
Abstract
Regioselective tritylation and carbonate aminolysis were employed in this work to synthesize cellulose 2,3-bis(3,5-dimethylphenyl carbamate)-6-(α-phenylethyl carbamate)-type chiral selectors. We evaluated and optimized the critical aspects of regioselective tritylation and detritylation at C6 of the glucopyranose units of the polysaccharide backbone. The advantage of using cellulose II in comparison to cellulose I for tritylation was analyzed and the detritylation time was determined by a fast and simple thin-layer chromatography method. Optimization of both tritylation and detritylation was accompanied by a combination of analytical techniques. Oxycarbonylation with phenyl chloroformate was used to introduce a reactive phenyl carbonate moiety at C6 of the intermediate cellulose 2,3-bis(3,5-dimethylphenyl carbamate), which was subsequently converted to the respective cellulose 6-(α-phenylethyl carbamate) derivative by aminolysis with enantiopure (R)- or (S)-α-phenylethylamine. The starting material, intermediates, and target cellulose derivatives were comprehensively analytically characterized by ATR-FTIR, solid- and liquid-state 13C NMR, GPC, and elemental analysis. With the optimized protocol, it became possible to obtain cellulose carbamate-type chiral selectors through carbonate aminolysis with simple and commercially available primary amines instead of reaction with isocyanate reagents. The enantioseparation performance of the obtained chiral selectors was evaluated against cellulose tris(3,5-dimethylphenyl carbamate) as a reference selector with a selection of chiral analytes.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
High-performance liquid chromatography (HPLC) on chiral stationary phases (CSPs) is one of the most powerful techniques for the direct separation of enantiomeric compounds, or generally, stereoisomers, both on analytical and preparative scales (Ahuja 1997; Subramanian 2008). Polysaccharide derivatives are the most versatile chiral selectors (CSs) due to their superior chiral recognition ability in comparison to other macromolecular selectors, such as proteins, macrocyclic polyethers, macrocyclic antibiotics, etc. (Teixeira et al. 2019). In the 1980s, Okamoto et al. successfully introduced polysaccharide derivatives for enantioseparation by HPLC and a number of polysaccharide derivatives carrying different substituents have been screened since then (Shen and Okamoto 2016; Chankvetadze 2020; Wang et al. 2021), e.g., aryl and alkyl carbamates, alkyl esters, benzoates, and cycloalkyl substituents (Chankvetadze 2012). Among them, polysaccharide phenyl carbamates and benzoates were shown to be the most powerful (Minguillón et al. 1996; Fanali et al. 2019). Up to 90% of the tested enantiomeric compounds could be separated by these polysaccharide derivative-based CSs (Ikai and Okamoto 2009; Shen and Okamoto 2016; Yin et al. 2019a). Amylose and cellulose are the most common polysaccharide matrices for CSs (Okamoto et al. 1986; Okamoto et al. 1998; Okamoto 2009; Ali et al. 2009; Shen and Okamoto 2016), but also chitosan and chitin are being used (Ribeiro et al. 2017). Hetero-substituted polysaccharides (such with different moieties) were shown to possess, in certain cases, even higher chiral discrimination than their homo-substituted counterparts (Acemoglu et al. 1998; Felix 2001; Katoh et al. 2011; Yin et al. 2019b).
One central part of the regioselective synthesis of such hetero-substituted cellulose derivatives is the use of, e.g., trityl protecting groups for C6-OH (Kaida and Okamoto 1993) as it allows regioselective distinction of this hydroxy group from those in C2- and C3-positions. This strategy has been applied to generate a series of hetero-substituted polysaccharide derivatives as CSs (Chassaing et al. 1996; Chassaing et al. 1997; Zheng et al. 2009; Shen et al. 2018). Heterogeneous tritylation of regenerated cellulose (= cellulose II) obtained by deacetylation of cellulose acetate (Hearon et al. 1943b) or homogenous tritylation of celluloses in a mixture of N,N-dimethylacetamide (DMAc)/LiCl/pyridine at 80 °C have been used to yield cellulose derivatives with high DS (Gömez et al. 1996; Shen et al. 2018; Ganske and Heinze 2018). The degradation of cellulose in the mixture of DMAc/LiCl at elevated temperatures needs to be considered here (Potthast et al. 2002; Chrapava et al. 2003).
Tritylation of mercerized and regenerated celluloses in pyridine as a reaction medium has also been described (Kern et al. 2000; Li et al. 2008). Recently, also ionic liquids were exploited as the reaction media for homogeneous tritylation (Erdmenger et al. 2007; Granström et al. 2008). With regard to later deprotection, treatment with concentrated aqueous HCl (Kaida and Okamoto 1993; Kern et al. 2000; Li et al. 2008; Shen et al. 2018) or gaseous HCl (Acemoglu et al. 1998) are the most common methods to cleave off the protecting group. Also here, cellulose degradation by hydrolytic cleavage of the glycosidic bonds under strongly acidic conditions has to be taken into account (Kontturi et al. 2016).
The substituent at C6 of the glucopyranose units of the polysaccharide backbone was shown to have a significant influence on the overall chiral recognition (Kaida and Okamoto 1993; Chassaing et al. 1996; Acemoglu et al. 1998; Felix 2001). Besides the substituents and their pattern, the enantioseparation performance of a certain CS significantly depends on the nature of the polysaccharide, its degree of polymerization (DP) (Okada et al. 2016; Zhang et al. 2020), the amount of selector coated onto the silica support, and the used coating/immobilization procedures (Yashima et al. 1996; Wei et al. 2019). Evidently, there is also an influence of the dimensions of the silica gel (Yashima et al. 1996; Qin et al. 2010; Bezhitashvili et al. 2017; Kohout et al. 2019), the analytes, and of course the HPLC conditions (Yashima et al. 1996; Bui et al. 2021) as it is also in non-chiral separations.
Carbonate aminolysis was shown to be an efficient path towards the introduction of carbamate functionalities onto cellulose (Elschner et al. 2013; Ganske and Heinze 2018; Bui et al. 2022c), besides the routinely used isocyanate chemistry (Hearon et al. 1943a; Hearon et al. 1943c; Heard and Suedee 1996; Khan et al. 2008; Labafzadeh et al. 2014). However, the former way offers higher flexibility due to the greater availability of amines compared to the rather limited choice of available isocyanates. Different phenyl chloroformates have been evaluated as reagents for cellulose aryl carbonate synthesis (Ganske and Heinze 2018; Bui et al. 2022c), which were shown to be more reactive than the respective alkyl carbonates (Elschner and Heinze 2015). For details on carbonate aminolysis for obtaining cellulose carbamates see the comprehensive studies by Heinze et al. and others (Pourjavadi et al. 2011; Elschner et al. 2013; Elschner and Heinze 2015; Ganske and Heinze 2018; Bui et al. 2022c).
In the present work, we describe the regioselective synthesis and testing of hetero-substituted mixed cellulose 3,5-dimethylphenyl and α-phenylethyl carbamate chiral selectors with a special focus on the critical aspects of tritylation and detritylation.
Materials and methods
Materials
Cellulose I (Avicel® PH-101, MCC), p-cymene (99%), and acetic acid (> 99%) were purchased from Sigma-Aldrich (Schnelldorf, Germany). The MCC was dried at 40 °C in a vacuum oven for at least 2 days before use. 4-Methoxytrityl chloride (97%) was purchased from ABCR GmbH (Karlsruhe, Germany). 3,5-Dimethylphenyl isocyanate (> 98%), (3-aminopropyl)triethoxysilane (> 98%), phenyl chloroformate (> 98%), (R)-(+)-α-methylbenzylamine (> 99%), and (S)-(−)-α-methylbenzylamine (> 98%) were purchased from TCI Europe N.V. (Zwijndrecht, Belgium). All organic solvents for the synthesis, such as N,N-dimethylacetamide (DMAc), N,N-dimethylformamide (DMF), tetrahydrofuran (THF), pyridine, etc., were of reagent grade and dried with 3 Å molecular sieves (Sigma-Aldrich, Schnelldorf, Germany) for at least 3 days before use. Ethanol (EtOH) for precipitation and washing was of technical grade and obtained from Carl Roth GmbH + Co. KG or Fisher Scientific (Vienna, Austria). HPLC silica gel (7 μm, 1000 Å) was obtained from Daisogel Osaka Soda Co., Ltd. (Japan). TLC silica gel 60 RP-18 F254s was purchased from Merck KGaA (Germany). Empty stainless steel HPLC columns (150 × 4 mm, i.d.) and column hardware were purchased from Bischoff Analysentechnik u. -geräte GmbH (Leonberg, Germany). The HPLC solvents n-hexane (95%), 2-propanol (99.9%), and methanol (98%) were obtained from Fisher Scientific. A commercial CHIRALCEL® OD column (10 μm silica gel, 250 × 4.6 mm, i.d.) from Chiral Technologies Europe SAS (Illkirch Cedex, France) was used for comparative purposes. α-Methyl-D,L-phenylalanine methyl ester (98%, a) and Tröger’s base (98%, b) were purchased from Sigma-Aldrich (Schnelldorf, Germany). 2-Phenylcyclohexanone (> 98%, c), benzoin (> 98%, d), Pirkle’s alcohol (> 99%, e), and trans-stilbene oxide (98%, f) were purchased from TCI Europe N.V. (Zwijndrecht, Belgium).
Instrumentation
Solid-state 13C CP/MAS (12 kHz) and liquid-state 13C NMR experiments were carried out on Avance III HD and Avance II 400 instruments (Bruker, Rheinstetten, Germany) with resonance frequencies of 100.68 MHz and 100.62 MHz, respectively. Data processing was performed with TopSpin 3.6.2 and ACD/NMR Processor Academic Edition 12.01 software. Chemical shifts (δ) are given in ppm. ATR-FTIR spectra were recorded on a Frontier IR Single-Range spectrometer (PerkinElmer, Waltham, Massachusetts, US) equipped with a diamond/ZnSe crystal, LiTaO3 detector, and KBr windows. FTIR spectra were evaluated using SpectraGryph software (version v1.2.15). UV/Vis measurements were carried out on a PerkinElmer Lambda 35 spectrophotometer (PerkinElmer, Waltham, Massachusetts, US). GPC analyses were performed according to a standard procedure (Henniges et al. 2011; Jusner et al. 2022).
An Agilent Technologies, Inc. (Santa Clara, CA, USA) 1100 HPLC apparatus equipped with a quaternary pump (G1311A), autosampler (G1313A), TCC (G1316A), and DAD (G1315A) was used to evaluate the enantioseparation performance of the chiral columns. OpenLab CDS software (Agilent) was used for chromatographic data processing and evaluation.
Elemental analyses were carried out at the microanalytical laboratory of the University of Vienna on a EURO EA 3000 CHNS-O instrument (HEKAtech, Wegberg, Germany) with halide contents being determined by argentometry.
Syntheses
General information
In the case of the homogeneous reactions, the reactants were slowly added after the cellulose derivatives have been intensively mixed with the respective organic solvent mixtures at room temperature (RT) until a clear solution was formed. All reactions were performed under an inert atmosphere of dry nitrogen at the specified conditions. The precipitation of the crude products, vacuum filtration, and washing steps were repeated several times. Deionized water was used for washing. The purified cellulose derivatives were dried in a vacuum oven at 40 °C for at least 2 days before use. Any further variations or additional steps are described below.
Synthesis of the reference CS cellulose tris(3,5-dimethylphenyl carbamate) 1
Cellulose tris(3,5-dimethylphenyl carbamate) as a reference CS was synthesized according to the protocols of (Okamoto et al. 1984) and (Miaomiao et al. 2017). Yield: 5.73 g, 77 wt%.
Synthesis of the CSs 6 R and 6 S
Cellulose II preparation
The cellulose II allomorph was prepared based on the protocols of Li et al. (2008), Kern et al. (2000) with modifications. MCC (10 g) was dispersed and vigorously stirred in 25% aq. NaOH (180 mL) in a closed vessel at RT for 48 h. The mixture was then added to EtOH (240 mL) and stirred for 1 h. Cellulose II was collected by vacuum filtration and washed with a large excess of EtOH. Immersion in EtOH and filtration/washing were repeated. Cellulose II was collected by vacuum filtration, washed with a large excess of distilled water for quantitative removal of residual NaOH until neutral and dried in a vacuum oven at 40 °C for 2 days.
Synthesis of cellulose 6-(4-methoxytrityl ether) 2
Dried cellulose II (7 g) was immersed and vigorously stirred in anhydrous pyridine (140 mL). The suspension was stirred at 100 °C for 8 h. A solution of 4-methoxytrityl chloride (2.5 molar eq. relative to the glucopyranose repeating units of cellulose) in anhydrous pyridine (112 mL) was slowly added. The mixture was stirred at 100 °C for 14 h, and the clear, yellowish solution was allowed to cool to RT. A large excess of EtOH was used to precipitate crude cellulose derivative 2, which was collected by vacuum filtration, washed with a large excess of EtOH, and distilled water (two times each) and dried at 40 °C in a vacuum oven for 2 days. The crude product was purified by redissolution in pyridine, reprecipitation, filtration, and washing as described above. Yield: 18.07 g, 96 wt%.
Synthesis of cellulose 2,3-bis(3,5-dimethylphenyl carbamate)-6-(4-methoxytrityl ether) 3
Cellulose derivative 2 (15 g) was dissolved in anhydrous pyridine (150 mL). 3,5-Dimethylphenyl isocyanate (4 molar eq. relative to the repeating units of cellulose derivative 2) was added dropwise and the mixture was stirred at 110 °C for 18 h. The solution was allowed to cool to RT. The crude cellulose derivative 3 was precipitated in a large excess of EtOH and collected by vacuum filtration. The filter cake was washed with a large excess of EtOH and distilled water (two times each). The material was dried at 40 °C in a vacuum oven for 2 days and further purified by redissolution in pyridine, reprecipitation, filtration, and washing as described above. Yield: 24.54 g, 98 wt%.
Synthesis of cellulose 2,3-bis(3,5-dimethylphenyl carbamate) 4
Homogeneous detritylation was performed according to the protocols of Li et al. (2008) and Kern et al. (2000) with modifications. Cellulose derivative 3 (14 g) was dissolved in THF (280 mL). A mixture of concentrated aq. HCl (37 wt%, 63 mL) and THF (980 mL) was slowly added and the mixture was stirred at RT for 4 h. The reaction was then quenched by the addition of solid Na2CO3/K2CO3 until pH 7. Any formed salt was separated by vacuum filtration through a sintered glass frit. The THF in the filtrate was evaporated for the most part by rotary evaporation under reduced pressure. The crude cellulose derivative 4 was precipitated in a large excess of EtOH and collected by vacuum filtration, washed with a large excess of EtOH and distilled water (two times each), and dried at 40 °C in a vacuum oven for 2 days. The purification procedure was repeated after redissolution in THF. Yield: 8.34 g, 95 wt%.
Synthesis of cellulose 2,3-bis(3,5-dimethylphenyl carbamate)-6-(phenyl carbonate) 5
Cellulose derivative 4 (7 g) was dissolved in a mixture of anhydrous DMAc and pyridine (140 mL, 9:1, v/v). The solution was cooled to 0 °C by an ice/water bath. Phenyl chloroformate (2 molar eq. relative to the repeating units of cellulose derivative 4) was added dropwise. The reaction mixture was stirred for 12 h at 0 °C. A large excess of EtOH was employed to precipitate crude compound 5, which was collected by vacuum filtration, washed with a large excess of EtOH and distilled water (two times each), and dried at 40 °C in a vacuum oven for 2 days. Further purification of cellulose derivative 5 was carried out by repeating the above sequence after redissolution in DMAc. Yield: 8.74 g, 99 wt%.
Synthesis of cellulose 2,3-bis(3,5-dimethylphenyl carbamate)-6-(R/S-α-phenylethyl carbamate) 6 R and 6 S
The cellulose derivative 5 (2.5 g) was dissolved in anhydrous DMF (50 mL). Either (R)-(+)-α-methylbenzylamine or (S)-(−)-α-methylbenzylamine (5 molar eq. relative to the repeating units of cellulose derivative 5) was added dropwise, and the mixture was stirred at 50 °C for 24 h. The reaction mixture was allowed to cool to RT, and a large excess of EtOH was used to precipitate crude cellulose derivative 6R or 6S, which was then collected by vacuum filtration, washed with a large excess of EtOH and distilled water (two times each), and dried in a vacuum oven at 40 °C for 2 days. Purification of cellulose derivatives 6R and 6S was carried out by redissolution in DMF and repetition of the sequence above. Yields: 6R: 2.45 g, 94 wt%; 6S: 2.36 g, 90 wt%.
Synthesis of 3-aminopropyl-functionalized silica gel APS
Previously published procedures were adapted to synthesize 3-aminopropyl-functionalized silica gel (APS) as an inert carrier for the CSs (Engelhardt and Orth 1987; Yashima et al. 1996; Okada et al. 2016; Bui et al. 2022a). Silica gel (10 g) was immersed in toluene (200 mL) during mechanical stirring, and the mixture was dried by azeotropic distillation in a dry nitrogen atmosphere. After distilling off about half of the volume of toluene, the temperature of the suspension was reduced to 80 °C, and triethylamine (1 mL) was added as a catalyst, followed by the addition of (3-aminopropyl)triethoxysilane (APTES, 10 mL). The reaction mixture was mechanically stirred for 48 h. The suspension was then cooled to RT and the crude APS was collected by vacuum filtration through a sintered glass frit (DURAN®, porosity 4), washed with hot toluene (100 mL), EtOH (100 mL), and with distilled water (two times each with 200 mL). APS was then dried at 40 °C in a vacuum oven for 2 days. Yield: 9.96 g.
Silica coating and column packing
The coating amount of each CS (1, 6R, and 6S) on APS was 20 wt% (Zhang and Francotte 1995). The coating was performed according to a previously published protocol (Bui et al. 2022a). Column packing was carried out as described in Bui et al. (2022a, b).
HPLC method
The columns packed with modified silica gel—which was previously coated with cellulose derivatives/selectors 1 (CSP1), 6R (CSP6R), and 6S (CSP6S)—were rinsed with n-hexane/isopropanol in a stepwise gradient (30:70, 60:40, 90:10, v/v) before use. The concentration of the analytes was 1 mg/mL, the injection volume was 5 µL, and the flow rate was 1 mL/min. The detection wavelength was set to 254 nm. p-Cymene was used for dead time determination (t0). All measurements were carried out in triplicate.
Results and discussion
Synthesis part
Cellulose tris(3,5-dimethylphenyl carbamate) 1
The reference CS, i.e., cellulose tris(3,5-dimethylphenyl carbamate), was synthesized (see Fig. 1A) and analytically characterized by ATR-FTIR, solid-state 13C NMR, GPC, and EA. The solid-state 13C NMR spectra of cellulose derivative 1 versus the starting material (MCC) are shown in Fig. 2, the EA results of all reported cellulose derivatives are summarized in Table 1. The FTIR and solid-state 13C NMR spectra of cellulose derivative 1 were in full agreement with literature data (Liu et al. 2013; Wei et al. 2019; Bui et al. 2022a; Bui et al. 2022b). Cellulose derivative 1 is a well-known CS in chiral HPLC column materials and has been commercialized by several companies. However, we had to synthesize this reference CS in-house to compensate for differences in silica gel dimensions, cellulose starting material, column dimensions, coating procedure, and packing procedure—allowing for a fairer comparison between this reference and the novel CSs 6R and 6S described in this work. The DS of 3,5-dimethylphenyl carbamate substituents calculated based on the N content obtained by EA was 2.93 (98%). The molecular weight and dispersity of cellulose derivative 1 were 172.3 kDa (Mw) and 1.70 (Đ), respectively.
Influence of the cellulose allomorph on the tritylation reaction
In preliminary experiments, we observed that the heterogeneous tritylation of microcrystalline cellulose (= cellulose I) was less successful due to the high crystallinity of the starting material. Therefore, either the crystallinity had to be significantly reduced, or a homogeneous reaction was needed. For decreasing crystallinity and increasing accessibility, we chose mercerization and the resulting formation of cellulose II over extensive milling, to avoid tribochemical side reactions and cellulose degradation (Revol et al. 1987). Instead of dissolving cellulose I (MCC) in DMAc/LiCl at high temperature and accompanying degradation issues during heating (Potthast et al. 2002; Chrapava et al. 2003), the obtained cellulose II allomorph was used as the alternative starting material for tritylation.

A Chemical synthesis of cellulose tris(3,5-dimethylphenyl carbamate) and B synthesis pathway towards the novel chiral selectors cellulose 2,3-bis(3,5-dimethylphenyl carbamate)-6-(R/S-α-phenylethyl carbamate) 6R and 6S. Conditions: (a) pyridine, 110 °C, 24 h, N2; (b) pyridine, 100 °C, 14 h, N2; (c) pyridine, 110 °C, 18 h, N2; (d) THF, RT, 4 h, N2; (e) DMAc/pyridine (9:1, v/v), 0 °C, 12 h, N2; (f) DMF, 50 °C, 24 h, N2
Cellulose II was prepared according to the protocols of Li et al. (2008) and Kern et al. (2000) with adjustments towards the complete removal of NaOH. The material was characterized by solid-state 13C NMR (see Fig. S1). In the solid-state 13C NMR spectrum of cellulose II, the decrease in intensity of the resonance at 88.8 ppm assigned to the crystalline region at C4 indicated that the degree of crystallinity was significantly lower compared to the starting material (Park et al. 2009; Kim et al. 2013; Halonen et al. 2013; El-Kott et al. 2019).

Solid-state 13C NMR spectra of cellulose derivative 1 (red) versus microcrystalline cellulose (MCC, blue). The chemical structure of compound 1 is shown on the right
Cellulose 6-(4-methoxytrityl ether) 2
Several tritylating reagents can be used to protect C6-OH of cellulose: 4,4′-dimethoxytriphenylmethyl chloride, 4-methoxytrityl chloride, and trityl chloride. The methoxy-substituted trityl chlorides were shown to have a higher reactivity in comparison to unsubstituted trityl chloride. Under otherwise similar reaction conditions, a DS of 0.96 was achieved during 4 h of reaction time with 4-methoxytrityl chloride as a reactant, whereas 24 h were required in the case of trityl chloride. Furthermore, the detritylation time in the case of the methoxy-substituted trityl ether groups was only 5.5 h, while about 100 h were required for unsubstituted trityl ether groups. 4,4′-Dimethoxytriphenylmethyl chloride was less regioselective than the other two other trityl chlorides (Heinze et al. 1994; Gömez et al. 1996).
For tritylation, any residual NaOH from mercerization needed to be removed to ensure a high yield of tritylation, fewer by-products, and a short reaction time. A treatment of cellulose II in pyridine at 100 °C for 8 h was applied to additionally swell the starting material before its reaction with 4-methoxytrityl chloride (Mantanis et al. 1995). Through this pre-treatment, a comparably short reaction time (14 h) and high DS (0.98 by mean of UV/Vis measurement and 0.99 by EA, see also Supplementary Information) was achieved, which is an improvement compared to previously reported protocols (about 70–100 h) with DS of 0.97 and 0.93 (Kern et al. 2000; Li et al. 2008). The UV/Vis method to determine the DS of 4-methoxytrityl ether was modified based on the protocol of (Gömez et al. 1996) (see Method S1 in the Supplementary Information).
The FTIR and solid-state 13C NMR spectra of cellulose derivative 2 versus cellulose II are shown in Figs. S2 and S3. The FTIR spectrum of cellulose derivative 2 was consistent with literature data (Kern et al. 2000; Erdmenger et al. 2007) showing a decrease in the intensity of OH bands at 3467 cm− 1 and the presence of bands assigned to the aromatic ring, especially the vibrations of CH3–O–Ph at 1507 and 1251 cm− 1. In the solid-state 13C NMR spectrum of cellulose derivative 2, the resonances of the aromatic ring at 158.5, 147.8, 141.2, 127.5, 113.6, the quaternary carbon (C7) at 86.1, and the O–CH3 groups (C12) at 54.7 ppm confirmed successful tritylation. Cellulose derivative 2 was soluble in DMAc, DMF, 1,4-dioxane, THF, and acetone, which was also in agreement with the report of Gömez et al. (1996).
Cellulose 2,3-bis(3,5-dimethylphenyl carbamate)-6-(4-methoxytrityl ether) 3
Carbamoylation with isocyanates was employed to introduce 3,5-dimethylphenyl carbamate substituents at the free C2- and C3-OH groups of the glucopyranose units after tritylation/protection of C6-OH. The FTIR and solid-state 13C NMR spectra of the corresponding cellulose derivative 3 versus 2 are shown in Figs. S4 and S5. The absence of OH bands around 3467 and the presence of amide (N-H) vibrations at 3395, carbonyl (C=O) vibrations at 1723, aromatic ring signals at 1613, 1539, 1447, 836, and the C-O band at 1249 cm− 1 in the FTIR spectrum of cellulose derivative 3 indicated a successful reaction, supported by the solid-state 13 C NMR spectrum with the carbonyl group resonance (C17) at 151.9, CH3-Ph peak (C22) at 20.8, and aromatic carbon signals of 3,5-dimethylphenyl carbamate at 137.6 and 115.1 ppm. The DS of 3,5-dimethylphenyl carbamate calculated based on the N content of the EA results was 1.84 (92%).
Cellulose 2,3-bis(3,5-dimethylphenyl carbamate) 4
For subsequent detritylation, to liberate the protected C6-OH, different concentrations of HCl were suggested in the literature. The detritylation time had to be optimized and kept to a minimum, in order to limit cellulose degradation by acidic hydrolysis (Kontturi et al. 2016), and also (minor) decarbamoylation (Acemoglu et al. 1998). A homogeneous detritylation approach was used and the minimum reaction time—which has not been specified in previous works—was determined by a simple and fast thin-layer chromatography (TLC) method based on a protocol by Gömez et al. (1996) (Method S2). The TLC chromatograms are shown in Fig. S6. After 4 h of detritylation, the yellow color of the 4-methoxytrityl cations disappeared at the starting point of the TLC plate, indicating that the detritylation has been completed.
The FTIR and solid-state 13C NMR spectra of cellulose derivative 4 versus 3 are shown in Figs. S7 and S8, respectively. The absence of bands assigned to CH3–O–Ph of the 4-methoxytrityl ether at 1510, 1248, and the recurrence of OH bands at 3467 cm− 1 in the FTIR spectrum of cellulose derivative 4 supported the success of the detritylation. Also, the solid-state 13C NMR spectrum of compound 4 showed a nearly complete absence of the 4-methoxytrityl group resonances (C11 at 158.8, C7 at 86.1, and C12 at 54.5 ppm), even though a minute peak of the O-CH3 groups (C12 at 54.5 ppm) remained still visible (see Fig. S8), which indicated a minor number of 4-methoxytrityl groups still to be present. Therefore, a quantitative UV/Vis method was applied to determine the exact detritylation yield (Method S3), which was 96%, in agreement with the solid-state 13C NMR result. The DS of 3,5-dimethylphenyl carbamate based on the N content was still 1.84 (92%) —so the homogeneous detritylation during 4 h evidently did not affect the carbamate substituents at C2 and C3.
Cellulose 2,3-bis(3,5-dimethyl phenyl carbamate)-6-(phenyl carbonate) 5
Cellulose derivative 5 was synthesized by oxycarbonylation of C6-OH of cellulose derivative 4 with phenyl chloroformate to prepare a precursor, which subsequently was to be converted into cellulose derivatives 6R and 6S by carbonate aminolysis. Phenyl chloroformate was chosen as a reagent due to its higher reactivity compared to p-nitrophenyl chloroformate (Bui et al. 2022c; Bui et al. 2022a). FTIR and solid-state 13C NMR spectra of cellulose derivatives 5 versus 4 are shown in Fig. S9 and Fig. S10, respectively. The FTIR spectrum of cellulose derivative 5 again shows the absence of OH bands indicating full conversion of C6–OH. Also, the shift of the C=O band from 1723 (only carbamate vibrations) to 1747 (mixture of carbamate and carbonate), and an increase in the intensity of the C–O vibration at 1208 cm− 1 (carbamate and carbonate) indicated that oxycarbonylation was successful. In solid-state 13 C NMR, the additional resonances of C25 at 120.7 and C26 at 129.4 ppm, and the chemical shift of C6 moving from 61.4 to 67.2 ppm in the carbonate derivative are supportive as well (Bui et al. 2022a). The DS of phenyl carbonate was 0.97 (see Supplementary Information for the calculation).
Cellulose 2,3-bis(3,5-dimethylphenyl carbamate)-6-(R/S-α-phenylethyl carbamate) 6 R and 6 S
Aminolysis of the previously installed carbonate moieties with enantiopure (R)- and (S)-α-phenylethylamine was used to convert cellulose derivative 5 into the corresponding C6-(R/S-α-phenylethyl carbamates) yielding derivatives 6R and 6S as the final CSs (see Fig. 1B). With this synthesis sequence tritylation–carbamoylation–detritylation–oxycarbonylation–aminolysis in hand, it is now possible to increase the number of carbamate substituents for CSs significantly.

Solid-state 13C NMR spectra of cellulose derivative 6R (blue) versus 5 (red)
Aromatic amines cannot be used for the aminolysis of the carbonates due to their insufficient nucleophilicity (Elschner et al. 2013). However, primary and even secondary alkyl amines, being bulkier but usually also more nucleophilic, can be used in this way for obtaining carbamates by aminolysis, instead of the limited array of conventional, commercially available isocyanates.
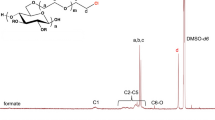
The FTIR spectra of cellulose derivatives 6R and 6S versus 5 are shown in Fig. S11 and Fig. S12, respectively, and the corresponding solid-state 13C NMR spectra in Fig. 3 and Fig. S13. The presence of a new band assigned to the N–H group at approx. 3390, the chemical shift of the C=O signal from 1747 (overlay of carbamate and carbonate vibrations) to approx. 1710 cm− 1 (only carbamate band), the decrease in the intensity of the C–O band at 1218 cm− 1 due to the loss of the phenyl carbonate moiety in the FTIR spectra of cellulose derivatives 6R and 6S support the expected structure, as do the additional resonances in the 13C solid-state NMR spectra, assigned to C23 at 154.8, C30 at 144.4, C32 at 127.5, C28 at 50.8 + 43.9, and C29 at 26.0 ppm. Liquid-state 13C NMR in pyridine-d5 as NMR solvent was also further employed to evaluate the chemical structure of cellulose derivatives 6R and 6S (see Figs. S14 and S15). Due to rotational isomerism at the amide bond, C28 appeared at two resonances at approx. 52 and 44 ppm in the solid-state 13C NMR spectra, while there was only one signal at 52.0 ppm in the solution-state 13C NMR spectra. Apparently, the rotational barrier was already overcome in solution at the measurement temperature (28.1 °C). The DS of R- and S-phenylethyl carbamate calculated based on the N content of the EA results were 0.84 and 0.87, respectively. The molecular weights and dispersity of cellulose derivatives 6R and 6S were 123.3 and 123.2 kDa, as well as 1.42 and 1.45 (Ɖ), respectively.
Enantioseparation evaluation
Six racemate pairs (α-methyl-D,L-phenylalanine methyl ester (a), Tröger’s base (b), 2-phenyl cyclohexanone (c), benzoin (d), Pirkle’s alcohol (e), and trans-stilbene oxide (f), Fig. 4) were used to exemplarily showcase the enantioseparation performance of the newly developed column materials (CSP6R and CSP6S) in comparison to a reference column (CSP1) and a commercial stationary phase (CHIRALCEL® OD). The last two were based on cellulose tris(3,5-dimethylphenyl carbamate) as a CS.

The chemical structures of the chiral analytes a–f
The chromatographic retention index (k1), selectivity (α), and resolution (Rs) data of the chiral analytes for all tested CSPs are summarized in Fig. 5 and Table S1. Exemplary HPLC chromatograms of the analytes b and d are shown in Fig. 6.

A Selectivity (α) and B resolution (Rs) data of chiral analytes a–f on CSP6R and CSP6S in comparison to CSP1 (an in-house prepared reference) and a commercially available CHIRALCEL® OD column (same chiral selector structure as CSP1)
All of the selected chiral analytes were (at least partially) separated on both CSP1 and CHIRALCEL® OD. Especially, the chiral analytes d, e, and f were baseline-separated with Rs > 1.5, while the analytes a, b, and c were partly separated. The selectivity of CSP1 was higher than that of CHIRALCEL® OD for the chiral analytes a, b, c, e, and f, while the resolution on CHIRALCEL® OD was higher for analytes d and f. Overall, the resolution values of the reference CSP1 and the commercial column were quite comparable. The CHIRALCEL® OD column was used in this study as a benchmark column. However, differences in silica gel dimensions (10 vs. 7 μm) and column dimensions (250 × 4.6 vs. 150 × 4 mm) with respect to the in-house prepared material have to be considered, especially in terms of column efficiency.

Comparison of chiral columns with different CSs. HPLC chromatogram for A chiral analyte b and B chiral analyte d
Regarding the overall enantioseparation performance of CSP6R and CSP6S, all of the selected chiral analytes were at least partially separated with Rs < 1.5 (see Fig. 5 and Table S1) except analyte f on CSP6S. Though the selectivity values (α) for all chiral analytes were rather similar, the resolution values of CSP6R were always higher than those of CSP6S. A somewhat higher resolution on CSP6R than on CSP1 was observed for chiral analytes a and b.
Conclusions and outlook
The critical synthetic aspects of tritylation and detritylation for obtaining hetero-substituted cellulose derivatives to be used as chiral selectors in HPLC after coating onto silica gel were thoroughly optimized. The benefit of using cellulose II as a starting material was also elaborated. In general, the newly developed CSPs have shown medium enantioseparation performance, although no clear trends can be identified at the moment and further evaluation with a bigger set of chiral analytes is necessary. Also, further tests with smaller silica particles and pore sizes for enhancing the column efficiency and overall resolution are needed (ongoing work) as well as the preparation of other CSs by using different chiral amines. The present work has laid the foundation for this endeavor, by providing an optimized synthesis methodology and improved protocols for tritylation/detritylation and cellulose carbonate aminolysis, which we hope will be found useful.
Data availability
Data and materials are available from the authors upon request.
References
Acemoglu M, Küsters E, Baumann J, Hernandez I, Pong-Mak C (1998) Synthesis of regioselectively substituted cellulose derivatives and applications in chiral chromatography. Chirality 10(4):294–306
Ahuja S (1997) Chiral separations and technology: an overview. In: Ahuja S (ed) Chiral separations: applications and technology. American Chemical Society, Washington
Ali I, Saleem K, Hussain I, Gaitonde VD, Aboul-Enein HY (2009) Polysaccharides chiral stationary phases in liquid chromatography. Sep Purif Rev 38(2):97–147
Bezhitashvili L, Bardavelidze A, Ordjonikidze T, Chankvetadze L, Chity M, Farkas T, Chankvetadze B (2017) Effect of pore-size optimization on the performance of polysaccharide-based superficially porous chiral stationary phases for the separation of enantiomers in high-performance liquid chromatography. J Chromatogr A 1482:32–38
Bui CV, Rosenau T, Hettegger H (2021) Polysaccharide-and β-Cyclodextrin-based chiral selectors for enantiomer resolution: recent developments and applications. Molecules 26(14):4322
Bui CV, Rosenau T, Hettegger H (2022a) Synthesis by carbonate aminolysis and chiral recognition ability of cellulose 2,3-bis(3,5-dimethylphenyl carbamate)-6-(α-phenylethyl carbamate) selectors. Cellulose. https://doi.org/10.1007/s10570-022-04898-8
Bui CV, Rosenau T, Hettegger H (2022b) Immobilization of a cellulose carbamate-type chiral selector onto silica gel by alkyne-azide click chemistry for the preparation of chiral stationary chromatography phases. Cellulose. https://doi.org/10.1007/s10570-022-04932-9
Bui CV, Rosenau T, Hettegger H (2022c) Synthesis of polyanionic cellulose carbamates by homogeneous aminolysis in an ionic liquid/DMF medium. Molecules 27(4):1384
Chankvetadze B (2012) Recent developments on polysaccharide-based chiral stationary phases for liquid-phase separation of enantiomers. J Chromatogr A 1269:26–51
Chankvetadze B (2020) Recent trends in preparation, investigation, and application of polysaccharide-based chiral stationary phases for separation of enantiomers in high-performance liquid chromatography. Trends Anal Chem 122:115709
Chassaing C, Thienpont A, Félix G (1996) Regioselective carbamoylated and benzoylated cellulose for the separation of enantiomers in high-performance liquid chromatography. J Chromatogr A 738(2):157–167
Chassaing C, Thienpont A, Soulard MH, Felix G (1997) Regioselectivity carbamoylated polysaccharides for the separation of enantiomers in high-performance liquid chromatography. J Chromatogr A 786(1):13–21
Chrapava S, Touraud D, Rosenau T, Potthast A, Kunz W (2003) The investigation of the influence of water and temperature on the LiCl/DMAc/cellulose system. Phys Chem Chem Phys 5(9):1842–1847
El-Kott A, Syef AFA, Alshehri MA, Al Dessouky SI, Keshk SMAS (2019) Suppression efficacy of lignosulfonate/mercerized cotton fiber composite against cancer cell’s activities. Adv Compos Lett 28:0963693519875974
Elschner T, Heinze T (2015) Cellulose carbonates: a platform for promising biopolymer derivatives with multifunctional capabilities. Macromol Biosci 15(6):735–746
Elschner T, Ganske K, Heinze T (2013) Synthesis and aminolysis of polysaccharide carbonates. Cellulose 20(1):339–353
Engelhardt H, Orth P (1987) Alkoxy silanes for the preparation of silica based stationary phases with bonded polar functional groups. J Liq Chromatogr 10(8–9):1999–2022
Erdmenger T, Haensch C, Hoogenboom R, Schubert US (2007) Homogeneous tritylation of cellulose in 1-butyl‐3‐methylimidazolium chloride. Macromol Biosci 7(4):440–445
Fanali C, D’Orazio G, Gentili A, Fanali S (2019) Analysis of enantiomers in products of food interest. Molecules 24(6):1119
Felix G (2001) Regioselectively modified polysaccharide derivatives as chiral stationary phases in high-performance liquid chromatography. J Chromatogr A 906(1–2):171–184
Ganske K, Heinze T (2018) Evaluation of the synthesis of soluble aromatic cellulose carbonates of low degree of substitution. Macromol Chem Phys 219(15):1800152
Gömez JAC, Erler UW, Klemm DO (1996) 4-methoxy substituted trityl groups in 6‐O protection of cellulose: homogeneous synthesis, characterization, detritylation. Macromol Chem Phys 197(3):953–964
Granström M, Kavakka J, King A, Majoinen J, Mäkelä V, Helaja J, Hietala S, Virtanen T, Maunu SL, Argyropoulos DS (2008) Tosylation and acylation of cellulose in 1-allyl-3-methylimidazolium chloride. Cellulose 15(3):481–488
Halonen H, Larsson PT, Iversen T (2013) Mercerized cellulose biocomposites: a study of influence of mercerization on cellulose supramolecular structure, water retention value and tensile properties. Cellulose 20(1):57–65
Heard CM, Suedee R (1996) Stereoselective adsorption and trans-membrane transfer of propranolol enantiomers using cellulose derivatives. Int J Pharm 139(1–2):15–23
Hearon WM, Hiatt GD, Fordyce CR (1943a) Carbamates of cellulose and cellulose acetate. I. Preparation. J Am Chem Soc 65(5):829–833
Hearon WM, Hiatt GD, Fordyce CR (1943b) Cellulose trityl ether. J Am Chem Soc 65(12):2449–2452
Hearon WM, Hiatt GD, Fordyce CR (1943c) Carbamates of cellulose and cellulose acetate. II. Stability toward Hydrolysis. J Am Chem Soc 65(5):833–836
Heinze T, Röttig K, Nehls I (1994) Synthesis of 2,3-O‐carboxymethylcellulose. Macromol Rapid Commun 15(4):311–317
Henniges U, Kostic M, Borgards A, Rosenau T, Potthast A (2011) Dissolution behavior of different celluloses. Biomacromolecules 12(4):871–879
Ikai T, Okamoto Y (2009) Structure control of polysaccharide derivatives for efficient separation of enantiomers by chromatography. Chem Rev 109(11):6077–6101
Jusner P, Bausch F, Schiehser S, Schwaiger E, Potthast A, Rosenau T (2022) Protocol for characterizing the molar mass distribution and oxidized functionality profiles of aged transformer papers by gel permeation chromatography (GPC). Cellulose 29(4):2241–2256
Kaida Y, Okamoto Y (1993) Optical resolution on regioselectively carbamoylated cellulose and amylose with 3,5-dimethylphenyl and 3,5-dichlorophenyl isocyanates. Bull Chem Soc Jpn 66(8):2225–2232
Katoh Y, Tsujimoto Y, Yamamoto C, Ikai T, Kamigaito M, Okamoto Y (2011) Chiral recognition ability of cellulose derivatives bearing pyridyl and bipyridyl residues as chiral stationary phases for high-performance liquid chromatography. Polym J 43(1):84–90
Kern H, Choi S, Wenz G, Heinrich J, Ehrhardt L, Mischnick P, Garidel P, Blume A (2000) Synthesis, control of substitution pattern and phase transitions of 2,3-di-O-methylcellulose. Carbohydr Res 326(1):67–79
Khan FZ, Shiotsuki M, Nishio Y, Masuda T (2008) Synthesis, characterization, and gas permeation properties of t-butylcarbamates of cellulose derivatives. J Membr Sci 312(1–2):207–216
Kim SH, Lee CM, Kafle K (2013) Characterization of crystalline cellulose in biomass: basic principles, applications, and limitations of XRD, NMR, IR, Raman, and SFG. Korean J Chem Eng 30(12):2127–2141
Kohout M, Hovorka Å, Herciková J, Wilk M, Sysel P, Izák P, Bartůněk V, von Baeckmann C, Pícha J, Frühauf P (2019) Evaluation of silica from different vendors as the solid support of anion-exchange chiral stationary phases by means of preferential sorption and liquid chromatography. J Sep Sci 42(24):3653–3661
Kontturi E, Meriluoto A, Penttilä PA, Baccile N, Malho JM, Potthast A, Rosenau T, Ruokolainen J, Serimaa R, Laine J (2016) Degradation and crystallization of cellulose in hydrogen chloride vapor for high-yield isolation of cellulose nanocrystals. Angew Chem Int Ed 55(46):14455–14458
Labafzadeh SR, Kavakka JS, Vyavaharkar K, Sievänen K, Kilpeläinen I (2014) Preparation of cellulose and pulp carbamates through a reactive dissolution approach. RSC Adv 4(43):22434–22441
Li Y, Liu R, Huang Y (2008) Synthesis and phase transition of cellulose-graft‐poly (ethylene glycol) copolymers. J Appl Polym Sci 110(3):1797–1803
Liu RQ, Bai LY, Zhang YJ, Zhang YP (2013) Green synthesis of a typical chiral stationary phase of cellulose tris(3,5-dimethylphenylcarbamate). Chem Cent J 7(1):1–5
Mantanis GI, Young RA, Rowell RM (1995) Swelling of compressed cellulose fiber webs in organic liquids. Cellulose 2(1):1–22
Miaomiao H, Xiaosa J, Han Y, Xiao L, Yadong L, Shengxiang J (2017) Controlled synthesis, immobilization and chiral recognition of carboxylic acid functionalized cellulose tris(3,5-dimethylphenylcarbamate). Carbohydr Polym 172:223–229
Minguillón C, Franco P, Oliveros L, López P (1996) Bonded cellulose-derived high-performance liquid chromatography chiral stationary phases I. Influence of the degree of fixation on selectivity. J Chromatogr A 728(1–2):407–414
Okada Y, Yamamoto C, Kamigaito M, Gao Y, Shen J, Okamoto Y (2016) Enantioseparation using cellulose tris(3,5-dimethylphenylcarbamate) as chiral stationary phase for HPLC: influence of molecular weight of cellulose. Molecules 21(11):1484
Okamoto Y (2009) Chiral polymers for resolution of enantiomers. Part A Polym Chem J Polym Sci 47(7):1731–1739
Okamoto Y, Kawashima M, Hatada K (1984) Chromatographic resolution. 7. Useful chiral packing materials for high-performance liquid chromatographic resolution of enantiomers: phenylcarbamates of polysaccharides coated on silica gel. J Am Chem Soc 106(18):5357–5359
Okamoto Y, Noguchi J, Yashima E (1998) Enantioseparation on 3,5-dichloro- and 3,5-dimethylphenyl carbamates of polysaccharides as chiral stationary phases for high-performance liquid chromatography. React Funct Polym 37(1–3):183–188
Okamoto Y, Sakamoto H, Hatada K, Irie M (1986) Resolution of enantiomers by HPLC on cellulose trans and cis-tris-(4-phenylazophenylcarbamate). Chem Lett 15(6):983–986
Park S, Johnson DK, Ishizawa CI, Parilla PA, Davis MF (2009) Measuring the crystallinity index of cellulose by solid state 13C nuclear magnetic resonance. Cellulose 16(4):641–647
Potthast A, Rosenau T, Sixta H, Kosma P (2002) Degradation of cellulosic materials by heating in DMAc/LiCl. Tetrahedron Lett 43(43):7757–7759
Pourjavadi A, Seidi F, Afjeh SS, Nikoseresht N, Salimi H, Nemati N (2011) Synthesis of soluble N-functionalized polysaccharide derivatives using phenyl carbonate precursor and their application as catalysts. Starch‐Stärke 63(12):780–791
Qin Q, Zhang S, Zhang WG, Zhang ZB, Xiong YJ, Guo ZY, Fan J, Run-Zheng S, Finlow D, Yin Y (2010) The impact of silica gel pore and particle sizes on HPLC column efficiency and resolution for an immobilized, cyclodextrin‐based, chiral stationary phase. J Sep Sci 33(17–18):2582–2589
Revol JF, Dietrich A, Goring DAI (1987) Effect of mercerization on the crystallite size and crystallinity index in cellulose from different sources. Can J Chem 65(8):1724–1725
Ribeiro J, Tiritan ME, Pinto MMM, Fernandes C (2017) Chiral stationary phases for liquid chromatography based on chitin-and chitosan-derived marine polysaccharides. Symmetry 9(9):190
Shen J, Okamoto Y (2016) Efficient separation of enantiomers using stereoregular chiral polymers. Chem Rev 116(3):1094–1138
Shen J, Wang F, Bi W, Liu B, Liu S, Okamoto Y (2018) Synthesis of cellulose carbamates bearing regioselective substituents at 2,3- and 6-positions for efficient chromatographic enantioseparation. J Chromatogr A 1572:54–61
Subramanian G (2008) Chiral separation techniques: a practical approach. Wiley, Weinheim
Teixeira J, Tiritan ME, Pinto MMM, Fernandes C (2019) Chiral stationary phases for liquid chromatography: recent developments. Molecules 24(5):865–903
Wang X, Li H, Quan K, Zhao L, Qiu H, Li Z (2021) Preparation and applications of cellulose-functionalized chiral stationary phases: a review. Talanta 225:121987
Wei Q, Su H, Gao D, Wang S (2019) HPLC with cellulose tris(3,5-dimethylphenyl carbamate) chiral stationary phase: influence of coating times and coating amount on chiral discrimination. Chirality 31(3):164–173
Yashima E, Sahavattanapong P, Okamoto Y (1996) HPLC enantioseparation on cellulose tris(3,5-dimethylphenylcarbamate) as a chiral stationary phase: influences of pore size of silica gel, coating amount, coating solvent, and column temperature on chiral discrimination. Chirality 8(6):446–451
Yin C, Chen W, Zhang J, Zhang M, Zhang J (2019) A facile and efficient method to fabricate high-resolution immobilized cellulose-based chiral stationary phases via thiol-ene click chemistry. Sep Purif Technol 210:175–181
Yin C, Zhang J, Chang L, Zhang M, Yang T, Zhang X, Zhang J (2019) Regioselectively substituted cellulose mixed esters synthesized by two-steps route to understand chiral recognition mechanism and fabricate high-performance chiral stationary phases. Anal Chim Acta 1073:90–98
Zhang GH, Xi JB, Chen W, Bai ZW (2020) Comparison in enantioseparation performance of chiral stationary phases prepared from chitosans of different sources and molecular weights. J Chromatogr A:461029
Zhang T, Francotte E (1995) Chromatographic properties of composite chiral stationary phases based on cellulose derivatives. Chirality 7(6):425–433
Zheng J, Bragg W, Hou J, Lin N, Chandrasekaran S, Shamsi SA (2009) Sulfated and sulfonated polysaccharide as chiral stationary phases for capillary electrochromatography and capillary electrochromatography–mass spectrometry. J Chromatogr A 1216(5):857–872
Acknowledgments
Peter Frühauf (University of Vienna) is gratefully acknowledged for his support with column packing.
Funding
Open access funding provided by University of Natural Resources and Life Sciences, Vienna (BOKU). The University of Natural Resources and Life Sciences, Vienna (BOKU) and the County of Lower Austria are acknowledged for their financial support in the framework of the “Austrian Biorefinery Center Tulln” (ABCT) and the BOKU doctoral school “Advanced Biorefineries: Chemistry & Materials” (ABC&M). The authors gratefully acknowledge the financial support by an Ernst Mach Grant (C.V.B., ASEA-UNINET, OeAD, ICM-2019-13801) and the Gesellschaft für Forschungsförderung Niederösterreich m.b.H. (H.H., Project LSC20-002).
Author information
Authors and Affiliations
Contributions
CVB, TR, and HH contributed to the study conception and design. Material preparation, data collection, and analysis were performed by CVB. The original draft of the manuscript was written by CVB. Review & editing by TR & HH Supervision, project administration, and funding acquisition by TR & HH. All authors commented on previous versions as well as read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors agreed to the publication in the submitted form.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bui, C.V., Rosenau, T. & Hettegger, H. Optimization of the regioselective synthesis of mixed cellulose 3,5-dimethylphenyl and α-phenylethyl carbamate selectors as separation phases for chiral HPLC. Cellulose 30, 2337–2351 (2023). https://doi.org/10.1007/s10570-022-05007-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-022-05007-5