
Abstract
Modern neuropathology serves a key function in the multidisciplinary management of brain tumor patients. Owing to the recent advancements in molecular neurooncology, the neuropathological assessment of brain tumors is no longer restricted to provide information on a tumor’s histological type and malignancy grade, but may be complemented by a growing number of molecular tests for clinically relevant tissue-based biomarkers. This article provides an overview and critical appraisal of the types of genetic and epigenetic aberrations that have gained significance in the molecular diagnostics of gliomas, namely deletions of chromosome arms 1p and 19q, promoter hypermethylation of the O6-methylguanine-methyl-transferase (MGMT) gene, and the mutation status of the IDH1 and IDH2 genes. In addition, the frequent oncogenic aberration of BRAF in pilocytic astrocytomas may serve as a novel diagnostic marker and therapeutic target. Finally, this review will summarize recent mechanistic insights into the molecular alterations underlying treatment resistance in malignant gliomas and outline the potential of genome-wide profiling approaches for increasing our repertoire of clinically useful glioma markers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gliomas are the most frequent primary brain tumors and include a variety of different histological tumor types and malignancy grades. Although the cellular origin of gliomas is still unknown, experimental data in mice suggest an origin from neoplastically transformed neural stem or progenitor cells. However, histological classification of gliomas essentially relies on morphological similarities of the tumor cells with non-neoplastic glial cells and the presence of particular architectural features; thereby, most gliomas can be classified as astrocytic, oligodendroglial, mixed oligo-astrocytic or ependymal tumors according to the criteria of the World Health Organization (WHO) classification of central nervous system tumors [81]. The astrocytic tumors are most common and include the most malignant type of glioma, the glioblastoma. Although ependymal tumors are often relatively circumscribed, most astrocytic, oligodendroglial and oligoastrocytic tumors in adults diffusely infiltrate the adjacent brain tissue [24]. Therefore, these latter neoplasms are often designated as ‘diffuse gliomas’. In contrast, the most frequent glioma in children, the pilocytic astrocytoma, is a low-grade (WHO grade I) neoplasm that usually shows limited infiltrative growth and does not progress to malignancy. In addition to these common glioma types, a number of rare, mostly low-grade malignant glioma entities and variants as well as several types of mixed glial and neuronal tumors may be observed, in particular in children and young adults. However, detailed discussion of these rare tumors is beyond the scope of this article, also because information about molecular alterations in these tumors is still limited and does not yet contribute to their clinical management.
Diffuse gliomas are one of the most devastating cancers because they often show locally aggressive behavior and cannot be cured by current therapies. Moreover, low-grade (WHO grade II) diffuse gliomas have a strong tendency for malignant progression to anaplastic (WHO grade III) gliomas and eventually secondary glioblastomas (WHO grade IV). An accurate distinction between the different glioma entities is important because of its strong prognostic and therapeutic implications. So far, histopathology is the gold standard for the typing and grading of gliomas. However, histological classification of gliomas is not trivial and associated with significant interobserver variability. Furthermore, the clinical behavior of individual tumors of a specific histopathological entity may substantially differ. Thus, additional markers are needed for a refined and more objective glioma classification, a better prediction of prognosis and a tailored therapeutic decision-making.
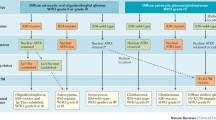
Like cancer in general, gliomas develop as a result of genetic alterations that accumulate with tumor progression. Knowledge of the genetic alterations in the various types and malignancy grades of gliomas has drastically increased over the past years (Fig. 1). Briefly, mutation of the tumor suppressor gene TP53 (located at 17p13.1) and loss of heterozygosity on chromosome arm 17p are found in more than half of the WHO grade II diffuse astrocytomas. In addition, gains on the long arm of chromosome 7 are often present. In contrast, oligodendroglial tumors frequently show combined losses of the short arm of chromosome 1 and of the long arm of chromosome 19. Oligoastrocytic neoplasms are genetically either related to oligodendroglial or to astrocytic tumors. Only recently, mutation of the isocitrate dehydrogenase 1 (IDH1) gene, or less commonly of the related IDH2 gene, have been identified in the vast majority of WHO grades II and III astrocytic, oligodendroglial, and oligoastrocytic gliomas [2, 137] suggesting a common initiating event in these histologically and biologically diverse glioma types. Anaplastic (WHO grade III) astrocytomas often carry additional, progression-associated genetic changes, such as losses of the tumor suppressor genes CDKN2A, CDKN2B, and p14 ARF on 9p21 and deletions on chromosomes 6, 11p, 22q, and others. Moreover, CDK4 or CDK6 amplification or inactivating alterations of RB1 are detectable in a subset of anaplastic gliomas, mainly anaplastic astrocytomas. Glioblastomas show complex chromosomal and genetic alterations that lead to inactivation of various tumor suppressor genes, as well as aberrant activation of proto-oncogenes [18, 120]. The vast majority of glioblastomas present de novo in elderly patients with a short clinical history. These primary glioblastomas are characterized by a distinct pattern of genetic aberrations when compared with the less common secondary glioblastomas, which develop by progression from pre-existing lower grade gliomas [93]. In particular, primary glioblastomas show frequent EGFR amplification and PTEN mutation but lack IDH1 mutation, while secondary glioblastomas are characterized by frequent mutations in the TP53 and IDH1 genes, but lack EGFR amplification [73, 93] (Fig. 1). At the chromosomal level, primary glioblastomas are distinct from secondary glioblastomas by the frequent trisomy of chromosome 7 and monosomy of 10, as well as frequent gains of chromosome arms 12p, 19q, and 20q [120]. Despite these differences, however, most of the genetic alterations in primary and secondary glioblastomas can be assigned to a common set of functional pathways [18] (Fig. 1; see also below).

Summary of most frequent molecular alterations in astrocytic, oligodendroglial, and oligoastrocytic gliomas. Primary glioblastomas, the most common gliomas in adults, show complex chromosomal, genetic, and epigenetic alterations targeting genes involved in important cellular pathways, namely the receptor tyrosin kinase/mitogen-activated protein kinase/phosphoinositol 3-kinase pathway (e.g. EGFR, MET, PDGFRA, ERBB2, NF1, PTEN, PIK3R1, PI3KCA, CTMP), the p53 pathway (e.g. TP53, p14 ARF , MDM2, MDM4), and the pRb1 pathway (e.g. CDKN2A, CDKN2B, CDK4, CDK6, RB1). In addition, primary glioblastomas frequently show monosomy 10, trisomy 7 and gains of 19q and 20q. Diffuse WHO grade II and III astrocytic, oligodendroglial and oligoastrocytic gliomas and secondary glioblastomas frequently carry mutations in IDH1 or IDH2, suggesting that they share a common, yet to be defined cell of origin. Diffuse astrocytic gliomas often carry additional TP53 mutations, while oligodendroglial tumors are characterized by 1p/19q deletion. Most oligoastrocytomas have either of these alterations. Molecular changes associated with progression to anaplastic glioma include 9p losses and inactivation of the CDKN2A, p14 ARF and CDKN2B genes on 9p21 as well as other changes, while progression to secondary glioblastoma is associated with frequent loss of 10q and DCC loss of expression among others. The majority of pilocytic astrocytomas are characterized by duplication/fusion or point mutation of the BRAF gene on 7q34, while other genomic aberrations are rare
From the identification of molecular changes to novel diagnostic, prognostic, and predictive biomarkers
Several of the molecular alterations detected in gliomas may have diagnostic and/or prognostic implications, as they are associated with histologically defined tumor types or malignancy grades. However, for most of the molecular changes this does not justify a designation as glioma biomarker, because biomarkers should provide unique diagnostic, prognostic, or predictive information exceeding that reached by mere histological classification. A meaningful diagnostic biomarker should be helpful in the classification of tumors with ambiguous histological features or allow for a clinically useful subdivision of tumors within a given histological tumor type. A useful prognostic biomarker should correlate with disease-free and overall survival, ideally providing information beyond that obtained by established prognostic parameters, such as patient age, clinical performance status, extent of resection, and WHO grade. A predictive biomarker, finally, should provide valuable information on the response to a given therapy, which will help to stratify patients into distinct therapeutic groups to allow for the optimal (“personalized”) treatment. In this regard, the number of molecular biomarkers in neurooncology to date is limited to a few alterations, namely combined deletions of the chromosome arms 1p and 19q in oligodendroglial tumors, MGMT hypermethylation in glioblastomas and anaplastic gliomas, IDH1 and IDH2 mutations in diffuse gliomas, as well as BRAF aberrations in pilocytic astrocytomas (Table 1). More detailed information on the respective testing methods and the information conveyed by these markers is provided in the subsequent paragraphs of this review. In addition, we will review some of the recent advancements concerning the understanding of the biological mechanisms of treatment resistance in high-grade gliomas, such as treatment-associated somatic mutations of the mismatch repair gene MSH6 [15, 57, 139]. We will also discuss the potential role of predictive testing for responsiveness towards targeted therapies, such as the assessment of the EGF receptor status in adult high-grade gliomas or the BRAF status in pilocytic astrocytomas.
Combined deletion of chromosome arms 1p and 19q as prognostic marker for oligodendroglial tumor patients
Loss of 1p and 19q is the genetic hallmark of oligodendroglial tumors [68, 81]. Frequencies reported vary due to interobserver variability in the distinction of oligoastrocytomas from oligodendrogliomas on the one hand and astrocytomas on the other, as well as the techniques used and the loci investigated [68]. Overall, losses of 1p and 19q are detected in up to 80% of oligodendrogliomas (WHO grade II) and approximately 60% of anaplastic oligodendrogliomas (WHO grade III), whereas 30–50% of oligoastrocytomas, 20–30% of anaplastic oligoastrocytomas, and <10% of diffuse astrocytic gliomas, including the glioblastomas, carry this aberration. The observation that usually both 1p and 19q are completely lost is explained by an unbalanced translocation t(1;19)(q10;p10), of which the 1p–19q derivative is lost, whereas the 1q–19p derivative is retained during cell replication [42, 62]. There is a strong association between 1p/19q codeletion and classical oligodendroglial features on histology (e.g., perinuclear halo and chicken-wire vascular pattern). However, morphology alone cannot predict the 1p/19q status [40, 109] and up till now the diagnosis of oligodendroglial neoplasms is based on the morphological criteria [81].
In 1998, it was first reported that 1p (and combined 1p/19q) loss predicts better response to chemotherapy and longer survival in anaplastic oligodendroglioma patients [17]. Since then, many subsequent studies have been performed, including three prospective randomized phase III trials that corroborated 1p/19q deletion as a powerful prognostic marker in patients with WHO grade III gliomas. Importantly, these studies also indicated that the prognostic power was independent of the type of adjuvant therapy, that is radiotherapy, chemotherapy or combined radio-/chemotherapy [16, 122, 135]. Nevertheless, discussion remains regarding the predictive (response to therapy) versus prognostic (independent of therapy) nature of this marker. Retrospective data on oligodendroglial tumor patients not receiving any radio- or chemotherapy after their initial surgical treatment revealed that 1p/19q loss was not associated with longer progression-free survival [131]. These data would suggest that 1p/19q loss characterizes a group of gliomas that is more sensitive to genotoxic therapy in general, i.e., radio- and alkylating chemotherapy, and, when treated, are associated with significantly longer survival.
Given the undisputed prognostic significance of 1p/19q loss in oligodendroglial tumor patients receiving adjuvant therapy, many institutions have now established diagnostic testing for this aberration. However, while 1p/19q loss is associated with more favorable prognosis of patients receiving adjuvant treatment, it needs to be emphasized that this marker is of limited help for making treatment decisions, such as radio- versus chemotherapy. One should also be aware that the prognostic relevance of 1p/19q loss may be less pronounced in the presence of other, prognostically unfavorable genetic alterations [121]. In addition, different types of 1p losses have been identified that have distinct prognostic implications. For example, oligodendroglial tumors carrying partial terminal or interstitial 1p losses are associated with shorter patient survival when compared with tumors with combined complete 1p/19q losses [33, 125] (Figs. 2, 3). Thus, molecular testing for 1p loss alone by studying just distal markers on 1p36 may pick up cases with less favorable prognosis when compared with oligodendroglial tumors having a complete 1p/19q co-deletion.

Different types of 1p and 19q losses as detected by array-CGH. Genomic profiles were obtained using tiling-resolution arrays containing 32,447 human BAC clones. Profiles are shown for chromosomes 1 and 19. On the X axis, BACs are aligned according to their physical mapping positions from the 1p telomere to the 1q telomere (n = 2,365) and from the 19p telomere to the 19q telomere (n = 735). On the the Y axis, the log2-transformed and normalized test:reference intensity ratios [“2Log(T/R)”]) are represented. Centromeric and heterochromatic regions are not evaluated using array-CGH resulting in absence of ratios (i.e. spots) in these regions and thereby visually separating the p-arm from the q-arm. Losses detected are indicated by a bar shown on the bottom of the ratio profiles, representing a complete chromosomes 1p and 19q loss (a), a loss of 1p11-p31.1, and 19q13.31-19qter (b), and a loss of 1pter-1p31.2 and 19q13.32-19qter (c)

Distribution of the four clinically relevant molecular alterations according to glioma entity. Colored squares indicate that the particular aberration is frequent in the respective tumor entity, i.e., usually detectable in 40% or more of the cases. Uncolored squares indicate that the aberration is rare in the respective tumor entity, i.e., usually restricted to <10% of the cases (except for MGMT promoter methylation being reported in approximately 20% of the pilocytic astrocytomas). Note that oligodendrogliomas and anaplastic oligodendrogliomas are characterized by the frequent coincidence of 1p/19q deletions, IDH1 or IDH2 mutation and MGMT promoter methylation (red squares). The same applies for oligoastrocytic tumors, although the frequency of 1p/19q deletion is less common when compared with “pure” oligodendrogliomas, also depending on the stringency of the histological classification used for mixed gliomas. IDH1 mutation and MGMT promoter methylation is frequent in diffuse astrocytomas, anaplastic astrocytomas and secondary glioblastomas (green squares). In contrast, primary glioblastomas rarely carry 1p/19q deletions and IDH1 mutations, while MGMT promoter methylation is found in approximately 40% of the cases (blue square). Pilocytic astrocytomas are uniquely characterized by BRAF alterations in more than 60% of the cases (orange square)
Techniques used and the loci investigated for 1p/19q testing differ widely among institutions, depending primarily on the local expertise, existing laboratory equipment and preferences of the neuropathologist and molecular biologist involved (Table 2). Most commonly, loss of heterozygosity (LOH) [59, 102] or (fluorescent) in situ hybridization ((F)ISH) [47, 114] analyses are used. FISH may be preferred by pathologists as tissue characteristics are retained and no corresponding blood sample is required. LOH analysis is most commonly available, but requires comparative evaluation of the same set of loci in DNA extracted from normal cells of the patient, usually peripheral blood leukocytes. In addition, one should be aware that “pseudo-LOH” may be detected in some tumors displaying allelic imbalances due to copy number gain rather than loss of one allele, e.g. the frequent gain of 19q in primary glioblastomas. On the other hand, LOH analysis may be scaled up more easily than FISH to the analysis of multiple loci along each chromosome, which would help to avoid detection of false-positive cases with partial 1p loss or isolated 19q loss, and may even (partly) be automated in larger molecular diagnostic units. Multiplex ligation-dependent probe amplification (MLPA) is another methodical option, as it allows detection of copy number changes of up to 45 loci in a single experiment [63, 64, 111]. Similar to LOH analysis, MLPA only requires standard laboratory facilities for PCR and capillary gel electrophoreses that are widely available in molecular laboratories, but has the advantage that a blood sample of the patient is not required.
MGMT hypermethylation as a prognostic or predictive marker in malignant gliomas
The MGMT (O 6-methylguanine-DNA methyltransferase) gene at 10q26 is frequently silenced by promoter hypermethylation in diffuse gliomas and this has been pinpointed as an epigenetic mechanism reducing MGMT expression levels. Importantly, an association between MGMT promoter methylation and the response of malignant gliomas to alkylating chemotherapy using nitrosourea compounds [32], temozolomide [48], or a combination of both [53] has been observed. Based on MGMT promoter methylation studies in a subpopulation of patients involved in the EORTC/NCIC 22981/26981 trial [48, 117], Hegi et al. reported that patients treated with radiotherapy and temozolomide, and whose tumors had a methylated MGMT promoter (which is seen in approximately 40% of primary glioblastomas) survive significantly longer when compared with patients whose tumors lacked MGMT promoter methylation [48]. In this land-mark paper, MGMT promoter methylation did not significantly influence survival in patients treated with radiotherapy alone, suggesting that the MGMT hypermethylation is predictive for favorable response to chemotherapy.
Mechanistically, a predictive power of the MGMT promoter status can be explained by the fact that MGMT encodes a DNA repair protein that removes alkyl groups from the O 6 position of guanine, which are introduced by alkylating chemotherapeutic agents, such as temozolomide [39]. Chemotherapy-induced DNA alkylation triggers cytotoxicity and apoptosis, while high MGMT expression in the tumor cells counteracts the cytotoxic effects and thereby may cause treatment failure. MGMT promoter methylation, however, impedes the transcription of the gene resulting in a lack of MGMT mRNA and protein expression. Glioblastoma cells with MGMT promoter hypermethylation thus respond better to temozolomide, as they lack the ability to repair the therapy-induced DNA damage.
Because the current standard of care for glioblastoma patients involves radiotherapy plus concomitant and adjuvant temozolomide treatment [117], testing for MGMT promoter methylation by means of methylation-specific polymerase (MSP) chain reaction analysis or other methods is now increasingly performed, not only for patients in clinical trials but also in the routine diagnostic setting. The advantage of MGMT methylation testing is that—if detected—a methylation signal specifically results from the neoplastic glial cells. In contrast, attempts to replace MGMT promoter methylation testing by a technically more simple immunohistochemical analysis of MGMT protein expression did not prove reliable for diagnostic purposes in most studies, as the immunohistochemical assays may be disturbed by a variable content of contaminating non-neoplastic cells, such as microglial cells, lymphocytes, reactive astrocytes and vascular cells, which retain MGMT expression also in MGMT-hypermethylated gliomas [34]. The same applies for expression analyses based on Western blotting or reverse transcription-PCR, as well as biochemical assays detecting enzymatic activity. All these methods may be confounded by contaminating non-neoplastic cells in the investigated tissue homogenates. Nevertheless, several studies reported on significant prognostic associations using such assays on glioma tissues [7, 22, 61, 78].
Although data from the EORTC/NCIC 22981/26981 trial [80] and another large prospective patient cohort [132] found that MGMT promoter methylation was predictive for longer survival only in those patients who received temozolomide, a recent paper reported that MGMT promoter methylation is also predictive of response to radiotherapy and linked to longer survival in the absence of adjuvant chemotherapy [104]. Although this association is somewhat debatable in glioblastomas, prospective trials on anaplastic (WHO grade III) gliomas clearly indicated a prognostic role of MGMT promoter methylation independent from the type of adjuvant treatment, i.e., alkylating chemotherapy or radiotherapy [123, 135]. The reason for a prognostic role of MGMT promoter methylation in patients just receiving radiotherapy remains unclear, and is rather unexpected from a functional point of view. One may speculate that the strong association of MGMT hypermethylation with 1p/19q codeletion [9, 88, 124] and IDH1 mutation [108] (Fig. 3) may reflect a global molecular constellation in anaplastic gliomas that per se is associated with higher sensitivity to cytotoxic therapy and more favorable outcome. Even further, the MGMT-hypermethylated anaplastic gliomas may belong to a group of gliomas characterized by multiple hypermethylated genes [91], potentially including yet unknown genes mediating radioresistance.
Similar to the findings in adult glioma patients, MGMT promoter methylation [110], and reduced MGMT protein expression [100] have been reported to be associated with longer survival of pediatric malignant glioma patients. In ependymal tumors, MGMT promoter methylation is less common when compared with diffuse astrocytic and oligodendroglial tumors [13, 74]. Other malignant brain tumors, such as medulloblastomas [31] and anaplastic meningiomas [12] rarely demonstrate MGMT promoter methylation.
Technical aspects of MGMT promoter methylation testing
A number of different methods are currently in use to assess the MGMT promoter methylation status in patient samples, for review see [133]; (Table 2). The most commonly employed method, and also the technique originally described to convey the relevant clinical information, is methylation-specific PCR analysis [48, 52]. This technique makes use of primers that specifically amplify fragments from either the methylated or the unmethylated sodium bisulfite-modified DNA sequence. To make the primers discriminative between both sequences, they are designed to contain a maximum number of CpG sites that differ in their sequence between methylated and unmethylated bisulfite-modified DNA. PCR products can then subsequently be evaluated on an agarose gel for the presence or absence in terms of an all or nothing signal. Alternatively, MSP can be performed quantitatively using real-time or TaqMan® PCR assays that allow for a higher level of standardization and the definition of cut-offs for methylation [127]. However, such cut-offs are just technically substantiated to date, and there is a need to validate cut-off points prospectively to establish clinically relevant methylation thresholds.
Another DNA-based methods is methylation-specific pyrosequencing [87], which certainly is one of the most sensitive methods to quantitatively detect even low methylation signals. The COBRA (combined bisulfite restriction analysis) method employs sodium bisulfit treatment of DNA followed by digestion of PCR products with restriction enzymes that differentially cut at methylated versus unmethylated sites [87]. Finally, methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) has the advantage that it provides semi-quantitative information on the percentage of methylated DNA but does not depend on a prior conversion of DNA by sodium bisulfite [65]. This may be an advantage because the bisulfite conversion step is particularly critical and every single reaction should be controlled for complete conversion. Otherwise, unmethylated cytosins that are not adequately converted by sodium bisulfite may be mistaken for methylated cytosins.
A comprehensive discussion of advantages and disadvantages of the individual methods to assess MGMT promoter methylation is beyond the scope of this review and has been addressed elsewhere [133]. However, it should be pointed out that the MGMT promoter methylation pattern is very heterogeneous from tumor to tumor, and the relevant CpG sites (or combinations of CpG sites) that need to be methylated to silence transcription and provide favorable outcome are not yet defined. Therefore, one has to be aware that the use of distinct primer combinations, even when the same method such as MSP is used, may result in different results, which may cause uncertainties when the same tumor is tested at different laboratories. In this respect, establishment of a consensus test method that assesses experimentally verified, most relevant CpG sites for transcriptional regulation, and is clinically validated in comparative analyses of the various techniques would be most desirable. In addition, quality control measures to ensure the sensitivity and specificity of the test across different laboratories need to be established.
IDH1 and IDH2 mutations as novel molecular markers for diffuse glioma
Mutations in the gene encoding the human cytosolic NADPH-dependent isocitrate dehydrogenase (IDH1), an enzyme that participates in the citric acid cycle, were originally identified in 2008 employing large scale sequencing analysis of 22 glioblastomas [95]. All mutations were found in the evolutionarily conserved residue R132 that is located in the substrate-binding site of IDH1. The mutations were detected preferentially in glioblastomas of young patients and in secondary glioblastomas [95]. Soon after, multiple studies corroborated these findings and additionally revealed that somatic IDH1 mutations are present in the vast majority of low-grade diffuse (WHO grade II) and anaplastic (WHO grade III) astrocytic, oligodendroglial, and mixed oligodendroglial neoplasms [2, 90, 137]. In a small subset of WHO grade II and III, diffuse gliomas and secondary glioblastomas that lack IDH1 mutations, the gene of the mitochondrial isoform IDH2 was found to harbor mutations affecting the analogous amino acid (R172) [137]. In contrast, IDH1 or IDH2 mutations are rare in primary glioblastomas and restricted to only individual cases of other primary brain tumors [2, 90, 137].
Already the original paper of Parsons et al. [95] recognized that IDH1 mutations were associated with prolonged overall survival in glioblastoma patients. Subsequent studies analyzing the whole spectrum of diffuse gliomas underscored the association between IDH1 mutation and better outcome, multivariate analyses often revealing IDH1 mutation as a strong and independent favorable prognostic marker [30, 108, 123, 132, 135]. So far, there is no evidence that the type of IDH1 mutation (R132H vs. non-R132H) affects patient survival [41]. In contrast to the strong prognostic significance, several studies reported a lack of predictive significance of IDH1 mutations in gliomas for response to (chemo)therapy [30, 123].
At the genetic level, the presence of IDH1 mutations in diffuse gliomas is strongly correlated with TP53 mutation or 1p/19q deletions [58, 123, 129]. Analysis of multiple glioma biopsies from the same patient revealed that IDH1 mutations do not occur after the acquisition of either a TP53 mutation or loss of 1p/19q [129]. In another study, all 128 gliomas with a complete co-deletion of 1p/19q harbored an IDH1 or IDH2 mutation [77]. These mutations thus seem to represent a very early event, affecting a common glial precursor cell, and may in fact form a prerequisite for the t(1;19) translocation leading to 1p/19q co-deletion and oligodendroglial tumor development. The presence of IDH1 mutations in gliomas was also reported to be tightly associated with MGMT promoter methylation and inversely correlated with the loss of chromosome 10 and EGFR amplification [108, 123]. This latter finding further underscores the notion that primary and secondary glioblastomas are genetically distinct entities, despite their histological similarities [129].
In up to 90% of the diffuse gliomas with mutated IDH1, the mutation is of the R132H (G395A) type [2, 90, 95, 137]. One study of more than 1,000 diffuse WHO grade II and III gliomas showed that R132C IDH1 mutations are associated with astrocytic neoplasms, while IDH2 mutations predominantly occur in oligodendroglial tumors [46]. Another study revealed that non-R132H IDH1 mutations are rare in classic oligodendrogliomas with 1p/19q loss and occur at significantly higher frequency in other grade II and III gliomas, including those with TP53 mutations [41]. Interestingly, all five examined astrocytomas of patients with Li-Fraumeni syndrome carried R132C IDH1 mutations, indicating that glial/glioma precursor cells with a germline TP53 mutation carry an increased risk to acquire such a non-R132H mutation [130].
IDH1 mutations have now also been reported in a subset of (anaplastic) gangliogliomas [56, 116], supratentorial primitive neuroectodermal tumors [2] and in gliomatosis cerebri [112]. IDH mutations are rare or absent in other glial tumors, such as pilocytic astrocytomas [75, 129] and ependymomas [129]. Importantly, the vast majority of high-grade (WHO grade III and IV) gliomas in the pediatric age group lacks IDH1 mutation as well, corroborating the notion that these pediatric neoplasms are fundamentally different from their adult counterparts [1, 2, 96]. IDH1 mutations are reported to occur in some non-central nervous system tumors, e.g., a subset of acute myeloid leukemias with a normal karyotype [23, 103], and occasionally in prostate cancer, B-ALL [23], and paraganglioma [37]. In the vast majority of non-glial tumors, however, such mutations are absent [8].
Under physiological conditions, IDH1 and IDH2 are involved in multiple metabolic processes, such as lipid synthesis, cellular defense against oxidative stress, oxidative respiration, and signal transduction. There are different hypotheses on the role of IDH1 and IDH2 mutations in gliomagenesis, including an effect on the stabilization of hypoxia inducible factor 1 (HIF-1), upregulation of (other) genes involved in angiogenesis, glucose transport and glycolysis, and inhibition of developmental apoptosis [5, 103, 141]. However, recent studies suggest that the heterozygous IDH1 and IDH2 mutations in gliomas do not just result in a loss of function, that is a reduced ability to catalyze conversion of isocitrate to α-ketoglutarate [137], but also confer an enzymatic gain of function, in particular the ability to catalyze the NADPH-dependent reduction of α-ketoglutarate to 2-hydroxyglutarate (2HG). Indeed, gliomas harboring IDH1 mutations demonstrate markedly elevated levels of 2HG, and this ‘onco-metabolite’ may contribute to the oncogenesis of gliomas [26]. Similarly, IDH1 and IDH2 mutations dramatically increase 2HG in acute myeloid leukemia [43, 128]. Because of the specificity of the IDH1 and IDH2 mutations in gliomas and of their metabolic effects, there is hope that these aberrations provide new avenues for anti-cancer therapies [36]. However, further study of the (exact effect of) IDH1 and IDH2 mutations is needed to seize this opportunity.
Various methods are applied for the detection of IDH1 and/or IDH2 mutations in clinical glioma specimens (Table 2), including single-strand conformation polymorphism analysis, direct sequencing [71, 77, 96], PCR-restriction fragment length polymorphism-based assays [14, 56, 86], DNA pyrosequencing and real-time PCR with post-PCR fluorescence melting curve analysis assays [46, 55]. Especially, these latter assays allow for rapid, inexpensive, and sensitive analysis of IDH1 and IDH2 mutations in routinely processed (i.e. formalin-fixed, paraffin-embedded) tumor tissue, even in samples with a low tumor cell content [14, 35, 55]. Only recently, specific, and robust monoclonal antibodies were established that can be used for immunohistochemical analysis of gliomas bearing the IDH1 R132H mutation [19, 72]. Testing for mutations in IDH1 or IDH2 can now easily and effectively be performed in a clinical setting and thereby enhance the diagnostic accuracy, especially, in cases where traditional methods are insufficient to reach a definitive diagnosis. For example, IDH1 and IDH2 mutation analysis might be helpful in case of a differential diagnosis of diffuse glioma versus pilocytic astrocytoma or ependymoma, and for discrimination between primary and secondary glioblastoma [56, 75]. Using immunohistochemistry with the anti-IDH1R132H antibody, even individual cells of gliomas with the IDH1 R132H mutation (e.g. in the periphery of diffuse infiltrative gliomas) can be detected and differentiated from non-neoplastic glial cells [19]. Of note, lack of staining with this antibody does not rule out the presence of an IDH1 mutation (about 10% of the IDH1 mutated gliomas carry a non-R132H mutation) nor of an IDH2 mutation. Given the diagnostic and prognostic importance as well as the robustness and relative ease of IDH1 and IDH2 mutation testing, it is very likely that determination of these mutations will soon become a part of the routine diagnostic assessment of gliomas. Furthermore, it needs to be investigated to which extent the IDH1 and IDH2 status may influence the classification and subsequent treatment of diffuse gliomas. For example, one may address interesting questions like (1) should IDH1 and IDH2 wild-type anaplastic gliomas be biologically considered as glioblastomas that consequently would require more aggressive treatment when compared with IDH1 or IDH2 mutant anaplastic gliomas, or (2) should the rare, prognostically favorable IDH1 mutant primary glioblastoma be regarded as a separate entity distinct from the “ordinary” IDH1 wild-type primary glioblastoma.
Role of testing for EGF receptor aberrations in gliomas
The epidermal growth factor receptor gene (EGFR) at 7p12 is the most frequently amplified and overexpressed gene in primary glioblastomas, affecting approximately 40% of these tumors [81, 138]. EGFR rearrangements are also frequent, the most common variant being EGFRvIII consisting of an 801-bp in-frame deletion of exons 2–7 that results in a constitutively activated truncated receptor protein lacking the ligand-binding domain [118]. EGFRvIII represents about half of the rearrangements and is identified in 20–30% of unselected primary glioblastomas and 50–60% of the EGFR-amplified glioblastomas [38]. Identification of EGFR amplification and rearrangements, such as EGFRvIII, are highly indicative for high-grade malignancy and, therefore, may provide diagnostic as well as prognostic information [64, 81]. In fact, detection of EGFR amplification/EGFRvIII in anaplastic or low-grade gliomas strongly suggests that these tumors are more malignant than indicated by their histopathology and an unfavorable impact on the prognosis has been described for these patients [66, 67, 115]. In primary glioblastoma such prognostic association is less obvious [132] although other studies reported on EGFR/EGFRvIII aberrations as poor prognostic factor [27, 89, 97, 113].
Detection of EGFR aberrations also may be relevant from a therapeutic point of view as inhibition of the EGFR pathway bears the potential of restoring apoptosis, thereby increasing the sensitivity to adjuvant therapies. Increased EGFRvIII signaling was indeed associated with a generally poor response to radiation and chemotherapy [4, 20, 134]. However, a benefit resulting from the combined treatment by EGFR inhibition with standard therapies (temozolomide and radiation therapy) is disputed and as yet the clinical benefit of the use of EGFR inhibitors in glioblastomas has been rather disappointing, that is progression-free survival was not prolonged and only a small subset of individual patients responded [45, 101]. Attempts to identify additional biomarkers predictive of response to EGFR-related therapies suggested that tumors with EGFRvIII and intact PTEN [85] or with EGFR amplification (but not EGFRvIII) and low levels of phosphorylated AKT [44] were more likely to respond to the small molecule tyrosine kinase inhibitors erlotinib or gefitinib. The EGFRvIII mutant, as not being present in non-neoplastic tissues, also may serve as an attractive target for immunotherapy [79, 107]. Recent studies reported that the anti-EGFRvIII peptide vaccine CDX-110 increased progression-free and overall survival in EGFRvIII-positive glioblastoma patients when added to radiochemotherapy [49–51]. Unfortunately, although an antibody specifically recognizing EGFRvIII has been developed for immunohistochemical application, existing patents currently prohibit the use of this antibody for clinical purposes [136, 140]. EGFRvIII analysis alternatively can be performed by reverse transcription-PCR analysis using primers located in exons 1 and 9 [79]. Furthermore, MLPA analysis allows for the detection of EGFR rearrangements by the simultaneous and semi-quantitative copy number analysis of multiple small DNA fragments encompassing different EGFR exons [64, 111], with available assays detecting EGFRvIII and different other types of rearrangements and being applicable to routinely processed formalin-fixed and paraffin-embedded tissue samples [64].
Special aspects of pediatric gliomas
Aberrant activation of the BRAF proto-oncogene at 7q34, most commonly by gene duplication and fusion or less frequently by point mutation, has only recently been identified as the characteristic genetic aberration in pilocytic astrocytomas. BRAF gene alterations are detectable in 60–80% of pilocytic astrocytomas, but are infrequent in diffusely infiltrating astrocytic gliomas [3, 69, 70, 98]. Thus, testing for BRAF gene alterations might be helpful in the sometimes difficult differential diagnosis between pilocytic astrocytomas and low-grade diffuse astrocytomas [75]. Detection of the BRAF–KIAA1549 fusion gene can be either accomplished by FISH analysis or specialized RT-PCR assays [69, 75] (Table 2). As mentioned above, low-grade diffuse astrocytoma—in contrast to pilocytic astrocytoma—would contain frequent mutations in the IDH1 gene and as such testing for both markers in combination could finally turn the scales for one of the two entities. However, IDH1 and IDH2 mutations are generally rare in pediatric astrocytomas, including the diffusely infiltrating tumors [2, 137], which implies that the significance of diagnostic testing for these mutations may be lower in pediatric glioma patients.
The frequent BRAF alterations in pilocytic astrocytomas may have additional clinical implications as a novel therapeutic target. Tumors with BRAF duplication or activating mutation show aberrant signaling via the BRAF pathway. In vitro studies revealed that both the stable silencing of BRAF through shRNA lentiviral transduction and pharmacological inhibition of MEK1/2, the immediate downstream phosphorylation target of BRAF, blocked proliferation and arrested growth of cultured glioma cells [98]. Thus, pharmacological inhibition of the MAPK pathway may serve as a potential treatment option in pediatric astrocytoma patients, as exemplified in a recent case report [106].
Ependymomas also occur relatively frequently in the pediatric age group. Histological grading of ependymomas is difficult and there appears to be a less-stringent association between tumor grade and prognosis in ependymoma when compared with astrocytoma patients. However, recent data suggest that in addition to age at diagnosis, gain of 1q and homozygous CDKN2A deletion are independent indicators of unfavorable prognosis, whereas gains of chromosomes 9, 15q, and 18 and loss of chromosome 6 are associated with excellent survival for pediatric and adult patients with intracranial ependymomas [76]. Based on these findings, the authors developed a molecular staging system comprising three genetic risk groups. Thus, the analysis of genetic markers in addition to established clinical and histopathologic variables may significantly improve outcome prediction of ependymoma patients and help to stratify patients into distinct risk groups.
Identification of novel biomarkers by genome-wide profiling approaches in gliomas
There are two main trends in glioma research that have already yielded and probably will yield further molecular biomarkers of clinical impact: the use of large-scale profiling techniques and, mainly driven by the need to work cost-effectively and to increase sample numbers, the formation of large research networks, such as The Cancer Genome Atlas Research Network (TCGA) or, more recently, the International Cancer Genome Consortium (ICGC) [60].
The success of large scale profiling approaches is exemplified by the first detection of IDH1 mutations in a study that sequenced 20,661 genes in 22 human glioblastoma samples [95]. In addition, this study confirmed a set of glioblastoma candidate genes that mainly functioned within the p53, pRb1, and Pi3k/Pten signaling pathways [95]. A publication that was launched contemporaneously by the TCGA consortium on the integrative analysis of DNA copy number, gene expression, and DNA methylation profiling in 206 human glioblastomas similarly reported that the most important pathways that are aberrant in gliomas are the pRb1/Cdk4/cyclin D/Cdkn2A/B pathway, the p53 pathway, and the receptor tyrosin kinase/Ras/Pi3k pathway [18]. Each of these pathways was confirmed to be disrupted in more than three quarters of glioblastomas, meaning that in most tumors two or all three of these pathways were involved. Sequencing of glioblastomas for mutations in 601 selected genes additionally revealed three previously unrecognized mutations that occurred with significant frequency, namely NF1 gene mutations in 14%, ERBB2 gene mutations in 8% and PIK3R1 mutations in nearly 10% of glioblastomas [18]. These novel findings may well have impact on future treatment strategies. As an example, PIK3R1 encodes the regulatory protein p85a subunit of Pi3k and the response to Pi3k inhibitors may depend on whether the tumors bear mutations in this specific gene or not.
Except for the identification of novel individual gene alterations, the signatures produced by high-throughput profiling techniques themselves might convey clinically relevant information. In this regard, it was shown that gene expression-based classification of morphologically ambiguous high-grade gliomas correlates better with prognosis than the histological classification [92]. Furthermore, molecular classification of gliomas on the basis of genomic profiles obtained by array-CGH closely parallels histological classification and is able to distinguish, with few exceptions, between different astrocytoma grades, as well as between primary and secondary glioblastomas [105]. Phillips et al. [99] first reported on three prognostic subclasses of high-grade astrocytomas, namely the proneural (PN), proliferative (Prolif) and mesenchymal (Mes) tumor subclasses that resemble distinct stages in neurogenesis. The proneural tumor signature displayed neuronal lineage markers and was associated with longer survival, while the proliferative and mesenchymal tumor signatures were both linked to shorter survival. Upon recurrence, malignant gliomas frequently shift toward the mesenchymal subclass. A recent study from the TCGA consortium reported on a robust gene expression-based molecular classification of glioblastomas into proneural, neural, classical, and mesenchymal subtypes [126]. Furthermore, it was found that genetic aberrations and expression of EGFR, NF1, and PDGFRA/IDH1 each define the classical, mesenchymal, and proneural subtypes, respectively. Response to aggressive therapy was found to differ by subtype, with the greatest benefit being observed in the classical subtype and no benefit in the proneural subtype. Interestingly, promoter DNA methylation profiling in 272 TCGA glioblastomas revealed that a subset of patients had concerted hypermethylation at a large number of loci, indicating the existence of a glioma-CpG island methylator phenotype (G-CIMP) [91]. Further investigations showed that G-CIMP tumors were more common among low-grade gliomas, displayed a proneural expression signature, frequently carried IDH1 mutations and were associated with significantly longer survival. Other authors also found a tight association between the IDH1 mutation status and gene expression profiles, suggesting two major pathomechanisms in diffuse astrocytic gliomas characterized either by IDH1 mutation and a proneural expression profile, found mostly in diffuse and anaplastic astrocytomas as well as secondary glioblastomas, or by lack of IDH1 mutation and a mesenchymal/proliferative expression profile [120]. In addition, IDH1 mutant glioblastomas have been reported to be frequently accompanied by telomerase-independent alternative lengthening of telomeres (ALTs), suggesting that such ALT + tumors belong to the less aggressive, ‘proneural’ glioblastomas [83].
Although these novel glioma signatures currently incur a lot of attention, their routine assessment by means of large-scale mRNA expression profiling is not suitable due to the limited availability and high costs of this approach. However, a recent study reported that immunhistochemical expression analysis of a nine gene signature, which is applicable to routinely processed tissue samples, may be sufficient to predict glioblastoma outcome [25]. Nevertheless, it remains to be proven that any of these prognostic gene signatures yields clinically relevant data beyond the information provided by the analysis of IDH1 mutation, MGMT promoter methylation and 1p/19q deletion.
A different approach by which large-scale profiling techniques may add in the identification of novel glioma biomarkers is the investigation of defined glioma cell subpopulations. Malignant gliomas are highly heterogeneous and their conventional light-microscopic diagnosis is based on the recognition of certain histological features, such as pathological vessel formation, presence/absence of necroses or an infiltrative growth pattern [81]. Bearing in mind that all these microscopic appearances are equivalents of defined biologic processes, it appears intriguing to dissect such tumor cell subpopulations and to assess their molecular signatures with two closely related goals: first, to foster understanding of the basic processes underlying glioma biology and secondly, to use these “subprofiles” to identify innovative biomarkers that are of relevance for the tumor’s clinical appraisal as a whole. In this regard, Dong et al. assessed the molecular profiles of perinecrotic palisades in comparison to non-palisading tumor cells distant from necrosis [29]. In conjunction with preceding studies, the authors found that the genes most commonly differentially expressed in these palisades conveyed response to hypoxic environmental conditions [10, 11]. Interestingly, a set of five RNAs (POFUT2, PTDSR, PLOD2, ATF5, and HK2) were not only differentially expressed in three micro-dissected glioblastomas, but also provided prognostic information in an independent set of glioblastoma patients. Thus, it appears feasible to derive tissue biomarkers that provide ancillary prognostic and predictive information from the study of defined subpopulations of tumor cells. Of course, this approach is not restricted to pseudopalisading tumor cells, but may be extended to other glioma cell subpopulations, e.g. tumor cells selected for their invasive or stemness properties [6, 28, 54].
Novel mechanistic insights into molecular alterations underlying treatment resistance of gliomas
Large-scale profiling approaches have also advanced the understanding of the molecular mechanisms that underlie treatment resistance in high-grade gliomas. Investigating gliomas that had recurred after treatment with alkylating agents, Hunter et al. identified somatic mutations of the mismatch repair gene MSH6 in a large-scale sequencing approach of the functional domains of 518 protein kinases [57]. These findings were further evaluated in an independent panel of 46 clinically well-documented glioblastomas [15]. Indeed, in recurrent glioblastomas, the rate of MSH6 mutations was significantly increased adding further evidence to a potential causal link between MSH6 deficiency and treatment resistance.
Interestingly, a subsequent publication reported that MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance [139]. In vitro modeling through exposure of a MSH6 wild-type glioblastoma line to temozolomide resulted in resistant clones with one clone showing an MSH6 mutation, namely Thr(1219)Ile, which had also been noted in two treated glioblastomas of the TCGA cohort [18]. Moreover, knockdown of MSH6 in the glioblastoma cell line U251 increased resistance to temozolomide cytotoxicity and reconstitution restored cytotoxicity in MSH6-null glioma cells. These findings indicate that MSH6 mutations and/or mutations in other DNA mismatch repair genes are selected in glioblastomas during temozolomide therapy and that patients who initially responded to a frontline therapy, i.e., particularly patients with MGMT-hypermethylated tumors, may develop treatment resistance by acquiring a hypermutator phenotype involving frequent mutations in DNA mismatch repair genes. As a perspective, combination of alkylating chemotherapy with molecular strategies targeting DNA mismatch repair-deficient cells may help in preventing or minimizing treatment resistance of gliomas.
Although undoubtedly representing a relevant novel discovery, MSH6 mutations may be just the tip of an iceberg of molecular changes that are associated with treatment response. Furthermore, these mutations appear not be linked to high-level microsatellite instability in gliomas [82]. Other studies have reported that alteration of the base excision repair pathway may sensitize glioma cells to temozolomide treatment and suggested inhibition of poly(ADP-ribose)polymerase as a promising therapeutic approach [21, 84, 119]. Furthermore, large-scale heterochromatin reorganization has been observed in glioma cells following treatment with temozolomide and carmustine, suggesting that treatment efficacy may implicate a first event characterized by changes in heterochromatin organization and, conversely, treatment failure may be associated with the aberrant euchromatinization of novel, yet to be identified chemotherapy resistance genes [94]. Another study aiming at identifying molecular profiles specific of treatment resistance to temozolomide identified a “glioma stem cell” or “self-renewal” expression signature as a predictor of poor survival [89]. This signature proved an independent prognostic factor also in multivariate analyses adjusted for the MGMT promoter methylation status and contained HOX genes as well as the putative glioma stem cell marker prominin-1 (CD133). Thus, in access to the identification of individual candidate genes like MSH6, large-scale profiling approaches might help to uncover more complex molecular profiles associated with treatment resistance. These profiles, although still preliminary, may help in identifying “pathways of therapy failure” that eventually could be specifically targeted.
Conclusions and perspectives
Molecular and translational glioma research has significantly advanced the understanding of glioma pathogenesis and identified a number of diagnostic, prognostic and/or predictive molecular markers that currently are on their way into clinical application. In fact, the antibody against the IDH1 R132H mutation is already used in many neuropathology laboratories, e.g. as a useful diagnostic marker for the differential diagnosis of diffusely infiltrating gliomas versus reactive astrogliosis [25]. Furthermore, both 1p/19q deletion and MGMT promoter methylation are presently being used to stratify patients into different clinical trials, each testing for the efficacy of different drugs or administration schemes in comparison to the respective standard protocol. In case that these studies will proof successful, it is to be foreseen that molecular assessment of the relevant markers will have to be implemented into the routine diagnostic setting outside of clinical trials. Clear cut-off levels for each molecular assay have to be developed and appropriate quality measures have to be established to ensure comparable sensitivity and specificity of molecular test results across different laboratories. Histological control of the tissue specimens used for molecular testing also is an important issue that requires an experienced neuropathologist to avoid false-negative test results due to inappropriate samples with too low tumor cell content.
In the near future, novel insights into the pathogenesis of gliomas are to be expected from ongoing, large-scale collaborative profiling studies addressing the complexity of genetic, epigenetic, transcriptomic, and miRNA changes by high-resolution array-based techniques or deep sequencing approaches. It appears very likely that these studies will uncover novel molecular markers that may further refine the diagnostic assessment of gliomas. However, the future role of molecular diagnostics in neurooncology, in particular concerning the value of predictive markers will also depend on the development and availability of novel therapeutic alternatives to allow for more sophisticated patient-tailored treatment choices based on molecular profiles.
References
Antonelli M, Buttarelli FR, Arcella A, Nobusawa S, Donofrio V, Oghaki H and Giangaspero F (2010) Prognostic significance of histological grading, p53 status, YKL-40 expression, and IDH1 mutations in pediatric high-grade gliomas. J Neurooncol [Epub ahead of print]
Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116:597–602
Bar EE, Lin A, Tihan T, Burger PC, Eberhart CG (2008) Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol 67:878–887
Barker FG 2nd, Simmons ML, Chang SM, Prados MD, Larson DA, Sneed PK, Wara WM, Berger MS, Chen P, Israel MA, Aldape KD (2001) EGFR overexpression and radiation response in glioblastoma multiforme. Int J Radiat Oncol Biol Phys 51:410–418
Bayley JP, Devilee P (2010) Warburg tumours and the mechanisms of mitochondrial tumour suppressor genes. Barking up the right tree? Curr Opin Genet Dev 20:324–329
Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, Aigner L, Brawanski A, Bogdahn U, Beier CP (2007) CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res 67:4010–4015
Belanich M, Pastor M, Randall T, Guerra D, Kibitel J, Alas L, Li B, Citron M, Wasserman P, White A, Eyre H, Jaeckle K, Schulman S, Rector D, Prados M, Coons S, Shapiro W, Yarosh D (1996) Retrospective study of the correlation between the DNA repair protein alkyltransferase and survival of brain tumor patients treated with carmustine. Cancer Res 56:783–788
Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP, Frattini M, Molinari F, Knowles M, Cerrato A, Rodolfo M, Scarpa A, Felicioni L, Buttitta F, Malatesta S, Marchetti A, Bardelli A (2009) IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat 30:7–11
Brandes AA, Nicolardi L, Tosoni A, Gardiman M, Iuzzolino P, Ghimenton C, Reni M, Rotilio A, Sotti G, Ermani M (2006) Survival following adjuvant PCV or temozolomide for anaplastic astrocytoma. Neuro Oncol 8:253–260
Brat DJ, Castellano-Sanchez AA, Hunter SB, Pecot M, Cohen C, Hammond EH, Devi SN, Kaur B, Van Meir EG (2004) Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res 64:920–927
Brat DJ, Van Meir EG (2004) Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab Invest 84:397–405
Brokinkel B, Fischer BR, Peetz-Dienhart S, Ebel H, Sepehrnia A, Rama B, Albert FK, Stummer W, Paulus W and Hasselblatt M (2010) MGMT promoter methylation status in anaplastic meningiomas. J Neurooncol [Epub ahead of print]
Buccoliero AM, Castiglione F, Rossi Degl’Innocenti D, Paglierani M, Maio V, Gheri CF, Garbini F, Moncini D, Taddei A, Sardi I, Sanzo M, Giordano F, Mussa F, Genitori L, Taddei GL (2008) O6-Methylguanine-DNA-methyltransferase in recurring anaplastic ependymomas: PCR and immunohistochemistry. J Chemother 20:263–268
Bujko M, Kober P, Matyja E, Nauman P, Dyttus-Cebulok K, Czeremszynska B, Bonicki W, Siedlecki JA (2010) Prognostic value of IDH1 mutations identified with PCR-RFLP assay in glioblastoma patients. Mol Diagn Ther 14:163–169
Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB, Batchelor TT, Futreal PA, Stratton MR, Curry WT, Iafrate AJ, Louis DN (2007) Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res 13:2038–2045
Cairncross G, Berkey B, Shaw E, Jenkins R, Scheithauer B, Brachman D, Buckner J, Fink K, Souhami L, Laperierre N, Mehta M, Curran W (2006) Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol 24:2707–2714
Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR, Silver JS, Stark PC, Macdonald DR, Ino Y, Ramsay DA, Louis DN (1998) Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 90:1473–1479
Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068
Capper D, Weissert S, Balss J, Habel A, Meyer J, Jager D, Ackermann U, Tessmer C, Korshunov A, Zentgraf H, Hartmann C, von Deimling A (2010) Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol 20:245–254
Chakravarti A, Chakladar A, Delaney MA, Latham DE, Loeffler JS (2002) The epidermal growth factor receptor pathway mediates resistance to sequential administration of radiation and chemotherapy in primary human glioblastoma cells in a RAS-dependent manner. Cancer Res 62:4307–4315
Cheng CL, Johnson SP, Keir ST, Quinn JA, Ali-Osman F, Szabo C, Li H, Salzman AL, Dolan ME, Modrich P, Bigner DD, Friedman HS (2005) Poly(ADP-ribose) polymerase-1 inhibition reverses temozolomide resistance in a DNA mismatch repair-deficient malignant glioma xenograft. Mol Cancer Ther 4:1364–1368
Chinot OL, Barrie M, Fuentes S, Eudes N, Lancelot S, Metellus P, Muracciole X, Braguer D, Ouafik L, Martin PM, Dufour H, Figarella-Branger D (2007) Correlation between O6-methylguanine-DNA methyltransferase and survival in inoperable newly diagnosed glioblastoma patients treated with neoadjuvant temozolomide. J Clin Oncol 25:1470–1475
Chou WC, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Ko BS, Wu SJ, Huang SY, Hsu SC, Chen YC, Huang YN, Chang YC, Lee FY, Liu MC, Liu CW, Tseng MH, Huang CF, Tien HF (2010) Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood 115:2749–2754
Claes A, Idema AJ, Wesseling P (2007) Diffuse glioma growth: a guerilla war. Acta Neuropathol 114:443–458
Colman H, Zhang L, Sulman EP, McDonald JM, Shooshtari NL, Rivera A, Popoff S, Nutt CL, Louis DN, Cairncross JG, Gilbert MR, Phillips HS, Mehta MP, Chakravarti A, Pelloski CE, Bhat K, Feuerstein BG, Jenkins RB, Aldape K (2010) A multigene predictor of outcome in glioblastoma. Neuro Oncol 12:49–57
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462:739–744
Dehais C, Laigle-Donadey F, Marie Y, Kujas M, Lejeune J, Benouaich-Amiel A, Pedretti M, Polivka M, Xuan KH, Thillet J, Delattre JY, Sanson M (2006) Prognostic stratification of patients with anaplastic gliomas according to genetic profile. Cancer 107:1891–1897
Delic S, Schmidt N, Lottmann N, Stefanski A, Stuhler K, Reifenberger G, Riemenschneider MJ (2009) In vivo and in vitro selection of infiltrating glioma cells: a comprehensive, multi-dimensional molecular analysis of the invasive phenotype of human glioblastomas. Acta Neuropathol 118:437–437 (Meeting Abstract)
Dong S, Nutt CL, Betensky RA, Stemmer-Rachamimov AO, Denko NC, Ligon KL, Rowitch DH, Louis DN (2005) Histology-based expression profiling yields novel prognostic markers in human glioblastoma. J Neuropathol Exp Neurol 64:948–955
Dubbink HJ, Taal W, van Marion R, Kros JM, van Heuvel I, Bromberg JE, Zonnenberg BA, Zonnenberg CB, Postma TJ, Gijtenbeek JM, Boogerd W, Groenendijk FH, Smitt PA, Dinjens WN, van den Bent MJ (2009) IDH1 mutations in low-grade astrocytomas predict survival but not response to temozolomide. Neurology 73:1792–1795
Ebinger M, Senf L, Wachowski O, Scheurlen W (2004) Promoter methylation pattern of caspase-8, P16INK4A, MGMT, TIMP-3, and E-cadherin in medulloblastoma. Pathol Oncol Res 10:17–21
Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG (2000) Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 343:1350–1354
Felsberg J, Erkwoh A, Sabel MC, Kirsch L, Fimmers R, Blaschke B, Schlegel U, Schramm J, Wiestler OD, Reifenberger G (2004) Oligodendroglial tumors: refinement of candidate regions on chromosome arm 1p and correlation of 1p/19q status with survival. Brain Pathol 14:121–130
Felsberg J, Rapp M, Loeser S, Fimmers R, Stummer W, Goeppert M, Steiger HJ, Friedensdorf B, Reifenberger G, Sabel MC (2009) Prognostic significance of molecular markers and extent of resection in primary glioblastoma patients. Clin Cancer Res 15:6683–6693
Felsberg J, Wolter M, Seul H, Friedensdorf B, Goppert M, Sabel MC, Reifenberger G (2010) Rapid and sensitive assessment of the IDH1 and IDH2 mutation status in cerebral gliomas based on DNA pyrosequencing. Acta Neuropathol 119:501–507
Fu Y, Huang R, Du J, Yang R, An N, Liang A (2010) Glioma-derived mutations in IDH: from mechanism to potential therapy. Biochem Biophys Res Commun 397:127–130
Gaal J, Burnichon N, Korpershoek E, Roncelin I, Bertherat J, Plouin PF, de Krijger RR, Gimenez-Roqueplo AP, Dinjens WN (2010) Isocitrate dehydrogenase mutations are rare in pheochromocytomas and paragangliomas. J Clin Endocrinol Metab 95:1274–1278
Gan HK, Kaye AH, Luwor RB (2009) The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci 16:748–754
Gerson SL (2004) MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer 4:296–307
Giannini C, Burger PC, Berkey BA, Cairncross JG, Jenkins RB, Mehta M, Curran WJ, Aldape K (2008) Anaplastic oligodendroglial tumors: refining the correlation among histopathology, 1p 19q deletion and clinical outcome in Intergroup Radiation Therapy Oncology Group Trial 9402. Brain Pathol 18:360–369
Gravendeel LA, Kloosterhof NK, Bralten LB, van Marion R, Dubbink HJ, Dinjens W, Bleeker FE, Hoogenraad CC, Michiels E, Kros JM, van den Bent M, Smitt PA, French PJ (2010) Segregation of non-p.R132H mutations in IDH1 in distinct molecular subtypes of glioma. Hum Mutat 31:E1186–E1199
Griffin CA, Burger P, Morsberger L, Yonescu R, Swierczynski S, Weingart JD, Murphy KM (2006) Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J Neuropathol Exp Neurol 65:988–994
Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin S, Schenkein DP, Su SM, Dang L, Fantin VR, Mak TW (2010) Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med 207:339–344
Haas-Kogan DA, Prados MD, Tihan T, Eberhard DA, Jelluma N, Arvold ND, Baumber R, Lamborn KR, Kapadia A, Malec M, Berger MS, Stokoe D (2005) Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst 97:880–887
Halatsch ME, Schmidt U, Behnke-Mursch J, Unterberg A, Wirtz CR (2006) Epidermal growth factor receptor inhibition for the treatment of glioblastoma multiforme and other malignant brain tumours. Cancer Treat Rev 32:74–89
Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, Weller M, Herold-Mende C, Unterberg A, Jeuken JW, Wesseling P, Reifenberger G, von Deimling A (2009) Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 118:469–474
Hatanpaa KJ, Burger PC, Eshleman JR, Murphy KM, Berg KD (2003) Molecular diagnosis of oligodendroglioma in paraffin sections. Lab Invest 83:419–428
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003
Heimberger AB, Crotty LE, Archer GE, Hess KR, Wikstrand CJ, Friedman AH, Friedman HS, Bigner DD, Sampson JH (2003) Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin Cancer Res 9:4247–4254
Heimberger AB, Hlatky R, Suki D, Yang D, Weinberg J, Gilbert M, Sawaya R, Aldape K (2005) Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin Cancer Res 11:1462–1466
Heimberger AB, Sampson JH (2009) The PEPvIII-KLH (CDX-110) vaccine in glioblastoma multiforme patients. Expert Opin Biol Ther 9:1087–1098
Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB (1996) Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 93:9821–9826
Herrlinger U, Rieger J, Koch D, Loeser S, Blaschke B, Kortmann RD, Steinbach JP, Hundsberger T, Wick W, Meyermann R, Tan TC, Sommer C, Bamberg M, Reifenberger G, Weller M (2006) Phase II trial of lomustine plus temozolomide chemotherapy in addition to radiotherapy in newly diagnosed glioblastoma: UKT-03. J Clin Oncol 24:4412–4417
Hoelzinger DB, Mariani L, Weis J, Woyke T, Berens TJ, McDonough WS, Sloan A, Coons SW, Berens ME (2005) Gene expression profile of glioblastoma multiforme invasive phenotype points to new therapeutic targets. Neoplasia 7:7–16
Horbinski C, Kelly L, Nikiforov YE, Durso MB, Nikiforova MN (2010) Detection of IDH1 and IDH2 mutations by fluorescence melting curve analysis as a diagnostic tool for brain biopsies. J Mol Diagn 12:487–492
Horbinski C, Kofler J, Kelly LM, Murdoch GH, Nikiforova MN (2009) Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol 68:1319–1325
Hunter C, Smith R, Cahill DP, Stephens P, Stevens C, Teague J, Greenman C, Edkins S, Bignell G, Davies H, O’Meara S, Parker A, Avis T, Barthorpe S, Brackenbury L, Buck G, Butler A, Clements J, Cole J, Dicks E, Forbes S, Gorton M, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Kosmidou V, Laman R, Lugg R, Menzies A, Perry J, Petty R, Raine K, Richardson D, Shepherd R, Small A, Solomon H, Tofts C, Varian J, West S, Widaa S, Yates A, Easton DF, Riggins G, Roy JE, Levine KK, Mueller W, Batchelor TT, Louis DN, Stratton MR, Futreal PA, Wooster R (2006) A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res 66:3987–3991
Ichimura K, Pearson DM, Kocialkowski S, Backlund LM, Chan R, Jones DT, Collins VP (2009) IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol 11:341–347
Ino Y, Betensky RA, Zlatescu MC, Sasaki H, Macdonald DR, Stemmer-Rachamimov AO, Ramsay DA, Cairncross JG, Louis DN (2001) Molecular subtypes of anaplastic oligodendroglioma: implications for patient management at diagnosis. Clin Cancer Res 7:839–845
International Cancer Genome Consortium (2010) International network of cancer genome projects. Nature 464:993–998
Jaeckle KA, Eyre HJ, Townsend JJ, Schulman S, Knudson HM, Belanich M, Yarosh DB, Bearman SI, Giroux DJ, Schold SC (1998) Correlation of tumor O6 methylguanine-DNA methyltransferase levels with survival of malignant astrocytoma patients treated with bis-chloroethylnitrosourea: a Southwest Oncology Group study. J Clin Oncol 16:3310–3315
Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M, Flynn H, Passe S, Felten S, Brown PD, Shaw EG, Buckner JC (2006) A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 66:9852–9861
Jeuken J, Cornelissen S, Boots-Sprenger S, Gijsen S, Wesseling P (2006) Multiplex ligation-dependent probe amplification: a diagnostic tool for simultaneous identification of different genetic markers in glial tumors. J Mol Diagn 8:433–443
Jeuken J, Sijben A, Alenda C, Rijntjes J, Dekkers M, Boots-Sprenger S, McLendon R, Wesseling P (2009) Robust detection of EGFR copy number changes and EGFR variant III: technical aspects and relevance for glioma diagnostics. Brain Pathol 19:661–671
Jeuken JW, Cornelissen SJ, Vriezen M, Dekkers MM, Errami A, Sijben A, Boots-Sprenger SH, Wesseling P (2007) MS-MLPA: an attractive alternative laboratory assay for robust, reliable, and semiquantitative detection of MGMT promoter hypermethylation in gliomas. Lab Invest 87:1055–1065
Jeuken JW, Sprenger SH, Boerman RH, von Deimling A, Teepen HL, van Overbeeke JJ, Wesseling P (2001) Subtyping of oligo-astrocytic tumours by comparative genomic hybridization. J Pathol 194:81–87
Jeuken JW, Sprenger SH, Wesseling P, Macville MV, von Deimling A, Teepen HL, van Overbeeke JJ, Boerman RH (1999) Identification of subgroups of high-grade oligodendroglial tumors by comparative genomic hybridization. J Neuropathol Exp Neurol 58:606–612
Jeuken JW, von Deimling A, Wesseling P (2004) Molecular pathogenesis of oligodendroglial tumors. J Neurooncol 70:161–181
Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, Collins VP (2008) Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68:8673–8677
Jones DT, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP (2009) Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene 28:2119–2123
Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Seo SI, Lee JY, Yoo NJ, Lee SH (2009) Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer 125:353–355
Kato Y, Jin G, Kuan CT, McLendon RE, Yan H, Bigner DD (2009) A monoclonal antibody IMab-1 specifically recognizes IDH1R132H, the most common glioma-derived mutation. Biochem Biophys Res Commun 390:547–551
Kleihues P, Ohgaki H (1999) Primary and secondary glioblastomas: from concept to clinical diagnosis. Neuro-oncol 1:44–51
Koos B, Peetz-Dienhart S, Riesmeier B, Fruhwald MC, Hasselblatt M (2010) O(6)-methylguanine-DNA methyltransferase (MGMT) promoter methylation is significantly less frequent in ependymal tumours as compared to malignant astrocytic gliomas. Neuropathol Appl Neurobiol 36:356–358
Korshunov A, Meyer J, Capper D, Christians A, Remke M, Witt H, Pfister S, von Deimling A, Hartmann C (2009) Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol 118:401–405
Korshunov A, Witt H, Hielscher T, Benner A, Remke M, Ryzhova M, Milde T, Bender S, Wittmann A, Schottler A, Kulozik AE, Witt O, von Deimling A, Lichter P, Pfister S (2010) Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol 28:3182–3190
Labussiere M, Idbaih A, Wang XW, Marie Y, Boisselier B, Falet C, Paris S, Laffaire J, Carpentier C, Criniere E, Ducray F, El Hallani S, Mokhtari K, Hoang-Xuan K, Delattre JY, Sanson M (2010) All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology 74:1886–1890
Levin N, Lavon I, Zelikovitsh B, Fuchs D, Bokstein F, Fellig Y, Siegal T (2006) Progressive low-grade oligodendrogliomas: response to temozolomide and correlation between genetic profile and O6-methylguanine DNA methyltransferase protein expression. Cancer 106:1759–1765
Li G, Wong AJ (2008) EGF receptor variant III as a target antigen for tumor immunotherapy. Expert Rev Vaccines 7:977–985
Linz U (2010) Commentary on Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial (Lancet Oncol. 2009;10:459-466). Cancer 116:1844–1846
Louis DN, Ohgaki H, Wiestler OD, CaveneeWK (eds) (2007) WHO classification of tumours of the central nervous system, 3rd edn. IARC Press, Lyon
Maxwell JA, Johnson SP, McLendon RE, Lister DW, Horne KS, Rasheed A, Quinn JA, Ali-Osman F, Friedman AH, Modrich PL, Bigner DD, Friedman HS (2008) Mismatch repair deficiency does not mediate clinical resistance to temozolomide in malignant glioma. Clin Cancer Res 14:4859–4868
McDonald KL, McDonnell J, Muntoni A, Henson JD, Hegi ME, von Deimling A, Wheeler HR, Cook RJ, Biggs MT, Little NS, Robinson BG, Reddel RR, Royds JA (2010) Presence of alternative lengthening of telomeres mechanism in patients with glioblastoma identifies a less aggressive tumor type with longer survival. J Neuropathol Exp Neurol 69:729–736
McEllin B, Camacho CV, Mukherjee B, Hahm B, Tomimatsu N, Bachoo RM, Burma S (2010) PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res 70:5457–5464
Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao PN, Beroukhim R, Peck TC, Lee JC, Sellers WR, Stokoe D, Prados M, Cloughesy TF, Sawyers CL, Mischel PS (2005) Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 353:2012–2024
Meyer J, Pusch S, Balss J, Capper D, Mueller W, Christians A, Hartmann C, von Deimling A (2010) PCR- and restriction endonuclease-based detection of IDH1 mutations. Brain Pathol 20:298–300
Mikeska T, Bock C, El-Maarri O, Hubner A, Ehrentraut D, Schramm J, Felsberg J, Kahl P, Buttner R, Pietsch T, Waha A (2007) Optimization of quantitative MGMT promoter methylation analysis using pyrosequencing and combined bisulfite restriction analysis. J Mol Diagn 9:368–381
Mollemann M, Wolter M, Felsberg J, Collins VP, Reifenberger G (2005) Frequent promoter hypermethylation and low expression of the MGMT gene in oligodendroglial tumors. Int J Cancer 113:379–385
Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven MC, Hainfellner JA, Heppner FL, Dietrich PY, Zimmer Y, Cairncross JG, Janzer RC, Domany E, Delorenzi M, Stupp R, Hegi ME (2008) Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol 26:3015–3024
Nobusawa S, Watanabe T, Kleihues P, Ohgaki H (2009) IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res 15:6002–6007
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522
Nutt CL, Mani DR, Betensky RA, Tamayo P, Cairncross JG, Ladd C, Pohl U, Hartmann C, McLaughlin ME, Batchelor TT, Black PM, von Deimling A, Pomeroy SL, Golub TR, Louis DN (2003) Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res 63:1602–1607
Ohgaki H, Kleihues P (2007) Genetic pathways to primary and secondary glioblastoma. Am J Pathol 170:1445–1453
Papait R, Magrassi L, Rigamonti D, Cattaneo E (2009) Temozolomide and carmustine cause large-scale heterochromatin reorganization in glioma cells. Biochem Biophys Res Commun 379:434–439
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, Bax DA, Coyle B, Barrow J, Hargrave D, Lowe J, Gajjar A, Zhao W, Broniscer A, Ellison DW, Grundy RG, Baker SJ (2010) Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol 28:3061–3068
Pelloski CE, Ballman KV, Furth AF, Zhang L, Lin E, Sulman EP, Bhat K, McDonald JM, Yung WK, Colman H, Woo SY, Heimberger AB, Suki D, Prados MD, Chang SM, Barker FG 2nd, Buckner JC, James CD, Aldape K (2007) Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol 25:2288–2294
Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, Toedt G, Wittmann A, Kratz C, Olbrich H, Ahmadi R, Thieme B, Joos S, Radlwimmer B, Kulozik A, Pietsch T, Herold-Mende C, Gnekow A, Reifenberger G, Korshunov A, Scheurlen W, Omran H, Lichter P (2008) BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 118:1739–1749
Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, Aldape K (2006) Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9:157–173
Pollack IF, Hamilton RL, Sobol RW, Burnham J, Yates AJ, Holmes EJ, Zhou T, Finlay JL (2006) O6-methylguanine-DNA methyltransferase expression strongly correlates with outcome in childhood malignant gliomas: results from the CCG-945 cohort. J Clin Oncol 24:3431–3437
Preusser M, Gelpi E, Rottenfusser A, Dieckmann K, Widhalm G, Dietrich W, Bertalanffy A, Prayer D, Hainfellner JA, Marosi C (2008) Epithelial growth factor receptor inhibitors for treatment of recurrent or progressive high grade glioma: an exploratory study. J Neurooncol 89:211–218
Reifenberger G, Louis DN (2003) Oligodendroglioma: toward molecular definitions in diagnostic neuro-oncology. J Neuropathol Exp Neurol 62:111–126
Reitman ZJ, Yan H (2010) Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst 102:932–941
Rivera AL, Pelloski CE, Gilbert MR, Colman H, De La Cruz C, Sulman EP, Bekele BN, Aldape KD (2010) MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neuro Oncol 12:116–121
Roerig P, Nessling M, Radlwimmer B, Joos S, Wrobel G, Schwaenen C, Reifenberger G, Lichter P (2005) Molecular classification of human gliomas using matrix-based comparative genomic hybridization. Int J Cancer 117:95–103
Rokes CA, Remke M, Guha-Thakurta N, Witt O, Korshunov A, Pfister S and Wolff JE (2010) Sorafenib plus valproic acid for infant spinal glioblastoma. J Pediatr Hematol Oncol
Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Bigner DD (2008) Tumor-specific immunotherapy targeting the EGFRvIII mutation in patients with malignant glioma. Semin Immunol 20:267–275
Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, El Hallani S, Boisselier B, Mokhtari K, Hoang-Xuan K, Delattre JY (2009) Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 27:4150–4154
Scheie D, Cvancarova M, Mork S, Skullerud K, Andresen PA, Benestad I, Helseth E, Meling T, Beiske K (2008) Can morphology predict 1p/19q loss in oligodendroglial tumours? Histopathology 53:578–587
Schlosser S, Wagner S, Muhlisch J, Hasselblatt M, Gerss J, Wolff JE, Fruhwald MC (2010) MGMT as a potential stratification marker in relapsed high-grade glioma of children: the HIT-GBM experience. Pediatr Blood Cancer 54:228–237
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G (2002) Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 30:e57
Seiz M, Tuettenberg J, Meyer J, Essig M, Schmieder K, Mawrin C, von Deimling A, Hartmann C (2010) Detection of IDH1 mutations in gliomatosis cerebri, but only in tumors with additional solid component: evidence for molecular subtypes. Acta Neuropathol 120:261–267
Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, Makino K, Saya H, Hirano H, Kuratsu J, Oka K, Ishimaru Y, Ushio Y (2003) Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res 63:6962–6970
Smith JS, Alderete B, Minn Y, Borell TJ, Perry A, Mohapatra G, Hosek SM, Kimmel D, O’Fallon J, Yates A, Feuerstein BG, Burger PC, Scheithauer BW, Jenkins RB (1999) Localization of common deletion regions on 1p and 19q in human gliomas and their association with histological subtype. Oncogene 18:4144–4152
Smith JS, Tachibana I, Passe SM, Huntley BK, Borell TJ, Iturria N, O’Fallon JR, Schaefer PL, Scheithauer BW, James CD, Buckner JC, Jenkins RB (2001) PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst 93:1246–1256
Sonoda Y, Kumabe T, Nakamura T, Saito R, Kanamori M, Yamashita Y, Suzuki H, Tominaga T (2009) Analysis of IDH1 and IDH2 mutations in Japanese glioma patients. Cancer Sci 100:1996–1998
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996
Sugawa N, Ekstrand AJ, James CD, Collins VP (1990) Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci USA 87:8602–8606
Tentori L, Leonetti C, Scarsella M, D’Amati G, Vergati M, Portarena I, Xu W, Kalish V, Zupi G, Zhang J, Graziani G (2003) Systemic administration of GPI 15427, a novel poly(ADP-ribose) polymerase-1 inhibitor, increases the antitumor activity of temozolomide against intracranial melanoma, glioma, lymphoma. Clin Cancer Res 9:5370–5379
Toedt G, Barbus S, Wolter M, Felsberg J, Tews B, Blond F, Sabel MC, Hofmann S, Becker N, Hartmann C, Ohgaki H, von Deimling A, Wiestler OD, Hahn M, Lichter P, Reifenberger G, Radlwimmer B (2010) Molecular signatures classify astrocytic gliomas by IDH1 mutation status. Int J Cancer
Trost D, Ehrler M, Fimmers R, Felsberg J, Sabel MC, Kirsch L, Schramm J, Wiestler OD, Reifenberger G, Weber RG (2007) Identification of genomic aberrations associated with shorter overall survival in patients with oligodendroglial tumors. Int J Cancer 120:2368–2376
van den Bent MJ, Carpentier AF, Brandes AA, Sanson M, Taphoorn MJ, Bernsen HJ, Frenay M, Tijssen CC, Grisold W, Sipos L, Haaxma-Reiche H, Kros JM, van Kouwenhoven MC, Vecht CJ, Allgeier A, Lacombe D, Gorlia T (2006) Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol 24:2715–2722
van den Bent MJ, Dubbink HJ, Marie Y, Brandes AA, Taphoorn MJ, Wesseling P, Frenay M, Tijssen CC, Lacombe D, Idbaih A, van Marion R, Kros JM, Dinjens WN, Gorlia T, Sanson M (2010) IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumors: a report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clin Cancer Res 16:1597–1604
van den Bent MJ, Dubbink HJ, Sanson M, van der Lee-Haarloo CR, Hegi M, Jeuken JW, Ibdaih A, Brandes AA, Taphoorn MJ, Frenay M, Lacombe D, Gorlia T, Dinjens WN, Kros JM (2009) MGMT promoter methylation is prognostic but not predictive for outcome to adjuvant PCV chemotherapy in anaplastic oligodendroglial tumors: a report from EORTC Brain Tumor Group Study 26951. J Clin Oncol 27:5881–5886
van den Bent MJ, Looijenga LH, Langenberg K, Dinjens W, Graveland W, Uytdewilligen L, Sillevis Smitt PA, Jenkins RB, Kros JM (2003) Chromosomal anomalies in oligodendroglial tumors are correlated with clinical features. Cancer 97:1276–1284
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110
Vlassenbroeck I, Califice S, Diserens AC, Migliavacca E, Straub J, Di Stefano I, Moreau F, Hamou MF, Renard I, Delorenzi M, Flamion B, DiGuiseppi J, Bierau K, Hegi ME (2008) Validation of real-time methylation-specific PCR to determine O6-methylguanine-DNA methyltransferase gene promoter methylation in glioma. J Mol Diagn 10:332–337
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, Sharp KA, Levine RL, Thompson CB (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17:225–234
Watanabe T, Nobusawa S, Kleihues P, Ohgaki H (2009) IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174:1149–1153
Watanabe T, Vital A, Nobusawa S, Kleihues P, Ohgaki H (2009) Selective acquisition of IDH1 R132C mutations in astrocytomas associated with Li-Fraumeni syndrome. Acta Neuropathol 117:653–656
Weller M, Berger H, Hartmann C, Schramm J, Westphal M, Simon M, Goldbrunner R, Krex D, Steinbach JP, Ostertag CB, Loeffler M, Pietsch T, von Deimling A (2007) Combined 1p/19q loss in oligodendroglial tumors: predictive or prognostic biomarker? Clin Cancer Res 13:6933–6937
Weller M, Felsberg J, Hartmann C, Berger H, Steinbach JP, Schramm J, Westphal M, Schackert G, Simon M, Tonn JC, Heese O, Krex D, Nikkhah G, Pietsch T, Wiestler O, Reifenberger G, von Deimling A, Loeffler M (2009) Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J Clin Oncol 27:5743–5750
Weller M, Stupp R, Reifenberger G, Brandes AA, van den Bent MJ, Wick W, Hegi ME (2010) MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nat Rev Neurol 6:39–51
Weppler SA, Li Y, Dubois L, Lieuwes N, Jutten B, Lambin P, Wouters BG, Lammering G (2007) Expression of EGFR variant vIII promotes both radiation resistance and hypoxia tolerance. Radiother Oncol 83:333–339
Wick W, Hartmann C, Engel C, Stoffels M, Felsberg J, Stockhammer F, Sabel MC, Koeppen S, Ketter R, Meyermann R, Rapp M, Meisner C, Kortmann RD, Pietsch T, Wiestler OD, Ernemann U, Bamberg M, Reifenberger G, von Deimling A, Weller M (2009) NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol 27:5874–5880
Wikstrand CJ, Hale LP, Batra SK, Hill ML, Humphrey PA, Kurpad SN, McLendon RE, Moscatello D, Pegram CN, Reist CJ et al (1995) Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res 55:3140–3148
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773
Yip S, Iafrate AJ, Louis DN (2008) Molecular diagnostic testing in malignant gliomas: a practical update on predictive markers. J Neuropathol Exp Neurol 67:1–15
Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL, Louis DN (2009) MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res 15:4622–4629
Yoshimoto K, Dang J, Zhu S, Nathanson D, Huang T, Dumont R, Seligson DB, Yong WH, Xiong Z, Rao N, Winther H, Chakravarti A, Bigner DD, Mellinghoff IK, Horvath S, Cavenee WK, Cloughesy TF, Mischel PS (2008) Development of a real-time RT-PCR assay for detecting EGFRvIII in glioblastoma samples. Clin Cancer Res 14:488–493
Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, Ding J, Lei Q, Guan KL, Xiong Y (2009) Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 324:261–265
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Riemenschneider, M.J., Jeuken, J.W.M., Wesseling, P. et al. Molecular diagnostics of gliomas: state of the art. Acta Neuropathol 120, 567–584 (2010). https://doi.org/10.1007/s00401-010-0736-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-010-0736-4