
Abstract
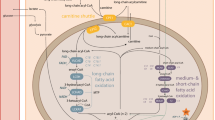
Short-chain 3-hydroxyacyl-CoA dehydrogenase (HADH, SCHAD) deficiency (OMIM #231530) represents a recently described disorder of mitochondrial fatty acid beta-oxidation, with less than ten cases described worldwide. The main clinical presentation of this metabolic disease is different from other inherited defects of fatty acid β−oxidation as the hypoglycemia is associated with hyperinsulinism. We present the clinical, biochemical and molecular findings of four new Caucasian patients with HADH deficiency. These new cases contribute to a more comprehensive description of the phenotype, diagnostic biomarkers and treatment options for this poorly defined disease.

Similar content being viewed by others

References
Bennett MJ, Russell LK, Tokunaga C et al. (2006) Reye-like syndrome resulting from novel missense mutations in mitochondrial medium- and short-chain L-3-hydroxy-acyl-CoA dehydrogenase. Mol Genet Metab 89:74–79
Bennett MJ, Spotswood SD, Ross KF et al. (1999) Fatal hepatic short-chain L-3-hydroxyacyl-coenzyme A dehydrogenase deficiency: clinical, biochemical, and pathological studies on three subjects with this recently identified disorder of mitochondrial beta-oxidation. Pediatr Dev Pathol 2:337–345
Bennett MJ, Weinberger M, Kobori J et al. (1996) Mitochondrial short-chain L-3-hydroxyacyl-coenzyme A dehydrogenase deficiency: A new defect of fatty acid oxidation. Pediatr Res 39:185–188
Clayton PT, Eaton S, Aynsley-Green A et al. (2001) Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of beta-oxidation in insulin secretion. J Clin Invest 108:457–465
Di Candia S, Gessi A, Pepe G et al. (2009) Identification of a diffuse forme of hyperinsulinemic hypoglycemia by 18-fluoro-L-3, 4 dihydroxyphenylalanine positron emission tomography/CT in a patient carrying a novel mutation of the HADH gene. Eur J Endocrinol 160:1019–1023
Eaton S, Chatziandreou I, Krywawych S et al. (2003) Short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency associated with hyperinsulinism: a novel glucose-fatty acid cycle? Biochem Soc Trans 31:1137–1139
Filling C, Keller B, Hirschberg D et al. (2008) Role of short-chain hydroxyacyl CoA dehydrogenases in SCHAD deficiency. Biochem Biophys Res Commun 368:6–11
Hardy O, Hohmeier H, Becker T et al. (2007) Functional genomics of the B-cell: short-chain 3-hydroxyacyl CoA dehydrogenase regulates insulin secretion independent of K currents. Mol Endocrinol 21:765–773
Hussain K, Clayton P, Krywawych S et al. (2005) Hyperinsulinism of infancy associated with a novel splice mutation in the SCHAD gene. J Pediatr 146:706–708
Kapoor R, James C, Flanagan S et al. (2009) Hydroxy-acyl-CoA dehydrogenase deficiency and hyperinsulinemic hypoglycemia: characterization of a novel mutation and severe dietary protein sensivity. J Clin Endocrinol Metab 94:2221–2225
Li C, Chen P, Palladino A et al. (2010) Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency involves activation of glutamate dehydrogenase. J Biol Chem 285(41):31806–31818
Molven A, Matre GE, Duran M et al. (2004) Familial hyperinsulinemic hypoglycemia caused by a defect in the SCHAD enzyme of mitochondrial fatty acid oxidation. Diabetes 53:221–227
Molven A, Rishaug U, Matre GE et al. (2002) Hunting for a hypoglycemia gene: severe neonatal hypoglycemia in a consanguineous family. Am J Med Genet 113:40–46
Schulze A, Lindner M, Kohlmüller D et al. (2003) Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics 111:1399–1406
Shekhawat P, Matern D, Strauss A (2005) Fetal fatty acid oxidation disorders, their effect on maternal health and neonatal outcome: Impact of expanded newborn screening on their diagnosis and management. Pediatr Res 57:78R–86R
Stanley C, Bennett MJ, Mayatepek E (2006) Disorders of mitochondrial fatty acid oxidation and related metabolic pathways. In: Fernandez J, Saudubray JM, van den Berghe G (eds) Inborn metabolic diseases – diagnosis and treatment, 4th edn. Springer, Berlin, pp 175–196
Strauss A, Andresen BS, Bennett MJ (2009) Mitochondrial fatty acid oxidation defects In: Sarafoglou K, Hoffmann G, Roth K. Pediatric Endocrinology and Inborn errors of Metabolism. McGraw-Hill. 51-70
Tein I, De Vivo D, Hale D et al. (1991) Short-chain L-3-hydroxy-acyl-CoA dehydrogenase deficiency in muscle: A new cause for recurrent myoglobinuria and encephalopathy. Ann Neurol 30:415–419
Vidnes J, Oyasaeter S (1977) Glucagon deficiency causing severe neonatal hypoglycemia in a patient with normal insulin secretion. Pediatr Res 11:943–949
Vilarinho L, Rocha H, Sousa C et al. (2010) Four years of expanded newborn screening in Portugal with tandem mass spectrometry. 2010 . J Inherit Metab Dis online doi:10.1007/s510545-010-9048-z
Vredendaal J, van den Berg I, Stroobants A et al. (1998) Structural organization of the human short-chain L-3-hydroxyacyl-CoA dehydrogenase gene. Mamm Genome 9:763–768
Wanders R, IJlst L, Van Gennip AH et al. (1990) Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: identification of a new inborn error of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis 13:311–314
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Carlo Dionisi-Vici
Competing interest: None declared.
Rights and permissions
About this article
Cite this article
Martins, E., Cardoso, M.L., Rodrigues, E. et al. Short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: the clinical relevance of an early diagnosis and report of four new cases. J Inherit Metab Dis 34, 835–842 (2011). https://doi.org/10.1007/s10545-011-9287-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-011-9287-7