
Abstract
Background
Aegilops tauschii Coss. as a donor of wheat D genome has an important role in wheat breeding programs. Genetic and phylogeographic diversity of 79 Ae. tauschii accessions collected from north and northwest of Iran were analyzed based on retroelement insertional polymorphisms using inter-retrotransposon amplified polymorphism (IRAP) and retrotransposon-microsatellite amplified polymorphism (REMAP) markers.
Results
In total, 306 and 151 polymorphic bands were amplified in IRAP and REMAP analyses, respectively. As a result, a high level of polymorphism was observed among the studied accessions as revealed by an average of 25.5 bands per primer/primer combination and mean PIC value of 0.47 in IRAP and an average of 25.16 bands per primer combination and mean PIC value of 0.47 in REMAP. Genetic relationships of the accessions were analyzed using distance- and model-based cluster analyses.
Conclusion
The result showed that genetic distance did not seem to be related to geographic distribution, and the accessions could be divided into three groups, which was further supported by principal coordinate analysis. These results on genetic diversity and population structure of Ae. tauschii in Iran should provide important knowledge on genetic resources and their applications in wheat breeding programs.
Similar content being viewed by others

Background
Approximately, 8000 years ago, a spontaneous hybridization between cultivated emmer wheat (T. turgidum; 2n = 4× = 28, AABB) and Aegilops tauschii (2n = 2× = 14, DD) in the Fertile Crescent resulted in hexaploid bread wheat (Triticum aestivum; 2n = 6× = 42, AABBDD) [1,2,3]. Ae. tauschii Coss. (2n = 2× = 14) as a donor of wheat D genome is a source of many favorable genes for important agronomic traits [4], bread-making quality [5], resistance to Ug99 [6], leaf rust [7], stripe rust [8, 9], grain yield [10], and cadmium (Cd) tolerance [11]. Among the triple genome of the hexaploid wheat and also compared with Ae. tauschii, D genome of wheat has the slightest diversity [12]. Therefore, Ae. tauschii with the widest geographical distribution and greater genetic variation compared with the corresponding homologous loci in the D genome of bread wheat is a promising genetic resource for broadening wheat genetic background. Considering the wide genetic variation in Ae. tauschii germplasm and its easy crossability with wheat, Ae. tauschii accessions have been exploited by various groups throughout the world for wheat improvement [13,14,15].
Based on spike morphology, two subspecies including ssp. tauschii with extended cylindrical spikelets and ssp. strangulata with moniliform spikes bearing quadrate spikelets have been identified in Ae. tauschii. Three distinct varieties, namely var. anathera, var. meyeri, and var. typica, have been recognized within ssp. tauschii, while ssp. strangulata contains only the var. strangulata. The description of Ae. tauschii variants followed the morphological characteristics revealed wide geographical distribution for ssp. tauschii around Eurasian continent, while ssp. strangulata has a confined dispersal [16]. The ssp. tauschii is spread across the entire Ae. tauschii geographical range, whereas ssp. strangulata is only present in Transcaucasia and around the Caspian Sea region of Iran. However, recently, one ssp. strangulata accession is reported from Uzbekistan, which is the first report for this subspecies out of their natural habitat [17]. The analysis of corresponding variants between the D genome of T. aestivum and Ae. tauschii showed a closer fit with ssp. strangulata than ssp. tauschii. According to earlier genetic investigations, Transcaucasia and southwestern Caspian Iran are the origins of bread wheat [18, 19]. Wang et al. [3] analyzed the genetic relationships among 477 Ae. tauschii and wheat accessions using the Ae. tauschii 10 K Infinium single-nucleotide polymorphism (SNP) array and identified 12 Ae. tauschii accessions, each closely related to a wheat D genome chromosome. All 12 accessions belonged to spp. strangulate collected from southwestern and southern Caspian Sea, Iran. Genetic diversity of Ae. tauschii’s germplasm has been extensively studied using various molecular techniques such as allozyme [20], RFLP [19], microsatellite [21,22,23,24,25], IRAP [26], SNPs [3, 27, 28], AFLP [29], and gene sequences [30].
Cereal genomes consisted of an extraordinary number of transposable elements, in particular, LTR retrotransposons, which is highly dynamic. It is reported that the LTR retrotransposons are often found in different densities or copy numbers among individuals of the same species [31,32,33]. Therefore, several DNA marker techniques have been devised based on LTR retrotransposons which are more common retrotransposon families in plants [32,33,34]. Owing to generating steady and great insertions in the genome, retrotransposons are known as one of the main creators of genetic diversity and tools for discovering the genomic changes related to their activity [35]. IRAP and REMAP as two retrotransposon-based markers have been widely used to analyze genetic diversity and population structure in various crop plants such as cotton [36], alfalfa [37], barley [38], sunflower [39], and wheat and its wild relatives [40, 41]. IRAP reveals retrotransposon insertional polymorphisms by amplifying the portion of DNA between two retroelements from the same or different families by using a single or paired primer. REMAP reveals polymorphism in the regions amplified between adjacent microsatellite loci and inserted retrotransposon using ISSR and retrotransposon-based primers [42]. IRAP and REMAP markers have been used to assess the genetic diversity and population structure in a collection of 48 Old Portuguese bread wheat cultivars. A high level of polymorphism was revealed by both marker systems; however, the power of IRAP markers was higher in detecting genetic variability at the individual level, but did not differentiate higher taxa. Based on REMAP marker data, botanical varieties were clustered together, and homonym bread wheat cultivars were identified [40]. Taheri et al. [41] assessed the genetic diversity of 14 populations of T. urartu and T. boeoticum from west and northwest of Iran by IRAP and REMAP markers. Grouping of the accessions using REMAP data could differentiate them according to their species and geographical origin, but grouping based on IRAP could not separate the accessions of two species. However, based on both marker systems, considerable diversity was observed among and within the studied populations.
Ae. tauschii’s genome contains at least 66% LTR [43]; therefore, insertional polymorphism could be useful for the detection and evaluation of the level of LTR retrotransposon intraspecific variability in Ae. tauschii accessions. Few studies have been utilized retrotransposon-based markers to evaluate genetic variability in collections of Ae. tauschii. Saeidi et al. [26] analyzed the genetic relationship of 57 accessions of Ae. tauschii from north and center of Iran, using IRAP markers, and reported high levels of genetic diversity in the studied accessions. However, IRAP data could not differentiate the accessions based on their geographical origins. Boyko et al. [44] mapped 80 retrotransposon markers in the high-density and unified cytological and genetic map of Ae. tauschii.
In the present study, we used IRAP and REMAP markers to analyze genetic diversity and population structure of a newly collected Ae. tauschii accessions from north and northwest of Iran.
Methods
Plant materials and DNA extraction
Seventy-nine accessions of Aegilops tauschii Coss. were collected from north and northwest of Iran. For each accession, 10–15 plants at least 2 m apart were randomly sampled. The name and geographical information of collection sites were described in the Supplementary table. From each accession, 10–15 seeds were grown in a greenhouse, and DNA was extracted from bulk leaves in a sample of 10–15 plants following the CTAB extraction protocol [45]. DNA quality and quantity were measured using a NanoDrop spectrophotometer at 260 nm and 280 nm wavelengths. The presence of high-molecular-weight DNA was also checked by agarose gel electrophoresis (0.8%).
IRAP and REMAP analyses
Seven LTR retrotransposon primers from Sukkula, Nikkita, and BARE-1 families of barley (Hordeum vulgare L.) genome including Sukkula, Nikkita, LTR6150, LTR6149, 5′LTR1, 5′LTR2, and 3′LTR (Table 1) and their combinations were used in IRAP analyses. REMAP analyses were performed using combinations of seven LTR primers used in IRAP and three ISSR primers. The PCRs were performed in a 10-μL reaction mixture containing 4-μL master mix 2× PCR (ready-to-use PCR master mix 2×; Ampliqon), 2 μL DNA template, and 2 μL distilled water. The amplification program consisted of 5 min of initial denaturation at 94 °C followed by 35 cycles of 94 °C for 60 s, annealing at each primer/primer combination specified Ta for 60 s, and 72 °C for 45 s followed with a final extension at 72 °C for 7 min. The amplification products were separated by 4% ultrathin (0.2 mm) non-denature polyacrylamide gel and detected by ethidium bromide staining using a Gel-Scan 3000 electrophoresis system (Corbett, Sydney, Australia).
Data analysis
The IRAP and REMAP amplification profiles were scored as the presence (1) or absence (0) at each polymorphic band position for each primer/primer combination in all accessions. Each polymorphic band was treated as a single locus with two alleles. Monographic bands were not scored and used in the analyses. The polymorphism information content (PIC), Shannonʼs information index, and Nei’s gene diversity (He) [46] were calculated by GenAlEx 6.503 software [47].
Pairwise, genetic distance between accessions was calculated based on the minimum evolution evolutionary distance coefficient [48] using IRAP and REMAP data, and the correction between two distance matrices was statistically tested by using the mantel test. The ability of IRAP and REMAP markers to reveal genetic relationships among all the Ae. tauschii accessions was evaluated by the neighbor-joining algorithm (NJ), for which the trees were constructed using MEGA 7.0 software [49]. Support for the tree was determined by performing 1000 bootstrap operations on the data set generated by distance analysis. Population structure analysis of 79 Ae. tauschii accessions was performed based on IRAP and REMAP by using the Bayesian Markov chain Monte Carlo model-based clustering implemented in the software package STRUCTURE v2.3.4 [50]. The model was run by varying the number of subpopulation (K) from 1 to 10 with 5 replications for each K and with a burn-in period of 10,000, followed by 100,000 Markov chain Monte Carlo replications. The optimum number of subpopulation (K) which best estimated the structure of the 79 accessions was predicted based on the log probability of the data [LnP(D)] and delta K (ΔK) [51], using online software STRUCTURE HARVESTER [52]. The accessions were assigned to subpopulations based on their probability of association of ≥ 60% to each of the two groups; accessions with a probability of association < 60% were considered as admixtures. Principal coordinate analysis (PCoA) was performed using GenAlex 6.5 [34] to further investigate the population structure of Ae. tauschii accessions. The Nei’ gene diversity (He), Shannon’s information index (I), and within-population variation (WP) were calculated for each geographical population. The analysis of molecular variance (AMOVA) was performed using GenAlex 6.5 [34] to calculate molecular variance components and their statistical significance levels for variation among and within the Aegilops tauschii geographical populations.
Results
Insertional polymorphism and genetic diversity
IRAP
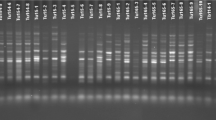
Banding patterns of IRAP amplicons for some Ae. tauschii accessions generated using 5′LTR primer are shown in Fig. 1a. In IRAP analysis, out of 28 single primers or primer combinations, five single primers (3′LTR, 5′LTR, Sukkula, Nikkita, and LTR6149) and seven primer combinations produced scorable bands. In total, 306 polymorphic bands were amplified with an average of 25.5 bands per primer/primer combination. The 5′LTR1 primer with 47 and Sukkula primer, and Nikkita + LTR6149 primer combination with 8 bands, produced the highest and lowest number of polymorphic bands in the studied accessions, respectively. The polymorphic information content (PIC) value ranged from 0.36 (LTR6149) to 0.50 (Sukkula + LTR6150) with an average of 0.47. Considering all polymorphic primers/primer combinations, the highest and lowest number of polymorphic loci were amplified in Mazandaran (218) and Azerbaijan (182) populations, respectively. The lowest and highest average number of polymorphic markers were also scored in the populations collected from Azerbaijan (15.17) and Mazandaran (18.17) populations, respectively (Table 2).

Polyacrylamide gel profile of Ae. tauschii accessions using a 5′LTR in IRAP and b ISSR6 + Sukkula in REMAP
The parameters relating to the genetic diversity of geographical groups including the number of polymorphic loci (NPL), Nei’s gene diversity (He), Shannon’s information index (I), and within-population diversity (WP) are shown in Table 4. The highest (0.40) and lowest (0.24) He values were observed in Guilan and Azerbaijan populations, respectively. Guilan and Azerbaijan populations showed the maximum (0.58) and minimum (0.36) values of Shannon’s index, and the highest and lowest within-population diversity were recorded in Guilan (54.48) and Mazandaran (41.13) populations, respectively.
REMAP
Figure 1b shows REMAP banding pattern of some Ae. tauschii accessions generated using Sukkula and ISSR6 primers combination. Out of 18 LTR and ISSR primer combinations examined in the REMAP analyses, six primer combinations produced scorable bans. A total of 151 polymorphic bands with an average of 25.16 markers per primer combination were amplified. The highest (44) and lowest (13) number of polymorphic markers were amplified using ISSR6 + 5′LTR1 and ISSR42 + Sukkula combinations, respectively. The PIC value varied from 0.42 (ISSR42 + Sukkula) to 0.49 (ISSR6 + Nikkita and ISSR6 + Sukkula) with an average of 0.47. Based on all polymorphic primer combinations, Mazandaran and Golestan with 88 and 81 loci had the highest and lowest number of polymorphic loci, respectively. The maximum and minimum average number of polymorphic loci were also scored in Mazandaran and Golestan populations with values of 14.67 and 13.50, respectively (Table 3).
Based on REMAP data, maximum and minimum values of He were recorded in the Guilan (0.38) and the Mazandaran (0.27) populations, respectively, and Shannon’s index ranged from 0.56 (Guilan) to 0.39 (Mazandaran). The highest level of within-population diversity was observed in the Golestan population (29.18), and the Mazandaran population revealed the lowest within-population diversity (23.40) (Table 2).
Pairwise Nei’s genetic distance among populations was calculated based on IRAP and REMAP data. Based on IRAP data, the highest (0.203) and lowest (0.064) distances were found between “Azerbaijan and Mazandaran” and “Golestan and Guilan,” respectively, which was in agreement with their geographical distances. Among the populations, the Azerbaijan population was far genetically relative to others. In REMAP analysis, the highest genetic distance (0.213) was also observed between Azerbaijan and Mazandaran populations and the lowest (0.043) between Golestan and Guilan populations.
Analysis of molecular variance (AMOVA) revealed high genetic variation within populations and low genetic differentiation among populations. The proportion of variation attributable to within-population differences was high, 90, 94, and 92% by IRAP, REMAP, and combined data, respectively.
Genetic relationships and population structure
The genetic relationships among 79 Ae. tauschii accessions were assessed using the minimum evolution distance coefficient based on IRAP, REMAP, and combined data. Mantel test revealed low and nonsignificant correlation between two matrices (r = 0.127, P = 0.265). The resulting distance matrices were used to construct dendrograms using the neighbor-joining clustering algorithm. The model-based hierarchical structure among the studied accessions was also conducted by STRUCTURE v2.3.4 software [50] using all three data sets.
Based on IRAP data, the 79 accessions were clustered into 3 groups (Fig. 2a). All the accessions in group 1 belong to Guilan province except one each from Azerbaijan and Golestan provinces. In group 2, most of the rest of Guilan accessions were co-clustered with accessions from Mazandaran and two of Golestan. The majority of accessions from Golestan and Azerbaijan along with some accessions from Guilan and one from Mazandaran constructed group 3. Based on the established phylogenetic lineages using model-based cluster analysis, the relevant population structure was captured at K = 2 (Fig. 2b). The 79 Ae. tauschii accessions are allocated into two subpopulations which were in agreement with two major groups and which identified using the neighbor-joining tree constructed from the genetic distances among Ae. tauschii accessions. The relationships between 79 accessions were also determined using PCoA. The biplot of accessions based on two first coordinates obtained by PCoA led to results comparable to those obtained using distance- and model-based cluster analyses (Fig. 2d). The two first coordinates explained 22.88 of the total IRAP data variation, where coordinates 1 and 2 accounted for 13.21 and 9.67% of the variation, respectively.

The genetic relationships and population structure of the 79 Ae. tauschii accessions based on IRAP data concluded from a distance and model-based cluster analyses and PCoA. a Neighbor-joining phylogenetic tree, b population structure on K = 2, c clusters number (K) plotted versus ΔK for determining ideal K-value, and d PCoA based on the first two coordinates
Distance-based cluster analysis using minimum evolution distance coefficient and N-J algorithm based on REMAP data resulted in a dendrogram assigning the accessions into three distinct groups (Fig. 3a). Group 1 consisted of all accessions from Azerbaijan, the majority of Golestan’s accessions, and some accessions from Guilan. The accessions in group 2 belong to Guilan except two from Mazandaran and two from Golestan. Group 2 was a mixed group including accessions from Guilan, Golestan, and Mazandaran. In the model-based cluster analysis, the maximum value for ΔK was observed when K = 4 (Fig. 3c), indicating the presence of four main population groups (Fig. 3b). The result of model-based cluster analysis does not fully support the grouping obtained by the N-J distance-based clustering. The distribution of the 79 Ae. tauschii accessions on the basis of REMAP data was explained by the first two principal coordinates, where the first and second coordinates explained 11.77 and 7.20% of the total variation among the accessions, respectively, and could not show a clear separation of accessions from different provinces, although the accessions from the single province were grouped closely (Fig. 3d).

The genetic relationships and population structure of the 79 Ae. tauschii accessions based on REMAP data concluded from a distance and model-based cluster analyses and PCoA. a Neighbor-joining phylogenetic tree, b population structure on K = 4, c clusters number (K) plotted versus ΔK for determining ideal K-value, and d PCoA plot based on the first two coordinates
Cluster analysis using the N-J algorithm based on combined IRAP and REMAP could not clearly separate accessions from different provinces. In the resulting phylogenetic tree, the accessions were assigned into two groups. Group 1 was a mixed group that consisted of accessions from the four provinces including all accessions of Azerbaijan and Mazandaran and the majority of Guilan’s accessions along with some from Colestan. In group 2, the rest accessions of Guilan and Golestan were co-clustered (Fig. 4a). In model-based clustering, to find out the ideal K-value, the number of clusters (K) was plotted versus ΔK, which presented a clear peak at K = 6 (Fig. 4c). With six groups, the majority of the accessions collected from Guilan province were clustered together, and Golestan’s accessions were also clustered closely (Fig. 4b). In PCoA using combined data, the first and second coordinates accounted for 5.82 and 4.60% of the total variation, respectively. Biplot derived from the PCoA of the 79 Ae. tauschii accessions could not display clear relationships among accessions according to their collected provinces, and the accessions from the same province did not closely group (Fig. 4d). However, the results of PCoA agreed with STRUCTURE analyses compared with N-J clustering.

The genetic relationships and population structure of the 79 Ae. tauschii accessions based on IRAP + REMAP data concluded from a distance and model-based cluster analyses and PCoA. a Neighbor-joining phylogenetic tree, b population structure on K = 6, c clusters number (K) plotted versus ΔK for determining ideal K-value, and d PCoA plot based on the first two coordinates
Discussion
Various DNA markers including AFLP [53], SSR [24], retrotransposon-based markers [26, 53], DArT [54], and SNP [28] have been extensively used to analyze the genetic diversity and population structure of Ae. tauschii collections. Among the various DNA fragments used as DNA markers, the transposable elements have been reported as drivers of structural and functional variations in the wheat genome. Transposable elements are major contributors to genome plasticity and thus are likely to have a dramatic impact on genetic diversity and speciation [55].
In the present study, the genetic diversity and population structure of 79 Ae. tauschii from north and northwest of Iran were assessed by insertional polymorphism using two retrotransposon-based markers: IRAP and REMAP. As a result, a high level of polymorphism was observed among the studied accessions as revealed by an average of 25.5 bands per primer/primer combination and mean PIC value of 0.47 in IRAP and an average of 25.16 bands per primer combination and a mean PIC value of 0.47 in REMAP analyses. Safiyar et al. [56] used 15 IRAP and REMAP markers to analyze the genetic diversity of Ae. tauschii collections from different regions of Iran. They reported a high level of polymorphism for both markers, but the level of polymorphism was higher for IRAP compared with REMAP as detected by the effective number of alleles, Nei’s gene diversity, and Shannon’s information index. Saeidi et al. [26] detected a high level of genetic diversity among 57 Ae. tauschii accessions from northern and Central Iran by means of retroelement insertional polymorphisms generated using IRAP markers. The REMAP markers were used for molecular diversity analysis of 45 genotypes from four Aegilops species, and a high number of polymorphic bands (96.09% of polymorphism) with a mean PIC value of 0.25 was produced [57].
Retrotransposons as major generators of genetic diversity and tools for detecting the genomic changes created by their large and stable insertions in the genome have been widely applied in plant genetic studies [34, 58]. The IRAP and REMAP methods are two common retrotransposon-based markers that were first implemented in barley for fingerprinting and biodiversity studies [50, 59]. Due to the evidence that the closely related retrotransposons are transcribed and translated in diverse grass species belonging to different subfamilies [60], we utilized seven LTR retrotransposon primers developed from Sukkula, Nikkita, and BARE-1 families of barley to analyze genetic relationships among Ae. tauschii accessions. Five out of the seven examined primers and seven primer combinations allowed the amplification of multiple and distinguishable fragments in the 79 Ae. tauschii accessions, and the degree of polymorphism was very high, 100% by either method. In addition to the detection of high IRAP and REMAP polymorphism among the 79 accessions, polymorphism within each geographical population was also high for most of the primers/primer combinations (Tables 2 and 3). In our study, the inability of some primers/primer combinations to amplify fragments from Aegilops genome may be the limited dispersal of the barley’s retrotransposon families that were used to design the primers in the Ae. tauschii genome.
The values of intrapopulation genetic diversity based on Nei’s gene diversity using IRAP data ranged from 0.24 among accessions from the Azerbaijan population to 0.48 among accessions from the Guilan population. Based on REMAP data, it ranged from 0.27 for Mazandaran’s accessions to 0.38 for Guilan’s accessions. The same trends within-population genetic diversity were observed based on Shannon’s information index (Table 4). The higher and lower within-population diversity could be due to the number of accessions sampled from each population. Singh et al. [17] used SNP markers to compute Nei’s diversity index for Ae. tauschii lineage 1 (L1), lineage 2 (L2), possible hybrids, wheat, and Ae. tauschii collection combined and reported the highest Nei’s diversity index for L2 = 0.1326 followed by L1 = 0.0872 and wheat of 0.0158. Higher values of the Nei’s index indicate greater allelic diversity in a given population.
The phylogenetic trees were constructed by two different clustering methods to analyze the genetic relationships of 79 Ae. tauschii accessions using IRAP, and REMAP data did not agree fairly well with the accession’s provinces. However, in most cases, the accessions from the same provinces were closely clustered (Figs. 2, 3, and 4). This could be due to the fact that the accessions from a province have been collected from locations with different geographical and ecological properties. In general, there was no clear relationship between the genetic distance calculated using marker data and the geographical distance for the populations, which may result from the high degree of polymorphism found within the populations. Closer genetic relationships among accessions which geographically located distantly may likely implicate long-distance seed distribution. Analysis of genetic relationships among 57 accessions of Ae. tauschii from northern, northwest, northeast, and Central Iran using retroelement insertional polymorphisms generated by the IRAP method revealed that the accessions from the northwest and central were co-clustered with the accessions from north and northeast, and no clear grouping was observed based on the geographical origins [26]. It is reported that the amplification of transposable elements particularly retroelements is under major environmental effect [61,62,63]; therefore, the conditions in the regions favor activity and dispersion of the elements.
Mantel test revealed a low and nonsignificant correlation between the two matrices (r = 0.127, P = 0.265). It could be due to different genomic regions amplified by these two marker systems. Using the IRAP, genomic regions are amplified by two nearby retrotransposons using outward-facing primers, whereas, in REMAP, amplification between retrotransposons proximal to simple sequence repeats (microsatellites) produces the marker bands (59, 60).
Conclusion
Wang et al. [3] used 7815 SNPs providing complete coverage of the genome to interrogate 402 accessions of Ae. tauschii, 75 hexaploid wheats, and seven tetraploid wheats conclude that southwestern Caspian Iran is the center of wheat genetic diversity and the center of origin of bread wheat. Therefore, studying Ae. tauschii accessions from the wheat center of origin using molecular markers has an essential role in wheat breeding programs. We analyze the genetic diversity of 79 accessions mainly collected from the Caspian area using two highly polymorphic markers: IRAP and REMAP. The results of the molecular genetic diversity analysis clearly showed that this collection of Ae. tauschii will be helpful for wheat breeding programs as revealed by high PIC and diversity indices and the number of polymorphic markers.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- Ae:
-
Aegilops
- T:
-
Triticum
- IRAP:
-
Inter-retrotransposon amplified polymorphism
- REMAP:
-
Retrotransposon-microsatellite amplified polymorphism
- RFLP:
-
Restriction fragment length polymorphism
- AFLP:
-
Amplified fragment length polymorphism
- SNP:
-
Single-nucleotide polymorphism
- DArT:
-
Diversity arrays technology
- SSR:
-
Simple sequence repeat
- ISSR:
-
Inter-simple sequence repeats
- PIC:
-
Polymorphism information content
- He:
-
Nei’s gene diversity
- I:
-
Shannon information index
- PB:
-
Number of polymorphic bands
- NJ:
-
Neighbor joining
- PCoA:
-
Principal coordinate analysis
References
Salamini F, Ozkan H, Brandolini A, Schafer-Pregl R, Martin W (2002) Genetics and geography of wild cereal domestication in the near east. Nat Rev Genet 3:429–441. https://doi.org/10.1038/nrg817
Dubcovsk J, Dvorak J (2007) Genome plasticity a key factor in the success of polyploid wheat under domestication. Science 316:186–1866. https://doi.org/10.1126/science.1143986
Wang J, Luo MC, Chen Z, You FM, Wei Y, Zheng Y, Dvorak J (2013) Aegilops tauschii single nucleotide polymorphisms shed light on the origins of wheat D-genome genetic diversity and pinpoint the geographic origin of hexaploid wheat. New Phytol 198:925–937. https://doi.org/10.1111/nph.12164
Gianibelli MC, Wrigley CW, MacRitchie F (2002) Polymorphism of low Mr glutenin subunits in Triticum tauschii. J Cereal Sci 35:277–286. https://doi.org/10.1006/jcrs.2001.0424
Li Y, Zhou R, Wang J, Liao X, Branlard G, Jia J (2012) Novel and favorable allele clusters for end use quality revealed by introgression lines derived from synthetic wheat. Mol Breed 29:627–643. https://doi.org/10.1007/s11032-011-9578-6
Olson EL, Rouse MN, Pumphrey MO, Bowden RL, Gill BS, Poland JA (2013) Introgression of stem rust resistance genes SrTA10187 and SrTA10171 from Aegilops tauschii to wheat. Theor Appl Genet 126:2477–2484. https://doi.org/10.1007/s00122-013-2148-z
Hiebert CW, Thomas JB, Somers DJ, McCallum BD, Fox SL (2007) Microsatellite mapping of adult-plant leaf rust resistance gene Lr22a in wheat. Theor Appl Genet 115:877–884. https://doi.org/10.1007/s00122-007-0604-3
Huang L, Zhang LQ, Liu BL, Yan ZH, Zhang B, Zhang HG, Zheng YL, Liu DC (2011) Molecular tagging of a stripe rust resistance gene in Aegilops tauschii. Euphytica 179:313–318. https://doi.org/10.1007/s10681-010-0330-9
Liu D, Zhang L, Yan Z, Lan X, Zheng Y (2011) Stripe rust resistance in Aegilops tauschii and its genetic analysis. Genet Resour Crop Evol 57:325–332. https://doi.org/10.1007/s10722-009-9510-7
Gororo NN, Eagles HA, Eastwood RF, Nicolas ME, Flood RG (2002) Use of Triticum tauschii to improve yield of wheat in low-yielding environments. Euphytica 123:241–254. https://doi.org/10.1023/A:1014910000128
Qin P, Wang L, Liu K, Mao S, Li Z, Gao S, Shi H, Liu X (2015) Genome wide association study of Aegilops tauschii traits under seedling-stage cadmium stress. The Crop J 3:405–415. https://doi.org/10.1016/j.cj.2015.04.005
Mirzaghaderi G, Mason AS (2019) Broadening the bread wheat D genome. Theor Appl Genet 132:1295–1307. https://doi.org/10.1007/s00122-019-03299-z
Dhaliwal HS, Singh H, Gupta S, Bagga PS, Gill KS (1991) Evaluation of Aegilops and wild Triticum species for resistance to leaf rust (Puccinia recondita f. sp. tritici) of wheat. Int J Trop Agric 9:118–122
Assefa S, Fehrmann H (2000) Resistance to wheat leaf rust in Aegilops tauschii Coss. and inheritance of resistance in hexaploid wheat. Genet Resour Crop Evol 47:135–140. https://doi.org/10.1023/A:1008770226330
Kalia B, Wilson DL, Bowden RL, Singh RP, Gill BS (2016) Adult plant resistance to Puccinia triticina in a geographically diverse collection of Aegilops tauschii. Genet Resour Crop Evol 64:913–926. https://doi.org/10.1007/s10722-016-0411-2
Kihara H, Tanaka M (1958) Morphological and physiological variation among Aegilops squarossa strains collected in Pakistan, Afghanistan and Iran. Preslia 30:241–251
Singh N, Wu S, Tiwari V, Sehgal S, Raupp J, Wilson D, Abbasov M, Gill B, Poland J (2019) Genomic analysis confirms population structure and identifies inter-lineage hybrids in Aegilops tauschii. Front. Plant Sci 10:9. https://doi.org/10.3389/fpls.2019.00009
Jaaska V (1981) Aspartate aminotransferase and alcohol dehydrogenase isoenzymes: intraspecific differentiation in Aegilops tauschii and the origin of the D genome polyploids in the wheat group. Plant Syst Evol 137:259–273. https://doi.org/10.1007/BF00982790
Dvorak J, Luo MC, Yang ZL, Zhang HB (1998) The structure of the Aegilops tauschii gene pool and the evolution of hexaploid wheat. Theor Appl Genet 97:657–670. https://doi.org/10.1007/s001220050942
Dudnikov AJ (2003) Allozymes and growth habit of Aegilops tauschii: genetic control and linkage patterns. Euphytica 129:89–97. https://doi.org/10.1023/A:1021558628874
Lelley T, Stachel M, Grausgruber H, Vollmann J (2000) Analysis of relationships between Aegilops tauschii and the D genome of wheat utilizing microsatellites. Genome 43:661–668. https://doi.org/10.1139/g00-036
Pestsova E, Korzun V, Goncharov NP, Hammer K, Ganal MW, Roder MS (2000) Microsatellite analysis of Aegilops tauschii germplasm. Theor Appl Genet 101:100–106. https://doi.org/10.1007/s001220051456
Saeidi H, Rahiminejad MR, Vallian S, Heslop-Harrison JS (2006) Biodiversity of diploid D-genome Aegilops tauschii Coss. in Iran measured using microsatellites. Genet Resour Crop Evol 53:1477–1484. https://doi.org/10.1007/s10722-005-7110-8
Naghavi MR, Hajikram M, Taleei AR, Aghaei MJ (2010) Microsatellite analysis of genetic diversity and population genetic structure of Aegilops tauschii Coss.in northern Iran. Genet Resou Crop Evol 57:423–430. https://doi.org/10.1007/s10722-009-9481-8
Yu H, Yang J, Cui H, Abbas A, Wei S, Li X (2021) Distribution, genetic diversity and population structure of Aegilops tauschii Coss. in major wheat-growing regions in China. Agriculture 11(4):311. https://doi.org/10.3390/agriculture11040311
Saeidi H, Rahimnejad MR, Helsop-Harrison JS (2008) Retroelement insertional polymorphisms, diversity and phylogeography within diploid, D-genome Aegilops tauschii (Triticeae, Poaceae) sub-taxa in Iran. Ann Bot 101:855–861. https://doi.org/10.1093/aob/mcn042
Iehisa JCM, Shimizu A, Sato K, Nishijima R, Sakaguchi K, Matsuda R, Nasuda S, Takumi S (2014) Genome-wide marker development for the wheat D genome based on single nucleotide polymorphisms identified from transcripts in the wild wheat progenitor Aegilops tauschii. Theor Appl Genet 127:261–271. https://doi.org/10.1007/s00122-013-2215-5
Su Y, Zou M, Zhu Y, Han X, Li Y, Zhang D, Li S (2020) Analysis of population structure and origin in Aegilops tauschii Coss. from China through SNP markers. Genet Resour Crop Evol 67:923–934. https://doi.org/10.1007/s10722-020-00890-y
Kakizaki A, Kawahara T, Zhuk MA, Smekalova TN, Sato K, Abe T, Sasanuma T (2017) Genetic characterization of genetic resources of Aegilops tauschii, wheat D genome donor, newly collected in North Caucasia. Am J Plant Sci 8:2769–2784. https://doi.org/10.4236/ajps.2017.811187
Kroupin PY, Chernook AG, Bazhenov KGI, Goncharov NP, Chikida NN, Divashuk MG (2020) Allele mining of TaGRF-2D gene 5’-UTR in Triticum aestivum and Aegilops tauschii genotypes. PLoS ONE 15(4):e0231704. https://doi.org/10.1371/journal.pone.0231704
Wicker T, Yahiaoui N, Guyot R, Schlagenhauf E, Liu ZD, Dubcovsky J, Keller B (2003) Rapid genome divergence at orthologous low molecular weight glutenin loci of the A and Am genomes of wheat. The Plant Cell 15:1186–1197. https://doi.org/10.1105/tpc.011023
Baucom RS, Estill JC, Leebens-Mack J, Bennetzen JL (2009) Natural selection on gene function drives the evolution of LTR retrotransposon families in the rice genome. Genome Res 19:243–254. https://doi.org/10.1101/gr.083360.108
Belyayev A, Kalendar R, Brodsky L, Nevo E, Schulman AH, Raskina O (2010) Transposable elements in a marginal plant population: temporal fluctuations provide new insights into genome evolution of wild diploid wheat. Mobile DNA 1:6. https://doi.org/10.1186/1759-8753-1-6
Kalendar R (2011) The use of retrotransposon-based molecular markers to analyze genetic diversity. Ratar. Povrt. / Field Veg. Crop Res. 48:261–274. https://doi.org/10.5937/ratpov1102261K.35
Kalendar R, Flavell AJ, Ellis THN, Sjakset T, Schulman AH (2011) Analysis of plant diversity with retrotransposon- based molecular markers. Heredity 106:520–530
Smykal P, Bacova- Kerteszova N, Kalendar R, Corander J, Schulman AH, Pavele M (2011) Genetic diversity of cultivated flax (Linum usitatissimum L.) germplasm assessed by retrotransposon-based markers. Theor Appl Genet 122:1385–1397
Abdollahi Mandoulakani B, Piri Y, Darvishzaded R, Bernoosi I, Jafari M (2012) Retroelement insertional polymorphism and genetic diversity in Medicago sativa populations revealed by IRAP and REMAP markers. Plant Mol Biol 30:286–296
Singh S, Nandha PS, Singh J (2017) Transposon-based genetic diversity assessment in wild and cultivated barley. The Crop J 5:296–304
Basirnia A, Darvishzadeh R, Abdollahi Mandoulakani B (2014) Retrotransposon insertional polymorphism in sunflower (Helianthus annuus L.) lines revealed by IRAP and REMAP markers. Plant Biosys. https://doi.org/10.1080/11263504.2014.970595
Carvalho A, Guedes-Pinto H, Martins-Lopes P, Lima-Brito J (2010) Genetic variability of Old Portuguese bread wheat cultivars assayed by IRAP and REMAP markers. Ann Appl Biol 156(3):337–345
Taheri MT, Alavi-Kia SS, Mohammadi SA, Moghaddam Vahed M (2018) Assessment of genetic diversity and relationships among Triticum urartu and Triticum boeoticum populations from Iran using IRAP and REMAP markers. Genet Resour Crop Evol 65:1867–1878
Kalendar R, Grob T, Regina M, Suoniemi A, Schulman A (1999) IRAP and REMAP: two new retrotransposon-based DNA fingerprinting techniques. Theore Appl Genet 98:704–711. https://doi.org/10.1007/s001220051124
Luo MC, Gu YQ, Puiu D et al (2017) Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 551:498–502. https://doi.org/10.1038/nature24486
Boyko E, Kalendar R, Korzun V, Fellers J, Korol A, Schulman AH, Gill BS (2002) A high-density cytogenetic map of the Aegilops tauschii genome incorporating retrotransposons and defense-related genes: insights into cereal chromosome structure and function. Plant Mol Biol 48:767–790. https://doi.org/10.1023/a:1014831511810
Saghai-Maroof MA, Soliman K, Torgensen RA, Allard RW (1984) Ribosomal DNA spacer length polymorphism in barley: Mendelian inheritance, chromosomal location and population dynamics. Proc Natl Acad Sci USA 81:8014–8018. https://doi.org/10.1073/pnas.81.24.8014
Nei M (1973) Analysis of gene diversity in suddivided populations. Proc Natl Acad USA 70:3321–3323. https://doi.org/10.1073/pnas.70.12.3321
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Swofford DL, Olsen GJ, Waddell PJ, Hillis DM (1996) Phylogenetic Inference. In: Hillis DM, Moritz C, Mable BK, Sunderland MA (eds) Molecular Systematics. Sinauer Associates, Sunderland, pp 407–514
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/genetics/155.2.945
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Earl DA, vonHoldt BM (2012) Structure harvester: a website and program for visualizing structure output and implementing the Evanno method. Consev Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Mizuno N, Yamasaki M, Matsuoka Y, Kawahara T, Takumi S (2010) Population structure of wild wheat D-genome progenitor Aegilops tauschii Coss.: implications for intraspecific lineage diversification and evolution of common wheat. Mol Ecol 19:999–1013. https://doi.org/10.1111/j.1365-294X.2010.04537.x
Sohail Q, Shehzad T, Kilian A, Eltayeb AE, Tanaka H, Tsujimoto H (2012) Development of diversity array technology (DArT) markers for assessment of population structure and diversity in Aegilops tauschii. Breed Sci 62:38–45. https://doi.org/10.1270/jsbbs.62.38
Bariah I, Keidar-Friedman D, Kashkush K (2020) Where the wild things are: transposable elements as drivers of structural and functional variations in the wheat genome. Front Plant Sci 11:585515. https://doi.org/10.3389/fpls.2020.585515
Safiyar S, Aalami A, Abdollahi Mandoulakani B, Rabiei B, Kordrostami M (2021) Genetic diversity of Aegilops tauschii accessions and its relationship with tetraploid and hexaploid wheat using retrotransposon-based molecular markers. Cereal Res Commun. https://doi.org/10.1007/s42976-021-00167-9
Leśniowska-Nowak J, Okoń S, Wieremczuk A (2020) Molecular diversity analysis of genotypes from four Aegilops species based on retrotransposon–microsatellite amplified polymorphism (REMAP) markers. Cereal Res Commun 49:37–44. https://doi.org/10.1007/s42976-020-00086-1
Ghobadi G, Etminan A, Mehrabi AM, Shooshtari L (2021) Molecular diversity analysis in hexaploid wheat (Triticum aestivum L.) and two Aegilops species (Aegilops crassa and Aegilops cylindrica) using CBDP and SCoT markers. J Genet Eng Biotechnol 19(56). https://doi.org/10.1186/s43141-021-00157-8
Kalendar R, Schulman AH (2007) IRAP and REMAP for retrotransposon-based genotyping and fingerprinting. Na Protoc 1:2478–2484. https://doi.org/10.1038/nprot.2006.377
Vicient CM, Kalendar R, Schulman AH (2001) Envelope-class retrovirus-like elements are widespread, transcribed and spliced, and insertionally polymorphic in plants. Genome Res 11:2041–2049. https://doi.org/10.1101/gr.193301
Li W, Zhang P, Fellers JP, Friebe B, Gill BS (2004) Sequence composition, organization, and evolution of the core Triticeae genome. Plant J 40:500–511. https://doi.org/10.1111/j.1365-313X.2004.02228.x
Altinkut A, KotserubaV KVM, Nevo E, Raskina O, Belyayev A (2006) Ac-like transposons in populations of wild diploid Triticeae species: comparative analysis of chromosomal distribution. Chromosome Res 14:307–317. https://doi.org/10.1007/s10577-006-1048-3
El Zayat MAS, Hassan AH, Nishawy E, Ali M, Amar MH (2021) Patterns of genetic structure and evidence of Egyptian citrus rootstock based on informative SSR, LTR-IRAP and LTR-REMAP molecular markers. J Genet Eng Biotechnol 19:29. https://doi.org/10.1186/s43141-021-00128-z
Acknowledgements
We would like to thank the Center of Excellence in Cereal Molecular Breeding, University of Tabriz, Tabriz, Iran, for providing laboratory facilities for this work.
Funding
This study was financially supported by the Iran National Science Foundation (INSF no. 95848959).
Author information
Authors and Affiliations
Contributions
SM conducted the experiment, collected and analyzed data, and wrote the original draft. SAM conceptualized and supervised the experiment, edited the manuscript, and provided fund. AS and ARD collected the plant materials. All the authors read and agreed to published the version of manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary table.
Name and geographical coordination of the studied Ae. tauschii accessions.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Minaei, S., Mohammadi, S.A., Sabouri, A. et al. High genetic diversity in Aegilops tauschii Coss. accessions from North Iran as revealed by IRAP and REMAP markers. J Genet Eng Biotechnol 20, 86 (2022). https://doi.org/10.1186/s43141-022-00363-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-022-00363-y