
Abstract
Despite improvements in extracranial therapy, survival rate for patients suffering from brain metastases remains very poor. This is coupled with the incidence of brain metastases continuing to rise. In this review, we focus on core contributions of the blood–brain barrier to the origin of brain metastases. We first provide an overview of the structure and function of the blood–brain barrier under physiological conditions. Next, we discuss the emerging idea of a pre-metastatic niche, namely that secreted factors and extracellular vesicles from a primary tumor site are able to travel through the circulation and prime the neurovasculature for metastatic invasion. We then consider the neurotropic mechanisms that circulating tumor cells possess or develop that facilitate disruption of the blood–brain barrier and survival in the brain’s parenchyma. Finally, we compare and contrast brain metastases at the blood–brain barrier to the primary brain tumor, glioma, examining the process of vessel co-option that favors the survival and outgrowth of brain malignancies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Brain metastases involve tumor cells from peripheral sites escaping their primary location, circulating in the blood, disrupting the blood–brain barrier (BBB), extravasating into the brain, and therein producing malignant secondary tumors [1]. Representing the majority of intracranial tumors, brain metastases occur frequently in patients with advanced malignancies, such as breast, lung, skin, prostate, ovarian, and colorectal cancer [2]. The occurrence of brain metastases is estimated at 10–40% in patients with solid malignant tumors, with the incidence of brain metastases having increased over the last several decades [2]. This increase is a product of both advances in brain tumor detection and improvements in systemic extracranial therapy, which by extending survival also increase the risk that brain metastases will eventually occur. Indeed, a significant number of cancer patients, free from brain metastases at initial diagnosis, will develop intracranial tumors during the course of their disease or, in some cases, years after treatment of the primary tumor [3, 4]. Importantly, brain metastases are particularly lethal, with a median survival estimated between ≤ 4 and ≤ 12 months across all species of primary tumor [5, 6]. A principle reason for this lethality is that despite improvements in extracranial chemotherapy, few successful chemotherapeutic options are available for brain metastases, with treatment often consisting of palliative local approaches such as stereotactic or large field radiation and neurosurgical resection.
A crucial aspect in the etiology of brain metastases is our understanding of how blood-borne circulating tumor cells (CTCs) are able to overcome the BBB. Clinically, it is observed that certain primary tumors preferentially metastasize to particular secondary sites, above and beyond what would be predicted by vessel connections and anatomical proximity [7]. This preferential metastatic colonization, known as organotropism, describes how CTCs possess attributes that allow them to act as seeds favored to become disseminated tumor cells (DTCs) at organ sites with the appropriate soil, that is, organ properties which favor metastatic invasion and development [8]. For example, of the primary tumors metastasizing to the brain and spinal cord, the majority of cases (67–80%) are made up of lung cancer, breast cancer, and melanoma [9]. For CTCs to extravasate into the brain they need properties which favor interaction with (and disruption of) the BBB. Additionally, the brain’s microenvironment requires DTCs to possess or develop characteristics that allow for survival in the unique parenchyma of the brain, significantly different from any peripheral site, including limited access to nutrients, region-specific immune processes, distinct resident cells, and hypoxic conditions. This process of colonization and survival often involves a bi-directional series of events: On the one hand, tumor cells are able to adapt to the microenvironment of the brain. On the other hand, tumor cells can also alter the surrounding extracellular matrix (ECM) and resident cells to form their own brain metastatic niche. Importantly, the vast majority of tumor cells that bridge the BBB fail to survive or grow macrometastases in the brain [10, 11], while those that do survive initially remain in the perivascular microenvironment [10]. This creates a profound selective pressure for those tumor cells that are best able to colonize the vasculature of the brain. In this review, we focus on core contributions of the BBB to the origin of brain malignancies. We first discuss the anatomy of the BBB under physiological conditions. We then turn our focus to the neurotropic mechanisms that tumors possess to facilitate survival and malignant outgrowth into the brain.
The blood–brain barrier: selective gatekeeper of the neurovascular unit
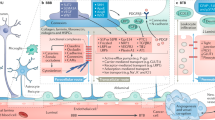
The neural networks of the CNS require that the parenchyma is constantly protected from toxins, pathogens, and ions leaking in via the circulating blood. At the same time, the brain is one of the most metabolically active organs in the body, receiving approximately 15–20% of the systemic cardiac output which provides a continuous supply of nutrients and energy substrates to maintain its activity. To achieve this delicate balance, the CNS has evolved a highly regulated, protective barrier between the blood vessel and brain tissue, the BBB (Fig. 1), which involves the complex structural and functional relationship between endothelial cells (ECs), pericytes, the neurovascular basal lamina (BL), and cells of the CNS (astrocytes, neurons, microglia, and perivascular macrophages). Indeed, the current literature discusses the BBB as a core anatomical and functional aspect of the neurovascular unit (NVU) [12]. The NVU describes the close developmental, anatomical, and functional relationships shared between the vasculature, cells of the CNS, and surrounding ECM. This includes their shared role in the regulation of the BBB and perivascular microenvironment, control of cerebral blood flow, and coordinated response to brain damage. The NVU, thus, describes the multifaceted responses of vascular and CNS cells acting as a unified biological interface.

The blood–brain barrier of the neurovascular unit. The molecular (glycocalyx, basal lamina, membrana limitans gliae perivascularis) and cellular (endothelial cells, pericytes, astrocytes, neurons, microglia) components of the blood–brain barrier. The basal lamina (produced by endothelial cells and pericytes) and membrana limitans gliae perivascularis (produced by astrocytes) form a continuous extracellular matrix under physiological conditions. Created with BioRender.com
Endothelial cells
The BBB exists at all levels of the vascular tree within the CNS, including the penetrating arteries and arterioles, dense capillary bed, post-capillary venules, as well as draining venules and veins. Particularly important to BBB structure and function are ECs, modified simple squamous epithelial cells derived from the mesoderm and covering the vessel walls. In larger brain vessels, ECs form a cylindrical lattice of numerous, tightly bound individual cells. As one moves into the cerebral microvasculature, the diameter of the vessels decreases and the boundary of the lumen consists of only a few ECs, or a single EC folded upon itself, with a lumen diameter of approximately 7 µm. In the CNS, ECs form continuous capillaries (with the exception of the circumventricular organs) and are particularly thin, with less than 0.25 µm between the cell’s luminal and abluminal surface [13]. Despite this limited distance, neurovascular ECs manifest several core properties of the BBB, including a unique membrane composition, rich in various lipid species, which contribute to a pronounced negative charge [14]. Significantly reduced transcellular movement and vesicular transport is also observed on the luminal EC surface [15], as well as a high quantity of efflux transporters working to eject lipid soluble substances back into the blood [16]. In addition, low constitutive expression levels of intercellular adhesion molecule-1 (ICAM-1) on the luminal EC surface limits immune cell extravasation into the CNS [17]. Indeed, the brain’s immune surveillance under physiological condition is significantly less than in other tissues [18]. To facilitate the constant supply of oxygen required to power the brain, the lipid membrane of neurovascular ECs allows for the unrestricted diffusion of small gaseous molecules (e.g., O2, CO2). At the same time, specialized blood-to-brain influx transport mechanisms are in place to provide nutrients like glucose and amino acids that are unable to diffuse freely into the brain. The influx of ions and charged molecules is further tightly regulated to limit disruption of the functional circuits of the CNS. Altogether, neurovascular ECs represent the most significant cellular component of the BBB, generating the primary structural and functional barrier between the blood and brain’s parenchyma.
Endothelial glycocalyx
Luminally, ECs are covered by a negatively charged layer, the endothelial glycocalyx, which is increasingly recognized as an important player in the structure and function of the BBB. Regarding chemical composition, the endothelial glycocalyx is principally composed of three main classes of molecule: proteoglycans, glycosaminoglycans (GAGs), and glycoproteins [19]. Proteoglycans consist of a core protein covalently bound to one or more GAG carbohydrate chains and exist as either membrane-bound (e.g., syndecans, glypican-1) or “free-floating” proteins (e.g., perlecan) suspended in the glycocalyx gel and capable of diffusing into the blood stream [20]. While a diverse array of membrane-associated proteoglycans are expressed on the EC luminal surface, the most prominent are the heparan sulfate proteoglycans (HSPG), specifically the syndecans and glypican-1. The structural and functional diversity of proteoglycans is extended by various combinations of the covalently attached GAGs (e.g., heparan sulfate), which are long, linear carbohydrate chains of repeating disaccharide subunits, which form dense branches extending out from the core protein. Another molecular component of the endothelial glycocalyx are membrane-bound glycoproteins, which are primarily cell adhesion molecules involved in intracellular signaling, inflammatory processes and immune cell extravasation. Similar to proteoglycans, a structurally and functionally diverse array of glycoproteins are present, including members of the selectin family, integrin family, and the immunoglobulin superfamily.
With respect to ultrastructure, the prevailing model argues that the endothelial glycocalyx is a bi-layer sugar–protein fiber matrix [21]. This bi-layer model describes two layers contributing to glycocalyx ultrastructure: (i) a thin (200–300 nm), stable inner layer forming a dense meshwork strongly anchoring to the EC membrane, and (ii) a much larger (460 nm–1 μm), robust but porous outer layer, consisting of mostly negatively charged GAGs and adsorbed plasma proteins, which is able to dynamically respond to the chemical and physical properties of the blood, including to shear stress and incorporating/exchanging components with the plasma.
From a functional perspective, the endothelial glycocalyx provides several contributions to the physiology of the BBB. It is an important regulator of vascular permeability and cell–cell interactions, with both the physical influence and negative charge of the glycocalyx limiting blood-borne cells and large molecules from perfusing across or interacting with the endothelial surface. Indeed, the large, dynamic outer layer of the glycocalyx (extending up to 1 μm into the lumen) works against the interaction of endothelial surface adhesion molecules and those of circulating cells (e.g., immune cells, tumor cells) by providing the surface adhesion molecules (extending out often only 10 nm) a physical and negatively charged shield, made up of GAGs, suspended proteoglycans, absorbed water, and plasma proteins. The endothelial glycocalyx is also involved in mechanotransduction and response to shear stress [22, 23], as well as contributing to anticoagulant pathways and fluid homeostasis [19], thus maintaining a steady-state between the blood and the vessel. Taken together, these properties of the endothelial glycocalyx provide a polar, chemical, and physical barrier on the luminal surface of ECs in the brain, regulating immune cell interactions, vascular permeability, and homeostasis which together enhance the functions of the BBB.
Endothelial cell junctions
In addition to the glycocalyx, ECs have evolved highly specialized and enriched cell junctions, creating tight adhesions between the lateral membranes of adjacent ECs. The junctional complexes of ECs form from multiple transmembrane proteins, consisting of tight junctions (TJs) and adherens junctions (AJs), involved in restricting and regulating the paracellular flux of immune cells and solutes from the blood into the brain parenchyma. Based on the organization of TJs and AJs on the lateral surface of neurovascular EC, the paracellular region is separated into apical and basolateral domains. At the BBB, highly specialized and unique TJs are the most apical EC junctional protein complex, often located around the paracellular cleft. They form from tight junction-associated MARVEL proteins (in particular occludin and tricellulin), proteins of the claudin family (in particular, claudin-5 and claudin-12) and junctional adhesion molecules (JAMs). The extracellular domains of these proteins connect to one another through homophilic binding, allowing for the connection of two adjacent EC membranes. Additionally, unique tricellular TJ molecules such as LSR and MARVELD have been described where three adjacent ECs come into contact [24]. Inside the EC, TJ transmembrane structures link to zonula occludens (ZO) proteins, cytoplasmic plaque proteins acting as a scaffold, providing intracellular connections to the actin fiber cytoskeleton. Crucially, this scaffolding is dynamic, responding to mechanical and chemical signaling as well as the physiological environment of the EC, such as inflammation [25]. Contributing to the specialization of the BBB, TJs regulate the diffusion of ions and solutes down concentration gradients across the paracellular space, while also restricting the free movements of proteins and lipids from the luminal and abluminal EC membrane, generating high transendothelial electrical resistance [26]. Importantly, TJ disruption is associated with numerous disease states [33], leading to increased permeability [27]. In contrast to TJs, AJs are located at the basolateral EC membrane. AJs are composed of transmembrane cadherins (in particular, epithelial cadherin and vascular endothelial cadherin) and intercellular catenins connecting to the cytoskeleton, again via actin, with platelet and endothelial cell adhesion molecule-1 (PECAM1) critically involved in regulating AJ formation [28]. While the manifold functions of AJs are not fully understood, evidence suggest that unlike TJs, AJs are less involved in establishing a paracellular barrier but are involved in supporting vessel integrity by managing tensile forces acting on ECs. They also are believed to facilitate cell–cell contacts, respond to diverse signaling pathways, establish cell polarity, and promote the maturation, maintenance, and plasticity of TJs [29]. In regard to this latter function, increasing evidence suggests that complex crosstalk occurs between the components of AJs and TJs generating and regulating the endothelial paracellular barrier [29].
Basal lamina
The abluminal surface of ECs is bound to the BL, a thin and selectively permeable membrane, formed by ECs and pericytes, adhering to these cells via integrins and enveloping the blood vessels and associated mural cells. The BL, measuring 50–200 nm, provides structural support, as well as creating an extracellular signaling interface for the cells of the NVU. With respect to the BBB, the BL provides an additional physical layer that resists the migration of cells and molecules towards—and separates ECs from—the brain’s tissue. Beyond the endothelial BL, a parenchymal boundary membrane termed the membrana limitans gliae perivascularis (MLGP) is intimate to the parenchyma of the brain. The MLGP is primarily produced by astrocyte end-feet processes, which tightly ensheath the vascular throughout the CNS, regulating and amplifying BBB properties [30, 31]. Under physiological conditions, these two extracellular membranes form an indistinguishable, continuous boundary, separated only by pericytes. However, a potential perivascular space exists between these two layers. In the case of pathology, leucocytes may congregate in this perivascular space, which acts as a regulatory checkpoint for further passage into the brain’s parenchyma [32, 33]. Regarding structure and composition, the BL is a highly organized, three-dimensional network primarily made of collagen IV proteins, nidogens, HSPG (in particular, perlecan and agrin) and laminins. The BL contains laminin-411 and laminin-511 derived from ECs, with low expression regions of laminin-511, acting as exit points for T-cell extravasation [32]. In larger penetrating arteries and arterioles, the MLGP includes laminin-111 (derived from pial cells) and laminin-211 (derived from astrocytes), whereas in the microvasculature laminin-111 is not present [34, 35]. Depending on physiological conditions, additional molecules are also found in the BL, including fibulins, fibronectin, various other collagen types, and thrombospondin [36]. Moreover, HSPGs are able to act as storage molecules, suspending growth factors and other bioactive compounds that can be released from the BL during vascular remodeling. Functionally, the BL is crucial to establishing the BBB, with knockout of HSPGs (perlecan/agrin) or collagen IV producing embryonic lethality [37, 38]. The BL and MLGP, thus, represent complex, dynamic ECM interfaces of the NVU which play a crucial role in generating and regulating the integrity of the BBB.
Pericytes
Pericytes are mural cells of microvessels, found intimate to the endothelium and involved in numerous supporting functions throughout the vasculature. In the CNS, pericytes are located between the BL and MLGP, and are, thus, suspended in the ECM of the neurovasculature. Unlike peripheral pericytes, which derive from the mesoderm, pericytes in the CNS derive from the neural crest [39], with the ratio of pericytes to ECs in the neurovasculature significantly greater than in the periphery. For instance, while muscle tissue vessels have a ratio of 1:100 pericytes to ECs, and lung vessels a ratio of 1:10, in the CNS this is estimated to be between 1:1 and 1:3 [40]. Morphologically, pericytes are flattened cells which extended multiple elongated processes along the abluminal surface of the endothelium. However, because these cells are suspended in the abluminal ECM they are rarely in direct contact with the EC membrane, instead forming discrete cellular adhesions such as peg-and-socket junctions, mediated by the adhesion molecule N-cadherin [41] as well as adhesion plaques, gap junctions, and tight junctions facilitating communication with ECs [42]. Regarding function, pericytes are involved in supporting angiogenesis, ECM deposition, endothelial proliferation, immune cell regulation, and inflammatory processes, as well as responding to neural activity to control blood flow [43]. In this latter respect, pericytes play a similar role in the brain’s microcirculation to smooth muscle cells as they possess contractile elements able to control the vessel diameter [42, 44], with pericytes being actively relaxed by the release of signaling molecules including prostaglandin E2 and NO via the neurotransmitter glutamate [44]. Finally, pericytes play a critical role in regulating the development of the BBB through interactions with ECs, as well as in preserving the barriers functionality across the lifespan [43, 45]. For example, in mouse models in which pericytes are ablated, endothelial hyperplasia, abnormal vasculogenesis [46], and increased BBB permeability are observed [45]. Overall, pericytes are a crucial supporting element in the homeostasis of the brain’s microcirculation, regulating numerous processes involved in BBB function and maintenance.
Astrocytes
Astrocytes, the most abundant cell type in the brain, are glial cells that extend end-foot membrane processes, involved in forming the MLGP. The polarized end-foot processes of astrocytes almost completely ensheath the EC layer, basal lamina, and associated pericytes. Limited gaps found between the end-foot processes allow for the contact of neuronal synapses directly with the MLGP, providing for neuronal communication with the blood vessels, which contributes to blood-flow regulation and BBB permeability [47,48,49]. Astrocyte end-foot processes are unique from the deeper, parenchymal portions of the cell in contact with neurons and other glia, possessing specific properties associated with the MLGP and BBB. These include dystroglycan–dystrophin complexes tethering the end-foot process to the MLGP [50], gap/tight junctions connecting adjacent processes [51], as well as region-specific K + (Kir 4.1 [52]), glucose (GLU1, [53]), and water channels (aquaporin-4, [54]), which are fundamental in maintaining the energy and ionic homeostasis of the brain’s perivascular microenvironment. Astrocytes are also implicated in regulating various signaling pathways associated with the BBB, monitoring innate immunity, as well as maintaining endothelial junctional complexes [55]. Astrocytes thus contribute to a unique covering of the vasculature found throughout the CNS, providing an additional protective and regulatory layer to the BBB.
Microglia
Microglia, the resident immune cells of the CNS, are myeloid cells [56] that derive from hematopoietic precursors migrating from the yolk sac into the CNS during development [57]. Making up approximately 10–15% of the total cells in the CNS [58], microglia play a vital role in the innate immunity of the brain and spinal cord. Structurally, microglia are small (5–10 μm) cells that extend radial processes into the ECM of the parenchyma. Functionally, the processes of microglia sense for pathogens and toxins, with these cells responsible for antigen presentation and neuronal development under physiological conditions [59]. In the case of pathology (e.g., infection, tissue damage), a prominent model argues that microglia are able to transition to two activated phenotypes with divergent functions, termed M1 and M2, which involve either pro-inflammatory/pro-killing (M1 phenotype) or immunosuppression and neural repair (M2 phenotype) functions [60]. However, this polarized differentiation during pathology has been challenged, suggesting that these functions represent a continuum rather than a dichotomy [61]. Regardless, the core aspect of microglia during brain trauma is a broad range of activated functions related to immunity and repair in the CNS. At the BBB, microglial end-feet extend and connect with the MLGP and, along with astrocytes, form part of the glial ensheathment of the neurovasculature [62]. Microglia are implicated in a host of consequences at the BBB in numerous disease states resulting from inflammatory processes including multiple sclerosis, Alzheimer’s disease, and ischemic stroke [63]. Specifically, microglia activation is associated with the release of pro-inflammatory factors that influence the permeability of the BBB by producing alterations in the integrity of endothelial junctional complexes [64]. Importantly, along with the innate immunity of the CNS, peripheral immune cells are also directly involved in the regulation and permeability of the BBB both in health and disease [65, 66]. Indeed, crosstalk between microglia and peripheral immune cells play an important role in neuroinflammatory processes, influencing the leakiness of the barrier [66].
Influence before invasion? Evidence suggesting a pre-metastatic niche at the blood–brain barrier
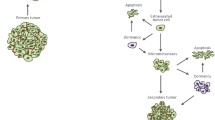
Important to our understanding of metastases is the realization that the microenvironment of distal organs play a profound role in the successful invasion and progression of CTCs. Indeed, the concept of the metastatic niche describes the alterations that occur in an organs microenvironment in the presence of successful DTCs, favoring the survival and progression of the metastatic lesion. More recently, growing evidence indicates that microenvironmental changes are able to occur even before CTCs arrive in distal organs, giving rise to the idea of a pre-metastatic niche. The concept of a pre-metastatic niche describes how primary tumors are able to actively and selectively modify a distal organ’s microenvironment through the release of factors and/or EVs, occurring before metastatic spread [67]. Put another way, the pre-metastatic niche describes the stepwise, complex molecular and cellular changes at a secondary site, induced by secreted or shedded factors by a primary tumor, prior to dissemination. Through this process, primary tumors gain the ability of action at a distance, priming secondary organs for metastatic invasion before CTCs arrive. Such changes in a distal organ microenvironment include disruption of the vascular barrier, alteration of local resident cells, ECM remodeling, release of pro-metastatic factors (e.g., growth factors, cytokines, chemokines), and alterations to immune function [67, 68]. Importantly, the pre-metastatic and metastatic niches both occur in the initial stages of metastatic development and although both niches conceptually follow one another, it is probable that their development and influence can occur both together and independently. As highlighted by Geissler and colleagues [69], the distinction between these niches is best viewed as a difference in function, with the pre-metastatic niche aiding in CTC access, anchorage, and early survival, while the metastatic niche promotes the survival, protection, and proliferation of DTCs. Pre-metastatic niche formation and its influence on metastatic invasion is described in detail for the liver [70], bone [71], lung [72], and lymph nodes [73]. However, far less is currently understood regarding the pre-metastatic niche at the BBB.
While findings are limited as to the exact nature of a pre-metastatic niche at the BBB, growing evidence suggest that such a priming event may occur in certain cancers. Recently, focus has been placed on tumor-derived EVs, in particular exomes, which have been demonstrated to shape the pre-metastatic niche of various organs [74,75,76]. Exosomes are tiny vesicles (50–100 nm in diameter) released by cells, able to contain small bioactive molecules, often contributing to paracrine signaling. However, exomes are also able to be shuttled through the blood and be taken up by distal cells. One such exome cargo molecule are microRNAs (miRs), which are tiny, non-coding stands of RNA, able to modulate gene expression [77].
With respect to the development of a brain pre-metastatic niche, exosome-mediated transfer of cancer-secreted miR-105 has been shown to suppress the tight junction protein ZO-1, is detected in the circulation at the pre-metastatic stage, and is associated with metastatic progression in breast cancer [78]. In a similar vein, exome-delivered miR-181c is demonstrated to facilitate disruption of the BBB both in vitro and in vivo by downregulating the gene PDPK1, which causes abnormal localization of actin [79]. Interestingly, Tominaga and colleagues [79] suggest that tumor-derived exomes likely contain multiple miRs that each alter and disrupt the BBB through different mechanisms, thus facilitating tumor migration. Long non-coding RNAs are also implicated in this processes, with Xu and colleagues [80] demonstrating that EV-delivered LINC00482 inhibits miR-142-3p in the perivascular microenvironment, upregulating the expression of TGF-β1 which promote microglial M2 polarization in brain metastatic lung cancer, thus contributing to the pre-metastatic niche in vivo. Hoshino and colleagues also observed that exomes isolated from organ-specific metastatic breast cancer cells travel exclusively to the associated organs in vivo [75]. This included exomes obtained from brain metastatic breast cancer cells traveling to the BBB, in this case with PECAM1-positive brain ECs representing 98% of the exome-containing cells [75]. Moreover, they show that the adhesion molecule integrin β3 was significantly expressed on those exomes that preferentially accumulated in the brain. These results, thus, suggest not only that exomes are able to possess integrin β3 facilitated neurotropic specification but also that the endothelium of the BBB plays an important role in the establishment of a brain pre-metastatic niche.
Finally, in addition to exomes, freely circulating miR-122 secreted by breast cancer cells has been shown to downregulate the glycolytic enzyme pyruvate kinase, suppressing glucose uptake by astrocytes, both in vitro and in vivo [81]. Moreover, inhibition of miR-122 in vivo restored glucose uptake in the brain, reducing the incidence of metastasis [81]. Given the importance of glucose metabolism for metastatic tumor cell survival and growth, these results suggest that an aspect of brain pre-metastatic niche formation may involve alterations in resident cell energy utilization to favor tumor cell access to energy substrates, facilitating progression. Thus, when taken together, emerging evidence suggests that alterations BBB and perivascular microenvironment through tumor-secreted factors may favor the creation of a pre-metastatic niche in the brain. In turn, such a niche has the potential to assist CTC invasion. Ultimately, the development of future techniques to experimentally define [69] and identify the development of a pre-metastatic niche in the brain could allow preemptive interventions to mitigate brain metastases [67].
Breaking and altering: mechanisms facilitating tumor cell extravasation and metastatic niche formation
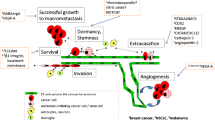
Before extravasation across the BBB can occur, tumor cells at a primary site must develop to a stage whereby they are able to exfiltrate their original location, enter the blood stream, and reach the brain. Core features of this development include (i) undergoing epithelial-to-mesenchymal transition (EMT) which enhances aggressiveness and stem-like characteristics, (ii) infiltrating and manipulating the ECM of the primary site, and (iii) intravasating into the blood steam. This path from primary tumor to brain metastases is a complex, multistep process, described in detail elsewhere [10, 82, 83]. Here, we focus on the mechanisms facilitating tumor cell extravasation across the BBB (see Fig. 2). The extravasation of tumor cells across the EC layer within the brain occurs predominantly in capillaries and post-capillary venules, particularly at capillary branches [10, 84]. As the lumen diameter is smallest at these locations, this maximizes the force of blood flow, which is able to distort and flatten CTCs against ECs and the associated glycocalyx, increasing the potential for arrest and adhesion [85, 86]. Once arrest has occurred, tumor cells have two potential means of extravasating across the BBB. First, the paracellular route between the lateral membranes of ECs or second, the transcellular route, migrating through the EC by establishing a transcellular pore [87]. Importantly, extravasation across the endothelium is a rate-limiting step in the development of brain metastases. In particular, arrest of CTCs along the neurovasculature is a non-trivial process, requiring specific mechanisms to facilitate adhesion and transmigration. Indeed, an important aspect of our understanding of metastatic development is the finding that a vast number of CTCs are often observed in patients’ blood in comparison to the actual number of extra- or intracranial metastatic lesions that develop [88]. Metastatic spread from a primary to a secondary site, therefore, appears to be a highly inefficient process, as few CTCs are ever able to establish metastases, including crossing the BBB and colonizing the brain [89]. This suggests that those CTCs that are able to successfully detect, arrest, and then cross the BBB must possess mechanisms that favor this series of events. In support of this, growing evidence indicates that neurotropic tumor cells display genetic changes that correlate with brain invasion. For example, patient-derived brain metastatic lesions across several cancers have been shown to possess mutations not observed in matched primary tumors or extracranial metastases [90]. However, within individuals, genetic homogeneity is observed across metastatic brain lesions regardless of spatial distribution or temporal onset, suggesting a conservation of neurotropic properties once they arise in metastatic cells [90]. Moreover, within different primary tumor molecular subtypes, neurotropic specificity also exists, with certain genetic changes enhancing dissemination into the brain. For example, brain metastatic triple-negative or basal-type breast cancers disrupt the BBB and colonize the brain, whereas BBB permeability remains unaltered by HER2/neu-positive breast cancer [91]. In addition, CNS metastases develop in around half of patients with mutant epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase-rearranged non-small-cell lung cancer, suggesting these genetic alterations are involved in neurotropic secondary tumors [92].

The origin of brain metastases at the blood–brain barrier. Tumor cell route to the neurovasculature. 1 Tumor cells at the primary site accumulate attributes that enable the manipulation of the surrounding tissue, allowing for cell migration and tissue invasion. 2 Tumor cells reach blood vessels at the primary site and intravasate across the vessel wall into the blood stream. 3 Circulating tumor cells reach the neurovasculature, arrest on the endothelium and extravasate across the blood–brain barrier. Tumor cell extravasation. A Tumor cells arrive in the neurovasculature, where chemokines are able to facilitate attraction to the brain endothelium. B Arrest on the brain endothelium is supported by vessel dynamics, as well as surface adhesion molecules located on both the endothelial and tumor cell membrane. C Various secreted and shedded factors released by the tumor cell disrupt the blood–brain barrier and facilitate extravasation across the brain endothelium. D After extravasation, disseminated tumor cells remain in the perivascular microenvironment where they continue to release factors that support the development of a metastatic niche. Created with BioRender.com
In a similar vein, how the brain is colonized appears to be influenced by tumor cell molecular subtype. For example, the molecular subtype of brain metastatic lung cancer has a pronounced influence on the spatial distribution of metastatic lesions in the cranial cavity [93]. Moreover, recent work by Basnet and colleagues indicates that genetic adaptation to specific organ microenvironments occurs in tumor cells by demonstrating different patterns of gene activity between metastasis locations using in situ transcriptomic profiling [94]. Specifically, using mouse xenograft breast cancer micrometastases, they show brain-specific and lung-specific transcriptome signatures in secondary tumors, differing substantially both from each another and from the initial tumor cell population. They also demonstrate that the brain metastatic variant had reduced oxidative stress and antioxidative response, suggesting alterations favoring survival in the hypoxic microenvironment of the brain.
Taken together, these findings suggest that those tumor cells capable of achieving early-stage metastatic extravasation into and colonization of the brain undergo specific changes in their genetic profile. These changes likely promote mechanisms that favor neurotropism, not only facilitating disruption of the BBB but also the capacity to survive in the nutrient-sparse and hypoxic conditions of the brain parenchyma. In the following sections, we focus on several prominent neurotropic molecular mechanisms (in particular, secreted factors and cell-surface molecules) that tumor cells have at their disposal to manipulate BBB permeability, facilitate extravasation, and form a metastatic niche in the brain.
Proteases
Various proteolytic enzymes are implicated in the formation of brain metastases by disrupting or remodeling the endothelial glycocalyx, junctional complexes and ECM of the BBB, thus facilitating tumor cell extravasation and survival (Table 1). To date, heparanase, an endoglycosidase able to cleave heparan sulfate from HSPGs, is one of the most investigated proteases involved in cancer, correlating with tumor angiogenesis, metastasis, and reduced survival across numerous cancers in various organ systems [95]. Regarding brain metastases, heparanase is implicated in the origin and proliferation of brain lesions arising from breast cancer [96,97,98,99] and melanoma [100,101,102,103,104,105,106,107]. With respect to mechanism, heparanase is involved in the proteolytic degradation of HSPGs at the BBB [104], leading to barrier disruption and ECM remodeling, which enhances tumor cell invasion of the brain. In addition, degradation of the ECM by cleavage of heparan sulfate has the potential to induce the release of numerous suspended factors, including growth factors, chemokines and other bioactive compounds that may further promote metastatic niche formation and cancer outgrowth [95]. Recent evidence also implicates heparanase in the regulation of angiogenesis [95], transcription [108], signaling pathways [109], and exosome generation [110], each of which may additionally enhance the invasion and survival of brain metastatic tumor cells. Given the abundance of HSPGs in the neurovascular glycocalyx, we further speculate that significant cleavage and sheading of heparan sulfate at the EC luminal surface may degrade glycocalyx integrity, potentially exposing underlying cell adhesion molecules, facilitating tumor cell extravasation.
In addition to heparanase, matrix metalloproteases (MMPs) are also implicated in the disruption of the BBB during the occurrence of brain metastases. MMPs are a class of calcium-dependent, zinc-containing endopeptidases able to hydrolyze and breakdown components of junctional complexes and the ECM [111]. Because of the widespread proteolytic function of MMPs, they are implicated in the formation and promotion of the tumor microenvironment [112]. For example, in brain metastatic lung cancer cells, significantly elevated levels of the aldo–keto reductase AKR1B10 are associated with MMP2 and MMP9 expression via MEK/ERK signaling, facilitating TJ degradation in vitro [113]. Silencing of AKR1B10 in these tumor cells downregulated MMP2 and MMP9 expression, suppressing both in vitro and in vivo tumor cell extravasation across the BBB [114]. Clinically, in patients with brain metastases, elevated serum levels of MMP9 (but not MMP2) has been observed [115], while both MMP2 and MMP9 were significantly increased in patients’ cerebrospinal fluid [116]. Experimental data further suggest that astrocyte activity is likely involved in the expression of MMP2 and MMP9 during brain metastases [84, 117]. MMP1 is also heavily implicated in the development of brain metastatic breast cancer [114, 118,119,120,121,122], being associated with both degradation of BBB TJs and brain-metastasizing potential. Recent studies also highlight the role of MMP2 and MMP9 in brain metastatic melanoma [123,124,125], while targeted knockdown of MMP1 has been shown to attenuate brain and lung metastasis formation in vitro and in vivo [114, 121].
Finally, several additional proteolytic enzymes are implicated in the formation of brain metastases. In patients with primary breast tumors, high levels of cathepsin S, a member of the cysteine cathepsin protease family, is associated with decreased brain metastasis-free survival. Cathepsin S is produced by both macrophages and tumor cells and facilitates BBB extravasation through proteolytic degradation of the junctional protein JAM-B, with depletion of Cathepsin S via inhibitors significantly reducing brain metastasis in vivo [126]. In non-small-cell lung cancer, ADAM9 (a member of the “a disintegrin and metalloprotease” family) expression was shown to be significantly greater in highly brain metastatic tumor cells compared to bone-metastatic or primary tumor cells, with higher invasive potential, increased adhesion capacities, and greater expression of integrin α3β1 [127]. Additionally, in breast cancer ADAM8 is implicated in regulating the expression of MMP9, with inhibition leading to reduced trans-endothelial migration [128]. Lastly, serine proteases in melanoma have been demonstrated to facilitate extravasation across an in vitro BBB model by disrupting junction complexes and causing apoptosis in ECs. Interestingly, the use of a serine protease inhibitor approximately halved the number of melanoma cells able to migrate across an endothelial monolayer [129]. Taken together, numerous cancers associated with brain metastases, including breast, lung and skin cancer, are associated with the release of proteolytic enzymes, which degrade and remodel the junctional complexes and ECM of the BBB, facilitating extravasation and metastatic niche formation in the brain.
MicroRNAs
MiRs, small non-coding RNAs involved in the post-transcriptional control of gene expression [130], are implicated in the formation of brain metastases (Table 2). Regarding miRs there are two promising avenues of investigation. First, miRs (either directly secreted or packaged in exomes) are implicated in mechanisms associated with BBB disruption and microenvironment alterations by tumor cells, representing a clinical target. Second, analysis of miR expression patterns in patients’ serum, cerebrospinal fluid, or tumor tissue, may offer a means of improving prognostic and diagnostic accuracy of brain metastases, acting as a potential biomarker. In both cases, miRs may be either upregulated or downregulated. Depending on the miR, this may either facilitate or mitigate a cancer’s metastatic potential.
Several miRs are identified in influencing the integrity of the BBB and metastatic niche formation in brain metastatic breast cancer [78, 79, 81, 131,132,133,134,135], lung cancer [80, 136, 137], and melanoma [138]. For example, a miR profile analysis of cancer stem-like cells derived from breast cancer revealed that significantly lower level of miR-7 was related to preferential organotropism for the brain, with reduced MiR-7 producing greater levels of Kruppel-like factor 4 [131]. Also in breast cancer, exosome-mediated miR-105 expression by brain metastatic tumor cells is associated with the reprograming of activated microglia, upregulating immune-suppressive cytokines and supporting brain metastatic niche formation [133]. In addition, MiRs derived from resident cells of the brain are also implicated in metastatic niche progression [134, 135]. For example, astrocyte-derived exosomes containing miR-19a target PTEN in brain metastatic breast cancer cells, activating the PI3K/Akt pathway, promoting invasion into the brain parenchyma [134].
Regarding prognosis and diagnostics, the landscape of miRs at the primary tumor site or circulating in the blood may allow for the mapping of specific signatures, which improve metastasis identification and risk assessment [139]. For example, in melanoma, miRs expression exhibits a high frequency of genetic modifications [140], as well as melanoma-specific patterns [141]. Regarding brain metastatic melanoma, a retrospective, cohort-based study analyzing genome-wide and targeted miR expression in primary melanoma tissue identified a prognostic signature of 4-miR (miR-150-5p, miR-15b-5p, miR-16-5p, and miR-374b-3p) that improved predictions for the development of brain metastasis [138]. Moreover, miR-150-5p was shown to predominantly occur from tumor-infiltrating lymphocytes, suggesting that the immune factors are also a marker for patient outcomes.
Overall, while there is limited overlap between the miRs in these findings, it is important to bear in mind that these are early investigations into the prognostic and diagnostic significance of miRs in brain metastases. The goal of future work will be to validate, extend, and specify these results. In doing so, specific patters of miRs may prove a useful prognostic and diagnostic tool in the identification of primary tumors with the potential to metastasize to the brain.
Growth factors
Growth factors are a superfamily of molecules, which are capable of promoting a diverse range of cellular processes related to growth and development. These include crucial roles in controlling cell proliferation, migration, and differentiation. Importantly, growth factors and their signaling pathways are implicated in the onset and progression of brain metastases across various cancers (Table 3). For example, hepatocyte growth factor (HGF) and angiopoietin-2 are involved in brain metastatic breast cancer [142, 143]; while HGF, placental growth factor (PLGF), and VEGF-A are associated with brain metastatic lung cancer [10, 144, 145]. Strongly implicated in brain metastases are a host of cytokines, shown to be involved in tumor dissemination across the BBB and metastatic outgrowth. Chemokines (or chemotactic cytokines) are an important subfamily of cytokines involved in stimulating the migratory behavior of leucocytes and are also implicated in attracting tumor cells to the brain endothelium. Importantly, cytokines and chemokines are also involved in promoting neuroinflammation, facilitating disruption of the BBB, altering immune cell behavior, and aiding the outgrowth of metastatic tumor cells into the brain parenchyma. Chemokines and cytokines are implicated in the formation of brain metastatic breast cancer [121, 146,147,148,149,150,151,152,153], lung cancer [80, 154,155,156,157,158,159,160,161,162], melanoma [123, 124, 163,164,165,166,167], and renal cell cancer [168]. For example, in brain metastatic breast cancer, Curtaz and colleagues demonstrated that BBB permeability in vitro was significantly increased after applying sera from breast cancer patients with brain metastases, but was not increased with sera from patients with bone or visceral metastases. Significantly increased levels of the chemokines CX3CL1 and CXCL13 were only detected in the sera from the brain metastatic breast cancer patients, which correlated with the tumor’s estrogen/progesterone receptor status [150]. Also in breast cancer, Chung and colleagues demonstrated that brain metastases-associated fibroblasts express significantly higher levels of CXCL12 and CXCL16 than fibroblasts from primary tumors or normal breast tissue, which increases tumor cell migration, with inhibition of CXCR4 or CXCL16 reducing tumor cell migration [147]. Moreover, recent findings indicate that resident cells of the CNS are also influenced by growth factors [121, 142, 146, 148, 153, 154, 166, 168,169,170,171]. In particular, several studies suggest that brain metastatic tumor cells are able to induce polarization of microglia towards a M2 (immunosuppressive) phenotype, increasing immune invasion and enhancing metastatic outgrowth [152, 158, 159, 172]. Taken together, a range of growth factors are implicated in the origin of brain metastases at the neurovasculature, influencing both the integrity of the BBB and manipulating the function of the immune system to create a more favorable metastatic niche.
Endothelial and tumor cell-surface molecules
Cell-surface adhesion molecules are involved in establishing tumor cell–EC interactions, and thus play an important role in arresting CTC in the cerebral microvasculature and facilitating extravasation across the BBB. Indeed, one of the first aspects of brain colonization is the adhesion of CTCs to brain ECs. While the exact steps and adhesion molecules involved are not well understood, a core feature of this process involves tumor cells mimicking aspects of the inflammatory leukocyte adhesion cascade, including arrest, adhesion and diapedesis [173, 174]. In addition to promoting extravasation, cell-surface molecules also play a diverse role in interactions with the brain’s resident cells and ECM, allowing for the formation of a favorable metastatic niche. Numerous cell-surface molecules located on the tumor cell or endothelial surface are implicated in the formation and outgrowth of brain metastases (Table 4). These include ALCAM [175,176,177], PECAM1 [178], L1CAM [179, 180], melanotransferrin [181], E-Selectin [155, 156, 182,183,184,185,186], and E-cadherin/ N-cadherin [187,188,189,190].
The integrin family of adhesion molecules are shown to play a particularly important role in establishing brain colonization across breast cancer [175, 191,192,193,194,195,196,197,198,199,200], lung cancer [201, 202] and melanoma [203]. For example, multiple studies implicate the integrin subunit αv in brain metastatic breast cancer, with human brain lesion specimens exhibiting significant expression of αv integrins [200]. Moreover, this αv expression is significantly higher in brain lesion specimens than in the primary tumor. In addition, the anti-integrin αv monoclonal antibody, intetumumab, is shown to decrease breast cancer brain metastases and increase survival in vivo [194]. Integrins may also allow CTCs to indirectly use platelets and leukocytes as a means of bridging the endothelium, overcoming the low expression of adhesion molecules on the neurovascular EC surface [204, 205]. For example, upregulation of αvβɜ integrin facilitates tumor cell interaction with platelets in vivo causing thrombus formation, which promotes arrest of CTCs in the blood vessel, as well as metastatic progression [206]. Integrins may additionally play a signal transduction role in brain metastases, along with adhesion functions. For instance, increased signaling by β4 integrin in breast cancer CTCs was shown to disrupt brain EC junctional complexes by inducing HER2-dependent expression of VEGF [195].
Beyond ECs, Carbonell and colleagues demonstrate that breast cancer micrometastases, both in vivo and in human brain lesion specimens, localize to the neurovascular BL, with integrin β1 implicated in this process. Blockade of β1 integrin-mediated ECM adhesion in tumor cells prevented binding to the neurovascular BL, decreasing metastatic colonization and outgrowth in vivo [191]. VCAM1, the binding partner of integrin α4ß1, is also strongly implicated in the development of brain metastases, with endothelial expression significantly upregulated early on in metastatic seeding and in the tumor-associated microvessels [207,208,209]. Recent work highlights the potential of VCAM1 as a highly sensitive diagnostic marker for brain metastases using MRI [207, 209, 210], while anti-VCAM1 antibodies are a promising avenue of treatment [185, 208, 211].
Finally, tumor cell adhesion to the endothelium not only requires the appropriate surface molecules to be present, but also that these molecules are in a receptive state for cell–cell interactions. Brain metastatic breast cancer cells are shown have enriched expression of the gene ST6GALNAC5, which encodes for α-2,6-sialyltranserase [120]. Normally exclusive to the brain, this sialyltransferase catalyzes the addition of sialic acid to cell-surface glycoproteins. In turn, this alteration to surface glycoproteins is able to stimulate tumor cell adhesion, migration, and invasion [120, 212]. Taken together, numerous cancers associated with brain metastases are associated with the increased expression of cell-surface adhesion molecules. These molecules, in turn, support interaction with the cerebral endothelium, the arrest of tumor cells in the brain, extravasation across the BBB and manipulation of the neurovascular niche.
Malignancies at the neurovascular niche: brain metastatic and glioma progression along the blood–brain barrier
The neurovascular niche plays a crucial role in the progression of brain metastases once CTCs have crossed the BBB. For DTC to survive after extravasation, they require continuous contact with the ECs and ECM associated with brain microvessels. Indeed, numerous studies now show that DTC attach to the abluminal BL, providing access to oxygen and metabolites, as well as positioning these tumor cells for vessel co-option and metastatic outgrowth [10, 84, 179, 191]. Indeed, this appears to be an important element in the initial survival of DTCs, as early-stage migration beyond the neurovascular niche into the brain parenchyma appears universally fatal [10]. An important additional aspect of metastatic development in the brain is the ability of DTCs to remain dormant in the perivascular microenvironment as micrometastases, sometimes for many years [213]. Recent work shows that multiple aspects of the BBB are involved in the perivascular dormancy of DTCs, including endothelial-derived thrombospondin-1 and astrocyte-deposited laminin-211 [214, 215].
While the precise mechanisms that awaken dormant micrometastases and activate tumor outgrowth remain unresolved, once outgrowth is underway metastatic progression can take on various invasion patterns. Interestingly, the primary tumor does not appear to be a strong predictor of which invasion pattern is observed [199]. The most prominent patterns of macrometastatic outgrowth in the brain are: (i) a displacing or non-infiltrating outgrowth pattern, producing a clear tumor boundary without infiltration into the adjacent tissue, (ii) a diffuse invasion, involving either single cells or small macrometastatic outgrowths infiltrating the brain parenchyma, and (iii) angiotropic invasion or vessel co-option, in which tumor cells sheath along adjacent blood vessels, protruding into the brain tissue [199, 216]. Here, we focus on the latter of these invasion patterns and discuss the mechanisms promoting brain metastatic vessel co-option. Interestingly, the role of the neurovascular niche in malignant outgrowth in the brain is not limited to metastases, as glioma, the most frequent primary brain tumor, can also adopt an invasive pattern modeled along the brain’s blood vessels. Because of this commonality, we also highlight the mechanisms facilitating vessel co-option in glioma and discuss possible implications for the treatment of brain malignancies.
Tumor angiogenesis involves tumor cells in the brain stimulating the sprouting and proliferation of new vessels to support a microenvironment favoring malignant outgrowth. Vessel co-option in the brain is defined as a non-angiogenic process in which tumor cells hijack existing blood vessels, either (i) by ensheathing pre-existing vessels without widespread disruption to the BBB, or (ii) by displacing pericytes and astrocytes, manipulating ECs, and remodeling the neurovascular ECM, often causing BBB disruption. In addition to these direct, perivascular forms of vessel co-option, tumor cells are also able to move in a diffuse, vascular adjacent manner through the brain parenchyma. These methods of co-opting pre-existing vessels aid in tumor cell survival, providing access to nutrients and oxygen, as well as supplying a ready-built path to further invasion. Vessel co-option is also implicated in circumvention of the BBB, as acute lymphoblastic leukemia cells have been shown to co-opt and transit along bridging vessels (facilitated by integrin α6) and enter the cerebral spinal fluid without having to extravasate into the brain [217].
Preclinical evidence indicates that across brain metastatic breast, lung, skin and colorectal cancer cell lines, vessel co-option is a frequent and important contributor to tumor vascularization and metastatic outgrowth, often together with angiogenesis [10, 123, 218, 219]. Interestingly, similar findings are observed for low-grade glioma, with vessel co-option being an important feature of early malignant outgrowth [220,221,222,223]. In contrast, tumor angiogenesis is widely understood to play a prominent role in disease progression in the high-grade, aggressive glioma variant glioblastoma multiforme (GBM), including hyper-vascularization, endothelial proliferation and blood vessel malformation [224, 225]. This has led to a broad focus on developing anti-angiogenic treatments that target the pathological mechanisms (e.g., enhanced VEGF signaling) facilitating blood vessel growth. However, even at this late stage of the disease, preclinical studies suggest that vessel co-option still occurs [223, 226]. Findings from human brain lesion specimens support this preclinical evidence in both brain metastases and in glioma. In brain metastases, clinical specimens from brain metastatic breast, lung, skin and colorectal cancer have shown the occurrence of vessel co-option [10, 191, 199, 216, 227]. Similarly, vessel co-option has been observed in human GBM lesions [226, 228]. However, despite this preclinical and clinical evidence for vessel co-option in both brain metastases and glioma, no comprehensive study has established the relative occurrence of vessel co-option and angiogenesis in either of these brain malignancies [229].
With respect to mechanism, vessel co-option in brain metastases has been shown to involve cell adhesion molecules on the tumor cell surface, including integrin β1 [191] and L1CAM [179, 180] establishing attachment and sheath formation along the abluminal vessel surface and creating an integrated growth front that uses the existing vasculature. With regards to GBM, several factors are implicated in promoting vessel co-option, including bradykinin [230], Mammary-derived growth inhibitor [231], CXCR4 [232], EGFRvIII [233], Angiopoietin-2 [222], IL8 [234, 235], IRE-1α [236], and Wnt7 [237]. Moreover, GBM vessel co-option has been shown to either involve complete ensheathment of the vessel without significant alteration to the BBB or direct attachment to the abluminal surface of EC, disrupting the BBB and displacing both astrocytes and pericytes. Indeed, in this latter instance GBM has been shown to adopt the attributes of pericytes and in doing so influence the function of the surrounding neurovasculature in a process termed pericyte mimicry [238]. Interestingly, pericyte-like spreading along blood vessels is also observed in brain metastases [180].
With regards to therapy, over the past several decades a strong focus has been placed on the development of anti-angiogenic drugs (such as VEGF inhibitors) to treat both brain metastases and glioma [239, 240]. However, a growing body of evidence indicates that anti-angiogenic therapy, although often initially successful in reducing tumor size and growth, creates a selective environment that can induce a shift to the vessel co-option invasion pattern [241]. This process, in turn, may facilitate the often observed therapeutic resistance to anti-angiogenic drugs in both types of brain malignancy. However, to date, no clear consensus exists regarding the exact role of vessel co-option in the development of anti-angiogenic therapy resistance. Thus, illuminating the mechanisms of anti-angiogenic resistance and the potential involvement of vessel co-option is an important strand of preclinical research. Indeed, the study of vessel co-option in brain malignancies is an emerging field and anti-vessel co-option therapies may prove to be an important treatment option, complementary to anti-angiogenic therapies for both glioma and for brain metastases. Moreover, an integrated understanding of the means by which glioma and brain metastases interact with the perivascular microenvironment can inform the development of such treatments. Ultimately, improvements in detecting vessel co-option, along with a better understanding of the underlying mechanisms may lead to the development of interventions that mitigate this form of invasive outgrowth in the brain.
Conclusion and perspective
Despite improvements in extracranial therapy, survival rates for patients suffering from brain metastases remain very poor. This is coupled with the fact that the incidence of brain metastases is continuing to rise. The evidence discussed in the current work indicates that metastatic brain invasion involves neurotropic mechanisms, which facilitate BBB disruption and crossing. Moreover, the neurovascular niche is demonstrated to play an important role in supporting brain malignancies, with vessel co-option an important step in promoting tumor outgrowth for both brain metastases and glioma. The neurotropic toolkit that metastatic tumor cells are able to develop involves a range of genetic alterations, secreted/shredded factors, and surface molecules, which manipulate the NVU, forming a neurovascular metastatic niche. This is mediated partially by altering the normal physiology of resident cells and remodeling the associated ECM to create a perivascular metastatic microenvironment favorable for tumor cell survival. Given that brain metastases are a frequent and deadly consequence of advanced peripheral primary tumors, the development of clinical methods to detect and target the neurotropic mechanisms underlying metastatic brain invasion are of clear practical importance.
In particular, preventive and predictive approaches to the management of brain metastases present a promising avenue of future research. With regard to preventing brain metastases, one current approach involves prophylactic intercranial radiation for cancers including non-small-cell and small-cell lung cancers [242]. However, despite some efficacy in this means of prevention, the profound negative sequelae following this approach, broadly termed radiation-induced cognitive dysfunction, limit the application of this method. In contrast, developing a better understanding, identification, and characterization of the neurotropic changes occurring in the primary tumor and CTCs may allow for a targeted approach to the prevention of brain metastases, both for initial lesion occurrence and for relapse after surgical resection. For example, a recent phase 3 trial in ALK-positive non-small-cell lung cancer showed that the use of the ALK tyrosine kinase inhibitor alectinib was associated with longer progression-free survival and worked activity against the development of brain metastases [243, 244]. Similarly, the EGFR tyrosine kinase inhibitor Lapatinib has shown promise in preventing lesion relapse in HER2-positive breast cancer [245]. In addition, immunotherapies aimed at targeting neurotropic chemokine axes in brain metastases may also represent a promising means of preventing brain metastases [246].
In a similar vein, methods to predict the likelihood or onset of brain metastases represent a further an avenue of valuable research, able to support methods of prevention. For example, several recent studies have examined the pattern of MiR in patients’ serum or primary tumor as a means of creating a brain metastases-specific biomarkers [136, 138]. Such biomarkers could, in turn, inform targeted prevention, treatment, and management. Ultimately, our ability to predict, prevent, and treat brain metastases is limited by the complex and adaptive heterogeneity of both the primary and secondary tumors. Furthering our early-stage understanding of the neurotropic mechanisms available to cancer cells may allow for targeted approaches that are able overcome the heterogeneity of these malignancies.
In conclusion, a growing body of evidence suggests that neurotropic mechanisms can facilitate the formation of both a pre-metastatic and metastatic niche at the BBB. Basic research is required to gain a better understanding of such events. This research, in turn, could support the development of preemptive interventions to mitigate brain metastases. Furthermore, accurate detection and interpretation of neurotropic patterns associated with CTCs in patients with extracranial malignancies may improve the diagnosis and prognosis of brain metastases, as well as inform early treatment. At the same time, understanding the perivascular conditions that promote the dormancy of micrometastases at the BBB, as well as the early-stage events that awaken and activate macrometastatic outgrowth is an essential avenue of research. Therapeutics that are able to maintain tumor cell dormancy may offer a long-term means of controlling the malignant outgrowth of micrometastases in the brain. Finally, a better understanding of the mechanisms underlying invasion in the perivascular microenvironment, including the potential of tumor cells to hijack existing vasculature, offer the potential to develop novel anti-vessel co-option therapies that could aid in the treatment of malignancies of the brain.
Availability of data and materials
Not applicable.
References
Lamba N, Wen PY, Aizer AA (2021) Epidemiology of brain metastases and leptomeningeal disease. Neuro-Oncol 23(9):1447–1456. https://doi.org/10.1093/neuonc/noab101
Aizer AA, Lamba N, Ahluwalia MS et al (2022) Brain metastases: A Society for Neuro-Oncology (SNO) consensus review on current management and future directions. Neuro-Oncol 24(10):1613–1646. https://doi.org/10.1093/neuonc/noac118
Pestalozzi BC, Holmes E, de Azambuja E et al (2013) CNS relapses in patients with HER2-positive early breast cancer who have and have not received adjuvant trastuzumab: a retrospective substudy of the HERA trial (BIG 1–01). Lancet Oncol 14(3):244–248. https://doi.org/10.1016/S1470-2045(13)70017-2
Davis FG, Dolecek TA, McCarthy BJ, Villano JL (2012) Toward determining the lifetime occurrence of metastatic brain tumors estimated from 2007 United States cancer incidence data. Neuro-Oncol 14(9):1171–1177. https://doi.org/10.1093/neuonc/nos152
Lamba N, Kearney RB, Catalano PJ et al (2021) Population-based estimates of survival among elderly patients with brain metastases. Neuro-Oncol 23(4):661–676. https://doi.org/10.1093/neuonc/noaa233
Cagney DN, Martin AM, Catalano PJ et al (2017) Incidence and prognosis of patients with brain metastases at diagnosis of systemic malignancy: a population-based study. Neuro-Oncol 19(11):1511–1521. https://doi.org/10.1093/neuonc/nox077
Nguyen DX, Bos PD, Massagué J (2009) Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 9(4):274–284. https://doi.org/10.1038/nrc2622
Fidler IJ (2003) The pathogenesis of cancer metastasis: the “seed and soil” hypothesis revisited. Nat Rev Cancer 3(6):453–458. https://doi.org/10.1038/nrc1098
Nayak L, Lee EQ, Wen PY (2012) Epidemiology of BRAIN METASTASES. Curr Oncol Rep 14(1):48–54. https://doi.org/10.1007/s11912-011-0203-y
Kienast Y, von Baumgarten L, Fuhrmann M et al (2010) Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 16(1):116–122. https://doi.org/10.1038/nm.2072
Heyn C, Ronald JA, Ramadan SS et al (2006) In vivo MRI of cancer cell fate at the single-cell level in a mouse model of breast cancer metastasis to the brain. Magn Reson Med 56(5):1001–1010. https://doi.org/10.1002/mrm.21029
Schaeffer S, Iadecola C (2021) Revisiting the neurovascular unit. Nat Neurosci 24(9):1198–1209. https://doi.org/10.1038/s41593-021-00904-7
Coomber BL, Stewart PA (1985) Morphometric analysis of CNS microvascular endothelium. Microvasc Res 30(1):99–115. https://doi.org/10.1016/0026-2862(85)90042-1
Worzfeld T, Schwaninger M (2016) Apicobasal polarity of brain endothelial cells. J Cereb Blood Flow Metab 36(2):340–362. https://doi.org/10.1177/0271678X15608644
Ayloo S, Gu C (2019) Transcytosis at the blood–brain barrier. Curr Opin Neurobiol 57:32–38. https://doi.org/10.1016/j.conb.2018.12.014
Campos-Bedolla P, Walter FR, Veszelka S, Deli MA (2014) Role of the blood-brain barrier in the nutrition of the central nervous system. Arch Med Res 45(8):610–638. https://doi.org/10.1016/j.arcmed.2014.11.018
Aird WC (2007) Phenotypic heterogeneity of the endothelium. Circ Res 100(2):158–173. https://doi.org/10.1161/01.RES.0000255691.76142.4a
Ousman SS, Kubes P (2012) Immune surveillance in the central nervous system. Nat Neurosci 15(8):1096–1101. https://doi.org/10.1038/nn.3161
Weinbaum S, Tarbell JM, Damiano ER (2007) The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng 9(1):121–167. https://doi.org/10.1146/annurev.bioeng.9.060906.151959
Purcell SC, Godula K (2019) Synthetic glycoscapes: addressing the structural and functional complexity of the glycocalyx. Interface Focus 9(2):20180080. https://doi.org/10.1098/rsfs.2018.0080
Curry FE (2017) Layer upon layer: the functional consequences of disrupting the glycocalyx-endothelial barrier in vivo and in vitro. Cardiovasc Res 113(6):559–561. https://doi.org/10.1093/cvr/cvx044
Koo A, Dewey CF, García-Cardeña G (2013) Hemodynamic shear stress characteristic of atherosclerosis-resistant regions promotes glycocalyx formation in cultured endothelial cells. Am J Physiol-Cell Physiol 304(2):C137–C146. https://doi.org/10.1152/ajpcell.00187.2012
Fransson L-Å, Belting M, Cheng F, Jönsson M, Mani K, Sandgren S (2004) Novel aspects of glypican glycobiology. Cell Mol Life Sci CMLS 61(9):1016–1024. https://doi.org/10.1007/s00018-004-3445-0
Sasson E, Anzi S, Bell B et al (2021) Nano-scale architecture of blood-brain barrier tight-junctions. eLife 10:e63253. https://doi.org/10.7554/eLife.63253
Greenwood J, Heasman SJ, Alvarez JI, Prat A, Lyck R, Engelhardt B (2011) Review: Leucocyte–endothelial cell crosstalk at the blood–brain barrier: a prerequisite for successful immune cell entry to the brain. Neuropathol Appl Neurobiol 37(1):24–39. https://doi.org/10.1111/j.1365-2990.2010.01140.x
Zihni C, Mills C, Matter K, Balda MS (2016) Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol 17(9):564–580. https://doi.org/10.1038/nrm.2016.80
Keaney J, Campbell M (2015) The dynamic blood–brain barrier. FEBS J 282(21):4067–4079. https://doi.org/10.1111/febs.13412
Privratsky JR, Newman PJ (2014) PECAM-1: regulator of endothelial junctional integrity. Cell Tissue Res 355(3):607–619. https://doi.org/10.1007/s00441-013-1779-3
Tietz S, Engelhardt B (2015) Brain barriers: crosstalk between complex tight junctions and adherens junctions. J Cell Biol 209(4):493–506. https://doi.org/10.1083/jcb.201412147
del Zoppo GJ, Milner R, Mabuchi T, Hung S, Wang X, Koziol JA (2006) Vascular matrix adhesion and the blood–brain barrier. Biochem Soc Trans 34(6):1261–1266. https://doi.org/10.1042/BST0341261
Sorokin L (2010) The impact of the extracellular matrix on inflammation. Nat Rev Immunol 10(10):712–723. https://doi.org/10.1038/nri2852
Wu C, Ivars F, Anderson P et al (2009) Endothelial basement membrane laminin α5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med 15(5):519–527. https://doi.org/10.1038/nm.1957
Hallmann R, Hannocks M-J, Song J et al (2020) The role of basement membrane laminins in vascular function. Int J Biochem Cell Biol 127:105823. https://doi.org/10.1016/j.biocel.2020.105823
Sixt M, Engelhardt B, Pausch F, Hallmann R, Wendler O, Sorokin LM (2001) Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol 153(5):933–946. https://doi.org/10.1083/jcb.153.5.933
Engelhardt B, Sorokin L (2009) The blood–brain and the blood–cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol 31(4):497–511. https://doi.org/10.1007/s00281-009-0177-0
Hallmann R, Horn N, Selg M, Wendler O, Pausch F, Sorokin LM (2005) Expression and function of laminins in the embryonic and mature vasculature. Physiol Rev 85(3):979–1000. https://doi.org/10.1152/physrev.00014.2004
Sarrazin S, Lamanna WC, Esko JD (2011) Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol 3(7):a004952. https://doi.org/10.1101/cshperspect.a004952
Pöschl E, Schlötzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U (2004) Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 131(7):1619–1628. https://doi.org/10.1242/dev.01037
Reyahi A, Nik AM, Ghiami M et al (2015) Foxf2 is required for brain pericyte differentiation and development and maintenance of the blood-brain barrier. Dev Cell 34(1):19–32. https://doi.org/10.1016/j.devcel.2015.05.008
Shepro D, Morel NML (1993) Pericyte physiology. FASEB J 7(11):1031–1038. https://doi.org/10.1096/fasebj.7.11.8370472
Gerhardt H, Wolburg H, Redies C (2000) N-cadherin mediates pericytic-endothelial interaction during brain angiogenesis in the chicken. Dev Dyn 218(3):472–479. https://doi.org/10.1002/1097-0177(200007)218:3%3c472::AID-DVDY1008%3e3.0.CO;2-#
Winkler EA, Bell RD, Zlokovic BV (2011) Central nervous system pericytes in health and disease. Nat Neurosci 14(11):1398–1405. https://doi.org/10.1038/nn.2946
Armulik A, Genové G, Betsholtz C (2011) Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21(2):193–215. https://doi.org/10.1016/j.devcel.2011.07.001
Hall CN, Reynell C, Gesslein B et al (2014) Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508(7494):55–60. https://doi.org/10.1038/nature13165
Daneman R, Zhou L, Kebede AA, Barres BA (2010) Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 468(7323):562–566. https://doi.org/10.1038/nature09513
Hellström M, Gerhardt H, Kalén M et al (2001) Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol 153(3):543–554. https://doi.org/10.1083/jcb.153.3.543
Cohen Z, Molinatti G, Hamel E (1997) Astroglial and vascular interactions of noradrenaline terminals in the rat cerebral cortex. J Cereb Blood Flow Metab 17(8):894–904. https://doi.org/10.1097/00004647-199708000-00008
Paspalas CD, Papadopoulos GC (1996) Ultrastructural relationships between noradrenergic nerve fibers and non-neuronal elements in the rat cerebral cortex. Glia 17(2):133–146. https://doi.org/10.1002/(SICI)1098-1136(199606)17:2%3c133::AID-GLIA5%3e3.0.CO;2-3
Kaplan L, Chow BW, Gu C (2020) Neuronal regulation of the blood–brain barrier and neurovascular coupling. Nat Rev Neurosci 21(8):416–432. https://doi.org/10.1038/s41583-020-0322-2
Noell S, Wolburg-Buchholz K, Mack AF et al (2011) Evidence for a role of dystroglycan regulating the membrane architecture of astroglial endfeet. Eur J Neurosci 33(12):2179–2186. https://doi.org/10.1111/j.1460-9568.2011.07688.x
Nagy JI, Rash JE (2000) Connexins and gap junctions of astrocytes and oligodendrocytes in the CNS. Brain Res Rev 32(1):29–44. https://doi.org/10.1016/S0165-0173(99)00066-1
Gordon GRJ, Mulligan SJ, MacVicar BA (2007) Astrocyte control of the cerebrovasculature. Glia 55(12):1214–1221. https://doi.org/10.1002/glia.20543
Kimelberg HK, Nedergaard M (2010) Functions of astrocytes and their potential as therapeutic targets. Neurotherapeutics 7(4):338–353. https://doi.org/10.1016/j.nurt.2010.07.006
Nagelhus EA, Ottersen OP (2013) Physiological roles of aquaporin-4 in brain. Physiol Rev 93(4):1543–1562. https://doi.org/10.1152/physrev.00011.2013
Abbott NJ, Rönnbäck L, Hansson E (2006) Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci 7(1):41–53. https://doi.org/10.1038/nrn1824
García-Culebras A, Durán-Laforet V, Peña-Martínez C et al (2018) Myeloid cells as therapeutic targets in neuroinflammation after stroke: Specific roles of neutrophils and neutrophil–platelet interactions. J Cereb Blood Flow Metab 38(12):2150–2164. https://doi.org/10.1177/0271678X18795789
Nayak D, Roth TL, McGavern DB (2014) Microglia development and function. Annu Rev Immunol 32:367–402. https://doi.org/10.1146/annurev-immunol-032713-120240
Salter MW, Stevens B (2017) Microglia emerge as central players in brain disease. Nat Med 23(9):1018–1027. https://doi.org/10.1038/nm.4397
Streit WJ, Conde JR, Fendrick SE, Flanary BE, Mariani CL (2005) Role of microglia in the central nervous system’s immune response. Neurol Res 27(7):685–691. https://doi.org/10.1179/016164105X49463a
Tang Y, Le W (2016) Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol 53(2):1181–1194. https://doi.org/10.1007/s12035-014-9070-5
Ransohoff RM (2016) A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 19(8):987–991. https://doi.org/10.1038/nn.4338
Lassmann H, Zimprich F, Vass K, Hickey WF (1991) Microglial cells are a component of the perivascular glia limitans. J Neurosci Res 28(2):236–243. https://doi.org/10.1002/jnr.490280211
Dudvarski Stankovic N, Teodorczyk M, Ploen R, Zipp F, Schmidt MHH (2016) Microglia–blood vessel interactions: a double-edged sword in brain pathologies. Acta Neuropathol (Berl) 131(3):347–363. https://doi.org/10.1007/s00401-015-1524-y
Su EJ, Cao C, Fredriksson L et al (2017) Microglial-mediated PDGF-CC activation increases cerebrovascular permeability during ischemic stroke. Acta Neuropathol (Berl) 134(4):585–604. https://doi.org/10.1007/s00401-017-1749-z
Marchetti L, Engelhardt B (2020) Immune cell trafficking across the blood-brain barrier in the absence and presence of neuroinflammation. Vasc Biol 2(1):H1–H18. https://doi.org/10.1530/VB-19-0033
Dionisio-Santos DA, Olschowka JA, O’Banion MK (2019) Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J Neuroinflammation 16(1):74. https://doi.org/10.1186/s12974-019-1453-0
Peinado H, Zhang H, Matei IR et al (2017) Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer 17(5):302–317. https://doi.org/10.1038/nrc.2017.6
Patras L, Shaashua L, Matei I, Lyden D (2023) Immune determinants of the pre-metastatic niche. Cancer Cell 41(3):546–572. https://doi.org/10.1016/j.ccell.2023.02.018
Geissler M, Jia W, Kiraz EN et al (2023) The brain pre-metastatic niche: biological and technical advancements. Int J Mol Sci 24(12):10055. https://doi.org/10.3390/ijms241210055
Houg DS, Bijlsma MF (2018) The hepatic pre-metastatic niche in pancreatic ductal adenocarcinoma. Mol Cancer 17(1):95. https://doi.org/10.1186/s12943-018-0842-9
Ren G, Esposito M, Kang Y (2015) Bone metastasis and the metastatic niche. J Mol Med 93(11):1203–1212. https://doi.org/10.1007/s00109-015-1329-4
Maru Y (2015) The lung metastatic niche. J Mol Med 93(11):1185–1192. https://doi.org/10.1007/s00109-015-1355-2
Gillot L, Baudin L, Rouaud L, Kridelka F, Noël A (2021) The pre-metastatic niche in lymph nodes: formation and characteristics. Cell Mol Life Sci 78(16):5987–6002. https://doi.org/10.1007/s00018-021-03873-z
Costa-Silva B, Aiello NM, Ocean AJ et al (2015) Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol 17(6):816–826. https://doi.org/10.1038/ncb3169
Hoshino A, Costa-Silva B, Shen T-L et al (2015) Tumour exosome integrins determine organotropic metastasis. Nature 527(7578):329–335. https://doi.org/10.1038/nature15756
Plebanek MP, Angeloni NL, Vinokour E et al (2017) Pre-metastatic cancer exosomes induce immune surveillance by patrolling monocytes at the metastatic niche. Nat Commun 8(1):1319. https://doi.org/10.1038/s41467-017-01433-3
Hammond SM (2015) An overview of microRNAs. Adv Drug Deliv Rev 87:3–14. https://doi.org/10.1016/j.addr.2015.05.001
Zhou W, Fong MY, Min Y et al (2014) Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 25(4):501–515. https://doi.org/10.1016/j.ccr.2014.03.007
Tominaga N, Kosaka N, Ono M et al (2015) Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood–brain barrier. Nat Commun 6(1):6716. https://doi.org/10.1038/ncomms7716
Xu W, Patel N, Deng Y, Ding S, Wang T, Zhang H (2023) Extracellular vesicle-derived LINC00482 induces microglial M2 polarization to facilitate brain metastasis of NSCLC. Cancer Lett 561:216146. https://doi.org/10.1016/j.canlet.2023.216146
Fong MY, Zhou W, Liu L et al (2015) Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol 17(2):183–194. https://doi.org/10.1038/ncb3094
Heerboth S, Housman G, Leary M et al (2015) EMT and tumor metastasis. Clin Transl Med 4(1):6. https://doi.org/10.1186/s40169-015-0048-3
Wrobel JK, Toborek M (2016) Blood-brain barrier remodeling during brain metastasis formation. Mol Med 22(1):32–40. https://doi.org/10.2119/molmed.2015.00207
Lorger M, Felding-Habermann B (2010) Capturing changes in the brain microenvironment during initial steps of breast cancer brain metastasis. Am J Pathol 176(6):2958–2971. https://doi.org/10.2353/ajpath.2010.090838
Végh AG, Fazakas C, Nagy K et al (2012) Adhesion and stress relaxation forces between melanoma and cerebral endothelial cells. Eur Biophys J 41(2):139–145. https://doi.org/10.1007/s00249-011-0765-5
Wilhelm I, Fazakas C, Molnár J et al (2014) Role of Rho/ROCK signaling in the interaction of melanoma cells with the blood–brain barrier. Pigment Cell Melanoma Res 27(1):113–123. https://doi.org/10.1111/pcmr.12169
Arvanitis CD, Ferraro GB, Jain RK (2020) The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat Rev Cancer 20(1):26–41. https://doi.org/10.1038/s41568-019-0205-x
Nagrath S, Sequist LV, Maheswaran S et al (2007) Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 450(7173):1235–1239. https://doi.org/10.1038/nature06385
Massagué J, Obenauf AC (2016) Metastatic colonization by circulating tumour cells. Nature 529(7586):298–306. https://doi.org/10.1038/nature17038
Brastianos PK, Carter SL, Santagata S et al (2015) Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov 5(11):1164–1177. https://doi.org/10.1158/2159-8290.CD-15-0369
Yonemori K, Tsuta K, Ono M et al (2010) Disruption of the blood brain barrier by brain metastases of triple-negative and basal-type breast cancer but not HER2/neu-positive breast cancer. Cancer 116(2):302–308. https://doi.org/10.1002/cncr.24735
Rangachari D, Yamaguchi N, VanderLaan PA et al (2015) Brain metastases in patients with EGFR-mutated or ALK-rearranged non-small-cell lung cancers. Lung Cancer 88(1):108–111. https://doi.org/10.1016/j.lungcan.2015.01.020
Takano K, Kinoshita M, Takagaki M et al (2016) Different spatial distributions of brain metastases from lung cancer by histological subtype and mutation status of epidermal growth factor receptor. Neuro-Oncol 18(5):716–724. https://doi.org/10.1093/neuonc/nov266
Basnet H, Tian L, Ganesh K et al (2019) Flura-seq identifies organ-specific metabolic adaptations during early metastatic colonization. eife 8:e43627. https://doi.org/10.7554/eLife.43627
Ilan N, Elkin M, Vlodavsky I (2006) Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int J Biochem Cell Biol 38(12):2018–2039. https://doi.org/10.1016/j.biocel.2006.06.004
Cohen-Kaplan V, Doweck I, Naroditsky I, Vlodavsky I, Ilan N (2008) Heparanase augments epidermal growth factor receptor phosphorylation: correlation with head and neck tumor progression. Cancer Res 68(24):10077–10085. https://doi.org/10.1158/0008-5472.CAN-08-2910
Zhang L, Sullivan P, Suyama J, Marchetti D (2010) Epidermal growth factor-induced heparanase nucleolar localization augments DNA topoisomerase I activity in brain metastatic breast cancer. Mol Cancer Res 8(2):278–290. https://doi.org/10.1158/1541-7786.MCR-09-0375
Zhang L, Ridgway LD, Wetzel MD et al (2013) The identification and characterization of breast cancer ctcs competent for brain metastasis. Sci Transl Med 5(180):180ra148–180ra48. https://doi.org/10.1126/scitranslmed.3005109
Zhang L, Sullivan PS, Goodman JC, Gunaratne PH, Marchetti D (2011) MicroRNA-1258 suppresses breast cancer brain metastasis by targeting heparanase. Cancer Res 71(3):645–654. https://doi.org/10.1158/0008-5472.CAN-10-1910
Murry BP, Greiter-Wilke A, Paulsen DP, Hiatt KM, Beltrami CA, Marchetti D (2005) Selective heparanase localization in malignant melanoma. Int J Oncol 26(2):345–352. https://doi.org/10.3892/ijo.26.2.345
Murry BP, Blust BE, Singh A, Foster TP, Marchetti D (2006) Heparanase mechanisms of melanoma metastasis to the brain: development and use of a brain slice model. J Cell Biochem 97(2):217–225. https://doi.org/10.1002/jcb.20714
Denkins Y, Reiland J, Roy M et al (2004) Brain metastases in melanoma: roles of neurotrophins. Neuro-Oncol 6(2):154–165. https://doi.org/10.1215/S115285170300067X
Herrmann JL, Menter DG, Hamada J, Marchetti D, Nakajima M, Nicolson GL (1993) Mediation of NGF-stimulated extracellular matrix invasion by the human melanoma low-affinity p75 neurotrophin receptor: melanoma p75 functions independently of trkA. Mol Biol Cell 4(11):1205–1216. https://doi.org/10.1091/mbc.4.11.1205
Marchetti D (1997) Specific degradation of subendothelial matrix proteoglycans by brain-metastatic melanoma and brain endothelial cell heparanases. J Cell Physiol 172(3):334–342. https://doi.org/10.1002/(SICI)1097-4652(199709)172:3%3c334::AID-JCP7%3e3.0.CO;2-P
Marchetti D, Menter D, Jin L, Nicolson GL, Nakajima M (1993) Nerve growth factor effects on human and mouse melanoma cell invasion and heparanase production. Int J Cancer 55(4):692–699. https://doi.org/10.1002/ijc.2910550430
Marchetti D, McQuillan DJ, Spohn WC, Carson DD, Nicolson GL (1996) Neurotrophin stimulation of human melanoma cell invasion: selected enhancement of heparanase activity and heparanase degradation of specific heparan sulfate subpopulations1. Cancer Res 56(12):2856–2863
Marchetti D, Li J, Shen R (2000) Astrocytes contribute to the brain-metastatic specificity of melanoma cells by producing heparanase1. Cancer Res 60(17):4767–4770
Parish CR, Freeman C, Ziolkowski AF et al (2013) Unexpected new roles for heparanase in Type 1 diabetes and immune gene regulation. Matrix Biol 32(5):228–233. https://doi.org/10.1016/j.matbio.2013.02.007
Masola V, Zaza G, Gambaro G, Franchi M, Onisto M (2020) Role of heparanase in tumor progression: molecular aspects and therapeutic options. Semin Cancer Biol 62:86–98. https://doi.org/10.1016/j.semcancer.2019.07.014
Thompson CA, Purushothaman A, Ramani VC, Vlodavsky I, Sanderson RD (2013) Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes. J Biol Chem 288(14):10093–10099. https://doi.org/10.1074/jbc.C112.444562
Visse R, Nagase H (2003) Matrix metalloproteinases and tissue inhibitors of metalloproteinases. Circ Res 92(8):827–839. https://doi.org/10.1161/01.RES.0000070112.80711.3D
Kessenbrock K, Plaks V, Werb Z (2010) Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141(1):52–67. https://doi.org/10.1016/j.cell.2010.03.015
Liu W, Song J, Du X et al (2019) AKR1B10 (Aldo-keto reductase family 1 B10) promotes brain metastasis of lung cancer cells in a multi-organ microfluidic chip model. Acta Biomater 91:195–208. https://doi.org/10.1016/j.actbio.2019.04.053
Liu H, Kato Y, Erzinger SA et al (2012) The role of MMP-1 in breast cancer growth and metastasis to the brain in a xenograft model. BMC Cancer 12(1):583. https://doi.org/10.1186/1471-2407-12-583
Ricci S, Guadagno E, Bruzzese D et al (2017) Evaluation of matrix metalloproteinase type IV-collagenases in serum of patients with tumors of the central nervous system. J Neurooncol 131(2):223–232. https://doi.org/10.1007/s11060-016-2297-4
Friedberg MH, Glantz MJ, Klempner MS, Cole BF, Perides G (1998) Specific matrix metalloproteinase profiles in the cerebrospinal fluid correlated with the presence of malignant astrocytomas, brain metastases, and carcinomatous meningitis. Cancer 82(5):923–930. https://doi.org/10.1002/(SICI)1097-0142(19980301)82:5%3c923::AID-CNCR18%3e3.0.CO;2-2
Mendes O, Kim H-T, Lungu G, Stoica G (2007) MMP2 role in breast cancer brain metastasis development and its regulation by TIMP2 and ERK1/2. Clin Exp Metastasis 24(5):341–351. https://doi.org/10.1007/s10585-007-9071-0
Cierna Z, Mego M, Janega P et al (2014) Matrix metalloproteinase 1 and circulating tumor cells in early breast cancer. BMC Cancer 14(1):472. https://doi.org/10.1186/1471-2407-14-472
Stark AM, Anuszkiewicz B, Mentlein R, Yoneda T, Mehdorn HM, Held-Feindt J (2007) Differential expression of matrix metalloproteinases in brain- and bone-seeking clones of metastatic MDA-MB-231 breast cancer cells. J Neurooncol 81(1):39–48. https://doi.org/10.1007/s11060-006-9207-0
Bos PD, Zhang XH-F, Nadal C et al (2009) Genes that mediate breast cancer metastasis to the brain. Nature 459(7249):1005–1009. https://doi.org/10.1038/nature08021
Wu K, Fukuda K, Xing F et al (2015) Roles of the cyclooxygenase 2 matrix metalloproteinase 1 pathway in brain metastasis of breast cancer. J Biol Chem 290(15):9842–9854. https://doi.org/10.1074/jbc.M114.602185
Shen C-J, Kuo Y-L, Chen C-C, Chen M-J, Cheng Y-M (2017) MMP1 expression is activated by Slug and enhances multi-drug resistance (MDR) in breast cancer. PLOS One 12(3):e0174487. https://doi.org/10.1371/journal.pone.0174487
Simonsen TG, Gaustad J-V, Rofstad EK (2015) Intertumor heterogeneity in vascularity and invasiveness of artificial melanoma brain metastases. J Exp Clin Cancer Res 34(1):150. https://doi.org/10.1186/s13046-015-0264-0
Rossi S, Cordella M, Tabolacci C et al (2018) TNF-alpha and metalloproteases as key players in melanoma cells aggressiveness. J Exp Clin Cancer Res 37(1):326. https://doi.org/10.1186/s13046-018-0982-1
Izraely S, Ben-Menachem S, Sagi-Assif O et al (2019) The metastatic microenvironment: melanoma–microglia cross-talk promotes the malignant phenotype of melanoma cells. Int J Cancer 144(4):802–817. https://doi.org/10.1002/ijc.31745
Sevenich L, Bowman RL, Mason SD et al (2014) Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol 16(9):876–888. https://doi.org/10.1038/ncb3011
Shintani Y, Higashiyama S, Ohta M et al (2004) Overexpression of ADAM9 in non-small cell lung cancer correlates with brain metastasis. Cancer Res 64(12):4190–4196. https://doi.org/10.1158/0008-5472.CAN-03-3235
Conrad C, Götte M, Schlomann U et al (2018) ADAM8 expression in breast cancer derived brain metastases: functional implications on MMP-9 expression and transendothelial migration in breast cancer cells. Int J Cancer 142(4):779–791. https://doi.org/10.1002/ijc.31090
Fazakas C, Wilhelm I, Nagyőszi P et al (2011) Transmigration of melanoma cells through the blood-brain barrier: role of endothelial tight junctions and melanoma-released serine proteases. PLOS ONE 6(6):e20758. https://doi.org/10.1371/journal.pone.0020758
Kim J, Yao F, Xiao Z, Sun Y, Ma L (2018) MicroRNAs and metastasis: small RNAs play big roles. Cancer Metastasis Rev 37(1):5–15. https://doi.org/10.1007/s10555-017-9712-y
Okuda H, Xing F, Pandey PR et al (2013) miR-7 suppresses brain metastasis of breast cancer stem-like cells by modulating KLF4. Cancer Res 73(4):1434–1444. https://doi.org/10.1158/0008-5472.CAN-12-2037
Xing F, Sharma S, Liu Y et al (2015) miR-509 suppresses brain metastasis of breast cancer cells by modulating RhoC and TNF-α. Oncogene 34(37):4890–4900. https://doi.org/10.1038/onc.2014.412
Xing F, Liu Y, Wu S-Y et al (2018) Loss of XIST in breast cancer activates MSN-c-Met and reprograms microglia via exosomal miRNA to promote brain metastasis. Cancer Res 78(15):4316–4330. https://doi.org/10.1158/0008-5472.CAN-18-1102
Zhang L, Zhang S, Yao J et al (2015) Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 527(7576):100–104. https://doi.org/10.1038/nature15376
Sirkisoon SR, Wong GL, Aguayo NR et al (2022) Breast cancer extracellular vesicles-derived miR-1290 activates astrocytes in the brain metastatic microenvironment via the FOXA2→CNTF axis to promote progression of brain metastases. Cancer Lett 540:215726. https://doi.org/10.1016/j.canlet.2022.215726
Arora S, Ranade AR, Tran NL et al (2011) MicroRNA-328 is associated with (non-small) cell lung cancer (NSCLC) brain metastasis and mediates NSCLC migration. Int J Cancer 129(11):2621–2631. https://doi.org/10.1002/ijc.25939
Chen L, Xu S, Xu H, Zhang J, Ning J, Wang S (2012) MicroRNA-378 is associated with non-small cell lung cancer brain metastasis by promoting cell migration, invasion and tumor angiogenesis. Med Oncol 29(3):1673–1680. https://doi.org/10.1007/s12032-011-0083-x
Hanniford D, Zhong J, Koetz L et al (2015) A miRNA-based signature detected in primary melanoma tissue predicts development of brain metastasis. Clin Cancer Res 21(21):4903–4912. https://doi.org/10.1158/1078-0432.CCR-14-2566
Cho WCS (2010) MicroRNAs: potential biomarkers for cancer diagnosis, prognosis and targets for therapy. Int J Biochem Cell Biol 42(8):1273–1281. https://doi.org/10.1016/j.biocel.2009.12.014
Zhang L, Huang J, Yang N et al (2006) microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci 103(24):9136–9141. https://doi.org/10.1073/pnas.0508889103
Gaur A, Jewell DA, Liang Y et al (2007) Characterization of MicroRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res 67(6):2456–2468. https://doi.org/10.1158/0008-5472.CAN-06-2698
Xing F, Liu Y, Sharma S et al (2016) Activation of the c-Met pathway mobilizes an inflammatory network in the brain microenvironment to promote brain metastasis of breast cancer. Cancer Res 76(17):4970–4980. https://doi.org/10.1158/0008-5472.CAN-15-3541
Avraham HK, Jiang S, Fu Y, Nakshatri H, Ovadia H, Avraham S (2014) Angiopoietin-2 mediates blood–brain barrier impairment and colonization of triple-negative breast cancer cells in brain. J Pathol 232(3):369–381. https://doi.org/10.1002/path.4304
Navab R, Liu J, Seiden-Long I et al (2009) Co-overexpression of met and hepatocyte growth factor promotes systemic metastasis in nci-h460 non-small cell lung carcinoma cells. Neoplasia 11(12):1292. https://doi.org/10.1593/neo.09622
Li B, Wang C, Zhang Y et al (2013) Elevated PLGF contributes to small-cell lung cancer brain metastasis. Oncogene 32(24):2952–2962. https://doi.org/10.1038/onc.2012.313
Xing F, Kobayashi A, Okuda H et al (2013) Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol Med 5(3):384–396. https://doi.org/10.1002/emmm.201201623
Chung B, Esmaeili AA, Gopalakrishna-Pillai S et al (2017) Human brain metastatic stroma attracts breast cancer cells via chemokines CXCL16 and CXCL12. NPJ Breast Cancer 3(1):1–9. https://doi.org/10.1038/s41523-017-0008-8
Gril B, Paranjape AN, Woditschka S et al (2018) Reactive astrocytic S1P3 signaling modulates the blood–tumor barrier in brain metastases. Nat Commun 9(1):2705. https://doi.org/10.1038/s41467-018-05030-w
Sayyad MR, Puchalapalli M, Vergara NG et al (2019) Syndecan-1 facilitates breast cancer metastasis to the brain. Breast Cancer Res Treat 178(1):35–49. https://doi.org/10.1007/s10549-019-05347-0
Curtaz CJ, Schmitt C, Herbert S-L et al (2020) Serum-derived factors of breast cancer patients with brain metastases alter permeability of a human blood–brain barrier model. Fluids Barriers CNS 17(1):31. https://doi.org/10.1186/s12987-020-00192-6
Guldner IH, Wang Q, Yang L et al (2020) CNS-native myeloid cells drive immune suppression in the brain metastatic niche through Cxcl10. Cell 183(5):1234-1248.e25. https://doi.org/10.1016/j.cell.2020.09.064
Wu S-Y, Sharma S, Wu K et al (2021) Tamoxifen suppresses brain metastasis of estrogen receptor-deficient breast cancer by skewing microglia polarization and enhancing their immune functions. Breast Cancer Res 23(1):35. https://doi.org/10.1186/s13058-021-01412-z
Motallebnejad P, Rajesh VV, Azarin SM (2022) Evaluating the role of IL-1β in transmigration of triple negative breast cancer cells across the brain endothelium. Cell Mol Bioeng 15(1):99–114. https://doi.org/10.1007/s12195-021-00710-y
Seike T, Fujita K, Yamakawa Y et al (2011) Interaction between lung cancer cells and astrocytes via specific inflammatory cytokines in the microenvironment of brain metastasis. Clin Exp Metastasis 28(1):13–25. https://doi.org/10.1007/s10585-010-9354-8
Rai S, Nejadhamzeeigilani Z, Gutowski NJ, Whatmore JL (2015) Loss of the endothelial glycocalyx is associated with increased E-selectin mediated adhesion of lung tumour cells to the brain microvascular endothelium. J Exp Clin Cancer Res 34(1):105. https://doi.org/10.1186/s13046-015-0223-9
Jassam SA, Maherally Z, Smith JR et al (2016) TNF-α enhancement of CD62E mediates adhesion of non–small cell lung cancer cells to brain endothelium via CD15 in lung-brain metastasis. Neuro-Oncol 18(5):679–690. https://doi.org/10.1093/neuonc/nov248
Li YD, Lamano JB, Lamano JB et al (2019) Tumor-induced peripheral immunosuppression promotes brain metastasis in patients with non-small cell lung cancer. Cancer Immunol Immunother 68(9):1501–1513. https://doi.org/10.1007/s00262-019-02384-y
Jin Y, Kang Y, Wang M et al (2022) Targeting polarized phenotype of microglia via IL6/JAK2/STAT3 signaling to reduce NSCLC brain metastasis. Signal Transduct Target Ther 7(1):1–12. https://doi.org/10.1038/s41392-022-00872-9
Wu S-Y, Xing F, Sharma S et al (2020) Nicotine promotes brain metastasis by polarizing microglia and suppressing innate immune function. J Exp Med 217(8):e20191131. https://doi.org/10.1084/jem.20191131
Wu D, Deng S, Li L et al (2021) TGF-β1-mediated exosomal lnc-MMP2-2 increases blood–brain barrier permeability via the miRNA-1207-5p/EPB41L5 axis to promote non-small cell lung cancer brain metastasis. Cell Death Dis 12(8):1–10. https://doi.org/10.1038/s41419-021-04004-z
Risolino M, Mandia N, Iavarone F et al (2014) Transcription factor PREP1 induces EMT and metastasis by controlling the TGF-β–SMAD3 pathway in non-small cell lung adenocarcinoma. Proc Natl Acad Sci 111(36):E3775–E3784. https://doi.org/10.1073/pnas.1407074111
Zhang L, Yao J, Wei Y et al (2020) Blocking immunosuppressive neutrophils deters pY696-EZH2–driven brain metastases. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aaz5387
Schwartz H, Blacher E, Amer M et al (2016) Incipient melanoma brain metastases instigate astrogliosis and neuroinflammation. Cancer Res 76(15):4359–4371. https://doi.org/10.1158/0008-5472.CAN-16-0485
Zubrilov I, Sagi-Assif O, Izraely S et al (2015) Vemurafenib resistance selects for highly malignant brain and lung-metastasizing melanoma cells. Cancer Lett 361(1):86–96. https://doi.org/10.1016/j.canlet.2015.02.041
Zhang C, Zhang F, Tsan R, Fidler IJ (2009) Transforming growth factor-β2 is a molecular determinant for site-specific melanoma metastasis in the brain. Cancer Res 69(3):828–835. https://doi.org/10.1158/0008-5472.CAN-08-2588
Doron H, Amer M, Ershaid N et al (2019) Inflammatory activation of astrocytes facilitates melanoma brain tropism via the CXCL10-CXCR3 signaling axis. Cell Rep 28(7):1785-1798.e6. https://doi.org/10.1016/j.celrep.2019.07.033
Klein A, Schwartz H, Sagi-Assif O et al (2015) Astrocytes facilitate melanoma brain metastasis via secretion of IL-23. J Pathol 236(1):116–127. https://doi.org/10.1002/path.4509
Wyler L, Napoli CU, Ingold B et al (2014) Brain metastasis in renal cancer patients: metastatic pattern, tumour-associated macrophages and chemokine/chemoreceptor expression. Br J Cancer 110(3):686–694. https://doi.org/10.1038/bjc.2013.755
Gong X, Hou Z, Endsley MP et al (2019) Interaction of tumor cells and astrocytes promotes breast cancer brain metastases through TGF-β2/ANGPTL4 axes. NPJ Precis Oncol 3(1):1–9. https://doi.org/10.1038/s41698-019-0094-1
Hajal C, Shin Y, Li L, Serrano JC, Jacks T, Kamm RD (2021) The CCL2-CCR2 astrocyte-cancer cell axis in tumor extravasation at the brain. Sci Adv. https://doi.org/10.1126/sciadv.abg8139
Priego N, Zhu L, Monteiro C et al (2018) STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat Med 24(7):1024–1035. https://doi.org/10.1038/s41591-018-0044-4
Louie E, Chen XF, Coomes A, Ji K, Tsirka S, Chen EI (2013) Neurotrophin-3 modulates breast cancer cells and the microenvironment to promote the growth of breast cancer brain metastasis. Oncogene 32(35):4064–4077. https://doi.org/10.1038/onc.2012.417
Strell C, Entschladen F (2008) Extravasation of leukocytes in comparison to tumor cells. Cell Commun Signal 6(1):10. https://doi.org/10.1186/1478-811X-6-10
Sökeland G, Schumacher U (2019) The functional role of integrins during intra- and extravasation within the metastatic cascade. Mol Cancer 18(1):12. https://doi.org/10.1186/s12943-018-0937-3
Soto MS, Serres S, Anthony DC, Sibson NR (2014) Functional role of endothelial adhesion molecules in the early stages of brain metastasis. Neuro-Oncol 16(4):540–551. https://doi.org/10.1093/neuonc/not222
Münsterberg J, Loreth D, Brylka L et al (2020) ALCAM contributes to brain metastasis formation in non-small-cell lung cancer through interaction with the vascular endothelium. Neuro-Oncol 22(7):955–966. https://doi.org/10.1093/neuonc/noaa028
Zarghami N, Soto MS, Perez-Balderas F et al (2021) A novel molecular magnetic resonance imaging agent targeting activated leukocyte cell adhesion molecule as demonstrated in mouse brain metastasis models. J Cereb Blood Flow Metab 41(7):1592–1607. https://doi.org/10.1177/0271678X20968943
Kim S-H, Redvers RP, Chi LH et al (2018) Identification of brain metastasis genes and therapeutic evaluation of histone deacetylase inhibitors in a clinically relevant model of breast cancer brain metastasis. Dis Model Mech. https://doi.org/10.1242/dmm.034850
Valiente M, Obenauf AC, Jin X et al (2014) Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 156(5):1002–1016. https://doi.org/10.1016/j.cell.2014.01.040
Er EE, Valiente M, Ganesh K et al (2018) Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat Cell Biol 20(8):966–978. https://doi.org/10.1038/s41556-018-0138-8
Rolland Y, Demeule M, Fenart L, Béliveau R (2009) Inhibition of melanoma brain metastasis by targeting melanotransferrin at the cell surface. Pigment Cell Melanoma Res 22(1):86–98. https://doi.org/10.1111/j.1755-148X.2008.00525.x
Kang S-A, Hasan N, Mann AP et al (2015) Blocking the adhesion cascade at the premetastatic niche for prevention of breast cancer metastasis. Mol Ther 23(6):1044–1054. https://doi.org/10.1038/mt.2015.45
Woodman N, Pinder SE, Tajadura V et al (2016) Two E-selectin ligands, BST-2 and LGALS3BP, predict metastasis and poor survival of ER-negative breast cancer. Int J Oncol 49(1):265–275. https://doi.org/10.3892/ijo.2016.3521
Jassam SA, Maherally Z, Ashkan K, Pilkington GJ, Fillmore HL (2019) Fucosyltransferase 4 and 7 mediates adhesion of non-small cell lung cancer cells to brain-derived endothelial cells and results in modification of the blood–brain-barrier: in vitro investigation of CD15 and CD15s in lung-to-brain metastasis. J Neurooncol 143(3):405–415. https://doi.org/10.1007/s11060-019-03188-x
Brayton J, Qing Z, Hart MN, VanGilder JC, Fabry Z (1998) Influence of adhesion molecule expression by human brain microvessel endothelium on cancer cell adhesion. J Neuroimmunol 89(1):104–112. https://doi.org/10.1016/S0165-5728(98)00127-1
Dimitroff CJ, Descheny L, Trujillo N et al (2005) Identification of leukocyte E-selectin ligands, P-selectin glycoprotein ligand-1 and E-selectin ligand-1, on human metastatic prostate tumor cells. Cancer Res 65(13):5750–5760. https://doi.org/10.1158/0008-5472.CAN-04-4653
Chao YL, Shepard CR, Wells A (2010) Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol Cancer 9(1):179. https://doi.org/10.1186/1476-4598-9-179
Grinberg-Rashi H, Ofek E, Perelman M et al (2009) The expression of three genes in primary non-small cell lung cancer is associated with metastatic spread to the brain. Clin Cancer Res 15(5):1755–1761. https://doi.org/10.1158/1078-0432.CCR-08-2124
Yoo JY, Yang S-H, Lee JE et al (2012) E-cadherin as a predictive marker of brain metastasis in non-small-cell lung cancer, and its regulation by pioglitazone in a preclinical model. J Neurooncol 109(2):219–227. https://doi.org/10.1007/s11060-012-0890-8
Ulasov I, Borovjagin A, Fares J et al (2020) MicroRNA 345 (miR345) regulates KISS1-E-cadherin functional interaction in breast cancer brain metastases. Cancer Lett 481:24–31. https://doi.org/10.1016/j.canlet.2020.03.025
Carbonell WS, Ansorge O, Sibson N, Muschel R (2009) The vascular basement membrane as “soil” in brain metastasis. PLOS One 4(6):e5857. https://doi.org/10.1371/journal.pone.0005857
Lorger M, Krueger JS, O’Neal M, Staflin K, Felding-Habermann B (2009) Activation of tumor cell integrin αvβ3 controls angiogenesis and metastatic growth in the brain. Proc Natl Acad Sci 106(26):10666–10671. https://doi.org/10.1073/pnas.0903035106
Lal S, Kersch C, Beeson KA, Wu YJ, Muldoon LL, Neuwelt EA (2015) Interactions between αv-Integrin and HER2 and their role in the invasive phenotype of breast cancer cells in vitro and in rat brain. PLOS One 10(7):e0131842. https://doi.org/10.1371/journal.pone.0131842
Wu YJ, Muldoon LL, Gahramanov S, Kraemer DF, Marshall DJ, Neuwelt EA (2012) Targeting αV-integrins decreased metastasis and increased survival in a nude rat breast cancer brain metastasis model. J Neurooncol 110(1):27–36. https://doi.org/10.1007/s11060-012-0942-0
Fan J, Cai B, Zeng M, Hao Y, Giancotti FG, Fu BM (2011) Integrin β4 signaling promotes mammary tumor cell adhesion to brain microvascular endothelium by inducing ErbB2-mediated secretion of VEGF. Ann Biomed Eng 39(8):2223–2241. https://doi.org/10.1007/s10439-011-0321-6
Howe EN, Burnette MD, Justice ME et al (2020) Rab11b-mediated integrin recycling promotes brain metastatic adaptation and outgrowth. Nat Commun 11(1):3017. https://doi.org/10.1038/s41467-020-16832-2
Malin D, Strekalova E, Petrovic V et al (2014) αB-crystallin: a novel regulator of breast cancer metastasis to the brain. Clin Cancer Res 20(1):56–67. https://doi.org/10.1158/1078-0432.CCR-13-1255
Ranjan A, Gupta P, Srivastava SK (2016) Penfluridol: an antipsychotic agent suppresses metastatic tumor growth in triple-negative breast cancer by inhibiting integrin signaling axis. Cancer Res 76(4):877–890. https://doi.org/10.1158/0008-5472.CAN-15-1233
Berghoff AS, Rajky O, Winkler F et al (2013) Invasion patterns in brain metastases of solid cancers. Neuro-Oncol 15(12):1664–1672. https://doi.org/10.1093/neuonc/not112
Vogetseder A, Thies S, Ingold B et al (2013) αv-Integrin isoform expression in primary human tumors and brain metastases. Int J Cancer 133(10):2362–2371. https://doi.org/10.1002/ijc.28267
Berghoff AS, Kovanda AK, Melchardt T et al (2014) αvβ3, αvβ5 and αvβ6 integrins in brain metastases of lung cancer. Clin Exp Metastasis 31(7):841–851. https://doi.org/10.1007/s10585-014-9675-0
Yoshimasu T, Sakurai T, Oura S et al (2004) Increased expression of integrin α3β1 in highly brain metastatic subclone of a human non-small cell lung cancer cell line. Cancer Sci 95(2):142–148. https://doi.org/10.1111/j.1349-7006.2004.tb03195.x
García-Martín AB, Zwicky P, Gruber T et al (2019) VLA-4 mediated adhesion of melanoma cells on the blood–brain barrier is the critical cue for melanoma cell intercalation and barrier disruption. J Cereb Blood Flow Metab 39(10):1995–2010. https://doi.org/10.1177/0271678X18775887
Konstantopoulos K, Thomas SN (2009) Cancer cells in transit: the vascular interactions of tumor cells. Annu Rev Biomed Eng 11(1):177–202. https://doi.org/10.1146/annurev-bioeng-061008-124949
Gay LJ, Felding-Habermann B (2011) Contribution of platelets to tumour metastasis. Nat Rev Cancer 11(2):123–134. https://doi.org/10.1038/nrc3004
Felding-Habermann B, O’Toole TE, Smith JW et al (2001) Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci 98(4):1853–1858. https://doi.org/10.1073/pnas.98.4.1853
Cheng VWT, Soto MS, Khrapitchev AA et al (2019) VCAM-1–targeted MRI enables detection of brain micrometastases from different primary tumors. Clin Cancer Res 25(2):533–543. https://doi.org/10.1158/1078-0432.CCR-18-1889
Sikpa D, Whittingstall L, Fouquet JP et al (2020) Cerebrovascular inflammation promotes the formation of brain metastases. Int J Cancer 147(1):244–255. https://doi.org/10.1002/ijc.32902
Cheng VWT, de Pennington N, Zakaria R et al (2022) VCAM-1–targeted MRI improves detection of the tumor-brain interface. Clin Cancer Res 28(11):2385–2396. https://doi.org/10.1158/1078-0432.CCR-21-4011
Serres S, Soto MS, Hamilton A et al (2012) Molecular MRI enables early and sensitive detection of brain metastases. Proc Natl Acad Sci 109(17):6674–6679. https://doi.org/10.1073/pnas.1117412109
Corroyer-Dulmont A, Valable S, Falzone N et al (2020) VCAM-1 targeted alpha-particle therapy for early brain metastases. Neuro-Oncol 22(3):357–368. https://doi.org/10.1093/neuonc/noz169
Dobie C, Skropeta D (2021) Insights into the role of sialylation in cancer progression and metastasis. Br J Cancer 124(1):76–90. https://doi.org/10.1038/s41416-020-01126-7
Boire A, Coffelt SB, Quezada SA, Heiden MGV, Weeraratna AT (2019) Tumour dormancy and reawakening: opportunities and challenges. Trends Cancer 5(12):762–765. https://doi.org/10.1016/j.trecan.2019.10.010
Ghajar CM, Peinado H, Mori H et al (2013) The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 15(7):807–817. https://doi.org/10.1038/ncb2767
Dai J, Cimino PJ, Gouin KH et al (2022) Astrocytic laminin-211 drives disseminated breast tumor cell dormancy in brain. Nat Cancer 3(1):25–42. https://doi.org/10.1038/s43018-021-00297-3
Blazquez R, Sparrer D, Wendl C et al (2020) The macro-metastasis/organ parenchyma interface (MMPI) - A hitherto unnoticed area. Semin Cancer Biol 60:324–333. https://doi.org/10.1016/j.semcancer.2019.10.012
Yao H, Price TT, Cantelli G et al (2018) Leukaemia hijacks a neural mechanism to invade the central nervous system. Nature 560(7716):55–60. https://doi.org/10.1038/s41586-018-0342-5
Bentolila LA, Prakash R, Mihic-Probst D et al (2016) Imaging of angiotropism/vascular co-option in a murine model of brain melanoma: implications for melanoma progression along extravascular pathways. Sci Rep 6(1):23834. https://doi.org/10.1038/srep23834
Küsters B, Leenders WPJ, Wesseling P et al (2002) Vascular endothelial growth factor-A165 induces progression of melanoma brain metastases without induction of sprouting angiogenesis1. Cancer Res 62(2):341–345
Sakariassen PØ, Prestegarden L, Wang J et al (2006) Angiogenesis-independent tumor growth mediated by stem-like cancer cells. Proc Natl Acad Sci 103(44):16466–16471. https://doi.org/10.1073/pnas.0607668103
Watkins S, Robel S, Kimbrough IF, Robert SM, Ellis-Davies G, Sontheimer H (2014) Disruption of astrocyte–vascular coupling and the blood–brain barrier by invading glioma cells. Nat Commun 5(1):4196. https://doi.org/10.1038/ncomms5196
Holash J, Maisonpierre PC, Compton D et al (1999) Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 284(5422):1994–1998. https://doi.org/10.1126/science.284.5422.1994
Baker GJ, Yadav VN, Motsch S et al (2014) Mechanisms of glioma formation: iterative perivascular glioma growth and invasion leads to tumor progression, VEGF-independent vascularization, and resistance to antiangiogenic therapy. Neoplasia 16(7):543–561. https://doi.org/10.1016/j.neo.2014.06.003
Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT (2007) Angiogenesis in brain tumours. Nat Rev Neurosci 8(8):610–622. https://doi.org/10.1038/nrn2175
Verhoeff JJ, van Tellingen O, Claes A et al (2009) Concerns about anti-angiogenic treatment in patients with glioblastoma multiforme. BMC Cancer 9(1):444. https://doi.org/10.1186/1471-2407-9-444
Seano G, Jain RK (2020) Vessel co-option in glioblastoma: emerging insights and opportunities. Angiogenesis 23(1):9–16. https://doi.org/10.1007/s10456-019-09691-z
Siam L, Bleckmann A, Chaung H-N et al (2015) The metastatic infiltration at the metastasis/brain parenchyma-interface is very heterogeneous and has a significant impact on survival in a prospective study. Oncotarget 6(30):29254–29267
Claes A, Idema AJ, Wesseling P (2007) Diffuse glioma growth: a guerilla war. Acta Neuropathol (Berl) 114(5):443–458. https://doi.org/10.1007/s00401-007-0293-7
Kuczynski EA, Vermeulen PB, Pezzella F, Kerbel RS, Reynolds AR (2019) Vessel co-option in cancer. Nat Rev Clin Oncol 16(8):469–493. https://doi.org/10.1038/s41571-019-0181-9
Montana V, Sontheimer H (2011) Bradykinin promotes the chemotactic invasion of primary brain tumors. J Neurosci 31(13):4858–4867. https://doi.org/10.1523/JNEUROSCI.3825-10.2011
Le Joncour V, Filppu P, Hyvönen M, et al (2019) Vulnerability of invasive glioblastoma cells to lysosomal membrane destabilization. EMBO Mol Med 11(6):e9034 https://doi.org/10.15252/emmm.201809034
Yadav VN, Zamler D, Baker GJ, et al (2016) CXCR4 increases in-vivo glioma perivascular invasion, and reduces radiation induced apoptosis: A genetic knockdown study. Oncotarget 7(50):83701–83719. https://doi.org/10.18632/oncotarget.13295
Lindberg OR, McKinney A, Engler JR, et al (2016) GBM heterogeneity as a function of variable epidermal growth factor receptor variant III activity. Oncotarget 7(48):79101–79116. https://doi.org/10.18632/oncotarget.12600
Infanger DW, Cho Y, Lopez BS et al (2013) Glioblastoma stem cells are regulated by interleukin-8 signaling in a tumoral perivascular niche. Cancer Res 73(23):7079–7089. https://doi.org/10.1158/0008-5472.CAN-13-1355
Sharma I, Singh A, Siraj F, Saxena S (2018) IL-8/CXCR1/2 signalling promotes tumor cell proliferation, invasion and vascular mimicry in glioblastoma. J Biomed Sci 25(1):62. https://doi.org/10.1186/s12929-018-0464-y
Jabouille A, Delugin M, Pineau R, et al (2015) Glioblastoma invasion and cooption depend on IRE103B1; endoribonuclease activity. Oncotarget 6(28):24922–24934. https://doi.org/10.18632/oncotarget.4679
Griveau A, Seano G, Shelton SJ et al (2018) A glial signature and wnt7 signaling regulate glioma-vascular interactions and tumor microenvironment. Cancer Cell 33(5):874-889.e7. https://doi.org/10.1016/j.ccell.2018.03.020
Cheng L, Huang Z, Zhou W et al (2013) Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153(1):139–152. https://doi.org/10.1016/j.cell.2013.02.021
Berghoff AS, Preusser M (2018) Anti-angiogenic therapies in brain metastases. Memo Mag Eur Med Oncol 11(1):14–17. https://doi.org/10.1007/s12254-018-0384-2
Norden AD, Drappatz J, Wen PY (2008) Novel anti-angiogenic therapies for malignant gliomas. Lancet Neurol 7(12):1152–1160. https://doi.org/10.1016/S1474-4422(08)70260-6
Kuczynski EA, Reynolds AR (2020) Vessel co-option and resistance to anti-angiogenic therapy. Angiogenesis 23(1):55–74. https://doi.org/10.1007/s10456-019-09698-6
Bovi JA (2018) Prevention of brain metastases. Front Neurol. https://doi.org/10.3389/fneur.2018.00758
Camidge DR, Dziadziuszko R, Peters S et al (2019) Updated efficacy and safety data and impact of the EML4-ALK fusion variant on the efficacy of alectinib in untreated ALK-positive advanced non-small cell lung cancer in the global phase III ALEX study. J Thorac Oncol 14(7):1233–1243. https://doi.org/10.1016/j.jtho.2019.03.007
Peters S, Camidge DR, Shaw AT et al (2017) Alectinib versus crizotinib in untreated ALK-positive non–small-cell lung cancer. N Engl J Med 377(9):829–838. https://doi.org/10.1056/NEJMoa1704795
Lin NU, Diéras V, Paul D et al (2009) Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin Cancer Res 15(4):1452–1459. https://doi.org/10.1158/1078-0432.CCR-08-1080
Maurya SK, Khan P, Rehman AU et al (2022) Rethinking the chemokine cascade in brain metastasis: preventive and therapeutic implications. Semin Cancer Biol 86:914–930. https://doi.org/10.1016/j.semcancer.2021.12.009
Klemm F, Maas RR, Bowman RL et al (2020) Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell 181(7):1643-1660.e17. https://doi.org/10.1016/j.cell.2020.05.007
Yano S, Shinohara H, Herbst RS et al (2000) Expression of vascular endothelial growth factor is necessary but not sufficient for production and growth of brain metastasis1. Cancer Res 60(17):4959–4967
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the German Research Foundation (DFG) via the collaborative research centers 1080, projects A03 and SFB1292/2—project number 318346496, projects TP09, as well as the DFG grant SCHM 2159/4–1 to MHHS.
Author information
Authors and Affiliations
Contributions
All authors contributed to the writing of the paper and illustrations.
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare no competing interests.
Ethical approval and consent to participate
Not applicable.
Consent for publications
All authors have read and approved the manuscript for submission.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
McDonald, B., Barth, K. & Schmidt, M.H.H. The origin of brain malignancies at the blood–brain barrier. Cell. Mol. Life Sci. 80, 282 (2023). https://doi.org/10.1007/s00018-023-04934-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-023-04934-1