
Abstract
Flaxseed (Linum usitatissimum L.) is renowned for having the highest concentration of lignans among all plant species worldwide. However, it also contains a notable amount of cyanogenic glycosides, prompting efforts to reduce their levels. MicroRNAs, recognized as significant epigenetic factors, hold the potential to serve as genetic markers in achieving this breeding goal. In this study, 44,885 mature microRNAs were utilized alongside one genome and four transcriptomes of flax. Twelve gene sequences were obtained for five lignan enzymes and three enzymes related to cyanogenic glycosides. Based on our in silico approach, we identified 15 microRNA families for the lignan metabolic pathway, 10 for the cyanogenic glycoside pathway, and 6 regulating both pathways. Additionally, we contributed to the annotation of the used transcriptomes and verified the functionality of the “Finding genes by keyword” algorithm available on Phytozome 13. The results obtained led to the design of a unique schema for microRNA mediated regulation in the biosynthetic pathways of lignans and cyanogenic glycosides. This research will enhance our understanding of the regulatory mechanisms of microRNA in these biosynthetic pathways, along with other specialized metabolites. The predicted microRNAs can be employed in marker-assisted selection with the breeding objective of optimizing the ratio of nutritional and antinutritional components in flaxseed while maintaining current physiological parameters.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Flax (Linum usitatissimum L.) is an annual plant that belongs to the Linaceae family (Fojnica et al. 2022). Its primary products are currently seeds that are rich in omega-3 fatty acids and fibre, with the amount of lignans being 800 times higher compared to other plants (Kajla et al. 2015). Among its main antinutrients are cyanogenic glycosides, for which the breeding program is set up to minimize in contrast to lignan content (Fofana et al. 2017b; Kazachkov et al. 2020; Sharma et al. 2021). For these purposes, microRNAs represent a valuable tool in agronomic trait analysis, and innovative in silico approaches are necessary for a successful breeding process (Chen et al. 2021; Tan et al. 2022).
Lignans are well known for their therapeutic potential, attributed to their antioxidant, anti-inflammatory, anticancer, antidiabetic, estrogenic, and antiestrogenic features (Ebrahimi 2021; Osmakov et al. 2022). Lignans present in Linum usitatissimum L. include the stereospecific compounds (+)-pinoresinol and (−)-pinoresinol, (+)-lariciresinol and (−)-lariciresinol, and (+)-secoisolariciresinol and (−)-secoisolariciresinol. The biosynthetic pathway of these lignans can continue through the synthesis of (−)-yatein, (−)-podophyllotoxin, or (−)-hinokinin (Marcotullio et al. 2014; Corbin et al. 2017; De Silva et al. 2019). The most common form of (+)-secoisolariciresinol in seeds is its diglucosidic form, known as (+)-secoisolariciresinol diglucoside, while (−)-matairesinol is predominantly found in the aerial parts (Corbin et al. 2017; Markulin et al. 2019; Prasad et al. 2020). The strict stereospecific biosynthetic pathway of (+)-secoisolariciresinol diglucoside begins with the binding of two units of coniferyl alcohol radical, catalysed by Dirigent protein 5 or 6, resulting in the production of (−)-pinoresinol (Corbin et al. 2017). The conversion involves (−)-pinoresinol-(−)-lariciresinol reductase 1, which processes it into (−)-lariciresinol and subsequently into (+)-secoisolariciresinol. This compound is then glycosylated to form (+)-secoisolariciresinol diglucoside by the Uridine glycosyltransferase UGT74S1 (Ghose et al. 2014; Kazachkov et al. 2020). Dirigent protein 1 operates with opposite stereospecificity and is responsible for the formation of (+)-pinoresinol from the same two coniferyl alcohol radical units (Markulin et al. 2019). The enzyme (+)-pinoresinol-(+)-lariciresinol reductase 2 is capable of synthesizing both (+)-lariciresinol and (−)-secoisolariciresinol (Corbin et al. 2017; Tashackori et al. 2021). (−)-secoisolariciresinol forms its diglucosidic state in minimal amounts and is predominantly dehydrogenated by (−)-secoisolariciresinol dehydrogenase to produce the lignan (−)-matairesinol (Schmidt et al. 2012; Shi et al. 2022). The content of lignans in flaxseed is genotype-specific, a factor often overlooked in the sale of flaxseeds as dietary supplements (Zhang et al. 2022). General enhancement of lignan levels in flax genotypes will enable the full utilization of its potential. The highly valued metabolite, (+)-secoisolariciresinol diglucoside, is metabolized in the human gastrointestinal system by intestinal flora into phytohormones enterodiol and enterolactone. These derivatives of estrogen can substitute for it in case of deficiency, reducing the risk of several illnesses, including cardiovascular diseases, cancer, diabetes, and many others (Aqeel et al. 2019; Yang et al. 2021).
Cyanogenic glycosides (CG) constitute another class of specialized metabolites that play a role in defence reactions against herbivores and other stresses by releasing hydrogen cyanide (HCN) (Appenteng et al. 2021). This ability is approximately 300 million years old and is present in more than 3000 plant species across 130 families (Van Ohlen et al. 2017; Lechtenberg 2021). From an evolutionary perspective, ferns and gymnosperms synthesize CG from the aromatic amino acids tyrosine or phenylalanine, whereas angiosperms also utilize aliphatic valine, leucine, or isoleucine (Mobot 2021). Several plants, such as passiflora, have the ability to synthesize them even from cyclopentenyl glycine (Sculfort et al. 2021). Although some plants have a high CG content, they are still considered edible (Mosayyebi et al. 2020; Jaszczak-Wilke et al. 2021). The biosynthetic pathway of flax cyanogenic monoglucosides linamarin and lotaustralin begins with the N-hydroxylation of L-valine or L-isoleucine by cytochrome P450 monooxygenase (CYP) from the enzyme family CYP79D. The process continues with isomerization, dehydration, and C-hydroxylation by enzyme family CYP71E. Finally, it is completed by acetone cyanohydrin β-glucosyltransferase from family UGT85K (Fang et al. 2016; Mohd Azmi 2019; Juma et al. 2022). Together with NADPH reductase, these enzymes can form a complex known as metabolon, which is attached to the membrane and operates very efficiently (Zhang and Fernie 2021; Del Giudice et al. 2022). In the case of linustatin and neolinustatin, the diglucosidic bond is mediated by another nonspecific UDP-glucosyltransferase (Hartanti and Cahyani 2020; Kazachkov et al. 2020; Yulvianti and Zidorn 2021). CG are stored in vacuoles, separated from their hydrolytic lyases—β-glucosidases (De Brito and Martinoia 2018). Within the plant, they are mostly located in the aerial parts but can also be found in the roots (Akatsuka and Ito 2022). During the seed maturation they are transformed into diglucosidic form and transported to seed (Deng et al. 2021). Among the other roles of CG in plant organisms are the supply of nitrogen and glucose, the regulation of dormancy, seed germination, buds opening, flower development, cell signalling, and the expression of regulatory genes (Del Cueto et al. 2017; Ritmejerytė et al. 2019; Nyirenda 2020). Cyanide inhibits the utilization of oxygen and increases anaerobic metabolism (Chongtham et al. 2022). The toxic threshold of HCN in the blood ranges from 0.5 to 1.0 mg L−1 (20–40 µM), and the first clinical signs, such as headache, hyperventilation, vomiting, weakness, or abdominal cramps, become evident within 30 min (Alitubeera et al. 2019; Schrenk et al. 2019; Kuliahsari et al. 2021). The release of HCN occurs after the mechanical disruption of the vacuole when β-glucosidases, present in the cytosol, break the bonds of cyanogenic glycosides (Sun et al. 2018). One gram of linamarin generates 109 mg of HCN, and the consumption of 30 g of flaxseed increases HCN in the blood by 5 µM (Abraham et al. 2016; Schrenk et al. 2019). Similarly to lignans, the quantity of CG is genotype-variable, with current knowledge indicating their mutual negative correlation (Zuk et al. 2020).
MicroRNAs are short, noncoding, single-stranded RNA molecules with lengths ranging from 20 to 24 nucleotides, and play an important role in the negative regulation of gene expression (Song et al. 2019). They participate in responses to both abiotic and biotic stresses, as well as in the growth and development of plants (Yu et al. 2019). In the research field, they represent a novel approach as effective markers for various processes within an organism (Tyagi et al. 2019). MicroRNAs belong to highly conserved elements in the genome, and their reliability in complementary binding with mRNAs enables their precise bioinformatic prediction (Tafrihi and Hasheminasab 2019). They are active throughout the entire life cycle, from germination to reproduction, and in all parts of the plant, including roots, stems, leaves, flowers, seeds, and callus (Gramzow and Theißen 2019; Pirrò et al. 2019). The biosynthesis of microRNA begins with the transcription of the MIR gene (Jeena et al. 2022). The promoter of microRNA is typically located in an intergenic, intronic, or noncoding 3′–5′ space and includes a TATA-box, as well as the transcription start site (Buch et al. 2020; Sun et al. 2021). Transcription is typically carried out by RNA polymerase II, producing the long loop structure pri-microRNA (primary microRNA), which includes the 5′ cap and 3′ polyadenylated tail (Lee et al. 2019). During the processing of pri-microRNA, mediated by the endonuclease RNase III, many transcription factors participate in the conversion to precursor pre-miRNA and double-stranded duplex microRNA/microRNA* with two-nucleotide overhangs (Kai et al. 2022). Posttranscriptional modifications include several methylations, one of which is the methylation of both ends of the duplex by the methyltransferase HUA-enhancer 1 (Chen and Ren 2019). The RNA-induced silencing complex (RISC) is activated by the protein Argonaut 2, which binds to the methylated duplex and retains only one of its strands (Camargo et al. 2018). Our previous study assumes the involvement of a large number of microRNA families within the phenylpropanoid pathway, including lignans (Ražná et al. 2022).
Until 2021, only 126 species of medicinal plants had their genomes sequenced, with a significant increase in sequencing activities observed in the year 2020 (Cheng et al. 2021). Currently, the GeneBank database of the National Center for Biotechnology Information provides four reference genomes and hundreds of nucleotide sequences, as well as BioProject, BioSample, and SRA records for the species Linum usitatissimum L. (NCBI 2022, https://ncbi.nlm.nih.gov/). Furthermore, the MedPlant RNA-Seq Database stores two processed and 12 raw sequencing datasets (MedPlant 2022, https://medplantrnaseq.org/). The Phytozome database provides comprehensive information on one fully annotated genome and transcriptome (Phytozome 13 2022, https://phytozome-next.jgi.doe.gov/) and the GENOLIN project includes 59,626 flax unigenes (Plant Bioinformatics facility 2022, https://urgi.versailles.inra.fr). Sequencing lays the foundation for emerging omics approaches such as genomics, transcriptomics, proteomics, metabolomics, epigenomics, and panomics (Singh et al. 2022). Moreover, the simple and often cost-free access to data repositories, bioinformatic algorithms, and computational systems enables the mutual comparison of genomes, transcriptomes, proteomes, and metabolomes (Li et al. 2022). The absence of knowledge regarding the biosynthesis of lignans and cyanogenic glycosides opens the door to new scientific opportunities for a successful breeding process (Tan et al. 2022; Qiang et al. 2022).
Material and methods
Identification of microRNAs in the genome of Linum usitatissimum L.
The primary source of microRNAs was the largest microRNA database, miRBase Release 22.1 (miRBase 2022, https://www.mirbase.org/). From this server, a compressed .gz file containing all available mature microRNA sequences in FASTA format, along with their accession IDs (MIMATXXXXXXX), was obtained. The accession ID for each stem-loop structure (MIXXXXXXX) and its sequence was automatically acquired using the mentioned mature microRNA accession ID. The entire dataset of stem-loop sequences was then aligned with the Linum usitatissimum v1.0 genome (Lusitatissimum_200_BGIv1.0) using the BLAST search with default algorithm parameters available on Phytozome 13 (Phytozome 13 2022, https://phytozome-next.jgi.doe.gov/). The exported results for stem-loop structures were sorted based on their percentage of positives with a query sequence (stem-loop structure). Each mature microRNA sequence was assigned with only the highest found percentage of its stem-loop structure. For further analysis, only microRNAs with a percentage of positives with the query sequence equal to or higher than 80%, indicating at least 80% aligning of their stem-loop structure with the Linum usitatissimum v1.0 genome, were selected.
Gene sequences of key enzymes in the metabolic pathways of lignans and cyanogenic glycosides
Within the metabolic pathway of lignans, a total of 12 gene sequences were selected for five key enzymes: Dirigent protein (DIR), (−)-pinoresinol-(−)-lariciresinol reductase 1 (PLR 1), (+)-pinoresinol-(+)-lariciresinol reductase 2 (PLR 2), (−)-secoisolariciresinol dehydrogenase (SDH), and Uridine glycosyltransferase UGT74S1 (Corbin et al. 2017). For the three key enzyme families of cyanogenic glycosides, Cytochrome P450 monooxygenases CYP79D, CYP71E, and Acetone cyanohydrin β-glucosyltransferase UGT85K, a total of 10 gene sequences were selected (Fang et al. 2016; Mohd Azmi 2019; Juma et al. 2022). These gene sequences are the most closely related to the synthesis of metabolites of interest—(+)-secoisolariciresinol diglucoside, linamarin and lotaustralin with their diglucosidic forms. All access codes and descriptions for the input sequences are provided in Table 1.
Collection of transcriptomic data
The processed transcriptomes (contigs) of Linum usitatissimum L. were obtained from Phytozome 13 (L. usitatissimum v1.0; Lus10000001–Lus10043484) (Phytozome 13 2022, https://phytozome-next.jgi.doe.gov/), MedPlant RNA Seq Database (Linum usitatissimum 1; linum_usitatissimum-20100629:000001–linum_usitatissimum-20100629:073195 and Linum usitatissimum 2; medp_linus_20101112|1–medp_linus_20101112|78323) (MedPlant 2022, https://medplantrnaseq.org/), and Plant Bioinformatics Facility (project GENOLIN; genolin_c1–genolin_s59626) (Plant Bioinformatics facility 2022, https://urgi.versailles.inra.fr/). Each transcriptome was characterized by the number of contigs, size of the smallest contig, size of the longest contig, and the average size of contigs.
Alignment of selected gene sequences with transcriptomic data
The selected gene sequences were aligned to verify their presence in the Linum usitatissimum L. transcriptomes. This alignment was conducted based on two assumptions. Firstly, to investigate the negative regulation mediated by microRNA it is essential to confirm the presence of complementary mRNA. Secondly, aligning the selected gene sequences with transcriptomes aimed at capturing the variability of expressed mRNAs. The abundance of single nucleotide polymorphisms within microRNA families enables the regulation of various gene forms (Anwar et al. 2018; Vasconcelos et al. 2021; Xu et al. 2021). Aligning gene sequences with transcriptomic data is also valuable for describing processed but not annotated transcriptomic data, where contig names are often represented only by numbers. To verify and demonstrate the annotation of enzyme families in the transcriptomic data, we employed the “Finding genes by keyword” algorithm within the Linum usitatissimum v1.0 genome available on Phytozome 13 (Phytozome 13 2022, https://phytozome-next.jgi.doe.gov/).
The alignment was performed using the blastn suite algorithm from the Nucleotide BLAST, optimized for “Somewhat similar sequences (blastn)”, with default algorithm parameters to compare two or more sequences (NCBI 2022, https://ncbi.nlm.nih.gov/). Each individual transcriptomic dataset was divided into separate FASTA files, each containing no more than 10 million characters to comply with the established limit of the BLAST server. These were then used as subject sequences. From the sequences that generated significant alignments, only those with a query cover value equal to or higher than 50% were exported as FASTA (aligned contigs).
Prediction of microRNA families involved in biosynthetic pathways of lignans and cyanogenic glycosides
FASTA files containing selected mature miRNAs (with aligning of their stem-loop structure with the Linum usitatissimum v1.0 genome equal to or higher than 80%) and selected gene sequences enriched with aligned contigs from transcriptomic data (sequences with a query cover value equal to or higher than 50%) were uploaded and analysed using psRNATarget: A Plant Small RNA Target Analysis Server (2017 Update) with default settings of Schema V2 (2017 release) (psRNATarget 2022, https://www.zhaolab.org/psRNATarget/). The predicted microRNAs were analysed on three levels: predicted for individual enzyme families, predicted for individual pathways, and predicted for both biosynthetic pathways. Applying an in silico approach for microRNA prediction, a schema illustrating the biosynthetic pathways of lignans and cyanogenic glycosides, including metabolites, enzymes, and involved microRNA families was designed.
Results
MicroRNAs in the genome of Linum usitatissimum L.
From miRBase Release 22.1 (miRBase 2022, https://www.mirbase.org/) were successfully obtained 44,885 mature miRNA sequencies along with their corresponding system-loop structures. Aligning the stem-loop structures through a BLAST search on Phytozome 13 confirmed the presence of 11,919 mature miRNAs in the Linum usitatissimum v1.0 genome (Phytozome 13 2022, https://phytozome-next.jgi.doe.gov/). Figure 1 illustrates the distribution of occurred microRNAs based on their percentage of aligning with the Linum usitatissimum L. genome. Subsequently, only microRNAs with a percentage of positives equal to or higher than 80% (441 mature microRNAs) were selected for further analysis.

Distribution of the 11,919 identified microRNAs based on their percentage of aligning with the Linum usitatissimum v1.0 genome
Distribution of 34 microRNA families (miR156, miR157, miR159, miR160, miR162, miR164, miR166, miR167, miR168, miR169, miR171, miR172, miR319, miR390, miR393, miR394, miR395, miR396, miR397, miR398, miR399, miR408, miR530, miR828, miR2916, miR3533, miR4426, miR4995, miR5219, miR5288, miR5523, miR8005, miR11602, miR11604) within the new dataset of 441 mature microRNAs, showed in the Fig. 2., proved as the most occurred microRNAs of family miR156 (72), miR160 (50), miR171 (43) and miR167 (41).

Distribution of 34 microRNA families within the new dataset of 441 microRNAs
In the Fig. 3. presenting distribution of 47 origins within the new dataset of 441 mature microRNAs is the most distributed species Linum usitatissimum L.—Lus (124). On the next places are Malus domestica Borkh.—Mdm (31), Manihot esculenta Crantz—Mes (30), Populus trichocarpa Torr. & A.Gray ex. Hook—Ptc (26) and Solanum tuberosum L.—Stu (26).

Distribution of 47 origins within the new dataset of 441 microRNAs. Aau, Acacia auriculiformis A.Cunn. ex Benth.; Aly, Arabidopsis lyrata L.; Ama, Avicennia marina Forssk.; Aqc, Aquilegia caerulea E. James; Ath, Arabidopsis thaliana L.; Bcy, Bruguiera cylindrica L.; Bgy, Bruguiera gymnorhiza L.; Bna, Brassica napus L.; Bra, Brassica rapa L.; Bta, Bos taurus L.; Cas, Camelina sativa L.; Cca, Cynara cardunculus L.; Cme, Cucumis melo L.; Cpa, Carica papaya L.; Csi, Citrus sinensis L.; Ctr, Citrus trifoliata L.; Dpr, Digitalis purpurea L.; Eun, Eugenia uniflora L.; Fve, Fragaria vesca L.; Ghr, Gossypium hirsutum L.; Gma, Glycine max L.; Gra, Gossypium raimondii Ulbr.; Han, Helianthus annuus L.; Hbr, Hevea brasiliensis Willd. ex A. Juss.; Hsa, Homo sapiens L.; Lja, Lotus japonicus L.; Lus, Linum usitatissimum L.; Mdm, Malus domestica Borkh.; Mes, Manihot esculenta Crantz; Mtr, Medicago truncatula Gaertn.; Nta, Nicotiana tabacum L.; Osa, Oryza sativa L.; Pab, Picea abies L.; Pde, Pinus densata Mast.; Peu, Populus euphratica Oliv.; Pla, Paeonia lactiflora Pall.; Ppe, Prunus persica L.; Ptc, Populus trichocarpa Torr. & A.Gray ex. Hook; Rco, Ricinus communis L.; Sbi, Sorghum bicolor L.; Sly, Solanum lycopersicum L.; Ssl, Salvia sclarea L.; Stu, Solanum tuberosum L.; Vca, Vriesea carinata Wawra; Vun, Vigna unguiculata L.; Vvi, Vitis vinifera L.; Zma, Zea mays L
The resulting matrix illustrating the occurrence and averaged percentage of alignment for microRNA families and their origins within the new dataset of 441 microRNAs is presented in Fig. 4. The most occurred microRNA family within the identified origins was miR156, which has been found in 22 origins, followed by miR167 (19 origins) and miR171 (18). The origins that most frequently occurred within the identified microRNA families was Linum usitatissimum L. (23 microRNA families), followed by Manihot esculenta Crantz (23), Glycine max L. (10), and Populus trichocarpa Torr. & A.Gray ex. Hook (10). The highest averaged percentage of alignment was reached by species Linum usitatissimum L. (100%), followed by Populus euphratica Oliv. (91%) and Cynara cardunculus L. (89%). The microRNA families with an average of 100% included miR168, miR397, miR398, miR530, and miR828.

The resulting matrix illustrating the occurrence and averaged percentage of alignment for microRNA families and their origins within the new dataset of 441 microRNAs
Lignan and cyanogenic glycoside key enzymes in transcriptomic data
The obtained transcriptomes L. usitatissimum v1.0, Linum usitatissimum 1, Linum usitatissimum 2 and project GENOLIN are characterized in the Table 2.
Verification of gene occurrence
The alignment of gene sequences with transcriptomic data aimed to verify their presence in various Linum usitatissimum L. transcriptomes revealed varying RNA sequencing quality and distribution of aligned contigs within the same plant genome. However, all gene sequences were successfully aligned with each transcriptome. The transcriptome with the highest average of aligned contigs was L. usitatissimum v1.0 (32), followed by Linum usitatissimum 2 (22), Linum usitatissimum 1 (12), and project GENOLIN (9). The averages of aligned contigs with query coverage equal to or higher than 50% were as follows: L. usitatissimum v1.0 (4), Linum usitatissimum 1 (1), Linum usitatissimum 2 (1), and project GENOLIN (0).
Increasing of gene variability
To enhance the variability of mRNA sequences, aligned sequencies of contigs with a query cover value equal to or higher than 50% were obtained. While all selected gene sequences were found in all transcriptomes, not all exhibited at least 50% query coverage—SDH (AF352734.1) and CYP79D4 (AY599896.1). The highest number of aligned contigs was observed for the sequences of cytochrome P450 monooxygenase CYP71E (MK172858.1), Dirigent protein DIR 3 (KM433755.1) and DIR 6 (KM433752.1), and Uridine glycosyltransferase UGT74S1 (JX011632.1) and (JN088324.1). The count of sequencies with equal to or more than 50% query coverage was significantly smaller and balanced in all cases. The detailed results are presented in Table 3.
Annotation of transcriptomic data
For better description and annotation of transcriptomic data, contigs that reached more than 50% query coverage have been sorted based on their origin and name/number, and assigned with NCBI records in Table 4. Names of some contigs are abbreviated by symbol of three dots. For example, contig linum_usitatissimum-20100629:013755 is abbreviated as “…:013755”; medp_linus_20101112|9111 as “…|9111” and genolin_c1251 “…c1251”. Differences within contigs of the same enzyme family were observed in Uridine glycosyltransferase UGT74S, Secoisolariciresinol dehydrogenase (SDH), Cytochrome P450 monooxygenase CYP71E, and Acetone cyanohydrin β-glucosyltransferase UGT85K. On the other hand, the same contigs were identified for enzyme family Dirigent protein (DIR), Pinoresinol-lariciresinol reductase (PLR), and Cytochrome P450 monooxygenase CYP79D.
The verification of the annotation within the L. usitatissimum v1.0 transcriptome, as proved by the algorithm "Finding genes by keyword", showed that many contigs are not well annotated. The discrepancy in results within very similar keywords or the same enzyme family, suggests that either annotations or the algorithm are not refined enough to yield the expected output. The number of hits does not correspond to the number of matches observed in our results. A complete match was observed in the enzyme families Dirigent protein (DIR) but only with the keyword "Dirigent" (6/6), Pinoresinol-lariciresinol reductase (PLR) with the keywords "Pinoresinol" and "Lariciresinol" (5/5), and Acetone cyanohydrin β-glucosyltransferase UGT85K with the keywords "UGT85" and "UGT" (4/4). The results are presented in Table 5.
MicroRNA families involved in biosynthetic pathways of lignans and cyanogenic glycosides
The prediction utilized selected gene sequences from Table 1, alignment sequences of contigs from Table 4, and the dataset of 441 mature microRNAs (Figs. 1, 2, 3, and 4). The results indicate that for Dirigent proteins (DIR1-DIR6), only the microRNA family miR160 was predicted. Both (−)-pinoresinol-(−)-lariciresinol reductase 1 and (+)-pinoresinol-(+)-lariciresinol reductase 2 (PLRs) could be regulated by microRNA families miR159, miR164, miR166, miR167, miR171, miR395, miR399, and miR5219. Uridine glycosyltransferase UGT74S1 exhibited complementarity with microRNA families miR156, miR157, miR159, miR164, miR167, miR319, and miR395. Within the secoisolariciresinol dehydrogenase (SDH), microRNA families miR172, miR396, and miR5523 were identified. In total, 15 microRNA families (miR156, miR157, miR159, miR160, miR164, miR166, miR167, miR171, miR172, miR319, miR395, miR396, miR399, miR5219, and miR5523) were predicted for the biosynthetic pathway of lignans, with miR159, miR164, miR167, and miR395 appearing as the most active, regulating PLR and UGT74S enzyme families.
In the first key enzyme family of cyanogenic glycosides, microRNA families miR160, miR171, miR319, miR2916, and miR11602 were predicted for cytochrome P450 monooxygenase CYP79D. For cytochrome P450 monooxygenase CYP71E, microRNA families miR168, miR171, miR319, and miR396 were identified. The enzyme family acetone cyanohydrin β-glucosyltransferase UGT85K is likely regulated by microRNA families miR159, miR160, miR393, and miR5219. In the biosynthetic pathway of cyanogenic glycosides, a total of ten microRNA families were identified (miR159, miR160, miR168, miR171, miR319, miR393, miR396, miR2916, miR5219, and miR11602), with the most active microRNA family miR160, which can regulate enzyme families CYP79D and UGT85K. Additionally, microRNA families miR171 and miR319 were found to regulate enzyme families CYP79D and CYP71E.
Out of the 19 identified microRNA families, six microRNA families were predicted for both biosynthetic pathways: miR159 (regulating UGTS4S and UGT85K), miR160 (regulating DIR, CYP79D, and UGT85K), miR171 (regulating PLR, CYP79D, and CYP71E), miR319 (regulating UGTS4S, CYP79D, and CYP71E), miR396 (regulating SDH and CYP71E), and miR5219 (regulating PLR and UGT85K). The results are presented in Fig. 5.

The resulting matrix of predicted microRNA families involved in biosynthetic pathway of lignans and cyanogenic glycosides. DIR, Dirigent protein; PLR 1, (−)-pinoresinol-(−)-lariciresinol reductase 1; PLR 2, (+)-pinoresinol-(+)-lariciresinol reductase 2; UGT74S, Uridine glycosyltransferase UGT74S; SDH, (−)-secoisolariciresinol dehydrogenase; CYP, Cytochrome P450 monooxygenase; UGT85K, Acetone cyanohydrin β-glucosyltransferase UGT85K
Discussion
The consumption of flaxseed is continually increasing, bringing new challenges in the fields of food production and breeding (CBI 2019). By optimizing nutritional properties, it is possible to achieve a significant improvement in effects on the human body (Parikh et al. 2019). Numerous studies reveal that lignans are particularly beneficial in the prevention and treatment of cancer, diabetes, and many other health issues associated also with female reproduction (Mueed et al. 2023; Sirotkin 2023; Xi et al. 2023). On the other hand, cyanogenic glycosides, as a fraction of antinutrients, may reduce these benefits and even act antagonistically to the synthesis of lignans (Kazachkov et al. 2020; Zuk et al. 2020). An important milestone will be achieving an optimal ratio of nutritional and antinutritional components while maintaining current physiological parameters (Harenčár et al. 2021). The scientific potential of microRNAs lies mainly in their posttranscriptional silencing, which can occur within both the maternal and host organism (Loreti and Perata 2022). The research of this regulatory mechanism opens possibilities for understanding the function of metabolic and signalling pathways, resistance against pathogens and the abiotic environment, or using them as genetic markers in breeding (Summanwar et al. 2020; Ding and Zhang 2023; Hu et al. 2023; Pei et al. 2023; Zhang et al. 2023). Additionally, genetic engineering provides methods for silencing, enhancing, or probing microRNAs, allowing for their precise localization and quantification (Siddika and Heinemann 2021; Ražná et al. 2022). High-throughput technologies, such as next-generation sequencing, in combination with high-performance computers and in silico algorithms, allow the processing of data from various sources meanwhile comparative genomics enables their alignment, annotation, and predictions (Fort et al. 2022). The subject of current research is understanding their upregulation and downregulation patterns. This article assumes only microRNA negative regulation of key enzyme families in the biological pathway of lignans and cyanogenic glycosides, which are simultaneously in negative correlation. It does not take into account their indirect regulation through other epigenetic factors. Therefore, microRNAs are one of the epigenetic factors involved in the regulation of secondary metabolism, contributing to the organism's development and resistance to environmental conditions. Since the activity of enzymes in the metabolic pathways of lignans as well as cyanogenic glycosides varies over time and space, it is desirable to gain a deeper understanding of the involvement of individual regulatory elements, including microRNAs, and apply them to the breeding process.
MicroRNAs predicted in the genome of Linum usitatissimum L.
Based on demonstrated in silico approach of microRNA prediction for biosynthetic pathways of lignans and cyanogenic glycosides were obtained 44,885 mature microRNA sequences. In the genome Linum usitatissimum v1.0, available on Phytozome 13, were identified in total of 11,919 microRNAs however only 441 met criteria about the percentage of positives with query sequence equal or more than 80% (Phytozome 13 2022, https://phytozome-next.jgi.doe.gov/). The significant reduction in microRNAs available for subsequent analyses highlights their presence in the genome of Linum usitatissimum L. Barvkar et al. (2013) identified 23 microRNAs (miR156, miR159, miR160, miR162, miR164, miR166, miR167, miR168, miR169, miR171, miR172, miR319, miR390, miR393, miR394, miR395, miR396, miR397, miR398, miR399, miR408, miR530, miR828) in genome-wide analysis. The same microRNAs were also identified by Zhang et al. (2020), who additionally discovered several novel, yet unnamed microRNAs. MiR157 has been confirmed by Xie et al. (2023). We did not find any record of the occurrence of miR2916, miR3533, miR4426, miR4995, miR5219, miR5288, miR5523, miR8005, miR11602, miR11604 in the genome of Linum usitatissimum L. Perhaps due to the novelty of miRNAs, which is characterized by a high number. Further genome or transcriptome-based research is required to investigate these microRNA families.
The analysis of the selected 441 microRNAs within 34 microRNA families and 47 origins revealed that microRNAs originating from Linum usitatissimum L. (Lus) had the highest averaged percentage of alignment with the Linum usitatissimum v1.0 genome (100%, 124 microRNAs within 23 families). The same percentage was reached in the case of microRNA families miR168 (2 microRNAs), miR397 (2), miR398 (6), miR530 (2), and miR828 (1). Although their occurrence in the Linum usitatissimum v1.0 genome was estimated as higher than 80% in all cases, these percentages represent averages for one or several individual microRNAs. It is important to note that the results in the tables and matrix should be interpreted as indicative, given their high dependence on available sequences within the miRBase Release 22.1 database. MicroRNA family miR156 is one of the most investigated family, for which 72 mature microRNAs within 22 origins were obtained. Similarly, for miR160, 50 microRNAs within 16 origins were found, and for miR171, 43 microRNAs within 18 families. Replicates of the same mature microRNA sequencies, however, from different origins were kept to provide research at the microRNA family level and to indicate their inter-origin conservancy. The identified microRNA families and origins point to areas of increased interest in the field of plant microRNAs (Acquadro et al. 2017; Lian et al. 2018; Ramesh et al. 2019; Yawichai et al. 2019; Huang et al. 2020; Jerome Jeyakumar et al. 2020; Li et al. 2020, 2021; Liu et al. 2020, 2021; Pompili et al. 2020; Rock 2020; Kang et al. 2021; Wang et al. 2021; Han and Zhou 2022; Kai et al. 2022; Pokhrel and Meyers 2022).
Key enzymes aligned to transcriptomic data
All gene sequences of key enzymes within the metabolic pathways of lignans and cyanogenic glycosides were successfully aligned with each transcriptome. However, there were variations in the average number of contigs, likely influenced by the averaged size of contigs within the transcriptomic data: L. usitatissimum v1.0—32 aligned contigs in average—average size per contig of 1200 bp; Linum usitatissimum 2–22 contigs in average—average size of 633 bp; Linum usitatissimum 1–12 contigs—average size of 329 bp; project GENOLIN—9 contigs—size of 483 bp.
The averages of aligned contigs with query coverage equal to or higher than 50% followed a pattern of the size of the smallest contig: L. usitatissimum v1.0—4 aligned contigs—smallest contig of 150 bp; Linum usitatissimum 1 and 2—1 contig—smallest size of 100 bp; project GENOLIN—0 contigs—size of 40 bp. The presence of enzyme families (Dirigent protein (DIR), Pinoresinol-lariciresinol reductase (PLR), Uridine glycosyltransferase UGT74S, Secoisolariciresinol dehydrogenase (SDH), Cytochrome P450 monooxygenase CYP79D, Cytochrome P450 monooxygenase CYP71E, and Acetone cyanohydrin β-glucosyltransferase UGT85K) in the genome and transcriptome of Linum usitatissimum L. was also confirmed by Hano et al. (2006), Ganjewala (2010), Dalisay et al. (2015), Fofana et al. (2017a), Corbin et al. (2018), Kezimana et al. (2018), Kazachkov et al. (2020), Mikac et al. (2021), Xiao et al. (2021).
The description and annotation of enzyme families within the transcriptome L. usitatissimum v1.0 suggested that the annotation or the algorithm "Finding genes by keyword" on Phytozome 13 may need further refinement. In several cases, hundreds of hits (contigs related to the keyword) were obtained, however, none of them were correct.
In the study by Corbin et al. (2018), 44 contigs from the transcriptome L. usitatissimum v1.0 were annotated based on their similarity to the dirigent domain PFAM PF03018. In our research, we identified 6 contigs within the transcriptome L. usitatissimum v1.0 that met the criteria for query coverage equal to or higher than 50%. These 6 contigs are the first 6 sequences with the highest similarity to the dirigent domain PFAM PF03018 among the 44 contigs annotated by the mentioned authors. Additionally, the activity of these 6 contigs was confirmed by Dalisay et al. (2015). We also identified contigs that were used for designing primers for the enzyme families (−)-pinoresinol-(−)-lariciresinol reductase 1 and Uridine glycosyltransferase UGT74S (Ahmad et al. 2019), and identified as Cytochrome P450 monooxygenase CYP79D1, Cytochrome P450 monooxygenase CYP71E, and Acetone cyanohydrin β-glucosyltransferase UGT85K (Dalisay et al. 2015). For the enzyme family Secoisolariciresinol dehydrogenase (SDH), we did not find any similar publication. The used settings of the BLAST algorithm were not able to detect differences between contigs and assign them to individual gene homologs. In cases of differences, the neighbouring contigs were usually included. This will require more precise study including transcriptomic analysis at least for genes without any evidence in other publications. A comparison of our results with another research is presented in Table 6. Contig names are abbreviated.
DIR—Dirigent protein, PLR 1—(−)-pinoresinol-(−)-lariciresinol reductase 1, PLR 2—(+)-pinoresinol-(+)-lariciresinol reductase 2, UGT74S1—Uridine glycosyltransferase UGT74S, SDH—(−)-secoisolariciresinol dehydrogenase, CYP—Cytochrome P450 monooxygenase, UGT85K—Acetone cyanohydrin β-glucosyltransferase UGT85K.
MicroRNA mediated regulation of lignan and cyanogenic glycoside biosynthesis
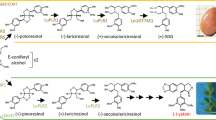
Based on abovementioned results, a unique schema of microRNA mediated regulation for biosynthetic pathways of lignans and cyanogenic glycosides has been designed (Fig. 6). Our previous research highlighted 51 microRNA families involved in the biosynthesis of lignans as products of the phenylpropanoid pathway (Ražná et al. 2022). There is no mention about miR319, miR395, and miR5219 that have been identified in this study. This is probably the first publication about microRNAs involved in the biosynthesis of cyanogenic glycosides, which requires further in-depth study on the epigenetics of flax cyanogenic glycosides.

Schema of microRNA mediated regulation for biosynthetic pathways of lignans and cyanogenic glycosides involving metabolites, enzymes and microRNA families. Underlined are microRNA families occurred in both pathways. DIR, Dirigent protein; PLR 1, (−)-pinoresinol-(−)-lariciresinol reductase 1; PLR 2, (+)-pinoresinol-(+)-lariciresinol reductase 2; UGT74S1, Uridine glycosyltransferase UGT74S1; SDH, (−)-secoisolariciresinol dehydrogenase; CYP, Cytochrome P450 monooxygenase; UGT85K, Acetone cyanohydrin β-glucosyltransferase UGT85K
Conclusion
In this article, we focused on 5 key enzymes of the lignan metabolic pathway and 3 enzymes of the cyanogenic glycoside pathway in flax (Linum usitatissimum L.). Based on the in silico approach, we identified 441 microRNAs within 34 microRNA families, of which 15 were predicted for the lignan metabolic pathway, 10 for the cyanogenic glycoside pathway, and 6 for both pathways. The sequences of these enzymes were identified and annotated across four transcriptomes, and we also pointed out the imperfections of the "Finding genes by keyword" algorithm available on Phytozome 13 (Phytozome 13 2022, https://phytozome-next.jgi.doe.gov/). From our findings, a unique schema of microRNA mediated regulation for the biosynthetic pathways of lignans and cyanogenic glycosides was designed. Moreover, this is probably the first publication about microRNAs involved in the metabolic pathway of cyanogenic glycosides. These results build upon our previous theoretical reviews (Harenčár et al. 2021; Ražná et al. 2022) and open the space for future transcriptomic (RT-qPCR and RNA-seq) and metabolomic (targeted and untargeted metabolomics) research.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Abraham K, Buhrke T, Lampen A (2016) Bioavailability of cyanide after consumption of a single meal of foods containing high levels of cyanogenic glycosides: a crossover study in humans. Arch Toxicol. https://doi.org/10.1007/s00204-015-1479-8
Acquadro A, Barchi L, Portis E et al (2017) Genome reconstruction in Cynara cardunculus taxa gains access to chromosome-scale DNA variation. Sci Rep. https://doi.org/10.1038/s41598-017-05085-7
Ahmad W, Zahir A, Nadeem M et al (2019) Enhanced production of lignans and neolignans in chitosan-treated flax (Linum usitatissimum L.) cell cultures. Process Biochem. https://doi.org/10.1016/j.procbio.2018.12.025
Akatsuka R, Ito M (2022) Content and distribution of prunasin in Perilla frutescens. J Nat Med. https://doi.org/10.1007/s11418-022-01654-x
Alitubeera PH, Eyu P, Kwesiga B et al (2019) Outbreak of cyanide poisoning caused by consumption of cassava flour—Kasese District, Uganda, September 2017. Morb Mortal Wkly Rep. https://doi.org/10.15585/mmwr.mm6813a3
Anwar N, Ohta M, Yazawa T et al (2018) miR172 downregulates the translation of cleistogamy 1 in barley. Ann Bot. https://doi.org/10.1093/aob/mcy058
Appenteng MK, Krueger R, Johnson MC et al (2021) Cyanogenic glycoside analysis in American elderberry. Mol. https://doi.org/10.3390/molecules26051384
Aqeel T, Gurumallu SCH, Hashimi SM et al (2019) Evaluation of protective efficacy of flaxseed lignan-Secoisolariciresinol diglucoside against mercuric chloride-induced nephrotoxicity in rats. Mol Biol Rep. https://doi.org/10.1007/s11033-019-05052-7
Barvkar VT, Pardeshi VC, Kale SM et al (2013) Genome-wide identification and characterization of microRNA genes and their targets in flax (Linum usitatissimum). Planta. https://doi.org/10.1007/s00425-012-1833-5
Buch DU, Sharma OA, Pable AA et al (2020) Characterization of microRNA genes from Pigeonpea (Cajanus cajan L.) and understanding their involvement in drought stress. J Biotech. https://doi.org/10.1016/j.jbiotec.2020.06.019
Camargo C, Wu K, Fishilevich E et al (2018) Knockdown of RNA interference pathway genes in western corn rootworm, Diabrotica virgifera virgifera, identifies no fitness costs associated with Argonaute 2 or Dicer-2. Pestic Biochem Phys. https://doi.org/10.1016/j.pestbp.2018.04.004
CBI (2019) Exporting linseeds to Europe. https://www.cbi.eu/market-information/grains-pulses-oilseeds/linseeds/europe
Chen J, Teotia S, Lan T et al (2021) MicroRNA techniques: valuable tools for agronomic trait analyses and breeding in rice. Front Plant Sci. https://doi.org/10.3389/fpls.2021.744357
Chen S, Ren G (2019) Analysis of methylation status of plant microRNAs. Plant microRNAs. https://doi.org/10.1007/978-1-4939-9042-9_21
Cheng QQ, Ouyang Y, Tang ZY et al (2021) Review on the development and applications of medicinal plant genomes. Front Plant Sci. https://doi.org/10.3389/fpls.2021.791219
Chongtham N, Bisht MS, Premlata T et al (2022) Quality improvement of bamboo shoots by removal of antinutrients using different processing techniques: a review. J Food Sci Technol. https://doi.org/10.1007/s13197-021-04987-9
Corbin C, Drouet S, Mateljak I et al (2017) Functional characterization of the pinoresinol–lariciresinol reductase-2 gene reveals its roles in yatein biosynthesis and flax defense response. Planta. https://doi.org/10.1007/s00425-017-2701-0
Corbin C, Drouet S, Markulin L et al (2018) A genome-wide analysis of the flax (Linum usitatissimum L.) dirigent protein family: from gene identification and evolution to differential regulation. Plant Mol Biol. https://doi.org/10.1007/s11103-018-0725-x
Dalisay DS, Kim KW, Lee C et al (2015) Dirigent protein-mediated lignan and cyanogenic glucoside formation in flax seed: integrated omics and MALDI mass spectrometry imaging. J Nat Prod. https://doi.org/10.1021/acs.jnatprod.5b00023
De Brito FR, Martinoia E (2018) The vacuolar transportome of plant specialized metabolites. Plant Cell Physiol. https://doi.org/10.1093/pcp/pcy039
De Silva SF, Alcorn J (2019) Flaxseed lignans as important dietary polyphenols for cancer prevention and treatment: chemistry, pharmacokinetics, and molecular targets. Pharm. https://doi.org/10.3390/ph12020068
Del Cueto J, Ionescu IA, Pičmanová M et al (2017) Cyanogenic glucosides and derivatives in almond and sweet cherry flower buds from dormancy to flowering. Front Plant Sci. https://doi.org/10.3389/fpls.2017.00800
Del Giudice R, Putkaradze N, Dos Santos BM et al (2022) Structure-guided engineering of key amino acids in UGT85B1 controlling substrate and stereo-specificity in aromatic cyanogenic glucoside biosynthesis. Plant J. https://doi.org/10.1111/tpj.15904
Deng P, Cui B, Zhu H et al (2021) Accumulation pattern of amygdalin and prunasin and its correlation with fruit and kernel agronomic characteristics during apricot (Prunus armeniaca L.) kernel development. Foods. https://doi.org/10.3390/foods10020397
Ding N, Zhang B (2023) microRNA production in Arabidopsis. Front Plant Sci. https://doi.org/10.3389/fpls.2023.1096772
Ebrahimi B, Nazmara Z, Hassanzadeh N et al (2021) Biomedical features of flaxseed against different pathologic situations: a narrative review. Iran J Basic Med Sci. https://doi.org/10.22038/IJBMS.2021.49821.11378
Fang Y, Mei H, Zhou B et al (2016) De novo transcriptome analysis reveals distinct defense mechanisms by young and mature leaves of Hevea brasiliensis (Para Rubber Tree). Sci Rep. https://doi.org/10.1038/srep33151
Fofana B, Ghose K, McCallum J et al (2017a) UGT74S1 is the key player in controlling secoisolariciresinol diglucoside (SDG) formation in flax. BMC Plant Biol. https://doi.org/10.1186/s12870-017-0982-x
Fofana B, Ghose K, Somalraju A et al (2017b) Induced mutagenesis in UGT74S1 gene leads to stable new flax lines with altered secoisolariciresinol diglucoside (SDG) profiles. Front Plant Sci. https://doi.org/10.3389/fpls.2017.01638
Fojnica A, Leis HJ, Murkovic M (2022) Identification and characterization of the stability of hydrophobic cyclolinopeptides from flaxseed oil. Front Nut. https://doi.org/10.3389/fnut.2022.903611
Fort RS, Chavez S, Trinidad Barnech JM et al (2022) Current status of regulatory non-coding RNAs research in the tritryp. ncRNA. https://doi.org/10.3390/ncrna8040054
Ganjewala D (2010) Advances in cyanogenic glycosides biosynthesis and analyses in plants: a review. Acta Biol Szeged. https://abs.bibl.u-szeged.hu/index.php/abs/article/view/2679
Ghose K, Selvaraj K, McCallum J et al (2014) Identification and functional characterization of a flax UDP-glycosyltransferase glucosylating secoisolariciresinol (SECO) into secoisolariciresinol monoglucoside (SMG) and diglucoside (SDG). BMC Plant Biol. https://doi.org/10.1186/1471-2229-14-82
Gramzow L, Theißen G (2019) Plant miRNA conservation and evolution. Plant MicroRNAs. https://doi.org/10.1007/978-1-4939-9042-9_3
Han H, Zhou Y (2022) Function and regulation of microRNA171 in plant stem cell homeostasis and developmental programing. Int J Mol Sci. https://doi.org/10.3390/ijms23052544
Hano C, Martin I, Fliniaux O et al (2006) Pinoresinol–lariciresinol reductase gene expression and secoisolariciresinol diglucoside accumulation in developing flax (Linum usitatissimum) seeds. Planta. https://doi.org/10.1007/s00425-006-0308-y
Harenčár Ľ, Ražná K, Nôžková J (2021) Cyanogenic glycosides—their role and potential in plant food resources. J Microbiol Biotech Food Sci. https://doi.org/10.15414/jmbfs.4771
Hartanti D, Cahyani AN (2020) Plant cyanogenic glycosides: an overview. Farmasains J Farm Il Kes. https://doi.org/10.22219/farmasains.v5i1.10047
Hu X, Yin G, Zhang Y et al (2023) Recent advances in the functional explorations of nuclear microRNAs. Front Immunol. https://doi.org/10.3389/fimmu.2023.1097491
Huang S, Zhou J, Gao L et al (2020) Plant miR397 and its functions. Funct Plant Biol. https://doi.org/10.1071/FP20342
Jaszczak-Wilke E, Polkowska Ż, Koprowski M et al (2021) Amygdalin: toxicity, anticancer activity and analytical procedures for its determination in plant seeds. Mol. https://doi.org/10.3390/molecules26082253
Jeena GS, Singh N, Shukla RK (2022) An insight into microRNA biogenesis and its regulatory role in plant secondary metabolism. Plant Cell Rep. https://doi.org/10.1007/s00299-022-02877-8
Jerome Jeyakumar JM, Ali A, Wang WM et al (2020) Characterizing the role of the miR156-SPL network in plant development and stress response. Plants. https://doi.org/10.3390/plants9091206
Juma BS, Mukami A, Mweu C et al (2022) Targeted mutagenesis of the CYP79D1 gene via CRISPR/Cas9-mediated genome editing results in lower levels of cyanide in cassava. Front Plant Sci. https://doi.org/10.3389/fpls.2022.1009860
Kai H, Yun W, Zhanpin Z et al (2022) miR160: an indispensable regulator in plant. Front Plant Sci. https://doi.org/10.3389/fpls.2022.833322
Kajla P, Sharma A, Sood DR (2015) Flaxseed—a potential functional food source. J Food Sci Technol. https://doi.org/10.1007/s13197-014-1293-y
Kang Y, Yang X, Liu Y et al (2021) Integration of mRNA and miRNA analysis reveals the molecular mechanism of potato (Solanum tuberosum L.) response to alkali stress. Int J Biol Macromol. https://doi.org/10.1016/j.ijbiomac.2021.04.094
Kazachkov M, Li Q, Shen W et al (2020) Molecular identification and functional characterization of a cyanogenic glucosyltransferase from flax (Linum usitatissimum). PLoS ONE. https://doi.org/10.1371/journal.pone.0227840
Kezimana P, Dmitriev AA, Kudryavtseva AV et al (2018) Secoisolariciresinol diglucoside of flaxseed and its metabolites: biosynthesis and potential for nutraceuticals. Front Gen. https://doi.org/10.3389/fgene.2018.00641
Kuliahsari DE, Sari INI, Estiasih T (2021) Cyanide detoxification methods in food: a review. IOP Conf Ser EES. https://doi.org/10.1088/1755-1315/733/1/012099
Lechtenberg M (2021) Cyanogenic glycosides and biogenetically related compounds in higher plants and animals. eLS. https://doi.org/10.1002/9780470015902.a0029316
Lee D, Park D, Park JH et al (2019) Poly (A)-specific ribonuclease sculpts the 3′ ends of microRNAs. RNA. https://doi.org/10.1261/rna.069633.118
Li J, Shen Y, Zhu J et al (2020) miR398 is involved in the relief of phenanthrene-induced oxidative toxicity in wheat roots. Environ Pollut. https://doi.org/10.1016/j.envpol.2019.113701
Li Y, Wang LF, Bhutto SH et al (2021) Blocking miR530 improves rice resistance, yield, and maturity. Front Plant Sci. https://doi.org/10.3389/fpls.2021.729560
Li W, Liu J, Zhang H et al (2022) Plant pan-genomics: recent advances, new challenges, and roads ahead. J Genet Genomics. https://doi.org/10.1016/j.jgg.2022.06.004
Lian C, Yao K, Duan H et al (2018) Exploration of ABA responsive miRNAs reveals a new hormone signaling crosstalk pathway regulating root growth of Populus euphratica. Int J Mol Sci. https://doi.org/10.3390/ijms19051481
Liu X, Tan C, Cheng X et al (2020) miR168 targets Argonaute1A mediated miRNAs regulation pathways in response to potassium deficiency stress in tomato. BMC Plant Biol. https://doi.org/10.1186/s12870-020-02660-5
Liu X, Huang S, Xie H (2021) Advances in the regulation of plant development and stress response by miR167. Front Biosci. https://doi.org/10.52586/4974
Loreti E, Perata P (2022) Mobile plant microRNAs allow communication within and between organisms. New Phytol. https://doi.org/10.1111/nph.18360
Marcotullio MC, Pelosi A, Curini M (2014) Hinokinin, an emerging bioactive lignan. Mol. https://doi.org/10.3390/molecules190914862
Markulin L, Corbin C, Renouard S et al (2019) Pinoresinol–lariciresinol reductases, key to the lignan synthesis in plants. Planta. https://doi.org/10.1007/s00425-019-03137-y
MedPlant (2022) RNA seq database. https://medplantrnaseq.org/
Mikac S, Markulin L, Drouet S et al (2021) Bioproduction of anticancer podophyllotoxin and related aryltretralin-lignans in hairy root cultures of Linum flavum L. Plant Cell Tissue Differ Second Metab Fundam Appl. https://doi.org/10.1007/978-3-030-11253-0_20-1
miRBase (2022) miRBase: the microRNA database. https://www.mirbase.org/
Mobot (2021) Evolution of land plants (under construction). http://www.mobot.org/moBot/research/apweb/orders/Sporing.html
Mohd Azmi AF (2019) Cyanogenic glycosides in cassava. https://etheses.whiterose.ac.uk/26731/1/Ahmad_Mohd_Azmi_Thesis_May_2019.pdf
Mosayyebi B, Imani M, Mohammadi L et al (2020) An update on the toxicity of cyanogenic glycosides bioactive compounds: possible clinical application in targeted cancer therapy. Mater Chem Phys. https://doi.org/10.1016/j.matchemphys.2020.122841
Mueed A, Deng Z, Korma SA et al (2023) Anticancer potential of flaxseed lignans, their metabolites and synthetic counterparts in relation with molecular targets: current challenges and future perspectives. Food Funct. https://doi.org/10.1039/D2FO02208G
NCBI (2022) The national center for biotechnology information. https://ncbi.nlm.nih.gov/
Nyirenda KK (2020) Toxicity potential of cyanogenic glycosides in edible plants. Med Toxicol. https://doi.org/10.5772/intechopen.91408
Osmakov DI, Kalinovskii AP, Belozerova OA et al (2022) Lignans as pharmacological agents in disorders related to oxidative stress and inflammation: chemical synthesis approaches and biological activities. Int J Mol Sci. https://doi.org/10.3390/ijms23116031
Parikh M, Maddaford TG, Austria JA et al (2019) Dietary flaxseed as a strategy for improving human health. Nutrients. https://doi.org/10.3390/nu11051171
Pei LL, Zhang LL, Liu X et al (2023) Role of microRNA miR171 in plant development. PeerJ. https://doi.org/10.7717/peerj.15632
Phytozome 13 (2022) The plant genomics resource. https://phytozome-next.jgi.doe.gov/
Pirrò S, Matic I, Guidi A et al (2019) Identification of microRNAs and relative target genes in Moringa oleifera leaf and callus. Sci Rep. https://doi.org/10.1038/s41598-019-51100-4
Plant Bioinformatics Facility (2022) Plant and fungi data integration. https://urgi.versailles.inra.fr/
Pokhrel S, Meyers BC (2022) Heat-responsive microRNAs and phased small interfering RNAs in reproductive development of flax. Plant Direct. https://doi.org/10.1002/pld3.385
Pompili V, Piazza S, Li M et al (2020) Transcriptional regulation of MdmiR285N microRNA in apple (Malus x domestica) and the heterologous plant system Arabidopsis thaliana. Hortic Res. https://doi.org/10.1038/s41438-020-0321-5
Prasad K, Khan AS, Shoker M (2020) Flaxseed and its components in treatment of hyperlipidemia and cardiovascular disease. Int J Angiol. https://doi.org/10.1055/s-0040-1709129
psRNATarget (2022) psRNATarget: a plant small RNA target analysis server (2017 Update). https://www.zhaolab.org/psRNATarget/
Qiang TY, Liu JS, Dong YQ et al (2022) Identification, molecular cloning, and functional characterization of a coniferyl alcohol acyltransferase involved in the biosynthesis of dibenzocyclooctadiene lignans in Schisandra chinensis. Front Plant Sci. https://doi.org/10.3389/fpls.2022.881342
Ramesh SV, Govindasamy V, Rajesh MK et al (2019) Stress-responsive miRNAome of Glycine max (L.) Merrill: molecular insights and way forward. Planta. https://doi.org/10.1007/s00425-019-03114-5
Ražná K, Harenčár Ľ, Kučka M (2022) The involvement of microRNAs in plant lignan biosynthesis—current view. Cells. https://doi.org/10.3390/cells11142151
Ritmejerytė E, Boughton BA, Bayly MJ et al (2019) Divergent responses of above-and below-ground chemical defence to nitrogen and phosphorus supply in waratahs (Telopea speciosissima). Funct Plant Biol. https://doi.org/10.1071/FP19122
Rock CD (2020) A role for MIR828 in pineapple fruit development. F1000Res. https://doi.org/10.12688/f1000research.21779.2
Schmidt TJ, Klaes M, Sendker J (2012) Lignans in seeds of Linum species. Phytochem. https://doi.org/10.1016/j.phytochem.2012.07.004
Schrenk D, Bignami M, Bodin L et al (2019) Evaluation of the health risks related to the presence of cyanogenic glycosides in foods other than raw apricot kernels. EFSA J. https://doi.org/10.2903/j.efsa.2019.5662
Sculfort O, Mcclure M, Nay B et al (2021) Assessing the role of developmental and environmental factors in chemical defence variation in Heliconiini Butterflies. J Chem Ecol. https://doi.org/10.1007/s10886-021-01278-7
Sharma P, Murigneux V, Haimovitz J et al (2021) The genome of the endangered Macadamia jansenii displays little diversity but represents an important genetic resource for plant breeding. Plant Direct. https://doi.org/10.1002/pld3.364
Shi X, Geng J, Feng J et al (2022) Identification and investigation of a novel NADP+-dependent secoisolariciresinol dehydrogenase from Isatis indigotica. Front Plant Sci. https://doi.org/10.3389/fpls.2022.1035121
Siddika T, Heinemann IU (2021) Bringing microRNAs to light: methods for microRNA quantification and visualization in live cells. Front Bioeng Biotechnol. https://doi.org/10.3389/fbioe.2020.619583
Singh R, Kusalik A, Dillon JAR (2022) Bioinformatics tools used for whole-genome sequencing analysis of Neisseria gonorrhoeae: a literature review. Brief Funct Genomics. https://doi.org/10.1093/bfgp/elab028
Sirotkin AV (2023) Influence of flaxseed (Linum usitatissimum) on female reproduction. Planta Med. https://doi.org/10.1055/a-2013-2966
Song X, Li Y, Cao X et al (2019) MicroRNAs and their regulatory roles in plant–environment interactions. Annu Rev Plant Biol. https://doi.org/10.1146/annurev-arplant-050718-100334
Summanwar A, Basu U, Rahman H et al (2020) Non-coding RNAs as emerging targets for crop improvement. Plant Sci. https://doi.org/10.1016/j.plantsci.2020.110521
Sun Z, Zhang K, Chen C et al (2018) Biosynthesis and regulation of cyanogenic glycoside production in forage plants. Appl Microbiol Biotechnol. https://doi.org/10.1007/s00253-017-8559-z
Sun J, Dong Y, Wang C et al (2021) Identification and characterization of melon circular RNAs involved in powdery mildew responses through comparative transcriptome analysis. PeerJ. https://doi.org/10.7717/peerj.11216
Tafrihi M, Hasheminasab E (2019) MiRNAs: biology, biogenesis, their web-based tools, and databases. MicroRNA. https://doi.org/10.2174/2211536607666180827111633
Tan YC, Kumar AU, Wong YP et al (2022) Bioinformatics approaches and applications in plant biotechnology. J Genet Eng Biotechnol. https://doi.org/10.1186/s43141-022-00394-5
Tashackori H, Sharifi M, Ahmadian Chashmi N et al (2021) Physiological, biochemical, and molecular responses of Linum album to digested cell wall of Piriformospora indica. Physiol Mol Biol Plants. https://doi.org/10.1007/s12298-021-01106-y
Tyagi S, Sharma S, Ganie SA et al (2019) Plant microRNAs: biogenesis, gene silencing, web-based analysis tools and their use as molecular markers. 3 Biotech. https://doi.org/10.1007/s13205-019-1942-y
Van Ohlen M, Herfurth AM, Wittstock U (2017) Herbivore adaptations to plant cyanide defenses. Herbivores. https://doi.org/10.5772/66277
Vasconcelos AM, Carmo MB, Ferreira B et al (2021) IsomiR_Window: a system for analyzing small-RNA-seq data in an integrative and user-friendly manner. BMC Bioinform. https://doi.org/10.1186/s12859-021-03955-6
Wang R, Reng M, Tian S et al (2021) Transcriptome-wide identification and characterization of microRNAs in diverse phases of wood formation in Populus trichocarpa. G3. https://doi.org/10.1093/g3journal/jkab195
Xi H, Zhou W, Sohaib M et al (2023) Flaxseed supplementation significantly reduces hemoglobin A1c in patients with type 2 diabetes mellitus: a systematic review and meta-analysis. Nutr Res. https://doi.org/10.1016/j.nutres.2022.12.008
Xiao Y, Shao K, Zhou J et al (2021) Structure-based engineering of substrate specificity for pinoresinol-lariciresinol reductases. Nat Commun. https://doi.org/10.1038/s41467-021-23095-y
Xie D, Yang X, He R et al (2023) Identification of regulatory pathways of miRna and phasiRna related to flax lignan synthesis. J Nat Fibers. https://doi.org/10.1080/15440478.2023.2185426
Xu P, Zhu Y, Zhang Y et al (2021) Global analysis of the genetic variations in miRNA-targeted sites and their correlations with agronomic traits in rapeseed. Front Genet. https://doi.org/10.3389/fgene.2021.741858
Yang X, Guo Y, Tse TJ et al (2021) Oral pharmacokinetics of enriched secoisolariciresinol diglucoside and its polymer in rats. J Nat Prod. https://doi.org/10.1021/acs.jnatprod.1c00335
Yawichai A, Kalapanulak S, Thammarongtham C et al (2019) Genome-wide identification of putative microRNAs in cassava (Manihot esculenta Crantz) and their functional landscape in cellular regulation. Biomed Res Int. https://doi.org/10.1155/2019/2019846
Yu Y, Zhang Y, Chen X et al (2019) Plant noncoding RNAs: hidden players in development and stress responses. Annu Rev Cell Dev Biol. https://doi.org/10.1146/annurev-cellbio-100818-125218
Yulvianti M, Zidorn C (2021) Chemical diversity of plant cyanogenic glycosides: an overview of reported natural products. Mol. https://doi.org/10.3390/molecules26030719
Zhang Y, Fernie AR (2021) Metabolons, enzyme–enzyme assemblies that mediate substrate channeling, and their roles in plant metabolism. Plant Commun. https://doi.org/10.1016/j.xplc.2020.100081
Zhang T, Li Z, Song X et al (2020) Identification and characterization of microRNAs in the developing seed of linseed flax (Linum usitatissimum L.). Int J Mol Sci. https://doi.org/10.3390/ijms21082708
Zhang J, Xie Y, Miao C et al (2022) Secoisolariciresinol diglycoside (SDG) lignan content of oil flax: genotypic and environmental variations and association with other traits. Oil Crop Sci. https://doi.org/10.1016/j.ocsci.2022.02.004
Zhang A, Zhang S, Wang F et al (2023) The roles of microRNAs in horticultural plant disease resistance. Front Genet. https://doi.org/10.3389/fgene.2023.1137471
Zuk M, Pelc K, Szperlik J et al (2020) Metabolism of the cyanogenic glucosides in developing flax: metabolic analysis, and expression pattern of genes. Metabolites. https://doi.org/10.3390/metabo10070288
Funding
Open access funding provided by The Ministry of Education, Science, Research and Sport of the Slovak Republic in cooperation with Centre for Scientific and Technical Information of the Slovak Republic. This publication was supported by the Operational program Integrated Infrastructure within the project: Demand-driven research for sustainable and innovative food, Drive4SIFood 313011V336, co-financed by the European Regional Development Fund.
Author information
Authors and Affiliations
Contributions
Both authors, Ľubomír Harenčár and Katarína Ražná, equally contributed to designing the research, preparing the methods, collecting and generating the data, evaluating the results, and writing the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Harenčár, Ľ., Ražná, K. In silico prediction of microRNA families involved in the biosynthesis of lignans and cyanogenic glycosides in flax (Linum usitatissimum L.). Plant Growth Regul (2024). https://doi.org/10.1007/s10725-024-01155-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10725-024-01155-9