
Abstract
Water plays a central role in wood research, since it affects all material properties relevant to the performance of wood materials. Therefore, experimental techniques for characterising water within wood are an essential part of nearly all scientific investigations of wood materials. This review focuses on selected experimental techniques that can give deeper insights into various aspects of water in wood in the entire moisture domain from dry to fully water-saturated. These techniques fall into three broad categories: (1) gravimetric techniques that determine how much water is absorbed, (2) fibre saturation techniques that determine the amount of water within cell walls, and (3) spectroscopic techniques that provide insights into chemical wood–water interactions as well as yield information on water distribution in the macro-void wood structure. For all techniques, the general measurement concept is explained, its history in wood science as well as advantages and limitations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Wood is a hygroscopic material which means that it absorbs and exchanges water molecules with the surroundings. Water molecules found within cell walls interfere with internal secondary wood–wood bonds, for example hydrogen bonds. Therefore, it is no surprise that absorbed water, often termed moisture, has a profound effect on wood properties dependent on these bonds, for example strength and stiffness (Tiemann 1906; Wagner et al. 2015). However, moisture also influences the performance of wood and wood products in other ways important for the structural application of these materials such as durability, i.e. resistance to biodegradation (Ammer 1963a; Meyer and Brischke 2015; Stamm and Baechler 1960; Stienen et al. 2014; Thybring 2013). The central role of moisture in the performance of wood materials makes the wood–water interaction an obvious target for enhancing material performance to specific applications. Thus, a wide range of physicochemical modification processes exist aimed at improving the performance of wood materials. Common targets for wood modification include improved durability and dimensional stability (Stamm and Baechler 1960). However, recent years have seen an increase in advanced modifications adding novel functionalities to wood such as stimuli-responsive properties, for example “sweating wood” for cooling purposes or stimuli-responsive filtration (Keplinger et al. 2016). For all these types of modifications, wood–water interactions within the modified material are important for the performance. For instance, the resistance to biological decay is directly linked to the water capacity of modified cell walls (Stamm and Baechler 1960; Thybring 2013), while advanced modifications such as stimuli-responsive hydrogel formation within the wood structure use water uptake or release for cooling purposes by water evaporation or for filtration (Keplinger et al. 2016). To advance the understanding of the fundamental mechanisms of wood performance as affected by water as well as for tailoring wood modifications to specific applications, characterisation of the amount, distribution, and physicochemical interactions of water within wood materials is important.
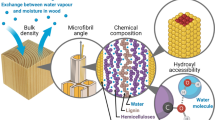
This review covers selected experimental techniques for characterising moisture in wood in the entire moisture content range from absolutely dry to fully water-saturated, i.e. where water takes up all available space in cell walls and voids (pits, lumina, vessels) in the wood structure. Furthermore, techniques which elucidate other fundamental aspects of wood–water interactions in untreated and modified wood are included. Experimental techniques which concern effects of water on, for example, mechanics or dimensions will not be discussed. The schematic sorption isotherm shown in Fig. 1 covering the entire relative humidity (RH) range provides an overview of the characterisation techniques covered in this review as well as the RH range to which each technique can be applied.

Schematic illustration of moisture uptake in wood in absorption from absolute dry state (lower black line) and desorption from fully water-saturated state (upper black line) in the entire relative humidity (RH) range from dry to water-saturated. Characterisation (gravimetric, fibre saturation, and spectroscopic) techniques described in the present review are indicated along with the RH range they cover
Terminology
One of the most fundamental and commonly used descriptors for moisture in wood is the moisture content (MC). In this review, it is described by water mass divided by dry wood mass. When dealing with physicochemically modified wood materials, it is often necessary to adjust MC for added dry mass due to the modification process (Hill 2008; Thybring 2013) in order to correctly compare a given modified material with untreated or differently modified materials. This is done by multiplying MC of the modified material, MCmod [%], with (1 + R mod) as shown in (1):
where MCR [%] is adjusted moisture content also termed “reduced moisture content”, m water [kg] is water mass, and m wood,dry [kg] and m mod,dry [kg] are dry masses of the wood itself and the modified wood material, respectively. R mod [−] is the modification ratio defined as relative mass gain due to modification
Mass change is commonly used in the literature to describe the intensity of modification either by the term “weight per cent gain” (WPG) [% WPG] in cases of increased mass due to modification, for example acetylation, furfurylation, etc., or by the term “mass loss” (ML) [% ML] in cases of decreased mass due to modification, for example thermal modification. It was chosen to describe the mass change by the common term “modification ratio” (R mod) and define it as a dimensionless fraction to avoid confusion with the relative mass gain given in per cent (WPG). Correction of MC in (1) and following equations should, however, only be done in cases where R mod is positive. Thus, MC should not be corrected for thermally modified wood where R mod < 0.
As water molecules absorbed by wood are continuously exchanged with the surroundings as well as meandering around within the material, MC is often characterised in the equilibrium state where there is no net change in water with the surroundings. This alleviates issues of comparing various samples with differences in moisture gradients depending on sample geometry, wood density, etc. In case of equilibrium, it is common to use the terms “equilibrium moisture content” (EMC) and “reduced equilibrium moisture content” (EMCR) in (1) instead of MC and MCR, respectively.
In this review, the term “absorption” is used to describe the process of moisture uptake in wood instead of the term “adsorption” more commonly used in the wood research literature. The IUPAC definition of “absorption” relates to the uptake of one material by another, regardless of the mechanism of uptake. On the other hand, the term “adsorption” in the terminology suggested by IUPAC relates to the uptake of one material on surfaces/interphases with another material and thus implies that water molecules attach to surfaces within the wood structure. As a result of the developments in the field of water uptake in wood cell walls in the last few decades, it was felt that the term “adsorption” is misleading as it relates to “internal surfaces”, a concept which can be questioned when water molecules enter the solid wood cell walls and are taken up by the constituent polymers. Therefore, the more general term “absorption” was adopted to include both water uptake in cell walls and capillary condensation in the wood structure.
This review covers the entire moisture range from dry to fully water-saturated in dependence of the surrounding climate. In line with common terminology in building materials research, the full RH range is divided into a hygroscopic and an over-hygroscopic range (Fig. 1). These two regimes intersect around 97–98% RH although the limiting RH is not well defined (Espinosa and Franke 2006). The differentiation reflects a change in water uptake in the material, where the hygroscopic range is dominated by cell wall absorption and the over-hygroscopic range is dominated by capillary condensation outside cell walls, i.e. in pits, lumina, vessels, and other voids in the wood structure.
Gravimetric techniques
Wood can absorb significant amounts of water in its structure, i.e. both within cell walls and in the void structure. The maximum amount depends on the wood density, which varies widely between species. As an example, a low-density wood species like balsa (Ochroma lagopus Sw.) with a bulk density around 100 kg/m3 can fit a total amount of water of around 960–980 kg/m3 in its bulk volume, equal to a maximum MC of 960–980%. On the other hand, a high-density wood species such as ironwood (Lophira alata Banks ex. Gaertn.) with a bulk density around 1000 kg/m3 can only fit about 630–780 kg/m3 water inside its bulk volume, yielding a maximum MC of just 63–78%. The noticeable change in mass associated with absorption of water makes gravimetric techniques ideal for determining the amount of water inside the bulk material.
Early-stage sorption balances, salt solutions, and climate chambers
In the early scientific investigations of wood materials, it is recognised that the MC of wood exposed to climatic conditions changes with changes in temperature and RH (Forest Products Laboratory 1919; Glass et al. 2014; Zeller 1920). In the early days of wood research, it was, however, a common practice to achieve a specific MC in wood samples by drying the material from green or water-saturated conditions (Carrington 1922; Record 1914; Tiemann 1906; Volbehr 1896) and then maybe leaving the wood for moisture to redistribute within the material before further testing, for example mechanical. Alternatively, wood samples were kept under laboratory conditions for a period of time to obtain an MC around 12% (Carrington 1921). The first published study in a scientific journal on wood MC in equilibrium with a controlled, stable climate appears to be the work of Zeller (1920), who used sulphuric acid solutions of various concentrations to generate various levels of RH in a specially designed climate chamber. Similar investigations performed at the Forest Products Laboratory in the USA might, however, predate this publication (Glass et al. 2014). In these early investigations, the now well-known effect of sorption history, i.e. that conditioning wood to a specific RH through moisture uptake (absorption) or through moisture release (desorption) will not produce similar MC, was not considered. Therefore, the relation between wood MC and RH at constant temperature was described by a single curve, i.e. sorption isotherm. The seemingly first absorption and desorption isotherms were reported by Larian and co-workers (Larian et al. 1930; Lavine and Gauger 1930) who conditioned birch shavings (about 1.5 g each) in desiccators over saturated salt solutions generating different RH levels. Initially, all samples were in the green condition, and after equilibrium with the specific RH had been obtained, they were dried over pure sulphuric acid (0% RH) followed by conditioning by absorption to the same level of RH as before. These data can therefore be regarded as the first desorption and absorption isotherms for wood, where the previous is initiated from a condition close to fully water-saturated. The same year, Pidgeon and Maass (1930) reported on absorption and desorption isotherms for white spruce and jack pine found by exposing small samples (150 mg) suspended from a quartz helix spring inside a vacuum tube system to various levels of RH. Desorption was, however, initiated from hygroscopic conditions (< 98% RH), the effect of which will be discussed later. RH in the vacuum tube system was controlled by tuning the vapour pressure above a water bath via controlling the bath temperature while keeping the rest of the system at constant temperature. Moisture uptake was measured by the spring elongation, and the system is a precursor to modern-day automated sorption balances. Several later studies on wood use this kind of technique for studying absorption and desorption in wood (Christensen 1959, 1960; Christensen and Kelsey 1959; Grace and Maass 1932; Kelly and Hart 1970; Spalt 1958). For modified wood, the first sorption isotherm appears to be due to Mörath (1931) who steamed beech at 150 and 190 °C and used sulphuric acid solutions for conditioning the material at different RH levels. The second reported sorption isotherm for modified wood seems to be the absorption and desorption isotherms reported by Spalt (1958) for acetylated (R mod = 0.32) white pine, although studies from the intervening period of time mention reduced hygroscopicities of modified wood (Buro 1954; Seborg et al. 1953) without showing sorption isotherms.
Of the different methods for generating specific RH levels detailed above, one of the most common methods is by saturated salt solutions (Ammer 1963b; Eriksson and Noren 1965; Forster 1998; Higgins 1957; Nearn 1955; Papadopoulos and Hill 2003; Wangaard and Granados 1967; Yasuda et al. 1995) since they are relatively simple to use and cover a wide range of RH levels (see, for example, Greenspan 1977) for solutions in the range 3–98% RH. Their use limits, however, which RH levels can be achieved. This problem is avoided when RH is generated by mixing of wet and dry air streams to produce any RH level desired in the range 0–97% RH. Such method has been employed in climate chambers (Strømdahl 2000; Thygesen et al. 2010) and has even be used below the freezing temperature of water (Hedlin 1968).
Whether using early-stage sorption balances, saturated salt solutions, or climate chambers for conditioning wooden samples, it is important to select appropriate sample masses taking into consideration the balance uncertainty and the stability of the conditioning climate in order to minimise the uncertainty in determined MC. The ASTM C1498 (ASTM 2016) and ISO 12571 (ISO 2013) standards which concern the determination of hygroscopic moisture sorption isotherms require the balance accuracy to be at least 0.01–0.1% of the sample mass. However, these standards also require a sample mass of 10 g or more, which is often not feasible or wanted in scientific studies, for example on sorption kinetics. To determine EMC, it is of course important that equilibrium has been obtained as marked by a constant sample mass after conditioning at constant RH for a given time. Many studies employ stability criteria adopted from the ASTM C1498 (ASTM 2016) and ISO 12571 (ISO 2013) standards to specify when the variation in sample mass is low enough for equilibrium to be assumed obtained. These standards require mass variations of < 0.1% over a period of 5 days after daily mass readings or over 7 days in two consecutive mass readings, respectively. Other stability criteria have been proposed in the literature, for example the one by Wadsö et al. (2004) which also takes into account the change in RH from the previous to the current climate.
One important thing to note about the use of early-stage sorption balances, saturated salt solutions, and climate chambers is that these methods are unsuitable for conditioning wood samples to saturation. Nonetheless, several studies condition wood samples in desiccators over liquid water as a way of saturating the material (Boonstra et al. 2007; Feist and Sell 1987; Harju et al. 2002; Kamdem et al. 2002; Meyer and Brischke 2015; Williams and Hale 2003), but the resultant EMC after such conditioning is questionable. This is due to the great difficulty with obtaining saturated water vapour as discussed by Strømdahl (2000). Differences in temperature between vapour and surrounding surfaces will result in either water condensation on surfaces if these are cooler or a non-saturated vapour if the surfaces are warmer. Even slight temperature fluctuations can give noticeable effects. Thus, fluctuations of 0.5 °C around room temperature cause a “saturated RH” around 97%, while even small changes of 0.2 °C limit the maximum RH to 99%. As shown by Hoffmeyer et al. (2011), prolonged conditioning of untreated wood in such presumably saturated water vapour will not give the same isotherm upon subsequent desorption as when the material is desorbing from the fully water-saturated state. Moreover, as discussed later under “Fibre saturation techniques”, it is highly uncertain that conditioning wood in presumably saturated water vapour will actually saturate cell walls.
Automated sorption balances
This type of equipment, often referred to as dynamic vapour sorption (DVS), combines automated climate control in the range 0–97% RH with continuous high-resolution mass measurements. RH is generated by mixing wet and dry gas streams, for example air or nitrogen. As only a small sample mass (typically in the 5–20 mg range) is needed like in the quartz helix spring systems, a faster approach to equilibrium is observed compared with conditioning larger samples over saturated salt solutions or in climate chambers. As automated sorption balances are becoming a common research tool in wood research laboratories, the literature is abundant with sorption isotherms determined with such equipment, also for various types of modified wood (Hill et al. 2013; Jalaludin et al. 2010a, b; Popescu and Hill 2013; Xie et al. 2010, 2011). While the low sample mass makes possible detailed studies of differences in water sorption between earlywood and latewood (Fredriksson and Thygesen 2017), even at the growth ring level (Hill et al. 2015; Song et al. 2014), it also requires the user to consider how to collect representative samples for the study in question. For generating representative samples of larger wood volumes, several studies have used milled wood (Hill et al. 2009; Himmel and Mai 2015; Xie et al. 2010; Zaihan et al. 2009, 2011), but it could be speculated if this gives a representative material or might produce unwanted side effects, for example a change in sample chemistry. Milling is not as intense a method for subdividing wood as ball milling which has been shown to change the material chemistry (Mao et al. 2006) and internal chemical bonding (Schwanninger et al. 2004). Nonetheless, different particle size fractions generated from milling might not have identical chemical composition if some anatomical fractions (e.g. lignin-rich middle lamella fragments) are over-represented in smaller size fractions that are more easily lost upon handling the milled material. Instead of milling, several studies have used wood slices cut with a microtome or razor blade to produce a large sample surface area while preserving the microscopic wood structure (Fredriksson and Thygesen 2017; Fredriksson et al. 2010; Glass et al. 2017; Hill et al. 2015; Hosseinpourpia et al. 2016; Song et al. 2014), while a few use small wood blocks (Fredriksson and Johansson 2016). All of these studies use the automated humidity control by which RH is automatically changed upon reaching a specified mass stability (dm/dt) criterion. If such dm/dt criterion is not selected carefully, however, it can lead to inaccurate results. If the stability criteria of the ASTM C1498 (ASTM 2016) and ISO 12571 (ISO 2013) standards are converted into %/min, i.e. per cent mass change per minute, employed in most automated sorption balances, the dm/dt is around 0.00001%/min. While the stability criteria of the standards are based on mass change relative to the previously observed mass, the dm/dt of automated sorption balances is typically based on mass change relative to the dry sample mass which is often determined by initial drying. Thus, it would seem that the latter gives stricter stability requirements. However, the dm/dt suggested by the producer of one of the most common types of automated sorption balances is 0.002%/min for 10–20 mg samples and 0.0005%/min for 100 mg samples, i.e. about 50–200 times higher than required by the standards for larger (> 10 g) samples. The other important parameter for establishing if equilibrium is obtained is the “stability window”, i.e. the time period over which the dm/dt needs to meet the stability criterion. In the ASTM C1498 (ASTM 2016) and ISO 12571 (ISO 2013) standards, the mass change is calculated over 5 and 7 days, respectively, whereas the default stability window in the most popular automated sorption balance in wood research is 15 min. The 0.002%/min over 15-min stability criterion is commonly employed for wood materials, but Glass et al. (2017) recently demonstrated that it gives too low EMC values in absorption and too high in desorption. The error was up to 0.7% MC for southern pine wood and up to 2.9% MC for isolated lignin for changes in the high-RH regime. Moreover, Glass et al. (2017) were unable to find literature data to support the claim that a dm/dt of 0.002%/min is sufficient for accurate determinations of EMC, even in publications often cited to document this claim. The use of a dm/dt criterion becomes critical when considering sample drying which in many studies is performed initially. The results of Christensen and Hergt (1969) show that the longer the time under constant climate before a given change in RH, the slower the rate of sorption. Thus, if a sample is initially dried out using a dm/dt criterion after experiencing stable conditions for a long while, it will exhibit a slower rate of desorption than in a subsequent second drying stage after re-wetting. This will affect the determined dry mass in the two drying stages as the dm/dt criterion is met at higher residual MC in the first stage, thus causing an overestimation of the dry mass. Examining the mass curves in the literature when a dm/dt criterion of 0.002%/min is used, it is often clearly seen that the sample material is far from having reached equilibrium with 0% RH before subsequent re-wetting is initiated. The overestimated dry mass is problematic as it shifts the entire sorption isotherm downwards, partly due to the increased dry mass but mainly due to decreased estimated water masses. Moreover, a second drying stage will yield a lower dry mass, perhaps leading to speculations about “trapped water” in the first drying stage (Hill et al. 2015). It is recognised that Hergt and Christensen (1965) did find that up to 1% MC is retained in the wood after vacuum-drying if the material is not rapidly dried from the water-saturated state, i.e. conditioning to any level of RH before drying results in incomplete drying even at a drying temperature of 65 °C. However, their experiments used thin, microtomed wood sections and drying times of 1–5 days which is considerably longer than the drying duration if using a dm/dt criterion of 0.002%/min. In the authors’ experience, drying for 6 h at 60 °C in dry nitrogen gas of a 20-mg wood block sample gives a reproducible dry mass within 0.05% even when several cycles of re-wetting to various RH (10–90%) and drying are used. For samples cut into thin slices, this drying protocol (6 h at 60 °C, 0% RH) should be more than enough to secure complete drying. Therefore, it is recommended that longer, fixed drying times, preferably at slightly elevated temperatures, are used in automated sorption balances in order to secure accurate dry mass determinations. For getting closer to the “true” EMC upon conditioning at a specific RH, it might be useful to lower the dm/dt as well as increase the stability window. However, further investigations are needed for establishing guidelines for the appropriate dm/dt settings for wood. Until such have been established, a degree of uncertainty exists as to the accuracy of reported EMC and sorption isotherms by the use of automated sorption balances.
The difference in MC in equilibrium with a given RH depends on whether equilibrium is reached by absorption or desorption (Urquhart 1929). This phenomenon is termed sorption hysteresis and can be seen as a difference between the absorption isotherm from dry state and desorption isotherm from water-saturated state (see Fig. 2). It has been speculated that it is linked to non-elastic mechanical responses during swelling, but the exact mechanism is not yet understood (Engelund et al. 2013). Nonetheless, wood modification can have different effects on sorption hysteresis. For instance, acetylation decreases the absolute hysteresis (Popescu et al. 2014), i.e. MC difference between the absorption and desorption isotherms, but this result appears to be correlated with the general reduction in MC at given RH. A similar general reduction in MC is found for thermal modification of wood; however, this treatment appears to increase the absolute sorption hysteresis (Olek et al. 2013). Automated sorption balances are useful for studies of sorption hysteresis, and several published studies have focused on this phenomenon. In most studies, absolute sorption hysteresis has a maximum around 70–75% RH and decreases for higher RH (Hill et al. 2009, 2012b; Himmel and Mai 2015; Hosseinpourpia et al. 2016; Popescu et al. 2014; Simon et al. 2017; Xie et al. 2011). The decreasing sorption hysteresis at high RH might, however, be an artefact due to desorption being initiated from a non-saturated state (often by conditioning to 95% RH). Thus, in recent results by Fredriksson and co-workers (Fredriksson and Johansson 2016; Fredriksson and Thygesen 2017), sorption hysteresis in untreated Norway spruce gradually increases up to at least 95% RH (see Fig. 2). Furthermore, as shown by Hoffmeyer et al. (2011), desorption from a non-saturated state will yield a lower desorption isotherm than for water-saturated wood until around 75% RH. Therefore, most desorption isotherms using automated sorption balances reported in the literature can be regarded as scanning curves above 70–75% RH produced by a reversal in sorption direction (absorption/desorption) at intermediate RH, i.e. away from the dry or saturated states.

a Sorption isotherms for desorption from water-saturated state and absorption from dry state for untreated and acetylated (R mod = 0.18) Norway spruce in the range of 0–97% relative humidity (RH). Based on data from Fredriksson and Johansson (2016), Fredriksson et al. (2013) at 20 °C and from authors at 25 °C (dm/dt = 0.0005%/min). Note that the desorption and absorption isotherms do not coalesce at 97% RH; b absolute sorption hysteresis of untreated wood from moisture content data acquired with automated sorption balances. Based on data for Norway spruce (Fredriksson and Johansson 2016; Fredriksson and Thygesen 2017; Källbom 2015; Sedighi-Gilani and Schwarze 2015) and Sitka spruce (Hill et al. 2009). Note that in all studies except (Fredriksson and Johansson 2016; Fredriksson and Thygesen 2017) desorption is initiated from a non-saturated state (conditioning to 95% RH)
Besides deriving MC in equilibrium with various climates, automated sorption balances have been used for studying the kinetics of sorption. In many polymeric materials, the sorption process exhibits two-stage behaviour (Christensen and Kelsey 1959; Crank 1953; Kelly and Hart 1970; Newns 1956) and cannot be explained by Fickian diffusion into the material. Thus, sorption is often a much slower process than expected for diffusion through the solid material which is also the case for wood (Christensen 1959; Kelly and Hart 1970). Instead, sorption might be controlled by relaxation of stresses arising from swelling of the dense and stiff wood cell walls (Christensen 1959, 1960; Christensen and Kelsey 1959). This also explains why samples conditioned for longer times after a change in RH exhibit slower rate of sorption as observed by Christensen and Hergt (1969). Due to the two-stage nature of the sorption process, it has often been analysed by the parallel exponential kinetics (PEK) model featuring two moisture sorption processes (fast and slow exponentials) using data for both untreated (Hill et al. 2010) and modified wood (Hill et al. 2012a; Himmel and Mai 2016; Jalaludin et al. 2010a; Popescu et al. 2014; Xie et al. 2010). It may be difficult to compare the results across studies due to differences in sample mass and geometry as diffusion into the sample at some point becomes relevant, at least in the low-RH regime (Christensen 1965). More importantly, however, the recent results by Glass et al. (2017) demonstrate that the PEK model is not capable of capturing the long-term kinetics of untreated wood if the data acquisition at a given RH is disrupted when a dm/dt stability criterion of 0.002%/min is met. This seriously questions whether results derived using PEK model fittings on sorption data with a 0.002%/min dm/dt criterion are correct. Adding to this uncertainty, the PEK model is quite sensitive to initial guesses of parameter values of the two moisture sorption processes (Himmel and Mai 2016).
More details about wood–water interactions can be gained from automated sorption balances using heavy water, i.e. deuterium oxide (2H2O or D2O), instead of normal water (1H2O or H2O) as conditioning vapour. Hereby, hydrogen of the hydroxyl (OH) groups interacting with heavy water will be exchanged for deuterium if the material is continuously exposed to 2H2O vapour for sufficient time. As deuterium (2H) is about 1 g/mol heavier than protium (1H), the material dry mass increases slightly upon deuteration (~ 1% for untreated wood), which can only be measured with enough accuracy using very sensitive balances. Therefore, vapour-phase deuteration has been used on untreated wood in an early-stage sorption balance (Taniguchi et al. 1978) as well as recently on untreated (Thybring et al. 2017) and modified wood (Popescu et al. 2014; Rautkari et al. 2013) using modern automated sorption balances, yielding information about how many hydroxyls are accessible for water. It should be noted, however, that not all accessible hydroxyls will have their hydrogen exchanged. For instance, Lindh et al. (2016) found that only two out of three surface hydroxyls on cellulose microfibrils are accessible for deuteration as O(3)H hydroxyls act solely as hydrogen bond acceptors, i.e. interacting exclusively with water via the O(3) (Lindh et al. 2016). Therefore, out of around 6 mmol/g accessible hydroxyls in wood cellulose (Engelund et al. 2013), only 4 mmol/g will contribute to the increased dry mass after deuteration. In order to determine the hydroxyl accessibility, the sample has to be dried initially and after deuteration for determining the change in dry mass. Hereafter, the hydroxyl accessibility, OHaccess [mol/g], can be calculated as
where Δm dry [g] is the change in dry mass due to deuteration, m 0,dry [g] is initial dry mass, ΔM hydrogen [g/mol] is molar mass difference between deuterium and protium (1.006 g/mol), and (1 + R mod)−1 [−] corrects for mass gain due to modification if present. The change in dry mass is dependent on not only the added deuterium on hydroxyls, but also the residual normal and heavy water in the first and last drying stages, respectively, i.e.
where Δm dry,true [g] is the “true” dry mass increase due to hydroxyl deuteration, m 1,water and m 2,water [g] are residual water masses after first and last drying stages, respectively, c [mol/g] is molar concentration of residual water per gram of wood, M [g/mol] is molar mass, and indices “D2O” and “H2O” refer to heavy water and normal water, respectively. Even if the concentrations are similar after both drying stages, the dry mass is still increased due to deuterium associated with the heavy water. Thus, a residual water content corresponding to 1% MC of normal water will add 0.11% mass. Compared with an expected dry mass increase due to deuteration in the range of 0.5–1.0%, this will yield an error of 11–22%. If only 0.1% MC residual water is left after drying, the error is one order of magnitude lower. However, this is only for similar concentrations of normal and heavy water after drying, which is an unlikely situation, even after similar drying conditions and durations in both first and last drying stages, since transport of heavy water is about 20% slower than that of normal water (Mills 1973). Therefore, if assuming a conservative estimate of 50% higher concentration of heavy water than of normal water after the drying stage, the measurement errors in determined hydroxyl accessibility for residual water contents of 1 and 0.1% MC would be 66–133 and 7–13%, respectively. This clearly demonstrates the need for sufficient drying of the material to eliminate residual water as a significant source of error. Therefore, the use of a dm/dt stability criterion for determining hydroxyl accessibility is not recommended due to the uncertainties it involves as discussed previously for generating sorption isotherm data. As mass differences involved in the determination of hydroxyl accessibility are much lower than those for sorption isotherms, the potential errors introduced are much higher.
The following is a short guideline for using heavy water in automated sorption balances. In general, it is advisable to use at least three but preferably more replicates for each sample batch due to measurement uncertainties. Only one conditioning stage in D2O vapour is needed as the determined accessibility depends on time of exposure and not on number of repeated conditioning cycles (Pönni et al. 2013). Moreover, the carrier gas (often nitrogen) should be dried out before the gas inlet to the automated sorption balance, e.g. with molecular sieves to get rid of tracer amounts of normal water. Even high-purity gas (like grade 5) has enough water to slightly reverse deuteration in the last drying stage. Lastly, one important issue should be mentioned concerning the RH level(s) used for D2O vapour conditioning or their evaluation by RH sensors. For automated sorption balances, the calibration of generated RH or built-in RH sensors is commonly done using saturated salt solutions which generate known equilibrium RH at a given temperature. However, two issues complicate the use of D2O in a similar way: the saturated vapour pressure of D2O is lower than of H2O, and saturated salt solutions with D2O generate a slightly (1–3% RH) lower RH than their H2O counterparts (Becker et al. 1969). These differences are important to remember if the deuteration technique is used to generate D2O sorption isotherms or deuterate a material at a specific RH or vapour pressure.
Pressure plate technique
As previously discussed, it is very difficult to condition samples close to saturation due to problems with keeping the temperature stable enough to achieve equilibrium in the over-hygroscopic RH range (98–100% RH). Therefore, in this range the pressure plate technique is often employed for conditioning, a method in which one or several samples are placed within an extractor where pressures up to 100 bar (10 MPa) can be applied. Typically, samples are cuboids or cylindrical discs with side length/diameter in the range 10–60 mm and a thickness of 2–20 mm (Almeida and Hernandez 2006, 2007; Cloutier and Fortin 1991; Fredriksson and Johansson 2016; Fredriksson et al. 2013; Thygesen et al. 2010; Zauer et al. 2016); however, Stone and Scallan (1967) used the pressure plate technique to condition microtome sections of 100 µm thickness and a cross-sectional area of 2 cm2. After water-saturating samples and the supporting substrate (ceramic plate or cellulose membrane, dependent on pressure range), pressure is applied to the extractor using dry gas, whereby water is pressed out of pores in which capillary forces are lower than the applied pressure. From a combination of the Young–Laplace and Kelvin equations, a correlation between applied pressure in the extractor and RH can be found:
where ΔP [Pa] is applied pressure (above atmospheric pressure), M w [kg/mol] is molar mass of water (0.018 kg/mol), R [J/mol K] is the universal gas constant (8.3145 J/mol K), T [K] is temperature, and ρ w [kg/m3] is density of water. By adjusting the applied pressure, it is possible to tune RH much more precisely in the 98–100% RH range than with other methods. For instance, an applied pressure of 15 bar corresponds to 98.90% RH, while 9 bar is equivalent to 99.34% RH. When equilibrium is reached as monitored on the amount of water pressed out over time, samples are taken out and weighed on a balance of sufficient mass resolution. According to standard ISO 11274 (ISO 1998) equilibrium is obtained when two consecutive weekly mass readings show < 0.1% variation in sample mass.
The technique has primarily been used to condition untreated wood (Almeida and Hernandez 2006, 2007; Cloutier and Fortin 1991; Fortin 1979; Fredriksson and Johansson 2016; Fredriksson et al. 2013; Griffin 1977; Stone and Scallan 1967), but some studies of modified wood have been published (Thygesen et al. 2010; Zauer et al. 2016) (see Fig. 3). The traditional use of the technique has been for conditioning in desorption from water-saturated state; however, Fredriksson and Johansson (2016) used a rebuilt extractor for absorption measurements which avoids problems of previous modifications of the technique to handle absorption (Cloutier and Fortin 1991; Fortin 1979).

a Desorption isotherms in the over-hygroscopic range determined after conditioning with the pressure plate technique and saturated salt solutions for untreated and modified Norway spruce (Thygesen et al. 2010; Zauer et al. 2016). Data points in the range of 94–98% RH from climate chamber measurements have been omitted from the results of Thygesen et al. (2010) for clarity. b Zoom of (a) in the 99.5–100.0% RH range
EMC in the over-hygroscopic RH range depends on the size of voids in the wood structure such as pits, lumina, vessels, etc., and the sorption isotherms therefore depend heavily on wood species due to the high variability of these anatomical characteristics. Moreover, while the over-hygroscopic absorption isotherms depend on water condensation in voids and pores of gradually increasing void sizes (Engelund et al. 2010), desorption isotherms are governed by the size of bottleneck pits connecting lumina/vessels to the surrounding climate (Fredriksson and Johansson 2016; Salin 2008). During absorption, lumina/vessels take up moisture when an RH corresponding to equilibrium with their lumen/vessel radius is reached, but during desorption they will empty at a lower RH corresponding to radius of their largest pits facing the surroundings. This is known as the “ink bottle effect” (Ravikovitch and Neimark 2002) and might explain the large sorption hysteresis in the over-hygroscopic regime. In an interesting study by Zelinka et al. (2016), mercury intrusion porosimetry is explored as a complementary technique for studying over-hygroscopic moisture sorption in wood. The technique is based on applying pressure to push liquid mercury into thinly cut transverse wood sections. The applied pressure correlates with the radius of voids filled with mercury as described by the Kelvin equation. Therefore, after adjusting for differences in contact angle and surface tension between mercury and liquid water, the volumetric uptake of mercury as a function of applied pressure can be used to calculate the capillary water in the wood void structure as a function of RH. The order of mercury intrusion into the wood structure, i.e. vessels-lumina-pits, makes the calculated capillary water curves correspond to the desorption isotherms of thin wood sections. For thicker wood sections without direct access to cell lumina, the order of emptying of voids as well as filling of voids with mercury changes. Mercury intrusion porosimetry would therefore need to be performed on such thicker samples in order to correctly represent over-hygroscopic desorption isotherms of capillary water.
Although wood modifications rarely cause significant changes to the void geometry unless modification agent fills up lumina or pits, they may considerably affect the over-hygroscopic sorption isotherms. This is due to potential changes after modification in the contact angle between wood surface and wetting liquid. Such change will affect EMC in the over-hygroscopic range as predicted by Engelund et al. (2010). The contact angle of liquids with modified wood has been investigated in several studies by the Wilhelmy and sessile drop methods. The first determines the force required during immersion and withdrawal from a probe liquid, for example water, while in the latter the contact angle is determined over time after dropping a liquid droplet onto the surface. Despite the shortcomings of both methods as discussed by Wålinder and co-workers (Wålinder and Johansson 2001; Wålinder and Ström 2001), they both show that modification can alter the contact angle with water. For instance, both thermal modification (Bakar et al. 2013; Hakkou et al. 2006; Kocaefe et al. 2008) and acetylation (Bryne and Wålinder 2010) appear to increase the contact angle with water, while furfurylation (Bryne and Wålinder 2010) slightly decreases it. The effect of these changes on the over-hygroscopic sorption isotherms has not been investigated.
Fibre saturation techniques
The original definition of the fibre saturation point (FSP) was suggested by Tiemann (1906) as the MC reached upon drying where lumina are empty of liquid water, cell walls begin to dry, and the mechanical properties start increasing. As shown by Stamm (1971), however, these three phenomena do not occur at the same MC. FSP was later suggested to be determined as MC at the first shrinkage upon drying from green or water-saturated states (Koehler and Thelen 1926). However, due to inconsistencies in the previous definitions, Stone and Scallan (1967) focused on the emptying of lumina in the over-hygroscopic RH range and defined FSP as the MC where lumina are empty, but cell walls are still water-saturated. Using the pressure plate technique, they found FSP for black spruce at 99.75% RH, while Griffin (1977) added his own experimental results and adjusted it to 99.93% RH. This definition is also problematic as liquid water may still be present in some cell lumina even at equilibrium conditions where cell walls in other parts of the same sample are not saturated (Passarini et al. 2015).
Although the traditional FSP concept originated more than a century ago, it is still widely used to describe the transition point imagined between cell wall saturation and (significant) moisture uptake in the wood void structure in the over-hygroscopic regime. For practical purposes, for example for structural engineering applications of wood with a strong focus on mechanical performance, the traditional concept of FSP works well. However, from a scientific standpoint the concept is problematic (Babiak and Kudela 1995; Feist and Tarkow 1967), and it mischaracterises the wood–water relations. Instead, in this review, FSP is regarded as the maximum cell wall water capacity measured in the fully water-saturated state (Hill 2008). The first part of this definition is in line with the intension behind the original definition by Tiemann (1906), i.e. the amount of water found within cell walls when these are water-saturated. By determining FSP in the fully water-saturated state, i.e. where water takes up all available space in cell walls and voids (pits, lumina, vessels) in the wood structure, uncertainties about partial drying of any cell walls are avoided. However, investigating FSP in the fully water-saturated state requires experimental techniques that can differentiate between water within and outside cell walls.
Differential scanning calorimetry
One technique which is capable of separating cell wall MC from the total MC is differential scanning calorimetry (DSC). This type of equipment measures the energy involved in changing the temperature of a small sample over a wide temperature range. The amount of sample depends on the geometry of the crucible of the specific DSC instrument, but typical sample masses and sizes are in the range 5–15 mg (Park et al. 2006; Weise et al. 1996) and 1-mm discs of 3–4 mm diameter (Simpson and Barton 1991; Zauer et al. 2014), respectively. The technique is particularly useful for determining the energy involved in exothermic or endothermic reactions occurring during an experiment, for example phase transitions such as freezing or thawing of water. As cell wall water is tightly associated with the chemical constituents, it will not exhibit a phase change at least down to − 70 °C (Berthold et al. 1994). This was first used by Magne et al. (1947) to determine the amount of cell wall water in water-saturated textile fibres (cotton, rayon, nylon, glass fibres) by measuring the energy for freezing water at − 4 °C. Since then, DSC techniques have been used to determine cell wall water within, for example, pulp and paper fibres (Maloney 2000; Nelson 1977; Park et al. 2006; Weise et al. 1996), untreated wood (Simpson and Barton 1991; Zauer et al. 2014; Zelinka et al. 2012), and modified wood (Repellin and Guyonnet 2005; Zauer et al. 2014). By cooling or quenching a wet sample to a given sub-zero temperature and ramping the temperature at constant rate to a specified temperature above the freezing point of water, the energy input (enthalpy) required to melt the ice formed can be measured. Temperature ranges and ramping rates employed vary between studies, but are most often found within the − 40 to 40 °C range (Park et al. 2006; Repellin and Guyonnet 2005; Simpson and Barton 1991; Weise et al. 1996; Zauer et al. 2014) and from 1 to 3 °C/min (Park et al. 2006; Repellin and Guyonnet 2005; Weise et al. 1996; Zauer et al. 2014) to 5 °C/min (Zelinka et al. 2012) or even 10 °C/min (Nelson 1977; Simpson and Barton 1991), respectively. The cell wall moisture content, MCcell-wall [%], can then be found as
where m water [g] is total mass of water, m tot [g] is total wet sample mass, m 0,dry [g] is dry sample mass, ΔH tot [J/g] is measured specific enthalpy of melting per total wet sample mass, H f [J/g] is melting enthalpy (often termed enthalpy of fusion) of pure water (333.6 J/g) (Lide 2013), and (1 + R mod)−1 [−] corrects for mass gain due to modification if present. If the sample is fully water-saturated, MCcell-wall corresponds to FSP, and using such samples avoids problems of non-saturation of any parts of the wood volume examined. The technique can, however, also be used to differentiate cell wall water from liquid water outside cell walls in the over-hygroscopic RH range as long as a sufficient amount of freezable water is present. Although the DSC technique removes the sample from its conditioning temperature and FSP has been reported to change with temperature (Babiak and Kudela 1995; Stamm and Loughborough 1935), it would be unreasonable to relate the determined FSP to a temperature around 0 °C since the experimental run in the DSC is much shorter than the time to equilibrate MC. Therefore, it can be assumed that the determined FSP relates to the conditioning temperature. Besides the above-described method for determining FSP, an additional one has been reported in several studies (Simpson and Barton 1991; Zauer et al. 2014; Zelinka et al. 2012) in which samples within a wide range of MC have been used and a linear regression is constructed between MC and specific enthalpy measured. The inflection point of this linear regression is then FSP, and the slope of the line is the enthalpy of water associated with the wood structure (Hager and Macrury 1980). In some studies (Simpson and Barton 1991; Zauer et al. 2014; Zelinka et al. 2012), it is stated that this method is more correct as it does not assume any specific value of the melting enthalpy. However, all studies show that the melting enthalpies determined are close to the generally accepted value of 333.6 J/g (Magne et al. 1947; Nelson 1977; Zauer et al. 2014) as shown in Fig. 4, considering the experimental uncertainties (small sample masses, high/low temperature ramping rates, heat flow accuracy, etc.) involved in the measurements. The only exception to this is the study by Zelinka et al. (2012) in which a very low melting enthalpy of 240 J/g is found, leading the authors to speculate about temperature buffering from the sample heat capacity. However, this effect should be mostly caught by baseline correction of the recorded energy flow. That the melting enthalpy can differ from that of bulk water is clear for water found in nano-pores (Jähnert et al. 2008; Shimizu et al. 2015). A vast majority of liquid water in wood is, however, found in voids larger than 0.1 µm. This is, for instance, seen in the negligible capillary water uptake at 98% RH (Engelund et al. 2010) corresponding to a void size of 0.1 µm. For voids larger than this size, the melting enthalpy reduction due to confinement is < 1% (Jähnert et al. 2008). If freezable water was found in wood nano-pores in detectable amounts, it would exhibit a significant freezing/melting point depression (Shimizu et al. 2015); however, no peak of appreciable lower melting point has been found for solid wood. Besides the effect of heavy confinement on melting enthalpy of liquids, there are no results found in the literature to suggest that surface chemistry or other factors should affect the melting enthalpy. Therefore, determining FSP by the DSC method using fully water-saturated samples is deemed appropriate, whereas conditioning to lower MC might not result in fully saturated cell walls and as a result yield a lower determined FSP.

Melting enthalpy as a function of reduced moisture content (corrected for added mass of modification) for untreated and variously modified Norway spruce samples. Some of the samples had excess water on surfaces, resulting in very high calculated moisture contents, even though this water was not physically inside the material. Authors’ own data
Solute exclusion technique
The amount of different pools of water in very small voids can be investigated by the solute exclusion technique (SET). The method was originally developed by Aggebrandt and Samuelson (1964) and modified by Stone and co-workers (Stone et al. 1969; Stone and Scallan 1967, 1968a, b) to characterise pore volumes and pore size distributions in pulps in the water-swollen state. Since then, SET has been used to characterise both untreated (Ahlgren et al. 1972; Farahani 2003; Feist and Tarkow 1967; Flournoy et al. 1991, 1993; Forster 1998) and modified wood (Farahani 2003; Forster 1998). In SET, a water-saturated sample with known mass of water is placed in an aqueous solution of molecular probe solutes of a known size and concentration. The typical sample is a cuboid of 20–30 mm side length and a thickness of 3–5 mm (Farahani 2003; Flournoy et al. 1991, 1993; Forster 1998), although several older studies have used shavings, pulp, or milled wood (Ahlgren et al. 1972; Feist and Tarkow 1967; Stone and Scallan 1968b). As solutes diffuse into the water within the sample, their concentration in the surrounding solution decreases. The change in concentration is related to the cell wall water volume that can be penetrated by solutes, and from this change in concentration the inaccessible moisture content, MCinaccess [% g/g], can be calculated:
where m water [g] is total mass of water in the sample before immersion, m sol [g] is mass of solution added, m dry [g] is dry mass of the sample, c initial [mol/g] is initial concentration of solutes in the solution, Δc [mol/g] is change in solute concentration after immersion of the sample, and (1 + R mod)−1 [−] corrects for mass gain due to modification if present. Concentration differences are small, and a HPLC or calibrated refractometer is needed for determining the concentration before and after immersion of the wet sample. To maximise the concentration difference, solution volumes used to immerse samples in are typically not more than 3–10 times the sample volume (Farahani 2003; Flournoy et al. 1991; Forster 1998; Stone and Scallan 1968b). It is important that equilibrium has been achieved before the change in concentration is determined. However, the incubation time employed in the literature varies dramatically from a couple of minutes for pulp samples (Stone and Scallan 1968b) to 1–14 days for solid wood samples (Farahani 2003; Flournoy et al. 1991; Forster 1998). Clearly, more research is needed concerning the approach to equilibrium and appropriate incubation times as a function of diffusion pathways into samples.
When selecting solutes for SET, it is important that they are chemically inert towards the sample and do not adsorb to surfaces within cell walls (Stone and Scallan 1967) as this would shift the equilibrium between pore solution and surrounding solution. The solutes most often used are small sugars, PEG, and dextrans. Their size is determined from the hydrodynamic diameter calculated from the Stokes–Einstein equation based on their diffusivity. The smallest solutes are saccharides such as glucose (0.8 nm) (Flournoy et al. 1991; Kuga 1981; Lin et al. 1987; Stone and Scallan 1968a; Wang et al. 2011), mannitol (0.8 nm) (Carpita et al. 1979), sucrose (1.0 nm) (Carpita et al. 1979; Kuga 1981; Van Dyke 1972), cellobiose (1.0 nm) (Lin et al. 1987), maltose (1.0 nm) (Flournoy et al. 1991; Stone and Scallan 1968a), raffinose (1.2 nm) (Flournoy et al. 1991; Kuga 1981; Stone and Scallan 1968a; Van Dyke 1972), and stachyose (1.4 nm) (Carpita et al. 1979; Stone and Scallan 1968a). PEGs range in size from 0.44 nm for ethylene glycol (Kuga 1981) to 13.0 nm (Lin et al. 1987; Van Dyke 1972), while dextrans have been reported in the literature from 1.8 nm in diameter (Ishizawa et al. 2007) up to 586 nm (Arond and Frank 1954). Figure 5 shows how the solute diameter correlates with molecular mass for both PEGs and dextrans.

a Diameter of dextran and PEG probe molecules as a function of molecular weight (based on data from Arond and Frank 1954; Carpita et al. 1979; Day et al. 1978; Flournoy et al. 1991; Granath and Kvist 1967; Hill et al. 2005; Ishizawa et al. 2007; Kuga 1981; Lin et al. 1987; Peters 1986; Stone et al. 1969; Stone and Scallan 1968a; Van Dyke 1972; Wang et al. 2011; Wong et al. 1988). b Inaccessible cell wall water volume as a function of size of different molecular probes. Data for untreated and acetylated (R mod = 0.21) Corsican pine from Forster (1998). Note that the fibre saturation point (FSP) can be determined from the plateau in inaccessible cell wall volume for probe sizes too large to enter cell walls
The inaccessible MC describes the mass of water inaccessible for solutes of a given size. Thus, by using several differently sized solutes it is in principle possible to determine the pore size distribution within cell walls as indicated in Fig. 5. However, the underlying assumption of (6) which allows calculation of probed water mass is that solute concentration within accessible pores is the same as in the surrounding solution (Lin et al. 1985). This assumption has been shown to be incorrect, since solute concentration within accessible pores decreases as the sizes of probe solutes and accessible pores get closer (Alince 1991; Day et al. 1978, 1979). Thus, for solutes about half as big as the accessible pore, the solute concentration within the pore is around 30% of that in the surrounding solution (Alince 1991), while it decreases to nearly zero for solute sizes close to the pore size. For this reason, SET does not provide absolute pore size distributions, but can only hint at it. However, for solutes too large to enter cell walls, the voids they occupy (pits, lumina, vessels, etc.) are much larger than the solute size. Therefore, the assumption of equal solute concentration in these voids to the ones in the surrounding solution is valid, and SET can offer a good estimate of total pore volume, i.e. FSP (Alince 1991). If several sizes of solutes are used which are too big to enter cell walls, a plateau corresponding to FSP is found for MCinaccess as a function of solute size (see Fig. 5).
Spectroscopic techniques
While the gravimetric and fibre saturation techniques are useful for determining total amount of water in wood or amount of cell wall water in the over-hygroscopic range, they are not able to provide a deeper insight into wood–water interactions or the distribution of water within the wood structure in the entire RH range. For this, spectroscopic techniques are needed that can distinguish water in different environments or provide more detail about molecular interactions and chemistry within cell walls. A wide range of spectroscopic techniques are available, but here two of the most important ones are highlighted.
Low-field nuclear magnetic resonance spectroscopy and magnetic resonance imaging
Low-field (LF) nuclear magnetic resonance (NMR) spectroscopy, also known as time-domain NMR (Thygesen and Elder 2008) or NMR relaxometry (Kekkonen et al. 2014), can distinguish between water-bound hydrogen located in different physical and chemical environments. Consequently, it is a good technique for determining the distribution of water within different parts of the wood structure (Araujo et al. 1992). The technique uses the very, very slight magnetic polarisation in a material caused by a permanent magnet within the spectrometer (typically up to a few tesla in strength). The polarisation arises because the permanent magnetic field B 0 gives rise to two energy states for the spins (along and against the field) of hydrogen nuclei within the sample with a tiny difference in abundance given by the Boltzmann distribution. This difference is what causes a polarisation vector M of the material aligned with B 0. The spectrometer is also equipped with an electromagnetic coil which can generate short-lived radiofrequency pulses that result in a magnetic field B 1 (≫ B 0) oriented perpendicular to B 0. These pulses will change the orientation of M, and the spectrometer will afterwards record the decay or relaxation of M back to its equilibrium value, realigned along B 0. This relaxation is characterised by two different relaxation times termed T 1 (longitudinal or spin–lattice relaxation) and T 2 (transverse or spin–spin relaxation). T 1 relaxation is studied as the increase in M in the direction along the permanent field B 0, while T 2 relaxation is studied in the transverse plane, i.e. perpendicular to B 0, as the decrease in the component of M in this plane. This latter process does not only depend on the realignment of M along B 0, but also on the dephasing of spins in the transverse plane due to transfer of spins between neighbouring nuclei. T 1 and T 2 relaxations are both first-order rate processes and can therefore be described by exponential decay functions with characteristic relaxation times T 1 and T 2, respectively. For fluids in porous materials, the relaxation times depend on the size of pores confining the fluid as well as interactions between pore wall and fluid (Mitchell et al. 2005). If a material contains fluid in micro- and macro-pores of various sizes, the fluid will exhibit several relaxation times characteristic of pore sizes and chemistry. By measuring the decay of the low-field (LF) NMR signal and deconvoluting it, the relaxation times of the different components, i.e. fluid in variously sized pores, can be determined and the distribution of fluid in different environments (pore sizes and chemistry) can be analysed. The deconvolution can be done by fitting discrete exponential functions to the decaying signal, for example to add components until residuals are randomly distributed. Another way is to fit continuous relaxation time distributions to the signal by non-negative least square fitting (Whittall et al. 1991) using algorithms often found in computer software for LFNMR data analysis, for example the CONTIN algorithm by Provencher (1982a, b). Before decay of the LFNMR signal can be measured, the spins of the sample need to be perturbed by one or more B 1 pulses. For this, several pulse sequences have been developed. For determining T 1 relaxation times, one of the pulse sequences termed “inversion recovery” and “saturation recovery” is used, while T 2 relaxation times are determined after free induction decay (FID) or the Carr–Purcell–Meiboom–Gill (CPMG) sequence (Carr and Purcell 1954; Meiboom and Gill 1958). In particular, CPMG is a commonly used sequence for determining T 2 relaxation as it reduces effects of magnet inhomogeneity by multiple refocusing of the spins in the transverse plane.
Several studies have employed LFNMR for characterising water in both untreated (Almeida et al. 2007; Araujo et al. 1992, 1994; Cox et al. 2010; Flibotte et al. 1990; Fredriksson and Thygesen 2017; Labbé et al. 2002, 2006; Menon et al. 1987; Passarini et al. 2015; Telkki et al. 2013; Thygesen and Elder 2008, 2009) and modified wood (Elder et al. 2006; Hietala et al. 2002; Javed et al. 2015; Kekkonen et al. 2014; Thygesen and Elder 2008, 2009). Based on a deconvolution of the decay curves, signals from water in different compartments within the wood structure have been analysed (Almeida et al. 2007; Araujo et al. 1992, 1993; Fredriksson and Thygesen 2017; Passarini et al. 2015) (see Fig. 6). A comparison of void geometries and volumes shows good correspondence between pore volumes of pits and lumina and the areas of respective T 2 peaks assigned to these voids for water-saturated samples (Fredriksson and Thygesen 2017). Even more details about the chemical environment of water in pores within wood can be gained from T 1 to T 2 2D LFNMR correlation spectroscopy as reported by Cox et al. (2010). In this method, T 1 and T 2 relaxations are determined simultaneously allowing an analysis of their correlation (Song 2009; Song et al. 2002). This is useful since T 2 relaxation occurs both as a result of realignment of M, like T 1 relaxation, and due to local interactions independent of T 1 relaxation. For a material with a spatially homogenous chemistry, relaxation times depend only on pore sizes which would result in similar T 1/T 2 ratios for the peaks observed (Song et al. 2002). On the other hand, if surface interactions vary between differently sized pores, this would show up as a markedly different T 1/T 2 ratio. In the results of Cox et al. (2010), the T 2 peak assigned to water in cell walls by using a regular CPMG splits up into two components in the T 1 T 2 correlation diagram implying two different environments for this water.

Low-field NMR characterisation of water in untreated and acetylated (R mod = 0.20) Norway spruce in a fully water-saturated state: a zoom of CPMG decay curves for three replicates of untreated and acetylated spruce, b average T 2 relaxation distribution curves for the three replicates derived from non-negative least square analysis with regularisation using Prospa version 3.1 (Magritek, NZ) software. Note that upon acetylation, the T 2 relaxation times increase for water in pits and lumina due to weakened wood–water interactions. The cell wall water appears to split into two distinct pools after acetylation. The amount of moisture (MCR) found in lumina remains the same, while the amounts in pits, small cavities, and cell walls are reduced. Original LFNMR data from Thygesen and Elder (2008)
There are a number of important issues to remember when using LFNMR to characterise water in wood. First of all, recorded decay curves should all be fully decayed within the duration of a scan, i.e. one decay measurement, in order to retrieve meaningful results from the data analysis algorithm. Secondly, temporal resolution should be adequate for capturing the shortest expected relaxation time. It is recommended that at least five data points are acquired before the shortest expected characteristic relaxation time in order to yield reasonable curve fits. Thus, the shortest expected relaxation time should be divided by five to find the maximum temporal spacing between equidistant pulses in a CPMG sequence. Since water in wood exhibits typical T 2 relaxation times in the range from milliseconds to seconds as shown in Fig. 6, each scan needs to capture a decaying signal over a range of > 1 s with a resolution of ~ 1/5 ms. As several scans are needed for adequate signal-to-noise ratio, each wood sample is exposed to a large number of radiofrequency pulses. Although the amount of energy put into the sample by each pulse is tiny, the accumulated number of pulses can heat up the sample considerably in extreme cases. This is problematic as isothermal conditions are required for LFNMR since the magnitude of M is temperature dependent (Dinesh and Rogers 1971). To limit this excessive heating, the waiting time between scans can be increased.
Magnetic resonance (MR) imaging uses the same principle as LFNMR although typically with considerably stronger permanent magnetic (2–14 tesla) and electromagnetic fields. By employing CPMG sequences, relaxation spectra from different parts of the wood structure can be analysed. This has been done on both annual ring level (Dvinskikh et al. 2011; Quick et al. 1990) and cell level (Almeida et al. 2008; Hernandez and Caceres 2010; Meder et al. 2003; Passarini et al. 2015), allowing a visualisation of water distributions in both the hygroscopic (Hernandez and Caceres 2010; Passarini et al. 2015) and over-hygroscopic RH range (Almeida et al. 2008). MR imaging is mainly appropriate for water with longer relaxation times than bound water (Hernandez and Caceres 2010).
Vibrational spectroscopy
Molecular bonds in a material vibrate at certain frequencies dependent on involved atomic masses and stiffness (elastic force constant) of the bond in question. For historic reasons, vibrational frequencies are often given in wavenumbers (cm−1) determined as frequency divided by speed of light (3 × 108 m/s). As an example, the covalent bonds C=C, C–C, and C–H all have different vibrational wavenumbers due to differences in either bond stiffness (single or double bond) or in masses of the atoms involved. In vibrational spectroscopy, this is used to probe the chemistry of materials as different chemical bonds are seen to vibrate at different wavenumbers. For instance, infrared (IR) spectroscopy detects the absorption of photon (light) energy at different wavelengths from an incoming infrared beam containing all wavelengths in the range 4000–400 cm−1, while near-infrared (NIR) spectroscopy uses a similar principle, but a different wavenumber range (14.000–4000 cm−1) in which mainly vibrational overtones and coupling of vibrations are found. Unlike these techniques, Raman spectroscopy detects the shift in scattered photon energy from a material which occurs for 10−6–10−8 of scattered photons from an incoming laser beam of fixed wavelength. This shift corresponds to a change in vibrational energy of molecular bonds in the material. Thus, Raman spectroscopy probes the same molecular bonds as IR and NIR spectroscopy, but is based on a different physical phenomenon.
Vibrational spectroscopy has been widely used to characterise chemical changes during wood modification (see, for example, González-Peña and Hale 2011; Kotilainen et al. 2000; Mitsui et al. 2008; Nuopponen et al. 2005; Stefke et al. 2008; Sun and Sun 2002; Temiz et al. 2007; Tserki et al. 2005; Venås et al. 2006; and Fig. 7). Moreover, spatial chemical distributions in advanced modified materials have been mapped on the cell wall level using confocal Raman microscopy (Cabane et al. 2014, 2016; Keplinger et al. 2015, 2016). For studying the relation with water, vibrational spectroscopy has been used to examine the presence of water or the sorption of water from the molecular vibrations of water (Christensen et al. 2006; Inagaki et al. 2008; Tsuchikawa and Tsutsumi 1998). One of the advantages of vibrational spectroscopy is, however, the insights it can provide into chemical interactions of wood materials with water. By deuteration of a material, i.e. exchanging protium (1H) for deuterium (2H) in chemical groups hydrogen-bonding with liquid D2O or D2O vapour (Lindh et al. 2016), a marked shift in vibrational wavenumbers of affected molecular bonds is seen. For instance, deuteration changes the fundamental stretching vibrations of O–H, N–H, and C–H by around 1000, 900, and 800 cm−1, respectively, which is due to the near-doubling of the vibrating hydrogen mass. If the extra neutron of deuterium instead was added to the heavier vibrating atom of the O–H, N–H, and C–H bonds, for example exchanging 16O for 17O, the shift in vibrational wavenumber would hardly be detectable with vibrational spectroscopy as the relative mass increase in O, N, and C would be significantly lower. Compared with gravimetric techniques using deuteration, the advantage of vibrational spectroscopy is that deuteration can be achieved without drying the material prior to exposure to D2O, unlike gravimetric techniques which require determination of initial dry mass.

Infrared vibrational spectra determined with ATR-FTIR equipment on untreated and acetylated (R mod = 0.13) Norway spruce after deuteration in liquid D2O. Average curves based on 4–5 replicates are shown. Note that for acetylated wood both O–H stretching (~ 3400 cm−1) and O–D stretching (~ 2400 cm−1) vibrations decrease, while the C=O stretching (~ 1750 cm−1) and CH3 deformations (~ 1350–1250 cm−1) increase. Authors’ own data
From vibrational spectroscopic studies, it can be seen that wood–water interactions are dominated by interactions with O–H groups (hydroxyls), since deuteration only affects O–H vibrations, while C–H stretching vibrations remain unchanged and no C–D stretching vibrations are observed (Hofstetter et al. 2006; Mann and Marrinan 1956a; Schmidt et al. 2006; Taniguchi et al. 1966; Watanabe et al. 2006). After deuteration and drying, the relative accessibility of hydroxyls to water can be determined as the ratio of integrated areas of O–D to the sum of O–H and O–D vibrations (Suchy et al. 2010b; Taniguchi et al. 1966) or alternatively as the ratio of weighted intensities of O–D to the sum of O–H and O–D vibrations (Mann and Marrinan 1956b; Sepall and Mason 1961). These calculations are based on the Beer–Lambert law which states that the absorbance of a given vibration is given by the product of the three following parameters: abundance of the vibration in the material, molar absorptivity of the vibration, and path length of the light beam through the material. For calculating the relative accessibility of hydroxyls to water, it is implicitly assumed that molar absorptivities of O–H and O–D vibrations are equal. This is not necessarily correct as O–D stretching vibrations in cellulose, starch, and wood have been reported to be about 10% more intense than O–H stretching vibrations (Mann and Marrinan 1956b; Nara et al. 1981; Thybring et al. 2017). From theoretical calculations, O–D stretching vibrations are expected to be less intense than O–H stretching vibrations (Crawford 1952; Swenson 1965), in line with studies on liquid normal and heavy water showing 29% lower intensity for O–D stretching vibrations (Venyaminov and Prendergast 1997). Moreover, it should be noted, as discussed for gravimetric techniques, that deuteration will not exchange hydrogens on all hydroxyls interacting with water as shown by Lindh et al. (2016), only those capable of acting as hydrogen bond donor, i.e. in which the hydrogen of hydroxyls is involved in a hydrogen bond with a water molecule. Water-interacting hydroxyls restricted to act as hydrogen bond acceptors, i.e. only interacting with water through their oxygen, are presumably only found on the surfaces of the stiff, aggregated cellulose microfibrils. Before the effect of these issues on the determined hydroxyl accessibility has been resolved for wood and plant materials, it is uncertain whether vibrational spectroscopy can yield quantitative results. Nonetheless, vibrational spectroscopy can be a useful technique for getting qualitative results for interactions between water and wood hydroxyls (see Fig. 7). Both IR and NIR spectroscopy have been applied to study untreated wood (Altaner et al. 2006; Chow 1972; Fackler and Schwanninger 2011; Fernandes et al. 2011; Suchy et al. 2010a, b; Taniguchi et al. 1966; Tsuchikawa and Siesler 2003a, b), while no published data on modified wood are available. Similarly, no known study uses Raman spectroscopy to probe the wood–water interactions by deuteration, whereas a few have been published on deuteration of proteins (Bolton and Scherer 1989; Sánchez-González et al. 2008) and lactose (Whiteside et al. 2008).
Even more information about hydroxyls can be gained from 2D correlation infrared spectroscopy (Noda 1990, 2006; Noda et al. 1993). By this technique, the IR spectrometer is coupled with a dynamic excitation which could be a change in climate (RH or temperature) or a mechanical excitation. This has, for instance, been done using dynamic mechanical analysis (Hinterstoisser and Salmén 2000; Hofstetter et al. 2006; Stevanic and Salmén 2006; Åkerholm and Salmén 2003). From a correlation analysis between obtained spectra and external excitation, information about which vibrations in the rather broad and unstructured O–H stretching region (3700–3000 cm−1) are affected by external stimulus can be gained, for example the location and orientation of hydroxyls in cellulose.
Conclusion
The interaction of water with wood is highly important for the performance of wood materials. This review describes a range of experimental techniques for characterising water within wood in the entire moisture domain from dry to fully water-saturated. The techniques fall into three broad categories: (1) gravimetric techniques that determine how much water is absorbed, (2) fibre saturation techniques that determine the amount of water within cell walls, and (3) spectroscopic techniques that provide insights into chemical wood–water interactions as well as yield information on water distribution in the macro-void wood structure. For each technique, advantages and limitations are discussed. The experimental data obtained from the various techniques show the many different aspects of wood–water interactions which can be probed. Thus, by combination of several experimental techniques, a wealth of information can be extracted about the state and distribution of water in wood, if the techniques are properly employed. The aim of this review is that more researchers become aware of the wide suite of available experimental techniques and by taking advantage of these techniques, will provide new insights into water interacting with wood materials.
References
Aggebrandt LG, Samuelson O (1964) Penetration of water-soluble polymers into cellulose fibers. J Appl Polym Sci 8:2801–2812
Ahlgren PA, Wood JR, Goring DAI (1972) Fiber saturation point of various morphological subdivisions of Douglas-fir and aspen wood. Wood Sci Technol 6:81–84
Åkerholm M, Salmén L (2003) The oriented structure of lignin and its viscoelastic properties studied by static and dynamic FT-IR spectroscopy. Holzforschung 57:459–465
Alince B (1991) Comments on porosity or swollen pulp fibers analyzed by solute-exclusion. Tappi 74:200–202
Almeida G, Hernandez RE (2006) Changes in physical properties of yellow birch below and above the fiber saturation point. Wood Fiber Sci 38:74–83
Almeida G, Hernandez RE (2007) Influence of the pore structure of wood on moisture desorption at high relative humidities. Wood Mater Sci Eng 2:33–44
Almeida G, Gagne S, Hernandez RE (2007) A NMR study of water distribution in hardwoods at several equilibrium moisture contents. Wood Sci Technol 41:293–307
Almeida G, Leclerc S, Perre P (2008) NMR imaging of fluid pathways during drainage of softwood in a pressure membrane chamber. Int J Multiph Flow 34:312–321
Altaner C, Apperley DC, Jarvis MC (2006) Spatial relationships between polymers in Sitka spruce: proton spin-diffusion studies. Holzforschung 60:665–673
Ammer U (1963a) Untersuchungen über das Wachstum von Rotstreifepilzen in Abhängigkeit von der Holzfeuchtigkeit [Investigations of the growth of brown-rot fungi in dependence of the wood moisture content]. Forstwiss Centralbl 82:360–391
Ammer U (1963b) Untersuchungen über die Sorption pilzbefallenen Holzes [Investigations of the sorption in fungally attacked wood]. Holz Roh Werkst 21:465–470
Araujo CD, MacKay AL, Hailey JRT, Whittall KP, Le H (1992) Proton magnetic-resonance techniques for characterization of water in wood—application to white spruce. Wood Sci Technol 26:101–113
Araujo CD, MacKay AL, Whittall KP, Hailey JRT (1993) A diffusion-model for spin-spin relaxation of compartmentalized water in wood. J Magn Reson Ser B 101(3):248–261
Araujo CD, Avramidis S, Mackay AL (1994) Behavior of solid wood and bound water as a function of moisture-content a proton magnetic-resonance study. Holzforschung 48:69–74
Arond LH, Frank HP (1954) Molecular weight distribution and molecular size of a native dextran. J Phys Chem US 58:953–957
ASTM (2016) ASTM C1498-04a Standard test method for hygroscopic sorption isotherms of building materials, ASTM International, West Conshohocken, PA, USA
Babiak M, Kudela J (1995) A contribution to the definition of the fiber saturation point. Wood Sci Technol 29:217–226
Bakar BFA, Hiziroglu S, Tahir PM (2013) Properties of some thermally modified wood species. Mater Des 43:348–355
Becker M, Schälike W, Zirwer D (1969) D2O-Dampfdrucke gesättigter Salzlösungen [D2O vapour pressures of saturated salt solution]. Z Naturforsch 24A:684–685
Berthold J, Desbrieres J, Rinaudo M, Salmén L (1994) Types of adsorbed water in relation to the ionic groups and their counterions for some cellulose derivatives. Polymer 35:5729–5736
Bolton BA, Scherer JR (1989) Raman spectra and water absorption of bovine serum albumin. J Phys Chem US 93:7635–7640
Boonstra MJ, Van Acker J, Pizzi A (2007) Anatomical and molecular reasons for property changes of wood after full-scale industrial heat treatment. In: Proceedings of the third European conference on wood modification, Bangor, UK, pp 343–358
Bryne LE, Wålinder MEP (2010) Ageing of modified wood. Part 1: wetting properties of acetylated, furfurylated, and thermally modified wood. Holzforschung 64:295–304
Buro A (1954) Untersuchungen über den Abbau von Kiefern- und Buchenholz durch holzzerstörende Pilze und deren Einfluß auf einige physikalische Eigenschaften des Holzes [Investigations of the degradation of pine and beechwood by wood decaying fungi and their influence on some physical properties of the wood]. Holz Roh Werkst 12:258–267
Cabane E, Keplinger T, Merk V, Hass P, Burgert I (2014) Renewable and functional wood materials by grafting polymerization within cell walls. Chemsuschem 7:1020–1025
Cabane E, Keplinger T, Künniger T, Merk V, Burgert I (2016) Functional lignocellulosic materials prepared by ATRP from a wood scaffold. Sci Rep 6:31287
Carpita N, Sabularse D, Montezinos D, Delmer DP (1979) Determination of the pore-size of cell-walls of living plant-cells. Science 205:1144–1147
Carr HY, Purcell EM (1954) Effects of diffusion on free precession in nuclear magnetic resonance experiments. Phys Rev 94:630–638
Carrington H (1921) The module of rigidity for spruce. Philos Mag 41:848–860
Carrington H (1922) The elastic constants of spruce as influenced by moisture content. Aeronaut J 26:462–471
Chow SZ (1972) Hydroxyl accessibility, moisture-content, and biochemical activity in cell-walls of Douglas-fir trees. Tappi 55:539–544
Christensen GN (1959) The rate of sorption of water vapour by wood and pulp. Appita J 13:112–123
Christensen GN (1960) Kinetics of sorption of water vapour by wood. Aust J Appl Sci 11:295–304
Christensen GN (1965) The rate of sorption of water vapor by thin materials. In: Winn PN (ed) Principles and methods of measuring moisture in liquids and solids. Reinhold Publishing Corporation, New York, pp 279–293
Christensen GN, Hergt HFA (1969) Effect of previous history on kinetics of sorption by wood cell walls. J Polym Sci A 1(7):2427–2430
Christensen GN, Kelsey KE (1959) The rate of sorption of water vapor by wood. Holz Roh Werkst 17:178–188
Christensen M, Frosch M, Jensen P, Schnell U, Shashoua Y, Nielsen OF (2006) Waterlogged archaeological wood—chemical changes by conservation and degradation. J Raman Spectrosc 37:1171–1178
Cloutier A, Fortin Y (1991) Moisture-content—water potential relationship of wood from saturated to dry conditions. Wood Sci Technol 25:263–280
Cox J, McDonald PJ, Gardiner BA (2010) A study of water exchange in wood by means of 2D NMR relaxation correlation and exchange. Holzforschung 64:259–266
Crank J (1953) A theoretical investigation of the influence of molecular relaxation and internal stress on diffusion in polymers. J Polym Sci 11:151–168
Crawford B (1952) Vibrational intensities II—the use of isotopes. J Chem Phys 20:977–981
Day JC, Alince B, Robertson AA (1978) Interaction of polymers in solution with porous solids. I. Penetration of porous glass by dextran. Can J Chem 56:2951–2958
Day JC, Alince B, Robertson AA (1979) The characterization of pore systems by macromolecular penetration. Cell Chem Technol 13:317–326
Dinesh, Rogers MT (1971) Temperature dependence of the proton spin-spin relaxation time in liquid chloroform. Chem Phys Lett 12:352–354
Dvinskikh SV, Henriksson M, Berglund LA, Furo I (2011) A multinuclear magnetic resonance imaging (MRI) study of wood with adsorbed water: estimating bound water concentration and local wood density. Holzforschung 65:103–107
Elder T, Labbé N, Harper D, Rials T (2006) Time domain-nuclear magnetic resonance study of chars from southern hardwoods. Biomass Bioenerg 30:855–862
Engelund ET, Thygesen LG, Hoffmeyer P (2010) Water sorption in wood and modified wood at high values of relative humidity. Part 2: appendix. Theoretical assessment of the amount of capillary water in wood microvoids. Holzforschung 64:325–330
Engelund ET, Thygesen LG, Svensson S, Hill CAS (2013) A critical discussion of the physics of wood–water interactions. Wood Sci Technol 47:141–161
Eriksson L, Noren B (1965) The effect of moisture changes on deformation of wood with tension in fibre direction. Holz Roh Werkst 23:201–209
Espinosa RM, Franke L (2006) Influence of the age and drying process on pore structure and sorption isotherms of hardened cement paste. Cement Concrete Res 36:1969–1984
Fackler K, Schwanninger M (2011) Accessibility of hydroxyl groups of brown-rot degraded spruce wood to heavy water. J Near Infrared Spectrosc 19:359–368
Farahani MRM (2003) Decay resistance of modified wood. Ph.D. thesis, University of Wales, Bangor, UK
Feist WC, Sell J (1987) Weathering behavior of dimensionally stabilized wood treated by heating under pressure of nitrogen gas. Wood Fiber Sci 19:183–195
Feist WC, Tarkow H (1967) A new procedure for measuring fiber saturation points. Forest Prod J 17:65–68
Fernandes AN, Thomas LH, Altaner CM, Callow P, Forsyth VT, Apperley DC, Kennedy CJ, Jarvis MC (2011) Nanostructure of cellulose microfibrils in spruce wood. P Natl Acad Sci USA 108:E1195–E1203
Flibotte S, Menon RS, MacKay AL, Hailey JRT (1990) Proton magnetic resonance of Western Red Cedar. Wood Fiber Sci 22:362–376
Flournoy DS, Kirk TK, Highley TL (1991) Wood decay by brown-rot fungi—changes in pore structure and cell-wall volume. Holzforschung 45:383–388
Flournoy DS, Paul JA, Kirk TK, Highley TL (1993) Changes in the size and volume of pores in sweetgum wood during simultaneous rot by Phanerochaete-Chrysosporium Burds. Holzforschung 47:297–301
Forest Products Laboratory (1919) Wood in aircraft construction. Aircraft Design Data, Note No. 12. US Department of Agriculture, Forest Service, Madison, WI, USA
Forster S (1998) The decay resistance of chemically modified softwood. PhD-thesis, University of Wales, Bangor, UK
Fortin Y (1979) Moisture content-matric potential relationship and water flow properties of wood at high moisture contents. Ph.D. thesis, University of British Columbia, Vancouver, Canada
Fredriksson M, Johansson P (2016) A method for determination of absorption isotherms at high relative humidity levels: measurements on lime-silica brick and Norway spruce (picea abies (l.) karst.). Dry Technol 34:132–141
Fredriksson M, Thygesen LG (2017) The states of water in Norway spruce (Picea abies (L.) Karst.) studied by low-field nuclear magnetic resonance (LFNMR) relaxometry: assignment of free–water populations based on quantitative wood anatomy. Holzforschung 71:77–90
Fredriksson M, Wadsö L, Ulvcrona T (2010) Moisture sorption and swelling of Norway spruce [Picea abies (L.) Karst.] impregnated with linseed oil. Wood Mater Sci Eng 5:135–142
Fredriksson M, Wadsö L, Johansson P (2013) Small resistive wood moisture sensors: a method for moisture content determination in wood structures. Eur J Wood Prod 71:515–524
Glass SV, Zelinka SL, Johnson JA (2014) Investigation of historic equilibrium moisture content data from the Forest Products Laboratory. General Technical Report FPL-GTR-229, US Department of Agriculture, Forest Service, Forest Products Laboratory, Madison, WI, USA
Glass SV, Boardman CR, Zelinka SL (2017) Short hold times in dynamic vapor sorption measurements mischaracterize the equilibrium moisture content of wood. Wood Sci Technol 51:243–260
González-Peña MM, Hale MDC (2011) Rapid assessment of physical properties and chemical composition of thermally modified wood by mid-infrared spectroscopy. Wood Sci Technol 45:83–102
Grace NH, Maass O (1932) The sorption of vapors on wood and cellulose. J Phys Chem 36:3046–3063
Granath KA, Kvist BE (1967) Molecular weight distribution analysis by gel chromatography on sephadex. J Chromatogr A 28:69–81
Greenspan L (1977) Humidity fixed-points of binary saturated aqueous-solutions. J Res NBS A Phys Chem 81:89–96
Griffin DM (1977) Water potential and wood-decay fungi. Annu Rev Phytopathol 15:319–329
Hager SL, Macrury TB (1980) Investigation of phase behavior and water binding in poly(alkylene oxide) solutions. J Appl Polym Sci 25:1559–1571
Hakkou M, Petrissans M, Gerardin P, Zoulalian A (2006) Investigations of the reasons for fungal durability of heat-treated beech wood. Polym Degrad Stabil 91:393–397
Harju AM, Kainulainen P, Venäläinen M, Tiitta M, Viitanen H (2002) Differences in resin acid concentration between brown-rot resistant and susceptible Scots pine heartwood. Holzforschung 56:479–486
Hedlin CP (1968) Sorption isotherms of twelve woods at subfreezing temperatures. Forest Prod J 17:43–48
Hergt HFA, Christensen GN (1965) Variable retention of water by dry wood. J Appl Polym Sci 9:2345–2361
Hernandez RE, Caceres CB (2010) Magnetic resonance microimaging of liquid water distribution in sugar maple wood below fiber saturation point. Wood Fiber Sci 42:259–272
Hietala S, Maunu SL, Sundholm F, Jämsä S, Viitaniemi P (2002) Structure of thermally modified wood studied by liquid state NMR measurements. Holzforschung 56:522–528
Higgins NC (1957) The equilibrium moisture content-relative humidity relationships of selected native and foreign woods. Forest Prod J 7:371–377
Hill CAS (2008) The reduction in the fibre saturation point of wood due to chemical modification using anhydride reagents: a reappraisal. Holzforschung 62:423–428
Hill CAS, Forster SC, Farahani MRM, Hale MDC, Ormondroyd GA, Williams GR (2005) An investigation of cell wall micropore blocking as a possible mechanism for the decay resistance of anhydride modified wood. Int Biodeter Biodegr 55:69–76
Hill CAS, Norton A, Newman G (2009) The water vapor sorption behavior of natural fibers. J Appl Polym Sci 112:1524–1537
Hill CAS, Norton A, Newman G (2010) Analysis of the water vapour sorption behaviour of Sitka spruce [Picea sitchensis (Bongard) Carr.] based on the parallel exponential kinetics model. Holzforschung 64:469–473
Hill CAS, Keating BA, Jalaludin Z, Mahrdt E (2012a) A rheological description of the water vapour sorption kinetics behaviour of wood invoking a model using a canonical assembly of Kelvin–Voigt elements and a possible link with sorption hysteresis. Holzforschung 66:35–47
Hill CAS, Ramsay J, Keating B, Laine K, Rautkari L, Hughes M, Constant B (2012b) The water vapour sorption properties of thermally modified and densified wood. J Mater Sci 47:3191–3197
Hill CAS, Ramsay J, Laine K, Rautkari L, Hughes M (2013) Water vapour sorption behaviour of thermally modified wood. Int Wood Prod J 4:191–196
Hill CAS, Ramsay J, Gardiner B (2015) Variability in water vapour sorption isotherm in Japanese Larch (Larix kaempferi Lamb.)—earlywood and latewood influences. Int Wood Prod J 6:53–59
Himmel S, Mai C (2015) Effects of acetylation and formalization on the dynamic water vapor sorption behavior of wood. Holzforschung 69:633–643
Himmel S, Mai C (2016) Water vapour sorption of wood modified by acetylation and formalisation—analysed by a sorption kinetics model and thermodynamic considerations. Holzforschung 70:203–213
Hinterstoisser B, Salmén L (2000) Application of dynamic 2D FTIR to cellulose. Vib Spectrosc 22:111–118
Hoffmeyer P, Engelund ET, Thygesen LG (2011) Equilibrium moisture content (EMC) in Norway spruce during the first and second desorptions. Holzforschung 65:875–882
Hofstetter K, Hinterstoisser B, Salmén L (2006) Moisture uptake in native cellulose—the roles of different hydrogen bonds: a dynamic FT-IR study using deuterium exchange. Cellulose 13:131–145
Hosseinpourpia R, Adamopoulos S, Mai C (2016) Dynamic vapour sorption of wood and holocellulose modified with thermosetting resins. Wood Sci Technol 50:165–178
Inagaki T, Yonenobu H, Tsuchikawa S (2008) Near-infrared spectroscopic monitoring of the water adsorption/desorption process in modern and archaeological wood. Appl Spectrosc 62:860–865
Ishizawa CI, Davis MF, Schell DF, Johnson DK (2007) Porosity and its effect on the digestibility of dilute sulfuric acid pretreated corn stover. J Agr Food Chem 55:2575–2581
ISO (1998) ISO 11274:1998 Soil quality—determination of the water-retention characteristic—Laboratory methods, ISO, Geneva, Switzerland
ISO (2013) ISO 12571:2013 Hygrothermal performance of building materials and products—determination of hygroscopic sorption properties, ISO, Geneva, Switzerland
Jähnert S, Chavez FV, Schaumann GE, Schreiber A, Schönhoff M, Findenegg GH (2008) Melting and freezing of water in cylindrical silica nanopores. Phys Chem Chem Phys 10:6039–6051
Jalaludin Z, Hill CAS, Samsi HW, Husain H, Xie YJ (2010a) Analysis of water vapour sorption of oleo-thermal modified wood of Acacia mangium and Endospermum malaccense by a parallel exponential kinetics model and according to the Hailwood-Horrobin model. Holzforschung 64:763–770
Jalaludin Z, Hill CAS, Xie Y, Samsi HW, Husain H, Awang K, Curling SF (2010b) Analysis of the water vapour sorption isotherms of thermally modified acacia and sesendok. Wood Mater Sci Eng 5:194–203
Javed MA, Kekkonen PM, Ahola S, Telkki VV (2015) Magnetic resonance imaging study of water absorption in thermally modified pine wood. Holzforschung 69:899–907
Källbom S (2015) Surface characterisation of thermally modified spruce wood and influence of water vapour sorption. Lic.tech. thesis, KTH Royal Institute of Technology, Stockholm, Sweden
Kamdem DP, Pizzi A, Jermannaud A (2002) Durability of heat-treated wood. Holz Roh Werkst 60:1–6
Kekkonen PM, Ylisassi A, Telkki VV (2014) Absorption of water in thermally modified pine wood as studied by nuclear magnetic resonance. J Phys Chem C 1184:2146–2153
Kelly MW, Hart CA (1970) Water vapor sorption rates by wood cell walls. Wood Fiber Sci 1:270–282
Keplinger T, Cabane E, Chanana M, Hass P, Merk V, Gierlinger N, Burgert I (2015) A versatile strategy for grafting polymers to wood cell walls. Acta Biomater 11:256–263
Keplinger T, Cabane E, Berg JK, Segmehl JS, Bock P, Burgert I (2016) Smart hierarchical bio-based materials by formation of stimuli-responsive hydrogels inside the microporous structure of wood. Adv Mater Interfaces 3:1600233
Kocaefe D, Poncsak S, Doré G, Younsi R (2008) Effect of heat treatment on the wettability of white ash and soft maple by water. Holz Roh Werkst 66:355–361
Koehler A, Thelen R (1926) Kiln drying of lumber. McGraw-Hill Book Co., London. Cited by Stevens WC (1963) The transverse shrinkage of wood. Forest Prod J 13:386–389
Kotilainen RA, Toivanen TJ, Alén RJ (2000) FTIR monitoring of chemical changes in softwood during heating. J Wood Chem Technol 20:307–320
Kuga S (1981) Pore size distribution analysis of gel substances by size exclusion chromatography. J Chromatogr A 206:449–461
Labbé N, De Jeso B, Lartigue JC, Daudé G, Pétraud M, Ratier M (2002) Moisture content and extractive materials in maritime pine wood by low field H-1 NMR. Holzforschung 56:25–31
Labbé N, De Jéso B, Lartigue JC, Daudé G, Pétraud M, Ratier M (2006) Time-domain 1H NMR characterization of the liquid phase in greenwood. Holzforschung 60:265–270
Larian M, Lavine I, Mann CA, Gauger AW (1930) II—Sorption of water vapor by lignite, peat, and wood. Ind Eng Chem 22:1231–1234
Lavine I, Gauger AW (1930) Studies in the development of Dakota lignite. Ind Eng Chem 22:1226–1231
Lide DR (2013) Enthalpy of fusion. In: Haynes WM (ed) CRC Handbook of Chemistry and Physics. CRC Press/Taylor and Francis, Boca Raton, pp 146–155
Lin KW, Ladisch MR, Voloch M, Patterson JA, Noller CH (1985) Effect of pretreatments and fermentation on pore-size in cellulosic materials. Biotechnol Bioeng 27:1427–1433
Lin JK, Ladisch MR, Patterson JA, Noller CH (1987) Determining pore-size distribution in wet cellulose by measuring solute exclusion using a differential refractometer. Biotechnol Bioeng 29:976–981
Lindh EL, Bergenstråhle-Wohlert M, Terenzi C, Salmén L, Furó I (2016) Non-exchanging hydroxyl groups on the surface of cellulose fibrils: the role of interaction with water. Carbohyd Res 434:136–142
Magne FC, Portas HJ, Wakeham H (1947) A calorimetric investigation of moisture in textile fibers. J Am Chem Soc 69:1896–1902
Maloney TC (2000) On the pore structure and dewatering properties of the pulp fiber cell wall. Ph.D. thesis, Helsinki University of Technology, Espoo, Finland
Mann J, Marrinan HJ (1956a) The reaction between cellulose and heavy water 1. A qualitative study by infra-red spectroscopy. T Faraday Soc 52:481–487
Mann J, Marrinan HJ (1956b) The reaction between cellulose and heavy water 3. A quantitative study by infra-red spectroscopy. T Faraday Soc 52:492–497
Mao JD, Holtman KM, Scott JT, Kadla JF, Schmidt-Rohr K (2006) Differences between lignin in unprocessed wood, milled wood, mutant wood, and extracted lignin detected by C-13 solid-state NMR. J Agric Food Chem 54:9677–9686
Meder R, Codd SL, Franich RA, Callaghan PT, Pope JM (2003) Observation of anisotropic water movement in Pinus radiata D. Don sapwood above fiber saturation using magnetic resonance micro-imaging. Holz Roh Werkst 61:251–256
Meiboom S, Gill D (1958) Modified spin-echo method for measuring nuclear relaxation times. Rev Sci Instrum 29:688–691
Menon RS, MacKay AL, Hailey JRT, Bloom M, Burgess AE, Swanson JS (1987) An NMR determination of the physiological water distribution in wood during drying. J Appl Polym Sci 33:1141–1155
Meyer L, Brischke C (2015) Fungal decay at different moisture levels of selected European-grown wood species. Int Biodeter Biodegr 103:23–29
Mills R (1973) Self-diffusion in normal and heavy water in the range 1°–45°. J Phys Chem US 77:685–688
Mitchell J, Stark SC, Strange JH (2005) Probing surface interactions by combining NMR cryoporometry and NMR relaxometry. J Phys D 38:1950–1958
Mitsui K, Inagaki T, Tsuchikawa S (2008) Monitoring of hydroxyl groups in wood during heat treatment using NIR spectroscopy. Biomacromol 9:286–288
Mörath E (1931) Beiträge zur Kenntnis der Quellungserscheinungen des Buchenholzes [Contributions to the knowledge of swelling phenomena in beechwood]. Kolloid-Beihefte 33:131–178
Nara S, Takeo H, Komiya T (1981) Studies on the accessibility of starch by deuteration. Starch Starke 33:329–331
Nearn WT (1955) Effect of water soluble extractives on the volumetric shrinkage and equilibrium moisture content of eleven tropical and domestic woods. Bulletin 598, Pennsylvania Agricultural Experiment Station, The Pennsylvania State university, PA, USA
Nelson RA (1977) The determination of moisture transitions in cellulosic materials using differential scanning calorimetry. J Appl Polym Sci 21:645–654
Newns AC (1956) The sorption and desorption kinetics of water in a regenerated cellulose. T Faraday Soc 52:1533–1545
Noda I (1990) 2-Dimensional infrared (2D IR) spectroscopy—theory and applications. Appl Spectrosc 44:550–561
Noda I (2006) Progress in two-dimensional (2D) correlation spectroscopy. J Mol Struct 799:2–15
Noda I, Dowrey AE, Marcott C (1993) Recent developments in 2-dimensional infrared (2D-IR) correlation spectroscopy. Appl Spectrosc 47:1317–1323
Nuopponen M, Vuorinen T, Jämsä S, Viitaniemi P (2005) Thermal modifications in softwood studied by FT-IR and UV resonance Raman spectroscopies. J Wood Chem Technol 24:13–26
Olek W, Majka J, Czajkowski L (2013) Sorption isotherms of thermally modified wood. Holzforschung 67:183–191
Papadopoulos AN, Hill CAS (2003) The sorption of water vapour by anhydride modified softwood. Wood Sci Technol 37:221–231
Park S, Venditti RA, Jameel H, Pawlak JJ (2006) Changes in pore size distribution during the drying of cellulose fibers as measured by differential scanning calorimetry. Carbohyd Polym 66:97–103
Passarini L, Malveau C, Hernández RE (2015) Distribution of the equilibrium moisture content in four hardwoods below fiber saturation point with magnetic resonance microimaging. Wood Sci Technol 49:1251–1268
Peters R (1986) Fluorescence microphotolysis to measure nucleocytoplasmic transport and intracellular mobility. BBA Biomembranes 864:305–359
Pidgeon LM, Maass O (1930) The adsorption of water by wood. J Am Chem Soc 52:1053–1069
Pönni R, Kontturi E, Vuorinen T (2013) Accessibility of cellulose: structural changes and their reversibility in aqueous media. Carbohyd Polym 93:424–429
Popescu CM, Hill CAS (2013) The water vapour adsorption–desorption behaviour of naturally aged Tilia cordata Mill. wood. Polym Degr Stabil 98:1804–1813
Popescu CM, Hill CAS, Curling S, Ormondroyd GA, Xie Y (2014) The water vapour sorption behaviour of acetylated birch wood: how acetylation affects the sorption isotherm and accessible hydroxyl content. J Mater Sci 49:2362–2371
Provencher SW (1982a) A constrained regularization method for inverting data represented by linear algebraic or integral equations. Comput Phys Commun 27:213–227
Provencher SW (1982b) CONTIN: a general purpose constrained regularization program for inverting noisy linear algebraic and integral equations. Comput Phys Commun 27:229–242
Quick JJ, Hailey JRT, MacKay AL (1990) Radial moisture profiles of cedar sapwood during drying: a proton magnetic resonance study. Wood Fiber Sci 22:404–412
Rautkari L, Hill CAS, Curling S, Jalaludin Z, Ormondroyd GA (2013) What is the role of the accessibility of wood hydroxyl groups in controlling moisture content? J Mater Sci 48:6352–6356
Ravikovitch PI, Neimark AV (2002) Experimental confirmation of different mechanisms of evaporation from ink-bottle type pores: equilibrium, pore blocking, and cavitation. Langmuir 18:9830–9837
Record SJ (1914) The mechanical properties of wood. Wiley, New York, p 165
Repellin V, Guyonnet R (2005) Evaluation of heat-treated wood swelling by differential scanning calorimetry in relation to chemical composition. Holzforschung 59:28–34
Salin JG (2008) Drying of liquid water in wood as influenced by the capillary fiber network. Dry Technol 26:560–567
Sánchez-González I, Carmona P, Moreno P, Borderías J, Sánchez-Alonso I, Rodríguez-Casado A, Careche M (2008) Protein and water structural changes in fish surimi during gelation as revealed by isotopic H/D exchange and Raman spectroscopy. Food Chem 106:56–64
Schmidt M, Gierlinger N, Schade U, Rogge T, Grunze M (2006) Polarized infrared microspectroscopy of single spruce fibers: hydrogen bonding in wood polymers. Biopolymers 83:546–555
Schwanninger M, Rodrigues JC, Pereira H, Hinterstoisser B (2004) Effects of short-time vibratory ball milling on the shape of FT-IR spectra of wood and cellulose. Vib Spectrosc 36:23–40
Seborg RM, Tarkow H, Stamm AJ (1953) Effect of heat upon the dimensional stabilization of wood. J Forest Prod Res Soc 3:59–67
Sedighi-Gilani M, Schwarze FWMR (2015) Hygric properties of Norway spruce and sycamore after incubation with two white rot fungi. Holzforschung 69:77–86
Sepall O, Mason SG (1961) Hydrogen exchange between cellulose and water 1. Measurement of accessibility. Can J Chem 39:1934–1943
Shimizu S, Agrawal KV, O’Mahony M, Drahushuk LW, Manohar N, Myerson AS, Strano MS (2015) Understanding and analyzing freezing-point transitions of confined fluids within nanopores. Langmuir 31:10113–10118
Simon C, Esteban LG, de Palacios P, Fernandez FG, Garcia-Iruela A (2017) Sorption/desorption hysteresis revisited. Sorption properties of Pinus pinea L. analysed by the parallel exponential kinetics and Kelvin–Voigt models. Holzforschung. https://doi.org/10.1515/hf-2016-0097
Simpson LA, Barton AFM (1991) Determination of the fibre saturation point in whole wood using differential scanning calorimetry. Wood Sci Technol 25:301–308
Song YQ (2009) A 2D NMR method to characterize granular structure of dairy products. Prog Nucl Magn Reson Spectrosc 55:324–334
Song YQ, Venkataramanan L, Hürlimann MD, Flaum M, Frulla P, Straley C (2002) T-1-T-2 correlation spectra obtained using a fast two-dimensional Laplace inversion. J Magn Reson 154:261–268
Song K, Yin Y, Salmén L, Xiao F, Jiang X (2014) Changes in the properties of wood cell walls during the transformation from sapwood to heartwood. J Mater Sci 49:1734–1742
Spalt HA (1958) The fundamentals of water sorption by wood. Forest Prod J 8:288–295
Stamm AJ (1971) Review of nine methods for determining the fiber saturation points of wood and wood products. Wood Sci 42:114–128
Stamm AJ, Baechler RH (1960) Decay resistance and dimensional stability of five modified woods. Forest Prod J 10:22–26
Stamm AJ, Loughborough WK (1935) Thermodynamics of the swelling of wood. J Phys Chem US 39:121–132
Stefke B, Windeisen E, Schwanninger M, Hinterstoisser B (2008) Determination of the weight percentage gain and of the acetyl group content of acetylated wood by means of different infrared spectroscopic methods. Anal Chem 80:1272–1279
Stevanic JS, Salmén L (2006) The primary cell wall studied by dynamic 2D FT-IR: interaction among components in Norway spruce (Picea abies). Cell Chem Technol 40:761–767
Stienen T, Schmidt O, Huckfeldt T (2014) Wood decay by indoor basidiomycetes at different moisture and temperature. Holzforschung 68:9–15
Stone JE, Scallan AM (1967) Effect of component removal upon porous structure of cell wall of wood. 2. Swelling in water and fiber saturation point. Tappi 50:496–501
Stone JE, Scallan AM (1968a) Effect of component removal upon porous structure of cell wall of wood. 3. A comparison between the sulphite and kraft processes. Pulp Pap Mag Can 69:69–74
Stone JE, Scallan AM (1968b) A structural model for the cell wall of water-swollen wood pulp fibres based on their accessibility to macromolecules. Cell Chem Technol 2:343–358
Stone JE, Scallan AM, Donefer E, Ahlgren E (1969) Digestibility as a simple function of a molecule of similar size to a cellulase enzyme. In: Hajny GJ, Reese ET (eds) Cellulases and their applications. American Chemical Society, Washington, DC, pp 219–241
Strømdahl K (2000) Water sorption in wood and plant fibres. Ph.D. thesis, Technical University of Denmark, Lyngby, Denmark
Suchy M, Kontturi E, Vuorinen T (2010a) Impact of drying on wood ultrastructure: similarities in cell wall alteration between native wood and isolated wood-based fibers. Biomacromol 11:2161–2168
Suchy M, Virtanen J, Kontturi E, Vuorinen T (2010b) Impact of drying on wood ultrastructure observed by deuterium exchange and photoacoustic FT-IR spectroscopy. Biomacromol 11:515–520
Sun RC, Sun XF (2002) Structural and thermal characterization of acetylated rice, wheat, rye, and barley straws and poplar wood fibre. Ind Crop Prod 16:225–235
Swenson CA (1965) Absolute infrared intensities of HDO in aqueous solution. Spectrochim Acta 21:987–993
Taniguchi T, Harada H, Nakato K (1966) Accessibility of hydroxyl groups in wood. Mokuzai Gakkaishi 10:215–220
Taniguchi T, Harada H, Nakato K (1978) Determination of water-adsorption sites in wood by a hydrogen–deuterium exchange. Nature 272:230–231
Telkki VV, Yliniemi M, Jokisaari J (2013) Moisture in softwoods: fiber saturation point, hydroxyl site content, and the amount of micropores as determined from NMR relaxation time distributions. Holzforschung 67:291–300
Temiz A, Terziev N, Eikenes M, Hafren J (2007) Effect of accelerated weathering on surface chemistry of modified wood. Appl Surf Sci 253:5355–5362
Thybring EE (2013) The decay resistance of modified wood influenced by moisture exclusion and swelling reduction. Int Biodeter Biodegr 82:87–95
Thybring EE, Thygesen LG, Burgert I (2017) Hydroxyl accessibility in wood cell walls as affected by drying and re-wetting procedures. Cellulose 24:2375–2384
Thygesen LG, Elder T (2008) Moisture in untreated, acetylated, and furfurylated Norway spruce studied during drying using Time Domain NMR. Wood Fiber Sci 40:309–320
Thygesen LG, Elder T (2009) Moisture in untreated, acetylated, and furfurylated Norway spruce monitored during drying below fiber saturation using time domain NMR. Wood Fiber Sci 41:194–200
Thygesen LG, Engelund ET, Hoffmeyer P (2010) Water sorption in wood and modified wood at high values of relative humidity. Part I: results for untreated, acetylated, and furfurylated Norway spruce. Holzforschung 64:315–323
Tiemann HD (1906) Effect of moisture upon the strength and stiffness of wood. US Department of Agriculture, Forest Service, Washington, DC
Tserki V, Zafeiropoulos NE, Simon F, Panayiotou C (2005) A study of the effect of acetylation and propionylation surface treatments on natural fibres. Compos Part A Appl S 36:1110–1118
Tsuchikawa S, Siesler HW (2003a) Near-infrared spectroscopic monitoring of the diffusion process of deuterium-labeled molecules in wood. Part I: Softwood. Appl Spectrosc 57:667–674
Tsuchikawa S, Siesler HW (2003b) Near-infrared spectroscopic monitoring of the diffusion process of deuterium-labeled molecules in wood. Part II: Hardwood. Appl Spectrosc 57:675–681
Tsuchikawa S, Tsutsumi S (1998) Adsorptive and capillary condensed water in biological material. J Mater Sci Lett 17:661–663
Urquhart AR (1929) Adsorption hysteresis. J Text Inst 20:T117–T124
Van Dyke BH (1972) Enzymatic hydrolysis of cellulose—a kinetic study. Ph.D. thesis, Massachusetts Institute of Technology, Cambridge, MA, USA
Venås TM, Thygesen LG, Barsberg S (2006) Chemical reactions involved in furfurylation of solid wood—an investigation by ATR-IR spectroscopy. International Research Group on Wood Protection, Tromsø, Norway, IRG/WP 06-40347
Venyaminov SY, Prendergast FG (1997) Water (H2O and D2O) molar absorptivity in the 1000–4000 cm−1 range and quantitative infrared spectroscopy of aqueous solutions. Anal Biochem 248:234–245
Volbehr BFKJ (1896) Untersuchungen über die Quellung der Holzfaser [Investigations of the swelling of wood fibres]. Ph.D. thesis, Universität Kiel, Kiel, Germany
Wadsö L, Svennberg K, Dueck A (2004) An experimentally simple method for measuring sorption isotherms. Dry Technol 22:2427–2440
Wagner L, Bos C, Bader TK, de Borst K (2015) Effect of water on the mechanical properties of wood cell walls—results of a nanoindentation study. Holzforschung 69:471–482
Wålinder MEP, Johansson I (2001) Measurement of wood wettability by the Wilhelmy method. Part 1. Contamination of probe liquids by extractives. Holzforschung 55:21–32
Wålinder MEP, Ström G (2001) Measurement of wood wettability by the Wilhelmy method. Part 2. Determination of apparent contact angles. Holzforschung 55:33–41
Wang QQ, He Z, Zhu Z, Zhang YHP, Ni Y, Luo XL, Zhu JY (2011) Evaluations of cellulose accessibilities of lignocelluloses by solute exclusion and protein adsorption techniques. Biotechnol Bioeng 109:381–389
Wangaard FF, Granados LA (1967) The effect of extractives on water-vapor sorption by wood. Wood Sci Technol 1:253–277
Watanabe A, Morita S, Kokot S, Matsubara M, Fukai K, Ozaki Y (2006) Drying process of microcrystalline cellulose studied by attenuated total reflection IR spectroscopy with two-dimensional correlation spectroscopy and principal component analysis. J Mol Struct 799:102–110
Weise U, Maloney T, Paulapuro H (1996) Quantification of water in different states of interaction with wood pulp fibres. Cellulose 3:189–202
Whiteside PT, Luk SY, Madden-Smith CE, Turner P, Patel N, George MW (2008) Detection of low levels of amorphous lactose using H/D exchange and FT-Raman spectroscopy. Pharm Res 25:2650–2656
Whittall KP, Bronskill MJ, Henkelman RM (1991) Investigation of analysis techniques for complicated NMR relaxation data. J Magn Reson 95:221–234
Williams FC, Hale MD (2003) The resistance of wood chemically modified with isocyanates: the role of moisture content in decay suppression. Int Biodeter Biodegr 52:215–221
Wong KKY, Deverell KF, Mackie KL, Clark TA, Donaldson LA (1988) The relationship between fiber porosity and cellulose digestibility in steam-exploded Pinus-radiata. Biotechnol Bioeng 31:447–456
Xie YJ, Hill CAS, Xiao ZF, Jalaludin Z, Militz H, Mai C (2010) Water vapor sorption kinetics of wood modified with glutaraldehyde. J Appl Polym Sci 117:1674–1682
Xie YJ, Hill CAS, Xiao ZF, Mai C, Militz H (2011) Dynamic water vapour sorption properties of wood treated with glutaraldehyde. Wood Sci Technol 45:49–61
Yasuda R, Minato K, Norimoto M (1995) Moisture adsorption thermodynamics of chemically-modified wood. Holzforschung 49:548–554
Zaihan J, Hill CAS, Curling S, Hashim WS, Hamdan H (2009) Moisture adsorption isotherms of Acacia mangium and Endospermum malaccense using dynamic vapour sorption. J Trop For Sci 21:277–285
Zaihan J, Hill CAS, Hashim WS, Dahlan JM, Sun DY (2011) Analysis of the water vapour sorption isotherms of oil palm trunk and rubberwood. J Trop For Sci 23:97–105
Zauer M, Kretzschmar J, Großmann L, Pfriem A, Wagenführ A (2014) Analysis of the pore-size distribution and fiber saturation point of native and thermally modified wood using differential scanning calorimetry. Wood Sci Technol 48:177–193
Zauer M, Meissner F, Plagge R, Wagenführ A (2016) Capillary pore-size distribution and equilibrium moisture content of wood determined by means of pressure plate technique. Holzforschung 70:137–143
Zelinka SL, Lambrecht MJ, Glass SV, Wiedenhoeft AC, Yelle DJ (2012) Examination of water phase transitions in Loblolly pine and cell wall components by differential scanning calorimetry. Thermochim Acta 533:39–45
Zelinka SL, Glass SV, Boardman CR, Derome D (2016) Moisture storage and transport properties of preservative treated and untreated southern pine wood. Wood Mater Sci Eng 11:228–238
Zeller SM (1920) Humidity in relation to moisture imbibition by wood and to spore germination on wood. Ann Missouri Bot Garden 7:51–74
Acknowledgements
EET gratefully acknowledges the financial support from the VILLUM FONDEN postdoc programme and Innovation Fund Denmark. KM and LR gratefully acknowledge financial support from European Regional Development Fund through South Savo Regional Council from Finland and industrial partners.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Thybring, E.E., Kymäläinen, M. & Rautkari, L. Experimental techniques for characterising water in wood covering the range from dry to fully water-saturated. Wood Sci Technol 52, 297–329 (2018). https://doi.org/10.1007/s00226-017-0977-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00226-017-0977-7