
Abstract
Numerous studies on nitro group properties are associated with its high electron-withdrawing ability, by means of both resonance and inductive effect. The substituent effect of the nitro group may be well described using either traditional substituent constants or characteristics based on quantum chemistry, i.e., cSAR, SESE, and pEDA/sEDA models. Interestingly, the cSAR descriptor allows to describe the electron-attracting properties of the nitro group regardless of the position and the type of system. Analysis of classical and reverse substituent effects of the nitro group in various systems indicates strong pi-electron interactions with electron-donating substituents due to the resonance effect. This significantly affects the pi-electron delocalization of the aromatic ring decreasing the aromatic character, evidenced clearly by HOMA values. Use of the pEDA/sEDA model allows to measure the population of electrons transferred from the ring to the nitro group.
Similar content being viewed by others

Introduction
The nitro group is one of the most interesting and important substituents in organic chemistry and related fields [1,2,3,4]. Many monographic articles are available in the literature on the various properties and applications of nitro compounds. For example, according to ISI Web of Science [5], almost 2000 articles and 55 reviews containing the term “nitro” in the title have been published in the last 5 years. Moreover, in the Cambridge Structural Database (CSD) [6], about 51,500 crystal structures of chemical compounds containing the NO2 group are reported, of which almost 48,500 structures with an R-factor equal to or less than 0.10. It should be emphasized that nitro compounds are widely used in medicinal and pharmaceutical chemistry [7], explosives [8, 9], fertilizers [10], etc.
The unusual interest in nitro compounds is associated with the strongly electron-attracting (EA) character of the nitro group. Its EA ability is a result of both strong inductive and resonance activities. Its inductive effect, estimated by σI at 0.76 [11], results from a high group electronegativity χNO2 [12] which in the Pauling scale is equal to 4.00 for coplanar and 4.19 for perpendicular orientation with respect to the benzene ring [13]. The EA property of the nitro group is associated with the fact that all three atoms of this group are strongly electronegative (χN = 3.04, χO = 3.44) [14]. In addition, the nitro group exhibits a substantial variability in EA abilities dependent on the type of moiety to which it is attached [15]. This is clearly seen in the changes in values of substituent constants, σp = 0.78 and σp− = 1.27, respectively for para-substituted benzoic acid and phenol derivatives [16, 17]. This EA changeability is due to the resonance effect caused by an intramolecular charge transfer between the NO2 group and an electron-donating (ED) substituted moiety. Resonance structures induced by the nitro group depending on the substitution site of the second ED substituent are shown in Scheme 1 [18]. For the para substitution (Scheme 1a), the number of bonds between C (NO2) and C (X) is odd, while the number is even for the meta substitution (Scheme 1b). In the last case, however, the quinoid-type structure requires double excitation; hence, the interaction is weaker. This shows that the nitro group can cause dramatic changes in the pi-electron structure of aromatic systems due to the influence of the second substituent.

Quinoid-like structures of para-nitroaniline (a) and meta-nitroaniline (b)
It should be stressed that the EA ability of the nitro group also depends strongly on its conformation. The rotation of the nitro group relative to the plane of the benzene ring from 0° to 90° in para-nitrophenolate causes a decrease of sigma values from 1.27 to 0.70 [19], which is a very similar value to that for the field parameter F = 0.65 [17].
The variability of EA power of the nitro group was also confirmed by experimental values of the polarographic half-wave potentials, E1/2, experimentally determined for substituted nitrobenzene derivatives in aprotic solvents. The less negative value of E1/2 indicates a stronger electron-attracting ability of the nitro group, for example − 0.800 V (for p-CN-nitrobenzene) and − 1.400 V (for p-nitroaniline) [20]. The electrochemical properties of nitro compounds strongly depend on both the medium and their structure [21].
Physicochemical properties of the nitro group, both from the viewpoints of structural and quantum chemical research, have been presented in many papers [2, 3, 22,23,24]. The structure of the nitro group undergoes deformations due to intramolecular or intermolecular interactions, as documented by numerous experimental data [25]. These deformations can undoubtedly cause changes in the sigma and pi electron structure of the whole molecule; details are presented later in this work.
In crystal structures, the NO2 groups are characterized as poor hydrogen bond acceptors and hardly form conventional hydrogen bonds. However, it is known that nitro groups play a key role in crystal packing by participating in C–H···O and dipole-dipole interactions. Additionally, nitro groups are involved in interacting through C–H···O and orthogonal N–O···N–O-type dipole-dipole interactions. These orthogonal N–O···N–O interactions are frequently observed in the crystal structures of many nitroarene compounds and exhibit high stability [26, 27].
The substituent effect (SE) is among the most important factors affecting the chemical, physicochemical, and biochemical properties of chemical compounds. The great interest in SE research confirms the widespread use of this term in the titles (261), abstracts (1383), and keywords (241) of publications that appeared last year. Traditionally, the substituent effects are described in terms of Hammett constants [1, 28], σ, which are defined by the ionization constants of para- and meta-substituted benzoic acids, according to the equation:
where K(X) and K(H) are equilibrium constants for substituted and unsubstituted benzoic acids in water under normal conditions.
The Hammett approach to SE allows not only to qualitatively describe its magnitude for a given substituent X using substituent constants σ(X) but also to determine the sensitivity of a specific chemical reaction or physicochemical property P(X) of X–R–Y systems (R—transmitting moiety and Y—reaction/process site) to SE, as described by Eq. 2:
where σ(X)p,m are the substituent constants for para and meta positions and ρ is the reaction constant describing the sensitivity of the reaction/process to the influence of the substituent X.
The introduction of the fundamental Hammett constants (σp,m) in 1937 [28, 29] initiated the development of ideas for quantitative description of SE. To date, substituent constants (SC) have been successfully used to interpret the influence of substituents on chemical and physicochemical properties of various organic compounds [2, 16, 17, 30,31,32,33,34,35,36,37,38,39]. Unfortunately, as Hammett noticed himself (proposing two values of constants for the nitro group, 0.778 and 1.27) [28], the original sigma substituent constants did not have universal application. In systems, where the electron-donating/attracting ability of the reaction site (Y) differs significantly from that of the COOH/COO− groups in the reference reaction, they did not work well and had to be modified [17, 32]. Therefore, various new modifications of the original sigma constants were introduced depending on the electronic properties of the reaction sites and different types of intramolecular interactions; for review, see Ref. [17, 32, 38, 40].
For example, Taft et al. [40, 41] came to the conclusion that SE consists of two types of interactions: inductive and resonance, as shown in Eq. 3 based on Ref. [32]:
Later, acid-base equilibrium constants of 4-X-substituted derivatives of bicyclo[2.2.2]octane-1-carboxylic acids were used to define inductive SCs, σI, and then σR from Eq. 3 [42]. A similar approach was used by Swain and Lupton [43], who proposed SC values named F (for inductive/field) and R (for resonance), indicating the ratio of F/R dependent on the reaction series studied. In addition, they showed that 30 years after Hammett introduced the substituent constants, the number of their variously defined analogues reached over 30 positions!
Undoubtedly, the values of σ constants may significantly depend on several factors, i.e., path of interaction between the substituent and the reaction site, type of the reaction, nature of the moiety to which the substituents are attached, and also on the given reference reaction [30] as well as the medium in which the reaction/process is carried out [44,45,46]. Therefore, different substituent positions (e.g., meta-, para-) or, in other cases, different reaction sites with strong ED/EA properties are considered, as shown for nitro and amino groups in Table 1. Substituent constants of the nitro group based on the use of modern experimental and theoretical techniques are well presented in Ref. [22].
Due to the large number of known scales of substituent constants [43], a dilemma has arisen as to which substituent constants should be used in a given case. Therefore, over the last decades, many physical approaches have been proposed that use different quantum chemistry models, presented below.
Studies of the substituent effect by means of quantum chemistry models can be realized using two types of approaches. One of them with analysis of changes resulting from replacement of one substituent by another. The second approach refers to studies on changes of some physicochemical or biochemical properties, considering a set of various substituents—by means of “reaction series.” Application of both traditional and quantum chemistry-based descriptors of SE allows to show the nature of substituent effect in various reactions. Numerous studies have appeared recently on interrelation between the electron properties of the fixed functional group Y (“reaction site”) and the ED/EA properties of the varying substituents X in a typical X-R-Y reaction series, where R is a transmitting moiety [24, 47,48,49,50,51,52,53,54].
Given the great interest in nitro compounds, and especially the properties of the NO2 group, this review presents the results of the latest research in this field.
Quantum chemical models to describe substituent effect
Application of the quantum chemistry modeling has allowed the development of many descriptors (independent of empirical approaches) of the physical significance of SE, which are available using standard computational packages [55]. It should be emphasized, however, that these characteristics have almost always been confronted with classical Hammett-like descriptors. In most cases, good correlations were found between the proposed theoretical approaches and substituent constants (or their modifications). Good examples are satisfactory correlations between the molecular electrostatic potential (MESP) topography of monosubstituted benzenes and their sigma constants [56]. MESP values in specific atoms, atoms of the reaction sites or in other particular sites of molecules, show their good correlations with sigma constants [57,58,59]. Also, the use of the energy decomposition analysis (EDA) [60] showed good correlations of strength of π-conjugation (ΔEπ) in meta- and para-substituted benzylic cations and anions with the Hammett constants [61].
The dynamic development of quantum chemistry methods still leads to the search for new tools characterizing the impact of a substituent and changes in its electron properties. In recent decade, the most popular are energetic and electronic descriptors of the SE, which can be successfully applied to investigate nature of SE in various cyclic (aromatic, unsaturated, or saturated) compounds.
The descriptors that are often used in SE and aromaticity studies of nitro-substituted derivatives are described in more detail below. The presented approaches belong to the modern way of describing the substituent effect and can also be applied to heterocyclic systems.
Substituent effect stabilization energy (SESE) descriptor
SESE model [62] is one of the first SE descriptor based on an isodesmotic or homodesmotic reaction (Eq. 4) [63, 64]. SESE values are calculated as the energy difference of products and substrates [64,65,66] and define the overall energy of the process (4), according to Eq. 5.
Usually, its values are well correlated with empirical σ constants or their modifications (F) [47, 48, 52, 53, 67, 68], which for nitro-substituted derivatives are shown in Table 2. SESE values characterize the energetic interaction between substituent X and reaction site Y in X-R-Y the system. The greater value of SESE (see Eq. 5) indicates the higher stabilization energy of substrates caused by the substituent effect.
Charge of the substituent active region (cSAR) descriptor
Another approach describing the SE is based on an electronic structure and has already been intuitively proposed by Hammett [1]. Unfortunately, the correlations between atomic charges at substituents, q(X), and Hammett substituent constants fail [69]. However, the use of atomic charges to investigate SE is effective when q(X) is replaced by the charge of the substituent active region approach, named cSAR, introduced by Sadlej-Sosnowska [69,70,71]. The cSAR parameter is defined as a sum of charges at all atoms of the substituent X and the charge at the ipso carbon atom:
An advantage of the cSAR parameter is that it can be used to describe both the electron-donating/accepting properties of a substituent and the reaction site, regardless of the type of system. A more negative value indicates a stronger electron-accepting ability of the substituent, while a more positive one denotes stronger electron-donating properties. In addition, the same substituent X placed in a different position, or studied in another system, may have a completely different cSAR value.
Good agreement of cSAR(X) with Hammett constants, compared with q(X), is due to very weak polarity changes in CC bonds cut for the cSAR(X) approach, in contrast to C–X bonds, which are usually very polar, as shown in Scheme 2.

Graphical presentation of q(X) (a) and cSAR(X) (b) definitions
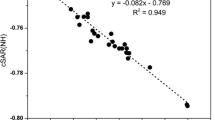
It is worth noting that cSAR(X) values exhibit good correlations with substituent constants independently of the method of atomic charge assessment (Mulliken [72], AIM [73], Voronoy [74], Hirshfeld [75], and NBO [76]), which has been documented for 12 para-substituted derivatives of nitrobenzene [15]. For example, the mutual correlations between cSAR(NO2) values calculated by the use of various methods of atomic charge assessment are presented in Fig. 1. The analogous linear regressions for cSAR(X) values also give good results [15].

Correlation between cSAR(NO2) calculated from VDD charges and data from Hirshfeld, Mulliken, Bader, and Weinhold methods for p-nitrobenzene X derivatives with X = NO2, CN, CHO, COOMe, COMe, Cl, H, Me, OMe, NH2, and NHMe (cc = 0.999, 0.998, 0.986, and 0.986, respectively). Reused from [15], this work is licensed under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/)
Pi and sigma electron donor-acceptor (pEDA and sEDA) model
Recently, Oziminski and Dobrowolski [67, 77] have proposed new models to characterize SE, describing a population of electrons at sigma and pi orbitals of planar pi-electron systems (or their fragments), named as sEDA and pEDA. Thus, these models allow to describe changes in sigma and pi electron structure of the substituent and pi-transmitting moieties. pEDA/sEDA denote pi and sigma electron donor-acceptor indices, respectively, and are calculated according to Eqs. 7 and 8 with the use of natural population analysis (NPA) based on the natural bond orbital (NBO) [76] theory:
where, for planar fragment (in XY plane) of the system in question, σiX means the sum of occupancies of 2s, 2px, and 2py sigma orbitals and πiX denotes the occupancy of the 2pz pi orbital of the ith atom in the structure; n is the number of atoms, for example for NO2: n = 3, for benzene ring (R): n = 6. Contrary to the original paper, pEDA and sEDA values do not need to be standardized by unsubstituted benzene as the reference molecule [77].
This approach was applied to determine the strength of SE on pi-electron population of the ring in mono- and disubstituted pentafulvene derivatives [78], in double bonded substituted pi-electron cyclic systems [79], and in meta and para series of nitrobenzene derivatives [49, 67]. Unlike previous descriptors, the found correlations of sEDA values with Hammett σ constants are not so clear, while pEDA values usually exhibit good correlations with R constants [49, 77].
Aromaticity indices: HOMA, NICS
The substituent effect on the aromatic properties of the transmitting moiety may be well investigated using aromaticity indices based on various criteria: geometric, magnetic, electronic, and energetic. The most popular aromaticity descriptor, the geometric aromaticity index HOMA (Harmonic Oscillator Model of Aromaticity) [80,81,82], is defined as a normalized sum of squared deviations of bond lengths from the values expected for a fully aromatic system. In the case of hydrocarbons an appropriate expression is given by Eq. 9:

where n is the number of bonds taken into account when carrying out the summation, i means the type of bond (e.g., CC or CN), αi is an empirical normalization constant (for CC and CN bonds, αCC = 257.7 and αCN = 93.52) chosen to give HOMA = 0 for non-aromatic system and HOMA = 1 for the system where all bonds are equal to the optimal value dopt,i assumed to be realized for full aromatic systems (for CC and CN bonds, dopt,CC = 1.388 Å and dopt,CN = 1.334 Å), and di,j are the experimental or computed bond lengths taken into calculation. For pi-electron systems with heteroatoms the parameters: dopt and α, can be found in a collection of papers [81, 83,84,85,86,87]. The utility of the HOMA approach is that it can be applied to describe pi-electron delocalization of any pi-electron part of a molecule.
Another way to study the SE on a transmitting moiety may come from magnetic studies of pi-electron systems. Introduced by Schleyer et al., NICS index (nucleus independent chemical shift) [88] is defined as a negative value of the absolute shielding calculated in the geometric center of the ring system. It can also be calculated at other points inside [89] or around molecules [90]. One of the possibilities is to estimate the NICS value 1 Å above the molecular plane, NICS(1), [91] or 1 Å along the principal axis (zz) perpendicular to the ring plane as NICS(1)zz [90]. It should be stressed that NICS values describe only changes in local aromaticity, i.e., of a given ring and depend on both the size of the ring as well as the neighboring parts of the ring.
In addition to the aromaticity indices mentioned above, there are other criteria that can be used to determine the SE on aromaticity of the transmitter. For example, based on electron delocalization, one can determine the MCI (multicenter bond index) [92] and the PDI (para delocalization index) [93] for six-member rings as measures of impact of the substituent as well the FLU (aromatic fluctuation index) [94]. Some help in understanding of the SE on aromatic systems can come from their reactivity properties estimated by means of the harmonic oscillator stabilization energy (HOSE) [95, 96].
Classification of the substituent effect
In general, the substituent effect can be considered in several possible ways of understanding this term, as shown in Scheme 3. The most commonly used way, already proposed by Hammett [1], is called the classical or traditional SE and describes the relationship of the properties of the “reaction site” Y (the fixed group in a given series) with various properties of substituents in the disubstituted X-R-Y system. Another way of using the SE term is the description of mutual dependences between various properties of the fixed group Y, caused by changes of substituents X. Another application of this term is based on the investigation of the influence of substituents X (or of both, X and Y) on the properties of the transmitting moiety R. Finally, one more aspect of the SE is observed, known as the reverse SE, when changes in electronic properties of the substituents X result from the nature of the “reaction site” Y (as well as the rest of the molecule R-Y). This type of interaction had already been noticed by Hammett [29] who proposed different values of substituent constants for the para-substituted nitro group in phenol and benzoic acid. In some systems where there is a problem of additivity or non-additivity of the SE, other classification models may be used [97, 98].

General model approach to the substituent effect. Graphical abstract reprinted from Phys Chem Chem Phys 18:11,711–11,721 (2016) [47] with permission from the PCCP Owner Societies. Copyright (2016) with permission from Royal Society of Chemistry
Classical substituent effect
The classical substituent effect in nitro derivatives allows to analyze the changes in structural and electronic properties of the nitro group (as the reaction site) caused by the substituents X, i.e., in X-R-NO2 systems. The strength of substituent effect on properties of the NO2 group depends strongly on a few factors: electronic character of substituent, type of transmitter, nature of interactions, medium in which the interactions are studied, as well as conformation of the nitro group with respect to the ring. The classical effect in nitro compounds has been studied for systems with different transmitting moieties R and various substituents X, the general scheme X-R-NO2.
Changes in properties of nitro group can be characterized by its structural parameters (ONO angle, φ, NO bond lengths, dNO, and the length of the linking CN bond, dCN), electronic densities, atomic charges, and reaction energies. As mentioned above, the properties of the nitro group also depend on its conformation. For example, interaction of the nitro group with a second substituent, LiO, in nitrophenolate, weakly affects dCN and dNO changes in comparison to unsubstituted nitrobenzene, as shown in Fig. 2 [3]. Electron density seems to be the more sensitive indicator, estimated by Bader’s bond ellipticity (ϵ) (marked in red). Substitution of LiO- leads to a significant increase in ϵ(CN), about ~ 60%, compared with the unsubstituted system, when the nitro group is coplanar with the benzene ring (Fig. 2). This indicates strong interactions in LiO-Ph-NO2 through resonance. The opposite is true in the case of the perpendicular conformation of the nitro group, when the resonance effect disappears, and thus ϵ (CN) values decrease.

Mayer bond orders and Bader’s bond ellipticities in percent of the values for H2C=CH2 (ϵ = 0.4181, H2C=NH (ϵ = 0.2261, and NH=O (ϵ = 0.383) for nitro and nitroso derivatives. Reprinted and modified from J Org Chem 60:6744–6755 [3]. Copyright (1995) with permission from the American Chemical Society
Furthermore, the results of the analysis of ϵ(CN) values in coplanar LiOPhNO2 and LiOPhNO systems show that increasing the resonance effect by introduction of LiO is stronger for the nitro than for the nitroso group. Although CN bond has more π-character in PhNO2 than in PhNO, as indicated by ϵ(CN) values, a stronger resonance effect is observed for the nitroso group, both for substituted (LiO) and unsubstituted systems [3]. However, a comparison of the variability of charge distributions on the N and O atoms of nitro and nitroso groups in LiO-R-NOX systems (marked in blue, Fig. 2) indicates a larger electronegativity and inductive effect of the nitro group than the nitroso group in intramolecular interactions with LiO. This confirms the higher negative oxygen charge and the large positive nitrogen charge in -NO2 compared with -NO.
Changes in the electronic character of the nitro group induced by the substituent are well characterized by cSAR(NO2) values. Attachment of the electron-donating amino group, or its anion NH−, to a nitrobenzene system leads to an increase of electron-accepting power of the nitro group. It is well evidenced by more negative values of cSAR(NO2) in para-nitroaniline (pNA) and para-nitroanilide anion (pNA anion) than in nitrobenzene, as shown in Table 3 [25]. A comparison of the absolute cSAR(NO2) values in pNA and nitrobenzene (coplanar conformation of NO2) gives a difference of 0.073 e, i.e., the addition of an amino group increases the electron-acceptor character of NO2 by 50%. Similar changes were found for the amino group in aniline and p-nitroaniline (0.069 e); the nitro group increases the electron-donating ability of the amino group. Moreover, in the p-nitroanilide anion, the electron-accepting character of the nitro group is about 2.5 times greater than in nitrobenzene. In addition, these EA changes of the nitro group depend strongly on its rotation relative to the ring. The nitro group loses as much as 0.049 e due to rotation in pNA.
The properties of the nitro group can be modified by introducing various substituents into the system under study as well as modifying the properties of a given substituent through its intermolecular interactions. For example, modification of the ED power of the amino group by intermolecular H-bonding with HF/F− (N···HF, H···FH, N−···HF, and NH···F− interactions) usually enhances the EA character of the nitro group, compared with an unmodified group, as shown in Fig. 3 (structures a–c) [15]. This enhancement clearly depends on both the strength and type of hydrogen bonds and the degree of twisting of the nitro group relative to the ring. The largest increase in the electron-accepting ability of the nitro group is observed for the “flat molecule,” while the smallest for the nitro group perpendicular to the plane of the ring; the latter case can be explained by a significant lowering of the resonance effect between the perpendicular NO2 group and the NH2 group or its modification by H-bonding. Only H2N···HF interactions (Fig. 3a) increase cSAR(NO2) (lower EA ability) due to the charge transfer from the lone pair of the NH2 group nitrogen atom to the region of intermolecular interactions; that is, this nitrogen atom is a hydrogen bond acceptor. In the case of p-NO2-anilinium cation, the EA power of the nitro group is the lowest (its cSAR value is − 0.019), since NH3+ is also an electron-accepting substituent, their σp constants are 0.78 and 0.60 [17], respectively. As can be seen in Fig. 3, modification of the properties of the amino group causes changes in EA power of the nitro group by about 0.5 cSAR units, which corresponds to 2.5 units of σ [15].

Graphical illustration of changes in cSAR(NO2) values for p-X-nitrobenzene derivatives (X = H, NH3+, NH2, and NH− and modified by the intermolecular H-bonding with HF/F−). Reused and modified from [15], this work is licensed under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/)
The nature of the transmitter and the substitution position have a significant impact on the strength of the substituent effect in nitro derivatives. Recently, the results of comprehensive studies on the effect of substituents on the properties of the nitro group in systems with different transmitters, R, have been presented, where R is aromatic, benzene (BEN) [48, 49]; olefinic, cyclohexa-1,3-diene (CHD) [54]; and aliphatic, bicyclo[2.2.2]octane (BCO) [53]; a wide range of substituents X (NMe2, NH2, OH, OMe, CH3, H, F, Cl, CF3, CN, CHO, COMe, CONH2, COOH, COCl, NO2, NO) in 3- (meta-type) or 4- (para-type) positions of R was used (as shown in Scheme 4).

General scheme of studied disubstituted X-R-NO2 systems: R = BEN (left), BCO (middle), and CHD (right) of nitro derivatives; X = NMe2, NH2, OH, OMe, CH3, H, F, Cl, CF3, CN, CHO, COMe, CONH2, COOH, COCl, NO2, NO
Electronic (cSAR, pEDA, sEDA) and geometrical parameters were used to describe the properties of the nitro group, while the substituents were characterized using both quantum chemistry based descriptors (cSAR, SESE) and Hammett constants (σ). It should be emphasized that cSAR is a universal “tool”—it can be used to describe features of both the reaction center (nitro group) and the substituent. Therefore, it is possible to easily compare changes in their properties (same scale). Figure 4 presents relationships between cSAR(NO2) and cSAR(X) for all systems shown in Scheme 4 [48, 53, 54]. The presented dependences are a good illustration for understanding and comparing the classical SE in the investigated systems. It is clearly seen that the ranges of variability of cSAR values on both axes and the slopes of the determined linear equations depend on the type of transmitter and the position of the substituent in the system. A significantly smaller range of cSAR(NO2) variability for 4-X-BCO-NO2 derivatives than in other systems indicates a different nature of the substituent effect compared with other cases. As already mentioned above, the slopes of linear equations (Hammett-type relations) inform about the sensitivity of the system to the substituent effect; in other words, they allow to measure intensity of the communication between X and NO2. The values of the determination coefficients for the relationships shown in Fig. 4 enable a deeper look at the presented results.

Dependence cSAR(NO2) on cSAR(X) for substituted BEN, CHD, and BCO nitro derivatives. Charges estimated using Hirshfeld method, B3LYP/6-311++G(d,p)
First, the obtained slope values of linear dependences reveal a different nature of intramolecular interactions between the nitro group and X in aromatic and olefinic series compared with aliphatic systems [48, 49, 53, 54]. For para-type substituted systems, the absolute slope values decrease in sequence from CHD (− 0.473), through BEN (− 0.400) to BCO (− 0.113), indicating the strongest SE on nitro group in CHD derivatives. The same sequence was found in the case of cSAR(NO2) dependence on SESE(X) [54]. These changes in slopes can be explained by different mechanisms of X···NO2 interactions in the investigated systems. In BEN and CHD systems, they are realized by a combination of resonance and inductive effects [54], whereas in BCO the interactions are mostly inductive [99]. This confirms the significant impact of the type of transmitting moiety on charge transfer between the nitro group and substituents.
Furthermore, the slopes of cSAR(NO2) to cSAR(X) correlations for 3-X-R-NO2-substituted CHD and BEN derivatives are about 60% lower than those obtained for the 4-X-R-NO2 system [48, 54]. This documents the stronger SE from para-type positions than from meta-type and thus confirms the important role of the substituent position in interactions with the nitro group.
A deeper look at the nature of the substituent effect on the nitro group comes from analysis of its interactions in pi-electron systems separated into two subgroups: with ED and EA substituents. For para-type BEN and CHD derivatives, when the dependences of cSAR(NO2) on SESE(X) or cSAR(X) and dCN on SESE(X) for EA and ED substituents are treated separately, the slopes of regression lines for ED-substitued systems are higher than for EA-substituted series [48, 54], as shown in Table 4. The range of variability of electronic properties of the nitro group is greater due to the presence of ED substituents than EA ones. Similar results are observed based on the correlations of changes in CN bond length caused by substituents, dCN on SESE (Table 4) [54]. Positive SESE values for para-type systems with ED substituents indicate that interactions between them and the nitro group stabilize these systems. In addition, according to the R2 values obtained, the similarity model is fulfilled much better for derivatives with ED substituents than EA ones. This indicates a stronger interaction of ED substituents than EA with nitro group, caused by the resonance effect and the formation of a quinoid-like structure (Scheme 1a). In the case of meta-type substituted derivatives, these interactions are significantly weaker. In general, the sensitivity of SE is again higher for CHD than for BEN systems, both for para- and meta-type substitutions.
A deeper analysis of the electronic structure of the nitro group leads to similar conclusions. Since the nitro group is planar, the pEDA/sEDA methods can be used to describe its sigma and pi electron structure [49]. Dependencies of pEDA(NO2) on cSAR(X) for derivatives of para- and meta-substituted nitrobenzene [49] and para-type cyclohexadi-1,3-ene systems, as presented in Fig. 5, provide information on the magnitude of the pi-charge transferred from substituents to the nitro group. The largest changes in the pi-charge transmission are observed for para-type substituted CHD systems. Furthermore, the transmission of pi-electrons from X in the para position to the nitro group through cyclohexa-1,3-diene is almost 30% higher than through benzene, which indicates high lability of pi-electrons in the diene part (Fig. 5b). Moreover, unlike benzene derivatives, there is hardly a distinction between the effect of EA and ED on the pi-electron structure of the nitro group.

Dependence of pEDA(NO2) on cSAR(X) for a meta- and para-substituted benzene and for b para-type substituted benzene and cyclohexa-1,3-diene nitro systems. Charges were calculated by NBO approach. Reprinted and modified with permission from J Phys Chem A 121:5196 (2017) [49]. Copyright 2017 American Chemical Society
Slopes of dependences of pEDA(NO2) on cSAR(X) once again confirm significantly greater changes in the pi-electron structure of the nitro group in para-substituted nitrobenzenes than in meta ones. This indicates that the nitro group attracts less pi-electron charge from substituents in the meta than in the para position. Considering the regressions separately for para-substituted ED and EA systems, and their slopes, it can be stated that the impact on the pi-electron structure of the nitro group of ED substituents is three times greater than that of EA substituents and even ~ 8 times stronger than that of meta ones. This strongly supports the importance of the resonance effect in para-substituted nitrobenzenes. A very similar picture is obtained when SESE is used as characteristic of the SE [49].
Additional information on the classical substituent effect on the nitro group may come from analysis of its sigma-electron structure, characterized by sEDA(NO2). Dependences of sEDA on σ constants for meta- and para-substituted nitrobenzene [49] and nitro-cyclohexa-1,3-diene systems are shown in Fig. 6. In the case of nitrobenzene derivatives, the relationships show a very characteristic picture. Despite the not sufficiently good determination coefficient (R2) for the para series, the substituent effect is stronger for the meta-type series (slope − 0.019) and almost five times weaker for the para ones (slope − 0.004). A similar conclusion can be drawn from the comparison of sEDA(NO2) for the meta-substituted series with respect to the para ones [49]. This is in line with a well-known fact that in meta-substituted systems the inductive effect, which is a rather short-range interaction, dominates. Less clear correlations (due to low R2 values) relate to nitro-cyclohexa-1,3-diene derivatives, which probably results from a strong pi-electron delocalization in these systems. In addition, in CHD derivatives, as already mentioned, the resonance effect is greater than in benzene systems.

As mentioned earlier, properties of the reaction center, the nitro group, also depend on the nature of transmitting moieties. This is well illustrated by comparing of the substituent effects on the charge of the nitro group in 4-X-substituted benzene and cyclohexadiene nitro derivatives, as shown in Fig. 7. Slope values of linear regression indicate an almost ~ 1.5 times stronger substituent effect for cyclohexadiene than for benzene nitro systems [100]. In addition, it was noticed that this effect is weaker when the reaction site is a strongly electron-donating group such as OH (Fig. 7). The results of NPA [76] Weinhold charge analysis on the nitro and hydroxyl group revealed greater sensitivity to the influence of the substituent by the electron-acceptor reaction center (NO2) than the electron donor (OH).

Correlation of the NPA charges at the NO2 (a) and OH (b) groups in cyclohexadiene derivatives on those of benzene derivatives, where X = BH2, B (OH)2, C, CH, CH3, CHO, Cl, CN, COCH3, COCl, CONH2, COOCH3, COOH, F, H, NH2, N (CH3)2, NO, NO2, OCH3, OH). Reused from [100]; this work is licensed under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/)
Substituent effect on various properties of the nitro group
Substituents also affect mutual dependences between various properties of nitro group, such as structural and electronic parameters. This is well shown by the changes in the pi and sigma electron structure of the nitro group in nitrobenzenes caused by substituents, that is, the dependence of sEDA(NO2) on pEDA(NO2), as presented in Fig. 8 [49]. In meta-substituted systems, the variability of pi and sigma electron structures is comparable (slope close to 1.0). In the case of para-substituted nitrobenzenes with EA substituents (negative R resonance constants), changes in the pi-electron structures of the nitro group are about 25% larger than in its sigma-electron population (the slope is 1.25). However, ED substituents (with constant positive resonance R values) cause more than 9 times greater variability of the pi-electron population than the sigma-electron population in the nitro group. This confirms again the important role of resonance effect in an intramolecular pi-electron transfer to the nitro group from substituents. A similar view results from the comparison of structural parameters of the nitro group [48].

Dependences of pEDA(NO2) on sEDA(NO2) with separation into three groups of nitrobenzene derivatives: para system with substituents characterized by R < 0 (point for Me is not taken into account) and R ≥ 0, and all meta series. Reprinted with permission from J Phys Chem A 121:5196 (2017) [49]. Copyright 2017 American Chemical Society
Experimentally, the variability of electron-accepting properties of the nitro group is well illustrated by measurements of polarographic half-wave potentials, E1/2, reversible one-electron reduction of substituted nitrobenzene derivatives carried out in aprotic solvents (to avoid subsequent kinetics) [21] and by their formal reduction potentials, E0, measured in dry acetonitrile with tetra-n-butylammonium hexafluorophosphate as supporting electrolyte [101]. It has been shown above that any modification of the electronic properties of a given X substituent changes cSAR(NO2), which means that X directly affects the properties of the nitro group. Qualitative equivalence between cSAR(NO2) and E0 for para-X-substituted nitrobenzene derivatives [15] is shown in Fig. 9. Firstly, it confirms again that the cSAR concept can be used to obtain quantitative characteristics of electronic properties of the nitro group. Moreover, the linear correlations between the experimental potentials E0 (or E1/2) and the theoretical cSAR(NO2) document that the electron-attracting ability of the nitro group significantly depends on the properties of the substituent. In addition, the results of a deeper analysis of the relationship between cSAR(NO2) and E0 (cc = − 0.957) again allows to distinguish two groups of substituted nitro derivatives: with electron-donating substituents (green) and with electron-accepting groups (blue). This confirms the different character of interactions of the nitro group with ED and EA substituents. Greater variability of cSAR(NO2) values is observed for ED substituents than for EA substituents, which means stronger interactions of the nitro group with the former.

Relationship between formal reduction potential (E0) and cSAR(NO2) values of the nitro group for para-nitrobenzene X derivatives (X = NO2, CN, CHO, COOMe, COMe, CF3, Cl, H, Me, OMe, NH2, and NHMe). Reused from [15]; this work is licensed under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/)
Reverse substituent effect
The reverse substituent effect works in the opposite direction to the classical one and describes the influence of the reaction site (Y) on the electronic properties of the substituent (X) in X-R-Y systems [15]. This effect may be well characterized using the cSAR(X) model, which allows to quantitatively measure changes in the electronic properties of a substituent, depending on substitution position, type of transmitter, and nature of the reaction site. In the case of nitro-substituted systems, the reverse SE refers to the effect of the nitro group (as a reaction site) on the electronic properties of each substituent at a given position of the transmitter.
The reverse SE of the nitro group can be well illustrated by cSAR(X) values for nitro-X-substituted benzene, cyclohexa-1,3-diene and bicyclo[2.2.2]octane derivatives and cSAR(X) values for monosubstituted systems, as shown in Table 5 [48, 53, 54, 102, 103]. For this purpose, substituents differing in electronic properties (X = NO2, COOH, H, NH2, NMe2) were selected. In addition, Hammett σp constants [17] also are given in Table 5. Comparison of the cSAR(X) values determined for disubstituted systems, X-R-NO2, with those calculated for monosubstituted systems, X-R, allows quantitative description of changes in electronic properties of a substituent due to the presence of the nitro group. It should be noted that for systems collected in Table 5 the variation range of σp constants is 1.61 and corresponds to the variation range for cSAR values from 0.259 to 0.345 for monosubstituted X-BEN and 4-X-CHD-NO2 derivatives, respectively. This means that the nitro group, as a reaction site, significantly changes the properties of substituents.
Moreover, taking into account the individual substituents, the results of the analysis of cSAR(X) values show that, regardless of the type of transmitter, greater variability of the cSAR values (Δ) is apparent for the ED substituents than for the EA substituents. For example, for nitrobenzene derivatives, for ED substituents the cSAR(X) values are ~ 78% and ~ 40% more positive for para and meta substitutions, respectively, than in monosubstituted systems. This shows that the presence of the nitro group leads to an increase in the donation ability of ED substituents by ~ 78% due to the strong resonance effect. The opposite phenomenon is observed in the case of EA substituents. Their cSAR(X) values range from ~ 30% (NO2-BEN-NO2) to ~ 50% (COOH-BEN-NO2) less negative in disubstituted systems than in monosubstituted ones, which is indicative of a reduction of the electron-attracting ability of EA substituents resulting from the presence of a nitro group. Moreover, the strength of the reverse substituent effect of the nitro group decreases in the sequence: CHD, BEN, and BCO, as confirmed by the ranges of cSAR(X) values, which is in line with aforementioned relationships. The above results confirm that the EA/ED ability of the substituent strongly depends on the presence of the nitro group, transmitting moiety as well as substitution position.
Analysis of the reverse substituent effect allows a deeper look at the properties of the nitro group as a substituent. In the CHD and BEN nitro derivatives considered, the absolute values of cSAR of the nitro substituent (in bold in Table 5) are smaller than the cSAR(NO2) determined for nitrobenzene. This means that in CHD and BEN nitro systems, the nitro substituent is less electron-attracting than in nitrobenzene. In the case of NO2-BEN-NO2 derivatives, its electron-attracting ability is reduced by about 33% for the para position and 27% for the meta position compared with that in nitrobenzene [48]. The opposite is true for aniline derivatives, where the presence of the amino group (as reaction site) causes an increase in the EA ability of the nitro group (up to 41%) for para substitution and a decrease (by 9%) for meta [47].
As already mentioned, the reverse substituent effect describes changes in the electronic properties of the substituent resulting from the position in the transmitting moiety, or due to the nature of the fixed group (reaction site). The first case can be represented by different substituent constants for para and meta positions, whereas the second case includes the substituent constants σ+ and σ− which describe substituent effects in molecules with positively and negatively charged reaction sites, respectively. It can therefore be said that the reverse substituent effect can also be documented experimentally. For this purpose, acidity constants (pKa) can be used. As mentioned above, Hammett used them to determine substituent constants [28, 29]. The pKa values in water at 25 °C of meta- and para-nitrobenzoic acid were 3.493 and 3.425, respectively, compared with 4.203 for benzoic acid itself [104]. Based on these values, Hammett proposed σ values of 0.710 for meta and 0.778 for para-nitro substituent [28]; these σ values are commonly used, rounded to 0.71 and 0.78, respectively (Table 1). This is the simplest way to show that the properties of the nitro group depend on the substitution position. Obviously, this is due to the different proportions of induction and resonance effects in these positions. The most fruitful treatments of the NO2 electronic effects on various systems (aliphatic and aromatic) and processes are presented by Shorter [22].
In addition, a clear impact of the nitro group on the rest of the molecule has been documented for 4- and 5-nitro-substituted salicylaldehyde derivatives [105].
Substituent effect on transmitting moiety in nitro derivatives
Substituent effect can significantly affect the geometry of the transmitting moiety. When the nitro group is attached to the benzene ring (nitrobenzene), changes in CC bond length are small [106, 107]. The presence of the nitro group in the aniline system in various positions affects CC distances, some lengthen by 0.01 ÷ 0.03 Å, and others shorten by about the same amount (in relation to those calculated for benzene), as shown in Table 6 [106]. Similar changes were observed in experimentally determined values of CC bond length for p-nitrophenol and p-nitrophenolate systems [38].
Interestingly, analysis of the ranges or standard deviations of the benzene ring bond length allows to describe the changes in the pi-electron structure of the benzene ring caused by strongly electron-accepting or electron-donating substituent. Comparison of experimental and theoretical data for nitrobenzene and aniline shows that the nitro group affects the pi-electron structure of the ring in its benzene derivative ca. 2.8 times more weakly than the amino group (see Table 6). This can be explained by the well-known fact that the pi-electron structure of benzene is very stable, so withdrawing electrons (NO2) is much more difficult than pushing electrons (NH2) on unoccupied molecular orbitals [107]. Furthermore, the joint interactions of amino and nitro groups at the meta and para positions in nitroaniline, compared with nitrobenzene, cause a decrease of delocalization in the benzene ring by ~ 4 and up to ~ 6.5 times, respectively [107].
The nitro group affects aromaticity of the transmitting moiety. The extent of pi-electron delocalization can be measured by ring reactivity. The effect of nitro and amino groups on the degree of activation or deactivation of benzene rings for executing an electrophilic attack has been demonstrated [106] for the geometries shown in Table 6. For this purpose, the electrostatic potential (calculated using SCF STO-5G wave functions) was used. It has been shown that the nitro group deactivates the ring, there is no negative electrostatic potential above or below the ring (see Fig. 4 in Ref. [106]), which confirms the electron withdrawing power of the nitro group. In aniline, the amino group has the opposite effect leading to the activation of the benzene ring. In nitroaniline, when both substituents are present simultaneously, the tendency to deactivate the ring is dominant; however, the directional properties of the amino group to the ortho and para positions are visible [106].
The substituent effect of nitro and amino groups on the benzene electronic system has also been studied using quantitative Kohn-Sham molecular orbital (KS-MO) theory and corresponding energy decomposition analyses (EDA) [24]. In nitrobenzene, the C–N bond formation is associated with a Pauli repulsive interaction between occupied orbitals on the Ph• and •NO2 fragment, as shown in Fig. 10. It has been documented that during C–N bond formation, the πLUMO of the NO2 group accepts electrons from the pi-system of the benzene ring which leads to lowering of the πHOMO energy on the phenyl ring and its deactivation. Interestingly, the πHOMO energy of nitrobenzene (− 7.0 eV) is almost 2.5 times lower than πHOMO in aniline (− 4.6 eV), which explains the lower reactivity of nitrobenzene. Furthermore, it has also been confirmed that the rotation of the nitro group by 90° relative to the benzene ring plane leads to a loss of donor-acceptor interactions with the ring.

MO diagrams for the C–N bond formation from Ph• and •NO2 into nitrobenzene. The energies (eV) of the molecular or fragment orbitals are given in black and the gross electron population of the fragment orbitals in red. Reused and modified from [24] this work is licensed under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/)
In addition, results of the Voronoi deformation density charge (VDD) analysis show that the C–N bond formation in nitrobenzene causes the largest deactivation in the ortho and para positions in nitrobenzene, which leads to the meta directing effect of the nitro group for electrophilic aromatic substitution, as shown in Fig. 11 [24].

VDD atomic charges (mili-electrons) of benzene and nitrobenzene. Reused and modified from [24]; this work is licensed under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/)
Changes in the pi-electron delocalization of the ring caused by a substituent are also measured using geometric, magnetic, and electron aromaticity indices. Addition of one substituent to the benzene ring very weakly influences its pi-electron structure, as shown by the small variability of aromaticity indices (geometric HOMA, magnetic NICS(1)zz, electronic PDI and FLU) for nitrobenzene, as presented in Table 7. This is in line with the tendency of aromatic systems to preserve the initial pi-electron structure during the substitution reaction [113]. More pronounced aromaticity changes are observed for nitro-substituted imidazole and pyrazole systems [115].
As shown, the nitro group, a strongly electron-attracting group, can significantly affect pi-electron delocalization of the aromatic ring in intramolecular interactions with substituents from different positions. The substituent effect of the nitro group on ring aromaticity was investigated for mono-, di-, tri-, and poly-substituted nitrobenzene and aniline derivatives [116]. The results of the analysis of HOMA and NICS values allowed to show how the number of nitro groups and their interplay can affect aromaticity of the ring, as shown in Table 8.
It should be noted that for mono- and di-nitro-substituted systems, such as 1,3- and 1,4-dinitro-benzenes, the nitro group is coplanar with the benzene ring, increasing conjugation with its pi-electrons. However, when the nitro groups are in adjacent position (1,2-dinitro-benzene and polynitrobenzenes), they exhibit steric repulsion, which is confirmed by torsion angle values between the plane of the ring and that of the NO2 group. The higher degree of rotation of the nitro group causes a reduction of its pi-conjugation with the ring. However, no clear changes in aromaticity were found between these two types of nitro derivatives (Table 8). In contrast, a significant reduction in the ring aromaticity of these compounds results from the presence of an amino group, especially when several such groups are added. This is due to both the strong interaction of the nitro group with ED substituents and intramolecular hydrogen bonds. In the latter case, the largest decrease in pi-electron delocalization is for 1,3,5-trinitro-2,4,6-triamino-benzene (HOMA = 0.2152).
The determination of the effect of intra- and intermolecular interactions on ring aromaticity in para-substituted nitrobenzene derivatives was performed using experimental (Cambridge Structural Database, CSD) [6] and calculated (BLYP-D3/TZ2P) structural data [25]; aromaticity was described by the HOMA index. Considering CSD data, it was found that the nitro group lies in the plane of the benzene ring only for a small number of molecules. The determined maximum twist angle (φ, relative to the ring planes) is ~ 40°, while its average value is 7.3° mainly due to intermolecular interactions in the crystals. The HOMA values obtained for this data set are in the range of 0.88 to 1.00. Dependences of HOMA on the rotation angle of the nitro group for para-substituted nitroaniline with intermolecular N···HF, H···FH, N···HF, and NH···F− interactions (quantum chemistry calculations results) are shown in Fig. 12. HOMA values increase with the rotation of the NO2 group for all types of H-bonded para-nitroaniline complexes. This means that weakening of the resonance effect by rotation of the NO2 group causes an increase of aromaticity. Furthermore, these results fully coincide with the observations found for the data set for crystal structures.

Dependences of HOMA values on rotation angle φ of NO2 group in para-nitroaniline complexes (for equilibrium structures, except for HNH···F− interactions). Reused from [25] Crystals 2016, this work is licensed under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/)
The aromaticity of 1-nitronaphthalene and its derivatives with secondary amino groups in para positions (4, 5) as well as 1-formyl analogues was also examined [117]; for this purpose, the HOMA index was used. The HOMA index of the ring with a substituent, calculated for the optimized structures of naphthalene and its 1-NO2 and 1-CHO derivatives is 0.7840, 0.7614, and 0.7322, respectively. However, it should be added that the nitro group is twisted by 35° with respect to the ring plane, while the formyl group is almost coplanar with the ring. This explains the weaker than expected resonance effect of the nitro group compared with that of the CHO group. Changes in ring aromaticity (HOMA) for different conformations of nitro and formyl groups (α, angle of rotation of the group in relation to the ring plane) are shown in Fig. 13.

Dependence of HOMA value on the angle between the naphthalene aromatic ring and the CHO (NO2) plane for a naphthalene-1-carbaldehyde (unfilled triangle, broken line) and 1-nitronaphthalene (filled circle, solid line) and for (b) 4,5-bis (dimethylamino)naphhtalene-1-carbaldehyde (filled triangle, broken dotted line) and 4,5-bis (dimethylamino-1-nitronaphthalene (unfilled circle, dotted line). Reprinted with permission from J Phys Chem A 121:2627 (2017) [117]. Copyright 2017 American Chemical Society
As in benzene derivatives (Fig. 12), HOMA values gradually increase as the α angle increases. In the case of small deviations from planarity, the nitro group (filled circle in Fig. 13a) affects the naphthalene ring more strongly (ΔHOMA = 0.05) than for angles greater than 40° (ΔHOMA = 0.02). This confirms once again that rotation of the nitro group decreases its pi-electron acceptor ability due to disappearance of the pi-conjugation with the ring. Thus, less effective changes in HOMA values are observed. In addition, the rotation of the nitro group by as little as 20° leads to its EA ability being weaker than in the CHO group (Fig. 13a). This is clearly related to a lower EA power of CHO than NO2. For para-substituted derivatives, a significant decrease in aromaticity is observed (Fig. 13b). This is due to resonance interactions between the nitro group and the NMe2 group in the para position. In optimal systems, the angle between the NO2 (or CHO) group and the ring plane is the result of competition between repulsive and attractive peri interactions on the one hand, and resonance interactions with para-amine groups on the other hand.
Systematic SE studies on the pi-electron structure, estimated by HOMA, were carried out for meta- and para-type disubstituted derivatives of benzene (BEN) and cyclohexa-1,3-diene (CHD) with different reaction sites [49, 54]. For the analyzed nitro derivatives, the impact of the nitro group on pi-electron structure can be represented by the linear dependence of HOMA on SESE, as shown in Fig. 14.

Dependences of HOMA on SESE for 4-X-CHD-NO2 and 4-X-BEN-NO2 series. Reused and modified from [54]; this work is licensed under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/)
The presented relationships confirm the different effects of electron-donating and electron-accepting substituents for both series, which have already been mentioned earlier. Moreover, independent of the type of transmitter, pi-electron delocalization is much stronger when substituents exhibit an opposite electronic nature to the nitro group (SESE > 0), which is confirmed by very good correlations between HOMA and SESE (R2CHD = 0.920; R2BEN = 0.965) in Fig. 14. In the case of derivatives with similar electronic properties of the substituent and nitro group (SESE < 0), the correlations are significantly worse. However, in the case of CHD derivatives, the removal of two points (X = NO2 and CF3; HOMA are 0.274 and 0.224, respectively) leads to a regression line with a slope of − 0.030, and the determination coefficient is 0.921. The greater pi-electron delocalization for CHD systems than for BEN ones is due to the greater lability of pi-electrons through the CHD diene part, while the benzene ring tends to preserve its pi-electron structure. In the case of meta-type BEN and CHD systems, the picture is not clear.
A deeper look at the electronic structure of the ring and nitro group affected by the SE can be derived from an analysis of changes in their sigma and pi electron population estimated as sEDA and pEDA, respectively. Nitro group is a strongly sigma and pi electron-attracting substituent; hence, it can substantially affect pi-electron structure of the ring.
The flow of pi-electrons between the nitro group and the aromatic ring in nitrobenzene derivatives is illustrated by dependences of pEDA(NO2) on pEDA(R) shown in Fig. 15 [49]. The use of these models allows to estimate how strongly pi-electrons are pulled out of the ring by a nitro group as a result of the resonance effect. The presented linear correlations (Fig. 15) indicate a stronger communication of the pi-electron structure between the NO2 group and the ring in para-substituted nitro derivatives (the higher slope values) than in the meta ones. Furthermore, in the case of para-nitrobenzene systems with ED substituents (σ ≤ 0), the changes in the pi-electron structure of the nitro group are approximately two times greater than in the case of derivatives with EA substituents (σ > 0). Such differences are not observed in 4-X-CHD nitro systems (unpublished results).

Dependence of pEDA(NO2) on pEDA(R) for all meta- and para-nitrobenzene derivatives with separated regression line for ED (σ ≤ 0) and EA (σ > 0) substituents. Reprinted with permission from J Phys Chem A 121:5196 (2017) [49]. Copyright 2017 American Chemical Society
In addition, the relationship between the pi-electron and sigma-electron population of the benzene ring for nitrobenzene derivatives, as shown in Fig. 16, is very interesting. It reveals that regardless of the position of the substituent and the properties of the substituent, the changes in pEDA(R) and sEDA(R) parameters of the ring are mutually correlated, but only in the subgroups with the same electronegativity of the atom linking the substituent with the benzene ring. The increase in sEDA(R) is always associated with a decrease in pEDA(R).

Dependence of pEDA(Ring) on sEDA(Ring) for meta- and para-substituted nitrobenzene derivatives. For red points, the sequence is Me, CN, CF3, CONH2, COOH, COMe, COCl, and CHO. Reprinted with permission from J Phys Chem A 121:5196 (2017) [49]. Copyright 2017 American Chemical Society
Considering the aromaticity of the nitrobenzene ring expressed by HOMA [49], the obtained slope of linear equation HOMAmeta vs HOMApara (equal to 0.561) shows that substituents in para positions have an almost two times stronger effect on the aromatic character of the ring than those in the meta positions. The results of research on nitronaphtholate systems (Fig. 17) also confirm that para-type systems usually show a greater variation in the aromaticity of individual rings than for meta-type interactions [118]. This confirms the stronger resonance interaction between NO2 and O− in the first case. Furthermore, the para-systems are characterized by a significantly higher mean SESE value than meta ones, showing a greater stability of para-type naphthalene derivatives.

Dependence of NICS(1)zz on HOMA for each ring in nitronaphtholates. Open circles stand for para-type and filled circles for meta-type. Reprinted and modified from J Phys Org Chem 20: 297–306 (2007) [118]. Copyright 2007 with permission from John Wiley and Sons
Analysis of the impact of the nitro group on aromaticity of the transmitting moiety has also been considered for heterocyclic compounds. For nitro carbazole derivatives, substitution of the nitro group causes pronounced changes in aromaticity (as shown by the HOMA index) in the pyrrole (middle) ring, but only weak changes in the benzene rings, as shown in Fig. 18 [119]. Moreover, aromaticity of the pyrrole ring is more significantly affected by the N-substitution than the C-substitution.

Calculated HOMA values for individual carbazole rings and its nitro derivatives. Reused and modified from [119]; this work is licensed under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/)
Furthermore, changes in the pi-electron structure of the pyrrole ring depend significantly on the spatial (planar or perpendicular) orientation of the nitro group. The strongest reduction in aromaticity of this ring occurs when the nitro group is in the same plane as the ring. This is due to the mesomeric (resonance) effect of the nitro substituent, as shown in Scheme 5, which is not observed on the rotation of the NO2 group.

Two canonical forms of N-nitro-substituted carbazole. Reused from [119]; this work is licensed under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/).
The introduction of further nitro groups (at C(3) and C(6) atoms) leads to a slight stabilization and increase in HOMA values for the pyrrole ring, as shown in Table 9.
Another heterocyclic compound is purine (Scheme 6a), a parent structure for many biologically important compounds, e.g., adenine and guanine. The effect of the nitro group on aromaticity of five- and six-membered rings in 9H, 7H, 3H, and 1H purine tautomers was studied in detail [120]. For this purpose, the HOMA index was used, the results obtained are shown in Table 10. The presence of the nitro group at the C6 position (Scheme 6b) leads to an increase in the range of variability of all HOMA values compared with unsubstituted purine. The opposite effect is observed in the case of the amino group, i.e., in adenine (Scheme 6c). In addition, unlike in adenine and purine, for 6-nitropurine the most stable tautomer is 7H, which is stabilized by intramolecular H-bond NO···H [120].

Structures, and numbering of atoms, of the most stable tautomers of purine (a), 9H 6-nitropurine (b), and 9H adenine (c)
Furthermore, the substituent effect of the nitro group in various positions (C2-, C8-, and N-) of adenine and purine tautomers was also investigated [121]. The obtained SESE values (relative to purine, SESEPU) for nitro and amine derivatives are shown in Table 11. Unlike the NH2 substituent, the NO2 group has been found to stabilize intramolecular interactions (positive SESE values). The substituent effect is more pronounced in the case of substitution C8-X than C2-X. Moreover, nitro substitution can change the stability of tautomers in the purine and adenine series, but 9H tautomers are the most stable in both cases.
Inductive and resonance substituent effects
As mentioned above, the nitro group’s ability to pull out electrons is due to two types of interaction: inductive and resonance. The inductive effect of the nitro group can be demonstrated by pKa values for CH4 and CH3-NO2 systems. Their values, as determined in water, are 48 and 10.2 [22], respectively. Further substitution of H by NO2 leads to a decrease in pKa value to 3.6 for CH2(NO2)2 and 0.2 for CH(NO2)3. For comparison, the acidity of C–H in PhCH (NO2)2 is 3.89, while the pKa values for its m- and p-nitro derivatives are 2.82 and 2.63, respectively (values in water, 20 °C) [22]. Thus, remote NO2 and phenyl play a lesser role than neighboring nitro groups. A very similar decrease in pKa value is observed for fluorinated CH2FCOOH, CHF2COOH, and CF3COOH acids (they are 2.6, 1.3, and 0.5, respectively), in which the strong inductive effect of fluorine manifests [122]. Changes in dissociation constants of a substituted organic acid are the simplest indicator of a substituent effect. For example, the use of pKa values of 4-X-substituted derivatives of bicyclo[2.2.2]octane-1-carboxylic acids enabled the definition of inductive substituent constants, σI [42].
The use of the cSAR and SESE concepts to describe SEs in 4-X-R-NO2 derivatives (Scheme 4) made it possible to study the nature of X···NO2 intramolecular interactions, i.e., their inductive and resonance contributions [53, 79].
The results of studies on mono X-substituted bicyclo[2.2.2]octane showed that the inductive effect is realized mostly via bonds; this was evidenced by the charge flow from the substituent to particular CH2 groups (measured as averaged cSAR(CH2) values), attenuated in a regular ratio, 3:2:1 for ortho, meta, and para-type positions [103]. This ratio is a typical relation assumed for the inductive effect [123]. In addition, it has been shown that differences in cSAR(X) values calculated for substituents in phenyl and BCO derivatives can be used as an approximate measure of the resonance effect of the X substituent.
In disubstituted 4-X-BCO-1-NO2 systems, the nitro group, as the reaction center, changes the influence of substituent X on particular CH2 groups [53]. The inductive effect in these systems is illustrated by dependences of cSAR(CH2) on cSAR(X) and cSAR (NO2) for the vicinal and the more distant CH2 group from X and NO2, respectively, as presented in Fig. 19. The influence of substituents X on the vicinal CH2 fragment (CH2[3]) is ~ 1.7 times stronger than on the more distant CH2 fragment (closer to nitro group, CH2[2]); the corresponding slope values are − 0.184 and − 0.110, respectively. For monosubstituted BCO derivatives [103], these slopes are − 0.190 and − 0.124, respectively, and therefore, the substituent effect on the vicinal part of CH2 is only about 1.5 times stronger than on the more distant one. However, a comparison of the slope values given above reveals a weakening of the inductive SE of the substituent X by the nitro group. With respect to mono-BCO series, this weakening by NO2 is greater for the CH2[2] fragment (~ 11%) than the CH2[3] (2.5%) which sits closer to X [53]. Due to the limited precision of data estimation, these conclusions should be considered qualitatively.

Dependence of cSAR(CH2)[2] (near NO2) and cSAR(CH2)[3] (near X) on cSAR(X) (a), on cSAR(NO2) (b) for 4-X-bicyclo[2.2.2]octane-1-NO2 derivatives, and on cSAR(CH) (c) for X-bicyclo[2.2.2]octane derivatives
Similar relationships considered from the perspective of the nitro group [cSAR(CH2) vs cSAR(NO2)] are shown in Fig. 19b. For the CH2 group closer to NO2 (CH2[2]), the slope of the regression line is ~ 1.5 less than for the CH2 closer to X (CH2[3]). This indicates, as expected, a stronger SE from X on the vicinal CH2 fragment (CH2[3]) than from the nitro group. Furthermore, in the case of disubstituted BCO derivatives, the obtained slopes are significantly smaller than those found for monosubstituted BCO (Y = H), where the slopes of the relationship between cSAR(CH2)[2] or cSAR(CH2)[3] and cSAR(CH) are 2.089 and 2.865, respectively, are shown in Fig. 19c [103]. This shows that substituents reduce the strength of inductive effect of the nitro group on cSAR(CH2) groups by as much as two times [53]. A similar effect was found for Y = COOH (4-X-BCO-1-COOH systems), while a smaller reduction in the inductive effect, compared with Y = H, was observed for the electron-donating reaction centers (Y = OH and NH2).
It is well-known that the nature of the substituent effect depends on the transmitting moiety. The substituent effect on the nitro group in bicyclo[2.2.2]octane differs from that observed in pi-electron systems, such as benzene, in which resonances work in addition to inductive effects. This is well illustrated by the dependences of cSAR(NO2) on cSAR(X) values for para-substituted BCO and BEN nitro derivatives (Fig. 4), which indicate a greater EA ability of the nitro group in the benzene system [54]. The same results from the comparison of linear dependences pEDA(NO2) on SESE for these systems (X = BF2, BH2, CF3, CH3, CHO, CN, F, NH2, NMe2, NO2, NO, OH, OMe) [67]. The slope of the regression line for BEN series (a = 0.0089) is much greater than for the BCO ones (a = 0.0012), which confirms stronger interactions of the nitro group with substituents in BEN systems. In addition, in the case of BEN derivatives, SESE values are well correlated with σp, while in the case of BCO systems—with F constants (see Table 2) [53]. Therefore, in para-X-nitrobenzene derivatives there are two types of interaction (resonance and inductive), whereas in 4-X-bicyclo[2.2.2]octane-1-nitro derivatives only the inductive/field effect is present.
Similar results surface from the analysis of SESE values for para-type X-substituted benzene and bicyclo[2.2.2]octane nitro derivatives, as presented in Table 12 [53]. The higher SESE values (in absolute terms) obtained for BEN systems denote stronger interactions between the nitro group and the substituents than in BCO systems. Furthermore, in nitro-BEN derivatives, ED substituents stabilize the system more strongly than in nitro-BCO ones, which is confirmed by positive SESE values for the former. Most importantly, the differences in SESE values for X-substituted BEN and BCO series, ΔSESE, can be interpreted as a result of a resonance effect for a particular X substituent. This is illustrated by the good correlation between ΔSESE and resonance constants R with a high determination coefficient R2 = 0.960.
A substantial difference of SEs between the para and meta positions in X-substituted nitrobicyclo[2.2.2]octane and nitrobenzene derivatives can be well described by estimated standard deviation (ESD) values of each parameter characterizing the SE, as shown in Table 13 [67]. For para-type (1,4) substituted systems, the ratios of ESD values (benzene/bicyclo[2.2.2]octane) for a given parameter are always greater for the nitrobenzene series than for the nitrobicyclo[2.2.2]octane ones. While for meta-type (1,3) substitution, the obtained ratios indicate similar strength of interactions between NO2 and substituents both for pi-BEN and sigma-BCO electron system.
In addition, the field/inductive effect in substituted nitrobenzene derivatives can be characterized by the NMR nitrogen chemical shifts. This has been documented for meta-substituted nitrobenzene systems (X = OH, OMe, NMe2, F, Cl, I, NO2) in DMSO as a solvent by the good correlations of the NMR nitrogen shielding on σm or F constants, as shown in Fig. 20. Analogous correlations for para-substituted series (with σp or R) show evident discrepancies in changes in the nitrogen NMR shielding induced by substituents. This means that the N shieldings of the nitro group are sensitive to the field/inductive effects of substituents, but do not respond to the resonance or conjugative effects that occur in para disubstituted benzenes [124].

A plot of substituent-induced changes in the nitrogen NMR shielding of meta- and para-substituted nitrobenzenes, in DMSO solvent, against the sigma (σ) constants. Reprinted and modified from Magn Reson Chem 31:916 (1993) [124]. Copyright 1993 from John Wiley and Sons
Solvent influence on the substituent effect in nitro derivatives
Solvent environment may substantially influence the strength of substituent effect. The use of quantum chemistry SE models makes it possible to compare the strength of substituent effects in both phases (gas phase and water solution, using polarizable continuum model method, PCM). Studies of the electrostatic interactions between water and three different types of 1,4-disubstituted X-R-Y systems—aromatic, benzene (BEN); olefinic, cyclohexa-1,3-diene (CHD); and aliphatic, bicyclo[2.2.2]octane (BCO)—revealed the enhancement of the substituent effect by water [125]. This was documented by the results of the analysis of linear relationships between the values of the same SE parameter determined in the gas phase and water solution.
For the nitro group as the reaction site, the stronger SE in solvent is clearly shown by dependences of cSAR(NO2) in water (PCM) on those in the gas phase (GP), as presented in Fig. 21, where the obtained slope values of linear regressions are significantly greater than 1.0. In addition, enhancement of the SE depends on type of the transmitting moiety and decreases in a sequence from CHD, through BEN to BCO. The similar picture results from dependences cSAR(X)PCM on cSAR(X)GP and CFIPCM on CFIGP (see Table 14).

Dependences of cSAR(NO2) in water on these in the gas phase for X-R-NO2 series. Reprinted with permission from J Phys Chem A 122:1896 (2018) [125]. Copyright 2018 American Chemical Society
Furthermore, systems with the nitro group as a stronger electron-accepting reaction site exhibit a greater solvent effect than the less electron-accepting COOH, as depicted by the slopes of the CFI dependences shown in Table 14. In general, the case when the amino group is the strongest electron-donating reaction site is similar. Moreover, the increase in electric permittivity significantly enhances the communication between X and Y for pi-electron systems.
Additionally, the solvent effect on the nitrogen NMR shielding of the nitro group was investigated. For nitrobenzene derivatives (X = OH, OMe, NMe2, F, Cl, I, NO2), the application of various solvents has revealed that the range of changes is about 6 ppm. A similar observation is made in nitroalkanes, where this changeability may span as much as 10 ppm in nitromethane [124].
Conclusion
The nitro group is the most versatile functional group used in organic synthesis, biochemistry, and related fields. The unusual interest in this group is associated with its strongly electron-attracting (EA) character, substantially dependent on the type of moiety to which the nitro group is attached. Additionally, the nitro group exhibits high electronegativity, χNO2, which results in strong inductive electron-accepting properties.
The understanding of the substituent effect of the nitro group on energetic and electronic properties of given compounds allows quantitative description of changes in electron structure of these systems, caused by the interaction of the nitro group with the rest of the molecule. Among the presented descriptors based on quantum chemistry, describing the substituent effect of the nitro group, particular attention should be paid to the cSAR approach. It allows to study both classical and reverse substituent effects. In addition, the cSAR(NO2) parameter is a reliable descriptor for studying changes in electron-attracting properties of the nitro group in each system, as well as its resonance and inductive interactions with other substituents. In the case of the nitro group, as a planar group, important also is the possibility to describe the changes in its pi and sigma electron structure using pEDA/sEDA model. The application of these models allows to show a quantitatively stronger influence of ED substituents on nitro group than EA ones (classical SE) in various systems. In this way, it may also describe how much the nitro group enhances the electron-donating power of ED substituents and weakens the electron-accepting ability of EA ones (reverse SE).
Intramolecular interactions of the nitro group with other substituents from different positions can strongly affect the pi-electron delocalization of the aromatic ring. These changes can be measured in aromatic systems using aromaticity indices, e.g., HOMA index. The charge transfer between the nitro group and aromatic ring is well illustrated by comparison of their pEDA and sEDA values. In all the abovementioned studies, the effect of the nitro group on the pi-electron structure is stronger when electron-donating substituents are attached to systems—in other words, when a strong resonance effect occurs.
References
Hammett LP (1940) Physical organic chemistry. McGraw-Hill, New York, p 184
Exner O, Krygowski TM (1996). Chem Soc Rev 25:71–75
Irle S, Krygowski TM, Niu JE, Schwarz WHE (1995). J Org Chem 60:6744–6755
Smith MB (2013) March’s advanced organic chemistry: reactions, mechanisms, and structure. 7th edn. Wiley, Hoboken
ISI Web of Science
Allen FH (2002). Acta Crystallogr Sect B: Struct Sci 58:380–388
Xu J (2012). Tetrahedron 68:10696–10747
Zhang C (2009). J Hazard Mater 161:21–28
Badgujar DM, Talawar MB, Asthana SN, Mahulikar PP (2008). J Hazard Mater 151:289–305
Rouchaud J, Neus O, Cools K, Bulcke R (2000). Toxicol Environ Chem 77:31–40
Charton M (1964). J Org Chem 29:1222–1227
Huheey JE (1966). J Phys Chem 70:2086–2092
Campanelli AR, Domenicano A, Ramondo F, Hargittai I (2004). J Phys Chem A 108:4940–4948
Pauling L (1960) The nature of the chemical bond. 3rd edn. Cornell Univ, USA
Stasyuk OA, Szatylowicz H, Fonseca Guerra C, Krygowski TM (2015). Struct Chem 26:905–913
Exner O (1978) In: Chapman NB, Shorter J (eds) Correlation analysis in chemistry—recent advances; Chapt. 10. Plenum Press, New York, pp 439–541
Hansch C, Leo A, Taft RW (1991). Chem Rev 91:165–195
Gleiter R, Haberhauer G (2012) Aromaticity and other conjugated effects. Wiley-VCH Verlag BmbH&Co, Weinheim, p 79
Dobrowolski MA, Krygowski TM, Cyrański MK (2009) Croatica Chim Acta 82:139–147
Kemula W, Krygowski TM (1979) In: Bard AJ, Lund H (eds) Encyclopedia of electrochemistry of the elements, vol XIII. Marcel Dekker Inc, New York, pp 77–130
Kemula W, Krygowski TM (1979) In: Bard AJ, Lund H (eds) Encyclopedia of electrochemistry of the elements; Chapt. XIII-2, Nitro compounds, vol 13. M. Dekker, pp 78–130
Shorter J (1996) In: Patai S (ed) The chemistry of amino, nitroso, nitro and related groups. J. Wiley & Sons, New York, pp 479–531
Boese R, Blaser D, Nussbaummer M, Krygowski TM (1992). Struct Chem 3:363–368
Stasyuk OA, Szatylowicz H, Krygowski TM, Fonseca Guerra C (2016). Phys Chem Chem Phys 18:11624–11633
Szatylowicz H, Stasyuk OA, Fonseca Guerra C, Krygowski TM (2016). Crystals 6:29
Wozniak K, He H, Klinowski J, Jones W, Grech E (1994). J Phys Chem 98:13755–13765
Tejender S, Thakur TS, Singh SS (2015). Cryst Growth Des 15:3280–3292
Hammett LP (1937). J Am Chem Soc 59:96–103
Hammett LP (1940) Physical organic chemistry. McGraw–Hill, New York, Table II, p 189
Hammett LP (1940) Physical organic chemistry. McGraw–Hill, New York, p 196
Jaffe HH (1953). Chem Rev 53:191–261
Exner O (1972) In: Chapman NB, Shorter J (eds) Advances in linear free energy relationships. Plenum Press, London, pp 1–70
Johnson CD (1973) The Hammett equation. Cambridge University Press, Cambridge
Zuman P (1967) Substituent effects in organic polarography. Plenum Press, New York
Charton M (1973). Progr Phys Org Chem 10:81–204
Katritzky AR, Topsom RD (1977). Chem Rev 77:639–658
Shorter J (1991) In: Zalewski RI, Krygowski TM, Shorter J (eds) Similarity models in organic chemistry, biochemistry and related fields. Elsevier, Amsterdam, p 77
Krygowski TM, Stępień BT (2005). Chem Rev 105:3482–3512
Exner O, Bohm S (2006) Theory of substituent effects: recent advances. Curr Org Chem 10:763–778
Taft RW, Lewis IC (1958). J Am Chem Soc 80:2436–2443
Taft RW, Lewis IC (1959). J Am Chem Soc 81:5343–5352
Roberts JD, Moreland WT (1953). J Am Chem Soc 75:2167–2173
Swain CG, Lupton EC (1968). J Am Chem Soc 90:4328–4337
Krygowski TM, Fawcett WR (1975). J Am Chem Soc 97:2143–2148
Fawcett WR, Krygowski TM (1976). Can J Chem 54:3283–3292
Krygowski TM, Wrona PK, Zielkowska U, Reichard C (1985). Tetrahedron 41:4519–4527
Szatylowicz H, Siodla T, Stasyuk OA, Krygowski TM (2016). Phys Chem Chem Phys 18:11711–11721
Szatylowicz H, Jezuita A, Ejsmont K, Krygowski TM (2017). Struct Chem 28:1125–1132
Szatylowicz H, Jezuita A, Ejsmont K, Krygowski TM (2017). J Phys Chem A 121:5196–5203
Shahamirian M, Szatylowicz H, Krygowski TM (2017). Struct Chem 28:1563–1572
Szatylowicz H, Siodla T, Krygowski TM (2017). J Phys Org Chem 30:e3694
Varaksin KS, Szatylowicz H, Krygowski TM (2017). J Mol Struct 1137:581–588
Szatylowicz H, Jezuita A, Siodla T, Varaksin KS, Domanski MA, Ejsmont K, Krygowski TM (2017). ACS Omega 2:7163–7171
Szatylowicz H, Jezuita A, Siodla T, Varaksin KS, Ejsmont K, Shahamirian M, Krygowski TM (2018). Struct Chem 29:1201–1212
Bachrach SM (2014) Computational organic chemistry. Wiley, New Jersey
Gadre SR, Suresh CH (1997). J Org Chem 62:2625–2627
Galabov B, Ilieva S, Schaefer III HF (2006). J Org Chem 71:6382–6387
Galabov B, Ilieva S, Hadijeva B, Atanasov Y, Schaefer III HF (2008). J Phys Chem 112:6700–6708
Remya GS, Suresh CH (2016). Phys Chem Chem Phys 18:20615–20626
te Velde G, Bickelhaupt FM, Baerends EJ, van Gisbergen SJA, Fonseca Guerra C, Snijders JG, Ziegler T (2001). J Comput Chem 22:931–967
Fernandez I, Frenking G (2006). J Org Chem 71:2251–2256
Krygowski TM, Zachara JE, Szatylowicz H (2004). J Org Chem 69:7038–7043
Pross A, Radom L, Taft RW (1980). J Org Chem 45:818–826
Hehre WJ, Radom L, Schleyer PvR, Pople JA (1986) Ab initio molecular orbital theory. John Wiley & Sons, New York
Hehre WJ, Ditchfield R, Radom L, Pople JA (1970). J Am Chem Soc 92:4796–4801
George P, Trachtman M, Bock CW, Brett AM (1976). J Chem Soc Perkin Trans 2:1222–1227
Krygowski TM, Oziminski WP (2014). J Mol Model 20:2352–2359
Siodla T, Oziminski WP, Hoffmann M, Koroniak H, Krygowski TM (2014). J Org Chem 79:7321–7331
Sadlej-Sosnowska N (2007). Polish J Chem 81:1123–1134
Sadlej-Sosnowska N (2007). Chem Phys Lett 447:192–196
Krygowski TM, Sadlej-Sosnowska N (2011). Struct Chem 22:17–22
Mulliken RS (1955). J Chem Phys 23:1833–1840, 1841–1846, 2338–2342, 2343–2346
Bader RWM (1990) Atoms in molecules: a quantum theory. Clarendon Press, Oxford
Bickelhaupt FM, van der Eikemma NIR, Fonseca Guerra C, Baerends EJ (1996). Organometallics 15:2923–2931
Hirshfeld FL (1977). Theor Chim Acta 44:129–138
Weinhold F, Landis CR (2005) Valency and bonding, a natural bond orbital donor-acceptor perspective. Cambridge University Press, Cambridge
Oziminski WP, Dobrowolski JC (2009). J Phys Org Chem 22:769–778
Krygowski TM, Oziminski WP, Palusiak M, Fowler PW, McKenzie AD (2010). Phys Chem Chem Phys 12:10740–10745
Mazurek A, Dobrowolski JC (2013). Org Biomol Chem 11:2997–3013
Kruszewski J, Krygowski TM (1972). Tetrahedron Lett 13:3839–3842
Krygowski TM (1993). J Chem Inf Comput Sci 33:70–78
Cyranski M (2005). Chem Rev 105:3773–3811
Madura ID, Krygowski TM, Cyrański MK (1998). Tetrahedron 54:14913–14918
Zborowski KK, Proniewicz LM (2009). Polish J Chem 83:477–484
Zborowski KK, Alkorta I, Elguero J, Proniewicz LM (2012). Struct Chem 23:595–600
Zborowski KK, Alkorta I, Elguero J, Proniewicz LM (2013). Struct Chem 24:543–548
Andrzejak M, Kubisiak P, Zborowski KK (2013). Struct Chem 24:1171–1184
Schleyer PvR, Maerker C, Dransfeld A, Jiao H, van Eikema Hommes NJR (1996). J Am Chem Soc 118:6317–6318
Cyrański MK, Krygowski TM, Wisiorowski M, van Eikema Hommes NJR, Schleyer PvR (1998). Angew Chem Int Ed 37:177–180
Chen Z, Wannere CS, Corminboeuf C, Puchta R, Schleyer PvR (2005). Chem Rev 105:3842–3888
Corminboeuf C, Heine T, Seifert G, Schleyer PvR, Weber J (2004). Phys Chem Chem Phys 6:273–276
Bultinck P, Rafat M, Ponec R, Van Gheluwe B, Carbó-Dorca R, Popelier P (2006). J Phys Chem A 110:7642–7648
Poater J, Fradera M, Duran M, Sola M (2003). Chem Eur J 9:400–406
Matito E, Duran M, Sola M (2005). J Chem Phys 122:14109
Wieckowski T, Krygowski TM (1981). Can J Chem 59:1622–1629
Krygowski TM, Anulewicz R, Kruszewski J (1983). Acta Cryst B39:732–739
Hęclik K, Dębska B, Dobrowolski JC (2014). RSC Adv 4:17337–17346
Hęclik K, Dobrowolski JC (2017). J Phys Org Chem 30:e3656
Szatylowicz H, Jezuita A, Ejsmont K, Krygowski TM (2019). J Mol Model 25:350–356
Krygowski TM, Dobrowolski MA, Cyrański MK, Oziminski WP, Bultinck P (2012). Comput Theor Chem 984:36–42
Kuhn A, von Eschwege KG, Conradie J (2012). J Phys Org Chem 25:58–68
Siodla T, Szatylowicz H, Varaksin KS, Krygowski TM (2016). RSC Adv 6:96527–96530
Szatylowicz H, Siodla T, Krygowski TM (2017). ACS Omega 2:1746–1749
Dippy JFJ (1939). Chem Rev 25:151–211
Jezierska-Mazzarello A, Szatyłowicz H, Krygowski TM (2012). J Mol Model 18:127–135
Politzer P, Abrahmsen L, Sjoberg P (1984). J Am Chem Soc 106:855–860
Jezuita A, Szatylowicz H, Krygowski TM (2020). Chem Phys Lett 753:137567–137570
Woinska M, Grabowsky S, Dominiak PM, Wozniak K, Jayatilaka D (2016). Sci Adv 2:e1600192
Boese R, Bläser D, Nussbaumer M, Krygowski TM (1992). Struct Chem 3:363–368
Funnell NP, Dawson A, Marshall WG, Parsons S (2013). CrystEngComm 15:1047–1060
Wójcik G, Holband J (2001). Acta Cryst B57:346–352
Ananyev IV, Lyssenko KA (2014). Russ Chem Bull Int Ed 63:1270–1282
Krygowski TM, Ejsmont K, Stepien BT, Cyrański M, Poater J, Sola M (2004). J Org Chem 69:6634–6640
Palusiak M, Domagała M, Dominikowska J, Bickelhaupt FM (2014). Phys Chem Chem Phys 16:4752–4763
Curutchet C, Poater J, Sola M, Elguero J (2011). J Phys Chem A 115:8571–8577
Omelchenko IV, Shishkin OV, Gorb L, Hill FC, Leszczynski J (2012). Struct Chem 23:1585–1597
Majerz I, Dziembowska T (2017). J Phys Chem A 121:2627–2635
Krygowski TM, Palusiak M, Płonka A, Zachara-Horeglad J (2007). J Phys Org Chem 20:297–306
Radula-Janik K, Kopka K, Kupka T, Ejsmont K (2014). Chem Heterocycl Compd 50:1244–1251
Stasyuk OA, Szatylowicz H, Krygowski TM (2014). Org Biomol Chem 12:456–466
Szatylowicz H, Jezuita A, Marek PH, Krygowski TM (2019). RSC Adv 9:31343–31356
Bégué J-P, Bonnet-Delpon D (2008) General remarks on structural, physical, and chemical properties of fluorinated compounds. Bioorganic and medicinal chemistry of fluorine. Wiley & Sons, New Jersey, p 13
Exner O, Böhm S (2003). Chem Eur J 9:4718–4723
Witanowski M, Sicinska W, Biedrzycka Z (1993). Magn Reson Chem 31:916–919
Szatylowicz H, Jezuita A, Siodla T, Varaksin KS, Ejsmont K, Madura ID, Krygowski TM (2018). J Phys Chem A 122:1896–1904
Acknowledgments
HS thanks the Warsaw University of Technology for supporting this work.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
This review is dedicated to our friend and mentor Professor Tadeusz Marek Krygowski of the Department of Chemistry of the Warsaw University in gratitude for his guidance, support, and engagement in our research.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jezuita, A., Ejsmont, K. & Szatylowicz, H. Substituent effects of nitro group in cyclic compounds. Struct Chem 32, 179–203 (2021). https://doi.org/10.1007/s11224-020-01612-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-020-01612-x