
Abstract
Drylands count among the most globally extensive biomes, and while many desert and dry rangeland ecosystems are under threat, genetic structures of dryland species are still rarely studied. Artemisia frigida is one of the most widely distributed plant species in the temperate rangelands of Eurasia and North America, and it also dominates in many habitats of Mongolia due to its tolerance to low temperatures, drought and disturbance. Local environmental conditions and grazing pressure can influence species performance and affect spatial patterns of genetic diversity in contrasting ways, and our study set out to evaluate such effects on the genetic diversity and structure of A. frigida. We first developed new species-specific Simple Sequence Repeats (SSRs) markers using whole genome sequencing. We then analysed 11 populations of A. frigida that had been sampled along a large climatic gradient in Mongolia, which were sub-structured according to three levels of grazing intensity. Estimates of genetic diversity at the population level were high (HO = 0.56, HE = 0.73) and tended to increase with higher precipitation and soil nutrient availability. Grazing had no effect on genetic diversity, however, a high number of grazing-specific indicator alleles was found at grazed sites. Genetic differentiation among populations was extremely low (global GST = 0.034). Analysis of Molecular Variance revealed 5% variance between populations along the climatic gradient, with 3% of the variance being partitioned among different grazing intensity levels. We found no relationship between geographic and genetic distances, and thus no isolation by distance in this widely distributed species. The relatively low genetic structuring suggests that considerable gene flow exists among A. frigida populations across the rangelands of Mongolia, in spite of the pervasive grazing in the region.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The study of genetic changes in natural populations in response to environmental conditions, land use and their respective interactions has become a major research topic (Grant and Grant 2002; Sork 2018). Populations’ and species’ responses to local selection pressures and associated long-term survival is strongly influenced by their prevailing level of genetic diversity and its partitioning between and within populations (Li et al. 2018; Pazouki et al. 2016). A population’s genetic diversity depends on a variety of factors including the species’ geographical distribution, reproductive system and ecological traits (Nybom 2004). Among more continuously distributed and outcrossing species, strong gene flow promotes genetic diversity and counteracts genetic differentiation among populations. For more isolated populations and/or self-fertilizing species where gene flow is more restricted, local genetic diversity can recede due to inbreeding and genetic drift as well as increased differentiation among populations (Kimura 2020). Moreover, the extent of gene flow between populations might not only depend on geographic distance but also on isolation patterns defined by environmental conditions (Sexton et al. 2014). Where gene flow occurs between populations from different environments, adaptation to local environmental conditions can be stalled; in contrast, where gene flow is prevalent across more similar environments, it may facilitate local adaptation by increasing population sizes and introducing new beneficial alleles (Sexton et al. 2014). Hence, climatic conditions can have a strong influence on natural selection among populations. In addition to such interactions with the local environment, human land use practices such as livestock grazing can have direct and compounding effects on a population’s genetic structure as well as its genetic diversity. For some species, land use can have positive effects on genetic diversity: either directly as a result of herbivore damage accelerating mutation rates (Marcotrigiano 2000); indirectly following diaspore transport across long distances (epizoochory) (Bläß et al. 2010); or by providing safe sites for germination due to grazing-induced gaps in vegetation (Frank 2005). On the other hand, adverse effects of grazing on genetic diversity can occur as a result of decreasing effective population sizes or restrictions in gene flow following animal consumption of whole plants or damage to flowers or fruits (Suoto and Tadey 2019), or by the spread of selectively favoured genotypes adapted to grazing. Thus, understanding a population’s genetic variability and sub-population structure with respect to both abiotic constraints and human land use can be critical to the successful conservation and management of its genetic resources. Drylands are particularly relevant in this respect, because they cover 30–40% of the global terrestrial surface, and they are subject to harsh climatic conditions and often intensively grazed. However, there is a definitive lack of studies on population genetics among dryland species (Greenville et al. 2017).
Central Asia hosts one of the world’s most extensive dryland regions. Climate, soil conditions and grazing are among the key variables controlling plant species occurrence and abundance in this region (von Wehrden and Wesche 2007; von Wehrden et al. 2009). Central Asia includes Mongolia, northern and western China and the Tibetan Plateau, which together comprise a total of ca. 5 million km2 of steppe habitat (Wesche et al. 2016), representing more than half of the entire Palaearctic steppe biome. Grasslands cover ~ 80% of Mongolia and have been subjected to a number of studies assessing the impact of its extreme continental climate (Fernández-Giménez and Allen-Diaz 1999; Shinoda et al. 2010; Sternberg 2008), its > 4000 year old history of nomadic pastoralism (Herrero‐Jáuregui and Oesterheld 2018; Hoshino et al. 2009), and the interacting effects of climate and grazing on vegetation (Ahlborn et al. 2020; Bat-Oyun et al. 2016; Hoshino et al. 2009; Khishigbayar et al. 2015; von Wehrden et al. 2012). However, only a few studies have investigated grazing effects on population genetics of Central Asian rangeland species, and they yielded contrasting results: Wang et al. (2004) reported that for Inner Mongolia, intensive grazing led to declining genetic diversity in populations of Artemisia frigida Willd. Due to plant loss and consequent reduced sexual reproduction, gene flow and size of gene pool. Likewise, Shan et al. (2006) reported that increased grazing pressure resulted in stronger divergence among populations and decreased genetic diversity in Stipa grandis P.A. Smirn as a result of the smaller effective population sizes. In contrast, for Stipa glareosa P.A. Smirn., overall genetic diversity was lower in less-grazed populations (Oyundelger et al. 2020), and moderate grazing even promoted genetic diversity in Stipa krylovii Roshev. and S. grandis (Peng et al. 2015) as a result of higher availability of free soil patches for seed germination.
Such contrasting results underline the possible interaction of different effects, particularly combinations of climatic and land use factors influencing the genetic constitution of rangeland species (Wang et al. 2006; Wu et al. 2010; Zhao et al. 2006). Some studies have indeed assessed the effect of environmental factors on population genetics. For instance, Yang et al. (2013) found that for Caragana Fabr. in Central Asia, variation in precipitation had stronger negative effects on genetic connectivity than isolation by distance. Likewise, Zhang et al. (2018) and Jiang et al. (2019) demonstrated that total annual and winter precipitation levels were crucial in shaping the population structure of Dactylis glomerata L. and Ammoipitanthus mongolicus (Komarov) Cheng respectively, since low precipitation restricts plant reproductive success.
Another counteracting factor for plant reproductive success in Central Asian drylands is soil nutrient availability (Baranova 2018; Ronnenberg and Wesche 2011). Only a few studies have demonstrated that soil nutrient availability affects genetic variation in plant populations: Stevens et al. (2007) found that high nutrient-soil conditions increased growth (i.e., diameter, height and total biomass) in Populus tremuloides Michx., which led to increased genotypic variability in growth. Elsewhere, Reisch et al. (2020) revealed a clear impact of soil nutrient conditions on clonal diversity and genetic variation in alpine populations of Carex nigra L., which increased with phosphorous concentration but decreased with potassium concentration.
Our study region of Mongolia is ideal for testing the contrasting effects of environmental conditions and grazing pressure. Mongolia experienced a strong revival of pastoralism at the beginning of the 1990s, when the privatization and growth of livestock rearing prompted discussions on potential degradation of communal rangelands. Since the nineties, the number of livestock has doubled to ca. 71 million (NSO 2020), which has had tremendous effects on both vegetation and soil (Jamsranjav et al. 2018). Besides grazing effects, climatic conditions such as annual precipitation, its variability and the occurrence of droughts have strongly influenced vegetation distribution and land degradation (Ahlborn et al. 2020; Dai 2011; Fatichi and Ivanov 2014). Intensive grazing in mesic steppes can lead to invasions by more drought-adapted species (xerophytisation) (Zemmrich et al. 2010). In temperate grasslands, intensive grazing has been shown to promote more drought-adapted Artemisia species such as A. frigida (Bai and Romo 1996; Jinhua et al. 2005), however, it is not clear whether certain ecotypes of this species were more successful than others. Artemisia frigida Willd. is a perennial, outbreeding and wind-pollinated species that is one of the most widely distributed rangeland plants (McArthur and Jeffrey 2004). While the species tolerates a large range of climatic conditions including cold and drought, seed production and germination of the species are largely dependent on climate, since in years of sufficient precipitation, plant growth and seed production can be promoted (Bai et al. 1995; Ronnenberg et al. 2007). As such, this drought-adapted species was chosen for the present study as it is well suited for studying the effects of environmental and grazing pressures on its population genetics, such that the results may inform rangeland management and conservation practice in Mongolia.
Recent developments in molecular science approaches have vastly enhanced the understanding of genetic variation and adaptation in many important, yet non-model, species. Non-coding DNA regions (introns) are highly variable and consequently widely used in population genetic and phylogeographic studies (Pleines et al. 2009). Microsatellites (i.e. SSRs) are commonly used as powerful markers in population genetics based on their high polymorphism and co-dominant inheritance (Kalia et al. 2011; Vieira et al. 2016). The SSRs still remain an important tool in the genomic era due to its cost-efficiency and the possibility to manually screen loci, which reduces error as the analysis relies on a relatively small number of loci compared to the high-throughput involved in sequencing fingerprints in e.g. RADseq (Hodel et al. 2016). As a neutral marker, SSR cannot trace positive or negative responses of genotypes to selection; however, it can provide some insight into the overall distribution of certain genotypes, which can be characterized by favourable suits of traits under unknown (multigenetic) control.
Our first aim was therefore to develop species-specific SSR markers for A. frigida using whole genome sequencing, and then to validate them. In order to investigate genetic variation from larger to smaller scales (i.e. between and within populations to between sites) along a local grazing gradient, we conducted SSR analysis on eleven populations, which represented the first genetic study of A. frigida in Mongolia. We hypothesized the following: (i) Genetic diversity in populations of this drought and disturbance adapted species is high, but, because of wind-facilitated genetic exchange in the vast steppe plains, we expect moderate genetic structuring in space; (ii) Genetic differentiation is higher in those populations where grazing pressure is more pronounced than in non-grazed (least-grazed) populations, as a result of adapted genotypes and/or accelerated genetic exchange; and (iii) Genetic differentiation among populations increases with increasing distance in our study area due to the large geographical distance and strong environmental gradient from north to south.
Materials and methods
Study species: Artemisia frigida Willd. (Asteraceae)
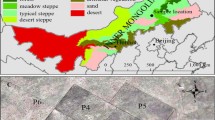
Prairie sagewort is an aromatic perennial sub-shrub that is 20 to 60 cm in height (McArthur and Jeffrey 2004) and occurs primarily on fixed and semi-fixed silty-sandy rangelands. The entire plant is covered in dense silvery pubescence (Fig. 1a), and the lower stems are often strongly branched and woody. The species most likely evolved during the Pleistocene-Holocene (Yurtsev 1987), and it currently has an Amphi-Beringian distribution range, growing mainly in the steppe and prairie rangelands of Alaska, western Canada, the United States, Siberia, Kazakhstan, Mongolia and northern China (Harvey 1981). Prairie sagewort is a prolific seed producer, with each 2.5 cm length of inflorescence containing approximately 1000 seeds (Harvey 1981). In addition to sexual reproduction, it also has a clonal growth strategy (Jinhua et al. 2005). The species was initially considered to be diploid (2n = 2x = 18; Garcia et al. 2004), however, Wan et al. (2011) identified a tetraploid cytotype (2n = 4x = 36) from an Inner Mongolian population, and Pellicer et al. (2010) confirmed the existence of tetraploid cytotypes in a Russian population. Artemisia frigida is one of the first perennials to become established on disturbed sites, and it tends to increase in abundance with overgrazing (Coupland 1950; Sarvis 1941) since it can tolerate harsh climatic conditions as well as mechanical disturbance. In Mongolia, it occurs throughout the country (Fig. 1b), and it serves as important forage for livestock (Jigjidsuren et al. 2015; Li et al. 2012).

a Artemisia frigida in its typical habitat in Mongolia, and b its distribution in Central Asia (samples were only collected from Mongolia; map drafted by Jan Treiber)
Study design
Our study was conducted in central and southern Mongolia between June and September 2018, covering eleven populations along a 600 km north to south gradient with 100–300 mm S/N differences in mean annual precipitation (Table 1; Fig. 2a). Population no.2 (hereafter Pop2) was sampled from Hustai National Park (NP), which lies slightly west of the gradient line and was chosen to avoid the capital city Ulaanbaatar (Fig. 2a). Nested within this gradient, sub-populations were sampled along local transects radiating away from herders’ camps or wells, which represented different levels of grazing intensity (150 m: heavily-grazed; 750 m: moderately-grazed; 1500 m: least-grazed) (Fig. 2b). Distances among sub-populations differing in grazing intensity were based on previous successful applications in the region (Earl 2012; Oyundelger et al. 2020; Sasaki et al. 2008) and elsewhere (Besnier and Glover 2013; Peper et al. 2011). However, grazing gradients reflect also small-scale spatial gradients, and therefore could not be directly distinguished from small-scale effects caused by isolation by distance. Pop2 was different because it was the only population where a fenced-off plot was available. The 10 m × 10 m large-mammal exclosure was built in 2003 and meets the criteria of a permanent sampling site for monitoring phenology (Tserendulam et al. 2018). In Hustai NP, the main grazers were Przewalski horse, marmot and red deer, while all other populations were grazed by goat, sheep, cattle or horse.

a Map of 11 study sites (corresponding to Table 1) showing mean annual precipitation totals; contour lines indicate elevation. Rainfall data were derived from CHELSA and elevation data from TanDEM-X—Digital Elevation Model (German Aerospace Center (DLR) 2018); map drafted by Jörg Lorenz. b Experimental design to study the relative effect of precipitation and grazing in Mongolian steppes. Eleven study sites were visited along a north to south gradient, and at each site sub-populations were sampled along local transect representing different grazing intensities (A–C, initial draft C. Römermann)
Sample collection and analyses
Field collection
Fresh leaf material was collected from a total of 361 individuals and stored in silica gel. For each population, representative herbarium specimens were deposited at Herbarium Senckenbergianum Görlitz. For the sub-populations, 12 individuals of A. frigida were sampled within a 10 m × 10 m plot, from which plant community composition and total vegetation cover of vascular plants were also recorded to characterize the community properties. Plots were then classified by steppe type according to a standard classification for Mongolian steppe vegetation (Tuvshintogtokh 2014). In addition, a sample of topsoil (0 ± 5 cm depth) with fine plant roots and some humic layer was taken at each plot to examine nutrient availability.
Soil preparation and analyses
Soil samples were initially dried in the laboratory for 18 h at 75 °C before being sieved (2 mm coarse screen) and analysed further. Soil pH and electrical conductivity (EC; as a proxy for salinity) were measured in the laboratory with a pH/EC meter (water: soil volume = 5:1). Plant available P, Mg, Ca, K, Al were extracted using the Olsen P method (22 °C, 200 rpm for 30 min; Sims 2000) followed by ICP determination. Nutrient contents in these extractions were measured by spectrometry (ICP-OES, Institute of Soil Science, Hannover University). By drying samples at 105 °C for 24 h, the rest water was determined. The carbonate content was first assessed using a simple test with 10% HCl, and samples showing a reaction were further analysed using a calcimeter following Scheibler’s method (ON L 1084-99, 1999). Total N and C concentrations were analysed using a CN analyser (Vario Pyro Cube; Elementar, Langenselbold, DE). The remaining water was used to calibrate the nutrient contents per gram of soil and the carbonate content to correct C/N measurements.
Climate data extraction
Twenty years (1994–2013) of meteorological data including mean annual temperature (MAT), mean annual precipitation (MAP), mean spring temperature (SpringT; March–May), mean summer temperature (SummerT; June–August) and mean summer precipitation (SummerP) were retrieved for each locality from the high resolution CHELSA_V1 dataset (Karger et al. 2017). The coefficient of variation of annual precipitation (cvP) was estimated based on the retrieved MAP data, and it was also used as predictor, since cvP is a critical driver of rangeland dynamics (von Wehrden et al. 2012).
Detection of SSRs, primer design and marker validation
Library preparation and quality control
Two individuals of A. frigida selected randomly from two distinct populations (collection ID: P8_15 and P9_17; BioSample accession numbers: SAMN16882070 and SAMN16882071) were used to develop new SSR markers by applying whole genome sequencing (WGS). High molecular weight DNA was extracted using the Mag-Bind® Plant DNA DS kit (Omega Bio-tek, Norcross, USA). Genomic DNA was quantified using a Qubit 4 Fluorometer (Life Technologies), and library preparation and sequencing were conducted in the Leibniz Institute of Plant Genetics and Crop Plant Research (IPK). As an input, 1.0 μg DNA (~ 20 ng/µl) per sample was used for covarizing (Covaris S220, Duty Factor: 8%, Peak Incident Power: 160, Cycles Per Burst: 200, Time: 60 s), and BluePippin (Sage Science) was used to produce fragments with an average size distribution of 450 base pair (bp) lengths. Library preparation was carried out as described by Meyer and Kircher (2010), and fragment size distribution was evaluated on an Agilent BioAnalyzer High Sensitivity DNA Kit (Aglient Technologies, Inc). DNA concentration was measured using the Qubit DNA Assay Kit in a Qubit 2.0 Fluorometer (Life Technologies). Finally, the DNA concentration of the library was checked by a quantitative polymerase chain reaction (qPCR) run, allowing for a targeted quantification of fragments with adaptor sequences on both ends before cluster generation, which was run on Illumina cBot following Illumina’s recommendation. Paired-end sequencing (2 × 250 bp) of the clusters was performed on the Illumina HiSeq 2500 platform, with a 1% Illumina PhiX library used as an internal control. The resulting 50 million raw reads were checked with FASTQC for quality control (Andrews 2010) and trimmed with Trimmomatic v0.39 (Bolger et al. 2014). WGS raw sequencing data were submitted to the NCBI Sequence Read Archive (SRA) and made publicly accessible under BioProject (accession number PRJNA680535; available at: https://www.ncbi.nlm.nih.gov/sra/PRJNA680535).
Bioinformatics and SSR development
In order to identify any low coverage contigs, the velvet v1.2.10 (Zerbino 2010) de novo genome assembler was utilized (kmer size: 45; maximum coverage parameter: 500; minimum contig length: 300 bp). Putative SSR markers were detected in the contigs of the assembled nuclear genome using the MISA tool with default parameters (Thiel et al. 2003). The contigs possessing SSR markers were compared between both A. frigida assemblies using blast + v2.7.1 (Camacho et al. 2009), and only contigs with differences in their repeat length were selected for further analysis. Back mapping of the raw reads against contigs containing di- and tri-nucleotide SSR motives were conducted on Bowtie2 v2.2.4 (Langmead et al. 2009), further examined by converting the mapping results and extracting the mapped contigs with SAM tools v1.2 (Li et al. 2009) and visualized in Geneious® v. 10.2.6 (https://www.geneious.com). Editing was not necessary since the reads were correctly mapped to the contigs. Primers were designed using the ThermoFisher tool using default parameters 18–22 nucleotides long, a GC content of ca. 40–50% (Tm between 58–60 °C) and, where possible, G/C-rich at the 3’-end. Heterodimers of the primers were checked and 40 primers that met the requirements were preferentially selected to produce a PCR product in the range of 100–600 bp, allowing for multiplex PCR. Primers were then synthesized by Metabion International AG, with the forward primer of each set being tailed with a M13 sequence (TGT AAA ACG ACG GCC AGT; according to Schuelke 2000) to facilitate dye-labelling. The nucleotide sequences of the primer pairs used for each microsatellite are shown in Table 2. The loci named ‘Arfi’ are the markers developed in this study, and the loci names starting with ‘Ch’ are from the master thesis of Wang (2011).
Application of SSR markers
Genomic DNA was extracted according to the ATMAB protocol (Doyle and Doyle 1987) with modifications by Ziegenhagen (1990). Developed SSR markers were tested with randomly chosen samples from the 11 populations. A total of 40 primer pairs were run as an initial screening for 8–16 samples. After PCR optimization, seven of these loci proved to be reproducible and were reused with the same set of samples to receive positive controls (for details about primers see Table 2). In addition, ten SSR markers published in the master thesis of Wang (2011) were tested with our samples in parallel. Potential duplications between both markers’ sources were checked beforehand. Based on reproducibility and polymorphism, four loci published by Wang (2011) were chosen in addition to our seven newly developed markers. Thus, a total of 11 SSR amplifications were performed in a total volume of 12.5 μl, and different PCR reaction mixtures and cycling programs were used (Suppl. 1 Table 1). Fragment sizes were determined by the central laboratory of the Senckenberg Biodiversity and Climate Research Center (SBiK-F, Germany) using an ABI3730 sequencer with LIZ600 size standard (Life Technology). The program Genographer version 2.1.4 (Benham 2001) was used for the initial screening of the microsatellite data, and Geneious® 10.2.6 (https://www.geneious.com) was used to score allele sizes. To estimate genotyping error, DNA amplification and fragment scoring were replicated for the randomly selected (8–16) samples (Hoffman and Amos 2005). In addition, each plate included two to three positive controls, which ensured that all the replications provided identical results. The number of microsatellite alleles per locus was compatible with the proposed tetraploidy, since individuals displayed a maximum of four alleles per locus. Due to a lack of information on allele dosage in the polyploids, only the allelic phenotypes (and no genotypes) could be assessed. Thus, deviations from the Hardy–Weinberg equilibrium could not be calculated, nor could the frequency of null alleles be estimated (Dufresne et al., 2014).
Analysis of genetic variation and population structure
Population genetic structure
Population structure was estimated by Bayesian clustering with STRUCTURE v. 2.3.4 conducted in R-package ParallelStructure (Besnier and Glover 2013). To choose the model with the best fitting number of clusters (K), Evanno’s method was employed with Structure Harvester (Earl 2012; Evanno et al. 2005). Results of Bayesian clustering were illustrated using Distruct v. 1.1 (Rosenberg 2004). To examine the distribution of genetic variation between populations and sub-populations, Analysis of Molecular Variance (AMOVA; Excoffier et al. 1992) was performed in R-package poppr (Kamvar et al. 2014) based on the individual level Bruvo distance matrix estimated in R-package Polysat v. 1.7 (Clark and Jasieniuk 2011; Suppl. 2 Table 1). The Bruvo metric was developed specifically to assess microsatellite data in polyploids and it considers allelic dosage in ambiguous polyploid genotypes using a step-wise mutation model (Bruvo et al. 2004; Dufresne et al. 2014). As such, we used the Bruvo distance wherever possible and estimated mean Bruvo distances between population and sub-population levels using the ‘meandist’ function in R-package vegan (Oksanen et al. 2007; Suppl. 2 Table 2 & 3). To facilitate comparisons with other studies, we also estimated pairwise GST (Nei and Chesser 1983) for (sub-) population levels in Polysat (Supl. 2 Tables 4 and 5). Principal Coordinate Analysis (PCoA) was conducted to assign individuals to populations based on (i) sub-population level mean Bruvo, and (ii) individual level Bruvo genetic distances using the R-package ape (Paradis et al. 2019). On the sub-population level ordination, environmental variables were fitted post hoc using vegan (Oksanen et al. 2007) and ggplot2 (Wickham 2011) to reveal environmental variables and grazing intensities that could be significantly associated with the population’s genetic structure. Environmental variables covered geography (latitude, longitude, and altitude), climate (MAT, SpringT, SummerT, MAP, SummerP and cvP), soil nutrient content (P, Mg, Ca, K, Al, C, and N) and vegetation type (mountain steppes, dry steppes, and desert steppes). Data on geographic and climatic variables are given in Table 1, and vegetation cover, plant species richness and analysed soil nutrient values are provided in Suppl. 1 Table 2.
In order to evaluate the number of alleles specific to certain grazing levels, data were exported from Polysat as a dominant marker matrix (presence/ absence of alleles; Suppl. 2 Table 6) and used for Indicator Species Analysis (ISA) implemented in R-package indicspecies (De Caceres and Jansen 2014). The analysis was based on: (i) the frequency of a given allele within a given groups (grazing level), and (ii) the frequency distribution across groups (represented by the share of overall occurrence found for a given group). The resultant frequencies for (i) and (ii) were multiplied and the result tested for significance using a permutation test, which examined whether alleles were significantly associated with a certain group. By taking the entire dataset into account, the ISA ensured that alleles potentially associated with grazing did not simply reflect small-scale isolation by distance effects.
Genetic diversity
Genetic diversity among and within populations was assessed using GenoDive v. 3.04 (Meirmans 2020) and the R-package Polysat v. 1.7 (Clark and Jasieniuk 2011; R Development Core Team 2018), both of which offer tools for handling microsatellite data at any ploidy level with allele dosage correction. First of all, diversity and polymorphism of the employed markers were analysed using G-statistics in GenoDive. Next, based on the individual level Bruvo distance, we developed a surrogate for gene diversity by calculating mean Bruvo distances among individuals for any given population using the ‘mrpp’ function in R-package vegan (Oksanen et al. 2007). As a result, we obtained a single value for each population (hereafter ‘Bruvo index’), which we used as a proxy for genetic diversity. In addition, allele diversity (AD, mean number of alleles per locus), effective number of alleles (EfN, number of alleles in a population weighted for their frequencies), percentage of polymorphic loci (PPL, %), observed heterozygosity (HO), expected heterozygosity (HE) and inbreeding coefficient (GIS; analogous to FIS) were estimated in GenoDive for all (sub-) populations such that comparisons with other studies could be made (Table 1). To overcome the problem of higher diversity in polyploids compared to diploids, we estimated the expected heterozygosity (HE, gene diversity) according to Nei (Meirmans 2020), representing a generic method that is independent of ploidy level. Observed heterozygosity (HO) was based on allelic phenotypes (e.g. AB), with both full and partial heteorozygosity being taken into account (possibilities of genotypes AAAB, AABB or ABBB) to allow for direct comparisons between HO and HE (for further information see Meirmans 2020). Correlations among different genetic diversity indices are given in Suppl. 1 Table 3.
Furthermore, we performed a Linear Mixed Model Analysis implemented in R-package lme4 (Bates et al. 2007) to contrast the effects of local environment and grazing pressure on genetic diversity. Based on the data distribution and variables, the Bruvo index was used as a response variable. Significant environmental variables revealed by PCoA (MAT, MAP, cvP, latitude soil P and Ca) were used as continuous predictors, while ‘grazing’ was included as a factor. The response variable was log-transformed, and all predictors were scaled to zero mean-unit-variance prior to modelling to make effect sizes comparable. Starting with the most comprehensive model, we conducted model simplification by dropping the least relevant models, and all complex models that did not result in significantly better results were rejected. We plotted residuals of the final parsimonious model to check for possible deviations from normality and reasonable distribution of variances.
Relationship between genetic and spatial distances
The relationships between genetic and geographical distances (Euclidean distances) were analysed for 11 populations of A. frigida using the Mantel test (10,000 randomizations) in the R-package vegan (Oksanen et al. 2007). An additional Mantel test was conducted between genetic distance and differences in plant community composition (Bray–Curtis distance of log-transformed vegetation cover). Correlation with geographical distance was based on population level mean Bruvo distance, while sub-population level mean Bruvo distance was used for correlation with plant community composition based on the available data for each grazing level within populations. All analyses were conducted in R 3.5.1 (R Development Core Team 2018).
Results
Microsatellite marker profile
The 11 SSR markers proved to be highly polymorphic, with an overall of 76% polymorphic information across all primers (Table 3). The locus Arfi16 was less polymorphic than others, however, it did not have a negative impact on analyses, according to pre-analyses excluding the locus (data not shown). Therefore, further analyses were based on all available 11 loci. The most polymorphic loci were Arfi2 and Arfi15, as shown by the number of effective alleles (EfN) and the expected heterozygosity (HE; Table 3). The 11 loci amplified in A. frigida yielded a total of 317 alleles across all samples. The total number of alleles (Num) was high in all the loci, albeit the effective number of alleles differed substantially.
Population genetic structure
The coefficient of genetic differentiation across all populations was low (Global GST = 0.034, p < 0.001), with genetic variation being partitioned to 5% among populations and 3% between sub-populations (among different grazing intensity levels), and the largest share resided between individuals (92%, p < 0.001, AMOVA; Table 4). The STRUCTURE analysis suggested the 11 populations form two genetic clusters (best fitting k = 2) that are not completely segregated, although Pop2 and Pop10 tended to show some genetic admixture (Suppl. 1 Fig. 1). The PCoA also revealed a similar pattern as the STRUCTURE analysis, with the first two axes explaining only about 14% of the genetic variation (Fig. 3). Furthermore, PCoA ordination with post hoc fitted predictor variables revealed significant correlations between genetic structure and environmental variables (MAT, MAP, cvP and latitude) and thus major habitats, but no main effect of grazing on genetic similarity was found (Fig. 3 & Suppl. 1 Fig. 2).

Principle Coordinate Analysis (PCoA) based on population level mean Bruvo distance using 11 SSR markers for the populations of A. frigida. Environmental predictors were fitted post hoc on the ordination plot (only those that passed p < 0.05 according to test with 1000 permutations are shown). MoS - mountain steppe, DryS - dry steppe and DeS - desert steppe
According to the ISA, some indicator alleles were associated with a certain grazing level across widely spaced populations, i.e. some alleles were prone to occur at only one grazing level, and/ or were absent at other levels, irrespective of their location along the main climatic gradient (Suppl. 1 Table 4). A total of 63 alleles were significantly indicative, which corresponds to 20% of all alleles. The heavily-grazed sites had 31 indicator alleles, which was more than three times that found in the least-grazed sites. In addition, 23 alleles were indicative of moderately grazed sites, indicating that certain alleles are associated with higher grazing intensity.
Genetic diversity
Populations along the main climatic gradient differed with respect to their genetic diversity (Table 1). The observed heterozygosity (HO) among populations ranged from 0.43 to 0.62 in Pop1 and Pop9 respectively; the expected heterozygosity (HE) within populations was highest in Pop10 (HE = 0.76). In contrast, allele diversity and number of effective alleles were relatively invariant among all populations. Across all populations, the Average Inbreeding Coefficient was low (GIS = 0.23) and Percentage of Polymorphic Loci was 48%, showing a moderate level of polymorphism. The highest Bruvo index was found in Pop1 (Bruvo index = 0.63) and the lowest was in Pop11 (Bruvo index = 0.51). This estimator was used for subsequent statistical tests involving genetic diversity.
Diversity within populations, i.e. among different grazing intensity levels, was moderate (Suppl. 1 Table 5). The highest genetic diversity was found in the heavily-grazed plot of Pop3 (Bruvo index = 0.67), while the lowest was found in the moderately-grazed plot of Pop11 (Bruvo index = 0.46). According to our linear mixed effect model, genetic diversity of A. frigida was affected by total summer precipitation and soil phosphorus content (Table 5). Estimates indicate that the Bruvo index increased with higher amounts of summer precipitation and soil phosphorus content. We further checked their interaction effect, but no interaction was found. Moreover, we found no significant grazing effect on genetic diversity or significant differences among different grazing intensity levels in the linear mixed effect model.
Relations between genetic and spatial distances
A Mantel test revealed no correlation of geographical and genetic distances between populations (r2 = 0.02, p = 0.1), as such, no indication of isolation by distance across the studied range of 600 km in southern central Mongolia was found. A further test with plant community composition also revealed a negligible correlation (r2 = 0.001, p = 0.2). Taken together, there was little evidence to suggest that genetic distances were strongly influenced by simple spatial trends.
Discussion
SSR marker development and application in polyploid Artemisia
In population genetics, several molecular markers are proposed and employed. In particular, co-dominant microsatellite markers facilitate the estimation of population genetic parameters in polyploids compared to dominant fingerprint systems (Clark and Jasieniuk 2011). Our newly developed SSR markers for the widespread polyploid species A. frigida has proved reliable and it provides a sound basis for further research, including on other species within the genus, as we could successfully cross-amplify three loci in the rather distantly related annual A. scoparia Waldst. et Kit. We also improved cost efficiency, because allele sizes and fluorescent dyes of each marker were adjusted to allow for multiplexing four to seven loci in one well for genotyping.
Polyploidy causes major obstacles for population genetic analysis. It matters whether a taxon is auto- or alloploid (Dufresne et al. 2014), however, this is often neither known nor easily detected. Since dosage information is generally missing, complete genotyping is still not feasable with SSR markers, and thus genetic diversity from polyploids may often be overestimated. Few software applications and statistical approaches have been proposed to correct possible bias in polyploids (see Dufresne et al. 2014 for a list). The R-package Polysat (Clark and Jasieniuk 2011) and the software GenoDive (Meirmans 2020) are specifically designed to handle microsatellite data with polysomic inheritance and the need for dosage correction. As such, for the present study, we used them to analyse the at least partly tetraploid samples of A. frigida. To ensure consistency, one measure of genetic diversity (Bruvo index) was employed through most of our analyses, with others being provided for cross comparisons with published studies (Table 1). However, it is acknowledged that such comparisons may not always be feasible due to the different marker systems being used. Moreover, in SSR analysis, choosing both diversity metrics and software packages may employ different dosage corrections and mutation models, even for the same index calculation (Dufresne et al. 2014).
Population genetic differentiaton
Artemisia frigida is a wide-spread, wind pollinated long-lived perennial that is valued as a highly nutritional rangeland plant in Mongolia (Undarmaa et al. 2015). Our results clearly demonstrated that the large plain steppe of Mongolia supports adequate gene flow between populations. The highest share of molecular variance was found between individuals within sub-populations (92%), while the variance between populations was 5% and among different grazing intensity levels was 3% (Table 4). The overall degree of differentiation across populations was low (Global GST = 0.034), which is consistent with another studies on A. frigida conducted in Inner Mongolia, China (DST = 0.05, Liu et al. 2010, and FST = 0.01–0.31, Liu et al. 2012).
In our study, the highest genetical distance was found between Pop2 and others (Hustai NP; GST = 0.027–0.042). The only exclosure of the study was located in Pop2 (Pop2C), however, this fenced site was not markedly genetically different from other sites, wheras the other two grazed sites from Hustai NP differed strongly (Pop2A > Pop2B). Given that the sites in Hustai NP were not different in terms of geography and climate (Table 1), other factors may have influenced the observed genetic structure. Hustai NP lies 100 km from the capital city Ulaanbaatar (UB), and every year, hundreds of herders with their livestock head to the capital city to purchase supplies. As such, due to the proximity of Hustai NP to UB, it is possible that the area experiences relatively higher levels of transiting herds, which may in turn lead to higher diaspore input from more distant sources via epizoochory. The potential for such has been demonstarted for Mongolian drylands, where diaspores can be transported in sheep and goat fur by up to 15 km per day (Bläß et al. 2010). While genetic diversity was lower in grazed sites compared to the exclosure site in Hustai NP (Suppl. 1, Table 5), grazed sites contained a higher number of indicator alleles (see below). It is therefore possible that genotypes introduced by herder movements may only have spread sucessfully on grazed sites, as the exclosure was devoid of bare soil patches where diaspores might otherwise have germinated (Frank 2005).
To reveal the presence of grazing-adapted genotypes, we conducted ISA analysis, which detected some indicator alleles characteristic of certain grazing intensity levels along the large-scale transect (Suppl. 1 Table 5). As a result, almost 50% of significant alleles were indicative of heavily-grazed sites, while 37% was attributed to moderately-grazed sites. Remarkably, most of these significant alleles were found in the Pop2 and Pop6 grazed sites, which further explains the higher genetical differentiation of the grazed sites in the Hustai NP. Given that SSR loci are expected to be evenly scattered throughout the genome and represent neutral non-coding sections of DNA, these data do not provide direct evidence of selection and adaptation. Nevertheless, the high number of indicator alleles found in the heavily- and moderately-grazed sites may point to the evolution of adapted genotypes and thus indicate local adaptive divergence among sub-populations.
Genetic diversity
Levels of genetic diversity of Mongolian populations of A. frigida were higher than those found in China based on SSR markers with 0.33–0.58 (HO) and 0.54–0.68 (HE; Liu et al. 2012). In our study, HO and HE varied respectively between 0.41–0.68 and 0.64–0.78 at the sub-population level (Suppl. 1, Table 5). A further three studies employed RAPD markers (Random Amplification of Polymorphic DNA) on A. frigida in Inner Mongolia, China (Wang et al. 2004; Tao et al. 2008; Liu et al. 2010), each of which detected higher overall shares of polymorphic loci (PPL: 83%, 95% and 79% respectively) across all populations, compared to ours (PPL: 48%) (Table 1). It is however noted that the data from China originate from profiles of RAPDs as a dominant and completely anonymous marker system. In any case, we revealed relatively higher potential levels of genetic diversity for A. frigida than that reported for other similarly widespread and long-lived perennials (e.g. Nybom 2004; Nybom and Bartish 2000).
Our results also reveal that higher amounts of summer precipitation and soil phosphorous promote genetic diversity in A. frigida (Table 5), suggesting that environmental conditions have a substantial impact on genetic diversity. In Mongolian rangelands, abiotic constraints exert strong controls on vegetation, where adequate precipitation and soil moisture during the growing season are vital to ensure seed germination of several species, including A. frigida (Ronnenberg et al. 2007). It has also been shown that soil carbon and phosphorous have significant positive effects on the establishment of seedlings and plant growth of Artemisia species, even under relatively dry conditions (Yang et al. 2015). Successful seed germination promotes reproductive success, resulting in more breeding individuals contributing to the gene pool and a consequent increase in genetic diversity. While previous population genetic studies on A. frigda did not specifically investigate the impact of environmental factors on genetic diversity, there are a few studies on other rangeland Artemisia shrub and sub-shrub species that indicate that several climatic factors affect genetic diversity. In particular, Huang et al. (2014) found that higher genetic diversity of A. halodendron Besser. was associated with a higher range of annual temperatures (intra-annual variation of temperature). Chaney et al. (2017) demonstrated that greater temperature seasonality and higher summer precipitation resulted in a greater probability of population survival of A. tridentata Nutt. These results support our findings that higher precipitation promotes genetic diversity in the sageworts.
While we found relatively limited grazing effects on the genetic diversity of A. frigida, this was somewhat expected, as the species is adapted to drought and disturbance and/or it may even benefit from mechanical disturbance. Artemisia frigida is characterized by a highly variable response to disturbance as it propagates horizontally via stolons (Li et al. 2004), which occupy more space to counteract grazing and the consequent suppression of its apical dominance (Jinhua et al. 2005). Our focus species also produces volatile organic compounds under heavy grazing pressure, which inhibits the growth of grass seedlings and can result in Artemisia dominance in heavily grazed areas (Zhao-Jiang et al. 2011). While such results may be interpreted as an adaptation of dryland plants to a long history of grazing, and our findings contrast with those of Wang et al. (2004), who found reduced genetic diversity of A. frigida under intensive grazing due to reductions in plant sexual reproduction by consumption of individuals.
Relations between genetic and spatial distances
With respect to geographical distance (600 km distance), climatic gradient (100–300 mm differences in mean annual precipitation along the S/N gradient) and differences in plant community composition, we had expected to find greater genetic differentiation with larger distance. However, our Mantel tests did not indicate any significant influence of spatial distance or simple spatial processes. Likewise, analysis of three other Artemisia species revealed no direct genetic relation with geographic distance (Badr et al. 2012), and similar findings were reported for Stipa species in Central Asia (Jing et al. 2013; Zhao et al. 2006).
Conclusions
Rangeland plant genetic resources have been rarely investigated and genetic parameters focusing on conservation are largely absent in land use policies on drylands. While dryland species are crucial for foraging herds and, consequently, the livelihoods and subsistence needs of herder communities, they are often threatened by over-grazing and drought. Our results on A. frigida revealed that genetic diversity of this cold-, drought- and disturbance-adapted species in Mongolia was relatively high and genetic differentiation low among eleven populations across a north to south gradient. Furthermore, we observed no severe consequences of grazing on genetic diversity, however, a high number of grazing-specific indicator alleles was found at grazed sites. This indicates that population differentiation may partly be driven by livestock grazing intensity, which may have beneficial effects on the genetic diversity of the species. In addition, sufficient precipitation and soil nutrient availability also promote genetic diversity of A. frigida in the water- and nutrient-limited arid lands. We therefore conclude that in spite of the degraded and overgrazed landscapes of Mongolia, sufficient genetic exchange persists among populations, even across larger distances, such that there is no clear indication of fragmentation. With regard to conservation genetics, while A. frigida populations in Mongolia are apparently genetically ‘healthy’, the same may not be concluded for the more intensively grazed rangelands of northern China.
Data availability
WGS raw sequencing data is available in the NCBI Sequence Read Archive (SRA) under BioProject accession number PRJNA680535, https://www.ncbi.nlm.nih.gov/sra/PRJNA680535. Further dataset generated and analyzed during the current study are available from the corresponding authors on reasonable request.
Code availability
Codes of R software used for statistical analyses are available upon request from the corresponding author.
References
Ahlborn J, von Wehrden H, Lang B, Römermann C, Oyunbileg M, Oyuntsetseg B, Wesche K (2020) Climate – land-use interactions in Mongolian rangelands: effects of grazing change along a largescale environmental gradient. J Arid Environ 173:104043. https://doi.org/10.1016/j.jaridenv.2019.104043
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. Babraham Bioinformatics, Babraham Institute, Cambridge, United Kingdom. http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Badr A, El-Shazly HH, Helail NS, El Ghanim W (2012) Genetic diversity of Artemisia populations in central and north Saudi Arabia based on morphological variation and RAPD polymorphism. Plant Syst Evol 298:871–886. https://doi.org/10.1007/s00606-012-0597-5
Bai YG, Romo JT (1996) Fringed sagebrush response to sward disturbances: seedling dynamics and plant growth. J Range Manag 49:228–233
Bai YG, Romo JT, Young JA (1995) Influence of temperature, light and water stress on germination of fringed sage (Artemisia frigida). Weed Sci 43:219–225
Baranova AN (2018) Grazing impacts on vegetation patterns in the Qilian Mountains, HeiHe River Basin, NW China. Dissertation, Staats- und Universitätsbibliothek Hamburg Carl von Ossietzky
Bates D, Sarkar D, Bates MD, Matrix L (2007) The lme4. R package version 2:74
Bat-Oyun T, Shinoda M, Cheng Y, Purevdorj Y (2016) Effects of grazing and precipitation variability on vegetation dynamics in a Mongolian dry steppe. J Plant Ecol 9:508–519. https://doi.org/10.1093/jpe/rtv083
Benham JJ (2001) Genographer. Montana State University, USA. https://wheat.pw.usda.gov/jag/papers99/paper399/1999_P3.html
Besnier F, Glover KA (2013) ParallelStructure: R package to distribute parallel runs of the population genetics program STRUCTURE on multi-core computers. PLoS ONE 8:e70651. https://doi.org/10.1371/journal.pone.0070651
Bläß C, Ronnenberg K, Tackenberg O, Hensen I, Wesche K (2010) The relative importance of different seed dispersal modes in dry Mongolian rangelands. J Arid Environ 74:991–997. https://doi.org/10.1016/j.jaridenv.2009.12.002
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Bruvo R, Michiels NK, D’Souza TG, Schulenburg H (2004) A simple method for the calculation of microsatellite genotype distances irrespective of ploidy level. Mol Ecol 13:2101–2106. https://doi.org/10.1111/j.1365-294X.2004.02209.x
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K (2009) BLAST+: architecture and applications. BMC Bioinform 10:1–9. https://doi.org/10.1186/1471-2105-10-421
Chaney L, Richardson BA, Germino MJ (2017) Climate drives adaptive genetic responses associated with survival in big sagebrush (Artemisia tridentata). Evolut Appl 10:313–322. https://doi.org/10.1111/eva.12440
Clark LV, Jasieniuk M (2011) POLYSAT: an R package for polyploid microsatellite analysis. Mol Ecol Resour 11:562–566. https://doi.org/10.1111/j.1755-0998.2011.02985.x
Coupland RT (1950) Ecology of mixed prairie in Canada. Ecol Monogr 20:271–315. https://doi.org/10.2307/1943568
Dai A (2011) Drought under global warming: a review. Wiley Interdiscp Rev 2:45–65. https://doi.org/10.1002/wcc.81
De Caceres M, Jansen F (2014) R package ‘indicspecies’: studying the statistical relationship between species and group of sites. CRAN. https://cran.r-project.org/web/packages/indicspecies/indicspecies.pdf. Accessed 31 Aug 2020
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Dufresne F, Stift M, Vergilino R, Mable BK (2014) Recent progress and challenges in population genetics of polyploid organisms: an overview of current state-of-the-art molecular and statistical tools. Mol Ecol 23:40–69. https://doi.org/10.1111/mec.12581
Earl DA (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Fatichi S, Ivanov VY (2014) Interannual variability of evapotranspiration and vegetation productivity. Water Resour Res 50:3275–3294. https://doi.org/10.1002/2013WR015044
Fernández-Giménez ME, Allen-Diaz B (1999) Testing a non-equilibrium model of rangeland vegetation dynamics in Mongolia. J Appl Ecol 36:871–885. https://doi.org/10.1046/j.1365-2664.1999.00447.x
Frank DA (2005) The interactive effects of grazing ungulates and aboveground production on grassland diversity. Oecologia 143:629–634. https://doi.org/10.1007/s00442-005-0019-2
Garcia S, Sanz M, Garnatje T, Kreitschitz A, McArthur ED, Vallès J (2004) Variation of DNA amount in 47 populations of the subtribe Artemisiinae and related taxa (Asteraceae, Anthemideae): karyological, ecological, and systematic implications. Genome 47:1004–1014. https://doi.org/10.1139/g04-061
German Aerospace Center (DLR) (2018) TanDEM-X - Digital Elevation Model (DEM) - Global, 90m. https://doi.org/10.15489/ju28hc7pui09
Grant PR, Grant BR (2002) Unpredictable evolution in a 30-year study of Darwin’s finches. Science 296:707–711. https://doi.org/10.1126/science.1070315
Greenville AC, Dickman CR, Wardle GM (2017) 75 years of dryland science: trends and gaps in arid ecology literature. PLoS ONE 12:e0175014. https://doi.org/10.1371/journal.pone.0175014
Harvey SJ (1981) Life history and reproductive strategies in Artemisia. Master thesis, Montana State University
Herrero-Jáuregui C, Oesterheld M (2018) Effects of grazing intensity on plant richness and diversity: a meta-analysis. Oikos 127:757–766. https://doi.org/10.1111/oik.04893
Hodel RG et al (2016) The report of my death was an exaggeration: a review for researchers using microsatellites in the 21st century. Appl Plant Sci 4:1600025. https://doi.org/10.3732/apps.1600025
Hoffman JI, Amos W (2005) Microsatellite genotyping errors: detection approaches, common sources and consequences for paternal exclusion. Mol Ecol 14:599–612. https://doi.org/10.1111/j.1365-294X.2004.02419.x
Hoshino A, Yoshihara Y, Sasaki T, Okayasu T, Jamsran U, Okuro T, Takeuchi K (2009) Comparison of vegetation changes along grazing gradients with different numbers of livestock. J Arid Environ 73:687–690. https://doi.org/10.1016/j.jaridenv.2009.01.005
Huang W, Zhao X, Zhao X, Li Y, Lian J, Yun J (2014) Relationship between the genetic diversity of Artemisia halodendron and climatic factors. Acta Oecologica 55:97–103. https://doi.org/10.1016/j.actao.2013.12.005
Jamsranjav C, Reid R, Fernández-Giménez M, Tsevlee A, Yadamsuren B, Heiner M (2018) Applying a dryland degradation framework for rangelands: the case of Mongolia. Ecol Appl 28:622–642. https://doi.org/10.1002/eap.1684
Jiang S, Luo MX, Gao RH, Zhang W, Yang YZ, Li YJ, Liao PC (2019) Isolation-by-environment as a driver of genetic differentiation among populations of the only broad-leaved evergreen shrub Ammopiptanthus mongolicus in Asian temperate deserts. Sci Rep 9:1–14. https://doi.org/10.1038/s41598-019-48472-y
Jigjidsuren S et al (2015) Handbook of key functional species of pastureland in Mongolia. TP Publisher, Ulaanbaatar
Jing Z, Yu J, Cheng J (2013) Genetic diversity of a dominant species Stipa bungeana and its conservation strategy in the Loess Plateau of China. Biochem Syst Ecol 47:126–131. https://doi.org/10.1016/j.bse.2012.10.004
Jinhua L, Zhenqing L, Jizhou R (2005) Effect of grazing intensity on clonal morphological plasticity and biomass allocation patterns of Artemisia frigida and Potentilla acaulis in the Inner Mongolia steppe. N Z J Agric Res 48:57–61. https://doi.org/10.1080/00288233.2005.9513631
Kalia RK, Rai MK, Kalia S, Singh R, Dhawan A (2011) Microsatellite markers: an overview of the recent progress in plants. Euphytica 177:309–334. https://doi.org/10.1007/s10681-010-0286-9
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. https://doi.org/10.7717/peerj.281
Karger DN, Conrad O, Böhner J, Kawohl T, Kreft H, Soria-Auza RW, Zimmermann NE, Linder HP, Kessler M (2017) Climatologies at high resolution for the earth’s land surface areas. Sci Data 4:170122. https://doi.org/10.1038/sdata.2017.122
Khishigbayar J, Fernández-Giménez ME, Angerer JP, Reid RS, Chantsallkham J, Baasandorj Y, Zumberelmaa D (2015) Mongolian rangelands at a tipping point? Biomass and cover are stable but composition shifts and richness declines after 20 years of grazing and increasing temperatures. J Arid Environ 115:100–112. https://doi.org/10.1016/j.jaridenv.2015.01.007
Kimura M (2020) Introduction to population genetics. In: My thoughts on biological evolution. Evolutionary studies. Springer, Singapore, pp 85–101
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. https://doi.org/10.1186/gb-2009-10-3-r25
Li JH, Li ZQ, Liu ZG (2004) Growth and resource allocation pattern of Artemisia frigida under different grazing and clipping intensities. Chin J Appl Ecol 15:408–412
Li H, Wysoker A, Durbin R (2009) 1000 Genome project data processing subgroup. The sequence alignment/map format and samtools. Bioinformatics 25:2078–2079
Li XH, Jiang DM, Alannusa ZQL, Oshida T (2012) Comparison of seed germination of four Artemisia species (Asteraceae) in northeastern Inner Mongolia, China. J Arid Land 4:36–42
Li S, Gan X, Han H, Zhang X, Tian Z (2018) Low within-population genetic diversity and high genetic differentiation among populations of the endangered plant Tetracentron sinense Oliver revealed by inter-simple sequence repeat analysis. Ann for Sci 75:74. https://doi.org/10.1007/s13595-018-0752-4
Liu Y, Pan N, Huang Y-h, Wen J, Zhou L (2010) Study on genetic diversity of Artemisia frigida by RAPD. Lishizhen Medicine and Materia Medica Res 21:1577–1579
Liu Y, Wang Z, Feng J, Gu YQ (2012) Analysis of genetic diversity in different Artemisia frigida Willd. populations by SSR method. In: Paper presented at the International Plant and Animal Genome Conference XX, San Diago, CA
Marcotrigiano M (2000) Herbivory could unlock mutations sequestered in stratified shoot apicies of genetic mosaics. Am J Bot 87:355–361. https://doi.org/10.2307/2656631
McArthur ED, Jeffrey RT (2004) Artemisia frigida Willd. In: Francis JK (ed) Wildland shrubs of the United States and its territories: Thamnic descriptions, vol 1. Gen. Tech. Rep. IITF-GTR-26. San Juan, PR: US Department of Agriculture, Forest Service, International Institute of Tropical Forestry; Fort Collins, CO: US Department of Agriculture, Forest Service, Rocky Mountain Research Station, p 63
Meirmans PG (2020) GenoDive version 3.0: easy-to-use software for the analysis of genetic data of diploids and polyploids. Mol Ecol Resour 20:1126–1131. https://doi.org/10.1111/1755-0998.13145
Meyer M, Kircher M (2010) Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc 6:5448. https://doi.org/10.1101/pdb.prot5448
Nei M, Cesser R (1983) Estimation of fixation indices andgene diversities. Ann Hum Genet 47:253–259. https://doi.org/10.1111/j.1469-1809.1983.tb00993.x
NSO (2020) Mongolain Statistical Yearbook. National Statistics Office of Mongolia. https://www.1212.mn/stat.aspx?LIST_ID=976_L10_1. Accessed 01 Dec 2020
Nybom H (2004) Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol 13:1143–1155. https://doi.org/10.1111/j.1365-294X.2004.02141.x
Nybom H, Bartish IV (2000) Effects of life history traits and sampling strategies on genetic diversity estimates obtained with RAPD markers in plants. Perspect Plant Ecol Evol Syst 3:93–114. https://doi.org/10.1078/1433-8319-00006
Oksanen J, Kindt R, Legendre P, O’Hara B, Stevens MHH, Oksanen MJ, Suggests M (2007) The vegan package. Community Ecol Package 10:631–637
Oyundelger K et al (2020) Climate and land use affect genetic structure of Stipa glareosa PA Smirn. in Mongolia. Flora 266:151572. https://doi.org/10.1016/j.flora.2020.151572
Paradis E et al (2019) Package ‘ape’. Analyses of phylogenetics and evolution, version 5.4. https://cran.stat.unipd.it/web/packages/ape/ape.pdf
Pazouki L, Shanjani PS, Fields PD, Martins K, Suhhorutšenko M, Viinalass H, Niinemets Ü (2016) Large within-population genetic diversity of the widespread conifer Pinus sylvestris at its soil fertility limit characterized by nuclear and chloroplast microsatellite markers. Eur J for Res 135:161–177. https://doi.org/10.1007/s10342-015-0928-5
Pellicer J, Garcia S, Canela MA, Garnatje T, Korobkov AA, Twibell JD, Vallès J (2010) Genome size dynamics in Artemisia L. (Asteraceae): following the track of polyploidy. Plant Biol 12:820–830. https://doi.org/10.1111/j.1438-8677.2009.00268.x
Peng J, Liang C, Niu Y, Jiang W, Wang W, Wang L (2015) Moderate grazing promotes genetic diversity of Stipa species in the Inner Mongolian steppe. Landsc Ecol 30:1783–1794. https://doi.org/10.1007/s10980-015-0227-z
Peper J, Jansen F, Pietzsch D, Manthey M (2011) Patterns of plant species turnover along grazing gradients. J Veg Sci 22:457–466. https://doi.org/10.1111/j.1654-1103.2011.01260.x
Pleines T, Jakob SS, Blattner FR (2009) Application of non-coding DNA regions in intraspecific analyses. Plant Syst Evol 282:281–294. https://doi.org/10.1007/s00606-008-0036-9
R Development Core Team (2018) R: a language and environment for statistical computing. 3.2.5 edn. R Foundation for Statistical Computing, Vienna, Austria
Reisch C, Meier S, Schmid C, Bartelheimer M (2020) Clonal diversity and genetic variation of the sedge Carex nigra in an alpine fen depend on soil nutrients. PeerJ 8:e8887. https://doi.org/10.7717/peerj.8887
Ronnenberg K, Wesche K (2011) Effects of fertilization and irrigation on productivity, plant nutrient contents and soil nutrients in southern Mongolia. Plant Soil 340:239–251. https://doi.org/10.1007/s11104-010-0409-z
Ronnenberg K, Wesche K, Pietsch M, Hensen I (2007) Seed germination of five mountain steppe species of Central Asia. J Arid Environ 71:404–410. https://doi.org/10.1016/j.jaridenv.2007.04.012
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138. https://doi.org/10.1046/j.1471-8286.2003.00566.x
Sarvis JT (1941) Grazing investigations on the northern Great Plains. Agricultural Experiment Station, North Dakota Agricultural College Fargo, North Dakota
Sasaki T, Okayasu T, Jamsran U, Takeuchi K (2008) Threshold changes in vegetation along a grazing gradient in Mongolian rangelands. J Ecol 96:145–154. https://doi.org/10.1111/j.1365-2745.2007.01315.x
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234. https://doi.org/10.1038/72708
Sexton JP, Hangartner SB, Hoffmann AA (2014) Genetic isolation by environment or distance: which pattern of gene flow is most common? Evolution 68:1–15. https://doi.org/10.1111/evo.12258
Shan D, Zhao M, Han B, Han G (2006) Examining the genetic diversity of Stipa grandis under various grazing pressures. Acta Ecol Sin 26:3175–3182. https://doi.org/10.1016/S1872-2032(06)60048-6
Shinoda M, Nachinshonhor G, Nemoto M (2010) Impact of drought on vegetation dynamics of the Mongolian steppe: a field experiment. J Arid Environ 74:63–69. https://doi.org/10.1016/j.jaridenv.2009.07.004
Sims JT (2000) Soil test phosphorus: Olsen P. In: Pierzynski GM (ed) Methods of phosphorus analysis for soils, sediments, residuals, and waters. North Carolina State University, Raleigh, pp 20–21
Sork VL (2018) Genomic studies of local adaptation in natural plant populations. J Hered 109:3–15. https://doi.org/10.1093/jhered/esx091
Souto CP, Tadey M (2019) Livestock effects on genetic variation of creosote bushes in patagonian rangelands. Environ Conserv 46:59–66. https://doi.org/10.1017/S0376892918000280
Sternberg T (2008) Environmental challenges in Mongolia’s dryland pastoral landscape. J Arid Environ 72:1294–1304. https://doi.org/10.1016/j.jaridenv.2007.12.016
Stevens MT, Waller DM, Lindroth RL (2007) Resistance and tolerance in Populus tremuloides: genetic variation, costs, and environmental dependency. Evolut Ecol 21:829–847. https://doi.org/10.1007/s10682-006-9154-4
Tao W, Ping C, Chenbo Z (2008) Analysis of genetic diversity of Artemisia frigida populations by RAPD in different synusiologic area of Inner Mongolia. J Northwest Sci-Tech Univ Agric for 36:166–169
Thiel T, Michalek W, Varshney R, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106:411–422. https://doi.org/10.1007/s00122-002-1031-0
Tserendulam TO, Bayarsaikhan U, Oyuntsetseg B, Wesche K (2018) The vascular plant flora of Hustai National Park, Mongolia: composition, life forms, ecological groups and geographical elements. Feddes Repertorium 129:137–160. https://doi.org/10.1002/fedr.201700006
Tuvshintogtokh I (2014) The steppe vegetation of Mongolia. Bembi san publisher, Ulaanbaatar
Undarmaa J, Toshiya O, Manibazar N, Yamanaka N (2015) Rangeland Plants of Mongolia: high mountain belt, Mountian forest-steppe belt, Steppe zone. vol 1. High mountain belt, Mountian forest-steppe belt, Steppe zone. Arid Land Research Center in Tottori University and Center for Ecosystem Study in Mongolian State University of Agriculture, Ulaanbaatar, Mongolia
Vieira ML, Santini L, Diniz AL, Munhoz CD (2016) Microsatellite markers: what they mean and why they are so useful. Genet Mol Biol 39:312–328. https://doi.org/10.1590/1678-4685-GMB-2016-0027
von Wehrden H, Wesche K (2007) Relationships between climate, productivity and vegetation in southern Mongolian drylands. Basic Appl Dryland Res 1:100. https://doi.org/10.1127/badr/1/2007/100
von Wehrden H, Wesche K, Miehe G (2009) Plant communities of the southern Mongolian Gobi. Phytocoenologia 39:331–376. https://doi.org/10.1127/0340-269X/2009/0039-0331
von Wehrden H, Hanspach J, Kaczensky P, Fischer J, Wesche K (2012) Global assessment of the non-equilibrium concept in rangelands. Ecol Appl 22:393–399. https://doi.org/10.1890/11-0802.1
Wan T, Sun Q, Cai P, Meng X, Yi W, Wang W (2011) Observation of chromosome karyotypes of Artemisia frigida in different ecological areas of Inner Mongolia. Acta Botan Boreali-Occiden Sin 31:456–461
Wang Z (2011) Study on genetic diversity of traditional Mongolian medicine Artemisia frigida. Master thesis, Minzu University of China
Wang J, Yang C, Yin J, Wang TJ, Liu PT (2004) Changes of the genetic diversity of Artemisia frigida population under the disturbance of grazing. Acta Ecol Sin 24:2465–2471
Wang JL, Zhao NX, Gai YB, Lin F, Ren AZ, Ruan WB, Chen L (2006) RAPD analysis of genetic diversity and population genetic structure of Stipa krylovii Reshov. in inner Mongolia steppe. Russ J Genet 42:468–475. https://doi.org/10.1134/S1022795406050024
Wesche K, Ambarlı D, Kamp J, Török P, Treiber J, Dengler J (2016) The Palaearctic steppe biome: a new synthesis. Biodivers Conserv 25:2197–2231. https://doi.org/10.1007/s10531-016-1214-7
Wickham H (2011) ggplot2. Wiley Interdiscip Rev Comput Stat 3:180–185. https://doi.org/10.1002/wics.147
Wu JB, Gao YB, Bao XY, Gao H, Jia MQ, Li J, Zhao NX (2010) Genetic diversity of Stipa grandis P. Smirn populations across the species’ range in the Inner Mongolia Plateau of China. Biochem Syst Ecol 38:471–477. https://doi.org/10.1016/j.bse.2010.04.008
Yang J, Cushman SA, Yang J, Yang M, Bao T (2013) Effects of climatic gradients on genetic differentiation of Caragana on the Ordos Plateau, China. Landsc Ecol 28:1729–1741. https://doi.org/10.1007/s10980-013-9913-x
Yang X, Huang Z, Zhang K, Cornelissen JH (2015) C: N: P stoichiometry of Artemisia species and close relatives across northern China: unravelling effects of climate, soil and taxonomy. J Ecol 103:1020–1031. https://doi.org/10.1111/1365-2745.12409
Yurtsev BA (1987) The Arctic flora of the USSR. Families Rubiaceae – Compositae [Arkticheskaya flora SSSR. Vyp. X. Semeystva Rubiaceae – Compositae]. vol 10. Nayuka, Leningrad
Zemmrich A, Hilbig W, Oyuunchimeg D (2010) Plant communities along an elevation gradient under special consideration of grazing in Western Mongolia. Phytocoenologia 40:91–115. https://doi.org/10.1127/0340-269X/2010/0040-0443
Zerbino DR (2010) Using the velvet de novo assembler for short-read sequencing technologies. Current Protoc Bioinform 31:11.5.1-11.5.2. https://doi.org/10.1002/0471250953.bi1105s31
Zhang C et al (2018) AFLP-based genetic diversity of wild orchardgrass germplasm collections from Central Asia and Western China, and the relation to environmental factors. PLoS ONE 13:e0195273. https://doi.org/10.1371/journal.pone.0195273
Zhao NX, Gao YB, Wang JL, Ren AZ (2006) Genetic diversity and population differentiation of the dominant species Stipa krylovii in the Inner Mongolia steppe. Biochem Genet 44:504–517. https://doi.org/10.1007/s10528-006-9054-x
Zhao-Jiang Z, Ru-Min Z, Pei-Jun G, Guo-Sheng W, Ping H, Yan G (2011) Allelopathic effects of Artemisia frigida Willd. on growth of pasture grasses in Inner Mongolia, China. Biochem Syst Ecol 39:377–383. https://doi.org/10.1016/j.bse.2011.05.010
Ziegenhagen B (1990) ATMAB DNA Extraction. Method Birgit Ziegenhagen. Philipps-Universität Marburg
Acknowledgements
The authors would like to thank Prof. Dr. Dr. h.c. Hermann Ansorge for his generous support that enabled our fieldwork collection, and students (Khaliun.U, Batzorig.G and Namuulin.T, Faculty of Biology, National University of Mongolia), who helped for the sampling. The sample collection was carried out as a part of a cooperation agreement between the National University of Mongolia (Faculty of Biology), Mongolia and the State Museum of Natural History Görlitz, Germany. We are also grateful to Jan Treiber for drawing the distribution map of the species (Fig. 1b), Jörg Lorenz for the map of study sites (Fig. 2a) and C. Römermann for an earlier draft of the design sketch. We thank to our technical assistants Michaela Schwager and Bettina Schlitt for their support during laboratory work, as well as our colleagues of the laboratory centres in Leibniz institute of Plant Genetics and Crop Plant Research (IPK) and Senckenberg Biodiversity and Climate Research Institute (SBiK-F) for technical supports. The authors also thank to Daniel McCluskey for his diligent proofreading and language editing of this paper. Oyundelger is funded by Scholarship program for the promotion of early-career female scientists of Technische Universität Dresden. Fieldwork was supported by both the German Academic Exchange Service (DAAD) and the German Ministry for Science and Education (BMBF, No. 01LC1820C – MoreStep project in the BioTip scheme).
Funding
Open Access funding enabled and organized by Projekt DEAL. Khurelpurev Oyundelger is funded by Scholarship program for the promotion of early-career female scientists of TU Dresden. Fieldwork was supported by both the German Academic Exchange Service (DAAD) and the German Ministry for Science and Education (BMBF, No. 01LC1820C—MoreStep project in the BioTip scheme).
Author information
Authors and Affiliations
Contributions
All authors contributed to this work, i.e., study conception and design, sample collection and vegetation surveys including species identification were performed by CMR, KW, BO and KO. Library construction and bioinformatics were done by DH and VH. DNA extractions, microsatellite analyses and statistics were done by KO. The first draft of the manuscript was written by KO and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Informed consent
The authors agree for publication of the paper and all data relevant to this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oyundelger, K., Herklotz, V., Harpke, D. et al. Contrasting effects of local environment and grazing pressure on the genetic diversity and structure of Artemisia frigida. Conserv Genet 22, 947–962 (2021). https://doi.org/10.1007/s10592-021-01375-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-021-01375-w