CH3, NH2, NC, N(CH3)2, OH, NHCH3, and CCH substituents) are higher in the excited states.
To gain better insight into the acidity differences we calculated other molecular characteristics which in the ground state are well correlated with the dissociation free energy ΔG, namely the NPA charge on the dissociating hydrogen atom, q H, and the electronic properties at the O–H bond critical points: the electron density ρc.p., and Laplacian L c.p.. A comparison of the ranges of the three characteristics in both states did not indicate whether the gaseous ΔG should be greater in the excited state or in the ground state: the q H, ρc.p., and L c.p. values were larger in the excited state than in the ground state. In the ground state the stronger an acid, the larger its q H and L c.p. and lower ρc.p.. Therefore, our conclusion was that according to V H and ρc.p. the acids in the first ππ* state should be weaker than in the S0 state, whereas according to q H and L c.p. they should be stronger.
These results indicate that none of the investigated parameters based on electron density can be used for a comparison of acid strength in both electronic states. However, all four characteristics (V H, q H, ρc.p., and Lc.p.) correlated fairly well with the Hammett constants. The corresponding correlation coefficients were 0.967, 0.950, −0.922, and 0.896 (whereas in the ground state they equaled 0.987, 0.979, −0.992, and 0.957). The excited state correlations were inferior to those in the ground state but still significant.
Apart from the electronic properties characterizing the functional group, the charges transferred between the active regions of substituents and of the functional group, which were found to correlate with the Hammett constants in the ground state [21], were also investigated in the excited states. For this purpose, the NPA charges of the two groups (COOH and substituent) were added to charges of their Cipso atoms. A comparison of the results for the excited and ground states is shown in Fig. 2a and b.

Plots of the charge transferred between the active regions of substituents and the COOH functional group, ΔqSAR, against the Hammett constants, for the series of p-substituted benzoic acids in the ground (triangle) and in the first excited ππ* state (plus): a optimized excited state, b vertical excited state
It can be seen in the optimized and vertical excited states, that the correlations for the charges transferred between active regions (ΔqSAR) versus Hammett constant were very good. Additionally, the plots of the calculated ΔG in the ground and optimized ππ* states versus ΔqSAR, shown in Fig. 3, indicate that the latter values were more successful in the comparison of the acidic properties in both states than other characteristics. For some substituents, with a positive σp (e.g., CN, CF3, Cl, and CHF2 at the right side of the intersection of the two lines) the plots point to larger values of ΔG in the ground state, whereas for the substituents with a negative σp (NH2, N(CH3)2, NHCH3, at the left side) they point to larger values of ΔG in the excited state.

Plots of the free energy of dissociation of p-substituted benzoic acids in the gas phase (ΔG, in kcal/mol) vs. ΔqSAR in the ground (triangle) and optimized ππ* (plus) states
The properties of p-substituted benzoic acids in the excited state were also calculated by the TD DFT method, with the B3LYP functional. Unfortunately we succeeded in the optimization of only 11 (out of 17) molecules. For the optimized set, the correlations analogous to those shown in Figs. 1 and 2 were similar.
p-Substituted nitrosobenzenes
For all 17 investigated compounds, the nature of the first excited state was nπ*, and of the second ππ*. The energy difference between the two states amounted to about 3 eV, so it was easy to optimize the two states separately, S1 (nπ*) and S2 (ππ*). The results of regressing the potentials at the nitrogen atom of the functional group against the Hammett constants are shown in Fig. 4a and b. The correlation coefficient values for V O plots in S1 and S2 are shown in Table 2.

Plots of the potential at the nitrogen atoms of the –NO group in the ground state (triangle), in the S1 (nπ*) state (plus) and in the S2 (ππ*) state (circle) of p-substituted nitrosobenzenes against the Hammett constants: a optimized excited states, b vertical excited states
The fact that potentials V N (and V O, not shown here) were less negative in S1 than in the ground state might indicate that the atomic electron density was more positive in S1, i.e., less electron density was transferred from other parts of the molecule to NO. This can be confirmed by a comparison of the NPA charges of the NO group in the ground, optimized, and vertical S1 (nπ*) state, which is shown in Fig. 5.

Plots of the charge of the NO functional group in the ground (triangle), in the S1 optimized excited (plus) state and in the S1 vertical state (circle) of p-substituted nitrosobenzenes, against the Hammett constants
On the other hand, V N and V O potentials in S2 were shifted in opposition to each other relative to their values in the ground state, and the trend in q NO in S2 could not be anticipated based on the V N and V O values. In fact, the NO charges in the S2 (ππ*) state were more negative than those in the ground state.
The modifications of the NO group charge as influenced by substituents are rendered by the plots of the charge transferred from the NO functional group active region to the substituent active region. The results are displayed in Fig. 6a and b, where the plots Δq(SAR) = qSAR(NO) − qSAR(X) are plotted versus σp. It can be seen that all the correlation coefficients for the plots were high, either for the optimized or vertical states. It can also be observed in S1, for all compounds, that the abscissa values are more positive or less negative in the optimized excited state. This means that for a given substituent, the NO group gains more negative charge in the ground state than in the excited state, which is in agreement with data in Fig. 5. This effect should impact on the reactivity involving the nitroso group in the first singlet excited states of nitrosobenzenes. The Δq(SAR) values in S2 (ππ*) are very similar to those in the ground state (Fig. 6).

Charge transferred between the active regions of substituents and the NO functional group, ΔqSAR, in the ground (triangle), S1 (nπ*) state (plus) and S2 (ππ*) state (circle) of p-substituted nitrosobenzenes, plotted against the Hammett constants: a optimized excited states, b vertical excited states
Conclusions
-
1.
Potentials at the individual atoms of the COOH and NO functional groups were linearly dependent on the Hammett constants similarly in the ground and excited states. This held for the optimized and vertical states as well.
-
2.
Plots of the charge transferred between the active regions of the COOH or NO functional group and the substituent active region also showed a linear dependence on the Hammett constants in the optimized and vertical ππ* states of p-substituted benzoic acids, and in nπ* and ππ* states of p-nitrosobenzenes.
-
3.
The correlation coefficients for the parameters calculated in the optimized excited states were always lower than those for the ground state. This fact may have a physical meaning or could be simply due to methods of calculating excited state properties, which are far from perfect.
-
4.
The correlation coefficients for the parameters calculated in the vertical excited state are higher than those for the optimized states for six out of seven lines shown in Figs. 1, 2, 4, 5, and 6.
-
5.
There was no systematic drift of the lines corresponding to excited states which could justify introduction of new substituent constants designed for the excited states.
-
6.
An attempt to find a single molecular parameter determining the molecule reactivity in the excited state in comparison to that in the ground state was performed based on the calculated free energies of dissociation of benzoic acids. The correlations with the potentials (V H), charges (q H), and topological properties of the O–H bond (electron density and Laplacian at the bond critical point) did not generate any univocal indication concerning the electronic state in which the acidic properties should be stronger. The calculated dissociation free energies were lower in the excited state for substituents with high positive Hammett constants (CN, CF3, CHF2) but the reverse was true for the substituents from the other end of the Hammett scale (NH2, N(CH3)2).
-
7.
The charge transferred between the active regions of the functional groups and substituents might perhaps be a candidate for a gauge of the molecular reactivity in the different electronic states, but this proposition should be tested for other groups of compounds.
References
Bartok W, Hartman RB, Lucchesi PJ (1965) Photochem Photobiol 4:499
Baldry PJ (1979) J Chem Soc Perkin 2:951
Ireland JF, Wyatt PAH (1976) In: Gold V, Bethell D (eds) Acid-base properties of electronically excited states of organic molecules in advances in physical organic chemistry, vol 12. Academic Press, London, New York, San Francisco
Shim SC, Park JW, Ham H-S (1982) Bull Korean Chem Soc 3:13
Shim SC, Park JW, Ham H-S, Chung J-S (1983) Bull Korean Chem Soc 4:45
Murugesan R, Rajasekar B, Lekshmana Thanulingam T, Shunmugasundaram A (1992) Proc Indian Acad Sci 104:431
Hammett LP (1970) Physical organic chemistry, 2nd edn. McGraw-Hill, New York, London
Smith MB, March J (2001) March’s advanced organic chemistry: reactions, mechanisms and structure, 5th edn. Wiley, New York, pp 681–685
Hansch C, Leo A, Taft RW (1991) Chem Rev 91:65
Papper V, Likhtenshtein GI (2001) J Photochem Photobiol A 140:39
Papper V, Pines D, Likhtenshtein G, Pines E (1997) J Photochem Photobiol A 111:87
Cordes T, Schadendorf T, Priewisch B, Rück-Braun K, Zinth W (2008) J Phys Chem A 112:582
Itoh M, Fujiwara Y, Sumitani M, Yoshihara K (1986) J Phys Chem 90:5672
Givens RS, Matuszewski B, Athey PS, Stoner MR (1990) J Am Chem Soc 112:6016
Jin R, Zhang J (2009) Theor Chem Acc 124:331
Cao C, Chen G, Yin Z (2008) J Phys Org Chem 21:808
Yamamoto H, Momiyama N (2005) Chem Commun 3514
Sadlej-Sosnowska N (2007) J Phys Chem A 111:11134
Sadlej-Sosnowska N (2009) Pol J Chem 83:2215
Sadlej-Sosnowska N (2007) Chem Phys Lett 447:192
Sadlej-Sosnowska N (2007) Pol J Chem 81:1123
Krygowski TM, Sadlej-Sosnowska N (2011) Struct Chem 22:17
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T, Kudin KN, Burant JC, Millam JM., Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M., Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, NakajimaT, Honda Y, Kitao O, Nakai H, Klene M., Li X., Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA Peng CY, Nanayakkara A, Challacombe M., Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2003) Gaussian 03, revision B.04. Gaussian, Pittsburgh
PC Spartan Pro 1.0.5 (2000) Wavefunction, Inc., 18401 Von Karman Avenue, Suite 370, Irvine, CA 92612, USA
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Menucci GA, Petersson H, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Danneneberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox JD (2009) Gaussian 09, Revision A.02. Gaussian Inc., Wallingford, CT
Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899
Cramer CJ (2003) Essentials of computational chemistry. Wiley, Chichester
Biegler König F, Schönbohm J (2002) AIM 2000, Version 2.0
Brown HC, McDaniel DH, Häfliger O (1955) In: Braude EA, Nachod FC (eds) Determination of organic structures by physical methods. Academic Press Inc., New York
Acknowledgment
We acknowledge the computing grant G44-16 from the Interdisciplinary Center for Mathematical and Computer Modeling (ICM) of the Warsaw University.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sadlej-Sosnowska, N., Kijak, M. Excited state substituent constants: to Hammett or not?. Struct Chem 23, 359–365 (2012). https://doi.org/10.1007/s11224-011-9878-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-011-9878-9