
Abstract
The incidence of brain metastasis is increasing, however, little is known about molecular mechanism responsible for lung cancer-derived brain metastasis and their development in the brain. In the present study, brain pathology was examined in an experimental model system of brain metastasis as well as in human brain with lung cancer metastasis. In an experimental model, after 3–6 weeks of intracardiac inoculation of human lung cancer-derived (HARA-B) cells in nude mice, wide range of brain metastases were observed. The brain sections showed significant increase in glial fibrillary acidic protein (GFAP)-positive astrocytes around metastatic lesions. To elucidate the role of astrocytes in lung cancer proliferation, the interaction between primary cultured mouse astrocytes and HARA-B cells was analyzed in vitro. Co-cultures and insert-cultures demonstrated that astrocytes were activated by tumor cell-oriented factors; macrophage migration inhibitory factor (MIF), interleukin-8 (IL-8) and plasminogen activator inhibitor-1 (PAI-1). Activated astrocytes produced interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α) and interleukin-1 β (IL-1β), which in turn promoted tumor cell proliferation. Semi-quantitative immunocytochemistry showed that increased expression of receptors for IL-6 and its subunits gp130 on HARA-B cells. Receptors for TNF-α and IL-1β were also detected on HARA-B cells but down-regulated after co-culture with astrocytes. Insert-culture with astrocytes also stimulated the proliferation of other lung cancer-derived cell lines (PC-9, QG56, and EBC-1). These results suggest that tumor cells and astrocytes stimulate each other and these mutual relationships may be important to understand how lung cancer cells metastasize and develop in the brain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Metastasis is the principal cause of the morbidity and death of cancer patients. The incidence of brain metastasis has been increasing in recent years, especially in breast cancer and lung cancer [1]. In the process of metastasis formation, the interaction between the metastatic tumor cells and host cells plays an important role in the microenvironment of the metastatic sites [2]. However, a molecular mechanism for brain metastasis is poorly understood to date. In the central nervous system, activated glial cells contribute to the innate immune response and produce a large variety of different inflammatory mediators as a chronic inflammatory reaction [3]. A similar mechanism could function in cell survival, growth, proliferation and colonization, invasion and motility of metastatic tumor cells in the microenvironment of brain metastases [4, 5]. Among the glial cells, astrocytes are the most abundant cell population and play an important role in maintaining homeostasis of the brain [6]. Astrocytes have been shown to produce a wide variety of cytokines including interleukin-1 (IL-1), interleukin-3 (IL-3), interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), transforming growth factor-β (TGF-β), insulin-like growth factor-1 (IGF-1) and platelet-derived growth factor (PDGF) [7–10]. Among them, it was suggested that IL-6, TGF-β and IGF-1 may contribute to the development of brain metastasis by breast cancer cells [11]. As for brain metastasis by lung cancer cells, it is not known whether or not the same cytokines are involved and what the difference between brain metastases derived by breast cancer cells and lung cancer cells.
Therefore, in the present study, we examined brain pathology in an experimental model system of brain metastasis, using HARA-B cells derived from human lung cancer cells, and assessed the effects of astrocytes on the growth of HARA-B cells as well as three other non-small cell lung cancer cell lines (PC-9, QG56, and EBC-1) in vitro. Furthermore, astrocytes-derived factors conducive to tumor cell growth and their receptor expression on tumor cells were investigated.
Materials and methods
Experimental model for brain metastasis
The study was approved by the Animal Care and Use Committee at Kyushu University and carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male 5-week-old nude mice (BALB/c nu/nu) (Kyudo, Kumamoto, Japan), kept in a specific pathogen-free environment, were used. A single suspension of human lung squamous cell carcinoma-derived cells (HARA-B) (2 × 105 cells/0.2 ml PBS) was inoculated into the left ventricle of the heart in nude mice according to the method described previously [12]. After 4–6 weeks, brains were subjected for immunohistochemical staining.
Human tissue samples
A total of 6 paraffin-embedded samples from patients with lung tumor brain metastasis were used. All sections were obtained from the National Hospital Organization Shikoku Cancer Center. Use of the human specimens was in accordance with the University Ethics Commission. The formalin-fixed, paraffin-embedded archival tissue blocks were retrieved, and matching hematoxylin and eosin (H & E)-stained slides were reviewed and screened for representative tumor regions by a neuropathologist.
Immunohistochemistry
Nude mice were perfused transcardially with 50 ml of 10 U/ml heparin and 0.5% procaine in PBS and 4% paraformaldehyde (PFA) in PBS prior to excision of the brain. Then the brain was removed, post-fixed for 3 h, and cryoprotected for 24 h in PBS containing 20% sucrose. The brain was cut into slices (30 μm thick) using a cryostat and the sections were placed on glass slides. In order to acquire the better immunoreactive images, sections were autoclaved with 0.01 M citrate buffer solution (pH 6.0), permeabilized with 0.3% TritonX-100 in PBS for 15 min, and then blocked in BlockAce (Dainippon Pharmaceutical, Japan) for 1 h at room temperature. Sections were incubated with mouse anti-human cytokeratin monoclonal antibody (AE1/AE3 pool of cytokeratin) (Dako, Glostrup, Denmark, 1:100) at 4°C overnight. Biotinylated anti-mouse IgG (Jackson, 1:200) were incubated for 2 h at room temperature, followed by the incubation with streptavidin Alexa488 (Molecular Probes, 1:500) for 2 h at room temperature. For double-staining of cytokeratin and GFAP, sections were incubated with Cy3-conjugated anti-GFAP antibody (Sigma, USA) (1:1000) at 4°C overnight after staining of cytokeratin. Every treatment was followed by washing three times with PBS containing 0.3% TritonX-100 for 5 min. The sections were mounted in the Perma Fluor Aqueous Mounting Medium (Thermo Shandon, Pittsburgh, PA, USA) and analyzed with a confocal microscope (LSM510 META, Carl Zeiss, Co. Ltd. Germany). Z-stack images were obtained from each section by LSM 510 META and total intensity were calculated by LSM image browser.
As for human tissue samples, after removal of paraffin in xylene and rehydration in a graded of alcohols (100%, 90%, 80%, 70%, 60%), sections were incubated for 30 min in 0.05 M phosphate buffer pH 7.6 containing tripsin and KCl for antigen retrieval. Then, the sections were incubated for 1 h in 0.3% H2O2, and blocked in PBS containing 1% BSA and 5% normal donkey serum (Jackson Immuno Research Laboratories Inc., West Grove, PA, USA) for 1 h at room temperature. Then, the sections were incubated with anti-GFAP antibody (ImmunoStar) (1:15) at 4°C overnight, goat anti-rabbit IgG Alexa 568 (Molecular Probes) (1:500) for 3 h at room temperature and FITC-conjugated anti-human cytokeratin antibody (CAM5.2) (Becton–Dickinson Biosciences, New Jersey, USA) (undiluted solution) for 1 h at room temperature. Every treatment was followed by washing three times with PBS containing 0.3% TritonX-100 for 5 min. The sections were mounted in the Perma Fluor Aqueous Mounting Medium (Thermo Shandon, Pittsburgh, PA, USA) and analyzed with a confocal microscope (LSM510 META, Carl Zeiss, Co. Ltd. Germany).
Cell culture
Primary glial cell cultures were performed according to the method described previously [13]. Briefly, the cerebral cortex obtained from 1-day-old C57BL/6 mice (Kyudo, Kumamoto, Japan) were isolated under a dissecting microscope and carefully separated from the choroid plexus and meninges. The isolated cerebral cortex were minced and treated with trypsin–EDTA solution (0.25% trypsin, 1 mM EDTA) and 1500 U DNase in Dulbecco’s modified Egle medium (DMEM; Nissui, Tokyo, Japan) at 37°C for 10 min. Cell suspensions were filtered through 70 μm pore size mesh (BD Falcon, Bedford, MA, USA) into DMEM containing 10% fetal calf serum (FCS; Hyclone, UT, USA), 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.37% NaHCO3, and 110 μg/ml pyruvic acid. After centrifugation, cells were filtered through 40 μm pore size-mesh (BD Falcon), plated into poly-L-lysine coated 75 cm2 cell culture flask at the density of two brains per flask in 10 ml of DMEM, and maintained at 37°C in 10% CO2–90% air with a change of the medium twice per week. Astrocytes were obtained after 28 days of mixed glial cell cultures as follows. After removing other glial cells by shaking the flasks, the astroglial layer was removed from the flasks by the treatment with trypsin–EDTA solution (0.06% trypsin, 0.25 mM EDTA in serum free DMEM) at 37°C for 45 min. Suspended astrocytes were filtered through 40 μm pore size-mesh and seeded. Astrocyte purity ranged from 90 to 95% as determined by immunostaining with anti-GFAP antibody (Sigma, St. Louis, MO, USA) (data not shown). Astrocytes were maintained in the same medium used for cell suspension from cerebral cortex at 37°C in 10% CO2–90% air. HARA-B cells and other lung cancer cell lines (QG56, EBC-1; squamous cell carcinoma) and PC-9 (non-small cell lung cancer cell) were maintained under the same condition. Cells were grown in 25 cm2 cell culture flask (Nalge Nunc International), and single-cell suspension of cells were obtained by trypsin treatment.
Cell proliferation assay
In the co-culture experiment, HARA-B cells (0.5 × 103 cells/well) and astrocytes (2.5 × 103 or 5 × 103 cells/well) were seeded into 8-well cell culture slide (BD Falcon) in DMEM for 24 h. Then, cells were rinsed twice with PBS and incubated in serum free DMEM. After 72 h of co-culture, cells were fixed with 4% PFA for 30 min at room temperature and permeabilized with 0.3% TritonX-100 in PBS for 15 min, followed with blocking solution containing 1% BSA and 5% normal donkey serum (Jackson) in PBS for 1 h at room temperature. Then cells were incubated with mouse anti-human cytokeratin monoclonal antibody (AE1/AE3 pool of cytokeratin) (Dako, Glostrup, Denmark) (1:100) at 4°C overnight, followed by the incubation with the secondary antibody (FITC-conjugated anti-mouse IgG; Sigma, 1:500) for 5 h at room temperature, and then, incubated with 300 nM 4′,6′-diamidino-2-phenylindole hydrochloride (DAPI, Sigma) for 30 min at room temperature. The number of HARA-B cells in each well, which were positively stained with an anti-human cytokeratin antibody, was counted using a digital camera system (Axio Cam, Carl Zeiss) mounted on a light and fluorescent microscope (Axopscope2 plus, Carl Zeiss). The results were expressed as the percentage of control (single cell culture of HARA-B cells).
Insert culture-medium was obtained as follows. Astrocytes (5 × 104 cells/well) were plated in 6-well cell culture plates (Falcon). Tumor cells derived from each lung tumor (EBC-1, PC9, QG56 and HARA-B cells) (5 × 103 cells/insert) were plated in cell culture-inserts (membrane pore size 0.4 μm; Becton–Dickinson), and then, placed in the well of astrocyte cultures. After 24 h of the insert-culture, cells were rinsed twice with PBS, incubated in serum-free DMEM for further 48 h. Then, the medium was collected. Each conditioned medium was centrifuged to remove debris (1500 rpm for 10 min at 4°C) before use. Tumor cells derived from each lung tumor (0.5 × 103 cells/well) were seeded into 8-well cell culture slide (BD Falcon) in DMEM for 24 h. Then, cells were rinsed twice with PBS and incubated in each Insert culture-medium. After 72 h, cells were fixed with 4% PFA for 30 min at room temperature and incubated with 300 nM DAPI for 10 min at room temperature. The number of tumor cells in each well, which were positively stained with DAPI, was counted as mentioned above. The results were expressed as the percentage of control (each tumor cells cultured in serum-free DMEM for 72 h).
The astrocyte-conditioned medium (ACM) was obtained from the primary culture of astrocytes at a density of 104 cells/well in serum free DMEM after 72 h-incubation. This ACM was centrifuged (1500 rpm for 10 min at 4°C), and then, added to HARA-B cells cultured for 1 day at a volume of 25% or 50%. The number of HARA-B cells were counted after the 72 h-incubation in the presence of ACM as described above.
HARA-B-stimulated astrocyte-conditioned medium (H-ACM) was obtained as follows. Culture medium of HARA-B cells (5 × 103 cells/well) were added to astrocytes cultures (5 × 104 cells/well) and incubated for 24 h. Then the medium was collected and centrifuged to remove debris (1500 rpm for 10 min at 4°C) before use.
In the proliferation assay using recombinant mouse cytokines, HARA-B cells 24 h after plating were rinsed twice with PBS and incubated in serum free DMEM with each cytokine (mIL-1β, 1 to 10 pg/ml; mTNF-α, 10 to 500 pg/ml; mIL-6, 10 to 500 pg/ml, Peprotech, Rocky Hill, NJ, USA). After 72 h, HARA-B cells were immunostained and counted as mentioned above.
In the experiments using neutralizing antibodies, cells were plated, rinsed twice with PBS after 24 h, and incubated in serum free DMEM with each specific neutralizing antibodies; mIL-1β antibody (ab), 0.2 to 2.0 μg/ml (R&D systems, Minneapolis, MN USA), mTNF-α ab, 0.2 to 2.0 μg/ml (R&D systems), mIL-6 ab, 2.0 to 20 ng/ml (Peprotech) for 48 h. As controls, rat IgG (Sigma) and rabbit IgG (R&D systems) were used. HARA-B cells were immunostained and counted as mentioned above.
SYBR green-based real-time quantitative RT–PCR
HARA-B cells (1 × 105 cells/insert) and astrocytes (1 × 106 cells/well) were cultured in cell culture-insert systems as mentioned above. Cells were collected by treatment with trypsin after 24, 48 and 72 h of insert-culture. As controls, single cell culture of HARA-B cells or astrocytes was used. Total RNA was isolated from each cell type by an extraction procedure using the RNA blood mini kit (QIAGEN in Japan). Contaminating DNA was removed by RNase-free DNase (QIAGEN). Single-strand cDNA was synthesized from cellular mRNA by using random 9 mer and RNA PCR kit (AMV) (Takara Bio Inc., Ootsu, Japan). PCR amplification was undertaken for plain SYBR Green I detection in using Light Cycler system (Roche Diagnostics GmbH, Mannheim, Germany). Each reaction was carried out in a total volume of 20 μl in glass capillary, containing 1 μl of cDNA, 2, 3 or 4 mM MgCl2, 10% LightCycler-DNA Master SYBR Green I buffer (Taq DNA polymerase, reaction buffer, deoxynucleotide triphosphate mix, 10 mM MgCl2 and SYBR Green I dye) and 0.5 μM of each primer (Table 1). After the PCR reaction, we confirmed that there was no primer dimer and non specific product in each PCR product by agarose gel electrophoresis and staining with ethidium bromide. The expression of all target genes was normalized to β-actin. Analysis was carried out with Light Cycler 3.5 software (Roche) and Microsoft Excel.
To see the reconstituted effects of HARA-B-derived substances on astrocytic expression of TNF-α, IL-1β and IL-6, the mouse primary astrocyte cells were treated with human IL-8 (R&D systems, Minneapolis, MN, U.S.A), MIF (R&D systems), or PAI-1 (Peprotech, Rocky Hill, U.S.A) for 72 h in serum-free medium. Total RNA was extracted using an RNeasy Plus Mini kit (QIAGEN, Hilden, Germany) and QIA shredder (QIAGEN). cDNA was synthesized using a SuperScript VILO cDNA synthesis kit (Invitrogen) SYBR-Green real-time PCR (Applied Biosystems, Foster City, CA) was performed on cDNA prepared from each sample using Platinum SYBR-Green qPCR Super-Mix-UDG (Invitrogen) and 0.5 μM each primer (Table 1). Thermal cycling condition were 10 min at 95°C, 45 cycles at 95°C for 15 s, followed by 1 min at 60°C. Data Analysis was completed using the ABI PRISM 7500HT Sequence detection software (Applied Biosystems). β-actin was used for normalization.
Cytokine ELISA assay
Co-cultures of HARA-B cells (0.5 × 103 cells/well) and astrocytes (5 × 103 cells/well) or astrocytes alone (5 × 103 cells/well) were seeded into 8-well cell culture slides in DMEM with FCS for 24 h. Then, cells were rinsed twice with PBS and incubated in serum free DMEM. After 48 or 72 h of culture, each conditioned medium was collected and centrifuged to remove debris (1500 rpm for 10 min at 4°C) before use. The amount of mouse IL-1β, TNF-α and IL-6 in each conditioned medium was measured with an ELISA kit for mouse cytokines (Biosource International). The absorbency at 450 nm was measured by a Microplate Reader (Immuno-Mini NJ-2300, Nalge Nunc International).
Cytokine proteome array
HARA-B cells (1 × 106 cells) were grown in 10 cm dish (BD, Franklin Lake, NJ, U.S.A) for 24 h and culture medium was collected. Protein array analysis was performed according to the manufacture’s instruction. Positive controls were located in the upper left-hand corner (two spots), upper right-hand corner (two spots) and the lower left-hand corner (two spots) of each array kit. Medium and culture medium were measured using the human cytokine array Panel A (Proteome Profiler) (R&D systems, Minneapolis, MN, U.S.A). Horseradish peroxidase substrate (Thermo scientific, Rockford, IL, U.S.A) was used to detect protein expression and captured by exposure to X-ray Film (FUJIFILM, Tokyo, Japan).
Immunocytochemistry
HARA-B cells (0.5 × 103 cells/well) with or without astrocytes (5 × 103 cells/well) were seeded into 8-well cell culture slides in complete medium. Cells were fixed with 4% PFA for 30 min at room temperature and permeabilized with 0.3% TritonX-100 in PBS for 15 min and blocked in PBS containing 1% BSA and 5% normal donkey serum (Jackson) for 1 h at room temperature. Cells were incubated with primary antibodies, containing monoclonal mouse anti-human cytokeratin antibody (AE1/AE3 pool of cytokeratin) (Dako, 1:100), rabbit anti-human IL-6Rα antibody (Santa Cruz, CA, USA) (1:200), rabbit anti-human gp130 antibody (Santa Cruz, 1:500), rabbit anti-human IL-1RtI antibody (Santa Cruz, 1:200), and goat anti-human TNFRI (Santa Cruz, 1:100) overnight at 4°C. Control cells were incubated without primary antibody (PBS containing 1% BSA) to test non-specific staining. The cells were then incubated for 5 h at room temperature with secondary antibody, containing FITC-conjugated anti-mouse IgG (Sigma, 1:500), Cy3-conjugated anti-rabbit IgG (Jackson, 1:500) and Cy3-conjugated anti-goat IgG (Jackson, 1:500) and then for 30 min at room temperature with 300 nM 4′,6′-diamidino-2-phenylindole hydrochloride (DAPI, Sigma). Slides were mounted in the Perma Fluor Aqueous Mounting Medium (Thermo) and were analyzed with a Zeiss LSM510 META confocal microscope.
Statistical analysis
One-way analysis of variance (ANOVA) and post-hoc Bonferroni/Dunn test were used to examine the statistical differences. Differences were considered significant at P < 0.05.
Results
Histological analyses of lung cancer cell-induced brain metastasis
Though one of the lung cancer cell line (HARA-B) induces bone metastasis [12], it was not known that HARA-B cells also induce brain metastasis. At 3 weeks after the inoculation of HARA-B cells into cardiac ventricle, metastatic foci were mainly found in midbrain-lateral cortex (data not shown). At 4–6 weeks, metastatic foci of various sizes were found throughout the brain. Bigger metastatic foci attracted more astrocytes (Fig. 1a), with the correlation factor of tumor size and GFAP intensity of 0.638 (Fig. 1b). Though the incidence of brain metastasis was different depending on each mouse, the highest incidence was generally observed in cerebral cortex and hippocampus (Fig. 1c). The correlation factor of tumor size and GFAP intensity was higher in hippocampus (0.716) than that in cortex (0.4927) (Fig. 1d). On the other hand, brain’s immune cells, including microglia and/or invaded macrophages, also showed accumulation around tumor cells but did not show stronger correlation between tumor size and immune cell population (data not shown). These results suggest that there are correlations between astrocytes and metastatic tumor cells in the microenvironment of brain metastasis.

Astrocyte accumulates around HARA-B cells in vivo. a Typical examples of immunostaining of astrogliosis (GFAP) around invaded tumor cells (human cytokeratin, CK). Accumulation of GFAP-positive astrocytes has relation to the size of the tumors. b Correlation between tumor size and astrogliosis. Accumulation of astrocytes was indicated as an intensity of GFAP fluorescence. c Typical examples of immunostaining indicating more accumulation of astrocytes in hippocampus than in cerebral cortex. d Correlation curve between tumor size and GFAP intensity in cortex (closed circle) and hippocampus (open circle). In hippocampus, astrogliosis around metastatic tumor foci increased logarithmically with correlation factor (R 2) of 0.72
Effects of astrocytes on the proliferation of HARA-B cells in vitro
In order to elucidate the relationship between astrocytes and HARA-B cells, interaction between 2 cell types was tested in vitro. Primary cultured mouse astrocytes were used and co-cultured with HARA-B cells. The proliferation of HARA-B cells was increased in co-culture with astrocytes in comparison to that in the control (in the absence of astrocytes). In addition, more astrocytes and longer incubation time yielded more proliferation of HARA-B cells (Fig. 2a). The relative increase of proliferation at a ratio of HARA-B cells to astrocytes of 1:5 and 1:10 were 285 ± 9.5% (n = 6) and 441 ± 11.2% (n = 6), respectively. Since the proliferation of cells in culture system depends on the cell number and physical contact, the effects of conditioned medium on the proliferation of HARA-B cells were examined. First, to avoid physical contact, HARA-B cells were cultured in insert-well with astrocytes in lower-well (ratio of HARA-B to astrocytes was 1:10). After 48 of insert-culture, the medium was collected and added to HARA-B cells and incubated for 48 or 72 h, and then the cell number was counted. Since the medium from insert-culture also increased the proliferation of HARA-B cells, it was suggested that some soluble factors were induced, presumably in astrocytes, without physical cell–cell contact between astrocytes and HARA-B cells (Fig. 2b). Second, it was investigated whether astrocyte-induced soluble factors are constitutive or inducible. Neither astrocyte-conditioned medium (ACM) nor HARA-B-conditioned medium (HCM) but medium from HCM-treated astrocytes (H-ACM) significantly increased the proliferation of HARA-B cells after 48 or 72 h of treatment (Fig. 2c). These results suggest that astrocytes could be stimulated by some soluble factors released from tumor cells, and then produce some growth-promoting factors for tumor cells in turn.

Astrocyte stimulated the proliferation of tumor cells via soluble factors. a The normalized number of HARA-B cells increased according to the ratio of astrocytes to HARA-B cells and incubation time in co-culture treatment (HARA-B cells : astrocytes = 1:5, A5; HARA-B cells : astrocytes = 1:10, A10). b Culture medium from insert-culture of astrocytes with HARA-B cells (HARA-B cells : astrocytes = 1:10, A10) significantly increased the proliferation of tumor cells compared to the one without insert-culture medium (control). c H-ACM (HARA-B-stimulated astrocyte-conditioned medium), but not HCM (HARA-B-conditioned medium) nor ACM (astrocyte-conditioned medium) significantly increased the proliferation of tumor cells. The incubation time was 48 h (gray bars) and 72 h (black bars). Each value represents the mean ± SEM (n = 6). ** P < 0.01
Identification of soluble factors produced by astrocytes
To identify growth-promoting soluble factors produced by astrocytes, mRNA expression of several cytokines and/or growth factors were examined. Activated astrocytes have been shown to produce a wide variety of cytokines including interleukin-1 (IL-1), interleukin-3 (IL-3), interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), transforming growth factor-β (TGF-β), insulin-like growth factor-1 (IGF-1) and platelet-derived growth factor (PDGF) [6–8]. To discriminate astrocytes-derived cytokines (mouse-origin) from HARA-B-derived cytokines (human-origin), primers for mouse cytokines which do not cross-react to human cytokines were used (Table 1). The amplification of mRNA shows that marked increases in the expression of IL-1β, TNF-α and IL-6 were found in astrocytes after 72 h in the insert-culture with HARA-B cells. The relative expression levels of IL-1β, TNF-α and IL-6 increased to 11.4 ± 2.2, 26.9 ± 1.9 and 15.4 ± 1.1 fold (n = 3 each), respectively. On the other hand, expressions of EGF, TGF-β, IGF-I and PDGF-B did not show significant change even after 72 h of the insert-culture with HARA-B cells (Fig. 3a). The RT–PCR for human-IL-1β, human-TNF-α and human-IL-6 in HARA-B cells with or without insert-culture with astrocytes was also performed but the fold increase in mRNA was not significant for either cytokine (data not shown). These results suggest that the origin of IL-1β, TNF-α and mouse-IL-6 were astrocytes but not HARA-B cells.

Expression of mRNA and release of cytokines and growth factors from activated astrocytes. (a) Quantitative RT–PCR of IL-1β, IL-6, TNF-α, transforming growth factor-β (TGF-β), insulin-like growth factor-1 (IGF-1), epidermal growth factor (EGF), and platelet-derived growth factor-B (PDGF-B) in astrocytes insert-cultured with HARA-B cells. The expression level of each cytokine or growth factor mRNA was normalized to the level of each cytokine in astrocytes cultured alone. The relative values of each cytokine mRNA in insert-cultured astrocytes for 72 h are shown. Each value represents the mean ± SEM (n = 3). Release of IL-1β (b), TNF-α (c) and IL-6 (d) into the culture medium of single-culture of astrocytes (Astro), insert-culture or co-culture of astrocytes and HARA-B cells for 48 and 72 h were detected by ELISA. Each value represents the mean ± SEM (n = 6). ** P < 0.01, ## P < 0.01
We also measured the protein levels of mouse-IL-1β, mouse-TNF-α and mouse-IL-6 in the conditioned medium obtained from single-cultured astrocytes (Astro), insert-cultured astrocytes (insert-culture), and co-cultured astrocytes (co-culture) with HARA-B cells after 72 h incubation. Significant increase in the amounts of IL-1β and TNF-α was observed after 48 and 72 h of insert-culture and co-culture (Fig. 3b, c), while the increase in IL-6 release was only observed after 72 h of insert-culture and co-culture (Fig. 3d). The amounts of each cytokine after 72 h in astrocyte-culture, co-culture, and insert-culture were 0 (not detectable), 8.8 ± 0.7 and 9.4 ± 0.8, pg/ml for IL-1β, 8.3 ± 1.3, 116 ± 5.5 and 121 ± 7.9 pg/ml for TNF-α, and 4.2 ± 1.3, 34.2 ± 1.9 and 40 ± 4.2 pg/ml for IL-6, respectively (n = 6).
Effects of recombinant IL-1β, TNF-α and IL-6 and their neutralizing antibodies on the proliferation of HARA-B cells
To confirm the effects of IL-1β, TNF-α and IL-6, recombinant mouse (m) IL-1β, mTNF-α and mIL-6 were applied to HARA-B cells. The concentrations of IL-1β, TNF-α and IL-6 used were employed according to the levels of these cytokines observed in the insert- or co-culture medium (Fig. 3b, c, d). Mouse-IL-1β in a range of 1-10 pg/ml, but not high concentration (50 pg/ml), promoted the proliferation of HARA-B cells (Fig. 4a). Mouse-TNF-α and mIL-6 (10–500 pg/ml each) showed growth-promoting effect on HARA-B cells in a dose-dependent manner (Fig. 4a). These results show that IL-1β, TNF-α and IL-6 released from mouse astrocytes could increase the proliferation of human-origin HARA-B cells.

Effects of recombinant cytokines on HARA-B cell proliferation and inhibitory effects of neutralizing antibodies. a Effects of recombinant mouse (m) IL-1β (1–50 pg/ml), mTNF-α (10–500 pg/ml), and mIL-6 (10–500 pg/ml). Data were given as the percentage of tumor cell proliferation without cytokines (without recombinant cytokines; 100%). HARA-B cells were cultured for 24 h in DMEM and then for 48 h in serum free DMEM with each cytokine. Each value represents the mean ± SEM (n = 6). b Effects of neutralizing antibodies. Anti-mIL-1β (1 μg/ml), anti-mTNF-α (1 μg/ml), anti-mIL-6 (10 ng/ml) neutralizing antibodies, all three antibodies (all ab) and corresponding control IgG were added to co-culture of HARA-B cells and astrocytes. Antibodies were added after 24 h of co-culture of HARA-B cells and astrocytes, and then maintained for 48 h with neutralizing antibodies or control IgG. Data were given as the percentage of control (single-culture of HARA-B cells; 100%) under the same condition without adding antibodies. Each value represents the mean ± SEM (n = 5). ** P < 0.01, # P < 0.05
In reverse, neutralizing antibodies against mIL-1β, mTNF-α and mIL-6 inhibited the effects of co-culture. The titrations of each antibody used (mIL-1β and mTNF-α antibody: 0.2–2.0 μg/ml, mIL-6 antibody: 2.0–20 ng/ml) were determined according to the maximal neutralizing concentration (data not shown). The proliferation of HARA-B cells was promoted to 449 ± 10.2% after co-culture with astrocytes for 72 h in comparison to that in the control (single-culture of HARA-B cells). The antibodies against mIL-1β (1 μg/ml), mTNF-α (1 μg/ml), mIL-6 (10 ng/ml) and all three antibodies were added at 24 h after the co-culture of HARA-B cells with astrocytes, and the co-culture were maintained for further 48 h in the presence of these antibodies. The proliferation of HARA-B cells was significantly attenuated to 149 ± 7.0, 217 ± 10.8, 253 ± 11 and 126 ± 5.9% in the presence of antibodies against IL-1β, TNF-α, IL-6 and all three antibodies, respectively (n = 5 each) (Fig. 4b).
Identification of tumor cell-derived factors which activate astrocytes
We then identified HARA-B-derived factors which activate astrocytes and promote expression of IL-1β, TNF-α, and IL-6. The factors in HARA-B cells culture medium was analyzed using the cytokine proteome profiler. The increased expression of cytokines in HARA-B conditioned medium compared to control (10% FBS DMEM) were IL-1ra, IL-2, IL-8, MIF, and SERPINE1 (PAI-1) (Fig. 5a). Among them, IL-8, MIF, and PAI-1 which showed greater expression were tested whether they really activate mouse astrocytes and stimulate the production of IL-1β, TNF-α, and IL-6. The expression of TNF-α mRNAs in astrocytes were significantly increased by recombinant human IL-8 (hIL-8, 10–100 ng/ml) and hMIF (10-100 ng/ml) (n = 3 each) (Fig. 5b). The TNF-α mRNA level was not detected with the application of hPAI-1 somehow (not shown). The expression of IL-1β mRNAs in astrocytes were also significantly increased by hIL-8 (100 ng/ml), hMIF (10–100 ng/ml), and hPAI-1 (100–1000 ng/ml) (n = 3 each) (Fig. 5c). The expression of IL-6 mRNAs in astrocytes were significantly increased by hMIF (10–100 ng/ml), and hPAI-1 (10–100 ng/ml) but not by hIL-8 (n = 3 each) (Fig. 5d). From these results, tumor-derived MIF would be the most potential candidate for stimulating astrocyte and IL-8 and PAI-1 may be less responsible.

HARA-B-derived factors which stimulate astrocytes and their effects on expression of mRNA of inflammatory cytokines in astrocytes. a Cytokine expression in HARA-B cells culture medium using the proteome profiler. The cytokine expression in medium (10% FBS DMEM) as negative control (upper panel) and in HARA-B conditioned medium (lower panel), showing the expression of IL-1ra, IL-2, IL-8, MIF, and SERPINE1 (PAI-1). b–d Expression of mRNA of inflammatory cytokines (TNF-α, IL-1β, IL-6) in astrocytes treated with each recombinant cytokines (IL-8, MIF, PAI-1). Quantitative RT–PCR of TNF-α (b), IL-1β (c), and IL-6 (d) in astrocytes treated with each cytokine released from HARA-B cells IL-8, MIF, and PAI-1. The expression level of each cytokine was normalized to the level of each cytokine in non-treated astrocytes. The relative values of each cytokine mRNA in astrocyte treated with each cytokine for 72 h are shown. Each value represents the mean ± SEM (n = 3). Data of PAI-1-treatment was not shown in TNF-α mRNA
Expression of cytokine receptors on HARA-B cells
The expression of receptors for IL-1β, TNF-α and IL-6 on HARA-B cells were examined by the immunocytochemical staining. Receptors and receptor subunit for these cytokines were detected on cytokeratin-positive HARA-B cells by using antibodies against human-IL-1RtI, human-TNFRtI, human-IL-6Rα and human-gp130. All of these receptors were detected in single-cultures of HARA-B cells (control) (Fig. 6a). To examine the time-dependent change in the expression level for each cytokine receptor and receptor subunit on HARA-B cells, the immunostaining was observed after 24, 48 and 72 h of co-culture with astrocytes. The semi-quantitative analyses showed that the expression level for IL-1RtI and TNFRtI decreased with time after co-culture, while the expression level for IL-6Rα and gp130 were up-regulated in co-culture with astrocytes (Fig. 6b). These results suggest that IL-6 receptors on HARA-B cells may be more functional when HARA-B cells were co-cultured with astrocytes and IL-6 may be the most important cytokine in the promotion of HARA-B cell proliferation in the brain.

Time-dependent expression of cytokine receptors on HARA-B cells. a Immunostaining of cytokine receptor and receptor subunit (IL-6Rα, gp130, TNFRtI and IL-1RtI) on HARA-B cells with or without co-culture with astrocytes for 24, 48, and 72 h. HARA-B cells were also immunostained with anti-cytokeratin (CK) antibody. b Quantification of fluorescent intensity for each receptor or receptor subunit per area of single cell. Data were given as the percentage of intensity in control HARA-B cells without co-culture (100%). Each value represents the mean ± SEM (n = 8). ** P < 0.01, ## P < 0.01
Effects of astrocytes on the growth of different lung tumor cell lines in vitro
To test if the mutual stimulation between astrocytes and lung cancer cells was general observation and not specific to HARA-B cells, three other cell lines derived from human squamous cell carcinoma (QG56, EBC-1) and non-small cell lung cancer (PC-9) were examined in vitro. Primary cultured astrocytes, which were prepared from C57BL/6 mice brain, were insert-cultured with other lung tumor cells in the ratio of 1:10 (lung tumor cells : astrocytes). The proliferation of each lung tumor cells increased to 210 ± 27% (QG56, n = 4), 480 ± 43% (EBC-1, n = 4), and 150 ± 12% (PC9, n = 4) after 72 h of incubation with insert-culture medium (ICM), respectively (Fig. 7). These results show that astrocytes, activated by the soluble contact with lung cancer cells, promote not only the growth of HARA-B cells but also that of other lung cancer cells, suggesting that mutual activation of astrocytes and lung tumor cells are common phenomena.

Increased proliferation of different lung cancer cell lines by astrocytes in vitro. The proliferation of HARA-B cells (a), QG56 (b), EBC-1 (c), PC9 (d) were enhanced when they were incubated with insert-culture medium of astrocytes for 72 h. Each value represents the mean ± S.E.M (n = 4). *** P < 0.005 (significance from control)
Astrogliosis around human brain metastasis of lung tumor
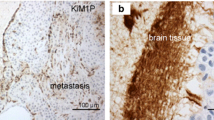
Since activated astrocytes gathered around brain metastasis in model mice, we examined whether the same pathology was observed in human tissue from brain with lung cancer metastasis. We observed brain metastasis of lung tumor in human tissue sections by Hematoxylin-Eosin staining (Fig. 8a). GFAP-positive astrocytes, which means activated astrocytes, accumulated around metastatic foci (Fig. 8b).

Astrocytes accumulate around metastasized lung cancer cells in human brain. a Hematoxylin and eosin (H & E)-staining of lung cancer cell metastasis (dark color) in the human brain section. b Immunostaining of astrocytes and tumor cells from human brain section. GFAP-positive astrocytes aggregated around cancer cells (CAM5.2), which looked similar to the brain metastasis of model mice
Discussion
Certain cancers, i.e. breast cancer and lung cancer, are liable to metastasize in the brain. The incidence of the brain metastasis has been increasing in recent years [1]. In the metastatic process, the microenvironment of the metastatic sites plays an important role for tumor cells to invade and proliferate in the target tissues [2]. Such a microenvironment contains many resident cell types in addition to tumor cells as well as migratory hematopoietic cells.
Though activated astrocytes and soluble factors produced by glial cells in vivo seem to play an important role in the development of brain metastases [5], mechanisms of brain metastases induced by lung cancer cells remained unclear.
In the present study, histological examination revealed that activated astrocytes accumulated around the metastatic foci of human lung cancer-derived cell line, HARA-B cells, in the brain. Similar accumulation of astrocytes around brain metastases was also observed in human brain section from patients with lung cancer metastasis (Fig. 8), as well as in autopsy cases [14]. In our animal models, brain metastases were observed not two but three weeks after intracardiac inoculation of HARA-B cells, mostly in lateral cortex including hippocampus where more GFAP-positive astrocytes were observed even in control condition (data not shown). In later phase of metastases, 4-6 weeks after inoculation, more metastases were observed in whole brain, especially in cerebral cortex. These informations might be useful to understand the process of brain metastasis and its diagnosis.
From our in vitro studies, it was suggested that astrocytes, activated by tumor cells even in the absence of physical contact, promote the proliferation of lung cancer cells by releasing trophic factors. Using one of the lung cancer cell lines, HARA-B cells, IL-1β, TNF-α and IL-6 were identified as astrocyte-oriented factors. It is known that activated astrocytes produce various inflammatory cytokines. IL-1, one of the inflammatory cytokines, has been shown to stimulate the growth of tumor cells in hepatic and/or lung metastases of melanoma tumor cells in vivo [15–17]. Sierra et al. [11] demonstrated that the growth of the breast cancer cell line, which was derived from a brain metastasis, was stimulated by the astrocytes through IL-6, TGF-β and/or IGF-1 in vitro. In brain metastasis of melanoma cells, it was reported that astrocytes produce neurotrophin-regulated heparanase [18, 19]. Recently, it was also reported that epidermal growth factor receptor (EGFR) and membrane type-1 matrix metalloproteinase (MT1-MMP) may be playing an important role in brain metastasis from lung adenocarcinoma and breast cancer [20]. All these results suggest that tumor-promoting factors released from astrocytes are different depending on the type of tumor cells and each type of tumor cells may play a different role in the microenvironment of metastatic sites.
So far, a factor produced by HARA-B cells was only a parathyroid hormone-related protein (PTHrP) [21]. However, application of PTHrP(1-34) (10−6 M) alone did not stimulate the production of IL-6 in astrocytes (supplementary figure) as was already reported [22]. It was reported that TNF-α stimulated the production of IL-6 in astrocytes and PTHrP acted in an additive fashion with TNF-α to astrocyte-induced expression of IL-6 [22]. In our case, however, PTHrP rather attenuated the effect of TNF-α (100 pg/ml) on astrocytic expression IL-6 (supplementary figure).
In the present study, we found that the production of IL-8, MIF, and SERPINE1 (PAI-1) were markedly increased in the medium of HARA-B cell, with less amount of IL-1ra, IL-2 (Fig. 5a), which increased expression of TNF-α, IL-1β, and IL-6 in astrocytes (Fig. 5b–d). It was recently reported that IL-8, as well as IL-6, were induced in nonsmall-cell lung carcinoma (non-SCLC) cells A549 [23]. In human lung adenocarcinoma cell line CPA-Yang2, which is highly metastasis cell line, quantitative RT–PCR showed that ESM1 (Endothelial cell-specific molecule 1), VEGF-C (Vascular Endothelial Growth Factor C), IL-6, IL-8, AR (androgen receptor) genes were overexpressed [24]. It was also found that IL-8 and matrix metalloproteinase-9 (MMP-9) are important cytokines which are closely related to the growth and metastasis of tumor [25]. Consistently, lung cancer patients had higher levels of serum and bronchoalveolar lavage fluid IL-6 and serum IL-8 compared to controls [26].
As for MIF, it is a multifunctional cytokine or an autocrine- and paracrine-acting cytokine/growth factor, being overexpressed in lung cancer, and therefore one of the biomarkers of non-SCLC [27]. The MIF receptor, CD74, was recently discovered and was found that CD74 and MIF were co-expressed in tumors in close proximity, and that co-expression of the MIF-CD74 pair was associated with both higher levels of tumor-associated angiogenic CXC chemokines and greater vascularity [28]. Importantly, expression level of MIF, together with CD147 proteins, in non-SCLC were related to the metastasis. Survival rate was markedly lower in patients with high expression level of MIF or CD147 [29]. It was also shown that MIF overexpression by adenovirus in human lung adenocarcinoma cells induces a dramatic enhancement of cell migration [30].
Urokinase plasminogen activator (uPA) and its inhibitor PAI-1 stimulate angiogenesis in non-SCLC [31] and a crucial role of PAI-1 in lung cancer invasiveness and influence on prognosis were reported [32–34]. Increased PAI-1 expression and stabilization of PAI-1 mRNA in human lung epithelial and carcinoma cells were regulated by tumor suppressor protein p53 [35]. Taken together, IL-8, MIF, and PAI-1 in lung cancer cells are important not only for invasiveness and prognosis but also as stimulants for astrocytes after brain metastasis.
Stimulated astrocytes by the factors mentioned above release IL-1β, TNF-α and IL-6. In our present study, expression of their receptors on HARA-B cells was confirmed immunocytochemically. Interestingly, the time-dependent change in the expression of receptors for each cytokine seemed different in vitro. IL-6 receptor and its subunit, gp130, were up-regulated with time, while expression of receptors for IL-1β and TNF-α were down-regulated. It was reported that leukocytes rapidly lose their surface receptors for TNF and IL-1 upon exposure to various stimuli in vitro and in fact lipopolisaccharide (LPS) induced down-regulation of monocyte and granulocyte receptors for TNF and IL-1 in humans in vivo [36]. In parallel, the release of IL-6 looked to be delayed compared to that of IL-1β and TNF-α (Fig. 3b, c, d). Taking account of these evidences, IL-1β and TNF-α might play an important role at the beginning, then IL-6 and its receptors would become more important functionally when brain metastases take place during long period. This might be the reason why the effects of neutralizing antibodies against IL-1β and TNF-α looked apparently robust when they were added during the first 24–72 h of co-culture. Considering the long-lasting effect of IL-6, blocking IL-6 may be useful not only for autoimmune and chronic inflammatory diseases [37] but also for brain metastasis. In vivo analyses on the effects of these cytokine blockages are now under investigation.
The role of activated microglia also needs to be investigated. It was reported that differential reactions of microglia to brain metastasis of lung cancer [38], showing an obvious increase in the number of microglia around metastatic lung cancer mass in the brain. However, only a few microglia expressed inducible nitric oxide synthase (iNOS) and TNF-α in the region where the tumor mass was situated. In vitro study, LPS-activated microglia showed both apoptotic effect and trophic effect, depending on the concentration of supernatant. Since the mechanism would be different between LPS- and metastasis-induced microglial activation, further investigation would be necessary.
In conclusion, the present results showed that the interaction between metastatic tumor cells and activated astrocytes are important in creating a favorable microenvironment for the tumor cells in the brain. They stimulate each other; first lung tumor cells stimulate astrocytes by releasing IL-8, MIF, and PAI-1, then activated astrocytes stimulate the proliferation of tumor cells by releasing cytokines such as TNF-α, IL-1β, and IL-6. These mutual relationships may be important to understand how lung cancer cells metastasize and develop in the brain.
Abbreviations
- ab:
-
Antibody
- ACM:
-
Astrocyte conditioned medium
- BSA:
-
Bovine serum albumin
- Cdna:
-
Complementary DNA
- DAPI:
-
4′,6′-diamidino-2-phenylindole hydrochloride
- DMEM:
-
Dulbecco’s modified Eagle medium
- EDTA:
-
Ethylenediaminetetraacetic acid
- EGFR:
-
Epidermal growth factor receptor
- FCS:
-
Fetal calf serum
- FITC:
-
Fluoresceinisothiocyanate
- GFAP:
-
Glial fibrillary acidic protein
- MT1-MMP:
-
Membrane type-1 matrix metalloproteinase
- HCM:
-
HARA-B conditioned medium
- H-ACM:
-
HARA-B-astrocytes conditioned medium
- ICM:
-
Insert culture medium
- IGF-1:
-
Insulin-like growth factor-1
- IL-1β:
-
Interleukin-1β
- IL-1ra:
-
Interleukin-1 receptor antagonist
- IL-3:
-
Interleukin-3
- IL-6:
-
Interleukin-6
- MIF:
-
Macrophage migration inhibitory factor
- PAI-1:
-
Plasminogen activator inhibitor
- PBS:
-
Phosphate buffer saline
- PDGF:
-
Platelet-derived growth factor
- PFA:
-
Paraformaldehyde
- PTHrP:
-
Parathyroid hormone-related protein
- SERPINE1:
-
Serpin peptidase inhibitor plasminogen activator inhibitor type 1)
- TGF-β:
-
Transforming growth factor-β
- TNF-α:
-
Tumor necrosis factor-α
References
Schouten LJ, Rutten J, Huveneers HA, Twijnstra A (2002) Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. Cancer 94:2698–2705
Fidler IJ, Yano S, Zhang RD et al (2002) The seed and soil hypothesis: vascularisation and brain metastases. Lancet Oncol 3:53–57
Aloisi F, Ria F, Adorini L (2000) Regulation of T-cell responses by CNS antigen-presenting cells: different roles for microglia and astrocytes. Immunol Today 21:141–147
Balkwill F, Mantovani A (2001) Inflammation and cancer: back to Virchow? Lancet 357:539–554
Fitzgerald DP, Palmieri D, Hua E et al (2008) Reactive glia are recruited by highly proliferative brain metastases of breast cancer and promote tumor cell colonization. Clin Exp Metastasis 25(7):799–810
Miller RH, Ffrench-Constant C, Raff MC (1989) The macroglial cells of the rat optic nerve. Annu Rev Neurosci 12:517–534
Aloisi F, Care A, Borsellino G et al (1992) Production of hemolymphopoietic cytokines (IL-6, IL-8, colony-stimulating factors) by normal human astrocytes in response to IL-1 beta and tumor necrosis factor-alpha. J Immunol 149:2358–2366
Hertz L, McFarlin DE, Waksman BH (1990) Astrocytes: auxiliary cells for immune responses in the central nervous system? Immunol Today 11:265–268
Lee SC, Liu W, Dickson DW et al (1993) Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 beta. J Immunol 150:2659–2667
Wang FW, Jia DY, Du ZH et al (2009) Roles of activated astrocytes in bone marrow stromal cell proliferation and differentiation. Neuroscience 160(2):319–329
Sierra A, Price JE, Garcia-Ramirez M et al (1997) Astrocyte-derived cytokines contribute to the metastatic brain specificity of breast cancer cells. Lab Invest 77:357–368
Iguchi H, Tanaka S, Ozawa Y et al (1996) An experimental model of bone metastasis by human lung cancer cells: the role of parathyroid hormone-related protein in bone metastasis. Cancer Res 56:4040–4043
Lyons S, Kettenmann K (1998) Oligodendrocytes and microglia are selectively vulnerable to combined hypoxia and hypoglycemia injury in vitro. J Cereb Blood Flow Metab 18:521–530
Zhang M, Olsson Y (1995) Reactions of astrocytes and microglial cells around hematogenous metastases of the human brain. Expression of endothelin-like immunoreactivity in reactive astrocytes and activation of microglial cells. J Neurol Sci 134:26–32
Giavazzi R, Garofalo A, Bani MR et al (1990) Interleukin 1-induced augmentation of experimental metastases from a human melanoma in nude mice. Cancer Res 50:4771–4775
Vidal-Vanaclocha F, Amezaga C, Asumendi A et al (1994) Interleukin-1 receptor blockade reduces the number and size of murine B16 melanoma hepatic metastases. Cancer Res 54:2667–2672
Vidal-Vanaclocha F, Alvarez A, Asumendi A (1996) Interleukin 1 (IL-1)-dependent melanoma hepatic metastasis in vivo; increased endothelial adherence by IL-1-induced mannose receptors and growth factor production in vitro. J Natl Cancer Inst 88:198–205
Marchetti D, Denkins Y, Reiland J et al (2003) Brain-metastatic melanoma: a neurotrophic perspective. Pathol Oncol Res 9(3):147–158
Denkins Y, Reiland J, Roy M et al (2004) Brain metastases in melanoma: roles of neurotrophins. Neuro Oncol 6(2):154–165
Yoshida S, Takahashi H (2009) Expression of extracellular matrix molecules in brain metastasis. J Surg Oncol 100(1):65–68
Iguchi H, Onuma E, Sato K et al (2001) Involvement of parathyroid hormone-related protein in experimental cachexia induced by a human lung cancer-derived cell line established from a bone metastasis specimen. Int J Cancer 94(1):24–27
Funk JL, Trout CR, Wei H et al (2001) Parathyroid hormone-related protein (PTHrP) induction in reactive astrocytes following brain injury: a possible mediator of CNS inflammation. Brain Res 915(2):195–209
Bauer M, Gräbsch C, Gminski R et al (2010) Cement-related particles interact with proinflammatory IL-8 chemokine from human primary oropharyngeal mucosa cells and human epithelial lung cancer cell line A549. Environ Toxicol. doi:10.1002/tox.20643
Yang S, Su J, Cao J et al (2009) Establishment of a novel Chinese human lung adenocarcinoma cell line CPA-Yang1 which produces highly bone metastases in immunodeficient mice. Zhongguo Fei Ai Za Zhi 12(7):753–759
Liu Z, Xu S, Xiao N et al (2010) Overexpression of IL-8 and MMP-9 confer high malignant phenotype in patients with non-small cell lung cancer. Zhongguo Fei Ai Za Zhi 13(8):795–802
Crohns M, Saarelainen S, Laine S et al (2010) Cytokines in bronchoalveolar lavage fluid and serum of lung cancer patients during radiotherapy—association of interleukin-8 and VEGF with survival. Cytokine 50(1):30–36
Khan N, Cromer CJ, Campa M, Patz EF Jr (2004) Clinical utility of serum amyloid A and macrophage migration inhibitory factor as serum biomarkers for the detection of nonsmall cell lung carcinoma. Cancer 101(2):379–384
McClelland M, Zhao L, Carskadon S, Arenberg D (2009) Expression of CD74, the receptor for macrophage migration inhibitory factor, in non-small cell lung cancer. Am J Pathol 174(2):638–646
Liu Q, Yang H, Zhang SF (2010) Expression and significance of MIF and CD147 in non-small cell lung cancer. Sichuan Da Xue Xue Bao Yi Xue Ban 41(1):85–90
Rendon BE, Roger T, Teneng I et al (2007) Regulation of human lung adenocarcinoma cell migration and invasion by macrophage migration inhibitory factor. J Biol Chem 282(41):29910–29918
Offersen BV, Pfeiffer P, Andreasen P, Overgaard J (2007) Urokinase plasminogen activator and plasminogen activator inhibitor type-1 in nonsmall-cell lung cancer: relation to prognosis and angiogenesis. Lung Cancer 56(1):43–50
Ramer R, Rohde A, Merkord J et al (2010) Decrease of plasminogen activator inhibitor-1 may contribute to the anti-invasive action of cannabidiol on human lung cancer cells. Pharm Res 27(10):2162–2174
Chorostowska-Wynimko J, Kedzior M, Struniawski R et al (2010) Cell phenotype determines PAI-1 antiproliferative effect—suppressed proliferation of the lung cancer but not prostate cancer cells. Pneumonol Alergol Pol 78(4):279–283
Di Bernardo MC, Matakidou A, Eisen T, Houlston RS (2009) GELCAPS Consortium. Plasminogen activator inhibitor variants PAI-1 A15T and PAI-2 S413C influence lung cancer prognosis. Lung Cancer 65(2):237–241
Shetty S, Shetty P, Idell S et al (2008) Regulation of plasminogen activator inhibitor-1 expression by tumor suppressor protein p53. J Biol Chem 283(28):19570–19580
van der Poll T, Coyle SM, Kumar A et al (1997) Down-regulation of surface receptors for TNF and IL-1 on circulating monocytes and granulocytes during human endotoxemia: effect of neutralization of endotoxin-induced TNF activity by infusion of a recombinant dimeric TNF receptor. J Immunol 158(3):1490–1497
Mihara M, Ohsugi Y, Kishimoto T (2009) Evidence for the role of Th17 cell inhibition in the prevention of autoimmune diseases by anti-interluekin-6 receptor antibody. Biofactors 35(1):47–51
He BP, Wang JJ, Zhang X et al (2006) Differential reactions of microglia to brain metastasis of lung cancer. Mol Med 12(7–8):161–170
Acknowledgments
We thank Prof. D. A. Brown (University College London, UK) for reading the manuscript. This work was supported by Grants-in Aid for Scientific Research of Japan Society for Promotion of Science.
Conflict of interest
All authors have no conflict of interest and no financial conflicts.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
The study was approved by the Animal Care and Use Committee at Kyushu University and carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Seike, T., Fujita, K., Yamakawa, Y. et al. Interaction between lung cancer cells and astrocytes via specific inflammatory cytokines in the microenvironment of brain metastasis. Clin Exp Metastasis 28, 13–25 (2011). https://doi.org/10.1007/s10585-010-9354-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-010-9354-8