
Abstract
A rye–wheat centric chromosome translocation 1RS.1BL has been widely used in wheat breeding programs around the world. Increased yield of translocation lines was probably a consequence of increased root biomass. In an effort to map loci-controlling root characteristics, homoeologous recombinants of 1RS with 1BS were used to generate a consensus genetic map comprised of 20 phenotypic and molecular markers, with an average spacing of 2.5 cM. Physically, all recombination events were located in the distal 40% of the arms. A total of 68 recombinants was used and recombination breakpoints were aligned and ordered over map intervals with all the markers, integrated together in a genetic map. This approach enabled dissection of genetic components of quantitative traits, such as root traits, present on 1S. To validate our hypothesis, phenotyping of 45-day-old wheat roots was performed in five lines including three recombinants representative of the entire short arm along with bread wheat parents ‘Pavon 76’ and Pavon 1RS.1BL. Individual root characteristics were ranked and the genotypic rank sums were subjected to Quade analysis to compare the overall rooting ability of the genotypes. It appears that the terminal 15% of the rye 1RS arm carries gene(s) for greater rooting ability in wheat.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Many present day bread-wheat cultivars carry a centric rye–wheat translocation 1RS.1BL in place of chromosome 1B (Braun et al. 1998). Originally the translocation was thought to have been fixed because the 1RS arm of rye (Secale cereale L., 2n = 2x = 14, genome formula RR) carries genes for resistance to various leaf and stem fungal diseases and insects (Zeller and Hsam 1984). However, the translocation increased grain yield even in the absence of pathogens (Villareal et al. 1991, 1995). It has been shown recently that this yield increase may be a direct consequence of a substantially increased root biomass (Ehdaie et al. 2003).
Studies by Ehdaie et al. (2003) showed a significant increase of root biomass in wheat lines with 1RS translocations and a positive correlation between root biomass and grain yield. In sand cultures, all three 1RS translocations on 1AL, 1BL, and 1DL in ‘Pavon 76’ genetic background showed clear position effects with more root biomass and root branching over Pavon 76 (a high-yielding spring wheat from CIMMYT). The root biomass among these translocation lines ranked as follows: Pavon 1RS.1AL > Pavon 1RS.1DL > Pavon 1RS.1BL > Pavon 76. On the other hand, in Colorado, the ‘Amigo’ 1RS.1AL translocation from a different rye source (‘Insave’) in wheat cv. ‘Karl 92’, showed 23% yield increase under field conditions over its winter wheat check, Karl 92 (Owuoche et al. 2003). In 1RS.1BL translocation wheats grown in acid soils, roots were thinner and there was a higher root length density, and this likely enhanced the root surface area (Manske and Vlek 2002). The yield advantage of 1RS translocation lines may be partly attributed to the increase in root biomass that increases uptake of water and nutrients from the soil (Ehdaie et al. 2003; Snape et al. 2007; Ehdaie and Waines 2008).
To elucidate the mechanisms responsible for increase in root biomass in 1RS wheats, it is necessary to genetically map and identify loci responsible for enhanced root traits. The objective of this study was to develop a consensus genetic map of the 1RS-1BS chromosome arms using a population of induced homoeologous recombinants, and subsequently use the genetic map to tag the 1RS chromosomal region responsible for increased root traits.
Molecular linkage maps of cereals are being improved rapidly by adding new types of markers, merging different species-specific maps and comparative mapping of markers between related genomes. Efficient use of resulting dense maps requires detailed insights into the relationship between genetic and physical distances (Künzel et al. 2000). Various types of markers have been mapped on the 1S arm of wheat (Röder et al. 1998; Peng et al. 2004) and rye (Mago et al. 2002). PCR-based markers were developed for 1RS.1AL and 1RS.1BL wheat–rye translocations in wheat (Weng et al. 2007). However, these translocations have not been used extensively to generate a consensus map of wheat and rye chromosomes, which would be a useful tool to study different agronomic characters influenced by the presence of rye chromatin. Recombination mapping has an advantage over deletion-bin mapping in generating higher resolution maps. In deletion mapping, the number of available breakpoints, hence the number of bins, limits the resolution, and after the set developed by Endo (Endo and Gill 1996) there have been no further efforts to generate new breakpoints. In genetic mapping, resolution is limited primarily by the number of available markers, and these increase steadily. Another advantage of recombination mapping is the ability to study the genes on rye chromosomes, where, because of diploidy, it is difficult, if not impossible, to practise deletion bin mapping. In this study, using a set of recombinants, we generated a consensus map of 1RS-1BS that integrates physical and molecular markers and attempted to narrow the regions containing major QTL for root characteristics.
Materials and methods
Plant material
Experimental material was provided by Dr. A. J. Lukaszewski, University of California, Riverside. It consisted of a set of rye–wheat recombinant lines in a near-isogenic background of bread wheat cv. Pavon 76. Pavon 76 is a spring hard wheat from the breeding program at Centro Internacional de Mejoramiento de Maíz y Trigo (CIMMYT), Mexico. The set includes centric translocation 1RS.1BL in Pavon 76, where the 1RS arm is from cv. Kavkaz and 1BL arm is from Pavon 76 (Lukaszewski 1993) and a set of 68 recombinants of the 1RS arm with 1BS in Pavon 76 (Lukaszewski 2000). The 1RS-1BS recombination was induced by the absence of the Ph1 locus. All recombinants are single breakpoints; therefore, the short arm of each recombinant chromosome contains one segment of 1RS (either terminal or proximal to the centromere) and a complementary segment of 1BS. Each recombinant has a normal 1BL long arm. Since they were produced by crossing over, they can be used to generate a genetic map. However, since recombination in wheat is predominantly in the distal portion of each arm (Gill et al. 1996), physically, these recombination breakpoints cover only the terminal 40% of the arm’s length.
Genetic mapping
The primary source of PCR-based markers were DNA sequences of cDNAs that had been used to define 763 expressed sequence tags (EST) allocated by low-resolution deletion mapping to wheat 1S chromosomes (Peng et al. 2004, http://wheat.pw.usda.gov/cgibin/westsql/map_locus.cgi). Based on posted Southern blot images, 91 EST loci allocated to 1BS were selected for primer design. The unigenes corresponding to these ESTs were identified from HarvEST: wheat assembly WK (www.harvest-web.org) and used for primer design. Primer pairs were designed using PRIMER 3 software http://frodo.wi.mit.edu/ (Rozen and Skaletsky 2000). It was expected that a majority of thus produced markers would be polymorphic in the wheat–rye context. Additionally, 16 1BS specific SSR/eSSR primer pairs were chosen from earlier studies (Röder et al. 1998; Peng and Lapitan 2005). Nine eSSR primer sequences were provided by Dr. N. Lapitan of Colorado State University, Fort Collins, CO, and information on seven SSR primers was from Röder et al. (1998). Pre-screening was done using five genotypes: Pavon 76, Pavon 1RS.1BL, Pavon Dt.1BL, T-1, and 1B + 5.
DNA extraction
DNA was extracted from young leaf tissue of rye–wheat recombinants and their parental lines using Plant DNAzol (Promega, Madison, WI) and further purified using a DNeasy plant kit (Qiagen, USA). Quality and quantity of the purified DNA were assessed on a 0.8% agarose gel by electrophoresis and using a UV spectrophotometer.
PCR, gel electrophoresis and scoring for EST-based markers
PCR was performed in a DNA Engine Dyad PELTIER Thermal Cycler System (MJ Research, USA). The 20 μl reaction mixtures consisted of 50 ng of template DNA, 2 μl of 10× PCR Buffer with 15 mM of MgCl2 (Qiagen, USA), 1 unit of HotStarTaq DNA Polymerase (Qiagen, USA), 0.2 mM dNTPs (Sigma Chemical Co., St. Louis, MO), and 2 pmole each of forward and reverse primer synthesized by Operon Biotechnologies, Inc. After 10 min of denaturation at 95°C, amplifications were performed for 35 consecutive cycles each consisting of 45 s at 95°C, 30 s at 60°C, 45 s at 72°C, followed by an 8 min extension step at 72°C. About 5 μl of PCR products were run on a 1.2% agarose gel. Gels were stained with ethidium bromide and gel images captured using a gel documentation system. Gel images were scored manually for presence or absence of bands/polymorphism.
PCR, gel electrophoresis and scoring for SSR-based markers
A tailed primer approach (Oetting et al. 1995) was used for simple sequence repeat (SSR) analysis. To facilitate labeling, unlabeled M13 tail (5′ CACGACGTTGTAAAACGAC 3′) was added to the 5′ end of forward SSR/eSSR primers (see Table 1) while reverse SSR primers did not contain a tail. An IRDye-labeled M13 forward primer (third primer) was included in the PCR, which generated labeled PCR product in the subsequent cycles of PCR for easy detection. PCR was performed in a DNA Engine Dyad PELTIER Thermal Cycler System (MJ Research, USA). The 10 μl reaction mixtures consisted of 10 ng of template DNA, 1× Thermophilic DNA polymerase buffer (50 mM KCl, 10 mM Tris–HCl (ph 9.0 at 25°C), and 0.1% Triton X-100, Promega, Madison, WI), 2.0 mM MgCl2, 0.5 unit of Taq DNA polymerase (Promega, Madison, WI), 0.2 mM dNTPs (Sigma Chemical Co., St. Louis, MO), 0.25 pmoles each of forward and reverse primers synthesized by Operon Biotechnologies, Inc., and 0.25 pmoles of M13F primer labeled at the 5′ end with an infrared dye IRD700 (LI-COR, Lincoln, NE). After 4 min of denaturation at 94°C, amplifications were performed with 35 cycles each consisting of 1 min at 94°C, 1 min at the specific annealing temperature (see Table 1), 90 s at 72°C, and then followed by a 15 min extension step at 72°C.
The PCR products were separated on 18 cm denaturing 7% Long Ranger (BMA, Rockland, ME) polyacrylamide gels using a LI-COR IR2 4200LR Global DNA sequencer dual dye system (Lincoln, NE). Formamide loading dye was added at an appropriate ratio to get sharp bands (Caruso et al. 2008). PCR products were denatured at 94°C for 3 min and transferred to ice before loading on the gel. Approximately 0.25 μl of diluted and denatured PCR product was loaded in each lane of the gel. At least three wells in each gel were loaded with 50–350 bp sizing standard (LI-COR, Lincoln, NE) to facilitate scoring. Gels were scored manually for presence (1) or absence (0) of wheat alleles.
Phenotyping of shoot and root traits
A phenotyping experiment to study root characters was set up in the glasshouse in sand-tube cultures (Champoux et al. 1995) in PVC tubes, 80 cm long and 10 cm in diameter during 2006, 2007 and 2008. The study involved five lines: Pavon 76 and Pavon 1RS.1BL as the parents, and recombinants T-14, 1B + 38, and 1B + 2 (Ehdaie and Waines 2006). The three recombinant lines were chosen from the set of 68 lines to subdivide the recombining portion of the arms into three segments of roughly equal lengths.
The seeds of these five lines were surface sterilized with 5% commercial bleach for 5 min, washed for 10 min in distilled water, soaked in water for 24 h and then germinated on wet filter paper in Petri dishes. 5-day-old seedlings were transplanted to 80 cm PVC tubes containing 1 m polythene tubing, closed at one end, with 8.5 kg of silica sand #30. Two small holes were made at the bottom of polythene tube to allow drainage of excess water. The PVC tubes were supported in metal frames and arranged in a randomized complete block design with four replicates. Plants were harvested 45 days after germination when the differences in root characters could be efficiently measured among different recombinant lines. The data for different shoot characters were recorded and the tubes containing roots were stored at 4°C until processing. The roots were washed and recovered without damage using a flotation technique (Böhm 1979). The shoot characters measured were longest leaf length (LLL), maximum width of the longest leaf (LLW), leaf area (LA), plant height (PH), number of tillers (NT), and dry shoot biomass (SB). The root characters measured were number of roots greater than 30 cm (NR > 30), longest root length (LRL), total length of roots greater than 30 cm (TRL), shallow root weight (SRW, root weight above 30 cm), deep root weight (DRW, root weight below 30 cm), dry root biomass (RB), and root biomass to shoot biomass ratio (R/S).
Statistical analysis
The shoot and root data were subjected to the analysis of variance (ANOVA) for each year (Steel et al. 1997). The combined ANOVA across years was performed for each measured and calculated trait. The overall rooting ability of each genotype was calculated by ranking each genotype for individual root traits in each replication in each of the 3 years. Genotypes with the highest values were ranked 5 and those with the lowest values were ranked 1. Subsequently, all the ranks of root characters for each genotype were summed providing a measure of the rooting ability index for each genotype at different replications (blocks). The genotypic rank sums averaged across the years were subjected to the non-parametric Quade analysis developed for randomized complete block designs (Conover 1980; Quade 1979) to differentiate genotypes for overall rooting ability.
Results
PCR-based 1BS-specific markers
To map locus specific markers, we targeted the 3′ UTR of ESTs for primer design due to its polymorphic nature. Primers were designed from the corresponding unigenes from the HarvEST assembly; 91 EST loci were selected from the 763 EST loci assigned to wheat 1S by Peng et al. (2004). These 91 primer pairs were screened against five genotypes: Pavon 76, Pavon 1RS.1BL, Pavon Dt.1BL, T-1, and 1B + 5. In the initial screening, eight primer pairs showed polymorphism for the presence/absence of a DNA band. These eight primer pairs were used for screening the population of 68 primary recombinant lines. Four of the primer pairs (see Table 1) showed reproducible polymorphism. The amplicon sizes ranged from 500 bp to 1.1 kb. Two were the expected fragment sizes and the other two primer pairs, Xucr_4 and Xucr_6, produced longer amplicons than the expected size due to amplification of introns (Table 1). The primer pair for Xucr_4 amplified two PCR products of 700 and 575 bp, but only the 575 bp band was polymorphic (Fig. 1a, b).

Polymerase chain reaction amplicon from 1BS-specific dominant marker Xucr_4 (575 bp) among different wheat lines on 1.2% agarose gel elctrophoresis. The numbers at the top of panels correspond to lines carrying different lengths of 1RS chromatin in wheat backgrounds. 1B + lines are 1BS arm with distal 1RS chromatin and T-lines are 1RS arm with distal wheat chromatin
SSR-based 1BS-specific markers
To add more markers, we selected 16 1BS-specific primer sequences from the lists of Röder et al. (1998) and Peng and Lapitan (2005). The pre-screening of these primers against the same five lines produced polymorphism for eight markers, but only four of these showed a convincing polymorphism for the entire population. Amplified PCR products were comparable to the expected size range of 150–200 bp (Table 1).
Genetic map based on the 1RS-1BS recombinant breakpoints
A calculative approach was used to generate a genetic map of the 1RS-1BS arms. The 1RS-1BS map from the previous study (Lukaszewski 2000), based on 12 markers and 103 recombinants, was enriched by eight additional molecular markers and recalculated using 68 lines. The population of recombinants was divided into two configuration groups, 1B + lines with distal 1RS, and T-lines with distal 1BS, comprising 34 lines each. Both groups were scored separately for the presence/absence of each of the 20 markers, with the presence of a marker denoted as a score of 1 and the absence as 0. Each line was then ranked according to its total score for all markers and each group was further divided into 11 subgroups on the basis of its ranking. Thus, the entire mapping population was divided into 22 subgroups (Fig. 2). In this fashion, the 20 markers subdivided the genetic maps of the arms into 15 intervals (Fig. 2). Since each of the 68 primary recombinants used in mapping was preselected, guaranteed recombining cells having single crossing-over events, the total genetic length of the map is 50 cM, and each breakpoint then equals 50/68 cM while the distance between any two given markers is calculated by the formula:

Diagrammatic representation of genetic positions of recombinant breakpoints of 1RS-1BS in Pavon 76 bread wheat. White bars are representing wheat 1BS and black bars represent rye 1RS. On the left side of the figure is the list of markers present starting from the centromere at the bottom (represented by circle; white = wheat and black = rye centromeres) to the top toward telomeres. Each black and white bar represents the 1RS-1BS arm showing the position of the markers with respect to recombinant breakpoints and these bars represent only the 40% distal end of the chromosome
Genetic distance = (50/total no. of lines) × no. of breakpoints per interval and no mapping functions needed to be applied.
Phenotyping of shoot and root traits
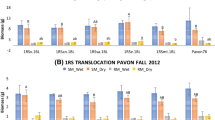
Phenotyping of recombinants was necessary to test the general applicability of the consensus 1RS-1BS map in locating a 1RS region showing better rooting ability. Various shoot and root traits were studied using two parents and three recombinants covering the whole 1RS-1BS map. There were significant differences among years for all shoot characters measured, except for the maximum width of the longest leaf (Table 2). Significant differences were found among the genotypes for shoot characters, except for maximum width of the longest leaf and leaf area. Genotype × year interaction was significant only for the number of tillers per plant (Table 2). This interaction was due to changes in the magnitude of the genotypic means across different years (non-crossover interaction) rather than changes in ranking of the means. Therefore, shoot characters in Table 2 are represented by means averaged across years. Pavon 1RS.1BL was taller, had longer leaf length and a greater root-to-shoot ratio than Pavon 76. Since Pavon 1RS.1BL and Pavon 76 had similar shoot biomass (Table 2), greater root-to-shoot biomass ratio in the former genotype indicated greater root biomass in 1RS.1BL than Pavon 76 (Table 3). Leaf area in 1B + 2 was the highest (30.1 cm2) followed by Pavon 1RS.1BL (29.7 cm2). Despite significant differences observed among the genotypes for shoot characters, the differences were relatively small, except for the shoot biomass in 1B + 2 in the third year (not shown) which was due to greater number of tillers per plant and plant height (Table 2). Otherwise, the rest of the genotypes did not show large differences for combined as well as for individual years for most of the shoot traits.
There were significant differences among years for all root characters measured (Table 3). Significant differences were found among the genotypes for all the root characters measured, except for longest root length. The genotype × year interaction was significant only for the number of roots greater than 30 cm. Therefore, means for root characters were averaged across 3 years (Table 3). The number of roots greater than 30 cm and root biomass in Pavon 1RS.1BL was greater than those in Pavon 76, which confirmed the results reported earlier (Ehdaie et al. 2003; Ehdaie and Waines 2006). The number of roots greater than 30 cm in Pavon 1RS.1BL, 1B + 2 and 1B + 38 was similar, but greater than those in Pavon 76 and T-14 (Table 3). A similar trend was observed for the total length of roots greater than 30 cm. Shallow root weight was highest in Pavon 1RS.1BL (267 mg plant−1) followed by 1B + 2 (255 mg plant−1), and 1B + 38 (227 mg plant−1). The lowest shallow root weight belonged to T-14 (193 mg plant−1). Deep root weight in Pavon 1RS.1BL, 1B + 2, and 1B + 38 were similar, but greater than those in Pavon 76 and T-14. The greater dry root biomass observed in Pavon 1RS.1BL as compared to Pavon 76 was because of a combination of greater shallow and deep root weight in the former than in the latter genotype (Table 3).
Quade analysis was used to compare the rooting ability index (RAI) of the examined genotypes based on the mean rank sums of root characters. The Quade statistic (Sj) ranged from −11.0 for T-14 to 11.5 for Pavon 1RS.1BL (Table 3). Differences among S j were statistically significant at P < 0.10 (Quade 1979). Pavon 1RS.1BL had the highest RAI (26.3) followed by 1B + 2 (22.0) and 1B + 38 (20.2). Pavon 76 (11.3) and T-14 (10.2) had the lowest RAI (Table 3).
Discussion
Genetic mapping in wide hybrids has been performed for many plant species, particularly in diploids including barley (Graner et al. 1991), chickpea (Winter et al. 2000), lentils (Eujayl et al. 1998), onion (van Heusden et al. 2000), Nicotiana species (Lin et al. 2001), and tomato (Bernatzky and Tanksley 1986; Helentjaris et al. 1986). In the early days of genetic mapping with molecular markers, wide hybrids were the approach of choice, for it guaranteed much higher levels of polymorphism than in intra-specific hybrids. Despite notable instances of non-Mendelian segregation and skewed distribution of recombination, wide hybrids produced useful genetic maps with higher marker saturation at considerably less cost and effort (van Heusden et al. 2000). The use of wide hybrids in allopolyploids is more complicated than in diploids as allopolyploids tend to have some kind of chromosome pairing control system in place that limits crossing over to homologues. Hence, homoeologous pairing may be low or even non-existent. In this study, 68 recombinants produced by crossing over were used. These recombinants were selected from a population of 103 such recombinants developed by Lukaszewski (2000). The entire recombinant population was selected from a population of ca. 17,000 progeny with the Ph1 system disabled. If the assumption is made that crossing over in the Ph1+ and Ph1− wheats is the same, and they appear to be, except for the absence of multiple crossovers per arm (Lukaszewski 2000), then the sample analyzed here would be equivalent to a population of 136 backcross progeny, a sensible number giving the maximum resolution level (spacing of breakpoints) of 0.7 cM. The physical distribution of 68 recombinant breakpoints used in the present study is shown in Fig. 3.

Physical distribution of recombinant breakpoints on the short arm of chromosome 1R-1B along the centromere (0), telomere (t) axis
With a total of 20 physical and molecular markers, we constructed the combined genetic map of 1RS-1BS recombinant breakpoints in Pavon 76 background. The genetic map produced here has an average density of 2.5 cM. The maps shown in Figs. 2 and 4 represent only the physical 40% of the distal ends of the 1S arms as no recombination took place in the proximal 60% of the arm. Any loci in this region would show complete linkage with the centromere.

Integrated genetic map of the rye–wheat 1RS-1BS arm showing the genetic distances (in cM) between markers. Genetic map represents 40% of the distal end of 1RS and 1BS chromosomes
The advantage of using these recombinant breakpoints in developing a genetic map is in their physical differences from one another. Here, we used two methods to map. Firstly, we used the recombinant breakpoints to develop a genetic map of the arm, and secondly, we used the map to classify the breakpoints. Each interval or genetic distance between two markers is represented by one or more recombinant breakpoints that most often include both reciprocal configurations of the chromosomes. This physical separation of the breakpoints further refines the genetic map to 0.7 cM level. The reciprocal nature of chromosomes with breakpoints in any given interval will permit allocation of identified genetic loci to very narrow physical segments of rye chromatin. This may be a useful tool in dissecting the genetic components of a particular gene of interest or an important agronomic trait present on 1RS.
Somers et al. (2003) detected an average of one single nucleotide polymorphism (SNP) per 540 bp ESTs in wheat and demonstrated the reliability of designing PCR primers for locus-specific amplicon production. In this study, we targeted a single chromosome arm, 1BS, and expected that most of the 91 EST loci allocated to this arm could be converted to 1BS locus-specific amplicon markers. Given that the recombination was intergeneric, we expected that most of these markers would be polymorphic between wheat and rye, and so would produce a highly saturated map. However, only four PCR-based markers showed reliable polymorphism, though we cannot rule out sequence-based polymorphisms that we could not detect. The sequences of the 91 EST loci on wheat 1S arms were used to search rice orthologs using TIGR rice version 4.0 gene models, and 54 of these sequences had rice blastx hits of e-20 or better, comprising 52 rice gene models. Of the 52 models, 27 were clustered on chromosome 5 of rice, which is known to be syntenic to chromosomes 1S of Triticeae (Sorrells et al. 2003; Peng et al. 2004; Gale and Devos 1998). In the present study, two gene models with genebank accessions viz; BE497177 and BF483588 were found to be represented as markers, Xucr_4 and Xucr_8, respectively (see Table 1). The rice annotation of Xucr_8 is described as a drought induced 19 protein (Di19) which can be helpful in studying drought. However, a high frequency of insertions and deletions in rice and maize make these genomes more fluid at the DNA sequence level than indicated by Southern analyses (Sorrells et al. 2003).
Testing a total of 16 SSR and eSSR markers yielded four polymorphic amplified products, which were comparable to the previous studies (Röder et al. 1998; Peng and Lapitan 2005). Röder et al. (1998) found different band sizes for cultivar and synthetic wheat, though the differences were comparable. Only 1 out of 9 eSSR markers could be mapped on 1BS-1RS. This low success rate may be due to low polymorphism between wheat and rye 1S arms. Whether this indicates high conservation of genic regions across species we cannot tell at this point. Peng and Lapitan (2005) showed amplification of 15 wheat eSSR markers in rye as well as barley. They detected polymorphism between wheat and barley but did not mention polymorphism between wheat and rye. A comparison of the current 1RS-1BS map with the SSR (Röder et al. 1998) and eSSR maps (Peng and Lapitan 2005) of the 1B chromosome shows a good agreement in marker order, location, and relative positions in its distal part, regardless of the projection mode used. A similar approach was used to generate a genetic map of ph1b-induced 2R-2B intergeneric recombinants, with a similar success (Lukaszewski et al. 2004), validating the wide-hybrid approach to mapping.
The integration of physical and molecular markers in the present 1RS-1BS map also provided the alignment of recombinant breakpoints over each map interval. This information offers great potential to study agronomic traits affected by the introduction of alien 1RS chromatin into wheat. Our working hypothesis was if a 1B + line shows some specific trait then this trait should be absent in its complementary T-line, and vice versa. To check the applicability of this concept, we looked at different root characters in wheat. Studies during the past few years showed an increased root biomass in wheat with the alien 1RS chromosome arm over standard spring wheat Pavon 76 and found a positive correlation of increased root biomass with grain yield (r = 0.90) (Ehdaie et al. 2003). Recent studies in rice (O. sativa L.) and maize (Zea mays L.) have also correlated the QTLs for root traits with QTLs for yield under field conditions (Yadav et al. 1997; Price et al. 1997, 2000; Tuberosa et al. 2002). A major limitation to study roots is the difficulty in making observations as the process is very laborious and time consuming. In the present study, we tested only three recombinant chromosomes with breakpoints selected to divide the recombining portion of the arm into three segments of roughly similar lengths. The experiments were conducted in 3 years at the same months of the year, in replicated trials, to determine the magnitude of the genotype × year interaction and the repeatability of results from the sand-tube technique. Significant variation was observed from year to year (Table 3), with considerably higher means in the third year. This might have been due to relatively lower temperatures in the third year providing better conditions for vegetative growth. The significant genotype × year interaction observed for number of roots greater than 30 cm (Table 3) was due to only one genotype, T-14, producing more roots only in year 2. Otherwise, other genotypes showed similar trends for this character across the 3 years. The lack of significant genotype × year interaction for most of the root characters examined (Table 3) indicated high repeatability for these root traits. Quade analysis used on rank sums separated the five genotypes in two groups viz., Pavon 1RS.1BL, 1B + 2 and 1B + 38 containing distal rye chromatin with higher rooting ability and Pavon 76 and T-14 lacking the distal rye segment with lower rooting ability (Table 2).
Overall, the five genotypes examined showed relatively small variation for shoot characters but they differed in root characteristics including root biomass. Based on the results, we propose the presence of quantitative trait locus or loci (QTL) for root traits in the distal 15% of the physical length of 1RS arm. In cereals, most of the gene rich regions for agronomic traits are concentrated in the distal ends of the chromosomes (Gill et al. 1996). Kim et al. (2004) conducted field studies for the agronomic performance of 1R from different sources of origin. They found 1RS increases the grain yield significantly, and interestingly, all the lines with 1RS did not show significant differences for shoot biomass. They did not look at the root traits, which could have also been useful. In a similar study, Waines et al. (2004) compared 1RS from different sources to study root biomass in hexaploid as well as tetraploid wheats. The translocated hexaploid wheats with 1RSAmigo and 1RSKavkaz showed 9 and 31% increase in root biomass than Pavon 76, respectively. Similar results were reported for the durum wheat ‘Aconchi’ (without 1RS) versus Aconchi with the 1RS arm. These studies point toward the definite presence of gene(s) for greater rooting ability on 1RS, and also the differential expression of alleles from different sources of 1RS in root traits. In a recent study on rice root anatomy, Uga et al. (2008) identified a QTL for metaxylem anatomy on the distal end of the long arm of chromosome 10. In another comparative study of rye DNA sequences with rice genome, the distal end of the long arm of chromosome 10 of rice was syntenic to 1RS (Hackauf et al. 2009). Both these studies provide evidence to support the general applicability of our mapping method to locate the probable region on 1RS, carrying gene(s)/QTL for root traits.
Our present finding on root studies prepares a platform to find gene(s)/QTL for root traits on 1RS. Future work will focus on use of a larger number of recombinant lines to narrow down the QTL region of 1RS responsible for increased root traits and find the molecular markers linked to these QTL. Ultimately, this would lead to our goal of physical mapping and then positional cloning of the root QTL.
References
Bernatzky R, Tanksley SD (1986) Toward a saturated linkage map in tomato based on isozymes and random cDNA sequences. Genetics 112:887–898
Böhm W (1979) Methods of studying root systems. Ecol Stud. pp 188
Braun HJ, Payne TS, Morgounov AI, Ginkel Mv, Rajaram S (1998) The challenge: one billion tons of wheat by 2020. In: Proceedings of the Ninth International Wheat Genetics Symposium, Saskatoon, Saskatchewan, Canada, 2–7 August 1998, pp 33–40
Caruso M, Federici CT, Roose ML (2008) EST-SSR markers for asparagus genetic diversity evaluation and cultivar identification. Mol Breed 21:195–204
Champoux MC, Wang G, Sarkarung S, Mackill DJ, O’Toole JC, Huang N, McCouch SR (1995) Locating genes associated with root morphology and drought avoidance in rice via linkage to molecular markers. Theor Appl Genet 90:969–981
Conover W (1980) Practical nonparametric statistics, 2nd edn. Wiley, New York
Ehdaie B, Waines JG (2006) Determination of a chromosome segment influencing rooting ability in wheat–rye 1BS-1RS recombinant lines. J Genet Breed 60:71–76
Ehdaie B, Waines JG (2008) Larger root system increases water—nitrogen uptake and grain yield in bread wheat. In: Appels et al. (eds) 11th International Wheat Genetics Symposium 2008. Sydney University Press, Brisbane, QLD, Australia, p 659
Ehdaie B, Whitkus RW, Waines JG (2003) Root biomass, water-use efficiency, and performance of wheat–rye translocations of chromosomes 1 and 2 in spring bread wheat ‘Pavon’. Crop Sci 43:710–717
Endo TR, Gill BS (1996) The deletion stocks of common wheat. J Hered 87:295–307
Eujayl I, Baum M, Powell W, Erskine W, Pehu E (1998) A genetic linkage map of lentil (Lens sp.) based on RAPD and AFLP markers using recombinant inbred lines. Theor Appl Genet 97:83–89
Gale MD, Devos KM (1998) Comparative genetics in the grasses. Proc Natl Acad Sci USA 95:1971–1974
Gill KS, Gill BS, Endo TR, Taylor T (1996) Identification and high-density mapping of gene-rich regions in chromosome group 1 of wheat. Genetics 144:1883–1891
Graner A, Jahoor A, Schondelmaier J, Siedler H, Pillen K, Fischbeck G, Wenzel G, Herrmann RF (1991) Construction of an RFLP map of barley. Theor Appl Genet 83:250–256
Hackauf B, Rudd S, JR Van der Voort, Miedaner T, Wehling P (2009) Comparative mapping of DNA sequences in rye (Secale cereale L.) in relation to the rice genome. Theor Appl Genet 118:371–384
Helentjaris T, Slocum M, Wright S, Schaefer A, Nienhuis J (1986) Construction of genetic linkage maps in maize and tomato using restriction fragment length polymorphisms. Theor Appl Genet 72:761–769
Heusden AWv, Ooijen JWv, Vrielink-van Ginkel R, Verbeek WHJ, Wietsma WA, Kik C (2000) A genetic map of an interspecific cross in Allium based on amplified fragment length polymorphism (AFLPTM) markers. Theor Appl Genet 100:118–126
Kim W, Johnson JW, Baenziger PS, Lukaszewski AJ, Gaines CS (2004) Agronomic effect of wheat–rye translocation carrying rye chromatin (1R) from different sources. Crop Sci 44:1254–1258
Künzel G, Korzun L, Meister A (2000) Cytologically integrated physical restriction fragment length polymorphism maps for the barley genome based on translocation breakpoints. Genetics 154:397–412
Lin TY, Kao YY, Lin S, Lin RF, Chen CM, Huang CH, Wang CK, Lin YZ, Chen CC (2001) A genetic linkage map of Nicotiana plumbaginifolia/Nicotiana longiflora based on RFLP and RAPD markers. Theor Appl Genet 103:905–911
Lukaszewski AJ (1993) Reconstruction in wheat of complete chromosomes 1B and 1R from the 1RS.1BL translocation of ‘Kavkaz’ origin. Genome 36:821–824
Lukaszewski AJ (2000) Manipulation of the 1RS.1BL translocation in wheat by induced homoeologous recombination. Crop Sci 40:216–225
Lukaszewski AJ, Rybka K, Korzun V, Malyshev SV, Lapinski B, Whitkus R (2004) Genetic and physical mapping of homoeologous recombination points involving wheat chromosome 2B and rye chromosome 2R. Genome 47:36–45
Mago R, Spielmeyer W, Lawrence J, Lagudah S, Ellis G, Pryor A (2002) Identification and mapping of molecular markers linked to rust resistance genes located on chromosome 1RS of rye using wheat–rye translocation lines. Theor Appl Genet 104:1317–1324
Manske GGB, Vlek PLG (2002) Root architecture—wheat as a model plant. Plant roots: the hidden half. Marcel Dekker Inc, New York, pp 249–259
Oetting WS, Lee HK, Flanders DJ, Wiesner GL, Sellers TA, King RA (1995) Linkage analysis with multiplexed short tandem repeat polymorphisms using infrared fluorescence and M13 tailed primers. Genomics 30:450–458
Owuoche JO, Sears RG, Brown-Guedira GL, Gill BS, Fritz AK (2003) Heterotic effects of wheat–rye chromosomal translocations on agronomic traits of hybrid wheat (Triticum aestivum L.) under an adequate moisture regime. Euphytica 132:67–77
Peng JH, Lapitan NLV (2005) Characterization of EST-derived microsatellites in the wheat genome and development of eSSR markers. Funct Integr Genomics 5:80–96
Peng JH, Zadeh H, Lazo GR, Gustafson JP, Chao S, Anderson OD, Qi LL, Echalier B, Gill BS, Dilbirligi M, Sandhu D, Gill KS, Greene RA, Sorrells ME, Akhunov ED, Dvorak J, Linkiewicz AM, Dubcovsky J, Hossain KG, Kalavacharla V, Kianian SF, Mahmoud AA, Miftahudin, Conley EJ, Anderson JA, Pathan MS (2004) Chromosome bin map of expressed sequence tags in homoeologous group 1 of hexaploid wheat and homoeology with rice and Arabidopsis. Genetics 168:609–623
Price AH, Tomos AD, Virk DS (1997) Genetic dissection of root growth in rice (Oryza sativa L.). I: a hydroponic screen. Theor Appl Genet 95:132–142
Price AH, Steele KA, Moore BJ, Barraclough PB, Clark LJ (2000) A combined RFLP and AFLP linkage map of upland rice (Oryza sativa L.) used to identify QTLs for root-penetration ability. Theor Appl Genet 100:49–56
Quade D (1979) Using weighted rankings in the analysis of complete blocks with additive block effects. J Am Stat Assoc 74:680–683
Röder MS, Korzun V, Wendehake K, Plaschke J, Tixier MH, Leroy P, Ganal MW (1998) A microsatellite map of wheat. Genetics 149:2007–2023
Rozen S, Skaletsky HJ (2000) Primer 3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Humana Press, Totowa, pp 365–386
Snape JW, Foulkes MJ, Simmonds J, Leverington M, Fish LJ, Wang Y, Ciavarrella M (2007) Dissecting gene × environmental effects on wheat yields via QTL and physiological analysis Euphytica. Springer Science + Business Media, Dordrecht Netherlands, pp 401–408
Somers DJ, Kirkpatrick R, Moniwa M, Walsh A (2003) Mining single-nucleotide polymorphisms from hexaploid wheat ESTs. Genome 46:431–437
Sorrells ME, Ml Rota, Bermudez-Kandianis CE, Greene RA, Kantety R, Munkvold JD, Miftahudin, Mahmoud A, Ma XF, Gustafson PJ, Qi LL, Echalier B, Gill BS, Matthews DE, Lazo GR, Chao S, Anderson OD, Edwards H, Linkiewicz AM, Dubcovsky J, Akhunov ED, Dvorak J, Zhang DH, Nguyen HT, Peng JH, Lapitan NLV (2003) Comparative DNA sequence analysis of wheat and rice genomes. Genome Res 13:1818–1827
Steel RGD, Torrie JH, Dickey DA (1997) Principles and procedures of statistics: a biometrical approach, 3rd edn. McGraw-Hill, Inc., New York 666
Tuberosa R, Sanguineti MC, Landi P, Giuliani MM, Salvi S, Conti S (2002) Identification of QTLs for root characteristics in maize grown in hydroponics and analysis of their overlap with QTLs for grain yield in the field at two water regimes. Plant Mol Biol 48:697–712
Uga Y, Okuno K, Yano M (2008) Qtls underlying natural variation in stele and xylem structures of rice root. Breed Sci 58:7–14
Villareal RL, Rajaram S, Mujeeb-Kazi A, Toro ED (1991) The effect of chromosome 1B/1R translocation on the yield potential of certain spring wheats (Triticum aestivum L.). Plant Breed 106:77–81
Villareal RL, Ed Toro, Mujeeb-Kazi A, Rajaram S (1995) The 1BL/1RS chromosome translocation effect on yield characteristics in a Triticum aestivum L. cross. Plant Breed 114:497–500
Waines JG, Ehdaie B, Sharma S (2004) Effect of origin of 1RS translocation on root biomass in wheats. Crop Science Society of America 49th annual meeting program, p 96
Weng Y, Azhaguvel P, Devkota RN, Rudd JC (2007) PCR-based markers for detection of different sources of 1AL.1RS and 1BL.1RS wheat-rye translocations in wheat background. Plant Breed 126:482–486
Winter P, Benko-Iseppon AM, Huttel B, Ratnaparkhe M, Tullu A, Sonnante G, Pfaff T, Tekeoglu M, Santra D, Sant VJ, Rajesh PN, Kahl G, Muehlbauer FJ (2000) A linkage map of the chickpea (Cicer arietinum L.) genome based on recombinant inbred lines from a C. arietinum × C. reticulatum cross: localization of resistance genes for Fusarium wilt races 4 and 5. Theor Appl Genet 101:1155–1163
Yadav R, Courtois B, Huang N, McLaren G (1997) Mapping genes controlling root morphology and root distribution in a doubled-haploid population of rice. Theor Appl Genet 94:619–632
Zeller FJ, Hsam SLK (1984) Broadening the genetic variability of cultivated wheat by utilizing rye chromatin. In: Sakomoto S (ed) 6th International Wheat Genetics Symposium, Plant Germplasm Institute, Koyoto University, Koyoto, Japan, pp 161–173
Acknowledgments
This research was supported by United States Department of Agriculture—Southwest Consortium on Plant Genetics & Water Resources, New Mexico, USA Project No. 04N09. SS would like to thank Dr. Claire Federici for providing training on SSR markers and useful discussions.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Kearsey.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sharma, S., Bhat, P.R., Ehdaie, B. et al. Integrated genetic map and genetic analysis of a region associated with root traits on the short arm of rye chromosome 1 in bread wheat. Theor Appl Genet 119, 783–793 (2009). https://doi.org/10.1007/s00122-009-1088-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-009-1088-0