
Abstract
For the purpose of screening putative anthracnose resistance-related genes of ramie (Boehmeria nivea L. Gaud), a cDNA library was constructed by suppression subtractive hybridization using anthracnose-resistant cultivar Huazhu no. 4. The cDNAs from Huazhu no. 4, which were infected with Colletotrichum gloeosporioides, were used as the tester and cDNAs from uninfected Huazhu no. 4 as the driver. Sequencing analysis and homology searching showed that these clones represented 132 single genes, which were assigned to functional categories, including 14 putative cellular functions, according to categories established for Arabidopsis. These 132 genes included 35 disease resistance and stress tolerance-related genes including putative heat-shock protein 90, metallothionein, PR-1.2 protein, catalase gene, WRKY family genes, and proteinase inhibitor-like protein. Partial disease-related genes were further analyzed by reverse transcription PCR and RNA gel blot. These expressed sequence tags are the first anthracnose resistance-related expressed sequence tags reported in ramie.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ramie (Boehmeria nivea L. Gaud), a perennial herbaceous plant grown for its fibers and a member of the Urticaceae family, is planted principally in China and other Asian countries including the Philippines, India, South Korea, and Thailand (Wang et al. 2008). Ramie fibers are long and strong and possess good durability and absorbency with excellent luster (Zheng et al. 2000). In China, about 130,000 ha are planted with ramie. Ramie crops are usually infected by diseases caused by fungi, viruses, or nematodes, which decrease the yield as well as the quality of the fibers. Anthracnose is a major disease of ramie caused by the fungus Colletotrichum, first described in 1831 by Corda. Its classification is phylum Deuteromycotina, class Coelomycetes, order Melanconiales, family Melanconiaceae. This important plant fungal disease is widespread in temperate zones, particularly in subtropical and tropical regions. It often damages fruit trees, vegetables, flowers, medicinal plants, and the fruit, stems, and leaves of field crops (Sutton 1992; Agostini et al. 1992), causing large losses in agricultural production. In China, Colletotrichum boehmeriae Sawada has been reported as a pathogen of ramie, but with no further investigations (Sawada 1914, 1919). In our former research, we have identified the disease caused by Colletotrichum gloeosporioides (Wang et al. 2010a, b) which can infect the stalk and foliage, thereby degrading not only the quantity but also the quality of the fiber. Generally, the disease causes yield losses of 20–40% and as high as 55% (Li and Ma 1993).
To study resistance genes in ramie, we constructed a suppression subtractive hybridization (SSH) (Diatchenko et al. 1996) library induced by C. gloeosporioides. The SSH technique enables specific cloning of expressed sequence tags (ESTs) representing genes that are differentially expressed in different mRNA populations and isolates genes without prior knowledge of their sequence or identity. It uses common molecular biological techniques that do not require specialized equipment or analyses (Moody 2001). This method was very useful in screening new genes and construction of cDNA library (Lin et al. 2010; Wang et al. 2011; Dipnarayan et al. 2011; Zhang et al. 2011a, b; Zhou et al. 2011; Prabu et al. 2011; Yang et al. 2011). Given the current understanding of ramie at the molecular level, this technique is a suitable method for the detection of low-abundance, differentially expressed transcripts that can lead to the isolation of putative disease resistance and defense-related genes.
Materials and Methods
Material Preparation and Isolation of Colletotrichum
Huazhu no. 4, an elite ramie cultivar in Hubei Province, China that shows high resistance to ramie anthracnose (C. gloeosporioides), was planted in the Ramie Germplasm Resources Garden of Huazhong Agricultural University. The plants used in this study were transplanted into the greenhouse under daylight with additional illumination provided by high-pressure sodium lamps to give a photoperiod of 12 h.
C. gloeosporioides samples were collected from anthracnose lesions on ramie, the Bast Fiber Crops Institute in Changsha, Hunan Province, China. After surface sterilization of lesions with 0.1% mercuric chloride for 40 s, followed by autoclaved water wash three times, small blocks (9 mm2) of diseased bark were aseptically transferred to potato dextrose agar (PDA) plates. Cultures were dark-cultured at 25°C incubator. C. gloeosporioides isolates were purified by single spore culturing (Choi et al. 1999). Spore masses were picked up with a sterilized wire loop and streaked on to the surface of water agar plates which were dark-cultured at 25°C incubator overnight. A single germinated spore was picked up with a sterilized needle and then transferred onto PDA plates. Pure cultures were stored at 4°C on PDA slants. Isolates were deposited in Huazhong Agricultural University, Hubei Province, China.
The ramie Huazhu no. 4 was sprayed with 20 ml of a solution of anthracnose spores (1 × 106/ml). The infected ramie plants and the control samples were cultured under conditions of 25°C and 80% humidity. Leaves were taken 6, 12, 24, 36, 48, and 72 h after inoculation. The samples were then quick-frozen in liquid nitrogen and stored at −80°C for RNA extraction.
Total RNA and mRNA Isolation
Total RNA was extracted from the samples by an optimized method (Wang et al. 2010a, b). The extracted total RNA was checked by electrophoresis on 1% agarose gels. mRNA was isolated using a PolyATract mRNA Isolation System kit (Promega, USA) according to the manufacturer’s instructions.
SSH Library Construction
An SSH library was constructed using a Clontech PCR-SelectTM cDNA Subtraction Kit (BD Biosciences, USA) based on the manufacturers’ instructions. The final PCR products were purified using an SBS Quick PCR Purification kit (SBS Genetech, Wuhan, China). The subtracted cDNA was inserted directly into pGEM®-T Easy Vector (Promega, USA) and then transformed into JM-109 Escherichia coli cells (Promega, USA) plated onto lysogeny broth (LB) agar containing 100 mg L−1 ampicillin, 1 mM isopropyl β-d-thiogalactopyranoside, and 80 mg L−1 5-bromo-4-chloro-3-indolyl β-d-galactopyranoside. Following incubation at 37°C overnight, positive transformants based on blue/white color selection were picked and arrayed into 96-well microplates and then cultured in LB medium containing 100 mg L−1 ampicillin. The resultant subtractive cDNA library was stored at −80°C with 15% glycerol.
Amplification of cDNA Inserts
The cDNA inserts were amplified by PCR in a 96-well plate (PTC-100, Germany) with nested PCR primer 1 and primer 2R, which were included in the PCR-SelectTM cDNA Subtraction Kit. PCR reactions (25 μL) contained 17.3 μL distilled water, 1 μL bacterial cultures, 1 μL each of nested PCR primer 1 and primer 2R (10 μM each), 2.5 μL 10× ExTaq buffer, 1.5 μL Mg2+ buffer (25 mM), 0.5 μL dNTPs (10 mM each), and 1 U ExTaq polymerase (SBS Genetech, Wuhan, China). Reaction samples were first denatured at 94°C for 5 min, followed by 30 cycles at 94°C for 20 s, 55°C for 45 s, and 72°C for 45 s, with a final extension at 72°C for 10 min. All PCR products were analyzed by agarose gel electrophoresis and clones of segments <200 bp or two or more segments eliminated.
Preparation of Probes and Differential Screening of the Subtracted Libraries
To exclude false-positive clones and to provide further data on the relative expression levels of the cloned cDNAs, initial screening of the library to remove false positives was performed by reverse northern analysis with total labeled cDNA from unsubtracted tester cDNA and unsubtracted driver cDNA as probes. Total RNAs were extracted from healthy ramie leaves and inoculated leaves. Then, they were reversed into cDNA separately. And then, they were labeled as tester cDNA and driver cDNA probes.
Five microliters of PCR products (about 100 ng) was mixed with 5 μL fresh 0.6 M NaOH for denaturation and 1.2 μL of the mixture was printed onto two Hybond-N+ nylon membranes (Boehringer, Mannheim, Germany). The membranes were neutralized in 0.5 M Tris–HCl (pH 7.5) for 5 min and rinsed in distilled water for 30 s. Samples were cross-linked to the membranes by baking for 2 h at 80°C and then stored at −4°C until use. Unsubtracted driver cDNA and tester cDNA probes were labeled using a DIG High Prime DNA Labeling and Detection Starter Kit II (Roche Diagnostics GmbH, Germany). Preparation of probes and hybridization were performed exactly according to the manufacturer’s instructions. The results of two hybridizations were recorded for each clone, and those showing the most marked differential expression were selected for sequencing (Sunny Biotechnology, Shanghai, China).
EST Sequencing and Analysis
Homology searches were conducted for all sequences using the GenBank database and the BLAST algorithms at the National Center for Biotechnology Information (NCBI) network service (http://www.ncbi.nlm.nih.gov/BLAST/). The cDNAs were classified according to the E-values generated in the BLAST searches. E-values <1e−10 were deemed to indicate significant homology and functional assignment. ESTs with E-values >1e−10 were deemed to have no significant homology to any known protein and assumed to be novel. EST sequences were assigned manually to functional categories based on a previous catalog system (Bevan et al. 1998).
RT-PCR Analysis
Total RNA was extracted from samples taken 6, 12, 24, 36, 48 and 72 h after inoculation. First-strand cDNAs were generated from 50 to 100 ng RNA samples using a reverse transcription PCR (RT-PCR) kit (Toyobo, Japan). To determine the expression of candidate genes, PCR was performed with one tenth of the first-strand cDNA template and the gene-specific primer pairs. Gene-specific RT-PCR primers were designed with Primer Premier 5.0 according to the cDNA sequences and were synthesized commercially (SBS Genetech, Wuhan, China). The following primer pairs were used: for biquitin-conjugating enzyme, fwd (5′-AAGCGACGATAATGGGTC-3′) and rev (5′-GCTATTGATGTTCGGGTGA-3′); for polypeptide for cytokinin-repressed mRNA, fwd (5′-AACACTATCGTCGGAGG-3′) and rev (5′-CAAGAAGGCACAGAGCAG-3′); for putative heat-shock protein 90, fwd (5′-AAGACCCTTAAACAACAAGA-3′) and rev (5′-AGTCGAGCCTAAACATCAG-3′); for actin factor, fwd (5′-GCCTGCTGCTTCCATTCC-3′) and rev (5′-TGGCTTACATTGCCCTTGA-3′); for pathogenesis-related protein, fwd (5′-CGGGCAACTACAATGGAGAA-3′) and rev (5′-AGCCATTTCAGAATCAAC-3′); for 3-hydroxyacyl-CoA dehydrogenase, fwd (5′-ATCCGTAACAGACCAAGA-3′) and rev (5′-GAGACAAACATCGCAAGT-3′); for Glycine max nodulin-26 mRNA, fwd (5′-GAGAAATAGCAAACCAACCC-3′) and rev (5′-CCCACCACGGACTACTGA-3′); for pathogenesis-related protein PR10, fwd (5′-GAAGGGCGGGCATGAGAT-3′) and rev (5′-TGTCGCTTTGTTTAGTTGTAGG-3′); and for stress and pathogenesis-related protein, fwd (5′-GGCATGAGATCAAGGAGG-3′) and rev (5′-TGTCGCTTTGTTTAGTTGTAGG-3′). The programs differed because of the different primers. The PCR products (8 μL) for each sample were electrophoresed in a 1.5% ethidium bromide agarose gel and viewed under ultraviolet light.
Northern Blot Analysis
Total RNA (20 μg) from samples taken at different times after inoculation were separated on a 1.0% agarose/formaldehyde gel and transferred to Hybond-N+ nylon membranes, by downward capillary transfer with 20× SSC. Specific probes were generated by purified PCR amplification of the relevant differentially expressed clones and labeled using a DIG High Prime DNA Labeling and Detection Starter Kit II (Roche Diagnostics GmbH, Germany). Labeling of probes and hybridization were performed exactly according to the manufacturer’s instructions.
Results and Discussion
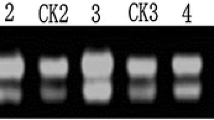
A subtracted cDNA library was constructed using the healthy ramie Huazhu no. 4 cultivar and Huazhu no. 4 inoculated with C. gloeosporioides. The library comprised approximately 1,000 clones, with insertions ranging from 50 to 1,100 bp. One hundred seventy-two clones of greater signal intensity were found to be regulated and were sequenced. The EST sequences obtained were identified by homology searches in BLASTN and BLASTX databases. The partial results are summarized in Table 1. One hundred thirty-two single ESTs were assigned to functional categories and classified into 14 putative cellular functions, basing on the functional categories established for Arabidopsis (Bevan et al. 1998). The distribution of the genes is illustrated in Fig. 1. In the 132 single ESTs, 70 had high similarity to database entries. The largest set of genes (28%) was assigned to disease/defense. Unclear classification and unclassified were the second (17%) and the third (11%) largest groups, respectively. Based on the results of northern hybridization and EST sequencing, RT-PCR was performed to evaluate the differential expression of selected disease-related genes (Fig. 2). As indicated by the SSH results (Table 1), we selected nine clones: ubiquitin-conjugating enzyme 2 mRNA, polypeptide for cytokinin-repressed, putative heat-shock protein 90 (Hsp90), actin factor, pathogenesis-related protein, 3-hydroxyacyl-CoA dehydrogenase, nodulin-26, pathogenesis-related protein PR10, and pathogenesis-related protein, all of which exhibited high homology in the BLASTX comparison with the NCBI database. RT-PCR showed high levels of the genes encoding stress and pathogenesis-related protein at the time of induction of pathology and in the control group (CK) (Fig. 2). The clone related to actin factor showed peak expression after 24 h. Expression of some genes was induced after the induction of pathology: polypeptide for cytokinin-repressed mRNA showed differences after 12 h and peaked at 24 h, after which expression was maintained. However, another clone related to the stress and pathogenesis-related protein was expressed only at 12–48 h. We conclude that expression of this gene was brought about the induction of pathology. The clone with high homology to putative heat-shock protein 90 was highly expressed in CK but showed a downward trend after the induction of pathology. The Hsp90 proteins might play a role either in stabilization of proteins involved in disease resistance or in assembly of multi-subunit complexes required for activation of R proteins and intracellular disease resistance signaling pathways (Scofield et al. 2005). In the RNA gel blot analysis, we used probes synthesized from clone 645 (polypeptide for cytokinin-repressed) and clone 696 (pathogenesis-related protein PR10). From Fig. 3, it can be seen that it is likely that the corresponding genes were expressed at similar levels as observed by RT-PCR.

Functional categories of genes included in the anthracnose-resistant subtractive cDNA library, based on the classification described in Bevan et al. (1998)

RT-PCR analysis of cDNAs representative of different types of gene expression patterns. CK, control leaves; 6h, 12h, 24h, 36h, 48h, 72h, 6, 12, 24, 36, and 72 h, respectively, after inoculation with C. gloeosporioides

Northern hybridization of total RNA from anthracnose-resistant ramie cultivar Huazhu no. 4 using clones 645 and 696. A, B, ECL detection of the 645 and 696 probes, separately; CK, control leaves; 12h, 24h, 12 and 24 h after inoculation with C. gloeosporioides
Breeding for stable anthracnose resistance is difficult because of the variation among C. gloeosporioides for pathogenicity and virulence. In addition, the host–pathogen interaction is poorly understood. Few reports on anthracnose and the cloning and characterization of ramie resistance gene homologs have been published. In this study, the transcription profiles of uninfected and infected leaves of the anthracnose-resistant Huazhu no. 4 ramie cultivar were compared by SSH analysis. The SSH library was not very extensive, which might be due to the efficiency of the adapters ligation, which plays a key role in the subtractive library construction. Additionally, the efficiency depends upon several factors such as the quality of cDNA synthesis, the completeness of enzyme digestion, and the quality of purification. However, we obtained many disease resistance- and defense-related ESTs. There were only about 200 ESTs of ramie in the GenBank database, so our results are meaningful for future research into disease resistance in ramie. We found several transcripts encoding proteins belonging to groups including pathogenesis-related proteins, metallothioneins, severe drought-stressed proteins, Hsp90, thaumatin-like protein, catalase (CAT1), the WRKY family, and oxalate oxidase.
Although PCR-selective cDNA subtraction is a powerful tool for identifying differentially expressed genes, and subtractive products may contain cDNAs that are common to or have similar levels in the two tissues, false-positive clones may occur (Zeng et al. 2006). As a result, we performed RT-PCR for further identification. According to the RT-PCR results, the resistant ramie cultivar had many resistance genes before being infected, including ubiquitin-conjugating enzyme, PR-1.2 protein, nodulin-26 mRNA, and mRNAs for stress and pathogenesis-related protein. These proteins maintained their high levels before and after inoculation and played an important role in the resistance process. The pathogenesis-related proteins included several proteins that are involved in the detoxification of reactive oxygen species (Mishra et al. 2007). Another aspect of our findings was the decline of Hsp90, a molecular chaperone responsible for the folding and functions of important cellular proteins including steroid hormone receptors, protein kinases, and proteins controlling the cell cycle and apoptosis (Schulte et al. 1998). Wicklow et al. (2009) demonstrated that monorden from Colletotrichum graminicola is a potent selective inhibitor of Hsp90. Monorden might be a competitive ligand for the ATP-binding site of Hsp90, thus preventing the ATPase activity necessary for its role as a chaperone (Roe et al. 1999). It could be confirmed by further experiments on our pathogen, C. gloeosporioides. 3-Hydroxyacyl-CoA dehydrogenase (HADH) deficiency is an autosomal recessive metabolic disorder, resulting from mutations in the HADH gene in chromosome 4q22-q26 (http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=231530; Vredendaal et al. 1998), which has so far only been described in a few patients (Martins et al. 2011). In plant of castor bean (Ricinus communis) endosperm and potato tubers, this enzyme had a high expression level (Courtois-verniquet and Douce 1993).
The conclusion of this study is that, in addition to the expression of several well-characterized pathogenesis-related proteins, high-level constitutive expression of genes encoding thaumatin-like protein, calmodulin, catalase, and proteinase inhibitor seems to be a major factor in rendering ramie resistant to infection by the fungus C. gloeosporioides. The recovery of many ESTs indicates that ramie quickly responds to C. gloeosporioides challenge and suggests that early transcriptional events can play an important role in determining whether ramie succumbs to or resists infection. This study is the first global analysis of genes in ramie with anthracnose. The ESTs identified following C. gloeosporioides challenge in this study should provide a useful genomic resource for biologists and plant breeders in developing new strategies for improving resistance to anthracnose in ramie.
References
Agostini JP, Timmer LW, Mitchell DJ (1992) Morphological and pathological characteristic of strains of Colletotrichum gloeosporiodes from citrus. Phytopathology 82:1377–1382
Bevan M et al (1998) Analysis of 1.9 Mb of contiguous sequence from chromosome 4 of Arabidopsis thaliana. Nature 391:485–488
Choi YW, Hyde KD, Ho WH (1999) Single spore isolation of fungi. Fungal Divers 3:29–38
Courtois-verniquet F, Douce R (1993) Lack of aconitase in glyoxysomes and peroxisomes. Biochem J 294:103–107
Diatchenko L, Lau YFC, Campbell AP, Chenchik A, Moqadam F, Huang B, Lukyanov S, Lukyanov K, Gurskaya N, Sverdlov ED, Siebert PD (1996) Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc Natl Acad Sci USA 93:6025–6030
Dipnarayan S, Vajinder K, Shripad RB, Ramamurthy S (2011) Characterization of upstream sequences of the LOJ gene leads to identification of a novel enhancer element conferring lateral organ junction-specific expression in Arabidopsis thaliana. Plant Mol Biol Rep 29:265–277
Li RM, Ma HG (1993) Studies on the occurrence and control of ramie anthracnose. J Plant Prot 20(1):83–89 (In Chinese)
Lin KH, Lin CH, Chan MT, Lo HF (2010) Identification of flooding-response genes in eggplant roots by suppression subtractive hybridization. Plant Mol Biol Rep 28:212–221
Martins E, Cardoso ML, Rodrigues E, Barbot C, Ramos A, Bennett MJ, Teles EL, Vilarinho L (2011) Short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: the clinical relevance of an early diagnosis and report of four new cases. J Inherit Metab Dis 34:835–842
Mishra RN, Reddy PS, Nair S, Markandeya G, Reddy AR, Sopory SK, Reddy MK (2007) Isolation and characterization of expressed sequence tags (ESTs) from subtracted cDNA libraries of Pennisetum glaucum seedlings. Plant Mol Biol 64:713–732
Moody DE (2001) Genomics techniques: an overview of methods for the study of gene expression. J Anim Sci 79:E128–E135
Prabu G, Kawar PG, Pagariya MC, Prasad DT (2011) Identification of water deficit stress upregulated genes in sugarcane. Plant Mol Biol Rep 29:291–304
Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH (1999) Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem 42:260–266
Sawada K (1914) Colletotrichum boehmeriae In: Taiwan Hakubutsu Gakkwai Kwaiho (J Formosan Nat Hist Soc) 17(2) (In Japanese)
Sawada K (1919) Taiwan agricultural research special report 19 (Taiwan Fungi Report 1), pp 571
Schulte TW, Akinaga S, Soga S, Sullivan W, Stensgard B, Toft D, Neckers LM (1998) Antibiotic radicicol binds to the N-terminal domain of Hsp90-binding agents. Cell Stress Chaperones 3:100–108
Scofield SR, Huang L, Brandt AS (2005) Development of a virus-induced gene-silencing system for hexaploid wheat and its use in functional analysis of the Lr21-mediated leaf rust resistance pathway. Plant Physiol 138(4):2165–2173
Sutton BC (1992) The genus Glomerella and its anamorph Colletotrichum. In: Colletotrichum-biology, pathology and control. CAB International, Wallingford, pp 1–26
Vredendaal J, van den Berg I, Stroobants A et al (1998) Structural organization of the human short-chain L-3-hydroxyacyl-CoA dehydrogenase gene. Mamm Genome 9:763–768
Wang B, Peng DX, Sun ZX, Zhang N, Gao SM (2008) In vitro plant regeneration from seedling-derived explants of ramie [Boehmeria nivea (L.) Gaud]. In Vitro Cell Dev Biol Plant 44:105–111
Wang XX, Wang B, Liu LJ, Chen J, Cui XP, Jiang H, Peng DX (2010a) First report of anthracnose caused by Colletotrichum gloeosporioides on ramie in China. Plant Dis 94:1508.1
Wang XX, Wang B, Liu LJ, Cui XP, Yang JY, Wang H, Jiang H, Luo BB, Long Z, Dou WX, Zhang N, Peng DX (2010b) Isolation of high quality RNA and construction of a suppression subtractive hybridization library from ramie (Boehmeria nivea L. Gaud.). Mol Biol Rep 37:2099–2103
Wang WK, Liu CC, Chiang TY, Chen MT, Chou CH, Yeh CH (2011) Characterization of expressed sequence tags from flower buds of alpine lilium formosanum using a subtractive cDNA library. Plant Mol Biol Rep 29:88–97
Wicklow DT, Jordan AM, Gloer JB (2009) Antifungal metabolites (monorden, monocillins I, II, III) from Colletotrichum graminicola, a systemic vascular pathogen of maize. Mycol Res 113(12):1433–1442
Yang Z, Peng ZS, Yang H, Yang J, Wei S, Cai SH (2011) Suppression subtractive hybridization identified differentially expressed genes in pistil mutations in wheat. Plant Mol Biol Rep 29:431–439
Zeng FC, Zhang XL, Zhu LF, Tu LL, Guo XP, Nie YC (2006) Isolation and characterization of genes associated to cotton somatic embryogenesis by suppression subtractive hybridization and macroarray. Plant Mol Biol 60:167–183
Zhang H, Hu YG, Wang CY, Ji WQ (2011a) Gene expression in wheat induced by inoculation with Puccinia striiformis west. Plant Mol Biol Rep 29:458–465
Zhang XD, Allan AC, Yi Q, Chen L, Li K, Shu Q, Su J (2011b) Differential gene expression analysis of Yunnan red pear, Pyrus Pyrifolia, during fruit skin coloration. Plant Mol Biol Rep 29:305–314
Zheng LS, Du YM, Zhang JY (2000) Biobleaching effect of xylanase preparation from an alkalophilic Bacillus on ramie fibers. Biotechnol Lett 22:1363–1367
Zhou MB, Yang P, Gao PJ, Tang DQ (2011) Identification of differentially expressed sequence tags in rapidly elongating Phyllostachys pubescens internodes by suppressive subtractive hybridization. Plant Mol Biol Rep 29:224–231
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Xuxia, W., Jie, C., Bo, W. et al. Characterization by Suppression Subtractive Hybridization of Transcripts That Are Differentially Expressed in Leaves of Anthracnose-Resistant Ramie Cultivar. Plant Mol Biol Rep 30, 547–555 (2012). https://doi.org/10.1007/s11105-011-0361-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-011-0361-y