
Abstract
Plant height is an important botanical feature closely related to yield. Two populations consisting of 118 and 262 accessions respectively were used to identify elite alleles for plant height and to validate their allelic effects. Plant height was measured from the early booting to the flowering stages. Simple sequence repeat markers for candidate quantitative trait locus (QTL) regions with large effects identified in a doubled haploid (DH) population (Hanxuan 10 × Lumai 14) were selected for further verification by association analysis. Nine loci significantly (P < 0.001) associated with plant height were detected 13 times in the population with 118 accessions. Three loci (Xgwm11-1B, Xwmc349-4B and Xcfd23-4D) were identified in three, two and two periods of plant height growth, respectively. Markers Xbarc168-2D, Xgwm249-2D, Xwmc349-4B, Xcfd23-4D and Xgwm410-5A located at or near additive QTL regions in the DH population proved to coincide with known Rht loci. The results showed a consistency between linkage analysis and association mapping, and also confirmed the value of fine mapping of QTL through combined linkage and association analyses. For final plant height, the alleles Xgwm11-1B 208 , Xwmc349-4B 103 and Xcfd23-4D 202 exhibited negative effects, i.e. reducing plant height; Xwmc349-4B 101 and Xcfd23-4D 205 showed significant positive effects. A second larger population (262 accessions) was used to validate the effects of these large-effect alleles and the efficacy of pyramiding in eight environments (year × site × water regime combinations). Strong correlations between final plant height and numbers of large-effect alleles indicated that the alleles contributed additively to plant height. The additive effects showed that pyramiding elite alleles for target traits has significant potential for wheat breeding.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plant height is an important botanical feature closely related to yield (Jiang et al. 2003). The use of dwarf and semi-dwarf wheat and rice varieties to increase crop yields was termed the “Green Revolution” (Hedden 2003). Most current leading cultivars in the northern China winter wheat region with heights of around 75–85 cm are even shorter than those with single semi-dwarfing genes (Zhou et al. 2007). However, the genetics of plant height are complex, with genes on 17 of the 21 wheat chromosomes having been reported (Börner et al. 1996). Twenty-two Rht genes with major effects have been described (McIntosh et al. 2008; Peng et al. 2011), but only Rht-B1b (formerly Rht1) and Rht-D1b (Rht2), carried by Norin 10, and Rht8 are widely used in wheat breeding programs; varieties with one or more of these genes accounted for >70 % of current commercial wheat cultivars worldwide (Ellis et al. 2007; Hedden 2003). The wide use of dwarfing sources based on a limited number of key parents leads to relatively narrow genetic diversity, which reduces adaptation to various environmental conditions (Evenson and Gollin 2003; Reif et al. 2005; Roussel et al. 2004). Therefore, it is essential to determine the genetic basis of plant height and exploit elite alleles, i.e. the alleles with favorable effects for breeding high-yielding varieties.
The rapid development of molecular markers has provided a basis for detailed genetic analyses of complex traits, such as plant height, which involves several genes, and particularly for understanding interactions with environmental factors. Quantitative trait loci (QTL) associated with plant height were detected on almost all 21 chromosomes by linkage analysis and association mapping (Cadalen et al. 1998; Cui et al. 2011; Huang et al. 2006; Keller et al. 1999; Klahr et al. 2007; McCartney et al. 2005; Wang et al. 2010; Wu et al. 2010; Zhang et al. 2011). However, most of these studies measured only the final plant height, and did not annotate the quantitative variation on a time scale. Zhu proposed statistical methods for analyzing conditional genetic effects (Zhu 1995). By analyzing developmental behavior within the period (t − 1) to t, quantitative genetic effects can be revealed at specific stages excluding the effect of previous times (t − 1). Plant height is not only easily measured, but also undergoes obvious changes along with plant development. It is therefore a popular model trait for the study of quantitative gene expression in developmental genetics (Wu et al. 2010). Several studies on conditional genetic effects of plant height development are reported in wheat (Cui et al. 2011; Wang et al. 2010; Wu et al. 2010), rice (Cao et al. 2001; Yan et al. 1998a), soybean (Sun et al. 2006) and maize (Yan et al. 2003). The overall research shows that conditional QTL mapping is a valid way of revealing dynamic gene expression for height development, especially the epistatic effects (Cao et al. 2001). Using the conditional QTL mapping method, genetic relationships between plant height and plant height components have been evaluated (Cui et al. 2011). Plant height development is a network of genes expressed selectively during the whole period of plant height growth (Wu et al. 2010).
Molecular marker-assisted selection (MAS) is a breeding approach increasingly adopted to eliminate defects in elite breeding lines (Kuchel et al. 2007). Many QTL for plant height have been identified by linkage analysis, but two fundamental limitations restrict the use of marker-assisted crop improvement practices. Firstly, there must be validation of previously reported QTL across time, space and genetic background. Only adequately verified markers are useful for MAS (Wheeler et al. 2005). In addition, the better of two alleles identified by linkage analysis may not represent the best genotype in the potential germplasm pool available for breeding programs (Jestin et al. 2011). Furthermore, many factors influence QTL detection and the true genetic effects of a QTL are influenced by environment, year, population and sample size (Li et al. 2010). Linkage mapping using segregating populations often detects broad chromosome regions that result in low resolution due to the limited polymorphism between two parents. Thus, it is necessary to verify the robustness of reported QTL and to fine-map chromosome regions to detect more closely linked markers in order to identify elite alleles for MAS. In a previous study, 25 additive QTL for developmental behavior of plant height were detected in a doubled haploid (DH) population derived from the cross Hanxuan 10 × Lumai 14 (Wu et al. 2010).
In the current study, two populations were used, one for conditional and unconditional association mapping, and another for verifying large-effect alleles of associated loci in eight environments (year × site × water regime combinations). Plant height was measured from the early booting to the flowering stages and selection of simple sequence repeat (SSR) markers was based on candidate QTL detected in the previous research. The purpose was to (1) verify large-effect QTL previously detected in the DH population and further identify markers closely linked to plant height; (2) identify the large-effect alleles of associated loci; and (3) assess the efficacy of pyramiding large-effect alleles with two populations in nine environments.
Materials and methods
Plant materials
Two hexaploid winter wheat populations were used as the plant materials. Population 1 (118 entries) was used for conditional and unconditional association mapping of plant height. Population 2 (262 accessions) was used to validate the large-effect alleles of associated loci. All materials were sown at the beginning of October and harvested in the following mid-June. Each experimental unit was a two-row plot of 2 and 0.3 m between two rows, with 40 seeds planted per row.
Population 1 consisted of 50 accessions and 68 homozygous lines with uniform plant height derived from BC2-3F2-5 progenies of the accessions (Supplementary Table S1). The recurrent parents were Jinmai 47, Lumai 14 and Yumai 18. The accessions were planted in Changping, Beijing (116°13′E; 40°13′N) in 2009 (named environment E0). These materials were managed under natural rainfall conditions of 192 mm during the growing season.
Population 2 consisted of 262 accessions (Supplementary Table S2), of which 254 were from China, three from USA, two from Australia, two from Italy, and one from Romania, including 209 modern varieties, 43 advanced lines and 10 landraces (Li et al. 2012). The cultivars from China were mainly planted in the Northern Winter Wheat Zone, and Yellow and Huai River Valleys Facultative Wheat Zone over recent decades. The accessions were grown at two sites over 2 years, viz. Changping and Shunyi (116°56′E; 40°23′N) in Beijing, and the planting years were 2009 and 2010. At each site, the field was managed under two water regimes, rain-fed (DS) and well-watered (WW). The total rainfalls in the growing seasons were 192 mm and 131 mm, respectively. The WW plots were irrigated with 750 m3/ha at the pre-overwintering, jointing, flowering and grain filling stages. E1, E2, E3, E4, E5, E6, E7 and E8 indicate the environments of Changping in 2009 under DS, Changping in 2009 under WW, Shunyi in 2009 under DS, Shunyi in 2009 under WW, Changping in 2010 under DS, Changping in 2010 under WW, Shunyi in 2010 under DS, and Shunyi in 2010 under WW, respectively.
Measurement of plant height at different growth stages
Plant height was measured every 7 days from the early booting stage until flowering, a total of four measurements, designated S1, S2, S3 and S4. S4 was the plant height at flowering, the final plant height. Based on development theory and QGAStation V1.0 software proposed by Zhu (Zhu 1995), conditional plant heights (\( {\text{S}}_{t} |{\text{S}}_{t - 1} \)) were obtained from the data for unconditional plant height (S1–S4). For conditional plant heights, \( {\text{S}}_{ 2} |{\text{S}}_{ 1} \) (PH1) revealed the net genetic effects of genes expressed during the first two stages (S1 and S2), and likewise for \( {\text{S}}_{3} |{\text{S}}_{2} \) (PH2) and \( {\text{S}}_{4} |{\text{S}}_{3} \) (PH3). All the phenotypic analyses were carried out using SAS V8.1 software.
Genotype detection
Based on the result of our previous research on the Hanxuan 10 × Lumai 14 DH population, QTL with large effects on chromosomes 1B, 2D, 4B, 4D, 5A and 7B were selected as candidate QTL for further verification by association analysis (Wu et al. 2010). Twenty-nine candidate SSR markers covering the respective chromosome regions were used for association mapping (Supplementary Table S3). In addition, the population structure was evaluated by another 29 unlinked loci evenly distributed across the entire wheat genome (Supplementary Table S4). The genetic locations of all the SSR markers were obtained from the consensus map Ta-SSR-2004 (Somers et al. 2004). The fluorescent primers were synthesized by ABI (Applied Biosystems, Foster City, CA, USA). Amplification products were separated by an ABI3730 DNA Analyzer (Applied Biosystems), and the outputs were analyzed by GeneMapper software (http://www.appliedbiosystems.com.cn/). The allele number, allele frequency and polymorphism information content (PIC) were calculated by PowerMarker V3.25 software (Liu and Muse 2005). Marker alleles with frequencies of <5 % were treated as rare alleles.
Population structure
Population structure was estimated by STRUCTURE v2.3.2 (Pritchard et al. 2000). The number of hypothetical subpopulations (K) was set from 2 to 9 with a burn-in period length of 50,000 iterations and a run of 500,000 replications of Markov Chain Monte Carlo (MCMC) after burn-in. Each K was duplicated five times. The admixture model of STRUCTURE allowed for a population mixture and correlated allele frequencies. The most appropriate K value was evaluated by lnP(D) in the STRUCTURE output (Evanno et al. 2005). According to the most appropriate K value, the Q-matrix of five repeats was integrated by using the CLUMPP software (Jakobsson and Rosenberg 2007).
Association mapping
For marker-trait association, a structured association approach was implemented by a general linear model (GLM) in TASSEL 2.1 (Bradbury et al. 2007). In order to correct for spurious associations, the Q-matrix was used in the model. The threshold (P value) for significant association between markers and traits was 0.001. The phenotypic variance explained (PVE) for each significantly associated locus was evaluated by R 2 values for the markers (Zhang et al. 2011). Allelic effects were evaluated in comparison to the “null allele” (missing data plus rare alleles) for each associated locus (Breseghello and Sorrells 2006). The large-effect alleles were confirmed with the final plant heights of population 2 by analysis of variance.
Results
Phenotypic variation
The mean unconditional plant height (S4) at the flowering stage in Population 1 was 82.4 cm, ranging from 60.0 to 118.0 cm, with a coefficient of variation (CV) of 16.0 %. The CVs of conditional plant height at the three growth periods (PH1, PH2 and PH3) were 9.2, 6.8 and 10.2 %, respectively (Supplementary Table S5). The high values of CV at various growth periods indicated wide phenotypic variation among accessions, which was suitable for association analysis. Compared to PH1 and PH2, the higher CV of PH3 showed that the third growth period (\( {\text{S}}_{4} |{\text{S}}_{3} \)) was more important for plant height development, leading to the obvious differences in plant height in Population 1. The phenotypic data statistics for plant height in Population 2 in eight environments at maturity are presented in Supplementary Table S6.
Allelic diversity and population structure
A total of 422 alleles were detected by 58 SSR markers in Population 1. The numbers of alleles per locus varied from 3 to 16, and PIC ranged from 0.033 to 0.818, with averages of 7 and 0.519, respectively. A key issue for association mapping is estimation of population structure, which can result in spurious associations between phenotypes and markers. In previous research, about 60 traits were measured at up to 10 environments in the diverse maize germplasm set of 302 inbred lines. Overall, population structure accounts for an average of 9.3 % of the phenotypic variation (Flint-Garcia et al. 2005). The Q-matrix from STRUCTURE can help to reduce the risk of false positives arising from population structure (Bradbury et al. 2007). Twenty-nine whole-genome SSR markers were selected to estimate the population structure of Population 1. The average lnP(D) value for each K (from 2 to 9) is visualized in Supplementary Fig. S1a and the inflection point appeared at K = 3 (marked with an asterisk). In addition, the second-order likelihood (ΔK) was also calculated. We found that the highest ΔK value occurred at K = 3, and was much less at K = 4–8 (Supplementary Fig. S1b). According to lnP(D) and ΔK, Population 1 was classified into three subpopulations, containing 52, 25 and 41 accessions, respectively (Supplementary Fig. S1c).
Verification of previous QTL for dynamic developmental plant height by association analysis
Candidate SSR markers for plant height, located in large-effect QTL regions, were used for association mapping in Population 1. Nine loci significantly (P < 0.001) associated with plant height were detected 13 times (Table 1). Three loci (Xgwm11-1B, Xwmc349-4B and Xcfd23-4D) were identified in three, two and two periods of plant height growth, respectively. The phenotypic variation explained (PVE) ranged from 14.06 to 28.34 % (Table 1). There were one, two, and seven loci significantly associated with plant height in PH1, PH2 and PH3, respectively; three loci were significantly associated with S4. In total, two markers Xgwm18-1B and Xgwm11-1B were detected four times, i.e. Xgwm18-1B was detected in PH3, Xgwm11-1B was significantly associated with PH2, PH3 and S4, and the genetic distance between them was only 0.7 cM in Ta-SSR-2004 (Somers et al. 2004).
Marker Xbarc168-2D detected in the first period (PH1) was responsible for 14.06 % of the variation in plant height. Two loci on chromosomes 1B and 4B were significantly associated with PH2, with PVEs of 23.55 and 15.55 %, respectively. Seven loci identified in PH3 were distributed on chromosomes 1B, 2D, 4B, 4D, 5A and 7B. Of them, Xgwm213-7B showed the largest effect, explaining 28.34 % of the phenotypic variation. At the flowering stage, Xgwm11-1B, Xwmc349-4B and Xcfd23-4D were significantly associated with the plant height. Of them, Xcfd23-4D, accounting for 25.82 % of the total variation, displayed the largest effect on plant height. Xgwm11-1B was identified in three periods, with PVEs of 23.55, 18.24 and 19.34 %, respectively (Table 1).
Allelic effects of associated loci verified in multiple environments
Detecting loci associated with dynamic developmental plant height was not the final objective, but was a prerequisite for its application. It is more significant to search for elite alleles, a major advantage of association mapping compared to biparental family-based linkage mapping. Allelic effects were estimated through comparison with the “null allele” for each associated locus (Breseghello and Sorrells 2006), and large-effect alleles and their phenotypic effects are presented in Table 2. The frequencies of all large-effect alleles were higher than 5 %, ranging from 5.08 to 86.44 %. As for effects at the flowering stage, Xgwm11-1B 208 and Xwmc349-4B 101 showed the largest negative (−10.7 cm) and positive (15.1 cm) effects on plant height, respectively. Shorter plant height (74.1 cm) was associated with Xgwm11-1B 208 (13 accessions), whereas 12 accessions with Xwmc349-4B 101 were clearly taller (97.5 cm) at flowering. As with previous reports, alleles at one locus could have opposite functions at the same stage (Wen et al. 2008). For Xgwm18-1B, 12 accessions carrying the 194-bp allele (negative effect −2.6 cm in PH3) contributed to shorter plants (77.4 cm), whereas nine accessions with the 189-bp allele (positive effect 9.5 cm) contributed to taller plants (89.5 cm). At flowering, 55 accessions carrying the allele Xcfd23-4D 202 with a negative effect (−4.2 cm) produced shorter plants (78.1 cm), and 50 accessions with a positive effect (5.1 cm) from the 205-bp allele were taller (87.5 cm).
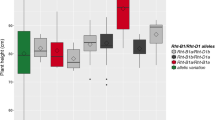
Population and environment are also critical factors influencing the results of association analysis. In molecular breeding programs, breeders require elite marker alleles associated with target traits that can be repeatedly verified across genetic backgrounds under different environmental conditions. In Population 1, Xgwm11-1B 208 , Xwmc349-4B 103 and Xcfd23-4D 202 were detected as having obvious negative effects; Xwmc349-4B 101 and Xcfd23-4D 205 showed positive effects at flowering. We then used Population 2 to confirm the large effect alleles of the associated loci in eight environments. The average final plant heights of accessions carrying the large effect alleles (with obvious negative or positive effects) were significantly (P < 0.05) or highly significantly (P < 0.01, P < 0.001) shorter or taller than those without the large-effect alleles in almost all environments (Fig. 1).

Verification of phenotypic effects of large-effect alleles in two populations in nine environments. Population 1 was planted in E0; Population 2 was planted in E1–E8. Bars indicate 2 standard errors. *, **, ***Significant at P = 0.05, 0.01 and 0.001, respectively
Efficacy of pyramiding large-effect alleles of associated loci
The objective of gene pyramiding in molecular breeding is to combine a series of target alleles in a specific line or variety (Servin et al. 2004). We examined the efficacy of pyramiding multiple alleles with large unidirectional effects in one individual. Three alleles, Xgwm11-1B 208 , Xwmc349-4B 103 and Xcfd23-4D 202 , with negative effects, and two alleles, Xwmc349-4B 101 and Xcfd23-4D 205 , exhibiting positive effects on plant height, were identified at the flowering stage. The plant height of genotypes with the three “pyramided” negative alleles was 72.8 cm in Population 1 in E0, and 60.8–77.7 cm in Population 2 in E1–E8. In contrast, the plant height of accessions carrying the two positive alleles was 113.6 cm in Population 1 in E0, and 96.8–123.7 cm in Population 2 in E1–E8 (Table 3). Consistent results indicated that pyramiding of elite alleles associated with plant height produced significantly shorter or taller plants. The linear correlations were highly significant (Fig. 2).

Linear regressions of final plant heights in Populations 1 (118 accessions) and 2 (262 accessions) in nine environments (E0–E8). a Plant height and number of alleles with negative effects on height; b plant height and number of alleles with positive effects on height
Discussion
The developmental genetics of plant height
Plant height is controlled by both Mendelian and quantitative genetics (Wang et al. 2010). In this research unconditional and conditional association mapping were employed to dissect the developmental genetics of plant height. Previous studies inferred that alleles at the same genomic location could have opposite genetic effects at different growth stages (Yan et al. 1998b). We obtained similar results. For example, shorter plant height was associated with Xgwm11-1B 199 (negative effect: −5.8 cm) in the second period (\( {\text{S}}_{3} |{\text{S}}_{2} \), PH2), but in contrast the effect of the same allele was 1.4 cm in the third period (\( {\text{S}}_{4} |{\text{S}}_{3} \), PH3). This further indicated that (1) gene expression occurs selectively (even with opposite effects) at different developmental stages; and (2) several genes with opposite genetic effects may express simultaneously in the same or adjacent genomic regions. However, unconditional analysis considering only final plant height in this research perhaps unavoidably excluded some loci associated with plant height development. This may be one reason why ten loci were detected by conditional analysis, whereas only three loci were detected for final plant height (Table 1).
There were one, two and seven loci significantly associated with PH1, PH2 and PH3, respectively. The participation of different numbers of loci revealed that plant height was a typical quantitative trait, and different genetic systems were responsible for plant height development during ontogeny (Wu et al. 2010). Moreover, seven loci were identified in the third period (\( {\text{S}}_{4} |{\text{S}}_{3} \), PH3), much more than the number of loci found associated with PH1 and PH2, showing that genes controlling plant height were actively expressed in the third period. Thus, final plant height is the accumulation of genetic effects that occur during the whole growth period, and conditional analysis detects the net effects of gene expression at specific stages.
Verification of previous QTL and fine mapping
Family-based linkage analysis and association mapping have been widely used in research on quantitative traits. Association mapping based on linkage disequilibrium (LD) can provide high resolution that may contribute to the full use of a potentially large range of allelic variation in natural populations (Remington et al. 2001). Both association mapping and linkage analysis are required to avoid detection of false-positive loci. Therefore, linkage analysis can be used for genomic scans, and association mapping can verify candidate QTL regions and fine mapping. In our research, SSR markers located at candidate QTL regions with large effects in the Hanxuan 10 × Lumai 14 DH population (Wu et al. 2010) were selected for association analysis. Nine loci significantly associated with plant height were detected 13 times (Table 1). The results showed a consistency between linkage analysis and association mapping, and also displayed the fine mapping of QTL through combined linkage and association analysis. For example, an additive plant height QTL, QPh.cgb-1B.1, was detected in the DH population. The genetic distance between the flanking markers (Xgwm582–Xgwm273) was 3.9 cM in Ta-SSR-2004 (Somers et al. 2004). Both Xgwm18-1B (0.2 cM from Xgwm273) and Xgwm11-1B, in the same region, were detected by association mapping in our research, and the genetic distance between them was only 0.7 cM. Some other additive QTL were mapped at known Rht loci, such as QPh.cgb-2D.1 which maps to Rht8, QPh.cgb-4D.1 to Rht2 or Rht10, and QPh.cgb-5A.6 to Rht12 (Wu et al. 2010). By association mapping, Xbarc168-2D and Xgwm249-2D (in QPh.cgb-2D.1), Xcfd23-4D (1.6 cM near to one flanking marker of QPh.cgb-4D.1), and Xgwm410-5A (one of the flanking markers of QPh.cgb-5A.6) were detected in our research. Xwmc349-4B is closely linked with Rht-B1 and Xgwm410-5A is linked with Rht12 at a distance of 11.0 cM (Ellis et al. 2005; Korzun et al. 1997; Somers et al. 2004).
Flint-Garcia et al. (2003) proposed that if a marker is too close to the causal gene, the frequency of the major allele is so high that other alleles become null alleles (low frequencies); the very low diversity of the marker will then fail to be detected by association mapping (Flint-Garcia et al. 2003). The genetic locations of Xgwm498-1B, Xgwm18-1B and Xgwm11-1B were 31.1, 33.6 and 34.3 cM, respectively, in the consensus map Ta-SSR-2004 (Somers et al. 2004). Xgwm18-1B and Xgwm11-1B were detected in our research, but it was surprising that Xgwm498-1B was not detected. The frequencies of the major alleles of Xgwm18-1B and Xgwm11-1B were 63.56 and 68.64 %; the PIC values were 0.5410 and 0.4840, respectively. The frequency of the major alleles of Xgwm498-1B was 92.37 %, and the PIC was only 0.1341 (Tables 1, 2). Thus, it is inferred that if a causal gene for plant height was near the genomic region of Xgwm498-1B, it should have been selected in breeding. Because of the effect of “selection valleys” (close proximity to the selected gene, and consequently low genetic diversity at the locus) (Barrero et al. 2011), Xgwm498-1B was at the bottom of the valley, and failed to be detected. Xgwm18-1B and Xgwm11-1B, on the other hand, flanked the valley, and were thus detected in our research. The same “selection valleys” in this chromosome region were reported to associate with plant height (Barrero et al. 2011).
Stable molecular markers and enough elite alleles determine the future of gene pyramiding
New strategies of MAS are flourishing in the genomics era, and the explosive growth in the number of QTL reminds us to ask (1) how many previously reported QTL could be repeatedly detected or verified, and (2) are there enough elite alleles in germplasm resources? It has been proposed that the narrow genetic base of modern crop cultivars is a major problem for further improvement of crop productivity (Abdurakhmonov and Abdukarimov 2008). Genetic diversity in germplasm resources provides a good way to reduce the problem of the high degree of similarity among cultivars. It is therefore important to search for elite alleles at loci associated with important traits. Different alleles of a causal gene can lead to drastic phenotypic differences; for example, a single SNP caused loss of seed shattering during rice domestication (Konishi et al. 2006). Therefore, exploration of elite alleles is a critical region for germplasm managers and plant breeders looking for alleles of interest in germplasm collections rather than as sequences in GenBank (Famoso et al. 2011). Many studies show that marker-based strategies of pyramiding are effective (Sacco et al. 2013; Werner et al. 2005). However, an essential prerequisite for gene pyramiding is stable molecular markers (Brar et al. 2000); that is, the elite alleles of associated loci must be repeatedly detected or verified without being limited by interactions with genetic background or environment. In our research, five large-effect alleles with obvious negative or positive effects were identified in Population 1 in E0 and validated using Population 2 in eight environments (E1–E8) (Table 2; Fig. 1). We also observed that genetic background has a great influence on the effect of elite alleles. For example, varieties Jimai 32 and Jinguang carried the negative allele (Xwmc349-4B 103 ), but showed extremely tall final plant heights (118 cm and 108 cm) compared to the average of all accessions with Xwmc349-4B 103 in Population 1 in E0 (data not provided). Rare individuals like Jimai 32 and Jinguang can hide the effect of Xwmc349-4B 103 in small populations, but cannot hide the effect in large populations. Therefore, it is easy for us to understand why no significant difference in plant height was detected between accessions carrying Xwmc349-4B 103 and others in E0 in Population 1 (118 accessions), but there was extreme significant difference across E1–E8 in Population 2 (262 accessions, Fig. 1). Pyramiding elite alleles associated with plant height showed that the greater the number of elite alleles, the higher (or lower) the resulting plant height (Table 3; Fig. 2). The obvious dosage effect shows that pyramiding elite alleles for a target trait has great potential for wheat breeding.
Many important traits are controlled by QTL. The combination of linkage analysis and association mapping provides an efficient method of finding elite alleles in natural populations. But as QTL are based on statistical calculations, verification is needed to confirm the effectiveness of such alleles (Ashikari and Matsuoka 2006). In conclusion, stable molecular markers and a sufficient number of elite alleles will determine whether the pyramiding of elite alleles by MAS will or will not be effective in plant breeding.
References
Abdurakhmonov IY, Abdukarimov A (2008) Application of association mapping to understanding the genetic diversity of plant germplasm resources. Int J Plant Genomics. doi:10.1155/2008/574927
Ashikari M, Matsuoka M (2006) Identification, isolation and pyramiding of quantitative trait loci for rice breeding. Trends Plant Sci 11:344–350
Barrero RA, Bellgard M, Zhang X (2011) Diverse approaches to achieving grain yield in wheat. Funct Integr Genomics 11:37–48
Börner A, Plaschke J, Korzun V, Worland AJ (1996) The relationships between the dwarfing genes of wheat and rye. Euphytica 89:69–75
Bradbury PJ, Zhang ZW, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Brar D, Sanchez A, Khush G, Li Z, Huang N (2000) Sequence tagged site marker-assisted selection for three bacterial blight resistance genes in rice. Crop Sci 40:792–797
Breseghello F, Sorrells ME (2006) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172:1165–1177
Cadalen T, Sourdille P, Charmet G, Tixier M, Gay G, Boeuf C, Bernard S, Leroy P, Bernard M (1998) Molecular markers linked to genes affecting plant height in wheat using a doubled-haploid population. Theor Appl Genet 96:933–940
Cao G, Zhu J, He C, Gao Y, Yan J, Wu P (2001) Impact of epistasis and QTL× environment interaction on the developmental behavior of plant height in rice (Oryza sativa L.). Theor Appl Genet 103:153–160
Cui F, Li J, Ding A, Zhao C, Wang L, Wang X, Li S, Bao Y, Li X, Feng D (2011) Conditional QTL mapping for plant height with respect to the length of the spike and internode in two mapping populations of wheat. Theor Appl Genet 122:1517–1536
Ellis M, Rebetzke G, Azanza F, Richards R, Spielmeyer W (2005) Molecular mapping of gibberellin-responsive dwarfing genes in bread wheat. Theor Appl Genet 111:423–430
Ellis M, Bonnett D, Rebetzke G (2007) Borlaug, Strampelli and the worldwide distribution of Rht8. Wheat Prod Stress Environ 12:787–791
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Evenson RE, Gollin D (2003) Assessing the impact of the green revolution, 1960 to 2000. Science 300:758–762
Famoso AN, Zhao K, Clark RT, Tung CW, Wright MH, Bustamante C, Kochian LV, McCouch SR (2011) Genetic architecture of aluminum tolerance in rice (Oryza sativa L.) determined through genome-wide association analysis and QTL mapping. PLoS Genet 7:e1002221
Flint-Garcia SA, Thornsberry JM, Buckler ES (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54:357–374
Flint-Garcia SA, Thuillet AC, Yu J, Pressoir G, Romero SM, Mitchell SE, Doebley J, Kresovich S, Goodman MM, Buckler ES (2005) Maize association population: a high-resolution platform for quantitative trait locus dissection. Plant J 44:1054–1064
Hedden P (2003) The genes of the green revolution. Trends Genet 19:5–9
Huang X, Cloutier S, Lycar L, Radovanovic N, Humphreys D, Noll J, Somers D, Brown P (2006) Molecular detection of QTLs for agronomic and quality traits in a doubled haploid population derived from two Canadian wheats (Triticum aestivum L.). Theor Appl Genet 113:753–766
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jestin C, Lodé M, Vallée P, Domin C, Falentin C, Horvais R, Coedel S, Manzanares-Dauleux M, Delourme R (2011) Association mapping of quantitative resistance for Leptosphaeria maculans in oilseed rape (Brassica napus L.). Mol Breed 27:271–287
Jiang G, Sun J, Liu H, Qu C, Wang K, Guo R, Bai K, Gao L, Kuang T (2003) Changes in the rate of photosynthesis accompanying the yield increase in wheat cultivars released in the past 50 years. J Plant Res 116:347–354
Keller M, Karutz C, Schmid J, Stamp P, Winzeler M, Keller B, Messmer M (1999) Quantitative trait loci for lodging resistance in a segregating wheat × spelt population. Theor Appl Genet 98:1171–1182
Klahr A, Zimmermann G, Wenzel G, Mohler V (2007) Effects of environment, disease progress, plant height and heading date on the detection of QTLs for resistance to Fusarium head blight in an European winter wheat cross. Euphytica 154:17–28
Konishi S, Izawa T, Lin SY, Ebana K, Fukuta Y, Sasaki T, Yano M (2006) An SNP caused loss of seed shattering during rice domestication. Science 312:1392–1396
Korzun V, Röder M, Worland A, Börner A (1997) Intrachromosomal mapping of genes for dwarfing (Rht12) and vernalization response (Vrn1) in wheat by using RFLP and microsatellite markers. Plant Breed 116:227–232
Kuchel H, Fox R, Reinheimer J, Mosionek L, Willey N, Bariana H, Jefferies S (2007) The successful application of a marker-assisted wheat breeding strategy. Mol Breed 20:295–308
Li H, Zhang L, Wang J (2010) Analysis and answers to frequently asked questions in quantitative trait locus mapping. Acta Agron Sin 36:918–931
Li W, Zhang B, Zhang J, Chang X, Li R, Jing R (2012) Exploring elite alleles for chlorophyll content of flag leaf in natural population of wheat by association analysis. Acta Agron Sin 38:962–970
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
McCartney C, Somers D, Humphreys D, Lukow O, Ames N, Noll J, Cloutier S, McCallum B (2005) Mapping quantitative trait loci controlling agronomic traits in the spring wheat cross RL4452 × ‘AC Domain’. Genome 48:870–883
McIntosh RA, Yamazaki Y, Dubcovsky J, Rogers J, Morris C, Somers DJ, Appels R, Devos KM (2008) Catalogue of gene symbols for wheat. In: McIntosh RA (ed) Proceedings of the 11th international wheat genetics symposium. Sydney University Press, NSW, pp 30–35
Peng Z, Li X, Yang Z, Liao M (2011) A new reduced height gene found in the tetraploid semi-dwarf wheat landrace Aiganfanmai. Genet Mol Res 10:2349–2357
Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000) Association mapping in structured populations. Am J Hum Genet 67:170–181
Reif JC, Zhang P, Dreisigacker S, Warburton ML, van Ginkel M, Hoisington D, Bohn M, Melchinger AE (2005) Wheat genetic diversity trends during domestication and breeding. Theor Appl Genet 110:859–864
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci USA 98:11479–11484
Roussel V, Koenig J, Beckert M, Balfourier F (2004) Molecular diversity in French bread wheat accessions related to temporal trends and breeding programmes. Theor Appl Genet 108:920–930
Sacco A, Di Matteo A, Lombardi N, Trotta N, Punzo B, Mari A, Barone A (2013) Quantitative trait loci pyramiding for fruit quality traits in tomato. Mol Breed 31:217–222. doi:10.1007/s11032-012-9763-2
Servin B, Martin OC, Mézard M (2004) Toward a theory of marker-assisted gene pyramiding. Genetics 168:513–523
Somers DJ, Isaac P, Edwards K (2004) A high-density microsatellite consensus map for bread wheat (Triticum aestivum L.). Theor Appl Genet 109:1105–1114
Sun D, Li W, Zhang Z, Chen Q, Yang Q (2006) Analysis of QTL for plant height at different developmental stages in soybean. Acta Agron Sin 32:509–514
Wang Z, Wu X, Ren Q, Chang X, Li R, Jing R (2010) QTL mapping for developmental behavior of plant height in wheat (Triticum aestivum L.). Euphytica 174:447–458
Wen Z, Zhao T, Zheng Y, Liu S, Wang C, Wang F, Gai J (2008) Association analysis of agronomic and quality traits with SSR markers in glycine max and glycine soja in China: II. Exploration of elite alleles. Acta Agron Sin 8:1339–1349
Werner K, Friedt W, Ordon F (2005) Strategies for pyramiding resistance genes against the barley yellow mosaic virus complex (BaMMV, BaYMV, BaYMV-2). Mol Breed 16:45–55
Wheeler NC, Jermstad KD, Krutovsky K, Aitken SN, Howe GT, Krakowski J, Neale DB (2005) Mapping of quantitative trait loci controlling adaptive traits in coastal Douglas-fir. IV. Cold-hardiness QTL verification and candidate gene mapping. Mol Breed 15:145–156
Wu X, Wang Z, Chang X, Jing R (2010) Genetic dissection of the developmental behaviours of plant height in wheat under diverse water regimes. J Exp Bot 61:2923–2937
Yan J, Zhu J, He C, Benmoussa M, Wu P (1998a) Molecular dissection of developmental behavior of plant height in rice (Oryza sativa L.). Genetics 150:1257–1265
Yan J, Zhu J, He C, Benmoussa M, Wu P (1998b) Quantitative trait loci analysis for the developmental behavior of tiller number in rice (Oryza sativa L.). Theor Appl Genet 97:267–274
Yan J, Tang H, Huang Y, Shi Y, Li J, Zheng Y (2003) QTL mapping for the developing of the plant height traits in maize. Chin Sci Bull 48:1959–1964
Zhang J, Hao C, Ren Q, Chang X, Liu G, Jing R (2011) Association mapping of dynamic developmental plant height in common wheat. Planta 234:891–902
Zhou Y, He Z, Sui X, Xia X, Zhang X, Zhang G (2007) Genetic improvement of grain yield and associated traits in the northern China winter wheat region from 1960 to 2000. Crop Sci 47:245–253
Zhu J (1995) Analysis of conditional genetic effects and variance components in developmental genetics. Genetics 141:1633–1639
Acknowledgments
We thank Professor Robert A. McIntosh (Plant Breeding Institute, University of Sydney, NSW, Australia) for revising the manuscript. This study was supported by the National Natural Science Foundation of China (31271720) and the National High Technology Research and Development Program of China (2011AA100501).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table S1
Backcross introgression lines (BILs) in Population 1 (XLS 30 kb)
Supplementary Table S2
262 accessions of Population 2 and their origins (XLS 53 kb)
Supplementary Table S3
Candidate SSR markers for association mapping in Population 1 (XLS 26 kb)
Supplementary Table S4
SSR markers for evaluating population structure of Population 1 (XLS 26 kb)
Supplementary Table S5
Conditional phenotypic values for plant height of Population 1 during three growth periods and unconditional plant height at flowering (XLS 24 kb)
Supplementary Table S6
Unconditional plant height of 262 accessions at maturity stage (XLS 24 kb)
Supplementary Fig. S1
Population structure of Population 1 based on 29 genome-wide SSR markers. a, b: Population structure as determined by lnP(D) and ΔK over five repeats of STRUCTURE analysis; c: Structure analysis revealed three sub-populations. Each accession is represented by a vertical bar, and the colored segments within each bar reveal the proportion of each subpopulation (PPT 859 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Zhang, B., Shi, W., Li, W. et al. Efficacy of pyramiding elite alleles for dynamic development of plant height in common wheat. Mol Breeding 32, 327–338 (2013). https://doi.org/10.1007/s11032-013-9873-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-013-9873-5